

Abstract
Senescence, the molecular program that limits the finite proliferative potential of a cell, acts as an important barrier to protect the body from cancer. Techniques for measuring transcriptome changes and for modulating their expression suggest that it may be possible to dissect the transcriptional networks underlying complex cellular processes. HMF3A cells are conditionally immortalized human mammary fibroblasts that can be induced to undergo coordinated senescence. Here, we used these cells in conjunction with microarrays, RNA interference, and in silico promoter analysis to promote the dissection of the transcriptional networks responsible for regulating cellular senescence. We first identified changes in the transcriptome when HMF3A cells undergo senescence and then compared them with those observed upon replicative senescence in primary human mammary fibroblasts. In addition to DUSP1 and known p53 and E2F targets, a number of genes such as PHLDA1, NR4A3, and a novel splice variant of STAC were implicated in senescence. Their role in senescence was then analyzed by RNA silencing followed by microarray analysis. In silico promoter analysis of all differential genes predicted that nuclear factor-κB and C/EBP transcription factors are activated upon senescence, and we confirmed this by electrophoretic mobility shift assay. The results suggest a putative signaling network for cellular senescence.
INTRODUCTION
Fibroblasts isolated from normal human tissue have a limited replicative potential in vitro. After a certain number of divisions, they become “senescent” and are irreversibly arrested with a characteristic enlarged and irregular morphology (Bayreuther et al., 1988). Senescence also can be prematurely induced by DNA damage (Robles and Adami, 1998), oxidative stress (Chen et al., 1998), and some oncogenes (Serrano et al., 1997). Progressive shortening of the telomeres or changes in telomeric structure are thought to trigger replicative senescence in human cells (Allsopp et al., 1992; Masutomi et al., 2003). Although some studies have found that the catalytic subunit of human telomerase (hTERT) alone can apparently immortalize several different types of somatic cells (Bodnar et al., 1998), by itself it was not sufficient to immortalize adult human mammary fibroblasts (O'Hare et al., 2001). Similar findings have been shown for human epithelial cells (Kiyono et al., 1998), myoblasts (Di Donna et al., 2003), and melanocytes (Sviderskaya et al., 2003).
The simian virus 40 (SV40) large tumor antigen (LT) was required in addition to hTERT to immortalize human mammary fibroblasts (O'Hare et al., 2001). LT is a viral oncoprotein that binds and inhibits the activity of several proteins, including p53 and Rb (Ali and DeCaprio, 2001). Rb and its family members p107 and p130 affect the activation of the E2F family of transcription factors (DeGregori, 2002). However, inhibition of Rb and p53 is insufficient for the immortalization ability of LT, even in rodent cells that do not require hTERT (Powell et al., 1999; Sachsenmeier and Pipas, 2001).
Senescence involves proteins such as Rb, p53, and the kinase p38 (Davis et al., 2003; Iwasa et al., 2003). However, p53 and Rb also are involved in nonpermanent arrest such as that induced upon growth factor deprivation (Itahana et al., 2002), and it is not yet clear what differentiates the irreversible and reversible processes of cell cycle arrest.
The advent of DNA microarrays has allowed for a systematic examination of the effects on the transcriptome of a number of diverse conditions such as starvation (Iyer et al., 1999), stress (Park et al., 2002), immortalization with oncogenes (Schwarze et al., 2002), telomerase inhibition (Damm et al., 2001), and serial passaging of cells through to replicative senescence (Shelton et al., 1999; Zhang et al., 2003). Others have used microarrays to identify the targets of transcription factors such as p53 and E2F (Ishida et al., 2001; Wang et al., 2001). Currently, a major challenge is to identify the roles of the induced and/or repressed genes and to decipher the signaling networks that regulate the different groups of genes in cellular processes such as senescence.
By using a combination of mathematical modeling and experimental perturbation of selected genes, it is hoped that the architecture of the cellular gene regulatory networks can be deciphered (Tegner et al., 2003). Microarray studies have been used in conjunction with gene deletions in yeast (Wagner, 2002) in an attempt to do this. Another approach has been to examine the upstream sequences of genes coregulated in microarray studies to find common transcription factor binding sites (Tavazoie et al., 1999). The discovery that short 20-mer double-stranded RNA oligonucleotides can target mRNA for degradation in a gene-specific manner (Elbashir et al., 2001) provides a way of studying the contributions of a gene to a particular process. This is further facilitated by the availability of vectors that allow for permanent production of the double-strand RNAs within the cell (Brummelkamp et al., 2002b).
Cellular senescence is commonly studied by the serial cultivation of primary cells. However, studies with human cells are complicated by the genetic and epigenetic variation that can exist between different donors as well as phenotypic differences between cells in the original isolates. Furthermore, in such cultures there can be growth-arrested cells in early passages and dividing cells toward the later passages, and this proliferative heterogeneity may mask small changes in gene expression. To overcome some of these problems, we have a developed a cell system in which cells can be induced to undergo senescence synchronously. This was done by retrovirally introducing hTERT and a temperature-sensitive non-DNA binding mutant (U19tsA58) of SV40 LT into adult human mammary fibroblasts (O'Hare et al., 2001).
Four fibroblast lines (HMF3A, B, C, and D) were created from the same pool of donor cells, differing only in the order and timing of the sequential transductions. The phenotypes of the lines have been examined at intervals during 3 yr of continuous culture. Each has been longitudinally profiled for biological parameters that included morphology, temperature sensitivity, growth rates, growth factor and serum dependence, sensitivity to apoptosis, and anchorage-independent growth. An in depth analysis of their karyotypes was carried out concurrently (Fauth et al., 2004). In this context, the line HMF3A was the most “normal” of the lines in terms of overall phenotypic stability, remaining stringently temperature sensitive and showing no signs of transformation in >300 population doublings postexplantation.
Here, we present a study of the gene expression changes that occur upon senescence in HMF3A cells and how they relate to those seen in primary cells serially passaged through to senescence and in cells reversibly arrested by serum deprivation. Furthermore, using RNA interference (RNAi), we examine the role in senescence of several genes identified in the study. We also investigated in silico the proportional occurrence of known transcription factor binding sites in the coregulated genes, to determine whether this approach can be used to ultimately build a computational model of cellular senescence.
MATERIALS AND METHODS
Antibodies
Antibodies used were IL-1β, MKP-1, ID1, p21, p107, p130, GADD34, G-CSF, MEIS2, C/EBPα (C-18), C/EBPγ (E-19), and C/EBPβ (H-7) (Santa Cruz Biotechnology, Santa Cruz, CA); BUB1B (Exalpha Biologicals, Maynard, MA), HMGB2 (BD Biosciences PharMingen, San Diego, CA); and LT and p53 (PAb416 and 421(Harlow et al., 1981). Double-stranded oligonucleotides with binding site for nuclear factor-κB (NF-κB), AP-1 (both from Promega, Madison, WI), and Myc and C/EBP (Santa Cruz Biotechnology) were used in electrophoretic mobility shift assay (EMSA). Reverse transcription-polymerase chain reaction (RT-PCR), Western blotting, and EMSA were performed as published previously (Lassar et al., 1991; Benvenuti et al., 2002). The RT reactions were checked by PCR with primers for glyceraldehydes-3-phosphate dehydrogenase to check that levels were equal. ImageJ (http://rsb.info.nih.gov/ij/) was used for densitometry.
Retroviral Generation of Cell Cultures
Generation of the HMF3A culture from primary breast fibroblasts was described previously (O'Hare et al., 2001). The same primary fibroblasts were infected with the hTERT retrovirus in the pBabeHygro vector (at passage 4) and with the wild-type U19 LT in the pZipNeoSV(X)1 vector (at passage 6) to produce the HMF3Dwt cells.
Annealed 64mer-DNA oligonucleotides (Sigma-Genosys, Haverhill, United Kingdom) were ligated into pSUPERretro as described previously (Brummelkamp et al., 2002b). The regions targeted for silencing were GATGTTCTCTTGCCACTTG-STACβ, CTACGAACTCAAGCCTTCC-NR4A3, CGACATCAACATCCTGCGC-GADD45A, CACCACTGGTTTCCCGAAA-BTG2, AGTTACCAGGGCCCTTGAT-BTG3, TCCTGCCCTTTCTGTACCT-DUSP1, and GTGTTGCATCCTCACCGAG-PHLDA1.
HMF3A, HMF3Dwt, and the cultures derived from them were routinely cultured at 33°C, 5% CO2 in DMEM supplemented with 10% (vol/vol) fetal calf serum (FCS) and antibiotics. For the experiments presented here, cells were used between passages 19 and 63. Quiescence of HMF3A cells was achieved by washing twice in phosphate-buffered saline and incubating them in DMEM with 0.25% (vol/vol) FCS for 1 wk at which time they were confluent. For neutralizing transforming growth factor-β (TGF-β), anti-TGF-β antibody (0.5 μg/ml; R&D Systems Europe, Oxford, United Kingdom) was added to fresh medium every 3 d.
RNA Extraction and cDNA Microarrays
RNA was extracted using TRIzol reagent (Invitrogen, Paisley, Scotland) as per the manufacturer's instructions. cDNA was labeled with either Cy3 or Cy5 as per Sanger protocol 5 (www.sanger.ac.uk/Projects/Microarrays/arraylab/methods.shtml). The labeled cDNA was hybridized to Hver 1.1.1, 1.2.1 or 1.3.1 DNA microarrays (Wellcome Trust Sanger Institute, Cambridge, United Kingdom) as per Sanger protocol 6.1 or 6.2. Reciprocal (flip-fluor) labelings were always performed. These arrays have just under 10 000 DNA elements (because some genes are represented by two or more different elements, ∼6000 different genes are represented). At least four arrays were hybridized for each comparison with at least two biological replicates (except where primary cells were directly compared with immortalized cells). The two channels were normalized by Lowess normalization by using GeneSpring version 5 (Silicon Genetics, Redwood City, CA). Raw and normalized data are deposited in the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). Lists of regulated genes were made using SAM (Tusher et al., 2001). Unless otherwise stated a delta N value was chosen so that the median number of false positives was ∼5% of the total number of significant genes. Except for the RNAi lists, gene lists from SAM were filtered to have cut-off average ratios greater than 1.3, and thus the reported level of false positives are actually overstated. Gene clustering was performed using standard correlation in GeneSpring version 6.
EnsMART was used to convert Sanger clone IDs to GenBank accession numbers, and EZ-retrieve (Zhang et al., 2002) was used to detect the proportion of binding sites for the different transcription factors in the region 750 bp upstream and 250 bp downstream of the transcription start site. Duplicate genes were removed from these lists before submission to EZ-retrieve. Unless otherwise stated a cut-off score of 85 was used when searching for transcription factor binding sites. Accession numbers for the mouse and rat homologues for these genes also were obtained via EnsMART and submitted to EZ-retrieve.
RESULTS AND DISCUSSION
LT Inactivation in Conditionally Immortalized Cells
HMF3A cells cease proliferating by 4 d at 39°C, and after 7 d the cells are irreversibly arrested. We directly compared the gene expression profiles of cells at 33°C and cells shifted to 39°C for 7 d (10 arrays, 3 biological replicates; Figure 1). To eliminate genes that change in response to the temperature shift, we compared the profiles of HMF3Dwt cells at 33°C and those shifted up to 39°C for 7 d (4 arrays, 2 biological replicates; Figure 1). Because HMF3Dwt cells were immortalized with wild-type U19 LT antigen, they proliferate at both temperatures. They actually proliferated slightly faster at 39°C, but upon shift up to 39°C from 33°C they showed an increase in p21 and other genes associated with growth inhibition (Supplemental Table 1). Because some of these genes, such as p21 regulate cell proliferation (Lodygin et al., 2002), only those genes whose change in expression upon temperature shift in HMF3Dwt cells was 0.77-fold or greater than their change in HMF3A cells (for up-regulated genes) or 1.3-fold or less (for down-regulated genes) were excluded from the list.
Figure 1.
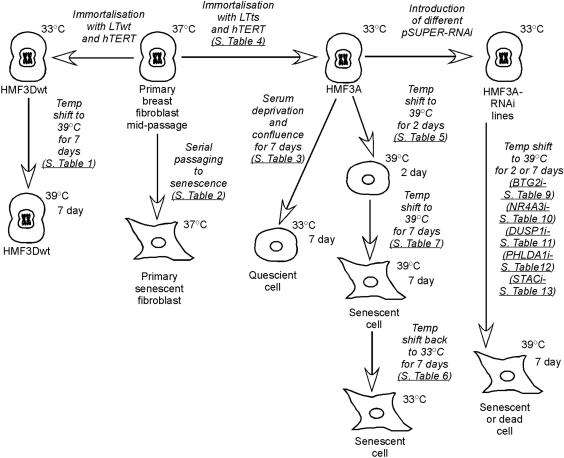
Outline of the experimental conditions used for microarray comparisons. Primary breast fibroblasts (HMF3) were either immortalized with the catalytic subunit of human telomerase (hTERT) and wild-type LT antigen (LTwt), conditionally immortalized with hTERT and temperature-sensitive LT antigen (LTts), or passaged through to senescence. The HMF3Dwt cells have LTwt and continue to proliferate when cultured at 39°C. The HMF3A cells have LTts and become senescent after 7 d at 39°C. RNAi lines were created by introducing pSUPER vector constructs into HMF3A lines.
We also performed several further comparisons on the arrays (indicated schematically in Figure 1). We first compared our model system to primary fibroblast senescence achieved through serially passaging cells until they cease proliferating. The primary fibroblasts (HMF3) from which HMF3A and HMF3Dwt were derived (O'Hare et al., 2001) were serially passaged twice through to senescence to yield two independent sets of RNA. A third set of RNA was prepared by passaging fibroblasts from a different female donor (F1068) through to senescence. The donor of HMF3 was 20 years of age, whereas the donor of F1068 was 26. For these experiments, the early passage proliferating cells were directly compared with senescent cells, and the three different sets of RNA were hybridized to four arrays each (GEO accession no. GSE687). To see how similar the primary cells were to HMF3A, HMF3A cells at 33°C were directly compared with the early passage-proliferating primary donor fibroblasts.
In regard to the changes after 7 d at 39°C, to determine which changes were occurring first, we compared HMF3A cells at 33°C to cells shifted to 39°C for 2 d. To determine which expression changes were not reversed when LT was reactivated, HMF3A at 33°C were compared with HMF3A cells shifted to 39°C for 7 d and then shifted back to 33°C for 3 d or 7 d. Furthermore, as a comparison with the irreversible process of senescence, we sought to identify genes that changed upon reversible growth arrest, quiescence. Thus, changes that occurred when HMF3A cells were grown to confluence and starved also were determined (2 biological replicate, 6 arrays; GEO accession no. GSE688).
These further comparisons provided a wealth of data on the changes that occur with primary cell senescence (Supplemental Table 2), reversible growth arrest (Supplemental Table 3), immortalization (Supplemental Table 4), 2-d inactivation of LT (Supplemental Table 5), and reactivation of LT (Supplemental Table 6). Here, we focus on the genes that change after LT inactivation for 7 d. We were able to divide the list of the genes that change in response to LT inactivation after 7 d into several different groups (Supplemental Figures 1 and 2 and Supplemental Table 7). The differences between proliferating primary and proliferating immortalized (HMF3A) cells are referred to as changes upon immortalization. The list generated here (Supplemental Table 4) is meant to be mainly used to see how genes that changed when LT was inactivated behaved when LT/hTERT was first introduced. It is interesting to compare the differences between primary cells and serially passaged senescent cells (Supplemental Table 2) and between primary cells and HMF3A cells (Supplemental Table 4), to see which changes occurred even in the presence of LT/hTERT. The difference in the temperature that the primary (37°C) and HMF3A (33°C) cells were cultured at also can be taken into account by examining the genes whose expression changed in the temperature-insensitive model (Supplemental Table 1).
Changes in Primary Fibroblast Senescence
Gene expression changes in common between primary cell senescence and LT inactivation (Supplemental Figures 1 and 2) include DUSP1, RGS3, NR4A3, growth arrest-specific (GAS) 6, PLOD2 and neprilysin (MME), and IGFBP4 (all up-regulated), and FOXM1, nuclear ribonucleoprotein A1 (HNRPA1), HMG17L1, CDC25B, CENPF, regulator of cytokinesis (PRC1), and ubiquitin-conjugating enzyme 2C (UBE2C) (all down-regulated). Genes that changed in primary cell senescence but not the model system include the metallothioneins, which were up-regulated. For all three clones of SERPINB2 and of AKR1B1, significant up-regulation was seen later than 7 d (Supplemental Table 6). Likewise, collagen 6A3 (COL6A3), collagen proteinase enhancer (COLCE), osteoblast-specific factor-2 (OSF2), and MGP were all significantly down-regulated in primary cells and only at a later time in HMF3A cells.
A large number of the changes detected in the model system (such as up-regulation of p21 [CDKN1A], BTG2, and collagenase MMP2 and down-regulation of MEST and cyclin A2 [CCNA2]) were not significant in the primary cells but have previously been found to change upon serial passaging of primary cells to senescence (Shelton et al., 1999; Lodygin et al., 2002; Schwarze et al., 2002). Many more changes were detected using the model system, partly due of course to the greater number of replicates. However, it also was clear that results obtained from the model system were more biologically reproducible, most likely to be due to the asynchronous nature of the growth arrest in serially passaged primary cells and that this added to the increased significance of the results. The proliferating primary fibroblasts used in this study were from adult tissue and had already undergone several divisions. The use of cells from different donors also added to the heterogeneity of the response. This highlights the importance of developing an inducible model system in which changes can be detected with greater sensitivity and which is then reproducible enough to be used to examine the importance of these changes.
Changes Associated with the Introduction of LT into Primary Cells
In the case of some of these genes, changes in RNA levels upon immortalization (and LT expression) preceded the opposite change observed upon inactivation of LT. An examination of the profile of CDKN1A reveals that its expression actually significantly decreases upon immortalization (Supplemental Table 4) and then increases upon inactivation of LT at 2 and 7 d. Other 7 d, up-regulated genes whose expression was decreased upon immortalization include quiescin (QSCN6), translocon-associated protein delta subunit (SSR4), and thioredoxin interacting protein (TXNIP) (Supplemental Figure 1C).
Conversely PRC1, FOXM1, TK1, and HMGB2 were up-regulated by immortalization and down-regulated with LT inactivation (Supplemental Figure 2C). Many of the down-regulated genes, such as PRC1, UBE2C, ANKT, HMGB2, Aurora-IPL1–related kinase (STK15), KIAA0101, uracil-DNA glycosylase (UNG), KNSL4, CCNA2, SYNGR2, NEK2, and CDC25C have been identified by microarray and other studies as being under the control of E2F1 and/or 4 (Ishida et al., 2001; Ren et al., 2002; Stanelle et al., 2002; Weinmann et al., 2002). However, they may not necessarily have been direct transcriptional targets in some of these studies.
Changes Associated with LT Inactivation
Some of the changes are consistent with reversal at 39°C of known effects of LT binding of p53 and pRb. An inspection of the genes up-regulated after 2 d of LT inactivation reveals that some of the gene expression changes observed after 7 d inactivation had already occurred by 2 d (Supplemental Figure 1C) and suggests that p53 had been activated. Known p53-responsive genes, including PIGPC1, CDKN1A, BTG2, PIG3, and CYFIP2 (Kannan et al., 2001; Mirza et al., 2003), were already increased. The p53 protein is unstable, and although LT inactivates its transcriptional activity, it also protects it from degradation. Western blotting revealed that both LT and p53 had decreased after a 2-d shift up (Figure 2A), indicating that p53 had been released by LT and was free to activate transcription of responsive genes.
Figure 2.
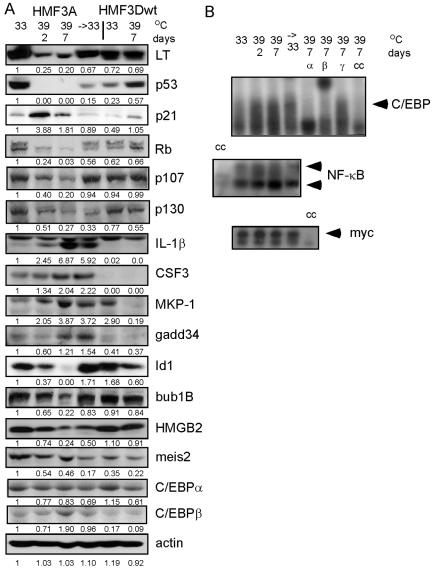
Changes in protein and DNA binding levels upon LT inactivation and reactivation (→ 33). Westerns were used to measure protein levels (A). EMSA were used to measure N-myc, C/EBP, and NF-κB DNA binding activity (B). The average ratios for the densitometry readings are given in relation to the level in HMF3A cells at 33°C. Supershifts using C/EBPα and β antibodies indicate that both of these proteins are involved. Western blots also were performed with these antibodies. Cold competition (cc) with unlabeled probe shows the specificity of the EMSA bands. Westerns were performed at least twice using different lysates. EMSA were performed three times using different lysates.
Likewise, many of the known and putative E2F targets down-regulated after 7 d also were down-regulated after 2 d (Supplemental Figure 2C), indicating that Rb had been released from LT and was free to sequester the E2F transcription factors. Furthermore, dephosphorylation of Rb (and hence activation) could be seen by 2 d (Figure 2A). However, the protein levels of Rb and family members p107 and p130 also decreased with LT inactivation (Figure 2A), which is consistent with their known regulation by E2F at the transcriptional level (DeGregori, 2002).
Many of the putative p53 and E2F targets could be responsible for the growth arrest. Up-regulation of p21 or BTG2 has been shown to halt the cell cycle as has inhibition of HMGB2, PRC1, or FOXM1 (Tirone, 2001). A cell lacking genes involved in cytokinesis or DNA synthesis also will be unlikely to proliferate. Increased expression levels of many of the down-regulated genes (such as those regulated by E2F) are associated with particular stages of the cell cycle such as mitosis and as such would not be expressed upon quiescence. Because LT inactivates p53 and Rb, the results obtained from quiescence of HMF3A cells are somewhat unexpected. However, several of the proposed E2F-regulated genes (including STK15 and UBE2C) were down-regulated (Supplemental Figure 2B). This may be due to an Rb-independent effect on E2F. Although the decrease in the level of the cell cycle-associated genes may explain the lack of proliferation, it does not reveal what makes senescence irreversible or what distinguishes senescence from quiescence at the molecular level.
Changes That Occur with Reactivation of LT
In this model system, when LT is reactivated by returning the cells to 33°C, some cells undergo apoptosis, whereas others do not proliferate. Interestingly, the expression profiles of HMF3A cells shifted back to 33°C after 7 d at 39°C reveal that many cell cycle-associated genes were no longer down-regulated. Also, p53 targets such as p21 and BTG2 are no longer up-regulated. In fact, many of the changes that occur upon LT inactivation do not remain “detectably” changed when the cells are switched back to 33°C for 7 d (Supplemental Figure 2A) even though the cells do not divide.
Therefore, the genes that remain unchanged when LT antigen is reactivated are particularly interesting. These include DUSP1, DUSP4, DUSP5, STAT1, BIRC3 and 2 (inhibitor of apoptosis protein 1 and 2), PRNP (prion protein), SOX9, BTG3, TSC22, GADD45A, and PPP1R15A (GADD34) and NR4A3 (Supplemental Figure 1E) and the down-regulated CALM3 (calmodulin 3), DPYSL3 (ULIP protein), PTMS (parathymosin), and MEIS2 (Supplemental Figure 2E). Some of the up-regulated genes such as TSC22 are TGF-β inducible. However addition of a pan-neutralizing anti-TGF-β antibody did not make a detectable difference to the gene expression profile or affect cell proliferation when cells were shifted back to 33°C for 3 d (our unpublished data).
Western blots confirmed that increased expression of DUSP1 (MKP-1), PPP1R15A (GADD34), IL1B, CSF3, and CDKN1A upon LT-inactivation led to increased protein levels (Figure 2A). The levels of several proteins such as CSF3, p53, meis2, and MKP-1 were different in HMF3A and HMF3Dwt cells at 33°C (Figure 2A). These differences were reproducible, and microarray comparisons (our unpublished data) between other lines derived from the HMF3 primary cells such as HMF3B, HMF3C, and HMF3D (O'Hare et al., 2001) show that there are differences between these cells at the gene expression level. A comparison of the gene profiles of these lines and how they relate to the phenotypes of the lines is underway, but due to space limitations is not discussed here.
As predicted, MEIS2, Id1, HMGB2, and BUB1B were down-regulated (Figure 2A). Interestingly the level of HMGB2 did not increase once LT was reactivated as predicted by the arrays. The level of p130 also failed to return to that seen in unshifted cells, in contrast to p107 (Figure 2A). As mentioned below (Figure 3A), RT-PCR results for HMGB2 indicate that there was some increase in HMGB2 RNA when cells were returned to 33°C, and so the lack of an increase in protein may be the result of regulation at the level of translation.
Figure 3.
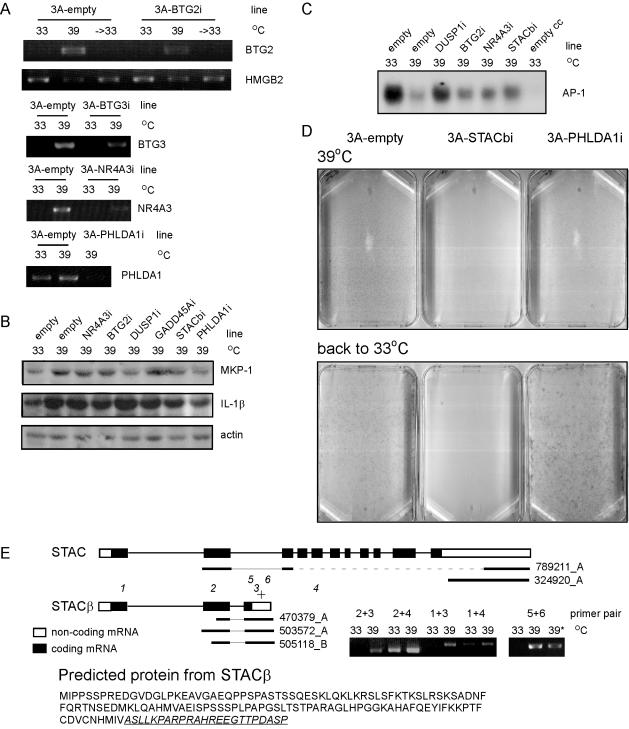
Differences in the RNAi lines. RT-PCR was used to determine the effectiveness of the stable RNAi vectors against BTG2, BTG3, NR4A3, and PHLDA1. (A) HMF3A cells containing the various p-SUPER vectors were shifted to 39°C for 7 d, and 3A-BTG2i cells also were shifted back to 33°C for 7 d. (B) The protein levels for MKP-1 (product of the DUSP1 gene) and IL-1β were examined for the various RNAi cultures after the cells were shifted to 39°C for 7 d. (C) The DNA binding activity of AP-1 was measured by EMSA after 7 d of LT inactivation in the DUSP1i and control cells. A cold competition (cc) control sample was used to assess the specificity of the band. (D) Staining of cells shifted to 39°C for 2 wk and then either shifted back to 33°C for 2 wk (back to 33°C) or left at 39°C for another 2 wk (39°C) was performed for 3A-STACβI, 3A-PHLDA1i, and control cells. (E) Comparison of the structure of STAC and the splice variant STACβ. RT-PCR were performed with various primer combinations (1–6). If STACβ has the same transcription start site as STAC, it would code for a shorter protein with the sequence given here, with the underlined area indicating where it would be different from STAC. For RNAi, a target sequence (+) was used that was only in STACβ. The resulting cells (39*) did have less STACβ mRNA than control cells, when compared after shifting of both cells to 39°C for 7 d. RT-PCR were performed three times with different RNA lysates. In addition to this samples were taken at three different times to ensure that the reaction was in the linear amplification stage. Westerns were performed at least twice by using different lysates. EMSA were performed three times by using different lysates.
Some genes such as GADD45A, CEBPG, and PPP1R15A were regulated in opposite directions in quiescence and senescence. Increases in p53 targets were not detected, although p53 has previously been shown to be activated upon starvation (Itahana et al., 2002). Such behavior is not typical of quiescence in normal cells and is likely to be due to LT inhibiting p53 in HMF3A cells. The increase of other genes such as PRNP and DUSP6 implies they are coregulated by another factor that also may be activated by LT inactivation.
Searching for Patterns in the Promoter Elements for Coregulated Genes
Transcription factors not only bind unique DNA sequences but also have a degree of nonspecificity in their sequence recognition. Thus, whether a transcription factor binding site exists in a promoter sequence cannot simply be determined by searching for an exact sequence. Instead, matrices are used that give the different nucleotides various weightings depending on their importance for transcription factor binding. A collection of these matrices exists in the TRANSFAC database (http://www.gene-regulation.com/pub/databases.html#transfac). A transcription factor matrix provides a score on how well a sequence (usually 5–20 base pairs long) matches a binding site. The relative occurrences in the lists of up- and down-regulated genes of a set of transcription factors (TRANSFAC matrices) was compared (Table 1 and Supplemental Table 8). We repeated the analysis using just the sequence upstream of the promoter (Supplemental Table 8) and observed similar patterns as when using the up- and downstream sequence. Many transcription factor motifs such as those for GATA-X, Pbx-1, and YY1 occurred in approximately equal proportions for the up- and down-regulated genes (Table 1).
Table 1.
Percentage of up- and down-regulated genes with one or more transcription factor matrices present in the region -750 and +250 bp of the transcription start site, as obtained from EZ-Retrieve, by using a cut-off of 85 for matrix matching
| Human
|
Mouse
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 d | 7 d | Back to 33C | 2 d | 7 d | Back to 33C | ||||||||
| Up | Down | Up | Down | Up | Down | Up | Down | Up | Down | Up | Down | ||
| M00051 | NF-kap | 28 | 20 | 21 | 16 | 16 | 17 | 9 | 21 | 15 | 17 | 17 | 11 |
| M00052 | NF-kap | 24 | 14 | 27 | 18 | 28 | 20 | 35 | 33 | 27 | 18 | 26 | 24 |
| M00054 | NF-kap | 37 | 24 | 39 | 23 | 32 | 29 | 34 | 33 | 31 | 22 | 38 | 31 |
| M00053 | c-Rel | 52 | 47 | 49 | 52 | 46 | 38 | 50 | 53 | 45 | 50 | 49 | 48 |
| M00116 | C/EBPa | 24 | 14 | 30 | 21 | 30 | 17 | 19 | 21 | 31 | 21 | 28 | 22 |
| M00117 | C/EBPb | 13 | 15 | 24 | 19 | 32 | 20 | 12 | 18 | 20 | 17 | 24 | 18 |
| M00001 | MyoD | 32 | 32 | 27 | 33 | 30 | 33 | 32 | 24 | 26 | 31 | 26 | 27 |
| M00002 | E47 | 24 | 11 | 17 | 15 | 19 | 18 | 24 | 15 | 21 | 17 | 20 | 20 |
| M00203 | GATA-X | 30 | 37 | 38 | 38 | 42 | 42 | 44 | 39 | 44 | 38 | 53 | 44 |
| M00131 | HNF-3b | 37 | 38 | 37 | 34 | 42 | 47 | 45 | 39 | 47 | 32 | 50 | 46 |
| M00087 | Ik-2 | 69 | 69 | 69 | 72 | 70 | 68 | 72 | 65 | 67 | 75 | 75 | 74 |
| M00063 | IRF-2 | 5 | 11 | 10 | 13 | 5 | 14 | 7 | 3 | 9 | 13 | 9 | 7 |
| M00173 | AP-1 | 43 | 37 | 43 | 33 | 42 | 39 | 54 | 33 | 43 | 36 | 40 | 37 |
| M00162 | Oct-1 | 20 | 18 | 25 | 15 | 24 | 26 | 22 | 27 | 24 | 15 | 19 | 33 |
| M00096 | Pbx-1 | 32 | 31 | 31 | 34 | 33 | 32 | 35 | 39 | 35 | 33 | 39 | 35 |
| M00042 | Sox-5 | 28 | 21 | 29 | 26 | 25 | 23 | 19 | 33 | 29 | 27 | 22 | 22 |
| M00032 | c-Ets- | 58 | 71 | 62 | 71 | 66 | 74 | 62 | 80 | 60 | 71 | 69 | 57 |
| M00059 | YY1 | 18 | 16 | 16 | 17 | 16 | 18 | 15 | 21 | 18 | 15 | 14 | 16 |
| M00073 | deltaE | 66 | 71 | 58 | 68 | 68 | 72 | 74 | 65 | 66 | 67 | 65 | 77 |
| M00113 | CREB | 24 | 31 | 29 | 23 | 29 | 20 | 27 | 56 | 28 | 25 | 31 | 18 |
| M00003 | v-Myb | 7 | 14 | 14 | 13 | 12 | 9 | 14 | 24 | 15 | 12 | 13 | 22 |
| M00055 | N-Myc | 18 | 28 | 21 | 28 | 28 | 18 | 35 | 39 | 29 | 27 | 34 | 27 |
| M00024 | E2F | 0 | 5 | 0 | 3 | 0 | 2 | 0 | 0 | 0 | 2 | 0 | 0 |
| M00050 | E2F | 58 | 75 | 57 | 64 | 60 | 63 | 49 | 74 | 51 | 63 | 52 | 55 |
| M00108 | NRF-2 | 28 | 39 | 22 | 30 | 23 | 21 | 22 | 27 | 20 | 31 | 31 | 22 |
| M00007 | Elk-1 | 3 | 10 | 8 | 7 | 6 | 6 | 5 | 6 | 5 | 8 | 10 | 5 |
| M00025 | Elk-1 | 28 | 46 | 26 | 33 | 27 | 23 | 22 | 39 | 27 | 33 | 34 | 16 |
| cut off 90 | |||||||||||||
| M00053 | c-Rel | 26 | 16 | 28 | 14 | 23 | 14 | ||||||
| M00025 | Elk-1 | 11 | 18 | 6 | 13 | 6 | 9 | ||||||
| M00032 | c-Ets- | 24 | 39 | 22 | 28 | 25 | 20 | ||||||
| M00050 | E2F | 9 | 19 | 10 | 17 | 15 | 12 | ||||||
The same analysis was performed for the mouse homologues of the changed genes.
Two matrices (M00024 and M00050) can be used for E2F. The first matrix is more stringent and occurs with lower frequency. A greater proportion of down-regulated genes after 2 or 7 d, than up-regulated genes, had at least one match to M00050. This pattern was clearer when a greater matrice cut-off (90) was used (Table 1) or when the criterion was altered to include a promoter containing two or more matches (Supplemental Table 8). A similar pattern was seen for the corresponding mouse homologues. This suggests that this relatively simple approach could indicate changes in the activity of transcription factors.
Up-regulated genes were more likely to have binding sites for NF-κB (7 d), C/EBPα (7 d), or E47 (2 d) than the down-regulated genes. We therefore examined changes in DNA binding activity for NF-κB and C/EBP by EMSA. Increased DNA binding activity after LT inactivation was confirmed for NF-κB and C/EBP (Figure 2B). LT inactivation increased the intensity of the multiple bands typically seen in EMSA for NF-κB due to the presence of different dimer combinations. The specific band for C/EBP was supershifted by antibodies against C/EBPα and C/EBPβ but not C/EBPγ. Whereas C/EBPα is present, its levels do not increase, whereas those of C/EBPβ do increase (Figure 2B). C/EBPγ protein expression could not be detected with the antibody used (our unpublished data), even though the C/EBPγ RNA (CEBPG) does increase with LT inactivation and stays increased upon reactivation of LT (Supplemental Figure 1E). Due to the lack of a positive control, it cannot be determined whether the lack of C/EBPγ protein (and supershift) is real or a result of poor antibody binding. Overexpression of either C/EBPα or β or both can induce differentiation and/or growth arrest in different cell types (Parkin et al., 2002). C/EBP and NF-κB coregulate many genes involved in the immune response, and C/EBP/NF-κB composite elements were found in IL1B and CSF3 (Shelest et al., 2002), two of the genes for which protein up-regulation was confirmed here. Increased NF-κB activity also is associated with protection from apoptosis and when activated with p53 may help the cell favor growth arrest over apoptosis (Heinrichs and Deppert, 2003).
The possibility that E47 was activated was not followed up. Activation of E47 along with that of its family members is associated with differentiation, and it has been shown that Id-1 negatively modulates E47 family members (Liu et al., 2002). The down-regulation of Id-1 protein levels upon LT inactivation was confirmed (Figure 2A), and this down-regulation has previously been associated with senescence (Sikder et al., 2003).
A smaller proportion of the genes up-regulated after 2 or 7 d of LT inactivation have binding sites for N-Myc, than those down-regulated. However, this pattern was not seen in the mouse homologues. EMSA results show that a protein binds to the consensus Myc site but that its DNA binding activity does not change upon LT inactivation (Figure 2B).
The matrices for Elk-1 (especially the less stringent M00025) were similar to that for Nrf-2 (M00108), and a greater proportion of genes down-regulated after 2 d at 39°C have these matrices for the region studied in both the human genes and their mouse homologues (Table 1). Both proteins are members of the Ets family of transcription factors and bind similar sequences. Both are activated by extracellular signal-regulated kinase (ERK) and p38. However, although Elk is involved in the response of the cell to mitogens, Nrf2 plays a role in the stress response (Camps et al., 2000; Zipper and Mulcahy, 2000). Perhaps the best example of the redundancy of many of the gene changes is the number of inhibitors for ERK that are induced upon inactivation of LT. This included the direct inactivators DUSP1, 4, 5, and 6 and PEA-15 and other proteins such as RGS3 that act further upstream in the signaling pathway (Sun et al., 1994; Shi et al., 2001).
PEA-15 inhibits the action of ERK on nuclear targets and can promote its effects on cytoplasmic targets (Formstecher et al., 2001). Elk-1 is constitutively bound to DNA with its activity altered by phosphorylation and hence is not a good target for EMSA. A transcriptional target of Elk-1 that is involved in proliferation is c-Fos, which is a component of the transcription factor dimer AP-1 (Kortenjann et al., 1994). Although no pattern was seen for AP-1 binding motifs, AP-1 DNA binding activity was found to decrease with the shift to 39°C as presented below.
Thus, this approach does not always identify changes in transcription factor activation or at least does not with the matrices known at the present time. However, given the simplicity of the approach it is relatively effective and provides hope that with further investigation and refinement of both the matrices used and the statistical relevance of results obtained, it can be a powerful tool in organizing microarray results into signaling networks.
Selective Silencing of Genes by RNA Interference
Although information can be obtained through in silico promoter studies to construct models of the signaling networks involved, a complementary approach is to determine the effects of particular gene expression changes. To this end, the pSUPER-retro vector system (Brummelkamp et al., 2002a) was used to selectively silence genes to determine whether they had a role in the changes to the transcriptome upon LT inactivation (GEO accession no. GSE690).
In determining whether the RNAi had successfully decreased the level of the target mRNA, an interesting effect was noticed in that for all genes a decrease was only observed with PCR primers that spanned the targeted region. A primer set that amplified a part of the 3′ untranslated region usually indicated an increased level of the partial message. This effect also was reflected on the arrays that largely contain probes that favor the 3′ ends of genes and do not distinguish between full or partial RNAs. Thus, the mRNA does seem only to be cleaved around the target region and fragments of the mRNA remain. Possible biological effects of these fragments are unclear.
BTG2
BTG2 (B-cell translocation gene-2) is a direct transcriptional target of p53, and its expression has been shown to be important in negatively regulating cell proliferation (Tirone, 2001). In our model system, its expression was repressed upon immortalization of the cells and induced within 2 d of LT inactivation. We wanted to use RNAi to establish the contribution of BTG2, if any, to the expression changes observed in our model. The precise function of BTG2 is not clear. It has been linked to Hoxb9 and the protein-arginine methyltransferases PRMT1 and 2 (Tirone, 2001). Introduction of pSUPER-BTG2 (or any of the other RNAi vectors) did not affect the growth of HMF3A cells at 33°C as expected from the predicted absence or low level of BTG2 mRNA present at 33°C. A partial decrease in the level of BTG2 at 39°C was observed by RT-PCR (Figure 3A). No effect on the arrest seen upon LT inactivation was observed in the HMF3A-BTG2i cells. Moreover, no effect of BTG2i was seen on HMGB2 expression (Figure 3A), even though BTG2 has been suggested to regulate Rb activation (Tirone, 2001).
Direct array comparisons were performed between HMF3A-pSUPER and HMF3A-BTG2i after both cultures were shifted to 39°C for 48 h. This time was chosen because of the rapid induction of BTG2. The results from these arrays suggest that in the absence of BTG2, some of the genes normally induced at 39°C, such as CDNK1A, CSF3, RRAD, and CRIP2, were further induced (Supplemental Table 9). A culture of HMF3A-BTG3i cells was generated (Figure 3A), but no effect on cell proliferation was observed. In this model, the expression of BTG2 does not seem to be necessary for the induction of senescence. However, it is possible that a more complete abrogation of BTG2, BTG3, or all BTG family members together would have a more perceivable effect.
NR4A3
NR4A3 (MINOR) is an orphan nuclear receptor that has been implicated in regulation of both cell proliferation and death (Martinez-Gonzalez et al., 2003). Its expression is elevated after serial passaging and in our model of senescence. Nuclear receptors of the 4A subfamily are thought to act as homo- and heterodimers regulating gene transcription by binding the NGRI-B response element (Martinez-Gonzalez et al., 2003). Reduction of the increase in expression of NR4A3 in HMF3A cells by RNA interference (HMF3A-NR4A3i cells) at 39°C (Figure 3A) did not alter the cell arrest. Direct comparisons between HMF3A-NF4A3i and HMF3A-pSUPER cells were performed after cultures were shifted to 39°C for 7 d. As for BTG2, the absence of NR4A3 resulted in higher expression of some of the induced genes (but not necessarily the same ones), including IL1B, PRNP, and PLAU (Supplemental Table 10). There was some indication that the induction of GAS6 with LT inactivation was decreased in the absence of NR4A3 (Supplemental Table 10). However, this effect also was observed in other lines. In a more specific manner, the induction of the maternally imprinted gene MEG3 also was decreased by the absence of NR4A3 (Supplemental Table 10). MEG3 does not encode for a protein but may regulate the transcription of DLK1, which is involved in cell growth (Georges et al., 2003). If the microarray results are correct, this shows that the combination of a model system, microarrays, and RNAi can be used to reveal novel pathways. The most interesting result from this study of NR4A3 is that its level is increased in senescence, and this may have a role in senescence through affecting cell growth via DLK1 and/or MEG3.
DUSP1
DUSP1 was found to be up-regulated in both models of senescence, and the increase in the level of its protein (MKP-1) was confirmed in HMF3A cells. MKP-1 inhibits ERK (Sun et al., 1994). An increase in the protein levels of the related protein MKP-2 has been linked to the decreased nuclear ERK phosphorylation associated with senescence (Torres et al., 2003). Western blots showed that the increase in MKP-1 observed upon LT inactivation was inhibited in HMF3A-DUSP1i (Figure 3B). When the DNA binding activity of AP-1 was examined in cells shifted to 39°C, a down-regulation was observed in the control cells and this decrease was partly abrogated in the HMF3A-DUSP1i cells (Figure 3C). This supports the suggestion of a pathway whereby LT inactivation induces the expression of DUSP1, such that ERK (and hence Elk-1 activity) is inhibited, c-Fos protein is not produced and AP-1 activity decreases. Activation of the other mitogen-activated protein kinases, JNK and p38, also may be affected, and they also are involved in regulating AP-1 activity and protein levels.
No consistent effect on cell growth was observed in the HMF3A-DUSP1i cells upon shift up to 39°C. As with the other cell cultures, the lack of DUSP1 induction upon LT inactivation led to some induced genes being induced even more, including DUSP6 (Supplemental Table 11). However, some of the genes expressed at a higher level in the DUSP1i cells at 39°C went down with LT inactivation in normal HMF3A cells. Thus, the induction of DUSP1 combined with LT inactivation could be responsible for the decrease in these genes when LT is inactivated in HMF3A cells. These genes included histone H3.3 (H3F3B), histone H1.0 (H1F0), phorbolin 3 (APOBEC3), stathmin (STMN1), HNRPU, and STK15 (Supplemental Table 11). Interestingly, H3F3B transcription is positively regulated by AP-1 (Witt et al., 1998). Some of the other genes, including STK15, APOBEC3, and STMN1, have putative Elk-1 binding sites in their promoters and so can be tentatively linked to this pathway. Thus, DUSP1 seems to play a role in the induction of senescence through inhibition of AP-1 activity and the subsequent transcription of genes involved in DNA replication.
PHLDA1
PHLDA1 (Pleckstrin homology-like domain, family A, member 1, hs.82101, not currently in ENSEMBL) is a pleckstrin homology domain protein whose mRNA expression increases with LT inactivation and remains up even when LT is reactivated. It was identified as a regulator of Fas expression upon activation of T lymphocyte hybridomas. Although it was originally suggested to be a direct transcriptional regulator, other evidence suggests that it controls protein synthesis (Hinz et al., 2001). To examine its role in senescence, HMF3A-PHLDA1i RNAi lines were created and array comparisons were performed as for NR4A3i and DUSP1i, that is, after 7 d of LT inactivation. Of all the comparisons, PHLDA1 gave results that were the most variable between the different RNA isolates, with no genes significantly regulated. The one clone suggested by SAM analysis to be regulated differently in the PHLDA1 line was IL1B, and the level of this protein was indeed shown to be reduced in these cells at 39°C (Figure 3B). The most aberrant RNA isolate was the second and if this is removed from the analysis, more genes (all less abundant in the HMF3A-PHLDAi cells) changed significantly. These include PHLDA1 (Supplemental Table 12). Although the HMF3A-PHLDAi cells did not continue to proliferate when shifted to 39°C, increased cell survival was observed when the cells were returned to 33°C (Figure 3D). Constitutive expression of PHLDA1 has been associated with increased susceptibility to apoptosis (Neef et al., 2002). Thus, up-regulation of PHLDA1 in this system may be linked to the priming of the cell for apoptosis upon reactivation of LT. This result emphasizes that changes in gene expression in cellular processes such as senescence may not always be linked to the most apparent phenotype, which in this case is the lack of proliferation. On senescence, the cell may prime itself for apoptosis to avoid the risk of uncontrolled proliferation in the case of DNA modification. These results suggest that PHLDA1 may play a role in this process.
STAC
STAC codes for a cysteine-rich protein containing an SH3 domain and is predominantly expressed in neuronal cells (Suzuki et al., 1996). There are five clones on the arrays that have some homology to STAC: 324920_A, 789211_A, 503572_A, 505118_B, and 470379_A. The first two do not change in response to LT inactivation and seem to represent the STAC gene in ENSEMBL (ENSG00000144681), whereas the latter three increase in expression after 7 d at 39°C and remain increased despite LT reactivation. Resequencing of these three clones (Wellcome Trust Sanger Institute) as well as the sequences from the original clone resource for 503572_A (AA131521) and 470379_A (AA031284) indicate that they represent a variant that contains exon 2 of the STAC gene and part of the intron between exons 2 and 3 (Figure 3E). Here, this STAC variant is called STACβ. RT-PCR using primers for exon 1 and the STACβ unique exon resulted in a product of the expected size; thus, it seems that STACβ contains exon 1, which contains the STAC start codon. The predicted protein for STACβ (Figure 3E) only partly retains the protein kinase C binding motif and does not have the SH3 motif.
The sequence for RNAi was designed to target only the STACβ (it is located in the intron of STAC). RT-PCR shows a reduction of the STAC message in the HMF3A-STACβi cells (Figure 3E). The array results (done for a 7-d shift) from the HMF3A-STACβi cells yielded the most significant changes. The most significant result from the HMF3A-STACβi cells is the exacerbation of the increase in a subgroup of the genes that respond to LT inactivation, including GADD45A, DUSP5, BIRC3, CSF3, TSC22, SGK, and TNFAIP3 (Supplemental Table 13). This could be a compensatory effect and/or may indicate that STACβ plays a functional role in dampening the induction of these genes. However, the induction of genes such as NDRG, PLOD2, SERPINH1, CREG, MME, and SSR4 was abrogated in the HMF3A-STACβi cells. Although the HMF3A-STACβi cells proliferated to the same extent as HMF3A-empty vector cells at 33°C upon LT inactivation a greater level of cell death was observed (Figure 3D). Further studies and confirmation of the gene expression changes with other techniques are needed to determine the role of STACβ in cell senescence.
The p53 pathway is activated in both apoptosis and senescence, and as stated with PHLDA1, the senescent cells may be primed for apoptosis. The results for STAC suggest that it may be one of the genes that plays a role in protecting the cell from apoptosis, either through its dampening of genes such as GADD45A and BIRC3 or its putative induction of NDRG and/or CREG.
Higher false positive rates than normal have been used here, particularly for the RNAi comparisons. Similarly, the connections suggested by the presence of particular transcription factor binding sites in promoter regions have high error rates. However, in this type of analysis and modeling, it is the identities and functions of groups of genes rather than individual genes that are important. Consequently, as long as the error rate is considered and the regulation of important genes checked by other means, it is appropriate to consider as much information as possible.
The ultimate goal of our studies is to model the signaling and gene expression networks that regulate cellular senescence. It is increasingly apparent that these sorts of models will be highly complex and difficult, perhaps impossible, to display on a two-dimensional page. We have started to build a putative model by displaying down and up-regulated genes and by using the patterns suggested by our RNAi work, the literature, in silico promoter analysis, and alternative cell treatments such as quiescence, to loosely group the genes. At the moment many of the proposed mechanisms of regulation, such as the genes activated by C/EBP, are only suggested by the presence of binding sites for particular transcription factors that need to be confirmed experimentally.
The microarray data from our experiments does reveal great differences between the transcriptome of proliferating HMF3A and primary cells. However, it also shows that many of the changes that occur in the HMF3A model of senescence also occur in primary cell senescence and that the reproducibility of a model inducible system is needed for studies that seek not only to detect changes but to establish the role of particular genes in those changes.
The results presented here highlight several features of senescence: the importance of p53; the role of the Rb family members (and perhaps ERK inactivation) in inducing down-regulation of genes responsible for DNA synthesis, cytokinesis, and other aspects of proliferation; the induction of many genes that have overlapping functions (such as inhibition of ERK); and the presence of negative feedback loops that serve not only to stabilize the level of expression of genes but also perhaps to allow the cell to compensate by overexpressing other genes if a gene is not expressed as normal. It also illustrates the result that genes may be activated under various conditions by perhaps different transcription factors. For example PRNP, STC1, and WNT5A are increased in both senescence and quiescence.
With the rapid advancement of programming tools to manipulate data obtained from large-scale expression studies, patterns and trends in the signaling pathways should become more apparent and the gene targets to perturb more identifiable, until the ultimate computational signaling model is obtained.
Supplementary Material
Acknowledgments
We thank the staff of the Sanger Institute Microarray Facility (http://www.sanger.ac.uk/Projects/Microarrays/). We thank Rowena Martin and Angelo Theodoratos for critical reading of the manuscript. The microarray consortium is funded by the Wellcome Trust, Cancer Research UK, and the Ludwig Institute for Cancer Research.
Article published online ahead of print in MBC in Press on December 1, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-05-0392).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Ali, S. H., and DeCaprio, J. A. (2001). Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin. Cancer Biol. 11, 15-23. [DOI] [PubMed] [Google Scholar]
- Allsopp, R. C., Vaziri, H., Patterson, C., Goldstein, S., Younglai, E. V., Futcher, A. B., Greider, C. W., and Harley, C. B. (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA 89, 10114-10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayreuther, K., Rodemann, H. P., Hommel, R., Dittmann, K., Albiez, M., and Francz, P. I. (1988). Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc. Natl. Acad. Sci. USA 85, 5112-5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti, S., Cramer, R., Quinn, C. C., Bruce, J., Zvelebil, M., Corless, S., Bond, J., Yang, A., Hockfield, S., Burlingame, A. L., Waterfield, M. D., and Jat, P. S. (2002). Differential proteome analysis of replicative senescence in rat embryo fibroblasts. Mol. Cell Proteomics 1, 280-292. [DOI] [PubMed] [Google Scholar]
- Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., Harley, C. B., Shay, J. W., Lichtsteiner, S., and Wright, W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349-352. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T. R., Bernards, R., and Agami, R. (2002a). Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2, 243-247. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T. R., Bernards, R., and Agami, R. (2002b). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550-553. [DOI] [PubMed] [Google Scholar]
- Camps, M., Nichols, A., and Arkinstall, S. (2000). Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 14, 6-16. [PubMed] [Google Scholar]
- Chen, Q. M., Bartholomew, J. C., Campisi, J., Acosta, M., Reagan, J. D., and Ames, B. N. (1998). Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 332, 43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm, K., et al. (2001). A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 20, 6958-6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, T., Singhrao, S. K., Wyllie, F. S., Haughton, M. F., Smith, P. J., Wiltshire, M., Wynford-Thomas, D., Jones, C. J., Faragher, R. G., and Kipling, D. (2003). Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J. Cell Sci. 116, 1349-1357. [DOI] [PubMed] [Google Scholar]
- DeGregori, J. (2002). The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim. Biophys. Acta 1602, 131-150. [DOI] [PubMed] [Google Scholar]
- Di Donna, S., Mamchaoui, K., Cooper, R. N., Seigneurin-Venin, S., Tremblay, J., Butler-Browne, G. S., and Mouly, V. (2003). Telomerase can extend the proliferative capacity of human myoblasts, but does not lead to their immortalization. Mol. Cancer Res. 1, 643-653. [PubMed] [Google Scholar]
- Elbashir, S. M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494-498. [DOI] [PubMed] [Google Scholar]
- Fauth, C., O'Hare, M. J., Lederer, G., Jat, P. S., and Speicher, M. R. (2004). Order of genetic events is critical determinant of aberrations in chromosome count and structure. Genes Chromosomes Cancer 40, 298-306. [DOI] [PubMed] [Google Scholar]
- Formstecher, E., Ramos, J. W., Fauquet, M., Calderwood, D. A., Hsieh, J. C., Canton, B., Nguyen, X. T., Barnier, J. V., Camonis, J., Ginsberg, M. H., and Chneiweiss, H. (2001). PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 1, 239-250. [DOI] [PubMed] [Google Scholar]
- Georges, M., Charlier, C., and Cockett, N. (2003). The callipyge locus: evidence for the trans interaction of reciprocally imprinted genes. Trends Genet. 19, 248-252. [DOI] [PubMed] [Google Scholar]
- Harlow, E., Crawford, L. V., Pim, D. C., and Williamson, N. M. (1981). Monoclonal antibodies specific for simian virus 40 tumor antigens. J. Virol. 39, 861-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs, S., and Deppert, W. (2003). Apoptosis or growth arrest: modulation of the cellular response to p53 by proliferative signals. Oncogene 22, 555-571. [DOI] [PubMed] [Google Scholar]
- Hinz, T., Flindt, S., Marx, A., Janssen, O., and Kabelitz, D. (2001). Inhibition of protein synthesis by the T cell receptor-inducible human TDAG51 gene product. Cell Signal. 13, 345-352. [DOI] [PubMed] [Google Scholar]
- Ishida, S., Huang, E., Zuzan, H., Spang, R., Leone, G., West, M., and Nevins, J. R. (2001). Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21, 4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana, K., Dimri, G. P., Hara, E., Itahana, Y., Zou, Y., Desprez, P. Y., and Campisi, J. (2002). A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 277, 18206-18214. [DOI] [PubMed] [Google Scholar]
- Iwasa, H., Han, J., and Ishikawa, F. (2003). Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 8, 131-144. [DOI] [PubMed] [Google Scholar]
- Iyer, V. R., et al. (1999). The transcriptional program in the response of human fibroblasts to serum. Science 283, 83-87. [DOI] [PubMed] [Google Scholar]
- Kannan, K., Amariglio, N., Rechavi, G., Jakob-Hirsch, J., Kela, I., Kaminski, N., Getz, G., Domany, E., and Givol, D. (2001). DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20, 2225-2234. [DOI] [PubMed] [Google Scholar]
- Kiyono, T., Foster, S. A., Koop, J. I., McDougall, J. K., Galloway, D. A., and Klingelhutz, A. J. (1998). Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84-88. [DOI] [PubMed] [Google Scholar]
- Kortenjann, M., Thomae, O., and Shaw, P. E. (1994). Inhibition of v-raf-dependent c-fos expression and transformation by a kinase-defective mutant of the mitogen-activated protein kinase Erk2. Mol. Cell. Biol. 14, 4815-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassar, A. B., Davis, R. L., Wright, W. E., Kadesch, T., Murre, C., Voronova, A., Baltimore, D., and Weintraub, H. (1991). Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 66, 305-315. [DOI] [PubMed] [Google Scholar]
- Liu, C. J., Ding, B., Wang, H., and Lengyel, P. (2002). The MyoD-inducible p204 protein overcomes the inhibition of myoblast differentiation by Id proteins. Mol. Cell. Biol. 22, 2893-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodygin, D., Menssen, A., and Hermeking, H. (2002). Induction of the Cdk inhibitor p21 by LY83583 inhibits tumor cell proliferation in a p53-independent manner. J. Clin. Investig. 110, 1717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gonzalez, J., Rius, J., Castello, A., Cases-Langhoff, C., and Badimon, L. (2003). Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ. Res. 92, 96-103. [DOI] [PubMed] [Google Scholar]
- Masutomi, K., et al. (2003). Telomerase maintains telomere structure in normal human cells. Cell 114, 241-253. [DOI] [PubMed] [Google Scholar]
- Mirza, A., et al. (2003). Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene 22, 3645-3654. [DOI] [PubMed] [Google Scholar]
- Neef, R., Kuske, M. A., Prols, E., and Johnson, J. P. (2002). Identification of the human PHLDA1/TDAG51 gene: down-regulation in metastatic melanoma contributes to apoptosis resistance and growth deregulation. Cancer Res. 62, 5920-5929. [PubMed] [Google Scholar]
- O'Hare, et al. (2001). Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc. Natl. Acad. Sci. USA 98, 646-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, W. Y., et al. (2002). Identification of radiation-specific responses from gene expression profile. Oncogene 21, 8521-8528. [DOI] [PubMed] [Google Scholar]
- Parkin, S. E., Baer, M., Copeland, T. D., Schwartz, R. C., and Johnson, P. F. (2002). Regulation of CCAAT/enhancer-binding protein (C/EBP) activator proteins by heterodimerization with C/EBPgamma (immunoglobulin/EBP). J. Biol. Chem. 277, 23563-23572. [DOI] [PubMed] [Google Scholar]
- Powell, A. J., Darmon, A. J., Gonos, E. S., Lam, E. W., Peden, K. W., and Jat, P. S. (1999). Different functions are required for initiation and maintenance of immortalization of rat embryo fibroblasts by SV40 large T antigen. Oncogene 18, 7343-7350. [DOI] [PubMed] [Google Scholar]
- Ren, B., Cam, H., Takahashi, Y., Volkert, T., Terragni, J., Young, R. A., and Dynlacht, B. D. (2002). E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16, 245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles, S. J., and Adami, G. R. (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 16, 1113-1123. [DOI] [PubMed] [Google Scholar]
- Sachsenmeier, K. F., and Pipas, J. M. (2001). Inhibition of Rb and p53 is insufficient for SV40 T-antigen transformation. Virology 283, 40-48. [DOI] [PubMed] [Google Scholar]
- Schwarze, S. R., DePrimo, S. E., Grabert, L. M., Fu, V. X., Brooks, J. D., and Jarrard, D. F. (2002). Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J. Biol. Chem. 277, 14877-14883. [DOI] [PubMed] [Google Scholar]
- Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593-602. [DOI] [PubMed] [Google Scholar]
- Shelest, E., Kel, A. E., Goessling, E., and Wingender, E. (2002). Prediction of potential C/EBP/NF-kappaB composite elements using matrix-based search methods. In Silico Biol. 3, 0007. [PubMed] [Google Scholar]
- Shelton, D. N., Chang, E., Whittier, P. S., Choi, D., and Funk, W. D. (1999). Microarray analysis of replicative senescence. Curr. Biol. 9, 939-945. [DOI] [PubMed] [Google Scholar]
- Shi, C. S., Lee, S. B., Sinnarajah, S., Dessauer, C. W., Rhee, S. G., and Kehrl, J. H. (2001). Regulator of G-protein signaling 3 (RGS3) inhibits Gbeta1gamma 2-induced inositol phosphate production, mitogen-activated protein kinase activation, and Akt activation. J. Biol. Chem. 276, 24293-24300. [DOI] [PubMed] [Google Scholar]
- Sikder, H. A., Devlin, M. K., Dunlap, S., Ryu, B., and Alani, R. M. (2003). Id proteins in cell growth and tumorigenesis. Cancer Cell 3, 525-530. [DOI] [PubMed] [Google Scholar]
- Stanelle, J., Stiewe, T., Theseling, C. C., Peter, M., and Putzer, B. M. (2002). Gene expression changes in response to E2F1 activation. Nucleic Acids Res. 30, 1859-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H., Tonks, N. K., and Bar-Sagi, D. (1994). Inhibition of Ras-induced DNA synthesis by expression of the phosphatase MKP-1. Science 266, 285-288. [DOI] [PubMed] [Google Scholar]
- Suzuki, H., Kawai, J., Taga, C., Yaoi, T., Hara, A., Hirose, K., Hayashizaki, Y., and Watanabe, S. (1996). Stac, a novel neuron-specific protein with cysteine-rich and SH3 domains. Biochem. Biophys. Res. Commun. 229, 902-909. [DOI] [PubMed] [Google Scholar]
- Sviderskaya, E. V., et al. (2003). p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J. Natl Cancer Inst. 95, 723-732. [DOI] [PubMed] [Google Scholar]
- Tavazoie, S., Hughes, J. D., Campbell, M. J., Cho, R. J., and Church, G. M. (1999). Systematic determination of genetic network architecture. Nat. Genet. 22, 281-285. [DOI] [PubMed] [Google Scholar]
- Tegner, J., Yeung, M. K., Hasty, J., and Collins, J. J. (2003). Reverse engineering gene networks: integrating genetic perturbations with dynamical modeling. Proc. Natl. Acad. Sci. USA 100, 5944-5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirone, F. (2001). The gene PC3(TIS21/BTG2), prototype member of the PC3/BTG/TOB family: regulator in control of cell growth, differentiation, and DNA repair? J. Cell Physiol. 187, 155-165. [DOI] [PubMed] [Google Scholar]
- Torres, C., Francis, M. K., Lorenzini, A., Tresini, M., and Cristofalo, V. J. (2003). Metabolic stabilization of MAP kinase phosphatase-2 in senescence of human fibroblasts. Exp. Cell Res. 290, 195-206. [DOI] [PubMed] [Google Scholar]
- Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98, 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, A. (2002). Estimating coarse gene network structure from large-scale gene perturbation data. Genome Res. 12, 309-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Wu, Q., Qiu, P., Mirza, A., McGuirk, M., Kirschmeier, P., Greene, J. R., Wang, Y., Pickett, C. B., and Liu, S. (2001). Analyses of p53 target genes in the human genome by bioinformatic and microarray approaches. J. Biol. Chem. 276, 43604-43610. [DOI] [PubMed] [Google Scholar]
- Weinmann, A. S., Yan, P. S., Oberley, M. J., Huang, T. H., and Farnham, P. J. (2002). Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 16, 235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt, O., Albig, W., and Doenecke, D. (1998). cAMP/phorbol ester response element is involved in transcriptional regulation of the human replacement histone gene H3.3B. Biochem. J. 329, 609-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Pan, K. H., and Cohen, S. N. (2003). Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc. Natl. Acad. Sci. USA 100, 3251-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Ramanathan, Y., Soteropoulos, P., Recce, M. L., and Tolias, P. P. (2002). EZ-Retrieve: a Web-server for batch retrieval of coordinate-specified human DNA sequences and underscoring putative transcription factor-binding sites. Nucleic Acids Res. 30, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipper, L. M., and Mulcahy, R. T. (2000). Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem. Biophys. Res. Commun. 278, 484-492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.