

Abstract
Background: The Antarctic intertidal zone is continuously subjected to extremely fluctuating biotic and abiotic stressors. The West Antarctic Peninsula is the most rapidly warming region on Earth. Organisms living in Antarctic intertidal pools are therefore interesting for research into evolutionary adaptation to extreme environments and the effects of climate change.
Findings: We report the whole genome sequence of the Antarctic-endemic harpacticoid copepod Tigriopus kingsejongensi. The 37 Gb raw DNA sequence was generated using the Illumina Miseq platform. Libraries were prepared with 65-fold coverage and a total length of 295 Mb. The final assembly consists of 48 368 contigs with an N50 contig length of 17.5 kb, and 27 823 scaffolds with an N50 contig length of 159.2 kb. A total of 12 772 coding genes were inferred using the MAKER annotation pipeline. Comparative genome analysis revealed that T. kingsejongensis-specific genes are enriched in transport and metabolism processes. Furthermore, rapidly evolving genes related to energy metabolism showed positive selection signatures.
Conclusions: The T. kingsejongensis genome provides an interesting example of an evolutionary strategy for Antarctic cold adaptation, and offers new genetic insights into Antarctic intertidal biota.
Keywords: Copepoda, Genome, Antarctic, Adaptation, Tigriopus
Data description
Approximately 12 000 species have been described in the diverse copepod subclass [1, 2]. These species dominate the zooplankton community, contributing about 70% of total zooplankton biomass [3], and are an important link between phytoplankton and higher trophic levels in the marine meiobenthic food web [4]. Harpacticoid copepods of the genus Tigriopus Norman 1868 are dominant members of shallow supratidal rock pools, distributed worldwide among habitats that vary widely in salinity, temperature, desiccation risk, and UV radiation. They are a model system in investigations of osmoregulation [5], temperature adaptation [6, 7] and environmental toxicology [8]. With publically available copepod genome resources (e.g., Tigriopus californicus [9], T. japonicus [10], Eurytemora affinis [11] and salmon louse Lepeophtheirus salmonis [12]), it is now possible to explore their fundamental biological processes and physiological responses to diverse environments.
Antarctica is not only an extreme habitat for extant organisms, but also a model for research on evolutionary adaptations to cold environments [13, 14]. The Antarctic intertidal zone, particularly in the Western Antarctic Peninsula region, is one of the most extreme, yet fastest warming environments on Earth. Thus, it is a potential barometer for global climate change [15]. Antarctic intertidal species that have evolved stenothermal phenotypes through adaptation to year-round extreme cold may now face extinction by global warming. The response of these species to further warming in Western Antarctica is of serious concern; however, to date, few studies have focused on Antarctic intertidal zone species.
First described in 2014, T. kingsejongensis was recognized as a new species endemic to a rock pool in the Antarctic Peninsula. It is extremely cold-tolerant and can survive in frozen sea water [16]. Compared to the congener T. japonicus, which is found in coastal areas of the Yellow Sea, morphological differences of this species include increased numbers of caudal setae in nauplii, an optimal growth temperature of approximately 8 °C, and differing developmental characteristics. Tigriopus kingsejongensis has evolved to overcome the unique environmental constraints of Antarctica, therefore providing an ideal experimental model for extreme habitat research. This species may represent a case of rapid speciation, since the intertidal zone on King George Island and the surrounding areas did not exist 10 000 years ago [17]. Tigriopus kingsejongensis likely evolved as a distinct species within this relatively short time period. Thus, interspecies and intraspecies comparative analyses of Antarctic Tigriopus species will help to define the trajectory of adaptation to the Antarctic environment, and also provide insights into the genetic basis of Tigriopus divergence and evolution.
Library construction and sequencing
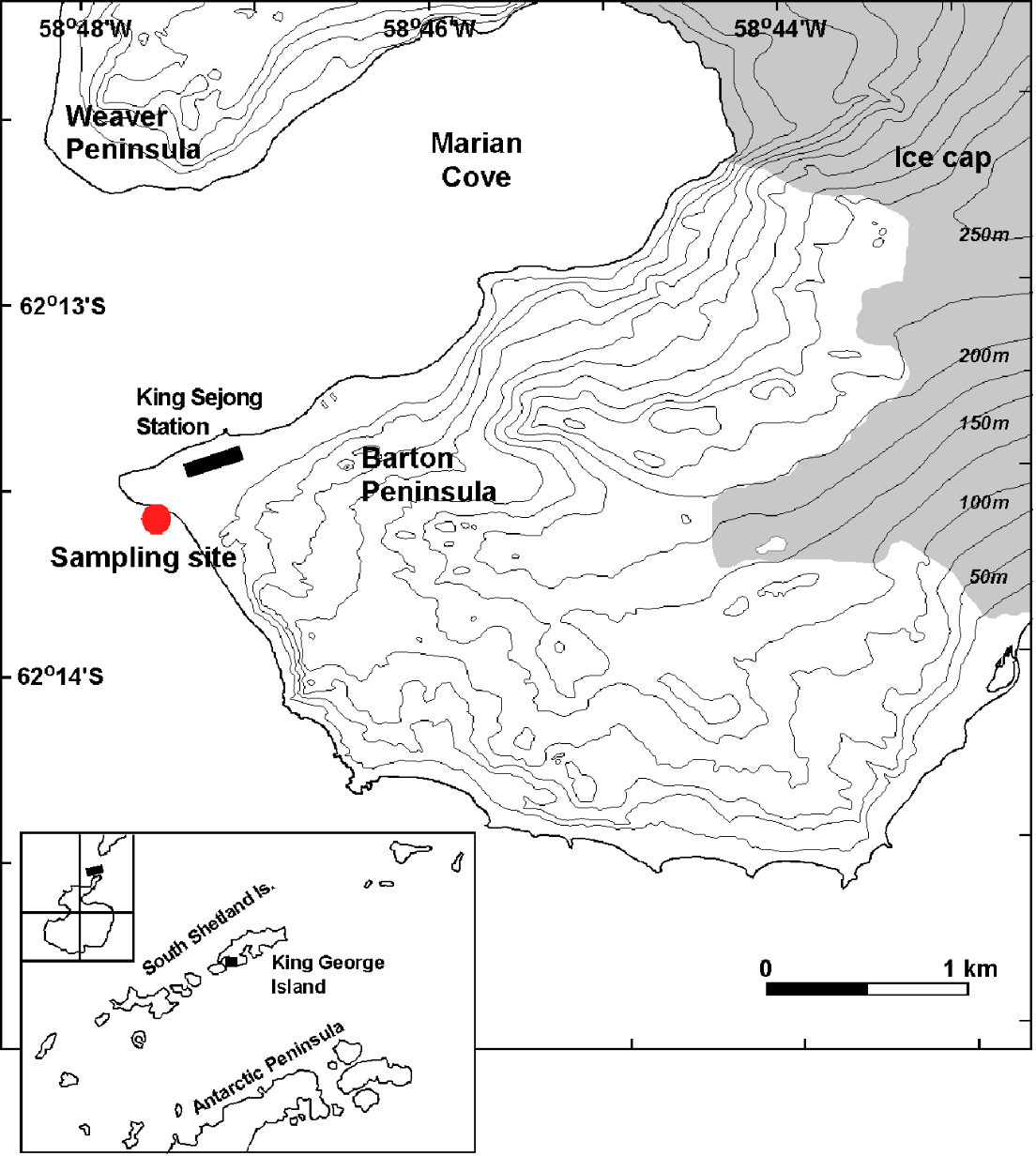
Tigriopus kingsejongensis specimens were collected using hand-nets from tidal pools in Potter Cove, near King Sejong Station, on the northern Antarctic Peninsula (62°14΄S, 58°47΄W) (Fig. 1 and Fig. S1) in January 2013. The water temperature was 1.6 ± 0.8 °C during this month. High molecular weight genomic DNA from pooled T. kingsejongensis was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Venlo, The Netherlands). For Illumina Miseq sequencing, four library types were constructed with 350, 400, 450, and 500 bp for paired-end libraries, and 3 kb and 8 kb for mate-pair libraries, prepared using the standard Illumina sample preparation methods (Table 1). All sequencing processes were performed according to the manufacturer's instructions (Illumina, Carlsbad, USA).
Figure 1.

Photograph of an adult Tigriopus kingsejongensis specimen (scale bar = 200 μm)
Table 1.
DNA library statistics
| Library | Reads (n) | Average | Sequences | Reads Average | Sequences | ||
|---|---|---|---|---|---|---|---|
| length | (bp) (n) | (trimmed) (n) | length | (trimmed) (n) | |||
| Paired-end | Sum | 99 710 266 | 29 271 916 613 | 65 644 374 | 14 668 956 871 | ||
| 350S1 | 6 661 392 | 300 | 2 005 078 992 | 4 446 394 | 233 | 1 034 231 244 | |
| 350S2 | 4 933 058 | 265 | 1 311 700 122 | 4 618 711 | 211 | 975 471 763 | |
| 400S1 | 65 668 598 | 300 | 19 766 247 998 | 36 863 154 | 228 | 8 397 426 481 | |
| 450S1 | 3 418 988 | 300 | 1 029 115 388 | 2 812 455 | 230 | 646 302 159 | |
| 450S2 | 8 009 162 | 245 | 1 968 652 020 | 7 660 814 | 199 | 1 527 566 312 | |
| 500S1 | 11 019 068 | 289 | 3 191 122 093 | 9 242 846 | 226 | 2 087 958 911 | |
| Mate-Paired | Sum | 103 373 998 | 7 753 049 850 | 73 515 391 | 5 169 006 268 | ||
| 3KS1 | 8 374 238 | 75 | 628 067 850 | 6 745 546 | 73 | 493 099 413 | |
| 3KS2 | 9 250 994 | 75 | 693 824 550 | 5 281 513 | 65 | 344 618 723 | |
| 3KS3 | 51 349 594 | 75 | 3 851 219 550 | 39 147 167 | 72 | 2 816 638 666 | |
| 3KS4 | 3 063 232 | 75 | 229 742 400 | 1 740 986 | 65 | 112 554 745 | |
| 8KS1 | 9 847 636 | 75 | 738 572 700 | 7 887 612 | 73 | 572 246 251 | |
| 8KS2 | 16 322 038 | 75 | 1 224 152 850 | 9 653 293 | 65 | 630 842 698 | |
| 8KS3 | 5 166 266 | 75 | 387 469 950 | 3 059 274 | 65 | 199 005 774 | |
| Total | 203 084 264 | 37 024 966 463 | 139 159 765 | 19 837 963 139 | |||
| Coverage (folds) | 120.7 | 64.7 | |||||
RNA was prepared from pooled T. kingsejongensis and T. japonicus specimens from two different temperature experiments (4 °C and 15 °C) using the RNeasy Mini Kit (Qiagen). For Illumina Miseq sequencing, subsequent experiments were carried out according to the manufacturer's instructions (Illumina). The de novo transcriptome assembly was performed with CLC Genomics Workbench (Qiagen), setting the minimum allowed contig length to 200 nucleotides. The assembly process generated 40 172 contigs with a maximum length of 23 942 bp and an N50 value of 1093 bp. Generated contigs were used as reference sequences to map trimmed reads, and fold-changes in expression for each gene were calculated with a significance threshold of P ≤ 0.05 using the CLC Genomics Workbench (Tables 2 and 3).
Table 2.
Transcriptome sequencing and assembly analysis for Tigriopus japonicus
| Sequencing | |
|---|---|
| Total reads (n) | 37 956 160 |
| Total bases (n) | 7 714 415 316 |
| Trimmed reads (n) | 35 577 636 |
| Trimmed bases (n) | 5 989 188 343 |
| Assembly | |
| Contigs (n) | 40 172 |
| Total contig length (bases) | 28 850 726 |
| N50 contig length (bases) | 1093 |
| Max scaffold length (bases) | 23 942 |
| Annotation | |
| With BLAST results | 20 392 |
| Without BLAST hits | 7090 |
| With mapping results | 8172 |
| Annotated sequences | 4518 |
Table 3.
RNA-seq statistics analysis for Tigriopus kingsejongensis
| Temperature | ||
|---|---|---|
| 4 °C | 15 °C | |
| Total reads (n) | 15 786 118 | 16 417 072 |
| Total bases (n) | 3 567 662 668 | 3 763 295 032 |
| Trimmed reads (n) | 14 845 103 | 15 388 513 |
| Trimmed bases (n) | 2 761 189 158 | 2 833 805 442 |
Genome assembly
First, k-mer analysis was conducted using jellyfish 2.2.5 [18] to estimate the genome size from DNA paired-end libraries. The estimated genome size was 298 Mb, with the main peak at a depth of ∼39× (Fig. 2). Then, assemblies were performed using a Celera Assembler with Illumina short reads [19]. Prior to assembly, Illumina reads were trimmed using the FASTX-Toolkit [20] with parameters −t 20, −l 70 and −Q 33, after which a paired sequence from trimmed Illumina reads was selected. Finally, trimmed Illumina reads with 65-fold coverage (insert sizes 350, 400, 450, and 500 bp) were obtained and converted to the FRG file format (required by the Celera Assembler) using FastqToCA. Assembly was performed on a 96-processor workstation with Intel Xeon X7460 2.66 GHz processors and 1 Tb random access memory (RAM) with the following parameters: overlapper = ovl, unitigger = bogart, utgGraphErrorRate = 0.03, utgGraphErrorLimit = 2.5, utgMergeErrorRate = 0.030, utgMergeErrorLimit = 3.25, ovlErrorRate = 0.1, cnsErrorRate = 0.1, cgwErrorRate = 0.1, merSize = 22, and doOverlapBasedTrimming = 1. The initial Celera assembly was 305 Mb, had an N50 contig size of 17 566 bp, and a maximum contig size of 349.5 kb. Scaffolding was completed using the SSPACE 2.0 scaffolder using mate-paired data [21]. Subsequently, we closed gaps using Gapfiller version 1.9 with 65× trimmed Illumina reads with default settings [22]. De novo assembly of 203 million reads from paired-end and mate-paired libraries yielded a draft assembly (65-fold coverage) with a total length of 295 Mb, and contig and scaffold N50 sizes of 17.6 kb and 159.2 kb, respectively (Table 4 and Fig. 3).
Figure 2.
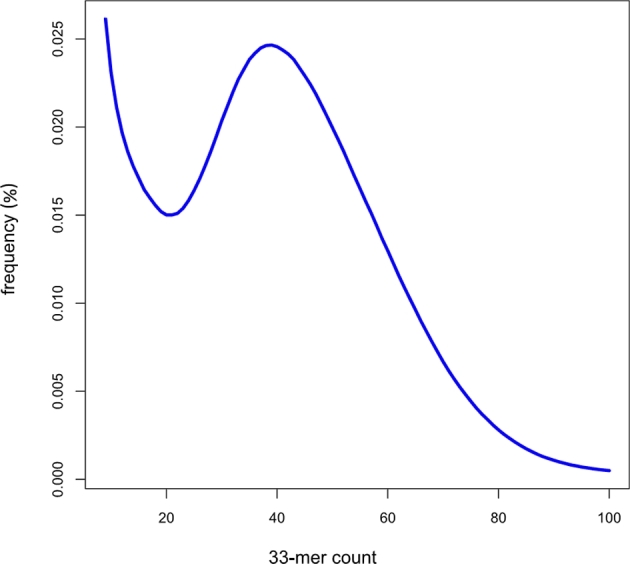
Estimation of the Tigriopus kingsejongensis genome size based on 33-mer analysis. X-axis represents the depth (peak at 39×) and the y-axis represents the proportion. Genome size was estimated to be 298 Mb (total k-mer number/volume peak)
Table 4.
Genome assembly statistics
| Type | Parameter | Assembly size according to Celera Assembler |
|---|---|---|
| Scaffold | Total scaffold length (bases) | 295 233 602 |
| Gap size (bases) | 10 474 460 | |
| Scaffolds (n) | 11 558 | |
| N50 scaffold length (bases) | 159 218 | |
| Max scaffold length (bases) | 3 401 446 | |
| Contig | Total contig length (bases) | 305 712 242 |
| Contigs (n) | 48 368 | |
| N50 contig length (bases) | 17 566 | |
| Max contig length (bases) | 349 507 |
Figure 3.
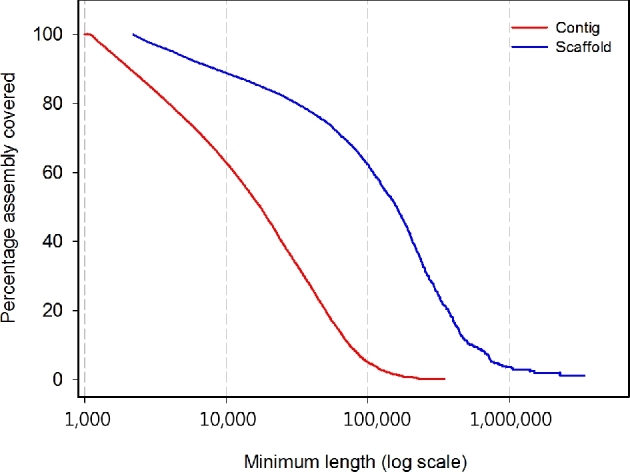
Scaffold and contig size distributions of Tigriopus kingsejongensis. The percentage of the assembly included (y-axis) in contigs or scaffolds of a minimum size (x-axis, log scale) is shown for the contig (red) and scaffold (blue)
Annotation
MAKER, a portable and easily configurable genome annotation pipeline, was used to annotate the genome [23]. Repetitive elements were identified using RepeatMasker [24]. This masked genome sequence was used with SNAP software [25] for ab initio gene prediction, after which alignment of expressed sequence tags (ESTs) with BLASTn [26] and protein information from tBLASTx [26] were included. The de novo repeat library of T. kingsejongensis from RepeatModeler was used for RepeatMasker; proteins from five species with data from Drosophila melanogaster, Daphnia pulex, T. japonicus, and T. californicus were included in the analysis. RNA-seq-based gene prediction, data were aligned against the assembled genome using TopHat [27], and Cufflinks [28] was used to predict cDNAs from the resultant data. Next, MAKER polished the alignments using the program Exonerate [29], which provided integrated information to synthesize SNAP annotation. Considering all information, MAKER then selected and revised the final gene model. A total of 12 772 genes were predicted in T. kingsejongensis using MAKER. Annotated genes contained an average of 4.6 exons, with an average mRNA length of 1090 bp. Additionally, 12 562 of 12 772 genes were assigned preliminary functions based on automated annotation using Blast2GO (Ver. 2.6.0) [30] (Figs. S2 and S3) with homology sequences from the SwissProt [31], TrEMBL, National Center for Biotechnology Information (NCBI) non-redudant protein databases [32] and REVIGO software was used to cluster related GO terms according to P-value [33]. Infernal version 1.1 [34] and covariance models (CMs) from the Rfam database [35] were used to identify other non-coding RNAs in the T. kingsejongensis scaffold. Putative tRNA genes were identified using tRNAscan-SE [36] (Table S1), which uses a CM that scores candidates based on their sequence and predicted secondary structures.
Non-gap sequences occupied 284.8 Mb (96.5%), and simple sequence repeats (SSRs) amounted to 1.2 Mb (0.4%) (Table S2). Transposable elements (TEs) comprised 6.5 Mb; roughly 2.3% of the assembled genome (Tables S2 and S3). On the basis of homology and ab initio gene prediction, the T. kingsejongensis genome contained 12 772 protein-coding genes (Table 5). By assessing the quality of the 12 772 annotated gene models, 11 686 protein-coding genes (91.5%) were supported by RNA-seq data, of which 7325 (63%) were similar to proteins from other species. To estimate genome assembly and annotation completeness, Core Eukaryotic Genes Mapping Approach (CEGMA) [37] and Benchmarking Universal Single-Copy Orthologs (BUSCO) [38] analysis was used (Table 6). The CEGMA report revealed that 193 of 248 CEGMA score genes were fully annotated (77.8% completeness), and 206 of 248 genes were partially annotated (83% completeness). BUSCO, a similar approach used for lineage-specific profile libraries such as eukaryotes, metazoans, and arthropods, revealed 71% complete and 6% partial Metazoan orthologous gene sets in our assembly; using an arthropod gene set, only 61.1% complete and 10.7% partial genes were assigned. CEGMA and BUSCO gene sets largely comprised insects; other non-insect arthropod genomes obtained similarly low assignment scores. Overall, the T. kingsejongenesis genome was moderately complete in non-dipteran arthropod genomes.
Table 5.
Tigriopus kingsejongensis genes: general statistics
| Genes (n) | 12 772 |
| Gene length sum (bp) | 82 293 116 |
| Exons per genes (n) | 4.6 |
| mRNA length sum (bp) | 43 306 342 |
| Average mRNA length (bp) | 1090 |
| Number of tRNA | 1393 |
| Number of rRNA | 215 |
Table 6.
Tigriopus kingsejongensis genome completeness reports with the other arthropod genomes
| Tigriopus | Daphnia | Ixodes | Mesobuthus | Strigamia | Tetranychus | Drosophila | Aedes | |
|---|---|---|---|---|---|---|---|---|
| Species | kingsejongensis | pulex | scapularis | martensii | maritima | urticae | melanogaster | aegypti |
| Assembly | This study | GCA_000187875.1 | GCA_000208615.1 | GCA_000484575.1 | Smar1.22 | GCA_000239435.1 | Dmel_r5.55 | AaegL3 |
| Sample type | genome | genome | genome | genome | genome | genome | genome | genome |
| CEGMAa | 83/77.8 | 99.2/98.8 | 79.8/41.9g | 57.3/24.2g | 95.1f | 98.0/95.2g | 100/100 | 99.2/83.5 |
| BUSCOb | 61.1 [10.5], 10.7, 28.1 | 83 [3.9], 11, 5.1e | 68.9 [2.4], 21.0, 10.1g | 34.4 [4.0], 23.0, 42.7g | 84 [5.9], 12, 3.2e | 68.8 [5.8], 9.9, 21.3g | 98 [6.4], 0.6, 0.3e | 86 [13], 10, 3.2 e |
| BUSCOc | 70.9 [13.6], 6.0, 23.0 | |||||||
| BUSCOd | 67.1 [16.8], 5.1, 27.7 |
Gene families
Orthologous groups were identified from 11 species (T. kingsejongensis, Aedes aegypti, D. melanogaster, Ixodes scapularis, Mesobuthus martensii, Strigamia martima, Tetranychus urticae, D. pulex, Homo sapiens, Ciona intestinalis, and Caenorhabditis elegans) (Table 7) using OrthoMCL [40] with standard parameters and options; transcript variants other than the longest translation forms were removed. For T. kingsejongensis, the coding sequence from the MAKER annotation pipeline was used. The 1:1:1 single-copy orthologous genes were subjected to phylogenetic construction and divergence time estimation. Protein-coding genes were aligned using the Probabilistic Alignment Kit (PRANK) with the codon alignment option [41], and poorly aligned sequences with gaps were removed using Gblock under the codon model [42]. A maximum likelihood phylogenetic tree was constructed using RAxML with 1000 bootstrap values [43] and calibrated the divergence time between species with TimeTree [44]. Finally, the average gene gain/loss rate along the given phylogeny was identified using CAFÉ 3.1 [45].
Table 7.
Summary of orthologous gene clusters in 11 representative species
| Species | Source of data | No. of coding genes | No. of gene families | No. of genes in gene families | No. of orphan genes | No. of unique gene families | Average No. of genes in gene families |
|---|---|---|---|---|---|---|---|
| Aedes aegypti | Ensembl genome 25 | 15 797 | 7958 | 12 792 | 7839 | 854 | 1.61 |
| Caenorhabditis elegans | Ensembl gene 78 | 20 447 | 6536 | 13 737 | 13 911 | 1528 | 2.10 |
| Ciona intestinalis | Ensembl gene 78 | 16 671 | 7017 | 9058 | 9654 | 503 | 1.29 |
| Daphnia pulex | Ensembl genome 25 | 30 590 | 6710 | 8362 | 7208 | 368 | 1.25 |
| Drosophila melanogaster | Ensembl gene 78 | 13 918 | 9673 | 21 917 | 20 917 | 2408 | 2.27 |
| Homo sapiens | Ensembl gene 78 | 20 300 | 8696 | 17 186 | 11 604 | 1065 | 1.98 |
| Ixodes scapularis | Ensembl genome 25 | 20 486 | 8097 | 11 277 | 12 389 | 873 | 1.39 |
| Mesobuthus martensii | http://lifecenter.sgst.cn/main/en/scorpion.jsp | 32 016 | 8389 | 19 961 | 23 627 | 2276 | 2.38 |
| Strigamia maritima | Ensembl genome 25 | 14 992 | 7727 | 11 012 | 7265 | 583 | 1.43 |
| Tetranychus urticae | Ensembl genome 25 | 18 224 | 6602 | 11 788 | 11 622 | 939 | 1.79 |
| Tigriopus kingsejongensis | this study | 12 772 | 6205 | 8813 | 6567 | 649 | 1.42 |
Orthologous gene clusters were constructed using four arthropod species (Antarctic copepod, T. kingsejongensis; scorpion, M. martensii; fruit fly, D. melanogaster, and water flea, D. pulex) to compare genomic features and adaptive divergence. In total, 2063 gene families are shared by all four species, and 1028 genes are T. kingsejongensis-specific. T. kingsejongensis shares 4559 (73.5%) gene families with D. pulex, which belongs to the same crustacean lineage, Vericrustacea; 3531 (56.9%) with D. melanogaster; and 3231 (52.1%) with M. martensii (Fig. 4A). Gene Ontology (GO) analysis revealed the 1028 T. kingsejongensis-specific genes are enriched in transport (single-organism transport, GO:0044765; transmembrane transport, GO:0055085; ion transport, GO:0006811; cation transport, GO:0006812) and single-organism metabolic processes (GO:0044710) (Tables S4 and S5).
Figure 4.

Comparative genome analyses of the T. kingsejongensis genome. A. Venn diagram of orthologous gene clusters between four arthropod lineages. B. Gene family gain-and-loss analysis. The number of gained gene families (red), lost gene families (blue) and orphan gene families (black) are indicated for each species. Time lines specify divergence times between the lineages.
Subsequently, gene gain-and-loss was analyzed in 11 representative species: T. kingsejongensis gained 735 and lost 4401 gene families (Fig. 4B). This species exhibits a gene family turnover of 5136, the largest value among the eight arthropods. The second largest value was obtained from T. uticae and the third from M. martensii. Non-insect arthropod genomes were relatively poorly assigned with CEGMA or BUSCO sets (Table 6). The assignment reports of these largely insect-based gene sets tend to have low assignment scores in non-insect or non-dipteran genomes [38, 46, 47]. This implies that careful examination of gene family turnover is needed in non-insect arthropod genomes, as well as globally approved arthropod orthologous gene sets.
Analysis of gene family expansion and contraction in T. kingsejongensis (Tables S6–S9) revealed 232 significantly expanded gene families, which are significantly overrepresented in amino acid and carbohydrate metabolism pathways, according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) [48].
Genome evolution
Adaptive functional divergence caused by natural selection is commonly estimated based on the ratio of nonsynonymous (dN) to synonymous (dS) mutations. To estimate dN, dS, the average dN/dS ratio (w), and lineage-specific positively selected genes (PSGs) in T. kingsejongensis and T. japonicus, protein-coding genes from T. japonicus were added to define orthologous gene families among four species (T. kingsejongensis, T. japonicus, D. pulex, and D. melanogaster) using the program OrthoMCL with the same conditions previously described. We identified 2937 orthologous groups shared by all four species; single-copy gene families were used to construct a phylogenetic tree and estimate the time since divergence using the methods described above. Each of the identified orthologous genes was aligned using PRANK, and poorly aligned sequences with gaps were removed using Gblock. Alignments with less than 40% identity and genes shorter than 150 bp were eliminated in subsequent procedures. The values of dN, dS and w were estimated from each gene using the Codeml program implemented in the Phylogenetic Analysis by Maximum Likelihood (PAML) package with the free-ratio model [49] under F3×4 codon frequencies; orthologs with w ≤ 5 and dS ≤ 3 were retained [50]. To examine the accelerated nonsynonymous divergence in either the T. kingsejongensis or T. japonicus lineages, a binomial test [51] was used to determine GO categories with at least 20 orthologous genes. To define PSGs in T. kingsejongensis and T. japonicus, basic and branch-site models were applied, and Likelihood Ratio Tests (LRTs) were used to remove genes under relaxation of selective pressure. To investigate the functional categories and pathways enriched in PSGs, the Database for Annotation, Visualization and Integrated Discovery (DAVID) Functional Annotation [52] was used with Fisher's exact test (cutoff: P ≤ 0.05).
The average w value from 2937 co-orthologous genes of T. kingsejongensis (0.0027) is higher than that of T. japonicus (0.0022). GO categories that show evidence of accelerated evolution in T. kingsejongensis are: energy metabolism (generation of precursor metabolites and energy, GO:0006091; cellular respiration, GO:0045333) and carbohydrate metabolism (monosaccharide metabolic process, GO:0005996; hexose metabolic process, GO:0019318) (Fig. 5A, Table S10). Branch-site model analysis showed that genes belonging to these functional categories have undergone a significant positive selection process by putative functional divergence in certain lineages. There are 74 and 79 PSGs in T. kingsejongensis (Table S11) and T. japonicus (Table S12), respectively.
Figure 5.

Tigriopus kingsejongensis-specific adaptive evolution. A. Global mean w (ratio of nonsynonymous (dN) to synonymous mutations (dS)) distribution by GO categories of T. kingsejongensis and T. japonicus. GO categories showing supposedly accelerated nonsynonymous divergence (binomial test, test statistic <0.05) in T. kingsejongensis and T. japonicus are colored in red and blue, respectively. B. A total of seven enzyme-coding genes were positively selected genes (PSGs) involved in the four metabolic pathways (oval frame) of T. kingsejongensis: energy (purple), nucleotide (red), lipid (green), and carbohydrate (blue) metabolic pathways. The three genes belonging to the oxidative phosphorylation pathway (KEGG pathway map00190) (rectangular frame) are presented below the enzymes involved. Solid lines indicate direct processes and dashed lines indicate that more than one step is involved in a process.
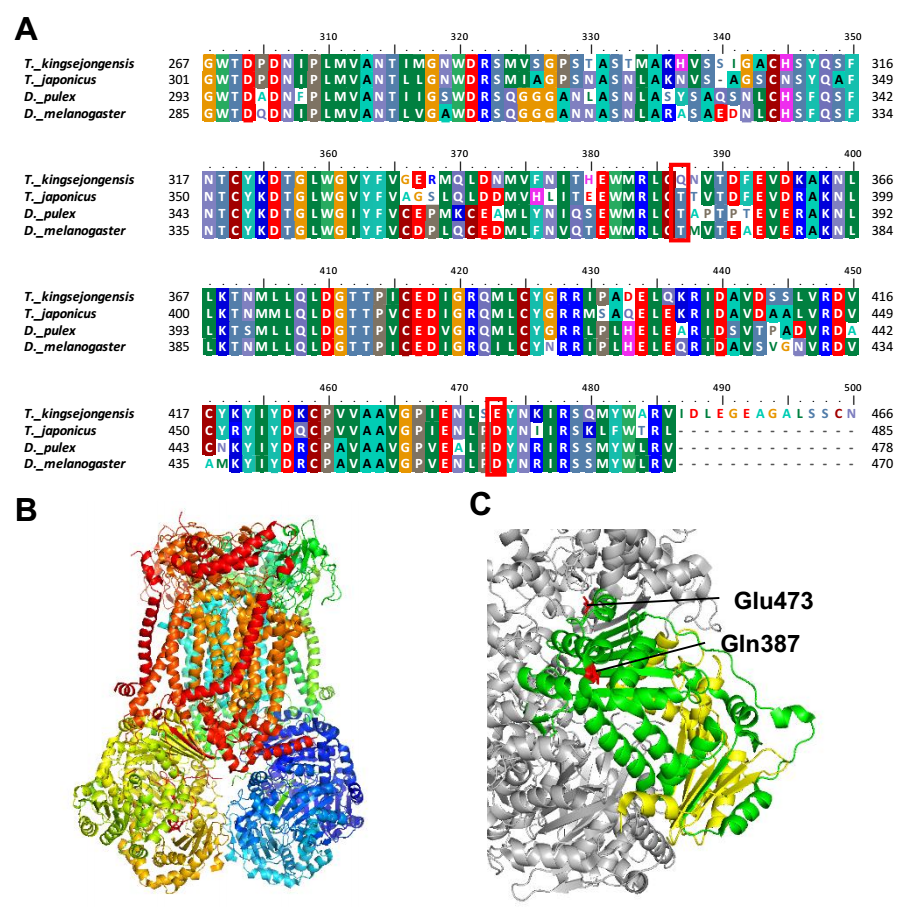
The functional categories enriched in T. kingsejongensis, when compared to T. japonicus, support the idea that functional divergence in T. kingsejongensis is strongly related to energy metabolism (oxidative phosphorylation, GO:0006119; energy-coupled proton transport down electrochemical gradient, GO:0015985; ATP synthesis-coupled proton transport, GO 0015986; generation of precursor metabolites and energy, GO:0006091) (Fig. 5B, Tables S13 and S14). In particular, three of the identified genes are involved in the oxidative phosphorylation (OxPhos) pathway, which provides the primary cellular energy source in the form of adenosine triphosphate (ATP). These three genes are nuclear-encoded mitochondrial genes: the catalytic F1 ATP synthase subunit alpha (ATP5A) (Fig. S4), a regulatory subunit acting as an electron transport chain such as ubiquinol-cytochrome c reductase core protein (UQCRC1) (Fig. S5), and an electron transfer flavoprotein alpha subunit (ETFA) (Fig. S6).
Availability of supporting data
T. kingsejongensis genome and transcriptome data are deposited in the Sequence Read Archive (SRA) as BioProjects PRJNA307207 and PRJNA307513, respectively. Other supporting data is available in the GigaScience repository, GigaDB [53].
Additional file
Supplementary data are available at GIGSCI online.
Figure S1. Map showing location of the Tigriopus kingsejongensis sampling site.
Figure S2. BLAST top-hit species distribution of Tigriopus kingsejongensis. Data obtained using BLASTx against the National Center for Biotechnology Information's (NCBI) non-redundant protein database with an E value cutoff of 1e−5.
Figure S3. Gene Ontology distribution of annotated genes. Gene Ontology (GO) annotation of predicted Tigriopus kingsejongensis genes was conducted using the GO annotation. The figure illustrates the number of genes from major GO modules of molecular function (MF), biological process (BP), and cellular component (CC).
Figure S4. Tigriopus kingsejongensis-specific amino acid changes in ATP synthase subunit alpha. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of the ATP synthase (PDB ID: 1BMF). C. The specific amino acid change Ala166 is colored in red (in stick form) and positioned within the external loop region of nucleotide-binding domain. The three domains of the ATP synthase subunit alpha illustrated in cartoon form are colored accordingly (blue, beta-barrel domain; green, nucleotide-binding domain; purple: C terminal domain).
Figure S5. Tigriopus kingsejongensis-specific amino acid changes in ubiquinol-cytochrome c reductase core protein I. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of ubiquinol-cytochrome c reductase (PDB ID: 1QCR). C. Positions of the specific amino acid changes in ubiquinol-cytochrome c reductase core protein I are colored red (stick form). The insulinase domain is yellow and the peptidase M16 domain is green.
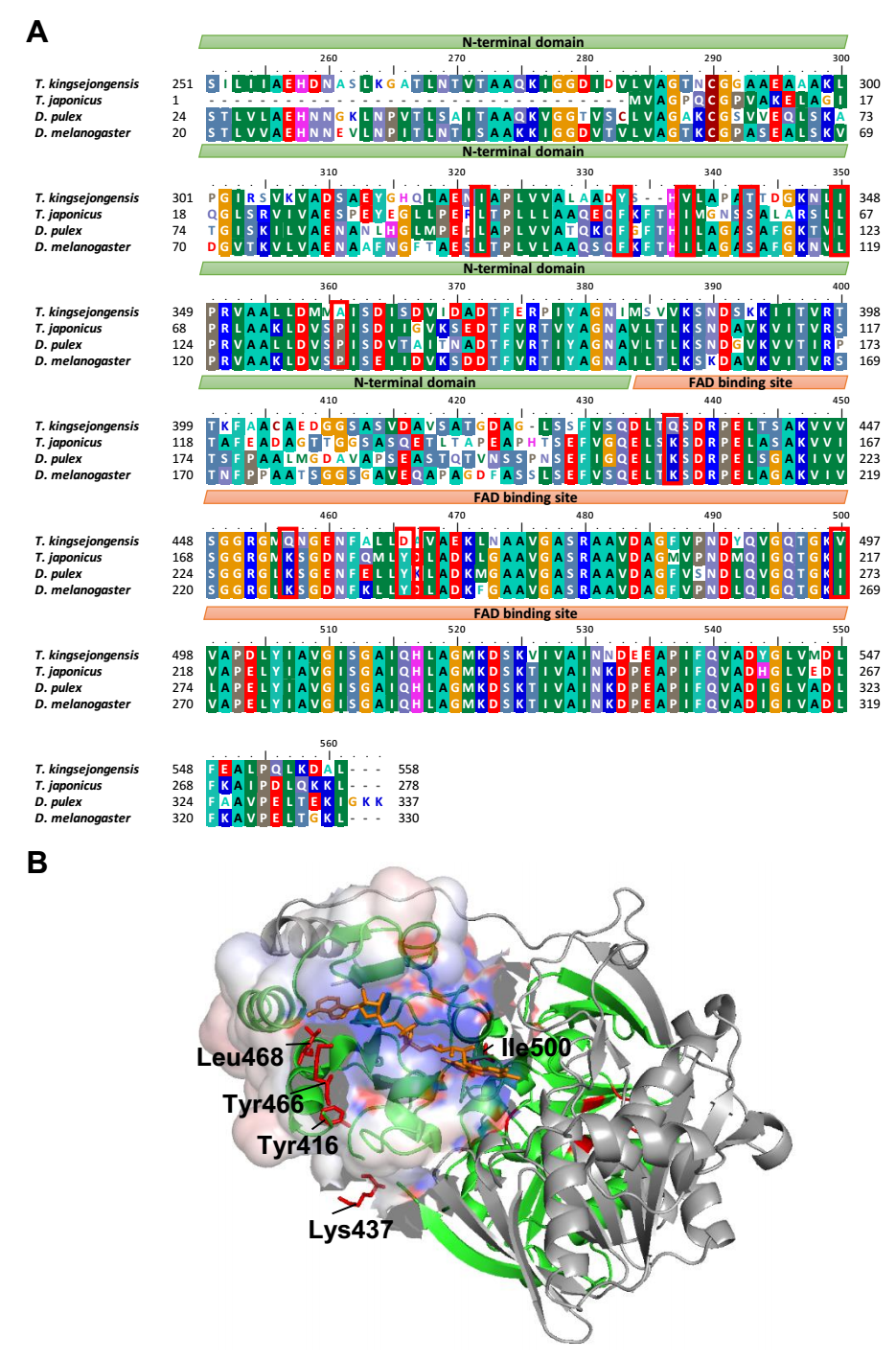
Figure S6. Tigriopus kingsejongensis-specific amino acid changes in electron-transferring flavoprotein. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. Among the ten amino acid changes, the five sites are located within the N-terminal domain and the other five are positioned within the FAD binding domain. B. Cartoon of the protein crystal structure of the electron-transferring flavoprotein (PDB ID: 1EFV). The five amino acid sites within the FAD binding domain are colored in red (stick form). Electron-transferring flavoprotein alpha subunit is green; FAD-binding domain is represented by color-coded electrostatic surface (blue, positive charge; red, negative charge; grey, neutral charge); FAD is orange (stick form). Notably, the Asp463 residue makes a salt bridge with Arg437 in the homology model structure of electron-transferring flavoprotein from T. kingsejongensis. In addition, Gln454 is located near the bound FAD co-factor and may form a hydrogen bond with the N7A atom of FAD in the model structure of electron-transferring flavoprotein from T. kingsejongensis.
Table S1. Number of tRNA in the Tigriopus kingsejongensis genome.
Table S2. Known repetitive and transposable elements in the Tigropus kingsejongensis genome.
Table S3. Transposable elements in the Tigriopus kingsejongensis genome.
Table S4. Gene Ontology (GO) of lineage-specific gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Table S5. Annotated domains of lineage-specific gene families in the Tigriopus kingsejongensis genome.
Table S6. Gene Ontology (GO) of expanded gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to p-value.
Table S7. Gene annotation of the expanded genes in the Tigriopus kingsejongensis genome.
Table S8. Gene Ontology (GO) of contracted genes in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Table S9. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of expanded genes in the Tigriopus kingsejongensis genome.
Table S10. Gene Ontology (GO) categories displaying w (ratio of nonsynonymous (dN) to synonymous mutations (dS)) in the genomes of Tigriopus kingsejongensis and T. japonicus.
Table S11. Lists and annotations of positively selected genes in the Tigriopus kingsejongensis genome.
Table S12. Lists and annotations of positively selected genes in the Tigriopus japonicus genome.
Table S13. Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Table S14. Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus japonicus genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
List of abbreviations
ATP: Adenosine triphosphate; BUSCO: Benchmarking Universal Single-Copy Orthologs; CEGMA: Core Eukaryotic Genes Mapping Approach; CM: Covariance model; DAVID: Database for Annotation, Visualization and Integrated Discovery; dN: Nonsynonymous mutations; dS: Synonymous mutations; EST: Expressed sequence tag; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; LRT: Likelihood Ratio Test; OxPhos: Oxidative phosphorylation; PAML: Phylogenetic Analysis by Maximum Likelihood; PRANK: Probabilistic Alignment Kit; PSG: Positively selected gene; RAM: Random access memory; SRA: Sequence Read Archive; SSR: Simple sequence repeat; TE: Transposable element; w: dN/dS ratio
Competing interests
The authors declare no competing interests.
Funding
This work was supported by the Korea Polar Research Institute-funded the grant ‘Antarctic organisms: cold-adaptation mechanism and its application’ (PE16070), and basic research program (PE14260).
Authors’ contributions
HP, S Kim and HWK conceived and designed experiments and analyses; S Kang, DHA, SGL, SCS, JL, GSM and HL performed experiments and conducted bioinformatics. Seunghyun Kang, HWK, S Kim and HP. wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Map showing location of the Tigriopus kingsejongensis sampling site.
{kind=link}
BLAST top-hit species distribution of Tigriopus kingsejongensis. Data obtained using BLASTx against the National Center for Biotechnology Information's (NCBI) non-redundant protein database with an E value cutoff of 1e−5.
{kind=link}
Gene Ontology distribution of annotated genes. Gene Ontology (GO) annotation of predicted Tigriopus kingsejongensis genes was conducted using the GO annotation. The figure illustrates the number of genes from major GO modules of molecular function (MF), biological process (BP), and cellular component (CC).
{kind=link}
Tigriopus kingsejongensis-specific amino acid changes in ATP synthase subunit alpha. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of the ATP synthase (PDB ID: 1BMF). C. The specific amino acid change Ala166 is colored in red (in stick form) and positioned within the external loop region of nucleotide-binding domain. The three domains of the ATP synthase subunit alpha illustrated in cartoon form are colored accordingly (blue, beta-barrel domain; green, nucleotide-binding domain; purple: C terminal domain).
{kind=link}
Tigriopus kingsejongensis-specific amino acid changes in ubiquinol-cytochrome c reductase core protein I. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of ubiquinol-cytochrome c reductase (PDB ID: 1QCR). C. Positions of the specific amino acid changes in ubiquinol-cytochrome c reductase core protein I are colored red (stick form). The insulinase domain is yellow and the peptidase M16 domain is green.
{kind=link}
Tigriopus kingsejongensis-specific amino acid changes in electron-transferring flavoprotein. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. Among the ten amino acid changes, the five sites are located within the N-terminal domain and the other five are positioned within the FAD binding domain. B. Cartoon of the protein crystal structure of the electron-transferring flavoprotein (PDB ID: 1EFV). The five amino acid sites within the FAD binding domain are colored in red (stick form). Electron-transferring flavoprotein alpha subunit is green; FAD-binding domain is represented by color-coded electrostatic surface (blue, positive charge; red, negative charge; grey, neutral charge); FAD is orange (stick form). Notably, the Asp463 residue makes a salt bridge with Arg437 in the homology model structure of electron-transferring flavoprotein from T. kingsejongensis. In addition, Gln454 is located near the bound FAD co-factor and may form a hydrogen bond with the N7A atom of FAD in the model structure of electron-transferring flavoprotein from T. kingsejongensis.
{kind=link}
Number of tRNA in the Tigriopus kingsejongensis genome.
Known repetitive and transposable elements in the Tigropus kingsejongensis genome.
Transposable elements in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of lineage-specific gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Annotated domains of lineage-specific gene families in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of expanded gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to p-value.
Gene annotation of the expanded genes in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of contracted genes in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of expanded genes in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) categories displaying w (ratio of nonsynonymous (dN) to synonymous mutations (dS)) in the genomes of Tigriopus kingsejongensis and T. japonicus.
Lists and annotations of positively selected genes in the Tigriopus kingsejongensis genome.
Lists and annotations of positively selected genes in the Tigriopus japonicus genome.
Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus japonicus genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Acknowledgements
We would like to thank Joseph A. Covi for comments and discussion.
References
- 1. Huys R, Boxshall GA. Copepod evolution. Ray Society; 1991. [Google Scholar]
- 2. Humes AG. How many copepods? Hydrobiologia 1994;292:1–7. [Google Scholar]
- 3. Wells P, Persoone G, Jaspers ECC. Marine ecotoxicological tests with zooplankton. In: Persoone G, Jaspers E, Claus C (Eds.), Ecotoxicological Testing for the Marine Environment. Bredene: Inst. Mar. Sci. Res.; 1984. [Google Scholar]
- 4. Ruppert E, Fox R, Barnes R. Invertebrate Zoology, A Functional Evolutionary Approach. Belmont, CA: Brooks/Cole-Thomson Learning; 2003. [Google Scholar]
- 5. Goolish E, Burton R. Energetics of osmoregulation in an intertidal copepod: Effects of anoxia and lipid reserves on the pattern of free amino accumulation. Funct Ecol 1989:81–9. [Google Scholar]
- 6. Lazzaretto I, Libertini A. Karyological comparison among different Mediterranean populations of the genus Tigriopus (Copepoda Harpacticoida). Boll Zool 2009;53:197–201. [Google Scholar]
- 7. Davenport J, Barnett P, McAllen R. Environmental tolerances of three species of the harpacticoid copepod genus Tigriopus. J Mar Biol Assoc UK 1997;77:3–16. [Google Scholar]
- 8. Raisuddin S, Kwok KW, Leung KM, Schlenk D, Lee J-S. The copepod Tigriopus: A promising marine model organism for ecotoxicology and environmental genomics. Aquat Toxicol 2007;83:161–73. [DOI] [PubMed] [Google Scholar]
- 9. Whole Genome Assembly of Tigriopus californicus provided by the Weizhong Li lab, UCSD Calit2 [ http://i5k.nal.usda.gov/Tigriopus_californicus]
- 10. Lee J-S, Rhee J-S, Kim R-O, Hwang D-S, Han J, Choi B-S, Park GS, Kim I-C, Park HG, Lee Y-M. The copepod Tigriopus japonicus genomic DNA information (574Mb) and molecular anatomy. Mar Environ Res 2010;69:S21–3. [DOI] [PubMed] [Google Scholar]
- 11. Whole genome assembly of Eurytemora affinis [ http://i5k.nal.usda.gov/Eurytemora_affinis]
- 12. The Salmon Louse Genome Project [ http://sealouse.imr.no/]
- 13. Thorne MAS, Kagoshima H, Clark MS, Marshall CJ, Wharton DA. Molecular analysis of the cold tolerant Antarctic Nematode, Panagrolaimus davidi. PLOS one 2014;9:e104526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Everatta MJ, Worlandb MR, Balea JS, Conveyb P, Hayward SAL. Pre-adapted to the maritime Antarctic? – Rapid cold hardening of the midge, Eretmoptera murphyi. J Insect Physiol 2012;58:1104–11. [DOI] [PubMed] [Google Scholar]
- 15. Bromwich DH, Nicolas JP, Monaghan AJ, Lazzara MA, Keller LM, Weidner GA, Wilson AB. Central West Antarctica among the most rapidly warming regions on Earth. Nature Geoscience 2013;6:139–45. [Google Scholar]
- 16. Park E-O, Lee S, Cho M, Yoon SH, Lee Y, Lee W. A new species of the genus Tigriopus (Copepoda: Harpacticoida: Harpacticidae) from Antarctica. Proc Biol Soc Wash 2014;127:138–54. [Google Scholar]
- 17. Birkenmajer K. Geology of Admiralty Bay, King George Island (South Shetland Islands). An outline. Pol Polar Res 1980;1:29–54. [Google Scholar]
- 18. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011;27:764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA et al. . A whole-genome assembly of Drosophila. Science 2000;287:2196–2204. [DOI] [PubMed] [Google Scholar]
- 20. Gordon A, Hannon G. Fastx-toolkit. FASTQ/A short-reads preprocessing tools (unpublished) http://hannonlab cshl edu/fastx_toolkit; 2010. [Google Scholar]
- 21. Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011;27:578–9. [DOI] [PubMed] [Google Scholar]
- 22. Nadalin F, Vezzi F, Policriti A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 2012;13:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 2011;12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smit AFA HR, Green P. RepeatMasker Open-3.0. 1996-2004 (http://www.RepeatMakser.org). [Google Scholar]
- 25. Korf I. Gene finding in novel genomes. BMC Bioinformatics 2004;5:59.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 2005;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005;21:3674–6. [DOI] [PubMed] [Google Scholar]
- 31. Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003;31:365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Acland A, Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, Bryant SH, Canese K, Church DM. Database resources of the national center for biotechnology information. Nucleic Acids Res 2014;42:D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one 2011;6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics 2009;25:1335–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res 2011;39:D141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997;25:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007;23:1061–7. [DOI] [PubMed] [Google Scholar]
- 38. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015:btv351. [DOI] [PubMed] [Google Scholar]
- 39. Chipman AD, Ferrier DE, Brena C, Qu J, Hughes DS, Schröder R, Torres-Oliva M, Znassi N, Jiang H, Almeida FC. The first myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol 2014;12:e1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003;13:2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A 2005;102:10557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000;17:540–52. [DOI] [PubMed] [Google Scholar]
- 43. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 2006;22:2971–2. [DOI] [PubMed] [Google Scholar]
- 45. Han MV, Thomas GW, Lugo-Martinez J, Hahn MW. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol 2013;30:1987–97. [DOI] [PubMed] [Google Scholar]
- 46. Rider SD, Morgan MS, Arlian LG. Draft genome of the scabies mite. Parasites & Vectors 2015;8:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoy M, Waterhouse R, Wu K, Estep A, Ioannidis P, Palmer W, Pomerantz A, Simão F, Thomas J, Jiggins F. Genome sequencing of the phytoseiid predatory mite Metaseiulus occidentalis reveals completely atomised Hox genes and super-dynamic intron evolution. Genome biology and evolution 2016;8:1762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2015:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 50. Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014;346:1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Consortium TCSaA Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005;437:69–87. [DOI] [PubMed] [Google Scholar]
- 52. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 2008;4:44–57. [DOI] [PubMed] [Google Scholar]
- 53. Kang S, Ahn D, Lee JH, Lee SG, Shin SC, Lee J, Min G, Lee H, Kim H, Kim S, Park H. Supporting data for “The genome of the Antarctic-endemic copepod, Tigriopus kingsejongensis”. GigaScience Database. 2016. http://dx.doi.org/10.5524/100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Map showing location of the Tigriopus kingsejongensis sampling site.
BLAST top-hit species distribution of Tigriopus kingsejongensis. Data obtained using BLASTx against the National Center for Biotechnology Information's (NCBI) non-redundant protein database with an E value cutoff of 1e−5.
Gene Ontology distribution of annotated genes. Gene Ontology (GO) annotation of predicted Tigriopus kingsejongensis genes was conducted using the GO annotation. The figure illustrates the number of genes from major GO modules of molecular function (MF), biological process (BP), and cellular component (CC).
Tigriopus kingsejongensis-specific amino acid changes in ATP synthase subunit alpha. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of the ATP synthase (PDB ID: 1BMF). C. The specific amino acid change Ala166 is colored in red (in stick form) and positioned within the external loop region of nucleotide-binding domain. The three domains of the ATP synthase subunit alpha illustrated in cartoon form are colored accordingly (blue, beta-barrel domain; green, nucleotide-binding domain; purple: C terminal domain).
Tigriopus kingsejongensis-specific amino acid changes in ubiquinol-cytochrome c reductase core protein I. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. B. Cartoon of the protein crystal structure of ubiquinol-cytochrome c reductase (PDB ID: 1QCR). C. Positions of the specific amino acid changes in ubiquinol-cytochrome c reductase core protein I are colored red (stick form). The insulinase domain is yellow and the peptidase M16 domain is green.
Tigriopus kingsejongensis-specific amino acid changes in electron-transferring flavoprotein. A. Clustal X alignment of the amino acid sequences between four species. Tigriopus kingsejongensis-specific amino acid changes representing positive selections are presented with red boxes. Among the ten amino acid changes, the five sites are located within the N-terminal domain and the other five are positioned within the FAD binding domain. B. Cartoon of the protein crystal structure of the electron-transferring flavoprotein (PDB ID: 1EFV). The five amino acid sites within the FAD binding domain are colored in red (stick form). Electron-transferring flavoprotein alpha subunit is green; FAD-binding domain is represented by color-coded electrostatic surface (blue, positive charge; red, negative charge; grey, neutral charge); FAD is orange (stick form). Notably, the Asp463 residue makes a salt bridge with Arg437 in the homology model structure of electron-transferring flavoprotein from T. kingsejongensis. In addition, Gln454 is located near the bound FAD co-factor and may form a hydrogen bond with the N7A atom of FAD in the model structure of electron-transferring flavoprotein from T. kingsejongensis.
Number of tRNA in the Tigriopus kingsejongensis genome.
Known repetitive and transposable elements in the Tigropus kingsejongensis genome.
Transposable elements in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of lineage-specific gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Annotated domains of lineage-specific gene families in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of expanded gene families in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to p-value.
Gene annotation of the expanded genes in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) of contracted genes in the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of expanded genes in the Tigriopus kingsejongensis genome.
Gene Ontology (GO) categories displaying w (ratio of nonsynonymous (dN) to synonymous mutations (dS)) in the genomes of Tigriopus kingsejongensis and T. japonicus.
Lists and annotations of positively selected genes in the Tigriopus kingsejongensis genome.
Lists and annotations of positively selected genes in the Tigriopus japonicus genome.
Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus kingsejongensis genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.
Enriched Gene Ontology (GO) categories identified by positively selected genes from the Tigriopus japonicus genome. REVIGO software was used to cluster related GO terms (in bold letters) according to P-value.