

Abstract

Polycomb repressive complex 2 (PRC2) has been shown to play a major role in transcriptional silencing in part by installing methylation marks on lysine 27 of histone 3. Dysregulation of PRC2 function correlates with certain malignancies and poor prognosis. EZH2 is the catalytic engine of the PRC2 complex and thus represents a key candidate oncology target for pharmacological intervention. Here we report the optimization of our indole-based EZH2 inhibitor series that led to the identification of CPI-1205, a highly potent (biochemical IC50 = 0.002 μM, cellular EC50 = 0.032 μM) and selective inhibitor of EZH2. This compound demonstrates robust antitumor effects in a Karpas-422 xenograft model when dosed at 160 mg/kg BID and is currently in Phase I clinical trials. Additionally, we disclose the co-crystal structure of our inhibitor series bound to the human PRC2 complex.
Introduction
It is well-established that trimethylation of lysine 27 on histone 3 (H3K27) contributes to the modification of chromatin structure, which serves to repress transcription.1−3 The addition of trimethyl “marks” on H3K27 is generally catalyzed by the multimeric protein complex polycomb repressive complex 2 (PRC2), through its enzymatic subunit enhancer of zeste homologue 2 (EZH2). EZH2 catalyzes the transfer of a methyl group from the cofactor S-adenosyl-l-methionine (SAM) to the ε-NH2 group of H3K27 culminating in trimethylation of H3K27 (H3K27me3) and subsequent silencing of targeted genes.
Dysregulation of mechanisms that alter chromatin structure has been implicated in a variety of disease processes, particularly oncogenesis.2 EZH2 is frequently overexpressed in a broad spectrum of solid and hematological cancers such as prostate, breast, kidney, lung, myeloma, and lymphoma.2,4 Elevated EZH2 transcript and protein levels in these cancers usually correlate with greater levels of H3K27me3, advanced stages of disease, and poor prognosis.5,6 Additionally, somatic recurrent mutations within the catalytic domain of EZH2 (the suppressor of variegation, enhancer of zeste, trithorax (SET) domain) have been identified in diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, and melanoma.7 These mutations alter the substrate specificity of EZH2 culminating in an increase in global levels of H3K27me3.4,8−10 Consequently, the increase in levels of H3K27me3, either by overexpression of EZH2 or its altered function through mutations, in cancer tissues may reinforce the silencing of target genes that promote differentiation and restrain proliferation.11 Alternatively, EZH2 may serve to silence genes not targeted in normal cells to afford growth and survival advantage in the malignant setting. Collectively, these observations offer a compelling argument for the inhibition of EZH2 as a potential therapeutic approach for the treatment of cancer.
The development of small molecule inhibitors of EZH2 has been an intense area of research. Several potent EZH2 inhibitors demonstrating promising antitumor activity in preclinical species have been disclosed. These EZH2 inhibitors include EPZ-005687,12 EPZ-6438,13,14 EPZ-011989,15 GSK-343,16 GSK-126,17 UNC-1999,18 EI1,19 and our previously disclosed probes CPI-36020 and 1 (CPI-169).21 Although these inhibitors originated from independent high-throughput screening efforts, they all share a common structural feature, a pyridone core. The prevalence of a pyridone motif embedded within these inhibitors highlights the importance of this functionality for high-affinity binding. While a novel series of 4-amino-2,2′,6,6′-tetramethylpiperidine analogues has also been identified as SAM-competitive inhibitors of EZH2, these analogues demonstrated inferior cellular potency when compared to the pyridone-based inhibitors.22,23 Currently, Phase I clinical trials of three different pyridone-based EZH2 inhibitors tazemetostat (EPZ-6438) (NCT01897571, NCT02601937, NCT02601950), GSK-126 (NCT02082977), and 13 (CPI-1205) (NCT02395601) have been reported (Figure 1).
Figure 1.

Reported EZH2 inhibitors.
Medicinal Chemistry and Structure–Activity Relationships
We recently disclosed 1, a potent indole based EZH2 inhibitor that showed robust antitumor activity and pharmacodynamic (PD) target engagement in a KARPAS-422 lymphoma xenograft model in mice.21 This compound, however, suffered from limited oral bioavailability (0.09% F and 0.12% F observed in rats and dogs, respectively).) As part of our ongoing drug discovery and development efforts, we continued to optimize the indole-based scaffold toward clinical candidate selection. Herein we report the optimization of the indole based EZH2 inhibitor series that led to the identification of 13, a potent and selective inhibitor of EZH2 currently under evaluation in Phase I clinical trials. In addition, we report the co-crystal structure of a similar pyridone containing inhibitor (10) bound to human PRC2. This structure has provided a context for the molecular nature of the interaction between our chemical series and its target.
In an attempt to improve upon the physical properties of 1, we began the investigation of our structure–activity relationship (SAR) with different N-substituents on the piperidine ring. We rationalized that the pyridone-indole core was previously optimized with respect to biochemical potencies (against both wild-type and mutant EZH2) as evidenced by N–H piperidine (2) and N–Me piperidine (3) analogues (Table 1). Unfortunately both 2 and 3 suffered from considerable loss in cellular potency in the HeLa H3K27me3 mechanism of action (MOA) assay. Derivatization of the N–H piperidine to a variety of amides, ureas, carbamates, and sulfonamides yielded biochemically potent analogues. However, these analogues displayed less than ideal cellular potencies when examined in the MOA assay. For example, substitution of the piperidine N-atom to give the corresponding isobutyl amide (4), isopropyl carbamate (5), and N–Me–urea (6) afforded approximately 5-, 2-, and 11-fold loss in cellular potencies relative to 1.
Table 1. Piperidine Structure–Activity Relationships.

Biochemical data generated via scintillation proximity assay (SPA) with PRC2, H3K27me3 activator peptide and biotinylated oligonucleosomes and 3[H]-SAM. IC50 values reported as an average ≥2 determinations.
The mechanism of action cellular assay measured global H3K27me3 levels in HeLa cells; see Supporting Information for further details.
pKa values generated via ChemDraw version 15.0
IC50 value from a single determination.
In addition to the disparate trends in potencies (i.e., poor translation into cellular assays), N-acylated derivatives (e.g., amides, ureas, carbamates, and sulfonamides) generally suffered from high microsomal clearance or rapid clearance in vivo (Figure 2). In contrast, a variety of basic amines derived from piperidine 2 showed acceptable ADME properties (low Clint and low CYP inhibition). As such, we subsequently sought to improve the physiochemical properties of piperdine 2 to attain the desired in vitro and in vivo potencies.
Figure 2.

General trends in structure–activity relationships.
We found that attenuation of the basicity and hence perturbation of the piperidine pKa had profound effects on the cellular potency, selectivity, toxicity, bioavailability, and PK properties.24 For example, oxetane 7 (ChemDraw calculated pKa was 7.6) demonstrated a 10-fold improvement in the cellular potency when compared to piperidine 2 (ChemDraw calculated pKa was 9.7).25 Other electron-withdrawing substituents, such as acetate 8 also afforded improvement in cellular potencies.
We synthesized n-trifluoropropyl piperidine analogue 9 (calculated pKa ∼ 8.2) and observed an over 3-fold increase in potency (EC50 of 0.29 μM) relative to piperidine 2 when tested in the cellular MOA assay (Table 1). While introduction of the n-trifluoropropyl motif was a step in the right direction, we believed that cellular potency could be further enhanced. To examine the effect of keeping the three-carbon linker fixed, and moving the fluorine atoms to the 2-position of the alkyl chain, we subsequently synthesized the 2,2-difluoropropyl containing analogue (10). The pKa was calculated to decrease by half a log unit relative to analogue 9 (Chemdraw calculated pKa 7.6 versus 8.2, respectively), but gratifyingly, even this modest decrease in basicity resulted in a 10-fold gain in cellular potency (EC50 of 0.020 μM) over the linear n-trifluoropropyl analogue. The addition of fluorine atoms in the β-carbon (with respect to the piperidine nitrogen) appeared optimal for attenuation of basicity of the piperidine and increase in cellular potency. As such, a series of analogues that embedded the β,β-difluoroethyl motif were synthesized and further examined. The tetrafluoropropyl (11), difluoroethyl (12), and trifluoroethyl (13) piperidines were prepared, and these fluorinated analogues were comparable in potency (EC50 of 0.038, 0.039, and 0.032 μM, respectively) to 10.
Co-crystal of Ligand Bound to PRC2 Complex
In order to understand the mechanism of action within this lead series, we were able to exploit the crystallization system developed for our recent structure of the human PRC2 catalytic complex.26 Co-crystals were obtained with the 2,2-difluoropropyl analogue (10) that diffracted to 3.5 Å and the structure determined by molecular replacement (PDB code: 5LS6). Electron density corresponding to compound 10 was identified in a pocket at the interface of the EZH2 SET domain, the SAL region of the EZH2 N-terminus and EED (Figure 3A). The electron density was improved by 4-fold averaging, so that even at 3.5 Å it is remarkably well-defined and the position and orientation of the inhibitor could be unambiguously determined (Figure 3B).
Figure 3.

(A) Surface representation of the co-crystal structure of 10 (yellow spheres) bound to the PRC2 complex (EED (gray), SUZ12 (pink), EZH2 (pale blue at N-terminus to blue at C-terminus) (PDB ID: 5LS6). (B) Electron density (NCS averaged Fourier Map (2Fo-Fc map contoured at 2σ)) surrounding compound 10. Main interactions are between pyridone of 10 and key residues from the SET-I and SAL domains of EZH2 and from EED (PDB ID: 5LS6). (C) The PRC2-compound 10 complex (colored as above) superimposed on the PRC2 complex with SAH cofactor and peptide (green). (PDB ID: 5HYN). (D) The pyridone and indole are constrained in a binding site composed of residues from both the SET domain (blue) and SAL region (pale blue) of EZH2 (PDB ID: 5LS6).
Key residues that define the inhibitor pocket include EZH2 SET domain Tyr661, Phe665, Tyr658, and Phe686, the EZH2 SAL region Tyr111 and Met110, and EED residues His213 and Asp237.27 Although the inhibitor binding site partially overlaps with the pocket for SAH (superimposed from PDB ID: 5HYN), it then extends in the opposite direction and is therefore distinct from both the substrate and cofactor binding sites (Figure 3B and 3C). The partial overlap between the 2,2-difluoropropyl piperidine 10 and the SAH carboxylic acid is consistent with a SAM competitive mechanism of inhibition.
Overall, 10 adopts a zigzag like shape, which is defined by both the constraints of the pocket, as well as the preorganized conformation induced by the chiral methyl proximal to the indole and the torsional angle defined by the C3-indole amide connecting to the pyridone. The indole and piperidine are tightly constrained by the narrow hydrophobic channel and by a putative hydrogen bond from the EED Asp237 side-chain to one of the fluorine atoms. Beyond the piperidine, the pocket widens considerably, which is consistent with the diverse array of substituents tolerated in this region (Table 1). At the opposite end of the molecule the pyridone is surrounded by the side chains of Phe665, Phe686, and Trp624 and has the potential to form two hydrogen bonds with the protein backbone of Trp624 (Figure 3D). Recently, a co-crystal structure of a human/chameleon hybrid PRC2 construct was reported that also contained a pyridone-based EZH2 inhibitor (PDB codes: 5IJ7 and 5IJ8).28 Both pyridone orientation and binding is consistent between the two structures.
A major difference between the inhibitor complex structure and that obtained with peptide/SAH occurs at the active site of the EZH2 SET domain (Figure 4). In the absence of substrate peptide, the C-terminus of the SET domain occupies the histone binding groove with the side chains of Tyr728 and Phe667 located to the target lysine channel. In the inhibitor complex beyond residue Ser729, the C-terminus is disordered. Similarly, in the structure of the isolated apo EZH2, there is a pronounced rearrangement of this region.29 The alternate conformation of the EZH2 C-terminus observed in these structures are indicative of its inherent flexibility. It is not clear to what extent the conformation observed in the inhibitor complex structure is due to the binding of inhibitor or reflects the absence of cofactor and/or histone substrate.
Figure 4.

Comparison of the EZH2 C-terminus with the SET domain active site. (A) Human PRC2 complex with compound 10. (PDB ID: 5LS6) (B) Equivalent region of the human PRC2-H3K27M-SAH complex, (PDB ID: 5HYN) with cofactor (orange) and histone H3 peptide (red). (C) Overlay of residues forming the SET domain lysine channel for both complexes, highlighting that the position of the C-terminus conformation observed in the compound complex is incompatible with substrate binding (red). (D) and (E) Surface representations showing that the lysine binding channel is closed in the PRC2-compound 10 complex (PDB ID: 5LS6) but accessible in the SAH/substrate complex (PDB ID: 5HYN). (F) Surface representation of EZH2 observed in the PRC2-compound 10 complex but with the H3 peptide overlaid from the PRC2-H3K27M-SAH complex, confirming that substrate binding is incompatible with the EZH2 conformation in the PRC2-compound 10 complex.
In Vitro ADME and In Vivo Pharmacokinetics of Fluorinated Analogues
Having established sufficient activity in the MOA assay, we subsequently profiled 9, 10, 12, and 13 in an ADME panel (Table 2). Relative to 1, these fluorinated analogues generally displayed higher plasma protein binding and in vitro clearance, which is presumably a function of their increased lipophilicity (cLogP). Interestingly, there were significant interspecies differences observed with regards to the in vitro microsomal clearances and PPB. The highest in vitro microsomal clearance (>100 μL/min/mg protein) and plasma protein binding (>97% bound) were uniformly observed in mice for 10, 12, and 13. Additionally, the in vitro clearance measured from rat-derived microsomes closely mirrored those derived from humans for these analogues.
Table 2. ADME of Fluorinated Analogues 9, 10, 12, and 13.
| compound | 9 | 10 | 12 | 13 | ||
|---|---|---|---|---|---|---|
| cLogPa | 3.39 | 3.71 | 3.19 | 3.41 | ||
| LiPEb | 3.1 | 4.0 | 4.2 | 4.1 | ||
| in vitro ADME | Clint (μL/min/mg protein)c | mouse | 61.3 | 193 | 131 | 159 |
| rat | 25.7 | 86.1 | 71.3 | 99.6 | ||
| dog | 35.3 | 68.9 | 52.4 | 73.9 | ||
| human | 35.4 | 83.8 | 73.6 | 104 | ||
| PPB (% bound)d | mouse | 99.7 | 99.4 | 97.8 | 97.9 | |
| rat | 97.4 | 92.4 | 83.1 | 89.9 | ||
| dog | 96.3 | 93.6 | 64.3 | 90.3 | ||
| human | 95.9 | 94.7 | 83.3 | 92.8 | ||
| CYP inh. (% at 10 μM)e | 3A4 | 5.0 | 0 | 25.2 | 0 | |
| 2D6 | 17.2 | 7.7 | 26.1 | 9.5 | ||
| 2C8 | 23.1 | 50.9 | 18.9 | 41 | ||
| 1A2 | 19.4 | 0 | 28.6 | 15 | ||
| 2C9 | 11.6 | 5.0 | 25.4 | 11.1 | ||
ClogP values were generated with ChemDraw Professional 15.0.
LiPE = lipophilic efficiency = pEC50 – cLogP (ChemDraw 15.0).
Intrinsic clearance (Clint) of compounds in mouse/rat/dog/human liver microsomes (expressed in μL/min/mg protein).
Measured in vitro binding of compounds to mouse/rat/dog/human plasma protein bindings (expressed in % bound).
In vitro inhibition of cytochrome P450 isoforms at single concentration (10 μM).
Based on their combination of potency and in vitro profile 12 and 13 were subsequently evaluated for their in vivo metabolic profile and systemic exposure in mice in advance of planned pharmacodynamic (PD) and efficacy studies (Table 3). Both 12 and 13 showed excellent oral bioavailability demonstrating significant improvement over 1. Surprisingly, 12 displayed a poor overall PK profile when compared to 13. When dosed in mice at 1 mg/kg intravenous (iv) and 100 mg/kg per os (po), 12 displayed high clearance of 4.45 L/h/kg (82% liver blood flow), low volume of distribution (1.10 L/kg), short half-life (0.40 h), and excellent bioavailability (∼92% F). In contrast, 13 exhibited moderate clearance of 2.16 L/h/kg (40% liver blood flow), a half-life of ∼1.6 h, similar volume of distribution (1.4 L/kg), and excellent bioavailability (100% F). As part of the comparison, we also examined total exposure (AUC) and unbound exposure (AUCunbound) because we were interested in maintaining free levels of compound significantly above the measured HeLa EC50. Both 12 and 13 achieved unbound exposures well above their respective cellular potencies; however, only the unbound exposure for 13 remained well above the cellular EC50 up to 4 h (Figure 5). Overall, the in vivo metabolic profile of 13 provided a compelling argument for further evaluation in a mouse xenograft model.
Table 3. Mouse Pharmacokinetic Profiles for Fluorinated Analogues 12 and 13a.
| iv |
po |
in vitro | |||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | CL [CLunbound]d (L/h/kg) | %Qe | t1/2f (h) | Vssg (L/kg) | AUC0-inf [AUCunbound]h (μM·h) | Cmaxi [Cmax-unbound] (μM) | tmaxj (h) | Fk (%) | ful |
| 12b | 4.45 [202.3] | 82 | 0.402 | 1.10 | 42.01 [0.924] | 52.14 [1.15] | 0.250 | 91.6 | 0.022 |
| 13c | 2.16 [102.9] | 40 | 1.63 | 1.36 | 88.88 [1.87] | 67.36 [1.41] | - | 100 | 0.021 |
Based on an intravenous (iv) dose of 1 mg/kg and a per os (po) dose of 100 mg/kg in fasted male Balb/C mice (except where indicated).
Compound was formulated in 5:25:70 DMA/PEG400/20% SBECD for iv dosing and in 0.5% methyl cellulose (adjusted to pH 3) for po dosing.
Compound was formulated in 5:60:35 DMA/PEG400/20% SBECD for iv dosing and in 0.5% methyl cellulose (adjusted to pH 3–4) for po dosing.
CL = total clearance. CLunbound = CL/free fraction.
Q = percent of liver blood flow, based on 5.4 L/h/kg (mouse).
Plasma half-life.
Vss = volume of distribution at steady state.
Extrapolated total exposure following single dose. Calculated unbound exposure is in parentheses.
Maximum plasma concentration achieved.
Time at which maximum plasma concentration was achieved.
Oral bioavailability.
fu = fraction unbound to plasma protein.
Figure 5.

Mouse pharmacokinetic profile of 13.
Synthesis of Indole Piperidine Analogues
Many of the analogues profiled in Table 1 were prepared by similar synthetic routes, with appropriately substituted building blocks. We identified indole 14 as a key scaffold for the construction of N-substituted piperidines. Synthetically, the alkylation of 2-methyl-1H-indole-3-carboxylate with branched alkyl electrophiles under a variety of basic conditions failed to deliver desired indole 14. The lackluster behavior of 2-methyl-1H-indole-3-carboxylate toward various alkylation conditions was presumably a consequence of deactivation of the indole N-atom by the 3-carboxylate moiety and additional steric constraints imposed by the 2-methyl substitution. As such, we looked to devise an alternative strategy toward a more convergent, robust, and scalable synthesis of indole 14 (Scheme 1).
Scheme 1. Palladium Mediated Intramolecular C–N Arylation to Indole 14.

Reagents and conditions: (a) AcOH, t-BuOH, reflux (76% yield); (b) RuPhos precatalyst (generation III) (2 mol %), RuPhos (3 mol %), NaOMe (1.5 equiv), 1,4-dioxane, 100 °C, followed by HCl (4 M in 1,4-dioxane) (81% yield over two steps).
We were drawn to the possibility of utilizing halo-aryl enamines as latent precursors for a palladium-mediated intramolecular C–N bond construction of indoles.30 We rationalized that generation of enamines from β-keto esters would allow for the introduction of a variety of amines, and this modular approach would serve as a diversity generating element for our drug discovery efforts. To that end, the construction of indole 14, began with the condensation reaction between β-keto ester3015 and chiral amine31,3216 under mildly acidic conditions to deliver enamine 17, predominantly as the Z-isomer, in 76% yield. Paramount to the success of this intramolecular C–N arylation was a systemic investigation of palladium catalysts and reactions conditions.30 We were gratified to find that treatment of chiral enamine 17 with Buchwald’s RuPhos precatalyst system in the presence of sodium methoxide cleanly induced intramolecular C–N bond arylation to yield chiral indole piperidine 18 while maintaining the stereochemical integrity of the chiral center. Subsequent deprotection of N-Boc piperidine 18 with anhydrous hydrochloric acid produced piperidine 14 in 81% yield over two steps (Scheme 2).
Scheme 2. Synthesis of 10 and 13.

Reagents and conditions: (a) 2,2,2-trifluoroacetic anhydride (1.4 equiv), i-PrNEt2 (2.5 equiv), CH2Cl2, 0 °C; (b) BH3·THF (2.2 equiv), THF, 70 °C; (c) 6 N aq NaOH (6.0 equiv), EtOH, 85 °C; (d) CDI (1.3 equiv), THF, 60 °C
The piperidine 14, a common and versatile intermediate, was subsequently utilized for the construction of a variety of analogues, in particular 13, via a series of similar chemical transformations. The synthesis of 13 commenced with amidation of piperidine 14 (in free base form) with trifluoroacetic anhydride in the presence of Hünig’s base to provide amide 19 in near quantitative yield. Subsequent reduction of amide 19 with borane THF complex and heating delivered N-trifluoroethylpiperidine 20. Saponification of the pendant methyl ester (20) with 6 N sodium hydroxide gave the penultimate acid 21 in 99% yield. The requisite amide bond was forged in a CDI mediated union between acid 21 and pyridone amine 22(33) to yield the N-trifluoroethylpiperidine analogue (13) in 79% yield. The current synthetic scheme enabled generation of enantiomerically pure 13 on over 100 g scales for in vivo profiling.
In Vivo Efficacy Studies
Having sufficient quantities in hand, we evaluated the performance of inhibitor 13 for tumor pharmacodynamic effects and antitumor efficacy in a KARPAS-422 B-cell lymphoma xenograft model in mice. KARPAS-422 xenograft cells harbor a recurrent, monoallelic mutation (Y641N) within the EZH2 catalytic domain.17 These mutations alter the EZH2 substrate specificity and thus represent a context of constitutive EZH2 pathway activation. Informed by previous in vivo studies, 13 was dosed at 160 mg/kg orally twice daily (po BID) for 25 days in tumor bearing female CB-17 SCID mice (Figure 6). Upon treatment of tumor-bearing CB-17 SCID mice with 13, tumor regression was observed within 2 weeks. By the end of day 25, significant tumor growth inhibition was recorded (>97% TGI relative to vehicle, see Figure 6A). Inhibitor 13 was well-tolerated for repeat dosing as demonstrated by the absence of significant body weight loss (Figure 6B). To allow for analysis of tumor tissues at the end of the study, treatment was suspended at day 25. Tumor samples were harvested 1 h post last dose, and their analysis revealed considerable reduction of H3K27me3 (47% reduction in H3K27me3/global H3 ratio relative to vehicle control, see Figure 6C). Analysis of plasma and tumor PK at 1 h post last dose on day 25 shows sufficient plasma and tumor tissue concentrations of 13, 11 388 ng/mL [22 μM] versus 5286 ng/g [10 μM], respectively (Figure 6D).
Figure 6.

Tumor xenograft with 13. (A) Effect of dosing analogue 13 at 160 mpk BID for 25 days on KARPAS-422 tumor growth in mouse xenograft. (B) Corresponding body weight change during the course of the study. (C) Pharmacodynamic effect of 13 on reduction of H3K27me3 normalized to total H3. (D) Analysis of plasma and tumor concentrations of 13 at 1 h post last dose on day 25.
Selectivity and Additional Profiling
On the basis of the successful KARPAS-422 efficacy, we selected 13 for further in vivo and in vitro profiling. Additional PK data on 13 in rats and dogs was collected (Table 4). Analogue 13 shows relatively high clearance in both rats and dogs (3.19 L/h/kg and 1.41 L/h/kg, respectively) but demonstrates good oral bioavailability in both species (44.6% F in rats and 46.2% F in dogs). As part of the characterization of 13, its activity against a number of other targets were evaluated. Inhibitor 13 showed a clean selectivity profile when tested against 30 other histone or DNA methyltransferases.34 Additionally, compound 13 demonstrated modest selectivity (EZH1 IC50 of 52 ± 11 nM) when tested against enhancer of zeste homologue 1 (EZH1), a methyltransferase highly related to EZH2. Examination of the sequence similarity between EZH1 and EZH2 in the context of the co-crystal structure reveals that the residue positioned in close proximity to the inhibitor, Cys663, is one of only four residues within the EZH2 SET domain that are not conserved in EZH1. The equivalent EZH1 residue is Ser664. Cys663 makes van der Waals contact with the bound inhibitor and is predicted to be a key selectivity determinant with respect to EZH1 (Figure 3B). In order to test this hypothesis, we measured the potency of 10 and 13 in the context of an EZH2 version with a single amino acid substitution from cysteine to serine at position 663 (C663S) incorporated into reconstituted PRC2. As expected, the potency of the inhibitors decreased in the mutated EZH2 C663S, which is consistent with the reduction in potency observed in EZH1 enzymatic assays. The fact that this single mutation does not fully recapitulate the EZH2/EZH1 potency differences may reflect a subtle effect of other more distant residue differences between the two proteins.
Table 4. Pharmacokinetic Data of 13 in Rats and Dogs.
| iv |
po |
in vitro | |||||||
|---|---|---|---|---|---|---|---|---|---|
| species | CL [CLunbound]c (L/h/kg) | %Qd | t1/2e (h) | Vssf (L/kg) | AUC0-inf [AUCunbound]g (μM·h) | Cmaxh (μM) | tmaxi (h) | Fj (%) | fuk |
| rata | 3.19 [31.6] | 97 | 0.653 | 2.22 | 7.06 [0.713] | 5.88 [0.593] | 0.50 | 44.6 | 0.098 |
| dogb | 1.41 [14.5] | 76 | 1.84 | 1.60 | 3.19 [0.309] | 1.24 [0.120] | 1.67 | 46.2 | 0.089 |
Based on an intravenous (iv) dose of 1 mg/kg and a per os (po) dose of 25 mg/kg in male Sprague–Dawley rats. Compound was formulated in 5:60:35 DMA/PEG400/20% SBECD for iv dosing and in 0.5% methyl cellulose (adjusted to pH 3–4) for po dosing.
Based on an intravenous (iv) dose of 1 mg/kg and a per os (po) dose of 5 mg/kg in male beagle dogs. Compound was formulated in 5:25:70 DMA/PEG400/20% SBECD for iv dosing and in 0.5% methyl cellulose for po dosing.
CL = total clearance. CLunbound = CL/free fraction.
Q = percent of liver blood flow, based on 3.3 L/h/kg (rat) and 1.85 L/h/kg (dog).
Plasma half-life.
Vss = volume of distribution at steady state.
Extrapolated total exposure following single dose. Calculated unbound exposure is in parentheses.
Maximum plasma concentration achieved.
Time at which maximum plasma concentration was achieved.
Oral bioavailability.
fu = fraction unbound to plasma protein.
Further in vitro profiling of 13 showed no time-dependent inhibition (TDI) of the cytochrome P450 enzymes 1A2, 2C9, 2C8, 2D6, and 3A4. Additionally, 13 was evaluated for secondary pharmacology against a panel of fifty-four physiologically relevant receptors, transporters, and ion channels at 10 μM. As such, 13 did not inhibit any target more than 50%. Finally, 13 was examined for any potential cause of cardiac arrhythmias associated with delayed ventricular repolarization (QT interval prolongation). When tested within an in vitro hERG binding assay at concentration ranges of 45 nM to 100 μM, 13 showed an IC50 of 21.3 μM. This concentration is well above the free concentration predicted to be achieved in patients.
Toxicology Study of 13
To establish a safety window for repeat dosing of 13 in human clinical trials, a preclinical safety study was conducted in two separate species. Compound 13 was orally administered in a GLP compliant toxicity study for 4 weeks to both Sprague–Dawley rats and beagle dogs followed by a 4-week recovery period. The compound was administered by oral gavage at single daily doses (QD) of 100, 300, and 600 mg/kg to rats for 28 days and at twice daily doses (BID) of 50, 150, and 500 mg/kg for 28 days to dogs. In general, 13 was well-tolerated in the 28-day GLP toxicology studies, and any findings were reversible over the recovery period. The GLP toxicology studies of 13 demonstrated an acceptable safety profile and enabled selection of clinical doses.
Conclusions
In summary, we have reported the discovery of 13, a highly potent and selective small molecule inhibitor of EZH2, suitable for introduction into Phase I clinical trials for the treatment of B-cell lymphomas (NCT02395601). Using 1, our previously disclosed EZH2 chemical probe as a starting point, we embarked on a focused campaign to optimize the cellular potency and physiochemical properties of our pyridone-indole scaffold. Removal of the sulfonamide functionality (embedded within 1) yielded analogues that retained biochemical potency but lacked sufficient cellular activity in the HeLa H3K27me3 assay. Observing that attenuation of the pKa of the piperidine N-atom has a profound effect on the correlation between biochemical and cellular potency, we ultimately discovered a series of fluorinated analogues with improved cellular activity and good oral bioavailability.
During the course of our investigations, we successfully determined the co-crystal structure of compound 10 bound to human PRC2. The present crystal structure illustrates several distinct binding features. The overall density reveals a zigzag-like shape of the inhibitor, likely a consequence of conformational preorganization induced by the chiral methyl and C3-amide substituents that conforms to the enzyme’s pocket. Additionally, the pyridone motif forms two hydrogen bonds with the protein backbone of Trp624 and is constrained in an aromatic environment created by Phe665, Phe686, and Trp624. These crucial interactions with the pyridone motif explain the importance of this functional group for its high affinity binding, its prevalence in a vast majority of other reported EZH2 inhibitors, and the difficulty in finding suitable pyridone replacements.
After triaging these analogues through in vitro and in vivo experiments, N-trifluoroethylpiperidine analogue 13 was evaluated in a KARPAS-422 lymphoma xenograft model. Gratifyingly, 13 was well-tolerated, proved efficacious, and achieved >97% TGI after treatment for 25 days. After further in vitro/in vivo characterization and safety studies, compound 13 was advanced into human clinical trials. The clinical impact of 13 on EZH2 inhibition in oncology will be described in due course.
Experimental Methods
All commercial reagents and anhydrous solvents were purchased and used without purification, unless specified. Column chromatography was performed using a Biotage chromatography system on Biotage or Silicycle normal phase silica gel columns. NMR spectra were recorded on a Varian Unity Inova (400 MHz) or an Oxford (Varian, 300 MHz) instrument. LC-MS were recorded on an Agilent 1200 series LC connected to an Agilent 6120 MS or Agilent 1100 series LC connected to an Agilent 1956B MS or a Shimadzu LC-MS-2020 system. Preparatory HPLC was performed using a Gilson GX-281 or P230 Gradient System (Elite). Chiral preparatory HPLC were performed using Elite P230 Preparative Gradient System, Thar Prep-80 and Thar SFC X-5 systems. The purity of the final products was >95% as determined by HPLC/MS and 1H NMR.
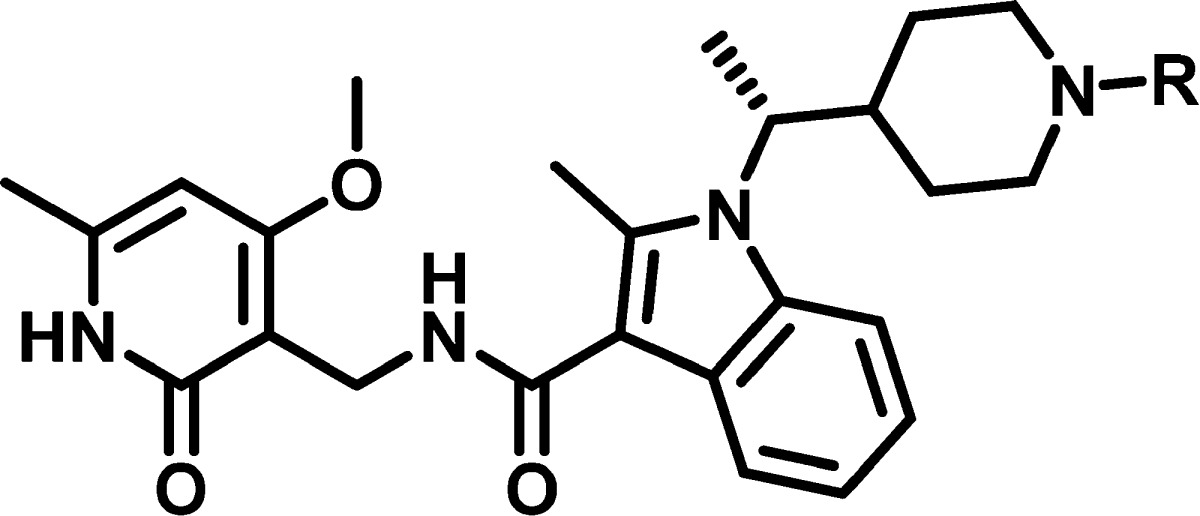
(R)-1-(1-(1-(Ethylsulfonyl)piperidin-4-yl)ethyl)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1H-indole-3-carboxamide (1)
Prepared as previously reported.21 LC-MS m/z 529 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 7.82–7.66 (m, 2H), 7.62 (d, J = 7.8, 1H), 7.17–6.99 (m, 2H), 6.15 (s, 1H), 4.32 (d, J = 5.1 Hz, 2H), 4.25–4.15 (m, 1H), 3.84 (s, 3H), 3.73–3.65 (m, 1H), 3.45–3.36 (m, 1H), 2.98 (q, J = 7.4 Hz, 2H), 2.87–2.77 (m, 1H), 2.60 (s, 3H), 2.54–2.45 (m, 1H), 2.42–2.30 (m, 1H), 2.20 (s, 3H), 2.06–1.97 (m, 1H), 1.58–1.48 (m, 3H), 1.42–1.31 (m, 1H), 1.17 (t, J = 7.4 Hz, 3H), 1.13–1.00 (m, 1H), 0.83–0.73 (m, 1H).
tert-Butyl (R)-4-(1-((3-(2-Bromophenyl)-4-methoxy-4-oxobut-2-en-2-yl)amino)ethyl)piperidine-1-carboxylate (17).30
A 2-L three-neck round-bottom flask (fitted with a magnetic stir bar, thermocouple, reflux condenser, and rubber septa) was charged with methyl 2-(2-bromophenyl)-3-oxobutanoate (116.25 g, 428.80 mmol), EtOH (850 mL, ∼7 mL/g), (R)-tert-butyl 4-(1-aminoethyl)piperidine-1-carboxylate (121.00 g, 529.93 mmol), and AcOH (29.50 mL, 515.31 mmol). The reaction vessel was heated over a heating mantle to 80 °C–85 °C for 18 h. After 18 h, the reaction mixture was cooled to ambient temperature, and the tert-butanol was removed in vacuo. The resultant oil was diluted with EtOAc and subsequently poured over saturated aqueous NaHCO3 while vigorously stirring. Once the evolution of CO2 (g) ceased, the biphasic solution was transferred to a separatory funnel, and the phases were separated. The aqueous phase was extracted with additional EtOAc (2×). The combined organic phase was dried over MgSO4, filtered, and concentrated in vacuo to afford crude product. This material was preabsorbed onto silica gel (∼100 g) and filtered through a fritted funnel with 20% EtOAc to 80% hexanes afford (R)-tert-butyl 4-(1-((3-(2-bromophenyl)-4-methoxy-4-oxobut-2-en-2-yl)amino)ethyl)piperidine-1-carboxylate (155.95 g, 76% yield). The material was used without further purification. LC-MS m/z 481 [M + H]+.
Methyl (R)-2-Methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxylate hydrochloride (14)
Step 1. To a 2 L 3-neck round-bottom flask (fitted with a magnetic stir bar, thermocouple, reflux condenser, and rubber septa) containing RuPhos precatalyst (generation III) (4.38 g, 5.24 mmol) and dicyclohexyl(2′,6′-diisopropoxy-[1,1′-biphenyl]-2-yl)phosphine (3.67 g, 7.86 mmol) was transferred (R)-tert-butyl 4-(1-((3-(2-bromophenyl)-4-methoxy-4-oxobut-2-en-2-yl)amino)ethyl)piperidine-1-carboxylate (126.11 g, 261.95 mmol) in 1,4-dioxane (800 mL, ∼ 6.4 mL/g). The resultant dark solution was evacuated and purged with N2 (g). To the mixture was added NaOMe (21.23 g, 392.93 mmol) in four portions, followed by addition of 1,4-dioxane (50 mL, total amount of 850 mL, ∼8 mL/g) to rinse the powder funnel. The resultant heterogeneous mixture was subjected to three cycles of evacuation and purging with N2 (g). The resultant dark brown/red suspension was heated over a heating mantle to 100 °C for 4–6 h. As the temperature reached 30–40 °C, a visible change in the reaction color was observed, and the mixture turned dark green. After 6 h, LC-MS analysis indicated complete conversion of the enamine to desired indole. The reaction mixture was cooled to ambient temperature and filtered over a pad made of a bed of silica gel over a bed of Celite. The filter cake was washed with EtOAc (3×), and the resultant brown filtrate was concentrated to provide methyl (R)-1-(1-(1-(tert-butoxycarbonyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (104.00 g, 99% yield) as an orange-colored foam. LC-MS m/z 401 [M + H]+.
Step 2. To a cooled (0 °C) solution of (R)-methyl 1-(1-(1-(tert-butoxycarbonyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (104.0 g, 259.67 mmol) in MeOH (300 mL, ∼3 mL/g) was added hydrogen chloride (80.0 mL, 320.0 mmol) in a dropwise manner. After complete addition of HCl, the reaction was allowed to gradually warm to ambient temperatures. After 24 h, LC-MS analysis indicated complete conversion of the carbamate to desired product. The reaction mixture was filtered over a pad of Celite and subsequently concentrated in vacuo. (Note: The reaction mixture contained minor quantities of insoluble black solids assumed to be Pd(0)) The resultant dark brown oil was further concentrated from acetone (2 × 50 mL). The oil was diluted in acetone (∼200 mL). The resultant solids were stirred for 2 h and filtered to give 59.58 g (68% recovery after first round) of (R)-4-(1-(3-(methoxycarbonyl)-2-methyl-1H-indol-1-yl)ethyl)piperidin-1-ium chloride as off-white solids. The filtrate was concentrated and subsequently triturated again with acetone (50 mL). The second crop of solids yielded an additional 10 g of (R)-4-(1-(3-(methoxycarbonyl)-2-methyl-1H-indol-1-yl)ethyl)piperidin-1-ium chloride (total mass: 70.0 g, 80% yield after two rounds of trituration) as off-white solids. LC-MS m/z 301 [M + H]+.
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (2)
Step 1. To a solution of (R)-methyl 1-(1-(1-(tert-butoxycarbonyl)piperidim-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (5.0 g, 12.5 mmol) in methanol (20 mL) and water (10 mL) was added sodium hydroxide (5.0 g, 125 mmol). The reaction mixture was stirred at 80 °C for 12 h. The mixture was cooled to room temperature and acidified to pH 5 with hydrochloric acid (2 N). The acidic solution was extracted with EtOAc. The combined organics layer was washed with brine, dried over Na2SO4, filtered, and concentrated to afford (R)-1-(1-(1-(tert-butoxycarbonyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylic acid (4.5 g, 94% yield) as an off-white solid. LC-MS m/z 409 [M + Na]+.
Step 2. A 250 mL round-bottom flask was charged with a magnetic stir bar, (R)-1-(1-(1-(tert-butoxycarbonyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylic acid (1.950 g, 5.05 mmol), 3-(aminomethyl)-4-methoxy-6-methylpyridin-2(1H)-one hydrochloride (2.065 g, 10.09 mmol), DMF (25.2 mL, 5.05 mmol), Hunig’s base (3.52 mL, 20.18 mmol). The reaction mixture was cooled to 0 °C, and COMU (2.16 g, 5.05 mmol) was added. The reaction was allowed to stir overnight to room temperature. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic extract was washed with brine, dried with MgSO4, filtered and concentrated in vacuo to afford the crude material, which was purified via silica gel chromatography (120 g) using MeOH/ethyl acetate (1:5) as eluent to afford tert-butyl (R)-4-(1-(3-(((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-2-methyl-1H-indol-1-yl)ethyl)piperidine-1-carboxylate (1.86 g, 65% yield). LC-MS m/z 537 [M + H]+ 1H NMR (400 MHz, DMSO-d6) δ 11.83–11.71 (m, 1 H), 7.80 (br. s., 1 H), 7.73 (d, J = 7.6 Hz, 1 H), 7.62 (d, J = 7.8 Hz, 1 H), 7.06 (td, J = 7.1, 14.4 Hz, 2 H), 6.21 (s, 1 H), 4.32 (br. s., 2 H), 4.16 (br. s., 1 H), 4.02 (br. s., 1 H), 3.85 (s, 3 H), 3.75 (br. s., 1 H), 2.70 (br. s., 1 H), 2.58 (s, 3 H), 2.37 (br. s., 1 H), 2.21 (s, 3 H), 1.90 (d, J = 12.9 Hz, 1 H), 1.53 (d, J = 6.9 Hz, 3 H), 1.35 (s, 10 H), 1.21 (br. s., 1 H), 0.89 (d, J = 8.7 Hz, 1 H), 0.67 (d, J = 11.8 Hz, 1 H).
Step 3. A 250 mL round-bottom flask was charged with a magnetic stir bar, (R)-tert-butyl 4-(1-(3-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methylcarbamoyl)-2-methyl-1H-indol-1-yl)ethyl)piperidine-1-carboxylate (1.850 g, 3.45 mmol), MeOH (13.79 mL, 3.45 mmol), and HCl (2.59 mL, 10.34 mmol) (4 N in dioxane). The reaction was allowed to stir at room temperature for 6 h before being concentrated in vacuo to afford 1 (1.65 g, 91% yield). LC-MS m/z 437 [M + H]+ 1H NMR (400 MHz, methanol-d4) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.52 (d, J = 7.7 Hz, 1 H), 7.08–6.96 (m, 2 H), 6.19 (s, 1 H), 4.43 (s, 2 H), 4.15 (dd, J = 10.4, 7.1 Hz, 1 H), 3.85 (s, 3 H), 3.40 (d, J = 12.8 Hz, 1 H), 3.07 (d, J = 12.6 Hz, 1 H), 2.96 (dt, J = 13.0, 2.8 Hz, 1 H), 2.68–2.50 (m, 5 H), 2.23 (s, 3 H), 2.17 (d, J = 14.3 Hz, 1 H), 1.56 (d, J = 6.8 Hz, 3 H), 1.14–1.05 (m, 1 H), 0.94 (d, J = 13.5 Hz, 1 H), 0.80 (t, J = 6.8 Hz, 1 H).
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-methylpiperidin-4-yl)ethyl)-1H-indole-3-carboxamide (3)
To a solution of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (0.1 g, 0.229 mmol) in THF (3 mL) was added formaldehyde (0.051 mL, 0.687 mmol). This solution was mixed for 1 h; subsequently, sodium triacetoxyborohydride (0.146 g, 0.687 mmol) was added, and the reaction was mixed at ambient temperature for 2.5 h. The crude reaction was deposited onto silica gel and purified by silica gel chromatography (90:10:1 DCM:MeOH:NH4OH) to afford 2 (68 mg, 66% yield). LC-MS m/z 451 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1 H), 7.76–7.65 (m, 2 H), 7.59 (d, J = 7.6 Hz, 1 H), 7.11–6.99 (m, 2 H), 6.14 (s, 1 H), 4.31 (d, J = 5.1 Hz, 2 H), 4.13 (br. s., 1 H), 3.83 (s, 3 H), 2.83 (d, J = 10.0 Hz, 1 H), 2.61–2.52 (m, 5 H), 2.19 (s, 3 H), 2.09 (s, 4 H), 1.88 (d, J = 10.7 Hz, 2 H), 1.53 (d, J = 6.7 Hz, 3 H), 1.34 (br. s., 1 H), 1.02 (d, J = 8.2 Hz, 1 H), 0.66 (br. s., 1 H).
(R)-1-(1-(1-Isobutyrylpiperidin-4-yl)ethyl)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1H-indole-3-carboxamide (4)
To a round-bottomed flask was added (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (60 mg, 0.137 mmol) and DCM (2 mL). The solution was cooled to 0 °C before addition of Et3N (21.07 μL, 0.151 mmol) and isobutyryl chloride (15.84 μL, 0.151 mmol). After stirring for 1 h at 0 °C, the reaction mixture was quenched by addition of a solution of 1 N NaOH (aqueous) and MeOH (1 mL). The solution was further stirred at 0 °C for 1 h, and subsequently diluted with MeOH (2 mL) followed by addition of lithium hydroxide monohydrate (11.54 mg, 0.275 mmol). This reaction was diluted with water and extracted with EtOAc. The combined organics layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude residue was purified via silica gel chromatography to afford 3 (27.7 mg, 39.8% yield). LC-MS m/z 507 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1 H), 7.75 (d, J = 7.4 Hz, 1 H), 7.72–7.67 (m, 1 H), 7.64 (d, J = 8.0 Hz, 1 H), 7.14–7.01 (m, 2 H), 6.15 (s, 1 H), 4.58–4.46 (m, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.09–3.99 (m, 1 H), 3.84 (s, 3 H), 3.81–3.72 (m, 1 H), 3.08–2.97 (m, 1 H), 2.92–2.81 (m, 1 H), 2.78–2.65 (m, 3 H), 2.59 (br. s., 3 H), 2.20 (s, 3 H), 2.03–1.90 (m, 1 H), 1.59–1.47 (m, 4 H), 1.02–0.86 (m, 6 H), 0.78–0.69 (m, 1 H).
Isopropyl (R)-4-(1-(3-(((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-2-methyl-1H-indol-1-yl)ethyl)piperidine-1-carboxylate (5)
Prepared in a similar manner to 4. LC-MS m/z 523 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.59 (br. s., 1 H), 7.74 (d, J = 7.8 Hz, 1 H), 7.69 (t, J = 4.9 Hz, 1 H), 7.62 (d, J = 7.8 Hz, 1 H), 7.13–7.01 (m, 2 H), 6.15 (s, 1 H), 4.78–4.67 (m, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.23–4.12 (m, 1 H), 4.12–4.02 (m, 1 H), 3.84 (s, 3 H), 3.82–3.74 (m, 1 H), 2.79–2.66 (m, 1 H), 2.58 (s, 3 H), 2.46–2.34 (m, 2 H), 2.20 (s, 3 H), 1.96–1.88 (m, 1 H), 1.58–1.46 (m, 4 H), 1.15 (d, J = 6.0 Hz, 6 H), 0.95–0.89 (m, 1 H), 0.74–0.65 (m, 1 H).
(R)-1-(1-(1-(Dimethylcarbamoyl)piperidin-4-yl)ethyl)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1H-indole-3-carboxamide (6)
Prepared in a similar manner to 4. LC-MS m/z 508 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.70–11.51 (m, 1 H), 7.80–7.54 (m, 3 H), 7.17–6.98 (m, 2 H), 6.15 (s, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.22–4.13 (m, 1 H), 3.84 (s, 3 H), 3.68–3.59 (m, 1 H), 3.40–3.33 (m, 1 H), 2.68 (s, 7 H), 2.60 (s, 3 H), 2.41–2.29 (m, 1 H), 2.20 (s, 3 H), 1.98–1.85 (m, 1 H), 1.55 (d, J = 6.9 Hz, 4 H), 1.35–1.19 (m, 1 H), 1.05–0.92 (m, 1 H), 0.75–0.61 (m, 1 H)
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(oxetan-3-yl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (7)
Prepared in a similar manner to 3. LC-MS m/z 493 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1 H), 7.76–7.65 (m, 2 H), 7.59 (d, J = 7.8 Hz, 1 H), 7.10–6.99 (m, 2 H), 6.14 (s, 1 H), 4.49 (t, J = 6.4 Hz, 1 H), 4.43 (t, J = 6.5 Hz, 1 H), 4.37 (t, J = 6.1 Hz, 1 H), 4.34–4.28 (m, 3 H), 4.21–4.10 (m, 1 H), 3.83 (s, 3 H), 3.30–3.23 (m, 1 H), 2.75 (br. s., 1 H), 2.71–2.64 (m, 1 H), 2.60 (s, 3 H), 2.19 (s, 4 H), 1.90 (br. s., 1 H), 1.75 (br. s., 1 H), 1.53 (d, J = 6.9 Hz, 3 H), 1.42 (br. s., 2 H), 1.11–0.98 (m, 1 H), 0.72–0.63 (m, 1 H).
Ethyl (R)-2-(4-(1-(3-(((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)carbamoyl)-2-methyl-1H-indol-1-yl)ethyl)piperidin-1-yl)acetate (8)
Prepared in a similar manner to 3. LC-MS m/z 523 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.59 (br. s., 1 H), 7.81–7.65 (m, 2 H), 7.60 (d, J = 7.4 Hz, 1 H), 7.16–6.98 (m, 2 H), 6.15 (s, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.23–4.11 (m, 1 H), 4.04 (q, J = 7.0 Hz, 2 H), 3.84 (s, 3 H), 2.95–2.86 (m, 1 H), 2.60 (s, 5 H), 2.20 (s, 4 H), 1.94–1.79 (m, 2 H), 1.54 (d, J = 6.9 Hz, 4 H), 1.41–1.32 (m, 1 H), 1.15 (t, J = 7.1 Hz, 3 H), 1.04 (d, J = 6.0 Hz, 2 H), 0.71–0.61 (m, 1 H).
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(3,3,3-trifluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (9)
Step 1. A sealed tube was charged with a magnetic stir bar, (R)-methyl 2-methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxylate (2.45 g, 8.16 mmol), DMF (20 mL), 1,1,1-trifluoro-3-iodopropane (1.92 mL, 16.32 mmol), and potassium carbonate (7 g, 50 mmol). The vessel was sealed and heated to 70 °C with stirring for 4 h before being allowed to cool to room temperature. The reaction was filtered through a bed of Celite which was rinsed with ethyl acetate (2 × 100 mL). The filtrate was washed with water, dried with MgSO4, filtered, and concentrated in vacuo to afford the crude product which was purified via silica gel chromatography (50 g) using ethyl acetate/hexanes (1:1) as eluent to afford the methyl (R)-2-methyl-1-(1-(1-(3,3,3-trifluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxylate (2.98 g, 92% yield). LC-MS m/z 533 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.60 (br. s., 1 H), 7.78–7.66 (m, 2 H), 7.60 (d, J = 8.2 Hz, 1 H), 7.13–7.00 (m, 2 H), 6.15 (s, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.22–4.09 (m, 1 H), 3.84 (s, 3 H), 3.03–2.91 (m, 1 H), 2.73–2.64 (m, 1 H), 2.60 (s, 3 H), 2.48–2.31 (m, 5 H), 2.20 (s, 3 H), 2.01–1.85 (m, 2 H), 1.58–1.46 (m, 4 H), 1.36–1.29 (m, 1 H), 1.08–0.98 (m, 1 H), 0.73–0.62 (m, 1 H).
Step 2. A 200 mL round-bottom flask was charged with a magnetic stir bar, (R)-methyl 2-methyl-1-(1-(1-(3,3,3-trifluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxylate (1.15 g, 2.90 mmol), ethanol (10 mL), water (2 mL), and sodium hydroxide (0.46 g, 11.60 mmol) . The reaction was heated to 80 °C with stirring for 12 h before being allowed to cool to room temperature. The ethanol was removed in vacuo, and the resulting mixture was adjusted to pH ∼ 6.5 with 10% HCl. A precipitate formed, which was collected via vacuum filtration using a Buchner funnel. The cake was washed with additional water (∼50 mL) and dried in vacuo to afford (R)-2-methyl-1-(1-(1-(3,3,3-trifluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxylic acid (1.02 g, 92% yield) as an off-white solid.
Step 3. A 100 mL round-bottom flask was charged with a magnetic stir bar, (R)-2-methyl-1-(1-(1-(3,3,3-trifluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxylic acid (1.02 g, 2.67 mmol), DMF (9 mL), 3-(aminomethyl)-4-methoxy-6-methylpyridin-2(1H)-one hydrochloride (0.82 g, 4.00 mmol), Hunig’s base (2 mL). The reaction was cooled to 0 °C with an ice bath, and COMU (2.29 g) was then added. After stirring for 24 h, the reaction was diluted with water (200 mL) and extracted with ethyl acetate (2 × 200 mL). The combined organic extract was washed with water (200 mL), brine (200 mL), was collected, dried with MgSO4, filtered, and concentrated in vacuo to afford the crude compound. This material was preabsorbed onto silica gel (∼10 g) and purified via silica gel chromatography (100 g) using DCM/MeOH (10:1) with 0.1% NH4OH to afford 8 (859 mg, 60% yield) as an off white solid. LC-MS m/z 533 [M + H]+; (400 MHz, DMSO-d6) δ 11.60 (br. s., 1 H), 7.78–7.66 (m, 2 H), 7.60 (d, J = 8.2 Hz, 1 H), 7.13–7.00 (m, 2 H), 6.15 (s, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.22–4.09 (m, 1 H), 3.84 (s, 3 H), 3.03–2.91 (m, 1 H), 2.73–2.64 (m, 1 H), 2.60 (s, 3 H), 2.48–2.31 (m, 5 H), 2.20 (s, 3 H), 2.01–1.85 (m, 2 H), 1.58–1.46 (m, 4 H), 1.36–1.29 (m, 1 H), 1.08–0.98 (m, 1 H), 0.73–0.62 (m, 1 H).
(R)-1-(1-(1-(2,2-Difluoropropyl)piperidin-4-yl)ethyl)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1H-indole-3-carboxamide (10)
Step 1. To a 500 mL round-bottom flask charged with a magnetic stir bar was added 2,2-difluoropropanoic acid (7.5 g, 68.14 mmol) and DCM (250 mL). The mixture was cooled to 0 °C, and oxalyl dichloride (5.48 mL, 8.22 g, 64.73 mmol) was added over 1 min. To this solution was added DMF (500 μL, 6.43 mmol), and the solution was warmed to room temperature with stirring until bubbling ceased (about 1 h). The solution is used as is in the subsequent step without further purification.
Step 2. To a 1 L round-bottom flask equipped with a magnetic stirrer was added (R)-methyl 2-methyl-1-(1-(piperidin-4-yl)ethyl)-1H-indole-3-carboxylate (7.5 g, 24.97 mmol), DCM (200 mL), and N-ethyl-N-isopropylpropan-2-amine (8.07 g, 62.43 mmol). The mixture was cooled to 0 °C in an ice bath and purged with nitrogen. To this stirred mixture of 2,2-difluoropropanoyl chloride (7.79 g, 60.62 mmol) in DCM through via an addition funnel over 15 min. The reaction was allowed to stir for an additional 30 min while warming to room temperature. The mixture was carefully quenched with saturated aqueous NaHCO3, the organic phase separated, washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The resulting material was purified by column chromatography (120 g silica column, 10% to 30% EtOAc in hexanes) to afford methyl (R)-1-(1-(1-(2,2-difluoropropanoyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (9.1 g, 92% yield).
Step 3. A 1 L 3-necked flask was equipped with magnetic stirrer and was fitted with a reflux condenser and an oil-filled bubbler outlet. The vessel was purged and placed under an atmosphere of nitrogen and methyl (R)-1-(1-(1-(2,2-difluoropropanoyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (9.1 g, 23.19 mmol) was dissolved in THF (150 mL) and cannulated into the reaction flask. The reaction was cooled to 0 °C in an ice bath and borane (1.0 M THF solution, 55 mL, 55 mmol) was added over 10 min via syringe. When intense bubbling subsided, the reaction mixture was heated to reflux for 2 h. The reaction was then cooled to 0 °C followed by the careful addition of MeOH (80 mL) (caution: vigorous H2 gas evolution observed). The reaction was then stirred at 0 °C for 5 min, and then allowed to warm to room temperature. The mixture was then heated to 65 °C for 45 min, cooled to room temperature, and was transferred to a 1 L round-bottom flask. The volatiles were removed under reduced pressure. The material was purified by column chromatography (120 g silica column, 10% to 40% EtOAc in hexanes) to afford methyl (R)-1-(1-(1-(2,2-difluoropropyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (7.96 g, 90% yield).
Step 4. In a 1 L round-bottom flask equipped with a magnetic stirrer, (R)-methyl 1-(1-(1-(2,2-difluoropropyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylate (7.96 g, 21.03 mmol) was dissolved in ethanol (80 mL), and a sodium hydroxide solution (6 M aqueous, 16 mL, 96 mmol) was added. The reaction was then heated to reflux (85 °C) for 16 h and was then cooled to 0 °C. A hydrochloric acid solution (2 M aqueous) was added until a pH of 6 was obtained. A precipitate formed which was collected via vacuum filtration using a Buchner funnel. The cake was washed with additional water (∼100 mL) and dried in vacuo to afford the title compound (7.65 g, 99% yield) as an off-white solid.
Step 5. A 500 mL round-bottom flask was charged with a magnetic stir bar, (4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methanaminium chloride (7.3 g, 35.7 mmol), DMF (60 mL), (R)-1-(1-(1-(2,2-difluoropropyl)piperidin-4-yl)ethyl)-2-methyl-1H-indole-3-carboxylic acid (7.65 g, 21.0 mmol), and Hunig’s base (15.0 mL, 84.0 mmol). The mixture was sonicated for 5 min before being cooled to 0 °C. To the stirred reaction mixture was added COMU (13.5 g, 31.5 mmol). The solution was stirred at 0 °C for 15 min before the ice bath was removed. The reaction was allowed to warm to room temperature with stirring overnight. The reaction mixture was then diluted with saturated aqueous NaHCO3 (300 mL) and extracted with EtOAc (2 × 200 mL). The combined organic layer was washed with water, brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified via silica gel chromatography (330 g) with DCM/MeOH (10:1) with 1% NH4OH to afford the title compound as an off-white solid. (6.1 g, 11.85 mmol, 56% yield). LC-MS m/z 515 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1 H), 7.79–7.65 (m, 2 H), 7.60 (d, J = 8.0 Hz, 1 H), 7.16–6.99 (m, 2 H), 6.15 (s, 1 H), 4.38–4.25 (m, 2 H), 4.21–4.10 (m, 1 H), 3.84 (s, 3 H), 3.03–2.90 (m, 1 H), 2.75–2.54 (m, 6 H), 2.25–2.11 (m, 5 H), 1.88 (br. s., 2 H), 1.66–1.45 (m, 6 H), 1.43–1.26 (m, 1 H), 1.12–0.97 (m, 1 H), 0.70–0.61 (m, 1 H).
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,3,3-tetrafluoropropyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (11)
Prepared in a similar manner to 10. LC-MS m/z 551 [M + H]+; 1H NMR (400 MHz, Acetone-d6) δ 12.06 (br. s, 1 H), 7.89 (d, J = 7.9 Hz, 1 H), 7.70 (t, J = 4.9 Hz, 1 H), 7.62 (d, J = 8.1 Hz, 1 H), 6.99–7.12 (m, 2 H), 6.15–6.48 (m, 2 H), 4.50–4.61 (m, 2 H), 4.18–4.30 (m, 1 H), 3.93 (s, 3 H), 3.04 (d, J = 11.5 Hz, 1 H), 2.85–2.96 (m, 2 H), 2.73–2.83 (m, 2 H), 2.71 (s, 3H), 2.28–2.43 (m, 2 H), 2.23 (br. s., 3 H), 2.00 (d, J = 12.8 Hz, 1H), 1.62 (d, J = 7.0 Hz, 3 H), 1.46 (dq, J = 12.2, 3.9 Hz, 1 H), 1.11–1.23 (m, 1 H), 0.79 (d, J = 12.8 Hz, 1 H).
(R)-1-(1-(1-(2,2-Difluoroethyl)piperidin-4-yl)ethyl)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1H-indole-3-carboxamide (12)
Prepared in a similar manner to 10. LC-MS m/z 501 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 11.60 (br. s., 1 H), 7.77–7.66 (m, 2 H), 7.60 (d, J = 7.6 Hz, 1 H), 7.14–7.00 (m, 2 H), 6.15 (s, 1 H), 6.06 (t, J = 55.7 Hz, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.15 (br. s., 1 H), 3.84 (s, 3 H), 3.03–2.93 (m, 2 H), 2.73–2.62 (m, 3 H), 2.60 (s, 3 H), 2.26–2.10 (m, 4 H), 1.93–1.79 (m, 1 H), 1.59–1.46 (m, 4 H), 1.41–1.29 (m, 1 H), 1.11–0.97 (m, 1 H), 0.67 (br. s., 1 H).
(R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (13)
Prepared in a similar manner to 10. LC-MS m/z 519 [M + H]+; 1H NMR (500 MHz, DMSO-d6) δ 11.59 (s, 1 H), 7.74 (d, J = 7.6 Hz, 1 H), 7.71–7.66 (m, 1 H), 7.61 (d, J = 7.8 Hz, 1 H), 7.13–7.01 (m, 2 H), 6.15 (s, 1 H), 4.32 (d, J = 4.9 Hz, 2 H), 4.22–4.12 (m, 1 H), 3.84 (s, 3 H), 3.15–2.95 (m, 3 H), 2.75–2.66 (m, 1 H), 2.60 (s, 3 H), 2.39–2.31 (m, 1 H), 2.20 (s, 3 H), 2.05–1.98 (m, 1 H), 1.92–1.84 (m, 1 H), 1.56–1.46 (m, 4 H), 1.42–1.32 (m, 1 H), 1.11–1.01 (m, 1 H), 0.69–0.62 (m, 1 H).
Acknowledgments
The authors thank Dr. Julian Levell for editorial suggestions and comments during the preparation of this manuscript. We would like to thank Custom NMR Services for assistance in acquisition of all NMR spectra. Y.Z., N.J., S.C., J.R.W. and S.J.G. were supported by the Francis Crick Institute, which receives its core funding (FC001078) from Cancer Research U.K., the U.K. Medical Research Council, and the Wellcome Trust. We greatly acknowledge Diamond Light Source for access to synchrotron time under proposal MX9826.
Glossary
Abbreviations Used
- PRC2
polycomb repressive complex 2
- H3K27
histone 3 lysine 27
- EZH2
enhancer of zeste homologue 2
- SAM
S-adenosyl-l-methionine
- PD
pharmacodynamics
- tPSA
total polar surface area
- SAR
structure activity relationships
- MOA
mechanism of action
- ADME
absorption, distribution, metabolism, excretion
- PK
pharmacokinetic
- Clint
intrinsic clearance
- PPB
plasma protein binding
- LiPE
lipophilic ligand efficiency
- IV
intravenous
- Q
liver blood flow
- PO
per os
- Vss
volume of distribution at steady state
- AUC
area under curve
- F
bioavailability
- DMA
dimethylacetamide
- PEG
polyethylene glycol
- SBECD
sulfobutyl ether β-cyclodextrin
- iPrNEt2
hunig’s base
- CDI
carbonyldiimadazole
- THF
tetrahydrofuran
- TGI
tumor-growth inhibition
- BID
twice a day
- QD
once a day
- GLP
good laboratory practice
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01315.
Co-crystal structure of inhibitor 10 bound to PRC2 available (PDB code: 5LS6); 100 g scale synthesis of inhibitor 13; biochemical assay details; mechanism of action (moa) cell assay details, descriptions of KARPAS-422 efficacy and PK/PD experiments; methods for structural biology experiments (PDF)
Molecular formula strings and compound data (CSV)
Author Present Address
δ For Rishi G. Vaswani: Blueprint Medicines, 38 Sidney Street, Suite 200, Cambridge, MA 02139, United States.
Author Present Address
† For Christopher G. Nasveschuk and Martin Duplessis: C4 Therapeutics, 675 W Kendall Street, Cambridge, MA 02142, United States.
Author Present Address
‡ For Les Dakin: Pfizer, Inc., 610 Main Street, Cambridge, MA 02412, United States.
Author Present Address
§ For Robert Campbell: Blueprint Medicines, 38 Sidney Street, #200, Cambridge, Massachusetts 02139, United States.
Author Present Address
∥ For Priyadarshini Iyer: Agenus Inc., 3 Forbes Road, Lexington, MA 02421, United States.
Author Present Address
⊥ For Andy Good: Schrödinger Inc., 222 Third Street, Ste. 2230, Cambridge, MA 02142, United States.
Author Present Address
# For Christina Lee: Editas Medicine, 300 Third Street, Cambridge, MA 02142, United States.
Author Present Address
∇ For Andrew Cook: H3 Biomedicine, Inc., 300 Technology Square #5, Cambridge, MA 02139, United States.
Author Present Address
○ For Emmanuel Normant: Surface Oncology, 215 First Street, Suite 400-S, Cambridge, Massachusetts 02142, United States.
Author Present Address
∂ For Steven F. Bellon: Foghorn Therapeutics, 161 First Street, Suite 2A, Cambridge, Massachusetts 02142, United States.
Author Present Address
⧫ For Brian K. Albrecht and Jean-Christophe Harmange: Third Rock Ventures, 29 Newbury Street, #301, Boston, Massachusetts 02116, United States.
Author Present Address
≠ For Ying Zhang, Neil Justin, Shuyang Chen, Jon Wilson, and Steve Gamblin: The Francis Crick Institute, 1 Midland Road, London NW1 1AT, United Kingdom.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Swalm B. M.; Knutson S. K.; Warholic N. M.; Jin L.; Kuntz K. W.; Keilhack H.; Smith J. J.; Pollock R. M.; Moyer M. P.; Scott M. P.; Copeland R. A.; Wigle T. J. Reaction Coupling Between Wild-Type and Disease-Associated Mutant EZH2. ACS Chem. Biol. 2014, 9, 2459–2464. 10.1021/cb500548b. [DOI] [PubMed] [Google Scholar]
- Simon J. A.; Lange C. A. Roles of the EZH2 Histone Methyltransferase in Cancer Epigenetics. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2008, 647, 21–29. 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Margueron R.; Reinberg D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349. 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H.; Hung M.-C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. 10.4143/crt.2014.46.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S.; Dhanasekaran S. M.; Zhou M.; Barrette T. R.; Kumar-Sinha C.; Sanda M. G.; Ghosh D.; Pienta K. J.; Sewalt R. G.; Otte A. P.; Rubin M. A.; Chinnaiyan A. M. The Polycomb Group Protein EZH2 Is Involved In Progression of Prostate Cancer. Nature 2002, 419, 624–629. 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Kleer C. G.; Cao Qi; Varambally S.; Shen R.; Ota I.; Tomlins S. A.; Ghosh D.; Sewalt R. G. A. B.; Otte A. P.; Hayes D. F.; Sabel M. S.; Livant D.; Weiss S. J.; Rubin M. A.; Chinnaiyan A. M. EZH2 is a Marker of Aggressive Breast Cancer and Promotes Neoplastic Transformation of Breast Epithelial Cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11606–11611. 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J. P.; Trojer P. Targeting Histone Lysine Methylation In Cancer. Pharmacol. Ther. 2015, 150, 1–22. 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Bauge C.; Bazille C.; Girard N.; Lhuissier E.; Boumediene K. Histone Methylases as Novel Drug Targets: Developing Inhibitors of EZH2. Future Med. Chem. 2014, 6, 1943–1965. 10.4155/fmc.14.123. [DOI] [PubMed] [Google Scholar]
- Verma S. K.; Knight S. D. Recent Progress in the Discovery of Small-Molecule Inhibitors of the HMT EZH2 for the Treatment of Cancer. Future Med. Chem. 2013, 5, 1661–1670. 10.4155/fmc.13.136. [DOI] [PubMed] [Google Scholar]
- Keilhack H.; Smith J. J. Small Molecule Inhibitors of EZH2: The Emerging Translational Landscape. Epigenomics 2015, 7, 337–341. 10.2217/epi.15.14. [DOI] [PubMed] [Google Scholar]
- Beguelin W.; Popovic R.; Teater M.; Jiang Y.; Bunting K. L.; Rosen M.; Shen H.; Yang S. N.; Wang L.; Ezponda T.; Martinez-Garcia E.; Zhang H.; Zheng Y.; Verma S. K.; McCabe M. T.; Ott H. M.; Van Aller G. S.; Kruger R. G.; Liu Y.; McHugh C. F.; Scott D. W.; Chung Y. R.; Kelleher N.; Shaknovich R.; Creasy C. L.; Gascoyne R. D.; Wong K.-K.; Cerchietti L.; Levine R. L.; Abdel-Wahab O.; Licht J. D.; Elemento O.; Melnick A. M. EZH2 Is Required for Germinal Center Formation and Somatic EZH2Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692. 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; Scott M. P.; Jin L.; Smith J. J.; Olhava E. J.; Chesworth R.; Moyer M.; Richon V. M.; Copeland R. A.; Keilhack H.; Pollock R. M.; Kuntz K. W. A Selective Inhibitor of EZH2 Blocks H3K27 Methylation and Kills Mutant Lymphoma Cells. Nat. Chem. Biol. 2012, 8, 890–896. 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Scott M. P.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7922–7927. 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz K. W.; Campbell J. E.; Keilhack H.; Pollock R. M.; Knutson S. K.; Porter-Scott M.; Richon V. M.; Sneeringer C. J.; Wigle T. J.; Allain C. J.; Majer C. R.; Moyer M. P.; Copeland R. A.; Chesworth R. The Importance of Being Me: Magic Methyls, Methyltransferase Inhibitors, and the Discovery of Tazemetostat. J. Med. Chem. 2016, 59, 1556–1564. 10.1021/acs.jmedchem.5b01501. [DOI] [PubMed] [Google Scholar]
- Campbell J. E.; Kuntz K. W.; Knutson S. K.; Warholic N. M.; Keilhack H.; Wigle T. J.; Raimondi A.; Klaus C. R.; Rioux N.; Yokoi A.; Kawano S.; Minoshima Y.; Choi H.-W.; Scott M. P.; Waters N. J.; Smith J. J.; Chesworth R.; Moyer M. P.; Copeland R. A. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust In Vivo Activity. ACS Med. Chem. Lett. 2015, 6, 491–495. 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S. K.; Tian X.; LaFrance L. V.; Duquenne C.; Suarez D. P.; Newlander K. A.; Romeril S. P.; Burgess J. L.; Grant S. W.; Brackley J. A.; Graves A. P.; Scherzer D. A.; Shu A.; Thompson C.; Ott H. M.; Van Aller G. S.; Machutta C. A.; Diaz E.; Jiang Y.; Johnson N. W.; Knight S. D.; Kruger R. G.; McCabe M. T.; Dhanak D.; Tummino P. J.; Creasy C. L.; Miller W. H. Identification of Potent, Selective Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Della Pietra A. III; Diaz E.; LaFrance L. V.; Mellinger M.; Duquenne C.; Tian X.; Kruger R. G.; McHugh C. F.; Brandt M.; Miller W. H.; Dhanak D.; Verma S. K.; Tummino P. J.; Creasy C. L. EZH2 Inhibition as a Therapeutic Strategy for Lymphoma with EZH2-Aactivating Mutations. Nature 2012, 492, 108–112. 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X.-P.; Kuznetsova E.; Rougie M.; Jiang A.; Pattenden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; Gu J.; Ardayfio O.; Zhang J.-H.; Yan X.; Fang J.; Mi Y.; Zhang M.; Zhou T.; Feng G.; Chen Z.; Li G.; Yang T.; Zhao K.; Liu X.; Yu Z.; Lu C. X.; Atadja P.; Li E. Selective Inhibition of EZH2 by a Small Molecule Inhibitor Blocks Tumor Cell Proliferation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21360–21365. 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley W. D.; Arora S.; Busby J.; Balasubramanian S.; Gehling V.; Nasveschuk C. G.; Vaswani R. G.; Yuan C.-C.; Hatton C.; Zhao F.; Williamson K. E.; Iyer P.; Méndez J.; Campbell R.; Cantone N.; Garapaty-Rao S.; Audia J.; Cook A. S.; Dakin L. A.; Albrecht B. K.; Harmange J.-C.; Daniels D. L.; Cummings R. T.; Bryant B. M.; Normant E.; Trojer P. EZH2 Inhibitor Efficacy in Non-Hodgkin’s Lymphoma Does Not Require Suppression of H3K27 Monomethylation. Chem. Biol. 2014, 21, 1463–1475. 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Vaswani R. G.; Nasveschuk C. G.; Duplessis M.; Iyer P.; Balasubramanian S.; Zhao F.; Good A. C.; Campbell R.; Lee C.; Dakin L. A.; Cook A. S.; Gagnon A.; Harmange J.-C.; Audia J. E.; Cummings R. T.; Normant E.; Trojer P.; Albrecht B. K. Discovery, Design, and Synthesis of Indole-Based EZH2 Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3644–3649. 10.1016/j.bmcl.2015.06.056. [DOI] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Gagnon A.; Garapaty-Rao S.; Balasubramanian S.; Campbell R.; Lee C.; Zhao F.; Bergeron L.; Cummings R.; Trojer P.; Audia J. E.; Albrecht B. K.; Harmange J.-C. P. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5, 378–383. 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garapaty-Rao S.; Nasveschuk C.; Gagnon A.; Chan E. Y.; Sandy P.; Busby J.; Balasubramanian S.; Campbell R.; Zhao F.; Bergeron L.; Audia J. E.; Albrecht B. K.; Harmange J.-C.; Cummings R.; Trojer P. Identification of EZH2 and EZH1 Small Molecule Inhibitors with Selective Impact on Diffuse Large B Cell Lymphoma Cell Growth. Chem. Biol. 2013, 20, 1329–1339. 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Röder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- Wuitschik G.; Carreira E. M.; Wagner B.; Fischer H.; Parrilla I.; Schuler F.; Rogers-Evans M.; Müller K. Oxetanes in Drug Discovery: Structural and Synthetic Insights. J. Med. Chem. 2010, 53, 3227–3246. 10.1021/jm9018788. [DOI] [PubMed] [Google Scholar]
- Justin N.; Zhang Y.; Tarricone C.; Martin S. R.; Chen S.; Underwood E.; De Marco V.; Haire L. F.; Walker P. A.; Reinberg D.; Wilson J. R.; Gamblin S. J. Structural Basis of Oncogenic Histone H3K27M Inhibition of Human Polycomb Repressive Complex 2. Nat. Commun. 2016, 7, 11316–111326. 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibaja V.; Shen F.; Harari J.; Korn J.; Ruddy D.; Saenz-Vash V.; Zhai H.; Rejtar T.; Paris C. G.; Yu Z.; Lira M.; King D.; Qi W.; Keen N.; Hassan A. Q.; Chan H. M. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperate to confer resistance to EZH2 inhibitors. Oncogene 2016, 35, 558–566. 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooun A.; Gajiwala K. S.; Deng Y.-L.; Liu W.; Bolanos B.; Bingham P.; He Y.-A.; Diehl W.; Grable N.; Kung P.-P.; Sutton S.; Maegley K. A.; Yu X.; Stewart A. E. Polycomb Repressive Complex 2 Structure with Inhibitor Reveals a Mechanism of Activation and Drug Resistance. Nat. Commun. 2016, 7, 11384. 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Zeng H.; Dong A.; Li F.; He H.; Senisterra G.; Seitova A.; Duan S.; Brown P. J.; Vedadi M.; Arrowsmith C. H.; Schapira M. Structure of the Catalytic Domain of EZH2 Reveals Conformational Plasticity in Cofactor and Substrate Binding Sites and Explains Oncogenic Mutations. PLoS One 2013, 8, e83737. 10.1371/journal.pone.0083737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani R. G.; Albrecht B. K.; Audia J. E.; Cote A.; Dakin L. A.; Duplessis M.; Gehling V. S.; Harmange J.-C.; Hewitt M. C.; Leblanc Y.; Nasveschuk C. G.; Taylor A. M. A Practical Synthesis of Indoles via a Pd-Catalyzed C–N Ring Formation. Org. Lett. 2014, 16, 4114–4117. 10.1021/ol5018118. [DOI] [PubMed] [Google Scholar]
- Chiral amine 15 was synthesized according to procedure outlined by Albrecht B. K.; Audia J. E.; Cook A. S.; Dakin L. A.; Duplessis M.; Gehling V. S.; Harmange J.-C.; Nasveschuk C. G.; Vaswani R. G. PCT Int. Appl. WO 2013120104 A2, August 15, 2013.
- Chiral amine 15 was resolved according to the procedure outline by Mortimore M.; Young S. C.; Everitt S. R. L.; Knegtel R.; Pinder J. L.; Rutherford A. P.; Durrant S.; Brenchley G.; Charrier J. D.; O’Donnell M. PCT Int. Appl. WO 2008079346 A1, July 3, 2008.
- See Supporting Information section.
- See Supporting Information for further details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.