

Abstract
MOB1 is a multifunctional protein best characterized for its integrative role in regulating Hippo and NDR pathway signaling in metazoans and the Mitotic Exit Network in yeast. Human MOB1 binds both the upstream kinases MST1 and MST2 and the downstream AGC group kinases LATS1, LATS2, NDR1, and NDR2. Binding of MOB1 to MST1 and MST2 is mediated by its phosphopeptide-binding infrastructure, the specificity of which matches the phosphorylation consensus of MST1 and MST2. On the other hand, binding of MOB1 to the LATS and NDR kinases is mediated by a distinct interaction surface on MOB1. By assembling both upstream and downstream kinases into a single complex, MOB1 facilitates the activation of the latter by the former through a trans-phosphorylation event. Binding of MOB1 to its upstream partners also renders MOB1 a substrate, which serves to differentially regulate its two protein interaction activities (at least in vitro). Our previous interaction proteomics analysis revealed that beyond associating with MST1 (and MST2), MOB1A and MOB1B can associate in a phosphorylation-dependent manner with at least two other signaling complexes, one containing the Rho guanine exchange factors (DOCK6-8) and the other containing the serine/threonine phosphatase PP6. Whether these complexes are recruited through the same mode of interaction as MST1 and MST2 remains unknown. Here, through a comprehensive set of biochemical, biophysical, mutational and structural studies, we quantitatively assess how phosphorylation of MOB1A regulates its interaction with both MST kinases and LATS/NDR family kinases in vitro. Using interaction proteomics, we validate the significance of our in vitro studies and also discover that the phosphorylation-dependent recruitment of PP6 phosphatase and Rho guanine exchange factor protein complexes differ in key respects from that elucidated for MST1 and MST2. Together our studies confirm and extend previous work to delineate the intricate regulatory steps in key signaling pathways.
The Mps One Binder (MOB) 1 proteins comprise an evolutionarily conserved eukaryotic protein family (with sizes ranging from 210–314 amino acids), whose founding member was discovered in yeast through interaction screens with the kinase Mps1 (1) and shown to be critical for mitotic exit (reviewed in 2, 3). Two yeast MOB proteins, Mob1 and Mob2, exist in S. cerevisiae. They function as AGC group kinase regulators, with Mob1 facilitating the activation of the Dbf2 kinase in the Mitotic Exit Network pathway (4, 5) and Mob2 facilitating the activation of the Cbk1 kinase in the RAM (regulation of Ace2 and morphogenesis) network pathway (6).
Drosophila Mats (Mob as tumor suppressor; also known as dMOB1) was later identified through genetic screens in Drosophila eye development and found to encode a regulator of the Hippo pathway that controls cell proliferation and tissue growth (7). Consistent with the function of MOB proteins as kinase activators in yeast, Mats associates with and activates the Dbf2-related kinase Warts (8). However, unlike in the yeast system where Mob1 and Mob2 activate distinct kinases (Dbf2 and Cbk1), Mats, in addition to regulating Warts, also physically and functionally interacts with another AGC kinase, Tricornered, which is implicated in the morphogenesis of polarized cellular extensions and is a closer ortholog of Cbk1 (9). Additional complexity in insects arises through the presence of three additional MOB proteins, at least one of which (dMOB2) is also capable of physically and functionally associating with Tricornered, which binds Warts in a yeast two-hybrid assay (9); see Fig. 1A.
Fig. 1.
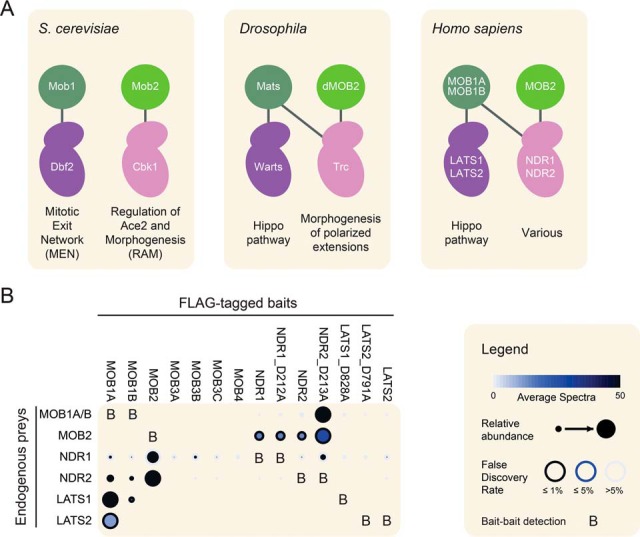
Specificity of interactions between MOB proteins and AGC kinases. A, Evolution of the interactions between MOB1, MOB2, and AGC family kinases. In S. cerevisiae, Mob1 and Mob2 contribute to the activation of different pathways through interactions with Dbf2 and Cbk1, respectively. In Drosophila and mammals, the Mob2 ortholog preferentially associates with Cbk1-related kinases while the Mob1 ortholog can associate with both Dbf2- and Cbk1- related kinases. MOB and kinases are colored to indicate their orthology to the yeast proteins; see text for details. B, Specificity of interaction between human MOB proteins and the indicated AGC kinases detected by affinity purification coupled to mass spectrometry from stable cell pools. Given the high identity between MOB1A and MOB1B, we could not uniquely assign peptides to either protein, and thus labeled them prey-wise as MOB1A/B. Also note that expression of wild-type LATS1 was not detected in the transfected pools and thus this data is not presented here. See Supplemental Tables 1 and 2 for the spectral count table and for the results from the HA-tagged experiment.
In mammals, the MOB family comprises seven members that can be grouped into 4 sub-families, each with orthology to one of the four Drosophila MOB proteins. Like Mats, the 96% identical MOB1A and MOB1B bind to and function to activate the AGC kinases LATS1 and LATS2 (large tumor suppressor) in the Hippo signaling pathway (10). This leads to the phosphorylation of the transcriptional co-activators YAP1 and TAZ (gene name WWTR1), and as a result, their cytosolic sequestration away from transcriptional targets (11, 12). Importantly, MOB1A and MOB1B also bind the Tricornered ortholog kinases NDR1 and NDR2 (nuclear Dbf2-related, gene names STK38 and STK38L, 13, 14), though the downstream signaling consequences of these events are less well understood. Like in flies, human MOB2 can also associate with the NDR1 and NDR2 proteins, though in an apparently different mode from MOB1A, and with opposing consequences as it acts as a negative regulator of these kinases (15). The other human MOB proteins (MOB3A, MOB3B, MOB3C and MOB4) do not appear to associate with LATS or NDR kinases (15, 16).
In addition to directly binding and allosterically activating their downstream AGC kinase partners, MOB proteins also facilitate their activation indirectly by bridging interactions with Ste20 related kinases, namely Cdc15 (17) and Kic1 (18) in yeast (through Mob1 and Mob2, respectively), Hpo (19) in flies, and MST1 and MST2 (gene names STK4 and STK3, 20) in mammalian cells. This bridging function of MOBs allows the Ste20 related kinases to phosphorylate specific AGC family kinases on positive regulatory sites within their C-terminal regulatory tails (17, 20–25).
The molecular basis by which MOB recognizes Ste20 related kinases was revealed by the discovery of Rock et al. that yeast Mob1 functions as a phospho-recognition module (26) and that human MOB1A and MOB1B exploit a conserved phospho-recognition infrastructure to bind MST1 and MST2 (27). Ni et al. (28) provided a structural view of MOB1B bound to a phosphorylated peptide derived from MST2 (pT378). The studies described in the accompanying manuscript (29) expand on these findings by defining the consensus binding preference (and binding mode) of MOB1 for phosphopeptides. Together with the elucidated MST1 phosphorylation consensus, this allows for a full rationalization of the redundancy of phosphosites present in MST1 that mediate interaction with MOB1 in a cellular context.
We note however, that MST1 and MST2 are not the only proteins that interact with MOB1 in a phosphorylation-dependent manner, and interaction proteomic analyses (27) showed apparently mutually exclusive and phosphorylation-dependent interaction with at least three independent protein modules (supplemental Fig. S1), namely: (1) the upstream Hippo kinase components including MST1 and MST2; (2) the phosphoprotein phosphatase 6, PPP6C, and its regulatory subunits (30, 31, here called the PP6 module); (3) a module comprised of the atypical guanine nucleotide exchange factors (DOCK6-8, ref 32) and poorly characterized associated proteins, including leucine rich repeats and calponin homology domain containing (LRCH1-4) proteins and cytokine receptor like factor 3 (CRLF3). Whether the structural elements uncovered by Rock et al. (26), Ni et al. (28), Kim et al. (33) and the accompanying paper (29) explain the phosphorylation-dependent recruitment of these additional complexes to MOB1 remains to be determined.
Importantly, in the context of the Hippo signaling pathway, MOB1A and MOB1B are also substrates for MST1 and MST2, with T12 and T35 acting as the predominant phosphorylation sites (34), though additional sites have been detected in phosphoproteomics studies (35). Recent structural studies have revealed a role for MOB1 phosphorylation in regulating MOB1 interaction with both upstream (Ste20) and downstream (AGC) kinases. Kim et al. reported the crystal structure of full-length murine Mob1b to reveal that in the absence of phosphorylation, the N terminus of Mob1b binds in an intramolecular manner to its Lats1 binding surface, thereby impeding Lats1 kinase interaction (33). As shown in an associated manuscript (29), this autoinhibitory feature is also employed by human MOB1A and is compatible with MOB protein binding to optimal phosphopeptide ligands. Ni et al. reported how the phosphorylated form of murine Mob1b binds to Lats1. In that structure, the phosphorylated N terminus of Mob1b engages its own phosphopeptide-binding infrastructure to expose the Lats1 binding surface, thus enabling Lats1 binding. Together, these studies shed light on the underlying binding modes and phospho-regulation mechanisms governing core Hippo pathway interactions. However, many questions of protein function and significance remain unaddressed, most notably because these studies relied heavily on in vitro models, with in vivo validation only provided in the context of the yeast Cbk1-Mob2 complexes (36).
In this study, we performed an in-depth characterization of the role of the MOB1 phosphorylation on its interaction with both the upstream MST1 and MST2 kinases and the downstream LATS and NDR family kinases. We show that MST kinase-generated phosphosites in MOB1 are suboptimal compared with its phosphopeptide-binding consensus. We also show that substitution of these suboptimal phosphosites with a sequence better matching the optimal binding consensus determined in the accompanying paper (29) has an impact on the ability of MOB1 to autoregulate its interactions in cells. Lastly, our proteomics cell-based studies reveal that MOB1 likely employs different modes of action to engage molecular complexes such as PP6 and DOCK6-8 as compared with MST1 and MST2, which hints at further complexities for future investigation.
EXPERIMENTAL PROCEDURES
Recombinant Protein Expression and Purification
NDR1 (amino acids 12–418, NP_009202.1), MOB1A (2–216, NP_060691) wild type and indicated MOB1A mutant proteins were expressed in E. coli BL21 (DE3) CodonPlus RIL cells as N-terminal dual 6xhistidine (HIS) and glutathione S-transferase (GST) Tobacco Etch Virus (TEV) cleavable fusion proteins using a modified pETM-30 vector. MOB1A wild type and mutants were purified in batch on glutathione-Sepharose resin and eluted by cleavage from the affinity tags with HIS tagged TEV protease. TEV protease was removed from the eluted protein by subtractive immobilized-metal affinity chromatography. Cleaved protein was then concentrated and buffer exchanged by size exclusion chromatography (SEC) using a Superdex 75 120 ml column (GE Healthcare, Uppsala, Sweden).
Crystallization and Data Collection
Crystals of purified MOB1A in the space group C2221 (a = 86.1, b = 86.2, c = 138.1, alpha = 90, beta = 90, gamma = 90) with two molecules in the asymmetric unit were obtained using the hanging drop method by mixing protein (at 7 mg/ml) and precipitant solution (0.1 m MES pH 5.5, 0.2 m NH4Cl, and 20% PEG 6000) in a 1:1 ratio.
For diffraction studies, all protein crystals were flash-frozen in mother liquors supplemented with 20–25% (v/v) ethylene glycol. Diffraction data collection was performed at the NE-CAT beamline 24-ID-E (Advanced Photon Source, Argonne National Laboratory, Argonne, IL).
Structure Solution and Refinement
X-ray data sets were processed using HKL2000 software (37). The MOB1A structure was solved by molecular replacement with Phaser - CCP4i using the MOB1A core domain (38, PDB ID: 1PI1) as a search model. Model autobuilding was carried out in Buccaneer to build the N-terminal extension of the MOB1A (39). The final structure model generated by iterative model building in COOT (40) was refined using Refmac (41). See supplemental Table S3 for data collection and model refinement statistics. The crystal structure of full-length human MOB1A in its apo state was deposited with the Protein Data Bank under the PBD code 5TWF.
Peptides for Biophysical Studies
FITC labeled nonphosphorylated and phosphorylated variants of MST1 T353 peptide (FITC-VASTMTDGAN(p)TMIEH), T367 peptide (FITC-DDTLPSQLG(p)TMVINA), MOB1A T12 peptide (RSSK(p)TFKPKKNIPEG), MOB1A T35 peptide (FITC-AEA(p)TLGSGNLRQAVM) and MOB1A12–38 peptide (FITC-RSSK(p)TFKPKKNIPEG) for fluorescence polarization binding experiments were purchased from Biomatik (Cambridge, ON).
Fluorescence Polarization Peptide Binding Assay
Twelve-point fluorescence polarization binding measurements were performed in triplicate in 384-well plate format using an Analyst HT (Artisan-Scientific, Sunnyvale, CA) reader. Each 20 μl measurement condition contained 20 nm FITC labeled MST1 peptide and the indicated concentration of MOB1 protein in 25 mm HEPES pH 7.5, 150 mm NaCl, 2 mm DTT, and 1 mg/ml bovine serum albumin (BSA). For competitive displacement assays, each 20 μl measurement condition contained 350 μM MOB1A core domain (residues 52–216), 20 nm FITC-MOB1A21–38 probe (giving rise to ∼80% saturated binding signal) and the indicated concentration of competitor protein, NDR112–418. Kd and IC50 values were obtained by fitting the results to a one-site specific binding model using GraphPad Prism.
Microscale Thermophoresis
MOB1A-NDR1 binding studies were performed in triplicate using a Monolith NT.115 (Nanotemper Technologies, Munich, Germany) according to manufacturer's instructions. Briefly, recombinant WT and mutant MOB1A proteins were labeled with the RED-NHS (amine reactive) protein labeling kit (Nanotemper Technologies). 50 nm of labeled WT or mutant MOB1A were mixed with the indicated concentrations of unlabeled NDR112–418 in 20 mm Tris pH 8.0, 150 mm NaCl, 1 mm TCEP, 0.05% Tween-20 and 0.1% BSA, incubated for 10 min at room temperature, and loaded into premium capillaries. Measurements were conducted at 20 to 30% laser power and 30% LED power. Kd values were obtained by fitting the results to a one site-specific binding model using GraphPad Prism.
Analytical ultracentrifugation (AUC)
Sedimentation-velocity AUC was performed with a Beckman ProteomeLab XL-1 at 169,000 r.c.m. Data were obtained after 7.5 h of centrifugation at 20°C by monitoring the relative refractive index between sample and blank. Various concentrations of MOB1AWT, ranging from 20 μM to 150 μM, were tested minimally in duplicate in AUC buffer (25 mm HEPES, pH 7.5, 100 mm NaCl, and 2 mm DTT). AUC data was processed using Sedfit software (NIH, Bethesda, MD).
Western blot
Samples were resolved on 10% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane, probed with the primary antibodies as indicated in legends, followed by fluorescent secondary antibodies for mouse (CW800, LICOR, Lincoln, NE) or rabbit (CW680, LICOR), and then directly imaged on an Odyssey scanner (LI-COR). LATS antibody was from Cell Signaling Technology (#9153), PPP6C antibody was previously described (27) and anti-tubulin was from the Developmental Studies Hybridoma Bank at the University of Iowa.
Stable Human Cell Line Generation for MOB1A mutants
3xFLAG tagged MOB1A constructs were generated via Gateway cloning into pDEST 5′ Triple FLAG pcDNA5 FRT TO. The accession number for the starting clone was BC003398. Point mutations were generated by polymerase chain reaction-directed mutagenesis (the position of the mutated amino acids is indicated on the basis of the reference sequence). All constructs were sequence verified. Stable cell lines were generated as Flp-In 293 T-REx cell pools as described (42), and expression was induced for 24 h with tetracycline (1 μg/ml). Okadaic acid was added at a concentration of 150 nm for 2.5 h unless otherwise indicated, and DMSO was used as a negative control.
FLAG Affinity Purification Coupled with Mass Spectrometry (AP-MS) for MOB1A Mutants
Cell pellets from one 150 mm plate were lysed in 50 mm Hepes-KOH (pH 8.0), 100 mm KCl, 2 mm EDTA, 0.1% Nonidet P-40, and 10% glycerol and affinity-purified with M2-FLAG magnetic beads and on-bead digestion as described (42). Peptides were analyzed by nano-LCMS using a home-packed 0.75 μm × 10 cm C18 emitter tip (Reprosil-Pur 120 C18-AQ, 3 μm). A NanoLC-Ultra HPLC system (Eksigent, Dublin, CA) was coupled to an LTQ Orbitrap Velos (Thermo Fisher Scientific, Waltham, MA) and samples were analyzed in data-dependent acquisition mode. A 60,000 resolution MS scan was followed by 10 CID MS/MS ion trap scans on multiple charged precursor ions with a dynamic exclusion of 20 s. The LC gradient was delivered at 200 nl/m and consisted of a ramp of 2–35% acetonitrile (0.1% formic acid) over 90 min, 35–80% acetonitrile (0.1% formic acid) over 5 min, 80% acetonitrile (0.1% formic acid) for 5 min, and then 2% acetonitrile for 20 min. This data set consisting of 26 raw files and associated peak list and results files has been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080331 and PXD005327, ftp://massive.ucsd.edu/MSV000080331.
MS Data Analysis For FLAG AP-MS For MOB1A Mutants
Raw files were converted to mzXML and mgf files using ProteoWizard 3.0.4468 (43) and analyzed using the iProphet pipeline (44) implemented within ProHits (45) as follows. The database consisted of the human and adenovirus sequences in the RefSeq protein database (version 57) supplemented with “common contaminants” from the Max Planck Institute (http://141.61.102.106:8080/share.cgi?ssid=0f2gfuB) and the Global Proteome Machine (GPM; http://www.thegpm.org/crap/index.html). The search database consisted of forward and reverse sequences (labeled “gi 9999” or “DECOY”); in total, 72,226 entries were searched. Spectra were analyzed separately using Mascot (2.3.02; Matrix Science) and Comet [2012.01 rev.3 (46)] for trypsin specificity with up to two missed cleavages; deamidation (Asn or Gln) and oxidation (Met) as variable modifications; the mass tolerance of the precursor ion was set at ±12 parts per million (ppm), the fragment ion tolerance at ± 0.6 amu. The resulting Comet and Mascot results were individually processed by PeptideProphet (47) and combined into a final iProphet output using the Trans-Proteomic Pipeline (TPP; Linux version, v0.0 Development trunk rev 0, Build 201303061711). TPP options were as follows: general options were -p0.05 -x20 -d “gi 9999,” iProphet options were -ipPRIME, and PeptideProphet options were -OpdP. All proteins with a minimal iProphet probability of 0.05 were parsed to the relational module of ProHits. For analysis with SAINT, only proteins with an iProphet protein probability of >0.95 were considered. Hits were also restricted to those detected with a minimum of two unique peptides.
Interaction Scoring For FLAG AP-MS MOB1A Mutants
For each bait protein, one plate per cell line was treated with 150 nm okadaic acid in DMSO or with DMSO alone for 2.5 h before harvesting. Samples were prepared for MS analysis, as described above, in biological duplicate from cells grown, treated, and processed at different times to maximize the variability and increase the robustness in the detection of true interactors. Cells expressing the FLAG tag alone were used for negative controls and processed in parallel to mitigate “batch effect” artifacts (48). The quality of each sample was assessed by manually aligning the runs for the biological replicates in ProHits. Samples of low quality were discarded, and additional biological replicates were acquired. SAINTexpress version 3.6.1 was used with default options (49). Six negative control experiments (three treated with DMSO and three treated with OA) were compressed to 4 virtual controls for SAINTexpress scoring to increase robustness (as in ref 49). Proteins with FDR of ≤1% were considered true positive interactions. Fold change was calculated for each prey protein as the ratio of average spectral counts from replicate bait purifications over the average spectral counts across all negative controls (peptide spectral counts were summed for each protein, ref 50). Visualization of the interactions as dot plots was through prohits-viz.lunenfeld.ca, and was first introduced in (51); once a particular prey passes the selected FDR threshold for at least one bait, all the quantitative data across all baits are retrieved and displayed. On these dot plots, the color intensity maps to the averaged spectral counts across both replicates (capped at a maximal value), whereas the size of the circles is proportional to the maximal spectral count value for the bait across all samples analyzed in parallel. The confidence score from SAINTexpress is mapped as the edge color.
Determination of the Specificity of Human MOB/AGC Kinase Interactions by AP-MS
Human MOBs and NDR/LATS kinases were cloned into pcDNA3-FLAG and pcDNA3–3HA (16) from cDNA collections and match following NCBI RefSeq entries: MOB1A (NP_060691), MOB1B (NP_001231695), MOB2 (NP_001165694), MOB3A (NP_570719), MOB3B (NP_079037), MOB3C (NP_660322), MOB4 (NP_056202). The NDR1 D212A, NDR2 D213A, LATS1 D828A, and LATS2 D791A kinase dead point mutations were generated by overlap extension PCR and constructs were sequenced.
For each biological replicate, stable pools of FLAG-tagged HEK293 (generated as in ref 42) were grown in five 15 cm plates to ∼80% confluency before harvesting in ice-cold PBS using a rubber spatula. After pelleting the cells by centrifugation and washing them once in ice-cold PBS, the cell pellets were frozen on dry ice. The frozen cell pellets were resuspended in ice-cold lysis buffer (50 mm HEPES-NaOH pH 8.0, 100 mm KCl, 2 mm EDTA, 10 mm NaF, 0.1% Nonidet P-40, and 10% glycerol), supplemented with 1 mm PMSF, 1 mm DTT and 1× protease inhibitor mixture (Sigma-Aldrich, St. Louis, MO) at a 1:4 pellet weight: volume ratio. Phosphatase inhibitors were freshly added to the lysis buffer (50 mm β-glycerolphosphate, 5 nm okadaic acid and 5 nm calyculin). Cells were lysed on a nutator at 4°C for 10 min. One freeze-thaw cycle was performed by incubating the tube on dry ice for 10 min, then transferring it to a 37°C water bath with agitation, then storing it on ice. The sample was spun down to remove cell debris from the sample (20 min at >16,000 × g, 4°C). The supernatant was transferred to a tube containing pre-washed anti-FLAG M2 agarose beads (A2220, Sigma-Aldrich; 7.5μl packed beads per experiment). Immunoprecipitation was performed for 3 h at 4°C with gentle agitation on a nutator. Beads were pelleted by centrifugation (500 × g for 1 min) and the supernatant was removed. The beads were transferred to a 1.5 ml disposable column and washed three times in 1 ml lysis buffer and three more times with FLAG rinsing buffer (50 mm NH4HCO3, pH 8.0 and 75 mm KCl) by forcing buffer through the column with a pipette bulb. Proteins were eluted 3 times by adding 150 μl of elution buffer (0.5 m NH4OH, pH 11.0–12.0), flicking the column to mix and incubating for 2–5 min at room temperature and the eluate was collected in a microfuge tube. The eluate was lyophilized in a centrifugal evaporator. To ensure removal of any remaining NH4OH, which may impede trypsin digestion, the sample was washed with 200 μl of water and lyophilized again.
Proteins were digested with 750 ng trypsin (T6567, Sigma-Aldrich) at 100 ng/μl in 50 mm NH4HCO3, pH 8.0 overnight at 37°C. The next day, 250 ng trypsin was added to continue the digestion for another 2–3 h. Formic acid was added to the sample to a final concentration of 2% (from 50% stock solution) and the mixture was lyophilized in a centrifugal evaporator. The lyophilized peptides were resuspended in 20 μl of HPLC reversed-phase buffer A (2% acetonitrile, 0.1% formic acid), spun down, and transferred to a fresh tube. The samples were either stored at −20°C or directly loaded (6 μl) onto a column for analysis by mass spectrometry.
Microcapillary reversed-phase columns (75-μm inner diameter, 363-μm outer diameter; Polymicro Technology, Phoenix, AZ) were cut to a final length of 15–20 cm, and spray tips were pulled in-house by hand. Columns were packed in-house (12 cm) with Magic C18 100-Å, 5-μm silica particles (Michrom, Auburn, CA) using a pressure bomb. Prior to loading the sample, columns were equilibrated in HPLC buffer A. Sample was applied to the column using a pressure bomb and then washed off line in buffer A for 10 min. The loaded column was then placed in-line with a LTQ mass spectrometer equipped with an Agilent 1100 pump with split flow and a Proxeon source. Buffer A is 2% ACN, 0.1% formic acid; buffer B is 98% ACN, 0.1% formic acid. The HPLC gradient program delivered an acetonitrile gradient (5–14% acetonitrile over 5 min, 14–40% over 60 min, and 40–80% over 10 min). The parameters for data dependent acquisition on the mass spectrometer were: 1 centroid MS (mass range 400–2000) followed by MS/MS on the 2 most abundant ions. General parameters were: activation type = CID, isolation width = 3, normalized collision energy = 32, activation Q = 0.25, activation time = 30 msec, wideband activation. For data dependent acquisition, minimum threshold was 1000, the repeat count = 1, repeat duration = 30 s, exclusion size list = 500, exclusion duration = 30 s, exclusion mass width (by mass) = low 1.2, high 1.5.
Mass spectrometry data was stored, searched and analyzed using the ProHits laboratory information management system (LIMS) platform, through conversion with ProteoWizard and analysis through Mascot, Comet and the Trans-Proteomic Pipeline against the modified RefSeq database (version 57) defined above, with the exception that the charges +2, +3, and +4 were considered, with the parent mass tolerance set at 3 amu and a fragment ion tolerance at ± 0.6 amu. Two unique peptides ions and a minimum iProphet probability of 0.95 were required for protein identification.
SAINTexpress version 3.3 used as a statistical tool to calculate the probability value of each potential protein-protein interaction from background contaminants (see above). Thirteen negative controls consisting of cells expressing the tag alone (please note that some of these controls were used for a previous publication, ref 52) were used for statistical assessment: they were compressed to five virtual controls (50). Spectral counts for MOB, LATS, and NDR proteins were extracted alongside the SAINT significance across both biological replicates and used in Fig. 1B (data visualized with tools first defined in ref 51) and supplemental Table S2. Because NDR1 (STK38) is frequently identified as a contaminant in FLAG affinity purifications (50), a separate set of stable cells expressing HA-tagged versions of the MOB proteins and the kinases were generated and used to profile interactions in one additional experiment as described above, except that the anti-HA beads (A2095; Sigma-Aldrich, St. Louis, MO) were used for affinity purification. Since a single control purification was included as part of this project, we supplemented this control with controls from the CRAPome (50, CC51–54 were used), compressed them to two virtual controls and performed SAINTexpress analysis as above. Spectral counts for MOB, LATS, and NDR proteins were extracted alongside the SAINT significance across both biological replicates and included in supplemental Table S3. Complete results have been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080570 and PXD005966, ftp://massive.ucsd.edu/MSV000080570.
The data is further available at ProHits-web.lunenfeld.ca (project: “phospho-dependent Hippo interactions”).
Intact Mass Determination by Mass Spectrometry
The phosphorylation status of wild type and mutant MOB1A proteins were determined using ESI-LC/MS at the AIMS Mass Spectrometry Laboratory in the Department of Chemistry at the University of Toronto. 20 μl of 200 μM protein samples in 25 mm HEPES pH 7.5, 100 mm NaCl, and 2 mm DTT were submitted for analysis. Samples were diluted in water 1000-fold prior to analysis using an Agilent 6538 Q-TOF interfaced to a 1290 ultrahigh pressure HPLC. A list of the ionic formulae was generated by Molecular Formula Generation Algorithm as implemented in the MassHunter data processing software. The Molecular Formulae Generation Score considers the best match for the mass accuracies and relative abundances of the isotopomers identified for the target isotopic envelop. The standard m/z acquisition range for biomolecules analysis was set between 200–2000 Da; however, the mass spectra shown were expanded to highlight the appropriate regions of interest. The reconstructed mass spectra were generated by the Maximum Entropy algorithm in a plot of neutral mass (Da) versus intensity. Special peaks were labeled with mass values rounded to one decimal place. The term “deconvolute” used in the figures refers to the process of spectral reconstruction employed in the MassHunter BioConfirm software package.
Experimental Design and Statistical Rationale
For the interaction studies using MOB1A mutants, biological duplicates were employed, and statistical scoring against six negative controls (compressed to four) was performed using Significance Analysis of INTeractome (SAINT) as described in “Interaction Scoring for AP-MS.” For the data presenting the specificity of interaction between MOB, LATS and NDR proteins, the thirteen negative controls were compressed to five virtual controls. Average SAINT score was used to determine the Bayesian FDR, which therefore requires a high confidence interaction across both biological replicates.
RESULTS
Interactions Between Human MOB Proteins and NDR and LATS Kinases Defined by Interaction Proteomics
Previous work in flies (9) and human cell lines (13, 14) revealed specificity in the association of specific MOB family members with AGC family kinases. However, these studies were often performed with overexpression of both partners or by yeast two-hybrid analysis. To verify in a cellular system the specificity of interactions between the seven human MOB proteins and the four related AGC group kinases LATS1, LATS2, NDR1 and NDR2, we generated stable cell lines that expressed a FLAG-tagged version of each protein, with the kinases in both wild type and kinase-dead forms. Affinity purification (using anti-FLAG coupled to agarose resin) was performed in biological duplicates, and the precipitates analyzed by mass spectrometry. The reciprocal nature of the experimental setup served to eliminate false positives in our dataset, whereby in one case the MOB protein is tagged and expressed in cells at relatively low levels and the endogenously expressed protein interactors are identified, and vice versa for the kinases. Consistent with previous data, MOB1A and MOB1B recovered both LATS and NDR family kinases, while MOB2 only recovered the two NDR kinases (Fig. 1B; supplemental Table S1). LATS only recovered peptides for MOB1A/B (though they did not pass our significance threshold), while the NDR kinases recovered MOB2 and the kinase-dead mutant of NDR2 recovered proportionally large amounts of MOB1A/B. None of the other MOB proteins precipitated significant amounts of LATS or NDR kinases nor were they themselves recovered through precipitation of the kinases (Fig. 1B and supplemental Table S1). Because NDR1 (STK38) is a frequent contaminant in FLAG purification experiments (50), we also analyzed one biological replicate of cells stably expressing HA-tagged versions of these proteins, and this confirmed our FLAG data (supplemental Table S2). Together, these results confirmed the specificity of the MOB1-LATS interaction and the MOB1 or 2-NDR interaction. To further investigate the interaction between MOB1 and AGC group kinases below, we employed NDR1 as a representative model for our in vitro MOB1 interaction studies, as it was producible in ample quantities in bacteria, in contrast to the human LATS kinases.
Nonphosphorylated Full-length Human MOB1A Adopts an Autoinhibitory Conformation that Occupies the NDR1 Kinase Binding Site
Kim et al. showed that the N-terminal extension of murine Mob1b in its dephophorylated state occludes the projected AGC group kinase-binding site and that this inhibition can be relieved in vitro by phosphomimetic mutations of T12 and T35 within the N-terminal extension or by deletions within this region (33). We observed the same autoinhibitory conformation of the N-terminal extension of human MOB1A while bound to high affinity phosphopeptide ligands from MST1 (see accompanying manuscript by 29), and also in a newly determined 3.1 Å X-ray crystal structure of human MOB1A in its apo state (Fig. 2A and supplemental Table S3).
Fig. 2.

Full-length nonphosphorylated MOB1A adopts an autoinhibitory conformation that impedes NDR family kinase binding. A, Crystal structure of full-length nonphosphorylated human MOB1A. N-terminal regulatory extension highlighted in red, linker region in orange, and core domain in green. B, Superimposition of full length nonphosphorylated human MOB1A and the yeast Cbk1251–756 (human NDR1 ortholog)-Mob245–287 (human MOB1 ortholog) complex (PDB 4LQS) (36). Note the steric clash between the nonphosphorylated N-terminal extension of MOB1A and the MOB1 binding domain (MBD) of CBK1. C, Domain architecture of wild-type MOB1A and the N-terminal truncation mutants used in the following panels. D, The N-terminal extension of MOB1A binds to the core domain of MOB1A in trans. Fluorescence polarization measurement of the binding affinities between MOB1A N-terminal truncation mutants and a FITC-MOB1A21–38 peptide. Data is plotted as the mean ± S.E. (n = 3). E, The N-terminal extension of MOB1A competes for the same surface on the MOB1A core domain as NDR1. Fluorescence polarization measurement of the displacement of FITC-MOB1A21–38 peptide from MOB1AΔ52 (core domain) by NDR112–418. Data is plotted as the mean ± S.E. (n = 3).
In brief, in the apo crystal structure of nonphosphorylated full length human MOB1A, the N-terminal extension preceding the core domain forms a 4-turn α-helix (residues 24 to 38 denoted helix α-1) followed by a short 1-turn helix (residues 41 to 45 denoted helix α0). These elements buttress against the MOB1A core in cis on a surface shown previously to mediate binding to the AGC group kinases (Note that residues 1 to 22 of MOB1 are disordered, also see ref 28, 36). Although the conformation of the N-terminal region of human MOB1A is in agreement with that reported for murine Mob1b (Fig. 2A, 2B and supplemental Fig. S2A, also see 33), it differs greatly from that reported for the yeast protein (53), where the corresponding N-terminal region participates in the homodimerization of yeast Mob1 (supplemental Fig. S2B). Consistent with the conformation in the apo crystal structure (Fig. 2A) and the behavior of isolated murine Mob1b in solution (33), human MOB1A in isolation was a monomer in solution as assessed by analytical ultracentrifugation (supplemental Fig. S3).
We predicted that the observed intramolecular mode of interaction between the human MOB1A core and the nonphosphorylated N-terminal extension would sterically impinge on the binding of MOB1A to NDR1 (as inferred in Fig. 2B and Fig. 3A), a prediction Kim et al. also made for murine Mob1b binding to murine Lats1 (28). To investigate the influence of the N-terminal extension of MOB1A on protein function, we first synthesized a FITC-labeled peptide corresponding to helix α-1 residues 21–38 (denoted FITC-MOB1A21–38), and tested it for binding to a series of MOB1A deletion constructs in trans using a fluorescence polarization assay (Fig. 2C, 2D). Consistent with expectations, the FITC-MOB1A21–38 peptide bound to MOB1A deletion mutants lacking the first 38 and 51 N-terminal residues. Binding, however, was appreciably weaker (> 5-fold) for MOB1A proteins harboring the full length N-terminal extension or a deletion mutant lacking the first 13 N-terminal residues, both of which could satisfy N-terminal binding to the core domain of MOB1A in cis.
Fig. 3.

Phosphorylation on T12 and T35 induces switch-like conformational changes in MOB1A. A, Superimposition of full-length nonphosphorylated human MOB1A onto the human pMOB1B10–212 - LATS1602–704 complex (PDB 5BRK) (28). Colors for nonphosphorylated MOB1A are as in Fig. 2A. The N-terminal extension of pMOB1B10–212 is shown in blue; LATS1 in gray. B, Phosphorylation of MOB1A reduces its binding affinity for MST1 phosphopeptides. Fluorescence polarization analysis of MOB1A and phospho-MOB1A binding to MST1 pT353 and pT367 peptides. Data is plotted as the mean ± S.E. (n = 3). Note that the measurements for the unphosphorylated MOB1 protein are from (29). C, Phosphorylation of T12 and T35 sites in MOB1A collaborate to reduce binding affinity for MST1 phosphopeptides. Fluorescence polarization analysis of the binding of MOB1A mutants to MST1 phosphopeptides. Data is plotted as the mean ± S.E. (n = 3).
Next, we took advantage of FITC-MOB1A21–38 binding to the MOB1A core domain in trans to monitor the ability of the MOB1A core to bind downstream NDR1 kinase using a competitive displacement assay. For this, we titrated unlabeled NDR1 (12–418) into the preformed complex of FITC-MOB1A21–38 and MOB1A52–216. We observed that increasing concentrations of NDR1 could effectively displace the FITC-MOB1A21–38 peptide, consistent with our inference that the N-terminal extension of MOB1A occupies the same surface on the core domain as NDR1 (Fig. 2E). Thus, we concluded that the crystal structure of full-length MOB1A was reflective of a protein conformation sampled in solution wherein the N-terminal extension engages the core domain of MOB1A in a manner that can competitively impede binding of NDR1.
Phosphorylation of MOB1 by MST1 on T12 and T35 Facilitates Binding to NDR1 and Represses MOB1 Binding to MST1 Phosphopeptides
We determined by intact mass spectrometry that recombinant MST1 could efficiently and stoichiometrically phosphorylate bacterially expressed MOB1Ain vitro on two sites. We confirmed the sites as being T12 and T35 within the N-terminal extension by mutational analysis followed by in vitro kinase reactions and intact mass measurements (supplemental Fig. S4A). Both T12 and T35 sites in MOB1A closely match the MST1 phosphorylation consensus determined in the accompanying paper (29; supplemental Fig. S5A). We next tested the effect of MOB1A phosphorylation on the ability of full length MOB1A to interact with FITC-MOB1A21–38. Strikingly, the binding affinity of phosphorylated full length MOB1A for the FITC-MOB1A21–38 peptide was enhanced relative to the nonphosphorylated form, to a level comparable to MOB1AΔ51 (Fig. 2D). This finding suggested that MOB1A phosphorylation could drive a change in the conformation of the N-terminal extension to increase the interaction potential of the core domain to nonphosphorylated ligands (in this case FITC-MOB1A21–38, but by inference targets such as NDR1 which engage the same surface).
Comparison of the crystal structure of nonphosphorylated human MOB1A presented here to the crystal structure of the T12 and T35 doubly phosphorylated MOB1B solved previously (28) hinted at the basis for the underlying phosphorylation-dependent conformational change (Fig. 3A). Engagement of the T12 phosphosite by the phospho-binding pocket on one surface of MOB1A would partially displace downstream portions of the N-terminal extension from their autoinhibitory position on the opposite surface of MOB1A due to the shortness of the intervening sequence (i.e. there are only eight residues available to span a path greater than > 28Å). Phosphorylation of the T35 site in contrast would likely destabilize the autoinhibitory conformation of the N-terminal tail both by steric effects arising from the bulkiness of the phosphate moiety and by disruption of hydrophobic interactions (supplemental Fig. S5B).
We next investigated the consequence of MOB1 phosphorylation on the ability of MOB1 to bind phosphopeptide ligands derived from MST1 in vitro. Consistent with the ability of the T12 site to occupy the MOB1B phospho-binding pocket in cis in response to phosphorylation, phospho-MOB1A displayed a 50-fold reduction in binding affinity for MST1 phosphopeptides in trans relative to nonphosphorylated MOB1A (Fig. 3B; please note that the measurements for the unphosphorylated MOB1 protein are from 29). Thus, the phosphorylation of MOB1A on the T12 and T35 sites appeared to both potentiate binding of the downstream NDR1 kinase while attenuating interaction with the upstream MST1 and MST2 kinases.
Relative Importance of T12 and T35 Site Phosphorylation in Downregulating MOB1 Phosphopeptide Binding
The relative importance of T12 and T35 site phosphorylation in regulating the phosphopeptide binding function of MOB1 has not been extensively explored. However, the crystal structure of phosphorylated human MOB1B (28) suggested that phosphorylation of the T12 site would exert a more pronounced effect as this residue directly occupies the phosphopeptide binding pocket of MOB1B (28). To investigate the relative importance of site phosphorylation on the ability to regulate MOB1 binding to MST1 phosphopeptides, we generated and functionally characterized alanine mutations at each site. In contrast to MOB1AWT protein, which displayed a 50-fold loss of binding to MST1 phosphopeptides in response to MOB1A phosphorylation by MST1, the T35A MOB1A mutant (denoted MOB1AT35A) with the T12 site intact displayed no loss in binding whereas the T12A MOB1A mutant (denoted MOB1AT12A) with the T35 site intact displayed an intermediate 6-fold loss of binding (Fig. 3C and supplemental Fig. S4A). These results indicated that the ability of MOB1A phosphorylation to maximally downregulate MST1 phosphopeptide binding was dependent on the concerted action of both T12 and T35 site phosphorylation.
The observation that the MOB1AT12A mutant retained some regulatory function whereas the MOB1AT35A mutant did not was surprising since this implied that the T35 phosphosite on its own was more competent at occupying the MOB1 phosphopeptide binding pocket than the phospho-T12 site, an apparent contradiction to expectations from the crystal structure (28). We reason that the displayed binding preference for the phospho-T12 site ‘in crystallo’ might reflect an artifact of crystallization, or alternatively, that the T35 phosphosite does engage the phosphopeptide-binding pocket of MOB1, but only in the absence of T12 phosphorylation (Fig. 3C). The contrasting behavior of the single site MOB1A mutants in vitro raised the intriguing possibility that differential phosphorylation of MOB1 could be exploited for graded signaling responses in a cellular context.
Relative Importance of N-terminal Extension Features and Phosphorylation in Regulating MOB1 Interaction with NDR1
We next investigated the effect of several N-terminal extension mutations in MOB1A on binding to the downstream kinase NDR1 using microscale thermophoresis. MOB1A proteins were labeled with RED-MALEIMIDE dye (NanoTemper) and thermophoretic changes reflective of binding were assessed in response to the titration of unlabeled NDR1 kinase (Fig. 4A). As expected, nonphosphorylated MOB1AWT bound weakly to NDR1 kinase, but this interaction could be potentiated nearly 10-fold by phosphorylation (Fig. 4A i). Addition of the predicted destabilizing mutations H24A/Q25A (small hydrophobic to large hydrophilic residues on the contact surface of helix α-1 with the core domain of MOB1A) partially increased binding to NDR1 in the case of nonphosphorylated MOB1A. However, full binding of the MOB1H24A/Q25A mutant to NDR1 could still be achieved via phosphorylation by MST1, consistent with the fact that the two regulatory sites remained intact (Fig. 4A ii).
Fig. 4.

Mutations in the N-terminal extension of MOB1A alter binding to upstream MST1 and downstream NDR1 kinases. A, Microscale Thermophoresis binding analysis of NDR112–418 to the indicated MOB1A mutants. Data is plotted as the mean ± S.E. (n = 3). B, Fluorescence polarization binding analysis of the indicated MOB1A mutants to MST1 pT367 (top) and MST pT353 (bottom) peptides. Data is plotted as the mean ± S.E. (n = 3). C, Microscale thermophoresis binding analysis of NDR112–418 to the indicated phosphorylated MOB1A mutants. Data is plotted as the mean ± S.E. (n = 3).
Deletion of the first 13 residues (encompassing the T12 site) had no effect on nonphosphorylated MOB1A binding to NDR1, as expected since the first 13 residues in MOB1A were disordered in the nonphosphorylated human MOB1A structure and hence were not expected to impinge on the NDR1 interaction (Fig. 4A iii). Unexpectedly, in its phosphorylated state, the MOB1Δ13 mutant displayed slightly enhanced binding to NDR1 relative to phosphorylated MOB1AWT. We hypothesized that in the absence of phospho-T12 site binding to the phosphopeptide binding pocket of MOB1A, the pT35 site accesses the phospho-binding pocket, leading to more efficient exposure of the NDR kinase binding site (Fig 4A iii). In contrast to the N-terminal 13 residue deletion, the more expansive 38-residue deletion increased binding of nonphosphorylated MOB1A to NDR1. However, consistent with the loss of both T12 and T35 phospho-regulatory sites, the MOB1AΔ38 mutant was no longer responsive to phosphorylation (Fig 4A iv). Deletion of the entire 51-residue N terminus (denoted MOB1AΔ51) decreased the binding affinity of nonphosphorylated MOB1A to NDR1, which was expected since residues 39 to 51 of the N terminus contribute to the NDR1 binding infrastructure of MOB1A (Fig. 2B, 3A and Fig. 4A v). As observed for the MOB1AΔ38 mutant, phosphorylation of the MOB1AΔ51 mutant had a marginal effect on NDR1 binding (Fig. 4A v). Taken together, the above results are consistent with the notion that in its nonphosphorylated state, the N-terminal region of MOB1A restricts binding to the downstream NDR family kinases and that mutations within this region and/or phosphorylation of the T12 and T35 regulatory sites promote NDR1 binding. As we observed for the phosphopeptide binding function of MOB1 above, the varied behavior of MOB1 deletion mutants in NDR1 binding function raised the possibility that differential phosphorylation of MOB1 could be exploited for graded signaling responses in a cellular context.
Functional Consequence of the Suboptimal Match Between the T12 Phosphosite Sequence of MOB1A and the MOB1B Phosphopeptide-binding Consensus
Inspection of the T12 phosphosite sequence in MOB1A revealed a suboptimal match to the MOB1B phosphopeptide binding consensus (supplemental Fig. S5A). As expected, a pT12 peptide of MOB1A in isolation bound only weakly to the MOB1A core domain in trans (318μM; supplemental Fig. S5A, S5C). This finding led us to test the effect of converting the low-affinity T12 sequence (TFKPK) in MOB1A to the high-affinity sequences observed in MST1 centered on the T353 and T367 sites (TMIEH & TMVIH respectively; ref 29). We reasoned that these substitutions would give rise to a MOB1A protein that would more potently downregulate binding to MST1 phosphopeptide ligands in trans in response to MOB1A phosphorylation, and indeed, this is what we observed (Fig. 4B). In their nonphosphorylated states, MOB1AT12→T353 and MOB1AT12→T367 bound equally well to phosphopeptide ligands in trans comparable to MOB1WT. However, in response to phosphorylation, we observed a more effective downregulation of phosphopeptide binding for the high affinity site MOB1A mutants (≫1000 fold downregulation of MST1 phosphopeptide binding versus 50-fold with WT; Fig. 4B and supplemental Fig. S4B).
We next examined how mutation of the T12 and T35 phospho-regulatory sites in MOB1A affected binding to the downstream NDR family kinases. As described above, nonphosphorylated MOB1AWT bound to NDR1 kinase weakly, while phospho-MOB1AWT showed an ∼9-fold increase in NDR1 kinase binding. Unexpectedly, rather than attenuating phospho-MOB1A binding to NDR1, mutation of T12 to alanine modestly enhanced binding (∼2-fold; Fig 4C). This finding indicated that in the absence of T12 phosphorylation, phospho-T35 may more effectively expose the NDR1 binding site, presumably by allowing pT35 unrestricted access (by lack of competition with pT12) to the MOB1A phosphopeptide binding site. This unexpected result agreed with the peculiar behavior of the MOB1AΔ13 mutant above. As expected, mutation of T35 to alanine decreased binding of phospho-MOB1A to NDR1 by 2-fold relative to phospho-MOB1AWT. This finding further supported the notion that the T35 site plays a more prominent regulatory role than the T12 site. In addition, conversion of the T12 site to better match to the MOB1A phosphopeptide binding consensus gave a more potent increase in the ability of phospho-MOB1A to bind the downstream NDR1 kinases (∼ 4-fold increase relative to phospho-MOB1AWT). This result further suggested that the T12 site is inferior to the T35 site in regulating MOB1A function, and that its potency can be increased by improving its binding affinity to the phosphopeptide binding site on MOB1A (Fig. 4C). We note that the T35 site is also an imperfect match to the MOB1B phospho-binding consensus (supplemental Fig. S5A; ref 29). However, we chose not to test the effect of enhancing the match due to the complication that the required mutations would disrupt the autoinhibitory conformation of helix α-1 in its nonphosphorylated state. The above results demonstrated that the phosphorylation status of T12 and T35 dictates (in a complicated but rationalizable manner) the ability of MOB1A to interact with both the upstream MST1 kinase and the downstream NDR1 kinase through modulation of two distinct protein interaction surfaces on MOB1A. In particular, the relief of autoinhibition of NDR family kinase binding by MOB1A phosphorylation is coupled to the inhibition of MST1 kinase binding.
Functional Characterization of the N-terminal Regulatory Tail of MOB1 in Cells Using Affinity Purification Coupled to Mass Spectrometry
To understand how the N-terminal extension of MOB1A affects protein interactions in cells, we generated stable cell lines with a series of MOB1A constructs used previously in our in vitro experiments: MOB1AΔ38, MOB1AT12→T353, MOB1AT12→T367, and a double-alanine substitution mutant at T12 and T35 (denoted MOB1AT12A/T35A). Immunoprecipitation of these proteins (anti-FLAG) followed by mass spectrometry showed that, as expected from our in vitro derived model, the MOB1AΔ38 and MOB1AT12A/T35A mutants, like MOB1AWT, efficiently interacted with upstream Hippo components following okadaic acid treatment (Fig. 5A; supplemental Tables S4 and S5). High-affinity MOB1AT12→T353 and MOB1AT12→T367 substitution mutants were impaired for MOB1A interaction with upstream Hippo components as would be expected if the N-terminal extension of MOB1A engaged its own phosphopeptide binding pocket more tightly (Fig. 5A). All MOB1A mutants retained the ability to interact with the downstream LATS1 and LATS2 and NDR2 kinases, however the interaction was reduced for the MOB1AT12A/T35A mutant (Fig. 5A), as would be predicted if these two sites could not be phosphorylated to efficiently expose the NDR family kinase binding site on MOB1A. LATS1 and LATS2 protein levels remained equal in the MOB1A stable cell lines (Fig. 5B) and thus any changes in protein interactions could not be attributed to changes in protein abundance. In summary, the behavior of MOB1A we observe in cells is consistent with a regulatory model derived from the structural and in vitro biophysical studies reported here.
Fig. 5.

Cell-based interaction analysis of MOB1A mutants. A, Interaction profiles of FLAG-MOB1A and the indicated MOB1A N-terminal extension mutants, affinity-purified from stable HEK293 cell lines after treatment with okadaic acid (OA) or DMSO vehicle controls. Statistical analysis of the interaction confidence was performed using SAINTexpress (49) and visualization was through prohits-viz.lunenfeld.ca (see Experimental Procedures). Interactions are listed for the upstream Hippo kinase module, LATS and NDR kinases, DOCK6-8 and PP6 phosphatase modules. See inset legend for details. All interactions are listed in supplemental Table S4 and the underlying protein identifications in supplemental Table S5. Also see ProHits-web.lunenfeld.ca (project: “phospho-dependent Hippo interactions”). B, Control Western blot analysis of FLAG-MOB1A and endogenous PPP6C, LATS1 and LATS2 expression levels.
Interestingly, our previous work also showed that phosphorylation-dependent association of the PP6 phosphatase module containing the catalytic subunit PPP6C with MOB1 was adversely affected by mutation of the MOB1A phosphopeptide-binding pocket (27). This result suggested that PP6 might engage MOB1A through a binding mechanism like that employed by MST1. However, our new data shows that the two MOB1A mutants harboring MST1 high-affinity binding sequences (which had the effect of abolishing MST1 interaction) instead resulted in an increase in PP6 module association, which was further enhanced by okadaic acid treatment (Fig. 5A). This result indicated a more complex mode of regulation for the MOB1-PP6 interaction (supplemental Fig. S6A) than originally hypothesized.
Lastly, as reported in (27), the DOCK6-8 module interacted with MOB1AWT, though the stimulatory effect of okadaic acid on the interactions was minimal here (Fig. 5A). Strikingly, in the absence of treatment with okadaic acid, the association of all components of this module was greatly enhanced for the MOB1AT12→T353 and MOB1AT12→T367 high-affinity substitution mutants. In contrast, interaction with the DOCK6-8 module was reduced and nearly abolished for the MOB1AΔ38 and the MOB1AT12A/T35A mutants, respectively. The lack of interaction detected with the MOB1AT12A/T35A mutant is consistent with the findings of Mou et al., which reported that DOCK8 binds the phosphorylated form of MOB1 (32). Regarding the stronger interactions displayed by the MOB1AT12→T353 and MOB1AT12→T367 mutants, okadaic acid treatment led to a reduction of association. In this sense, the interaction pattern with MOB1A was more similar to the interaction pattern of NDR and LATS family kinases (Fig. 5A and supplemental Fig. S6B), and importantly contrasted with the interaction pattern of MOB1A with the other two phosphorylation-dependent modules, namely the upstream Hippo components and the PP6 phosphatase (Fig. 5A). These surprising findings reveal that while our in vitro studies provide a framework for understanding the regulatory role of phosphorylation on the interaction of MOB1A with the core Hippo components (MST1 and MST2, MOB1A and MOB1B, and LATS1 and LATS2), additional structural and functional studies will be required to illuminate the regulated interaction mechanisms of MOB1 with the PP6 and DOCK modules.
DISCUSSION
Our comprehensive interaction proteomic study has revealed novel complexities in how MOB1A and MOB1B interact with the core Hippo pathway components MST1, MST2, LATS1, and LATS2 and with the LATS-related kinases NDR1 and NDR2 in a phospho-regulated manner to transduce signals. Our results and a great wealth of data recently published by other groups (26, 28, 33, 36; and the accompanying paper by ref 29) support the following sequential model for Hippo pathway signal propagation (Fig. 6), aspects of which are generalizable to paralogous signaling pathways across eukaryotes such as NDR kinase signaling in metazoans and MEN in yeast.
Fig. 6.

Model for the sequential signaling events in the core Hippo pathway. See text for details.
MOB1 functions as a central coordinator of signaling by virtue of possessing two physically distinct protein interaction surfaces: one for engaging linear phosphopeptide epitopes including those observed in MST1 and MST2 kinases, and a second for engaging the globular MOB1 interacting domain of the AGC family kinases LATS1, LATS2, NDR1, and NDR2.
MST1 and MST2 kinases, likely activated by upstream signals, have the potential to autophosphorylate on multiple sites in the linker region between the kinase domain and the SARAH domain. As the substrate consensus of MST1 and MST2 kinases is a close match to the MOB1 phosphopeptide-binding consensus, autophosphorylation of MST1 and MST2 could create docking sites that are efficiently recognized by the phosphopeptide binding infrastructure of MOB1 (See accompanying paper by ref 29, for details).
Once bound to MOB1, MST1, and MST2 kinases phosphorylate the N-terminal extension of MOB1, minimally on two key phospho-regulatory sites, T12 and T35, both of which closely match the substrate consensus of MST1 and MST2, but poorly match the phosphopeptide binding consensus of MOB1 itself (See accompanying paper by ref 29). These events induce a switch-like conformational change in the N-terminal extension of MOB1 that exposes the binding surface for NDR and LATS kinases while concomitantly blocking the phosphopeptide-binding infrastructure of MOB1. The latter causes the coordinated release of MST1 and MST2.
Prior to the release of MST1 and MST2 from MOB1, the transient binding of both upstream and downstream kinases allows MST1 and MST2 to phosphorylate the LATS and NDR kinases on the hydrophobic motif (HM) (as amply discussed in prior studies, ref 17, 20, 21–25), thereby facilitating their activation and the propagation of downstream signals, including through the phosphorylation of YAP1 and TAZ by LATS kinases in the Hippo pathway (Fig. 6).
Our results also highlight greater complexity than anticipated in the regulation of phospho-dependent interactions surrounding MOB1. We previously discovered that mutation of the basic residues in the MOB1B phosphopeptide binding pocket (MOB1KRR→3A) abrogated okadaic acid-induced interaction of MOB1B with MST1 and MST2 kinases, as well as with the PP6 regulatory subunit PPP6R3. This suggested a common binding mode in which PP6, MST1 and MST2 bind to the same phospho-recognition infrastructure on MOB1B (27). However, contradicting this inference, our new interaction proteomics results reveal that the hig-affinity MOB1AT12→T353 and MOB1AT12→T367 substitution mutants abolish the interaction with the MST1 and MST2 kinases (fully consistent with our predictions) while increasing the interaction with members of the PP6 complexes. This result supports the presence of a distinct and novel binding mode for PP6, the structural basis of which warrants future investigation. Similarly, based on the increase in interaction reported previously between MOB1 and the DOCK6-8 module following okadaic acid treatment (27) and the fact that the interaction of MOB1 with this module was mutually exclusive with its interaction with the upstream Hippo components and with the PP6 module, we hypothesized that the DOCK6-8 module may share the same mode of binding to MOB1 as MST1 and MST2. However, the results in Fig. 5 clearly indicate a dissimilar mode of binding of DOCK6-8 to MOB1 that instead resembles that of the LATS and NDR kinases. Whether either of the PP6 and DOCK6-8 modules feeds into the Hippo pathway or instead impart altogether novel and unrelated functions of MOB1 remains to be determined.
Why is the pathway constructed in a manner that apparently minimizes concurrent binding of the upstream MST1 kinase and the downstream NDR and LATS kinases to MOB1? Perhaps this design allows MOB1 to act in a catalytic manner whereby the forced release of MST1 from MOB1 can facilitate MST1 interaction with multiple MOB1 proteins, thereby enabling the activation of multiple LATS and NDR kinases. If true, then enhancing the concurrent binding of MST1 and LATS1 kinases to MOB1 would be predicted to inhibit Hippo pathway signal propagation. Alternatively, the enforcement of mutually-exclusive binding of MST1 and LATS and NDR kinases to MOB1 may serve as means to limit the propagation of signal. If this were the case, then an enhancement of concurrent binding of MST1 and LATS1 to MOB1 would be predicted to enhance Hippo pathway signaling. While not fully addressed in the current study, our results provide a framework to dissect this fundamental question of pathway design, with major implications for the development of therapeutic intervention strategies to restrain unbridled cell proliferation.
DATA AVAILABILITY
The crystal structure of full-length human MOB1A in its apo state was deposited with the Protein Data Bank housed at Rutgers, The State University of New Jersey (http://www.rcsb.org/pdb/home/home.do) under the PBD code 5TWF. The mass spectrometry data was deposited with the Center for Computational Mass Spectrometry housed at the University of California, San Diego. The MOB1A FLAG APMS data set consisting of 26 raw files and associated peak lists and results files have been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080331 and PXD005327, (ftp://massive.ucsd.edu/MSV000080331). The determination of the specificity of human MOB / AGC kinase interactions results have been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080570 and PXD005966, ftp://massive.ucsd.edu/MSV000080570. The mass spectrometry data is further available at ProHits-web.lunenfeld.ca (project: “phospho-dependent Hippo interactions”).
Supplementary Material
Acknowledgments
We thank Brett Larsen for help with mass spectrometry data analysis, and Gerald Gish for generating synthetic peptides for validation studies.
Footnotes
Author contributions: S.X., A.L.C., M.J.K., D.Y.M., S.G., A.-C.G., and F.S. designed research; S.X., A.L.C., M.J.K., D.Y.M. and S.G. performed research; I.K. collected X-ray diffraction data; S.X., A.L.C., D.Y.M., A.-C.G., and F.S. analyzed data; S.X., A.L.C., A.-C.G., and F.S. wrote the manuscript with input from all authors.
* This work was supported by a Cancer Research Society (CRS) grant to A.-C.G., a Terry Fox Research Institute team grant (to F.S. and A.-C.G.), a Canadian Cancer Society Research Institute Impact grant to FS, as well as by Canadian Institutes of Health Research Foundation Grants (FDN 143277 to F.S.; FDN 143301 to A.-C.G.). A.-C.G. and F.S. are the Canada Research Chairs (Tier 1) in Functional Proteomics and Structural Biology of Cell Signaling, respectively. Proteomics work was performed at the Network Biology Collaborative Centre at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, by the Ontarian Government and by Genome Canada and Ontario Genomics (OGI-088). Diffraction work conducted at the Northeastern Collaborative Access Team beamlines was funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403) and by an NIH-ORIP HEI grant (S10 RR029205). S.X. is supported by Natural Sciences and Engineering Research Council of Canada Doctoral Awards. S.G. was supported by postdoctoral fellowships from the Human Frontier Science Program (HFSP) and the European Molecular Biology Organization (EMBO). M.J.K. was supported by Canadian Institutes of Health Research through a Banting and Best Canada Graduate Scholarship.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- AP-MS
- Affinity purification coupled to mass spectrometry
- GST
- Glutathione S-transferase
- HM
- Hydrophobic motif
- LATS1
- 2 Large tumor suppressor kinases 1 and 2
- MST1
- 2 Mammalian STE20-like protein kinases 1 and 2, gene names STK4 and STK3
- MOB1A
- B Mps one binder kinase activator, 1A, 1B
- OA
- Okadaic acid
- WWTR1
- WW domain containing transcription regulator protein 1 / protein known as TAZ
- TAZ
- Transcriptional coactivator with PDZ-binding domain / gene name WWTR1
- YAP1
- Yes associated protein 1
- SAV1
- Protein Salvador homolog 1
- NDR1
- 2 Nuclear Dbf2 related kinases 1 and 2 / gene names STK38 and STK38L.
REFERENCES
- 1. Luca F. C., and Winey M. (1998) MOB1, an essential yeast gene required for completion of mitosis and maintenance of ploidy. Mol. Biol. Cell 9, 29–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiss E. L. (2012) Mitotic exit and separation of mother and daughter cells. Genetics 192, 1165–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hergovich A. (2011) MOB control: reviewing a conserved family of kinase regulators. Cell Signal 23, 1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee S. E., Frenz L. M., Wells N. J., Johnson A. L., and Johnston L. H. (2001) Order of function of the budding-yeast mitotic exit-network proteins Tem1, Cdc15, Mob1, Dbf2, and Cdc5. Curr. Biol. 11, 784–788 [DOI] [PubMed] [Google Scholar]
- 5. Komarnitsky S. I., Chiang Y. C., Luca F. C., Chen J., Toyn J. H., Winey M., Johnston L. H., and Denis C. L. (1998) DBF2 protein kinase binds to and acts through the cell cycle-regulated MOB1 protein. Mol. Cell. Biol. 18, 2100–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weiss E. L., Kurischko C., Zhang C., Shokat K., Drubin D. G., and Luca F. C. (2002) The Saccharomyces cerevisiae Mob2p-Cbk1p kinase complex promotes polarized growth and acts with the mitotic exit network to facilitate daughter cell-specific localization of Ace2p transcription factor. J. Cell Biol. 158, 885–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai Z. C., Wei X., Shimizu T., Ramos E., Rohrbaugh M., Nikolaidis N., Ho L. L., and Li Y. (2005) Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120, 675–685 [DOI] [PubMed] [Google Scholar]
- 8. Justice R. W., Zilian O., Woods D. F., Noll M., and Bryant P. J. (1995) The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546 [DOI] [PubMed] [Google Scholar]
- 9. He Y., Emoto K., Fang X., Ren N., Tian X., Jan Y. N., and Adler P. N. (2005) Drosophila Mob family proteins interact with the related tricornered (Trc) and warts (Wts) kinases. Mol. Biol. Cell 16, 4139–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bothos J., Tuttle R. L., Ottey M., Luca F. C., and Halazonetis T. D. (2005) Human LATS1 is a mitotic exit network kinase. Cancer Res. 65, 6568–6575 [DOI] [PubMed] [Google Scholar]
- 11. Zhao B., Wei X., Li W., Udan R. S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., Zheng P., Ye K., Chinnaiyan A., Halder G., Lai Z. C., and Guan K. L. (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., and Pan D. (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bichsel S. J., Tamaskovic R., Stegert M. R., and Hemmings B. A. (2004) Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J. Biol. Chem. 279, 35228–35235 [DOI] [PubMed] [Google Scholar]
- 14. Hergovich A., Bichsel S. J., and Hemmings B. A. (2005) Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol. Cell. Biol. 25, 8259–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohler R. S., Schmitz D., Cornils H., Hemmings B. A., and Hergovich A. (2010) Differential NDR/LATS interactions with the human MOB family reveal a negative role for human MOB2 in the regulation of human NDR kinases. Mol. Cell. Biol. 30, 4507–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goudreault M., D'Ambrosio L. M., Kean M. J., Mullin M. J., Larsen B. G., Sanchez A., Chaudhry S., Chen G. I., Sicheri F., Nesvizhskii A. I., Aebersold R., Raught B., and Gingras A. C. (2009) A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol. Cell. Proteomics 8, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mah A. S., Jang J., and Deshaies R. J. (2001) Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc. Natl. Acad. Sci. U.S.A. 98, 7325–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson B., Kurischko C., Horecka J., Mody M., Nair P., Pratt L., Zougman A., McBroom L. D., Hughes T. R., Boone C., and Luca F. C. (2003) RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell 14, 3782–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu S., Huang J., Dong J., and Pan D. (2003) hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456 [DOI] [PubMed] [Google Scholar]
- 20. Chan E. H., Nousiainen M., Chalamalasetty R. B., Schafer A., Nigg E. A., and Sillje H. H. (2005) The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086 [DOI] [PubMed] [Google Scholar]
- 21. Tamaskovic R., Bichsel S. J., Rogniaux H., Stegert M. R., and Hemmings B. A. (2003) Mechanism of Ca2+-mediated regulation of NDR protein kinase through autophosphorylation and phosphorylation by an upstream kinase. J. Biol. Chem. 278, 6710–6718 [DOI] [PubMed] [Google Scholar]
- 22. Harvey K. F., Pfleger C. M., and Hariharan I. K. (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467 [DOI] [PubMed] [Google Scholar]
- 23. Emoto K., He Y., Ye B., Grueber W. B., Adler P. N., Jan L. Y., and Jan Y. N. (2004) Control of dendritic branching and tiling by the Tricornered-kinase/Furry signaling pathway in Drosophila sensory neurons. Cell 119, 245–256 [DOI] [PubMed] [Google Scholar]
- 24. Millward T. A., Hess D., and Hemmings B. A. (1999) Ndr protein kinase is regulated by phosphorylation on two conserved sequence motifs. J. Biol. Chem. 274, 33847–33850 [DOI] [PubMed] [Google Scholar]
- 25. Stegert M. R., Tamaskovic R., Bichsel S. J., Hergovich A., and Hemmings B. A. (2004) Regulation of NDR2 protein kinase by multi-site phosphorylation and the S100B calcium-binding protein. J. Biol. Chem. 279, 23806–23812 [DOI] [PubMed] [Google Scholar]
- 26. Rock J. M., Lim D., Stach L., Ogrodowicz R. W., Keck J. M., Jones M. H., Wong C. C., Yates J. R. 3rd, Winey M., Smerdon S. J., Yaffe M. B., and Amon A. (2013) Activation of the yeast Hippo pathway by phosphorylation-dependent assembly of signaling complexes. Science 340, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Couzens A. L., Knight J. D., Kean M. J., Teo G., Weiss A., Dunham W. H., Lin Z. Y., Bagshaw R. D., Sicheri F., Pawson T., Wrana J. L., Choi H., and Gingras A. C. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal 6, rs15. [DOI] [PubMed] [Google Scholar]
- 28. Ni L., Zheng Y., Hara M., Pan D., and Luo X. (2015) Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev. 29, 1416–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Couzens A. L., Xiong S., Knight J. D. R., Mao D. Y., Guettler S., Picaud S., Kurinov I., Filippakopoulos P., Sicheri F., and Gingras A. C. (2017) MOB1 mediated phospho-recognition in the core mammalian Hippo pathway. Mol. Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stefansson B., and Brautigan D. L. (2006) Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IkappaBepsilon. J. Biol. Chem. 281, 22624–22634 [DOI] [PubMed] [Google Scholar]
- 31. Stefansson B., Ohama T., Daugherty A. E., and Brautigan D. L. (2008) Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry 47, 1442–1451 [DOI] [PubMed] [Google Scholar]
- 32. Mou F., Praskova M., Xia F., Van Buren D., Hock H., Avruch J., and Zhou D. (2012) The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J. Exp. Med. 209, 741–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim S. Y., Tachioka Y., Mori T., and Hakoshima T. (2016) Structural basis for autoinhibition and its relief of MOB1 in the Hippo pathway. Sci. Rep 6, 28488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Praskova M., Xia F., and Avruch J. (2008) MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gogl G., Schneider K. D., Yeh B. J., Alam N., Nguyen Ba A. N., Moses A. M., Hetenyi C., Remenyi A., and Weiss E. L. (2015) The Structure of an NDR/LATS Kinase-Mob Complex Reveals a Novel Kinase-Coactivator System and Substrate Docking Mechanism. PLos Biol. 13, e1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 38. Stavridi E. S., Harris K. G., Huyen Y., Bothos J., Verwoerd P. M., Stayrook S. E., Pavletich N. P., Jeffrey P. D., and Luca F. C. (2003) Crystal structure of a human Mob1 protein: toward understanding Mob-regulated cell cycle pathways. Structure 11, 1163–1170 [DOI] [PubMed] [Google Scholar]
- 39. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Winn M. D., Murshudov G. N., and Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 42. Kean M. J., Couzens A. L., and Gingras A. C. (2012) Mass spectrometry approaches to study mammalian kinase and phosphatase associated proteins. Methods 57, 400–408 [DOI] [PubMed] [Google Scholar]
- 43. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., and Nesvizhskii A. I. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111 007690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z. Y., Breitkreutz B. J., Ding Y., Colwill K., Pasculescu A., Pawson T., Wrana J. L., Nesvizhskii A. I., Raught B., Tyers M., and Gingras A. C. (2010) ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 28, 1015–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eng J. K., Jahan T. A., and Hoopmann M. R. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 47. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 48. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., Qin Z. S., Pawson T., Gingras A. C., Nesvizhskii A. I., and Tyers M. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teo G., Liu G., Zhang J., Nesvizhskii A. I., Gingras A. C., and Choi H. (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Knight J. D., Liu G., Zhang J. P., Pasculescu A., Choi H., and Gingras A. C. (2015) A web-tool for visualizing quantitative protein-protein interaction data. Proteomics 15, 1432–1436 [DOI] [PubMed] [Google Scholar]
- 52. Kean M. J., Ceccarelli D. F., Goudreault M., Sanches M., Tate S., Larsen B., Gibson L. C., Derry W. B., Scott I. C., Pelletier L., Baillie G. S., Sicheri F., and Gingras A. C. (2011) Structure-function analysis of core STRIPAK Proteins: a signaling complex implicated in Golgi polarization. J. Biol. Chem. 286, 25065–25075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mrkobrada S., Boucher L., Ceccarelli D. F., Tyers M., and Sicheri F. (2006) Structural and functional analysis of Saccharomyces cerevisiae Mob1. J. Mol. Biol. 362, 430–440 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure of full-length human MOB1A in its apo state was deposited with the Protein Data Bank housed at Rutgers, The State University of New Jersey (http://www.rcsb.org/pdb/home/home.do) under the PBD code 5TWF. The mass spectrometry data was deposited with the Center for Computational Mass Spectrometry housed at the University of California, San Diego. The MOB1A FLAG APMS data set consisting of 26 raw files and associated peak lists and results files have been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080331 and PXD005327, (ftp://massive.ucsd.edu/MSV000080331). The determination of the specificity of human MOB / AGC kinase interactions results have been deposited in ProteomeXchange through partner MassIVE as a complete submission and assigned the MassIVE ID MSV000080570 and PXD005966, ftp://massive.ucsd.edu/MSV000080570. The mass spectrometry data is further available at ProHits-web.lunenfeld.ca (project: “phospho-dependent Hippo interactions”).