

Abstract
The RecQ helicase Sgs1p forms a complex with the type 1 DNA topoisomerase Top3p that resolves double Holliday junctions resulting from Rad51-mediated exchange. We find, however, that Sgs1p functions independently of both Top3p and Rad51p to stimulate the checkpoint kinase Rad53p when replication forks stall due to dNTP depletion on hydroxyurea. Checkpoint activation does not require Sgs1p function as a helicase, and correlates with its ability to bind the Rad53p kinase FHA1 motif directly. On the other hand, Sgs1p's helicase activity is required together with Top3p and the strand-exchange factor Rad51p, to help stabilise DNA polymerase ɛ at stalled replication forks. In this function, the Sgs1p/Top3p complex acts in parallel to the Claspin-related adaptor, Mrc1p, although the sgs1 and mrc1 mutations are epistatic for Rad53p activation. We thus identify two distinct pathways through which Sgs1p contributes to genomic integrity: checkpoint kinase activation requires Sgs1p as a noncatalytic Rad53p-binding site, while the combined Top3p/Sgs1p resolvase activity contributes to replisome stability and recovery from arrested replication forks.
Keywords: DNA polymerase stabilisation, Rad53 activation, Sgs1p, S-phase checkpoint, Top3
Introduction
Genomic stability reflects both the ability to avoid DNA breakage and the promotion of efficient repair (Kolodner et al, 2002). In order to ensure complete repair, eukaryotic cells induce a checkpoint response that delays division and favours recovery from a range of genotoxic insults (reviewed in Nyberg et al, 2002). Many genes implicated in DNA checkpoint responses are found mutated in human disorders that lead to increased genetic instability and cancer. Among these are RecQ DNA helicases, a family of enzymes conserved from bacteria to man (reviewed in Khakhar et al, 2003). Of the five RecQ helicases in the human genome, mutations in three are responsible for genetic disorders (Bloom's, Werner's and Rothmund–Thomson syndromes) that correlate with enhanced frequency of chromosomal loss and rearrangement, leading to cancer or premature aging (reviewed in Mohaghegh and Hickson, 2001).
Both budding and fission yeast have but one RecQ helicase, encoded by SGS1 or Rqh1+, respectively (Gangloff et al, 1994; Murray et al, 1997; Stewart et al, 1997). Although neither gene is essential, ablation of SGS1 function leads to increased rates of mitotic and meiotic recombination, gross chromosomal rearrangements, chromosome loss and cellular senescence (Watt et al, 1996; Sinclair et al, 1997; Myung et al, 2001). In both yeast species, cells that lack the RecQ helicase show an increased sensitivity to DNA-damaging agents, such as ionising radiation, methylmethane sulphonate (MMS) and bleomycin (Murray et al, 1997; Stewart et al, 1997; Frei and Gasser, 2000). RecQ mutants are also hypersensitive to hydroxyurea (HU) and in budding yeast the absence of Sgs1p correlates with a reduction in the stability of DNA polymerases α and ɛ (pol α and pol ɛ) at stalled replication forks (Cobb et al, 2003).
Sgs1p/Rqh1p, like the human homologue BLM, interacts genetically and physically with DNA topoisomerase Top3p, a type IA enzyme that unlinks single-strand catenanes (Gangloff et al, 1994; Goodwin et al, 1999; Wu et al, 2000). In budding yeast, deletion of TOP3 results in slow growth and high levels of recombination and chromosome loss (Wallis et al, 1989; Myung et al, 2001), while gene disruption is lethal in fission yeast and mice (Li and Wang, 1998; Goodwin et al, 1999). Surprisingly, the slow growth phenotypes of top3 mutants can be suppressed by additionally ablating Sgs1p function (Gangloff et al, 1994). Forms of Sgs1p that are unable to bind Top3p are unable to suppress the hypersensitivity of the sgs1 mutant to MMS, nor do they suppress the synthetic lethality between sgs1 and the endonuclease slx4 (Mullen et al, 2000). Indeed, when double Holliday junction (HJ) structures form at strand breaks, Sgs1p/Blm and Top3p/hTop3α work together to reduce reciprocal exchange events (Ira et al, 2003; Wu and Hickson, 2003). In summary, extensive evidence suggests that Sgs1p and Top3p act as a complex when they counter reciprocal genetic exchange or unfavourable recombination events in the face of strand breaks.
On the other hand, Sgs1p also seems to fulfil unique replication fork-associated functions. Indeed, the levels of Sgs1p peak in S phase unlike Top3p, and the helicase colocalises with sites of DNA replication both in the absence and presence of damage (Frei and Gasser, 2000; Cobb et al, 2003). In fission and budding yeast, the intra-S-phase checkpoint is partially compromised by deletion of Sgs1p or Rqh1p (Frei and Gasser, 2000; Marchetti et al, 2002). Although the Saccharomyces cerevisiae top3 deletion was reported to impair Rad53p activation in response to MMS (Chakraverty et al, 2001), defects in cell cycle progression were not taken into account in this study, and our work argues against such a role for HU (see below). Indeed, XBlm or XTop3 depletion does not impair CHK1 or CHK2 kinase induction by replication arrest in Xenopus egg extracts (Li et al, 2004). From these data, we concluded that the relationship between Sgs1p, Top3p and S-phase checkpoint activation required further investigation.
The intra-S-phase checkpoint response in budding yeast is mediated largely by activation of the Rad53p kinase (equivalent to CHK2 in human cells). This occurs in response to fork stalling by high concentration of HU, or in response to strand breaks that arise from replication fork collision with MMS-induced alkylation (reviewed in Nyberg et al, 2002). Recent papers argue that both the HU- and MMS-induced S-phase checkpoint responses require a sufficient density of replication forks to achieve a threshold level of damage sufficient for Rad53p activation (Shimada et al, 2002; Tercero et al, 2003). It is therefore necessary to suppress defects in cell cycle progression in order to determine whether or not a protein contributes directly to checkpoint kinase activation. Keeping this in mind, we re-examine here the contributions of Top3p, Sgs1p and Rad51p to Rad53p checkpoint kinase activation in S-phase cells exposed to HU, under conditions that guarantee uniform S-phase progression. Their potential roles in checkpoint activation are compared with their roles in the stabilisation of DNA polymerases at stalled forks and the cellular recovery from arrest.
Using quantitative autophosphorylation and chromatin immunoprecipitation (ChIP) assays, we find that Sgs1p functions on at least two pathways when replication is blocked. First, it contributes to the appropriate checkpoint response by binding Rad53p. This function requires neither its helicase activity, Top3p nor Rad51p. Second, Sgs1p contributes to the stabilisation of DNA pol ɛ at stalled forks, acting together with Top3p and Rad51p. These mechanistically distinct functions for Sgs1p at replication forks contribute to genomic integrity.
Results
Sgs1p functions independently of Top3p to promote Rad53p activation by HU
Earlier studies based on a Rad53p mobility shift assay suggested that Sgs1p contributes to the activation of this checkpoint kinase in response to HU-induced fork arrest, although in the absence of Sgs1p, Rad53p could be stimulated through a Rad24p/Rad17p pathway (Frei and Gasser, 2000). In budding yeast, Rad24 protein (equivalent to RAD17Sp or RAD17Hs) forms a complex with the core Rfc complex to load a PCNA-like complex comprising Ddc1p, Rad17p and Mec3p (also called 9-1-1, after the homologues Rad9, Rad1 and Hus1), at strand breaks. Deletion of the Rad24p subunit compromises this pathway, reducing but not eliminating the checkpoint response mediated by Mec1p (ATRHs) and Rad53p (CHK2Hs; Paulovich et al, 1997; Pellicioli et al, 1999). It is assumed that in the absence of Sgs1p, strand breaks accumulate in HU-treated cells to stimulate Rad53p kinase activity through Rad24p and the 9-1-1 complex, as shown for XBlm-depleted Xenopus extracts (Li et al, 2004). Less clear, however, is the mechanism through which Sgs1p contributes to Rad53p activation in the absence of Rad24p.
Here we monitor Mec1p-stimulated, Rad53p autophosphorylation using an in situ autophosphorylation assay (ISA). ISA shows excellent correlation between [γ-32P]ATP incorporation and the Rad53p mobility shift, and monitors kinase activity quantitatively, due to the vast excess of substrate (Rad53p) present on the filter (Pellicioli et al, 1999). The kinetics of Rad53p activation can be compared among different mutants if one normalises for loading efficiency by probing an unrelated protein on the same filter (i.e. Rnase42), and includes an internal activity standard on each gel (described further in Materials and methods; Figure 1A and B).
Figure 1.
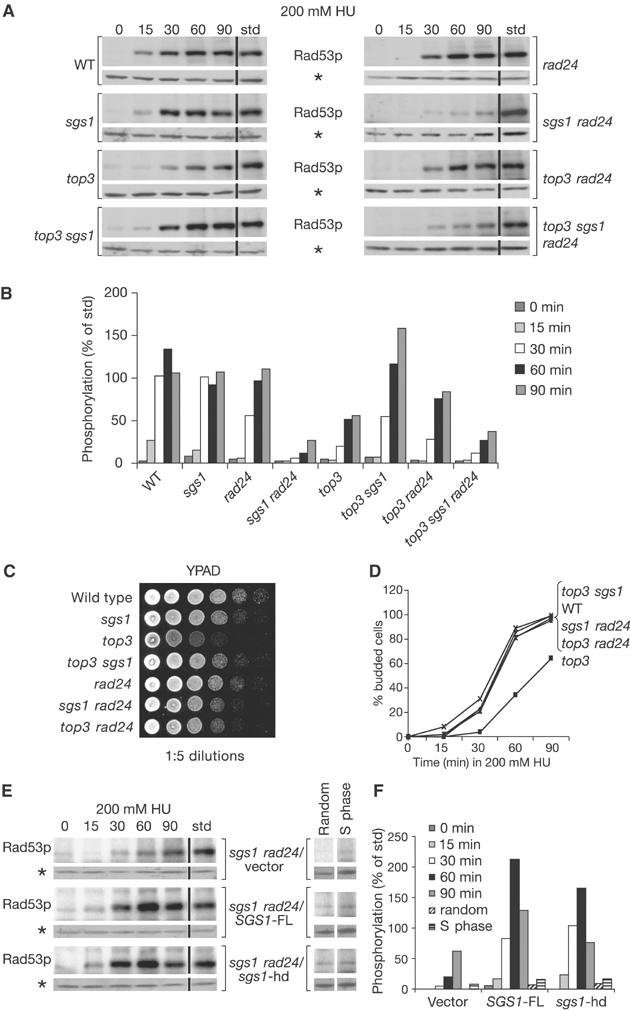
S-phase checkpoint response requires Sgs1p but not Top3p. (A) ISA analysis of Rad53p autophosphorylation was performed on GA-1020, GA-1748, GA-1761, GA-1799, GA-1800, GA-2047, GA-2056 and GA-2060 for which the relevant genotype is indicated beside each data set. For each strain, the upper box shows the incorporation of [γ-32P]ATP into Rad53p, and the bottom panel a Western for RnaseH42 on the same blot (*). Time (min) after α-factor release is indicated above each panel. std is 5 μl of a sample containing a fixed amount of an HU-activated Rad53p standard that is used to normalise all gels after identical exposure times (see Materials and methods). (B) Quantification of Rad53p autophosphorylation displayed as percentage of std. The quantification shown is an average of two experiments with standard derivations between 5 and 15%. (C) Five-fold serial dilutions for the indicated strains were plated onto YPAD and incubated at 30°C for 3 days. (D) Bud emergence was scored on 100 cells in cultures released into 0.2 M HU after α-factor block. (E) ISA analysis of Rad53p autophosphorylation was performed in GA-2057 bearing either pRS415 (vector), pRS415 with full-length SGS1 (SGS1-FL) or pRS415 expressing helicase-deficient protein (sgs1-hd) on HU, and on random and S-phase cultures without HU. (F) Quantitation of ISA for GA-2057 with vector, SGS1-FL or sgs1-hd as in (E).
Results from ISA agree with mobility shift assays (Frei and Gasser, 2000), and show that Rad24p and Sgs1p define parallel pathways that can both stimulate Rad53p activity as cells progress synchronously from a G1 arrest into 0.2 M HU (Figure 1A and B). In wild-type cells, the maximal level of Rad53p autophosphorylation is reached 60 min after release into HU. A similar level is achieved in an sgs1 or rad24 single mutant with a slight delay, while elimination of the two pathways significantly reduces maximal Rad53p activation (Figure 1A). Quantitation of Rad53p autophosphorylation is shown in Figure 1B. This supports the model that Sgs1p is essential on one of two pathways leading to Rad53p activation.
We next asked whether Sgs1p requires Top3p to activate the checkpoint on HU. If so, the deletion of top3 should similarly compromise Rad53p activation in cells lacking Rad24p. Although Rad53p activation is reduced in the top3 single mutant, we see that the Rad53p response is actually improved in a top3 rad24 double mutant (Figure 1A and B). The dissimilar response to HU for the double top3 rad24 and sgs1 rad24 mutants provides a first indication that Sgs1p and Top3p do not act comparably upstream of Rad53p stimulation.
Lack of Top3p impairs progression into and through S phase
Although the top3 single mutant appears to be checkpoint compromised and fails to reach wild-type levels of Rad53p activation after 90 min on HU (Figure 1A), the defect was suppressed rather than enhanced in a top3 rad24 strain. This required explanation. Recent studies have shown that the efficiency of S-phase checkpoint activation in yeast correlates with fork number and efficient entry into S phase (Shimada et al, 2002; Tercero et al, 2003). Because strains lacking Top3p have a pronounced slow growth phenotype, we suspected that their delayed entry into S phase might indirectly reduce Rad53p activation by HU in the top3 mutant cells.
Drop assays on rich media in the absence of damage confirmed that the top3 mutant is indeed slow growing and that slow growth is suppressed by deletion of either sgs1 or rad24 (Figure 1C; Gangloff et al, 1994; Chakraverty et al, 2001). This was confirmed by FACS analysis (data not shown). To show that the top3-associated slow growth reflects impaired entry into S phase, we monitored bud emergence in a synchronised population, when cells are released from an α-factor block into 0.2 M HU. We see that top3 cells delay entry into S phase, and again this delay can be suppressed by deleting either sgs1 or rad24 (Figure 1D).
Using this efficient suppression of the top3 growth phenotype, we could test whether slow growth per se is the source of Top3p's effect of Rad53p. Indeed, we find a complete restoration of the Rad53p response in the double top3 sgs1 mutant (Figure 1A and B). Importantly, we confirm the redundancy of Rad24p and Sgs1p pathways, and show that this redundancy is independent of Top3p, by further deleting rad24 in the top3 sgs1 strain: any coupling of rad24 and sgs1 mutations abolishes checkpoint activation on HU (Figure 1A). In summary, the combined deletion of sgs1 and rad24 in budding yeast systematically compromises the checkpoint response, while Top3p is implicated in neither pathway.
The checkpoint function of Sgs1p is independent of its helicase activity
Sgs1p has been shown by ChIP to localise to replication forks, where its helicase activity contributes to DNA polymerase stabilisation in the presence of HU (Cobb et al, 2003). We therefore next checked whether or not Sgs1p helicase activity contributes to checkpoint activation. A helicase-deficient mutant of Sgs1p (sgs1-hd) was introduced into the sgs1 rad24 background to see if it could suppress the observed reduction in Rad53p activation. As a control, we monitored Rad53p activity in the same background supplemented with the full-length SGS1 on a plasmid (SGS1-FL) or an empty vector. Intriguingly, levels of Rad53p autophosphorylation are nearly identical when expressing either full-length or the helicase-dead version of Sgs1p in the sgs1 rad24 mutant, while only low levels of autophosphorylation are detected in the empty vector control (Figure 1E and F). To ensure that expression of full-length or helicase-dead versions of Sgs1p does not artificially activate Rad53p, we also monitored Rad53p activity in plasmid bearing strains synchronised in S phase or in random culture without adding HU. As shown in Figure 1E and F, no checkpoint activation results from the protein expression alone. The fact that the Sgs1p is necessary but its helicase activity dispensable suggests that Sgs1p may promote checkpoint activation through protein–protein interactions at stalled forks. This stands in contrast to data showing that RecQ helicase activity is indeed required for DNA polymerase stabilisation and efficient cell recovery after HU treatment (Frei and Gasser, 2000; Cobb et al, 2003).
In vivo interaction between Sgs1p and Rad53p
Human BLM and WRN helicases, like Sgs1p, interact with a wide range of proteins including FANCD2, Rad51p, Top3p and the ATM/ATR kinases (reviewed in Mohaghegh and Hickson, 2001). Given that its helicase activity is dispensable for checkpoint activation, we next determined whether Sgs1p functions by directly recruiting Rad53p to the replication fork. Initial support for this hypothesis arose from a co-immunoprecipitation assay in cell extracts of a yeast strain that expresses cMyc-tagged Sgs1p and HA-tagged Rad53p under their endogenous promoters (Figure 2A). When Rad53p is immunoprecipitated by αHA-coated Dynabeads, we detect Myc-tagged Sgs1p in the precipitate, while it is not recovered in control precipitations (Figure 2A, upper panel). Surprisingly, this interaction is independent of the phosphorylation status of Rad53p, being unchanged by the addition of HU (note Rad53p shift on HU; Figure 2A).
Figure 2.
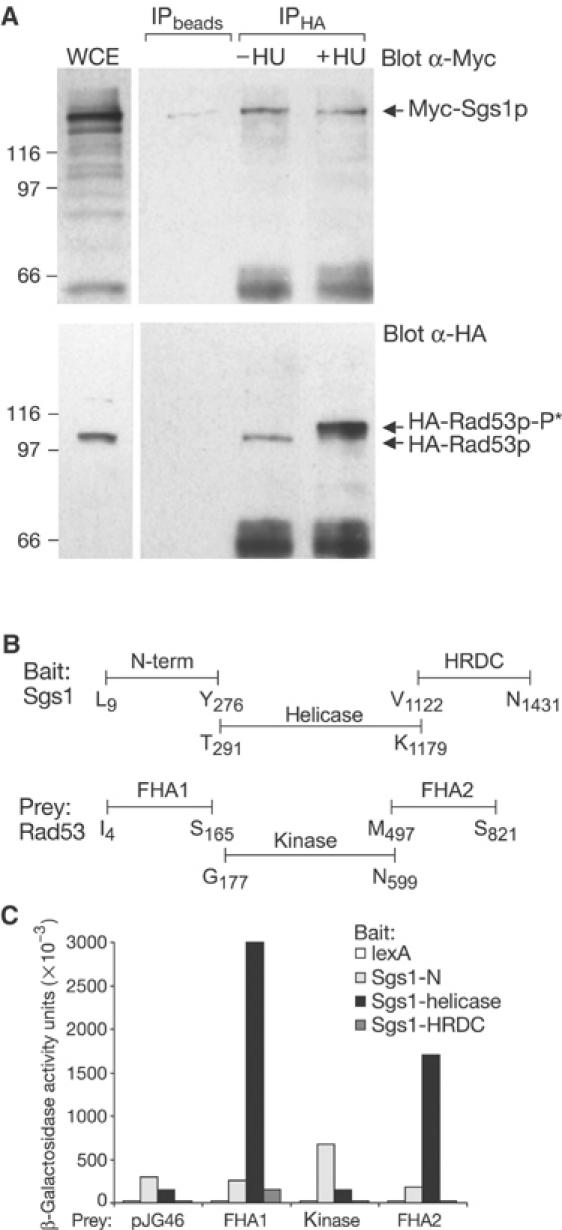
Specific interaction between Sgs1p and the Rad53p FHA1 domain. (A) Immunoprecipitation experiment performed with whole-cell extracts (WCEs) from GA-1142 cells (expressing Myc-tagged Sgs1p and HA-tagged Rad53p). WCEs were obtained from either random or HU-blocked cultures and α-HA (12CA5)-coupled Dynabeads were used for co-immunoprecipitation. Blots were probed with α-Myc (9E10) for Sgs1p (upper panel) or α-HA (Rad53, lower panel). (B) Scheme of Sgs1p and Rad53p domains used to fuse to the B42 activator domain in pJG46 or to the lexA DNA binding domain in pGAL-lexA. (C) The two-hybrid assay shows strong interactions between Sgs1-helicase and Rad53-FHA1 (20-fold), and Rad53-FHA2 (12-fold). Point mutations in FHA1 (R70A) or FHA2 (R605A) eliminate this interaction in pull-down assays (data not shown). β-Galactosidase units are described in Materials and methods.
To substantiate the interaction and map the domains of Sgs1p and Rad53p responsible for this interaction, subdomains of SGS1 and RAD53 were cloned for two-hybrid interaction analysis (Figure 2B). Results indicate a strong interaction between the helicase domain of Sgs1p and the FHA1 domain of Rad53p (20-fold increase over background) and a significant but weaker affinity for the FHA2 domain (12-fold increase; Figure 2C). Both interactions are compromised by point mutations in the FHA domains (e.g. FHA1R70A, data not shown). To see whether Top3p also binds Rad53p, we screened for interaction between endogenously tagged Top3p and either wild-type or mutant Rad53 FHA1 domains, using a GST pull-down assay (GST-FHA1 and GST-FHA1R70A). We detect interaction between FHA1 and Top3p, which is lost in the absence of Sgs1p (see Supplementary Figure S1). These data indicate that Sgs1p has the potential to recruit Rad53p to the fork directly, even if bound to Top3p, although Top3p has no essential role in Rad53p activation.
Sgs1p and Mrc1p work on the same pathway for checkpoint activation
Mrc1p and the vertebrate Claspin protein define a class of proteins that, like scRad9p, serve as mediators or adaptors for Rad53p activation, responding specifically to stalled replication forks (Kumagai and Dunphy, 2000; Alcasabas et al, 2001). Like Sgs1p, both are found associated with replication forks with or without damage (Katou et al, 2003; Lee et al, 2003; Osborn and Elledge, 2003). Because our data implicate Sgs1p in the HU checkpoint response, we next examined whether Sgs1p acts on the same pathway as Mrc1p for Rad53p activation.
As reported, Rad53p activation in response to HU is reduced but not eliminated in strains lacking the adaptor Mrc1p (Figure 3A and B). Combining sgs1 and mrc1 mutations is not synergistic and does not fully compromise Rad53p activation, as in the case of the sgs1 rad24 double mutant. Instead, we observe a very minor additive effect that correlates strictly with a reduction in growth rate. Figure 3C shows that sgs1 mrc1 cells have a significant increase in doubling time even in the absence of genotoxic stress. Since we know that slow growth can reduce checkpoint activation, it seems likely that sgs1 and mrc1 are not additive but are epistatic for Rad53p activation. If true, the mrc1 rad24 double mutant should show a pronounced defect in Rad53p activation, like the sgs1 rad24 strain.
Figure 3.
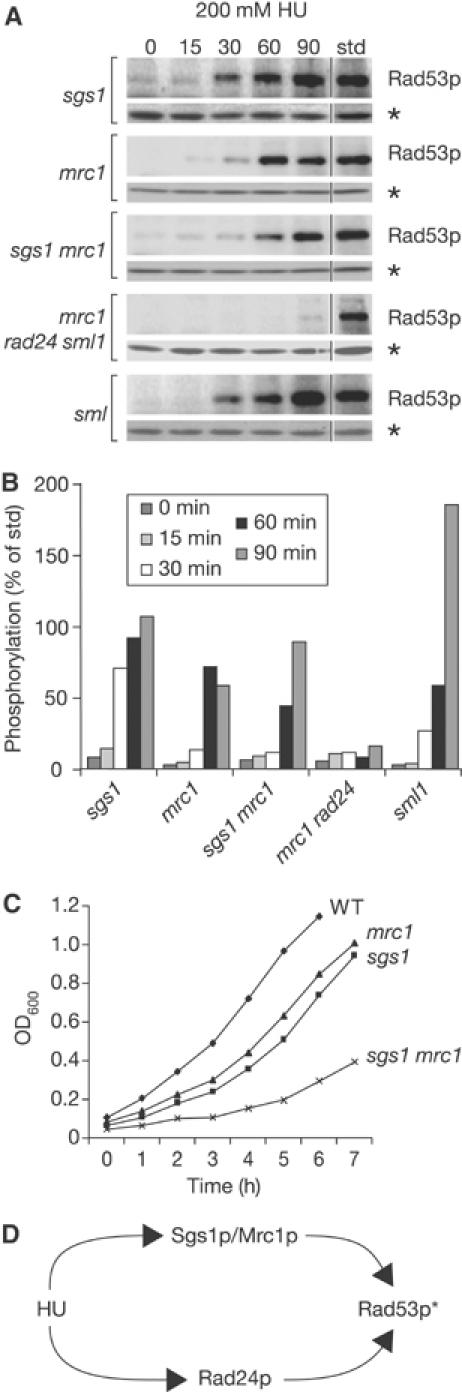
Mrc1p and Sgs1p work on the same pathway for checkpoint activation on HU. (A) Rad53p autophosphorylation was performed as in Figure 1A on isogenic strains GA-2071, GA-2135, GA-2223 and GA-2500. (B) Bar graph showing quantification of Rad53p autophosphorylation as in Figure 1B. (C) Growth curves for the indicated mutants. Cell density (OD600) was measured every hour for 7 h. (D) Schematic summary of the data presented in (A).
To create the mrc1 rad24 double mutant, appropriate rad24 and mrc1 haploid cells were mated. This, however, yielded no viable spores of mrc1 rad24 phenotype (Supplementary Figure S2), consistent with a previous report showing that mrc1 rad9 double mutants are inviable (Alcasabas et al, 2001). Because mrc1 rad9 lethality could be suppressed by upregulation of dNTP pools (i.e. RNR1 overexpression), we attempted to restore viability to the mrc1 rad24 mutant by upregulating Rnr1p through deletion of its repressor SML1 (Supplementary Figure S2). Indeed, the resulting mrc1 rad24 sml1 cells were viable and allowed us to test the epistasis of mrc1 and rad24 mutations with respect to Rad53p activation. As for sgs1 rad24, the double rad24 mrc1 mutation fully abrogates Rad53p activation on HU, unlike either single mutant (Figure 3A and B). Since the sml1 mutation alone has no effect on Rad53p activation, the simplest explanation is that Mrc1p and Sgs1p act in parallel to Rad24p (Figure 3D).
Sgs1p works independently of Rad51p in the checkpoint pathway
Yeast Sgs1p and human BLM protein both interact with Rad51p, a conserved strand-exchange protein (Wu et al, 2001), and the synthetic lethality observed for the coupling of sgs1 with various other mutations can be suppressed by rad51 deletion (Fabre et al, 2002). This places Rad51p upstream of many Sgs1p-mediated functions, and raises the question whether strand invasion events are necessary for Sgs1p-mediated activation of Rad53p. To test this, we monitored Rad53p activation in strains lacking rad51 alone, or coupled with sgs1 and rad24 deletions. We find that the rad51 deletion does not reduce the HU-induced activation of Rad53p, alone or when combined with rad24 or sgs1 (Figure 4A and B). Indeed, rather than reducing Rad53p activation, the rad51 mutation actually elevates the checkpoint response slightly (Figure 4A and B), probably reflecting reduced repair efficiency. In the rad24 background, kinase activation is identical irrespective of RAD51 status, and sgs1 deletion in either rad24 background compromises Rad53p activation. We conclude that on HU, Sgs1p contributes to Rad53p activation independently of Rad51p-mediated recombination events.
Figure 4.
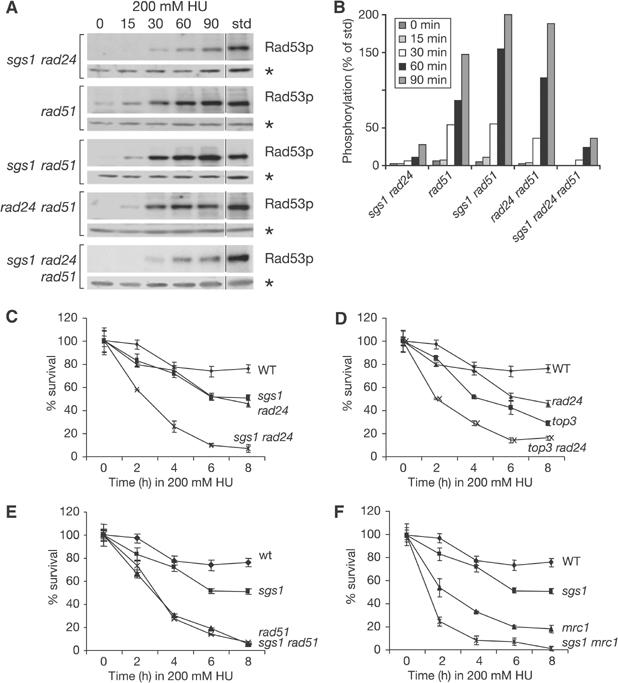
Sgs1p functions independently of Rad51p to achieve Rad53p induction on HU. (A) Rad53p autophosphorylation induced by 0.2 M HU was performed as in Figure 1A on isogenic strains GA-1762, GA-1911, GA-2049 and GA-2051. The sgs1 rad24 panel from Figure 1A is added to facilitate comparison. (B) Bar graph showing quantification of Rad53p autophosphorylation, as in Figure 1B. (C–F) The viability assays were performed on synchronised cultures released into 0.2 M HU for indicated times, as described in Materials and methods, using the following isogenic strains: GA-1020, GA-1748, GA-1761, GA-1762, GA-1799, GA-1911, GA-2047, GA-2056, GA-2135 and GA-2223.
Pathways for efficient recovery from HU arrest do not parallel checkpoint activation
We note that poor recovery after HU arrest is not necessarily correlated with the loss of checkpoint activation. This is documented for various mutations in the recombinational repair machinery, such as rad51, which impair recovery without diminishing checkpoint activation (Paulovich et al, 1997; Stewart et al, 1997; Marchetti et al, 2002). On the other hand, it is known that the checkpoint response contributes to recovery by preventing irreversible breakdown of stalled replication forks (Lopes et al, 2001; Tercero and Diffley, 2001; Osborn and Elledge, 2003; Tercero et al, 2003).
To examine the relative effects and epistatic nature of Sgs1p, Top3p and Rad24p for recovery after exposure to HU, single and double mutants were synchronously released into HU for increasing amounts of time. Unlike its effects on Rad53p activation, the top3 defect, like that of sgs1, is additive with rad24 in survival assays after transient HU exposure (Figure 4C and D), while top3 and sgs1 mutations are epistatic (data not shown). Although other interpretations are possible, the simplest explanation of these results is that Sgs1p and Top3p work together in a process that increases polymerase stability, or else promotes fork restart, which are essential for recovery from HU arrest. Thus the dependence of Sgs1p on Top3p for cellular recovery from HU arrest contrasts with its independent role in checkpoint activation.
Does Rad51p cooperate with Sgs1p in events that allow recovery? The survival of sgs1 and rad51 single mutants was compared with that of the double mutant. The rad51 cells were more severely impaired than those lacking sgs1; yet, combining the two mutations did not further reduce survival rates (Figure 4E). This suggests an epistatic relationship between SGS1 and RAD51. Finally, we examined the relationship between Mrc1p and Sgs1p for survival on HU. Intriguingly, although epistatic for Rad53p activation, the sgs1 and mrc1 defects are additive for the resumption of growth after HU arrest (Figure 4F). We conclude that Sgs1p is engaged in a recovery process that acts parallel to Mrc1p and that relies on the function of Top3p and Rad51p. This pathway is therefore clearly distinct from the role of Sgs1p in checkpoint activation.
Top3p is required for DNA pol ɛ stabilisation at stalled forks
Recovery from HU-induced arrest requires that DNA polymerases are engaged and can resume replication after a prolonged delay. Mutant phenotypes suggest that the maintenance of stable fork structures and suppression of strand breakage are active processes, and quantitative ChIP analysis implicates the helicase activity of Sgs1p in the preservation of DNA polymerases at stalled forks (Cobb et al, 2003). Furthermore, recent data in the Xenopus system suggest that XBlm acts together with XTop3 to suppress strand breaks upon fork arrest (Li et al, 2004). We therefore tested whether DNA polymerase stabilisation requires Top3p and/or recombination events.
Appropriate strains were synchronised with α-factor, released into 0.2 M HU and fixed at the indicated times. The Myc-epitope-tagged DNA pol ɛ was recovered by ChIP, and quantitative real-time PCR was used to analyse the enrichment of fragments at sites within 1 kb of early-firing origin ARS607, and at +4 and +14 kb from the origin (Figure 5A). We also measured the enrichment of DNA pol ɛ at a late-firing origin ARS501 (Figure 5A), which should be detected on HU only if the Rad53p checkpoint is compromised. Our ChIP data are presented as ratios of real-time PCR product accumulation rates, comparing DNA recovered through a specific antibody with the nonspecific association to control beads.
Figure 5.
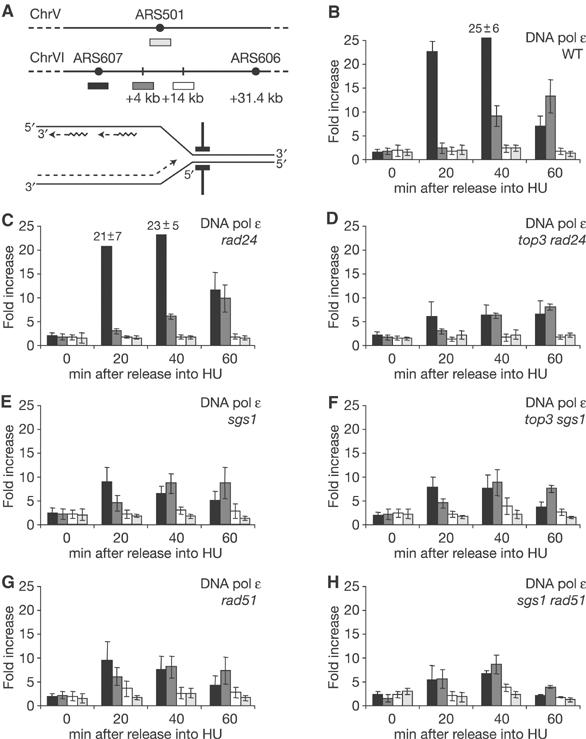
Top3p and Rad51p are required for DNA pol ɛ stabilisation. (A) Primers that amplify genome regions corresponding to an early-firing origin ARS607 (black bars), a nonorigin site at +4 kb (grey bars) and +14 kb (white bars) as well as a late-firing origin ARS501 (light grey bars) are shown. (B–H) ChIP was performed on Myc-tagged DNA pol ɛ as described in Materials and methods, on synchronised isogenic cultures of GA-2448, GA-2449, GA-2455, GA-2456, GA-2457, GA-2458 and GA-2796. The height of the bars represents the ratios of real-time PCR signals as fold increase of immunoprecipitation over beads alone, based on duplicate runs and multiple independent experiments. Standard deviation is calculated from all experiments.
To see if the loss of Top3p influences DNA pol ɛ stabilisation, it was necessary to assess a situation in which both the control and the test strains traverse S phase with similar kinetics. This could be achieved by using the double mutant top3 rad24 to eliminate the slow entry into S phase associated with the top3 mutant. The proper control for the top3 rad24 strain was then the rad24 single mutant. We show that the kinetics of S-phase progression are unchanged by the rad24 mutations, as is the stable association of DNA pol ɛ at forks stalled by HU (Figure 5B and C). In the double mutant top3 rad24, however, despite wild-type kinetics for entry into and progression through S phase, we observe a two- to three-fold reduction in the amount of DNA pol ɛ recovered at ARS607 and the +4 kb adjacent site (Figure 5D). This implicates Top3p in DNA polymerase stabilisation and is consistent with results showing a reduced survival for top3 mutants after exposure to HU (Figure 4D).
To examine whether Sgs1p and Top3p cooperate for DNA pol ɛ stabilisation, we performed ChIP experiments in a top3 sgs1 double mutant and compared this with the single sgs1 and double top3 rad24 mutants. As seen in the top3 sgs1 strain, DNA pol ɛ stabilisation is not further compromised beyond that observed in the sgs1 or top3 rad24 mutants (Figure 5D–F). This suggests an epistatic relationship between SGS1 and TOP3; that is, the two either act as a complex or separately on one pathway. We note that in these mutant backgrounds the late-firing origin ARS501 remains inactive on HU, consistent with our observations that Rad53p kinase is properly activated by either the Rad24p-mediated pathway in the sgs1-deficient strains or by the Sgs1p-mediated pathway in rad24 disrupt strains.
DNA pol ɛ stabilisation requires Rad51p
Stalling of replication forks has been proposed to induce homologous or illegitimate recombination in several organisms, even in the absence of accumulated double-strand breaks (DSBs; reviewed in Michel et al, 2001). Since Sgs1p and Top3p work downstream of Rad51p to resolve HJ, and since their activity as a complex is required for polymerase stability on HU, we next tested whether the contribution of Sgs1p/Top3p involves recombination at stalled forks.
DNA pol ɛ ChIP was performed in rad51 and sgs1 rad51 cells synchronously traversing S phase. In strains lacking Rad51p (Figure 5G), we see a reduction in the amount of DNA pol ɛ recovered at both the ARS607 and the +4 kb fragment, very similar to the pattern obtained in the sgs1 mutant (Figure 5E–G). Importantly, in the sgs1 rad51 double mutant, DNA pol ɛ stabilisation is not significantly lower until the latest time points (Figure 5H). While the latter may indicate some separation of function for Sgs1p and Rad51p, our data suggest that Rad51p helps maintain DNA pol ɛ stably associated at stalled replication forks by working together with the Sgs1/Top3 complex. This is further supported by the fact that the triple top3 sgs1 rad51 mutant shows a similar pattern of pol ɛ stabilisation (Supplementary Figure S3). It remains possible that the effect of rad51 on pol ɛ stabilisation and the epistatic relation between RAD51 and SGS1 in this activity simply reflect protein–protein interactions. We therefore tested whether the C-terminally truncated version of Sgs1p, which lacks the Rad51 interaction domain (Wu et al, 2001), can complement full-length Sgs1p for polymerase stabilisation. We see that the truncated version of Sgs1p behaves as full-length Sgs1p for pol ɛ stabilisation on HU (Supplementary Figure S4), suggesting that the destabilisation observed in the rad51 strain does not stem merely from a loss of Sgs1p–Rad51p interaction.
Sgs1p and Mrc1p act synergistically to stabilise DNA pol ɛ at stalled forks
Given the synergistic effect for recovery after HU treatment in the sgs1 mrc1 double mutant (Figure 4F), one might predict that Sgs1p and Mrc1p, unlike Sgs1p and Rad51p, work on parallel pathways to ensure stabilisation of DNA pol ɛ at HU-arrested forks. This was tested in the appropriate mutants on HU. In contrast to the wild-type control, yet similar to the sgs1 null allele, we see a two- to three-fold reduction in the amount of DNA pol ɛ recovered at ARS607 in the mrc1 mutant (Figure 6A–C). However, we recover more DNA pol ɛ at the +4 kb fragment at all time points (Figure 6C), which is consistent with results from chromosome-wide ChIP in mrc1 cells showing that DNA polymerases move along the chromosome beyond sites of nucleotide incorporation (Katou et al, 2003). We also note that the late-firing origin ARS501 is activated in mrc1 cells, unlike sgs1 cells, reflecting the more pronounced drop in Rad53p kinase activation associated with mrc1 deficiency (Figure 3A).
Figure 6.
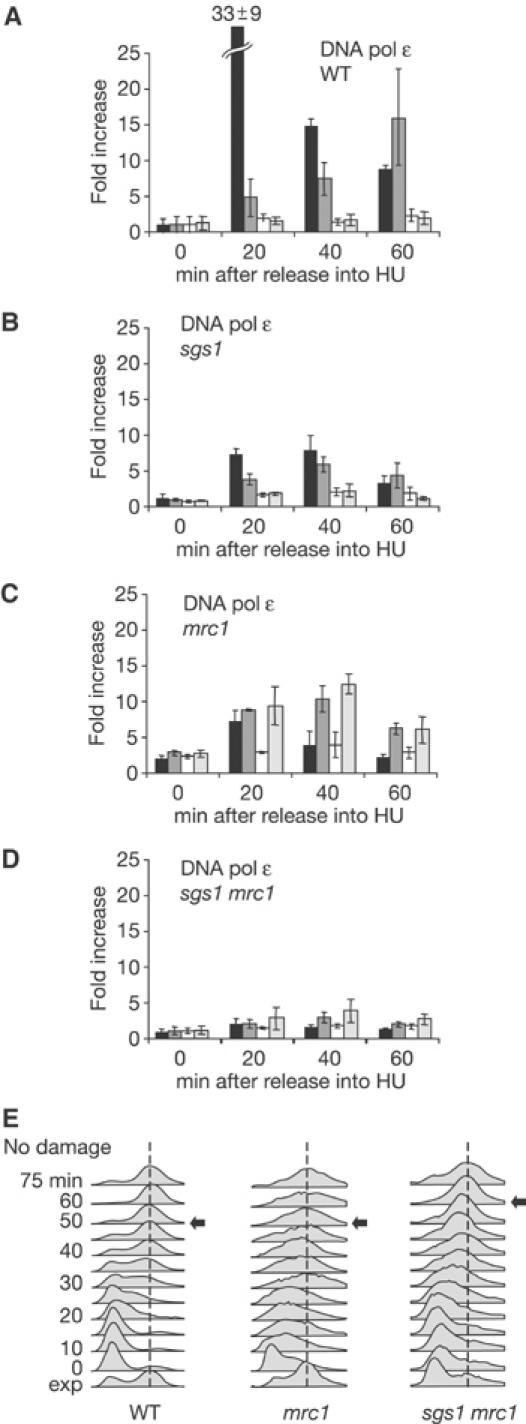
Sgs1p and Mrc1p act synergistically to stabilise DNA pol ɛ at stalled forks. (A–D) ChIP was performed on Myc-tagged DNA pol ɛ as described in Figure 5 on synchronised isogenic strains GA-2448, GA-2449, GA-2451 and GA-2452. (E) FACS analysis during S-phase progression in the absence of damage for GA-1020, GA-2135 and GA-2223.
In the double sgs1 mrc1 mutant, the destabilisation of DNA pol ɛ is clearly additive: very low levels of polymerase are recovered at ARS607 throughout the time course (Figure 6D). Again, it is not an artefact of cell cycle progression: FACS profiles for mrc1 and sgs1 mrc1 cells show similar rates of progression from G1 to G2 in the absence of damage, and similar budding indices (Figure 6E; data not shown), and, neither mutation affects origin firing efficiency (Cobb et al, 2003; Katou et al, 2003). This additive effect on DNA pol ɛ stabilisation at stalled forks suggests that Sgs1p and Mrc1p act on different pathways in the presence of HU, contributing to fork integrity and cell survival in an additive fashion.
Discussion
In this study, we have shown using a combined genetic and biochemical approach that the budding yeast RecQ helicase Sgs1p suppresses genomic instability resulting from HU-induced fork arrest in two independent ways. Extending earlier work, we show that Sgs1p requires the strand-cleaving enzyme Top3p to help stabilise DNA pol ɛ at stalled replication forks. The complex also requires the RecQ helicase activity, and acts epistatic to Rad51p, suggesting that the complex may reverse nonphysiological pairing events at stalled forks. This would restore or maintain single-stranded template, which is an essential prerequisite for maintaining engaged replication enzymes. We see a strong correlation between replisome maintenance and successful resumption of the cell cycle once HU is removed, and we predict that the stability of polymerases on HU will correlate with the suppression of fork-associated strand breaks. In this context, the role we propose for Sgs1p is conceptually similar to that ascribed to the mammalian Rad51-binding protein BRCA2 (Lomonosov et al, 2003). When cells are exposed to HU, fork-associated strand breaks are significantly more frequent in the absence of BRCA2, even though activation of the checkpoint kinases, CHK1 and CHK2, is normal. Similar to this, we find no consistent correlation between the loss of DNA polymerase stability in sgs1, top3 or rad51 mutants and an impaired activation of Rad53p. Indeed, DNA polymerases remain stably bound to HU-arrested forks both in rad24- (Figure 5C) and in rad53 sml1-deficient strains despite the strong reduction in checkpoint activation (Aparicio et al, 1999; Cobb et al, 2003). Similarly, the helicase Srs2p contributes to Rad53p activation in response to strand breaks (Liberi et al, 2000) but not to polymerase stability in the presence of HU (data not shown).
We further show that the contribution that Sgs1p makes to Rad53p activation at stalled forks is a mechanistically distinct pathway. Although the reduced checkpoint signal is only evident when a break-induced pathway is compromised (i.e. rad24-deficient cells), the Sgs1p-dependent activation occurs in the absence of Top3p and Rad51p, and with no need for RecQ helicase activity. We consider a direct recruitment mechanism to be the most likely, given the strong interaction between Sgs1p and Rad53p FHA1 domain (Figure 2). In vivo studies reinforce this conclusion by showing that mutation of the Rad53p FHA1 domain compromises its ability to respond to fork arrest, although it does not affect the G2/M Rad9-dependent damage response (Schwartz et al, 2003).
By being bound at the stalled fork, Sgs1p may help attract Rad53p, such that Mrc1p can contribute to its activation. This function of Sgs1p is independent of and parallel to Rad24p/9-1-1 (Rad17p, Mec3p, Ddc1p; Figures 4 and 6). We propose that Rad24p acts primarily at forks that have incurred breakage, while Sgs1p may help activate Rad53p at stalled but nonbroken forks. We cannot rule out, however, that both pathways recognise stalled forks as well as breaks, and that they are simply redundant. Arguing against simply redundancy is the fact that the checkpoint response to MMS (which induces replication-dependent strand breaks) relies almost entirely on Rad24p and not on Sgs1p (LB and SMG, unpublished observations). The different modes of checkpoint activation are also manifested when chronic (low level) and acute (high level) HU arrest responses are compared (Schollaert et al, 2004). We propose that this may also reflect breakage versus stalled fork signals.
Whereas sgs1 is epistatic to mrc1 for Rad53p activation, the two contribute to polymerase stabilisation on separate pathways. Their different modes of action are underscored by the observation that polymerases progress beyond the initial site of stalling in mrc1, but not in sgs1, strains (see Supplementary Figure S5; Katou et al, 2003). These pathways are summarised in Figure 7.
Figure 7.
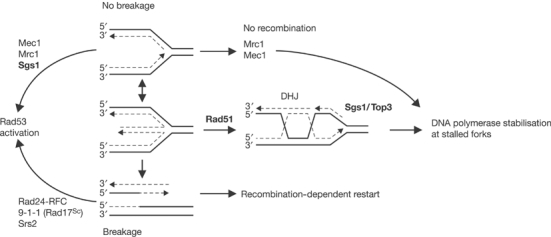
Different pathways use the RecQ helicase in response to replication stalling. Stalled replication forks activate Rad53p kinase in the absence of strand breaks in an Sgs1p-, Mrc1- and Mec1-dependent pathway that is independent of Top3p and Rad51p. Forks are stabilised by two mechanisms, including one that requires Rad51p and the Sgs1p/Top3p complex. We proposed that the Rad51p-dependent pathway leads to the formation of a four-way DNA junction due to fork reversal. DSBs will form at some of the stalled replication forks, and will activate the intra-S damage pathway (breakage pathway) for checkpoint activation, which relies on Rad24p and the 9-1-1 complex.
Efficient polymerase stabilisation correlates with efficient recovery from replicational stress
We see a strong correlation of cellular recovery from HU exposure with activities that ensure the initial stability of polymerases at forks, that is, during the first hour. These critical events occur prior to Rad53p activation. Support of this interpretation comes from the demonstration that Sgs1p acts together with Top3p to suppress both spontaneous and induced gross chromosomal rearrangements (Myung et al, 2001). Furthermore, XBlm and XTop3 act as a complex to suppress the accumulation of strand breaks in both normal and aphidicolin-impaired replication events, without affecting checkpoint kinase activation (Li et al, 2004).
While Rad53p-mediated checkpoint responses also contribute to cell survival in the face of genotoxic stress (Lopes et al, 2001; Tercero and Diffley, 2001, Tercero et al, 2003), we place events ensuing from Rad53p activation downstream of the DNA polymerase stabilisation that we monitor. Indeed, the accumulation of collapsed fork structures in rad53 mutants is observed after 2–3 h on HU, rather than during the first hour (Tercero and Diffley, 2001; Sogo et al, 2002). Although we rule out a role for Rad53p, the ATR homologue Mec1p is recruited to stalled forks and is essential for DNA pol ɛ stabilisation (Cobb et al, 2003).
Regulating recombination at stalled forks: essential for fork restart and fork protection
The impact of Rad51p on stalled fork stability was unexpected, yet it sheds light on the mechanism of Sgs1p/Top3p-dependent polymerase stabilisation. It has been suggested that the open helix at stalled replication forks can reanneal, reversing fork movement to form four-way DNA junction structures (Michel et al, 2001). These have been detected by electron microscopy (Sogo et al, 2002), and in bacteria, either RecG helicase or RecA can mediate this process (McGlynn and Lloyd, 2001; Robu et al, 2001). The resulting four-way junction can be resolved either with cleavage or without. Cleavage requires a helicase–endonuclease complex such as RuvABC in Escherichia coli, which then produces a DSB. The noncleaved pathway involves homologous recombination and formation of a double HJ (Figure 7). Together with Sgs1p/Top3p, Rad51p may promote the resolution of these structures without cleavage, thereby protecting replication forks from DSBs.
Cellular recovery or survival after acute HU treatment is strongly dependent on Rad51p, even though Rad51p has no role in checkpoint activation. No additive lethality is observed in the sgs1 rad51 strain, suggesting again that Sgs1p and Rad51p work together. Yet Rad51p has additional functions that are likely to reflect its role in homologous recombination, which in bacteria helps reinitiate replication at broken forks (discussed in Merrill and Holm, 1999; Michel et al, 2001). Here, Sgs1p/Top3p activity may avoid inappropriate exchanges mediated by Rad51p during a restart process (Ira et al, 2003). We conclude that Rad51p must be tightly regulated during DNA replication, both to avoid unfavourable types of DNA exchange and to provide a means to facilitate recombination-mediated repair. The control of recombination events during replication, a task partly attributable to Sgs1p, will indeed be key for maintaining genomic integrity in eukaryotic cells.
Materials and methods
Yeast strains and cell cultures
All strains used (Table I) are W303 derived, except the strain used for the Rad53p and Sgs1p immunoprecipitation experiment. Genomic deletions of SGS1 and RAD51 were performed with plasmid-borne sgs1-3::TRP1 (Lu et al, 1996) and rad51::URA3 (Aboussekhra et al, 1992), while all others are complete deletions using pFA6a PCR-based cassettes (Longtine et al, 1998), verified by PCR and phenotypic analyses. GA-2057 bearing empty plasmid pRS415, or pRS415 with full-length SGS1 (SGS1-FL) or a helicase-deficient allele sgs1-hd (K704A; Lu et al, 1996) were grown on selective media and then synchronised with α-factor in YPAD, prior to ISA described below. All other culture conditions were in YPAD at 30°C.
Table 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| GA-180 | MATa, ade2-1, trp1-1, his3-11, -15, ura3-1, leu2-3, -112, can1-100 | R Rothstein (W303-1A) |
| GA-181 | MATα, ade2-1, trp1-1, his3-11, -15, ura3-1, leu2-3, -112, can1-100 | R Rothstein (W303-1B) |
| GA-1020 | MATa, ade2-1, trp1-1, his3-11, -15, ura3-1, leu2-3, -112, can1-100, pep4∷LEU2 | R Rothstein (W303-1A) |
| GA-1142 | MATa, leu2, trp1, ura3-52, lys, SGS1-13MYC∷KanMX6, RAD53-3HA∷KanMX6 | This study |
| GA-1699 | GA-1020 with SGS1-13Myc∷HIS3 | Cobb et al, 2003 |
| GA-1748 | GA-180 with rad24∷TRP1 | This study |
| GA-1761 | GA-1020 with sgs1-3∷TRP1 | This study |
| GA-1762 | GA-1020 with rad51∷HIS3 | This study |
| GA-1799 | GA-1020 with top3∷HIS3 | This study |
| GA-1800 | GA-1020 with top3∷HIS3, sgs1-3∷TRP1 | This study |
| GA-1801 | GA-1020 with TOP3-13Myc∷HIS3 | This study |
| GA-1911 | GA-1020 with sgs1-3∷TRP1, rad51∷URA3 | This study |
| GA-1913 | GA-1020 with TOP3-13Myc∷HIS3, sgs1-3∷TRP1 | This study |
| GA-2047 | GA-1020 with top3∷HIS3, rad24∷URA3 | This study |
| GA-2049 | GA-1020 with sgs1-3∷TRP1, rad24∷URA3, rad51∷HIS3 | This study |
| GA-2051 | GA-1020 with rad24∷URA3, rad51∷HIS3 | This study |
| GA-2056 | GA-1020 with sgs1-3∷TRP1, rad24∷URA3 | This study |
| GA-2057 | GA-181 with sgs1-3∷TRP1, rad24∷URA3 | This study |
| GA-2060 | GA-1020 with top3∷HIS3, sgs1-3∷TRP1, rad24∷URA3 | This study |
| GA-2071 | GA-180 with sml∷KanMX6 | This study |
| GA-2127 | GA-1020 with sgs1-3∷TRP1, rad9∷HIS3 | This study |
| GA-2135 | GA-180 with mrc1-2∷HIS3 | S Elledge (Y1122) |
| GA-2223 | GA-1020 with sgs1-3∷TRP1, mrc1-2∷HIS3 | This study |
| GA-2448 | GA-1020 with POL2-13Myc∷KanMX6 | This study |
| GA-2449 | GA-1020 with sgs1-3∷TRP1, POL2-13Myc∷KanMX6 | This study |
| GA-2450 | GA-180 with sgs1-3∷TRP1, POL2-13Myc∷KanMX6 | This study |
| GA-2451 | GA-1020 with mrc1-2∷HIS3, POL2-13Myc∷KanMX6 | This study |
| GA-2452 | GA-1020 with sgs1-3∷TRP1, mrc1-2∷HIS3, POL2-13Myc∷KanMX6 | This study |
| GA-2455 | GA-180 with rad24∷URA3, POL2-13Myc∷KanMX6 | This study |
| GA-2456 | GA-180 with top3∷HIS3, rad24∷URA3, POL2-13Myc∷KanMX6 | This study |
| GA-2457 | GA-1020 with rad51∷URA3, POL2-13Myc∷KanMX6 | This study |
| GA-2458 | GA-180 with sgs1-3∷TRP1, rad51∷URA3, POL2-13Myc∷KanMX6 | This study |
| GA-2495 | GA-181 with rad24∷TRP1, sml1∷KanMx6 | This study |
| GA-2500 | GA-180 with rad24∷TRP1, sml1∷KanMx6, mrc1-2∷HIS3 | This study |
| GA-2504 | GA-181 with rad51∷URA3 | This study |
| GA-2796 | GA-1020 with sgs1-3∷TRP1, top3∷HIS3, POL2-13Myc∷KanMX6 | This study |
| GA-3033 | GA-180 with sgs1-3∷TRP1, top3∷HIS3, rad51∷URA3, POL2-13Myc∷KanMX6 | This study |
In situ autophosphorylation assay
Cultures at 0.5 × 107 cells/ml were synchronised with α-factor (Lipal Biochem, Zürich, Switzerland) during one generation, followed by release into prewarmed YPAD+0.2 M HU. All steps of ISA are as described (Pellicioli et al, 1999) except that 5 μCi/ml [γ-32P]ATP was used. For every sample, protein concentration was determined by Coomassie blue prior to equal loading on 10% SDS–polyacrylamide gels along with 5 μl of a standard (standard, std), containing a known amount of activated Rad53p. Dried filters were exposed for equal times on a Biorad Phosphorimager. After exposure, filters were reprobed with rat anti-RnaseH42 (kindly provided by U Wintersberger, Vienna, Austria) to check loading and allow comparison among different gels and mutants. By normalising exposures to have comparable autophosphorylation standard signal, one can directly compare Rad53p activation kinetics from gel to gel. All experiments were performed 2–3 times with similar results. Quantification of the Rad53p autophosphorylation was performed using the Quantity One software, by normalising to the loading control (Rnase42) and the kinase standard (std), which was taken as 100%. The quantifications shown are average over 2–3 experiments with standard derivations of 5–15%.
Survival and drop assays
Synchronised cultures (0.5 × 107 cells/ml) were released into fresh YPAD+0.2 M HU for indicated times, and survival ratios were determined as described (Frei and Gasser, 2000). Drop assays were a 1:5 dilution series of uniformly diluted cultures on YPAD plates.
Two-hybrid interaction
The two-hybrid analyses were performed as described previously (Aushubel et al, 1994). The lacZ reporter pSH1834, the bait and the prey plasmids were transformed into EGY191. Exponentially growing, glucose-depleted cells were exposed to 2% galactose for 6 h to induce the fusion proteins, and protein–protein interactions were detected by the quantitative β-galactosidase assay for permeabilised cells (Adams et al, 1997); β-galactosidase units are as defined therein. Four independent transformants were analysed for each interaction with a range of error of ∼10%.
Co-immunoprecipitation and ChIP
Co-immunoprecipitation was performed with GA-1142 cells and HA-coupled Dynabeads M-450 (Dynal AS, Norway) as described (Cobb et al, 2003). Stringent washes were performed (10 mM Tris pH 8.0, 250 mM NaCl, 1 mM EDTA, 1% NP-40, 2.5 mM deoxycholate). ChIP was performed as described by Cobb et al (2003) with the same sets of primers, and DNA was quantified by real-time PCR using the Perkin-Elmer ABI Prism 7700 Sequence Detector System and software. Dynabeads without antibody were used as background controls. ChIP data are averaged over three independent experiments with real-time PCR performed in duplicate (standard deviation is shown by error bars). The height of the bars represents the ratio of the difference of signal accumulation rates for immunoprecipitation versus input, divided by the difference in rates for Dynabeads alone.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Data
Acknowledgments
The Gasser laboratory acknowledges the Swiss Cancer League, the Swiss National Science Foundation, European RTN Checkpoints and Cancer and fellowships from the American Cancer Society to JAC (PF-01-142-01-CCG) and from the Danish Cancer Society to LB for support (DP00060). We thank colleagues in the Gasser laboratory for helpful discussions, U Wintersberger (Vienna, Austria) for antiserum and N Roggli for excellent artistic support.
References
- Aboussekhra A, Chanet R, Adjiri A, Fabre F (1992) Semidominant suppressors of Srs2 helicase mutations of S. cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol 12: 3224–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A, Gottschling DE, Kaiser CA, Stearns T (1997) Methods in Yeast Genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3: 958–965 [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Stout AM, Bell SP (1999) Differential assembly of Cdc45p and DNA polymerases at early and late origins of DNA replication. Proc Natl Acad Sci USA 96: 9130–9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aushubel FM, Brent R, Kinston R, Moore D, Seidman JJ, Smith J, Struhl K (1994) Current Protocols in Molecular Biology. New York: John Wiley and Sons [Google Scholar]
- Chakraverty RK, Kearsey JM, Oakley TJ, Grenon M, de La Torre Ruiz MA, Lowndes NF, Hickson ID (2001) Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol Cell Biol 21: 7150–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilisation at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S (2002) Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA 99: 16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei C, Gasser SM (2000) The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev 14: 81–96 [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14: 8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin A, Wang SW, Toda T, Norbury C, Hickson ID (1999) Topoisomerase III is essential for accurate nuclear division in S. pombe. Nucleic Acids Res 27: 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Khakhar RR, Cobb JA, Bjergbaek L, Hickson ID, Gasser SM (2003) RecQ helicases: multiple roles in genome maintenance. Trends Cell Biol 13: 493–501 [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Putnam CD, Myung K (2002) Maintenance of genome stability in S. cerevisiae. Science 297: 552–557 [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (2000) Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 6: 839–849 [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2003) Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell 11: 329–340 [DOI] [PubMed] [Google Scholar]
- Li W, Kim SM, Lee J, Dunphy WG (2004) Absence of BLM leads to accumulation of chromosomal DNA breaks during both unperturbed and disrupted S phases. J Cell Biol 165: 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang JC (1998) Mammalian DNA topoisomerase IIIα is essential in early embryogenesis. Proc Natl Acad Sci USA 95: 1010–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Chiolo I, Pellicioli A, Lopes M, Plevani P, Muzi-Falconi M, Foiani M (2000) Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. EMBO J 19: 5027–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR (2003) Stabilisation of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17: 3017–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in S. cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilises stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Lu JA, Mullen JR, Brill SJ, Kleff S, Romeo AM, Sternglanz R (1996) Human homologues of yeast helicase. Nature 383: 678–679 [DOI] [PubMed] [Google Scholar]
- Marchetti MA, Kumar S, Hartsuiker E, Maftahi M, Carr AM, Freyer GA, Burhans WC, Huberman JA (2002) A single unbranched S-phase DNA damage and replication fork blockage checkpoint pathway. Proc Natl Acad Sci USA 99: 7472–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG (2001) Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci USA 98: 8227–8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill BJ, Holm C (1999) A requirement for recombinational repair in S. cerevisiae is caused by DNA replication defects of mec1 mutants. Genetics 153: 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V (2001) Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci USA 98: 8181–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P, Hickson ID (2001) DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum Mol Genet 10: 741–746 [DOI] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Brill SJ (2000) Bipartite structure of the SGS1 DNA helicase in S. cerevisiae. Genetics 154: 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JM, Lindsay HD, Munday CA, Carr AM (1997) Role of S. pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol Cell Biol 17: 6868–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, Kolodner RD (2001) SGS1, the S. cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 27: 113–116 [DOI] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA (2002) Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 36: 617–656 [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17: 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH (1997) RAD9, RAD17, and RAD24 are required for S phase regulation in S. cerevisiae in response to DNA damage. Genetics 145: 45–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu ME, Inman RB, Cox MM (2001) RecA protein promotes the regression of stalled replication forks in vitro. Proc Natl Acad Sci USA 98: 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schollaert KL, Poisson JM, Searle JS, Schwanekamp JA, Tomlinson CR, Sanchez Y (2004) A role for S. cerevisiae Chk1p in the response to replication blocks. Mol Biol Cell 15: 4051–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MF, Lee SJ, Duong JK, Eminaga S, Stern DF (2003) FHA domain-mediated DNA checkpoint regulation of Rad53. Cell Cycle 2: 384–396 [PubMed] [Google Scholar]
- Shimada K, Pasero P, Gasser SM (2002) ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16: 3236–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Mills K, Guarente L (1997) Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science 277: 1313–1316 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Stewart E, Chapman CR, Al-Khodairy F, Carr AM, Enoch T (1997) rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J 16: 2682–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58: 409–419 [DOI] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ (1996) Sgs1, a homologue of the Blooms and Werners syndrome genes, is required for maintenance of genome stability in S. cerevisiae. Genetics 144: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Davies SL, Levitt NC, Hickson ID (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem 276: 19375–19381 [DOI] [PubMed] [Google Scholar]
- Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID (2000) The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem 275: 9636–9644 [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Data