

Abstract
Inflammation and microbiota are critical components of intestinal tumorigenesis. To dissect how the microbiota contributes to tumor distribution, we generated germ-free (GF) ApcMin/+ and ApcMin/+;Il10−/− mice and exposed them to specific-pathogen-free (SPF) or colorectal cancer-associated bacteria. We found colon tumorigenesis significantly correlated with inflammation in SPF housed ApcMin/+;Il10−/−, but not ApcMin/+ mice. In contrast, small intestinal neoplasia development significantly correlated with age in both ApcMin/+;Il10−/− and ApcMin/+ mice. GF ApcMin/+;Il10−/− mice conventionalized by an SPF microbiota had significantly more colon tumors compared to GF mice. Gnotobiotic studies revealed that while Fusobacterium nucleatum clinical isolates with FadA and Fap2 adhesins failed to induce inflammation and tumorigenesis, pks+ Escherichia coli promoted tumorigenesis in the ApcMin/+;Il10−/− model in a colibactin-dependent manner, suggesting colibactin is a driver of carcinogenesis. Our results suggest a distinct etiology of cancers in different locations of the gut, where colon cancer is primarily driven by inflammation and the microbiome, while age is a driving force for small intestine cancer.
Keywords: colorectal cancer, microbiota, colibactin, Fusobacterum nucleatum, gnotobiotic
Introduction
Colorectal cancer (CRC), the third most common type of malignancy and the third leading cause of cancer-related deaths in the United States (1), involves both genetic and environmental factors. Among the genomic changes associated with CRCs, loss-of-function mutations in the Apc (adenomatous polyposis coli) gene, a regulator of the WNT signaling pathway, are the most prevalent and are considered the initiating event in ~80% of CRCs (2). Of the environmental factors, the gut microbiota is increasingly appreciated as a key player in CRC pathogenesis. CRC patients often carry a distinct microbiota from the healthy population (3). Microbes can modulate CRC development directly by generating genotoxins, or indirectly and more commonly by mediating inflammatory and immune responses (4,5). Inflammation is not only a hallmark of CRC (5), but also an established risk factor for CRC as supported by epidemiological data from individuals with inflammatory bowel diseases (IBD) (6).
While human studies provide valuable correlation data on CRC, much of the mechanistic insight into the disease etiology is obtained from mouse models. Mouse CRC models can be categorized into two classes: spontaneous and chemical-induced (7). Spontaneous CRC mice carry mutations in genes frequently mutated in human CRCs. The multiple intestinal neoplasia (Min) mouse (referred to as ApcMin/+ hereafter), a commonly used animal model of intestinal carcinogenesis, carries a point mutation in one allele of the Apc gene and is susceptible to spontaneous intestinal adenoma formation, although predominantly in the small bowel, without exhibiting chronic intestinal inflammation (8). A general pro-tumorigenic role for the microbiota was demonstrated in the ApcMin/+ model, as the mice display reduced tumor load in the small and/or large intestine when derived under germ-free (GF) conditions (9,10). Noticeably, inflammation also enhances development of colon cancer in this model, as seen with the use of dextran sulfate sodium (DSS) (11), by specifically deleting the Apc gene in epithelial cells (12), and by genetically introducing defective IL-10 signaling (13–15). Inflammation and colonic polyposis in mice with Apc deficiency and T cell-specific deletion of Il10, ApcΔ468;CD4CreIl10f/f mice, can be attenuated by antibiotic treatment (14), suggesting that microbiota-driven inflammation underlies colitis-associated CRC.
The mechanisms by which microbes have been shown to promote development of CRC are diverse and somewhat specific to each microorganism. For example, enterotoxigenic Bacteroides fragilis promotes CRC through induction of the Th17 response (16), polyketide synthase (pks)+ island carrying E. coli via production of the genotoxin colibactin (17), and Fusobacterium nucleatum adhesins that either bind E-cadherin to promote tumor growth (FadA) or promote immune invasion and localization to tumors (Fap2) (18–20). Although genetics, inflammation, and microbes play a role in promoting intestinal carcinogenesis in preclinical models; it is unclear how these factors interact and how much each contributes to promotion of CRC.
To stringently evaluate the relationship between genetic susceptibility to inflammation, microbial status, and cancer, we utilized specific-pathogen-free (SPF) and gnotobiotic ApcMin/+ and ApcMin/+;Il10−/− mice. In this study, we found that inflammation and the microbiota is essential for colorectal but not small intestinal neoplasia in SPF ApcMin/+;Il10−/− mice. We also demonstrated that CRC-associated bacteria have differential abilities to promote CRC with colibactin-producing E. coli, but not F. nucleatum, inducing colon tumors in ApcMin/+;Il10−/− mice.
Materials and Methods
Animals
The University of Florida Institutional Animal Care and Use Committee approved all animal experiments (Protocol #201308038). 129/SvEv ApcMin/+ mice were derived GF and crossed to GF 129/SVEv Il10−/− mice to generate GF ApcMin/+;Il10−/− mice. GF ApcMin/+;Il10−/− and ApcMin/+ mice were transferred to the SPF breeding suite and bred for 2–3 generations. SPF ApcMin/+;Il10−/− and ApcMin/+ mice were either transferred to an SPF housing suite after weaning or remained in the breeding suite, mice transferred to the SPF housing suite were sacrificed at 12, 16 and 20 weeks of age. SPF ApcMin/+;Il10−/− and ApcMin/+ mice older than 20 weeks were retired breeders from the SPF breeding suite.
Bacterial strains and culture conditions
F. nucleatum strains were provided by Dr. Emma Allen-Vercoe (University of Guelph), including the IBD clinical isolate EAVG_016, and CRC isolates CC53, CC7/3JVN3C1, CC7/5JVN1A4, CC2/3Fmu1, CC2/3FmuA and CC7/4Fmu3 (used for the 20-week colonization experiment in ApcMin/+ mice). E. coli NC101 or NC101 ΔclbP were cultured from glycerol stocks in LB broth, then diluted 1:10 in fresh LB medium and cultured at 37°C before harvesting for gavage. F. nucleatum strains were cultured in Brain Heart Infusion Broth (BHI) (AS-872, Anaerobe Systems) statically at 37°C in an anaerobic chamber (type B Vinyl, Coy Laboratory). Enumeration of F. nucleatum was done by anaerobically plating serial dilutions of culture or fecal materials on fastidious anaerobic agar supplemented with 5% sheep blood.
PCR detection of F. nucleatum adhesins
All F. nucleatum strains were screened for the FadA and Fap2 adhesins by PCR using the following primers: Fn 16S_F GGATTTATTGGGCGTAAAGC, ; Fn 16S_R, GGCATTCCTACAAATATCTACGAA; fadA_F CAAATCAAGAAGAAGCAAGATTCAAT ; fadA_R, GCTTGAAGTCTTTGAGCTCT (18); fap2_F, AGCCTCTGAGGGTACAAGGT; fap2_R, TGAGCCCCTCCTTCTTCTGA. The screening revealed 4 F. nucleatum CRC isolate strains were fadA+, fap2− and 2 were fadA+, fap2+ (Supplementary Fig. S1).
E.coli NC01 clbP mutation
Inactivation of gene clbP was performed by using the lambda Red recombinase method (21) using primers clbP-P1 (TTCCGCTATGTGCGCTTTGGCGCAAGAACATGAGCCTATCGGGGCGCAAgtgtaggctggagctgcttc) and clbP-P2 (GTATACCCGGTGCGACATAGAGCATGGCGGCCACGAGCCCAGGAACCGCCcatatgaatatcctccttag). The allelic exchange was confirmed by PCR using primers IHAPJPN29 and IHAPJPN30 (22).
SPF microbiota preparation
Cecal and fecal contents were collected from wild type 129/SvEv mice that were housed under SPF conditions in the animal facility at the University of Florida. 1 gram of the contents was suspended in 10 ml sterile PBS, broken down using pipette tips, and vortexed. After settling for 2 min, the supernatant was transferred to a new tube, mixed with equal volume of sterile 20% glycerol, and frozen at −80°C.
Mouse colonization
7–12 week GF ApcMin/+ and ApcMin/+;Il10−/− were transferred to SPF conditions or gnotobiotic isolators as described above. SPF stock microbiota was diluted 1:106 and 200 ul of this mixture was gavaged to each mouse. E. coli NC101 or NC101 ΔclbP was gavaged at 108 CFU/mouse. For AOM/Il10−/− experiments, mice received 6 weekly intraperitoneal AOM (A5486, Sigma) injections (10 mg kg -1) starting 1 week post mono-association with E. coli and mice were sacrificed 20 weeks post mono-association. F. nucleatum was gavaged at 108 CFU/mouse when a single strain was used, or 108 CFU per strain per mouse when a mixture of strains were used. BHI medium weekly gavaged mice were used as control for F. nucleatum experiments.
Mice were euthanized at indicated time points. The small intestine, cecum and colon were cut open longitudinally and macroscopic tumors were counted. About 1 × 0.5 cm snips were taken from the proximal and distal colon, flash frozen in liquid nitrogen and stored at −80°C until analysis. The rest of the colon was Swiss rolled and fixed in 10% neutral buffered formalin solution. Swiss rolls were processed, paraffin-embedded, sectioned and H&E stained by the Molecular Pathology Core at the University of Florida. Histological scoring of inflammation was performed blindly as described previously (17) and calculated as the average between the proximal and distal colon region scores.
Immunohistochemistry (IHC)
IHC was performed as described previously (17). Briefly, Swiss roll sections were deparaffinized, rehydrated, and boiled in 10mM citrate buffer for antigen retrieval. For CTNBB1, the mouse anti-CTNNB1 antibody (1:300 overnight) (6101503, BD Transduction Laboratories) and mouse on mouse (M.O.M.) peroxidase kit (PK-2200, Vector Labs) were used. For PCNA, sections were blocked with 1% BSA, incubated with anti-PCNA clone PC10 (M087901-2, Dako) mouse monoclonal antibody (1:300, 30 minutes), followed by 1:1000 goat anti-mouse biotin secondary antibody (31800 Fisher), and then incubated with streptavidin-horseradish peroxidase (18–152, Millipore). Liquid DAB+ (K3467, Dako) was used according to manufacturer instructions for development.
Fecal DNA extraction and 16S qPCR
DNA was extracted using phenol:chloroform separation followed by DNeasy Blood & Tissue Kit (69506, Qiagen). qPCR was performed on CFX384 Touch Real-Time PCR Detection System (1855485, Bio-rad) using the SsoAdvanced™ Universal SYBR Green Supermix (1725274, Bio-rad). The following primers were used: Fuso_F GGATTTATTGGGCGTAAAGC, Fuso_R GGCATTCCTACAAATATCTACGAA; and Eubacteria_F GGTGAATACGTTCCCGG, Eubacteria_R TACGGCTACCTTGTTACGACTT.
16S rRNA sequencing
The V1–V3 region hypervariable region of the 16S rRNA gene was amplified using primer pair 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′). Both the forward and the reverse primers contained universal Illumina paired-end adapter sequences, as well as unique individual 4–6 nucleotide barcodes between PCR primer sequence and the Illumina adapter sequence to allow multiplex sequencing (Supplementary Table S1). PCR products were visualized on an agarose gel, before samples were purified using the Agencourt AMPure XP kit (A63881, Beckman Coulter) and quantified by qPCR with the KAPA Library Quantification Kit (KK4824, KAPA Biosystems). Equimolar amount of samples were then pooled and sequenced with an Illumina MiSeq. See Supplementary Data for description of 16S rRNA sequencing analysis.
qPCR examination of inflammatory cytokines
RNA was extracted from frozen tissue snips using Trizol reagent followed by phenol:chloroform separation. After DNA removal using the Turbo DNA-free Kit (AM1907, Ambion), 10–1000 ng of RNA was used for cDNA synthesis using iScript cDNA Synthesis Kit (1708891, Bio-rad). qPCR was performed on CFX384 Touch Real-Time PCR Detection System (1855485, Bio-rad) using the SsoAdvanced™ Universal SYBR Green Supermix (1725274, Bio-rad). The following primers were used: IL6_F CGGAGGCTTGGTTACACATGTT, IL6_R CTGGCTTTGTCTTTCTTGTTATC; TNFα_F ATGAGCACAGAAAGCATGATC, TNFα_R TACAGGCTTGTCACTCGAATT; IFNγ_F ACGCTTATGTTGTTGCTGATGG, IFNγ_R CTTCCTCATGGCTGTTTCTGG; IL-1β_F GCCCATCCTCTGTGACTCAT, IL-1β_R AGGCCACGGTATTTTGTCG; IL17A_F GCCCTCAGACTACCTCAACC, IL17A_R ACACCCACCAGCATCTTCTC; IL-22_F CATGCAGGAGGTGGTGCCTT, IL-22_R CAGACGCAAGCATTTCTCAG; 36B4_F TCCAGGCTTTGGGCATCA, 36B4_R CTTTATTCAGCTGCACATCACTCAGA; and GUSB_F CCGATTATCCAGAGCGAGTATG, GUSB_R CTCAGCGGTGACTGGTTCG. 36B4 and Gusb were used as references. Relative fold gene expression was calculated using the delta delta Ct method.
Statistical analysis
Data are expressed as mean +/− standard deviation. Significance thresholds of ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, or NS: not significant (p> 0.05) are shown. Statistics for all figures except for the 16S rRNA sequencing analysis were calculated with Graphpad Prism using the Mann-Whitney nonparametric unpaired two-tailed t-test. For correlational analyses, the Spearman nonparametric correlation analysis with a confidence interval of 95% and a two-tailed significance test with alpha= 0.05 was used.
Results
Inflammation promotes development of CRC in ApcMin/+;Il10−/− mice
To investigate the interaction between inflammation and CRC development, we interbred Il10−/− mice to ApcMin/+ mice (129SvEV background) to generate ApcMin/+;Il10−/− mice. Colon and cecal tumors increased dramatically in ApcMin/+;Il10−/− compared to ApcMin/+ mice (colon tumor mean= 5.03 vs 0.73, respectively p< 0.0001; cecal tumor mean= 0.51 vs 0, respectively p< 0.0005) (Fig. 1A and C), whereas small bowel lesions remained similar between the two genotypes (mean= 0.84 vs 2.23 p= 0.2914) (Fig. 1D). Histological assessment showed presence of colonic neoplastic lesions in ApcMin/+;Il10−/− mice (Fig. 1E) and as expected, increased inflammation in ApcMin/+;Il10−/− mice compared to ApcMin/+ mice (combined score mean = 1.32 vs 0.23, respectively p< 0.0001) (Fig. 1B). There was a significant correlation between development of CRC and extent of colon inflammation in ApcMin/+;Il10−/− mice (r= 0.7441 p< 0.0001) whereas no such correlation is observed in ApcMin/+ mice (r= −0.09608 p= 0.6135) (Fig. 1F). Furthermore, development of small intestinal neoplasia did not correlate with the state of colitis in either model (ApcMin/+;Il10−/− r= 0.2364; ApcMin/+ r= 0.2526; p> 0.05) (Fig. 1F). Interestingly, endpoint age (see methods for description of the chosen endpoint age range) was a significant contributor to tumorigenesis in the small bowel of both ApcMin/+;Il10−/− and ApcMin/+ mice (r= 0.6458, 0.8208, respectively p< 0.0001) but only weakly contributed to neoplasia in the large bowel of ApcMin/+;Il10−/− mice (r= 0.2561 p= 0.0256) (Fig. 1G). Thus genetic susceptibility to inflammation promotes colon but not small intestinal tumorigenesis in SPF ApcMin/+;Il10−/− mice, while age appears to be the primary factor contributing to small intestinal tumorigenesis in SPF ApcMin/+;Il10−/− and ApcMin/+ mice.
Figure 1.
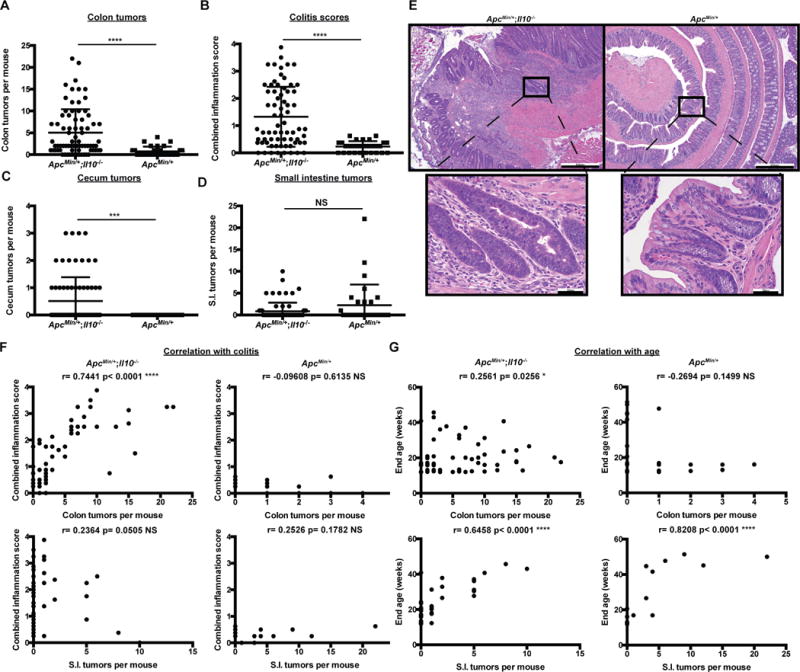
Inflammation fosters CRC development in genetically engineered mice.
A) Macroscopic colon tumor counts from 12–51 week old SPF ApcMin/+;Il10−/− and ApcMin/+ mice. B) Colon combined histological inflammation scores (the average of the proximal and distal inflammation scores) from SPF ApcMin/+;Il10−/− and ApcMin/+ mice. C-D) Cecum and small intestine macroscopic tumor counts from SPF ApcMin/+;Il10−/− and ApcMin/+ mice. E) Colon H&Es from 30–40 week old SPF ApcMin/+;Il10−/− and ApcMin/+ mice (5×, 40× magnification). F) Relationship between colon inflammation score and macroscopic colon or small intestine tumors in SPF ApcMin/+;Il10−/− and ApcMin/+ mice. G) Relationship between mouse endpoint age and macroscopic tumors in SPF ApcMin/+;Il10−/− and ApcMin/+ mice. Spearman correlation r values and corresponding p values are noted in each panel. Data are expressed as mean +/− standard deviation (SD). Two-tailed Mann-Whitney statistical analysis: ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, NS: not significant.
Due to its role in promoting cellular proliferation, we evaluated the distribution of nuclear Catenin Beta 1 (CTNNB1) and proliferating cell nuclear antigen (PCNA) in actively inflamed and neoplastic regions of ApcMin/+;Il10−/− and ApcMin/+ colons. Nuclear CTNNB1 and PCNA staining was mostly restricted to the crypt bases in ApcMin/+ mice (Fig. 2B, D and Supplementary Fig. S2B, D). In contrast, the colonic mucosa from ApcMin/+;Il10−/− mice showed areas of CTNNB1 and PCNA staining extending the full crypt length in dysplastic regions (Fig. 2A, C and Supplementary Fig. S2A, C). In addition, expression of proliferative and inflammatory IL-6, TNFα IFNγ, IL-1β, IL-22 and IL-17a mRNA increased in ApcMin/+;Il10−/− compared to ApcMin/+ proximal colon tissues (Fig. 2E). Furthermore, ApcMin/+;Il10−/− mice with a high number of tumors (>2) had significantly increased levels of TNFα, IFNγ and IL-1β mRNA compared to low tumor number (≤2) ApcMin/+;Il10−/− mice (Fig. 2F). Taken together, these data suggest that the heightened inflammatory and proliferative state observed in ApcMin/+;Il10−/− compared to ApcMin/+ mice increased propensity for colorectal tumor formation and progression.
Figure 2.
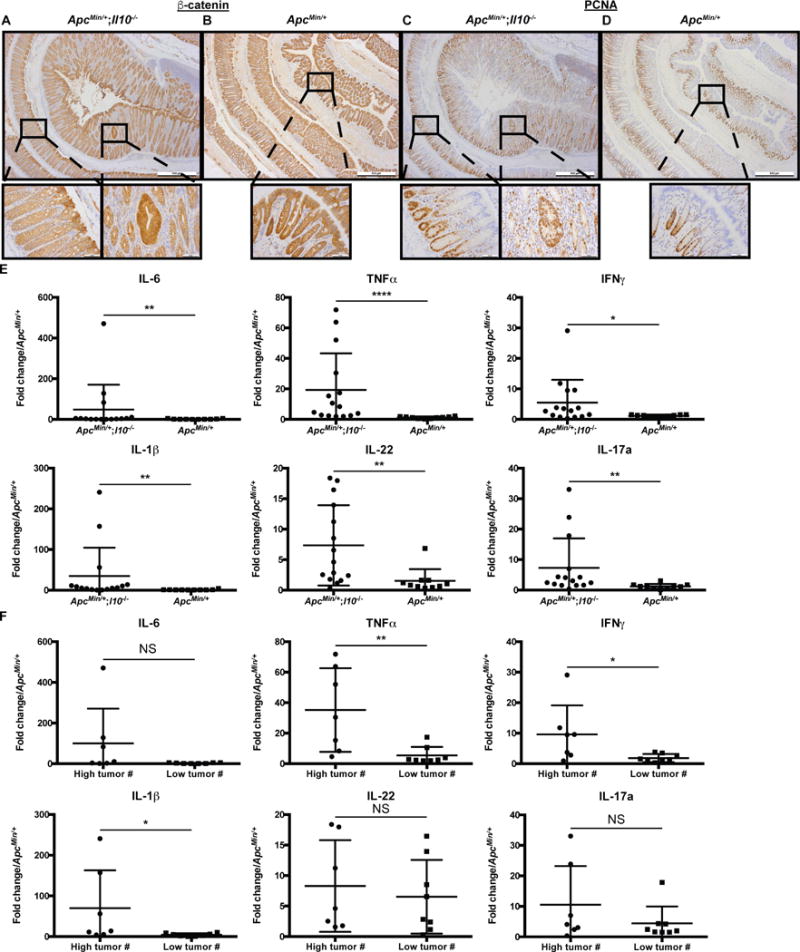
ApcMin/+;Il10−/− mice have increased colon proliferation and inflammation.
A-B) CTNNB1 immunohistochemistry (IHC) from ~16 week old SPF ApcMin/+;Il10−/− (A) and ApcMin/+ (B) colons. C-D) PCNA IHC from SPF ApcMin/+;Il10−/− (C) and ApcMin/+ (D) colons. Higher magnification of both dysplastic and normal regions are shown for SPF ApcMin/+;Il10−/− mice. E) IL-6, TNFα, IFNγ, IL-1β, IL-22 and IL-17a mRNA expression in 16–48 week old SPF ApcMin/+;Il10−/− and ApcMin/+ proximal colon tissue snips with relative fold expression compared to ApcMin/+ mice. F) IL-6, TNFα, IFNγ, IL-1β, IL-22 and IL-17a mRNA expression in 16–48 week old SPF ApcMin/+;Il10−/− stratified by tumor number (high: > 2 tumors or low: ≤ 2 tumors) with relative fold expression compared to ApcMin/+ mice. Data are expressed as mean +/− SD. Two-tailed Mann-Whitney statistical analysis: ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, NS: not significant.
Differential microbial composition within ApcMin/+;Il10−/− mice correlates with tumor multiplicity and inflammation status
We previously showed that inflammation altered microbial composition (17), a condition important for CRC development. Using 16S rRNA sequencing, we found that the SPF ApcMin/+;Il10−/− stool microbiome was associated with both colon inflammation score and tumor number (Fig. 3A), especially along the first Principal Coordinates Analysis (PCoA) axis (Fig. 3B). Using mixed linear models with either colon tumor number or colitis score as the independent variable, we identified 17 genera significantly affected by both colon tumor number and colitis score, including Allobaculum, Anaerotruncus, Butyrivibrio, Clostridium IV, Clostridium XI, Enterococcus, Oscillibacter, Pseudoflavonifractor, and Syntrophococcus (Fig. 3C, Supplementary Table S2). Only 5 genera were affected by colon tumor number but not combined inflammation score (Akkermansia, Coprobacillus, Escherichia Shigella, Marvinbryantia and Robinsoniella) while 4 genera were only affected by combined inflammation score and not colon tumor number (Alistipes, Enterorhabdus, Flavonifractor and Roseburia) (Fig. 3C). Interestingly, ~1/3 of these genera (10/26) were significantly increased in mice with colon tumors and/or inflammation while ~2/3 were decreased (16/26). Therefore, the microbial community composition is correlated with higher tumor numbers and inflammation, suggesting specific bacteria play a role in the development of colon inflammation and tumorigenesis in ApcMin/+;Il10−/ mice.
Figure 3.
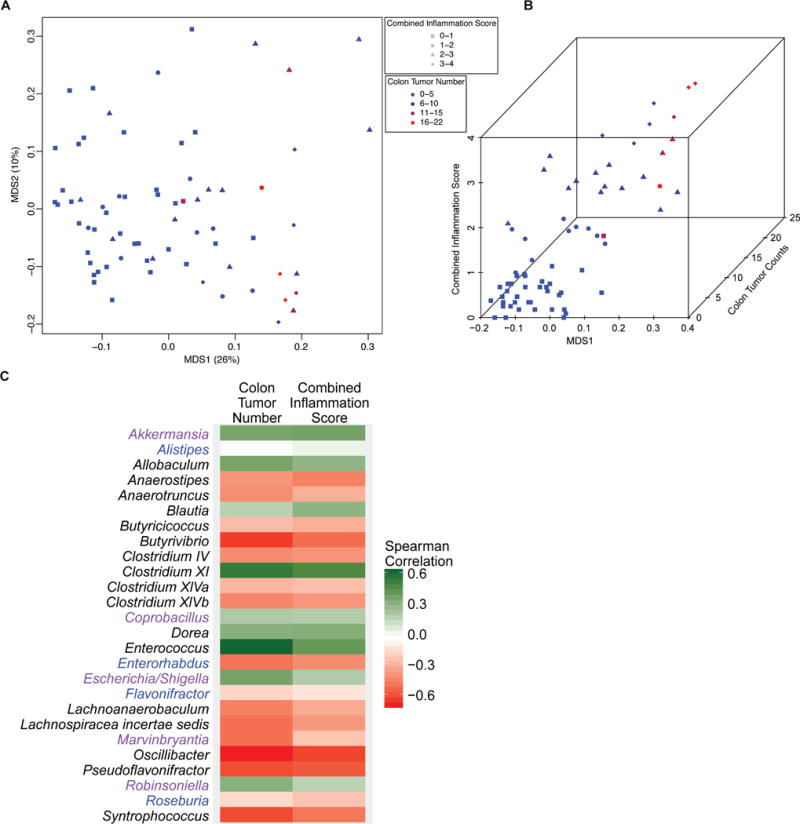
SPF ApcMin/+;Il10−/− with colon tumors and colitis have an altered stool microbiota.
A) PCoA comparing the stool microbial composition of SPF ApcMin/+;Il10−/− mice. Each symbol represents an individual mouse (n= 70) with symbol shape and color according to colon inflammation score and tumor number, respectively. B) 3D plot showing how MDS1 from the PCoA (x axis) varies with combined inflammation score (y axis) and colon tumor number (z axis). C) Heatmap depicting the Spearman correlations (positive correlations in green, negative correlations in red) for genera that are significantly associated with colon tumor number and/or colitis in ApcMin/+;Il10−/− mice. Genera significant for both colon tumor number and combined inflammation score are in black font, colon tumor number only in purple font, and combined inflammation score only in blue font.
Bacteria are essential for development of colon tumorigenesis in ApcMin/+;Il10−/− mice
To stringently evaluate the impact of bacteria on CRC development, we derived ApcMin/+;Il10−/− and ApcMin/+ mice in GF conditions and then performed microbial manipulation by either gavaging the mice with specific-pathogen-free (SPF) biota or allowing them to naturally acquire a microbiota in SPF conditions. Importantly, colon tumorigenesis was practically abolished in GF ApcMin/+;Il10−/− mice (mean= 0) compared to SPF conditions (Fig. 4A,D). Interestingly, SPF gavage enhanced colon tumor loads compared to passive SPF colonization of ApcMin/+;Il10−/− mice (mean= 3.86 vs 1 respectively p= 0.0126) (Fig. 4A,D), although colitis scores and most proliferative/inflammatory cytokine expression (IL-6, TNFα IFNγ, IL-22 and IL-17a) were not significantly different (colitis score means = 1.86 vs 2.36 respectively p=0.46) (Fig. 4B, 5D). Colon inflammation and tumors were negligible or absent in GF and SPF gavaged ApcMin/+ mice (Supplementary Fig. S3A, B), suggesting inflammation is a key component of bacteria-mediated colon tumorigenesis. Development of small bowel neoplasia in ApcMin/+;Il10−/− and ApcMin/+ mice (Fig. 5C, Supplementary Fig. S3C) was not significantly impacted by microbial colonization, suggesting a less pronounced role for the microbiota in the small intestine compared to the colon in this model. GF ApcMin/+;Il10−/− colons had reduced nuclear CTNNB1 and PCNA (Fig. 5A–C, Supplementary Fig. S4A–C) and decreased inflammatory cytokine expression (Fig. 5D) compared to SPF mice, indicating bacteria play a significant role in the increased inflammatory and proliferative state in SPF ApcMin/+;Il10−/− colons.
Figure 4.
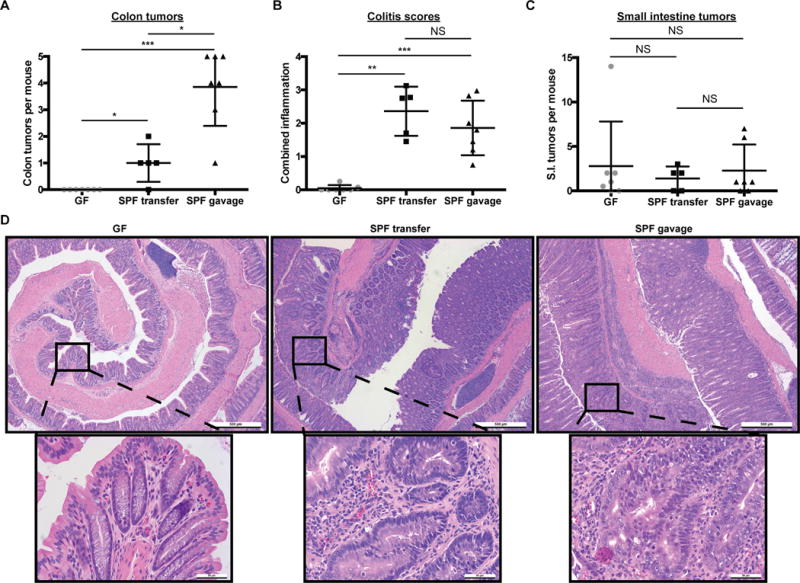
Bacteria promote distal colon inflammation and tumorigenesis in ApcMin/+;Il10−/− mice.
A–C) Colon macroscopic tumor counts (A), colitis scores (B), and small intestine tumor counts (C) from GF (n=7), SPF transferred (n=5) and SPF gavaged (n=7) ApcMin/+;Il10−/− mice. SPF transfer (transferred to SPF and allowed to naturally acquire their microbiota) and gavage (transferred to SPF and gavaged with the cecal and fecal contents from a wild type 129SvEv mouse) mice were sacrificed 16 weeks after transfer from GF. D) Colon H&Es from GF, SPF transferred, and SPF gavaged ApcMin/+;Il10−/− mice. Data are expressed as mean +/− SD. Two-tailed Mann-Whitney statistical analysis: ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, NS: not significant.
Figure 5.
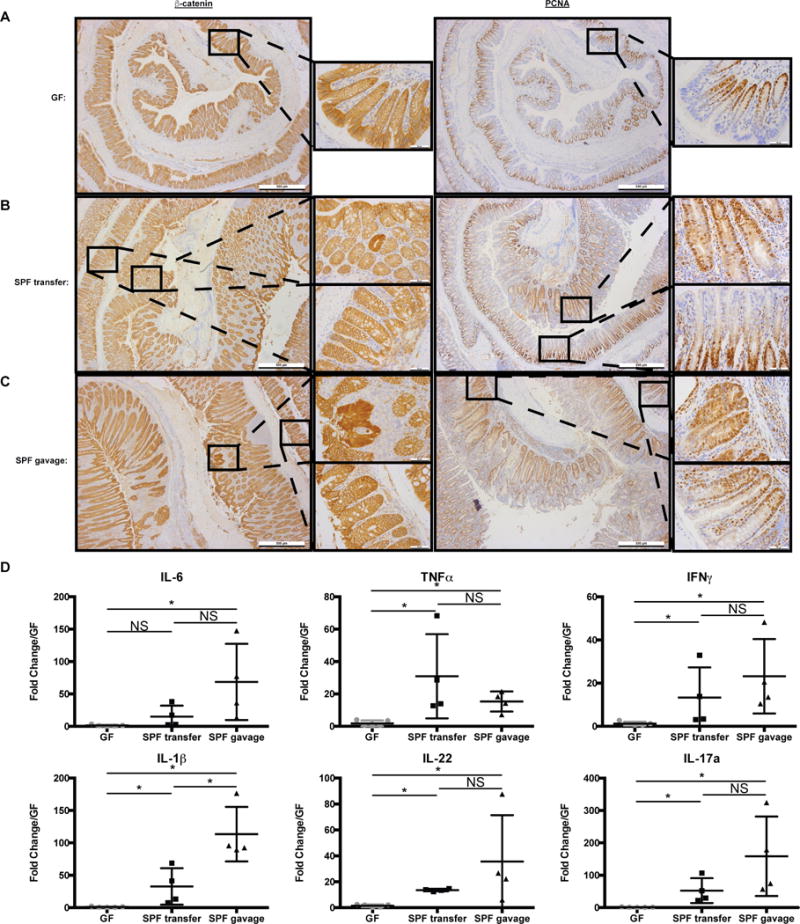
Microbiota promote colon inflammation and proliferation in ApcMin/+;Il10−/− mice.
A-C) β-catenin and PCNA IHC from GF (A), SPF transferred (B), and SPF gavaged (C) ApcMin/+;Il10−/− distal colons. Higher magnification of both dysplastic and normal regions are shown for SPF transfer and gavage ApcMin/+;Il10−/− mice. D) IL-6, TNFα, IFNγ, IL-1β, IL-22 and IL-17a mRNA expression in GF (n=5), SPF transfer (n=4), and SPF gavaged (n=4) ApcMin/+;Il10−/− proximal colon tissue snips with relative fold expression compared to GF. Data are expressed as mean +/− SD. Two-tailed Mann-Whitney statistical analysis: ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, NS: not significant.
Gnotobiotic experiments reveal specific microbial requirements for CRC development in ApcMin/+;Il10−/− mice
Fusobacterium spp. have been linked to the development of CRC (4) and recent studies showed increased carcinogenesis in F. nucleatum-colonized ApcMin/+ mice (23–25). To investigate the interplay between microbiota and F. nucleatum in CRC, we transferred GF ApcMin/+ to SPF conditions and gavaged them with SPF microbiota followed by weekly gavage with a fadA+, fap2− F. nucleatum CRC clinical isolate for 20 weeks. Interestingly, and in contrast to previous studies (23–25), presence of F. nucleatum failed to enhance carcinogenesis in these mice (Fig. 6A,C). We next transferred GF ApcMin/+;Il10−/− mice to SPF conditions, gavaged them with SPF microbiota and then introduced a mixture of 6 F. nucleatum strains (fadA+, fap2− or +) obtained from CRC patients by weekly gavage for 16 weeks. Although ApcMin/+;Il10−/− mice developed more inflammation and tumors than ApcMin/+ mice, presence of F. nucleatum species did not influence intestinal carcinogenesis or colitis (Fig. 6B,C). To rule out the possibility that the SPF biota down-modulates F. nucleatum carcinogenic properties, we transferred GF ApcMin/+ mice to a gnotobiotic isolator and associated these mice with a mixture of 6 F. nucleatum CRC clinical isolates (fadA+, fap2− or +; single gavage). Again, the presence of F. nucleatum isolates failed to enhance intestinal carcinogenesis in ApcMin/+ mice (Fig. 6D) despite the presence of high colony-forming unit (CFU) counts (mean= 107 CFU/g of stool). Although we did not see evidence of histological inflammation, to further confirm the effect of F. nucleatum on host inflammatory response, we examined inflammatory cytokine gene expression via qPCR. We found comparable levels of TNFα and IL1β in the mouse distal colon between germ-free and monoassociated ApcMin/+ mice, whereas IL-6, IFNγ, IL-22 and IL-17a levels were undetectable (data not shown).
Figure 6.
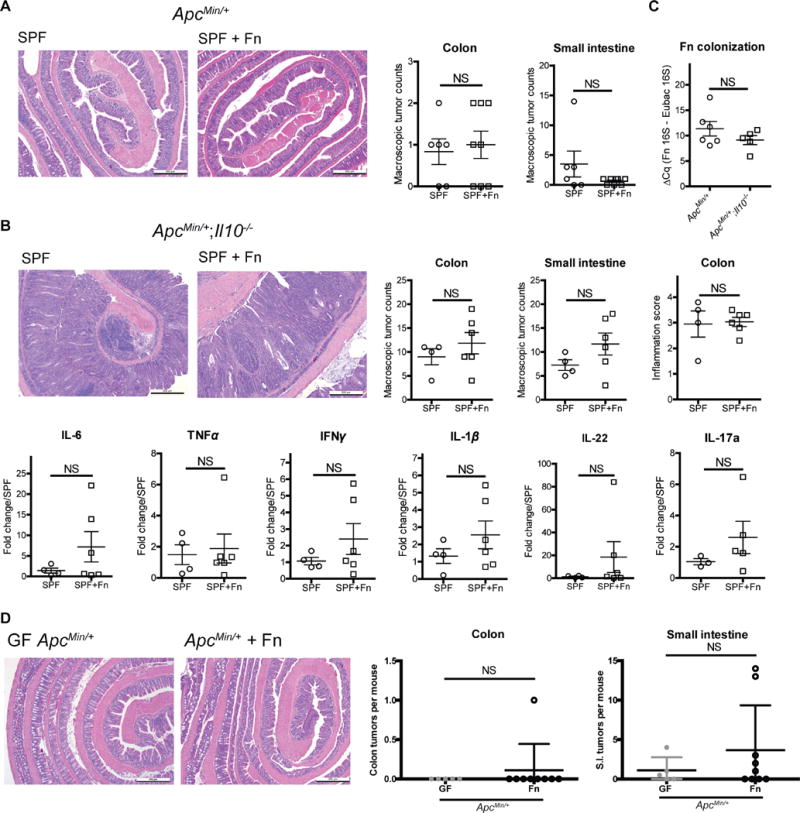
F. nucleatum (Fn) does not exhibit pro-inflammatory and pro-tumorigenic activities.
A) GF ApcMin/+ mice were transferred to SPF conditions and immediately gavaged with SPF microbiota (n= 6). SPF+Fn mice (n= 8) received Fn (a single strain human CRC isolate) via weekly gavage. SPF (control) mice received weekly gavage of BHI medium. Tumorigenesis was examined 20 weeks later. B) GF ApcMin/+;Il10−/− mice were transferred to SPF conditions and immediately gavaged with SPF microbiota (n= 4). SPF+Fn mice (n=6) received Fn (a mixture of 6 human CRC isolates) via weekly gavage. SPF (control) mice received weekly gavage of BHI medium. Tumorigenesis and inflammation were examined 16 weeks later (top panel). IL-6, TNFα, IFNγ, IL-1β, IL-22 and IL-17a mRNA expression in SPF and SPF+Fn ApcMin/+;Il10−/− distal colon snips (bottom panel). C) qPCR examination of fecal Fn levels in SPF+Fn ApcMin/+ and ApcMin/+;Il10−/−− mice. D) GF ApcMin/+ mice were transferred to a gnotobiotic isolator and gavaged with Fn (a mixture of 6 human CRC isolates). Tumorigenesis was examined 16 weeks later (GF n= 5 and Fn colonized n=9). In panels A-B, D, representative histology images of the colon are shown on the left. Macroscopic tumor counts are shown on the right. Data are expressed as mean +/− SD. Two-tailed Mann-Whitney statistical analysis: *p< 0.05, NS: not significant.
To further study the relationship between microbial status and carcinogenesis in gnotobiotic ApcMin/+;Il10−/− mice, we colonized these mice with E. coli NC101, a strain producing the colibactin genotoxin. We previously showed that removing pks from E. coli NC101 decreased development of CRC in the azoxymethane (AOM)/Il10−/− mouse model (17). We next mono-associated ApcMin/+;Il10−/− mice by oral gavage (108 CFU/mouse) with an E. coli NC101 mutant deficient for clbP, the pks gene necessary for colibactin activation (ΔclbP). We found that wild type NC101-colonized mice developed significantly more colon tumors than E. coli NC101 ΔclbP associated mice (mean= 1.71 vs 0.17 respectively p= 0.0023) (Fig. 7A). The finding that NC101 ΔclbP has diminished carcinogenic capacity compared to NC101 was confirmed in the AOM/Il10−/− model (mean= 2 vs 5 tumors respectively p= 0.039, Supplementary Fig. S5). Importantly, deletion of clbP did not compromise the ability of E. coli NC101 to induce inflammation (colitis score mean= 2.675, 2.5 respectively p= 0.76) (Fig. 7B, D). Presence of a functional pks did not influence development of small intestinal tumors in ApcMin/+;Il10−/− mice (mean= 0.29, 0.8 respectively p= 0.22) (Fig. 7C). Overall, these findings show that ApcMin/+;Il10−/− mice are sensitive to microbial status and develop site specific tumors, with colibactin-producing E. coli but not F. nucleatum FadA or Fap2 enhancing colon tumor development.
Figure 7.
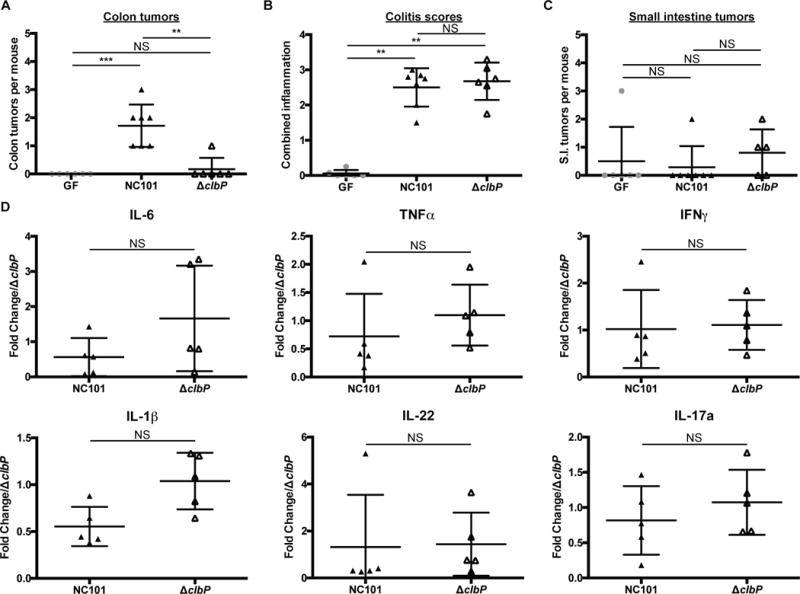
Colibactin promotes CRC development in ApcMin/+;Il10−/− mice.
A–C) Colon tumor counts (A), colitis scores (B), and small intestine tumor counts (C) from GF ApcMin/+;Il10−/− (n=6) and 16 week E. coli NC101 (n=7) or ΔclbP (n=6) mono-associated ApcMin/+;Il10−/− mice. D) IL-6, TNFα, IFNγ, IL-1β, IL-22 and IL-17a mRNA expression in NC101 (n=5) or ΔclbP (n=5) mono-associated ApcMin/+;Il10−/− distal colon snips with relative fold expression compared to ΔclbP mono-associated ApcMin/+;Il10−/− mice. Data are expressed as mean +/− SD. Two-tailed Mann-Whitney statistical analysis: ****p< 0.0001, ***p< 0.001, **p< 0.01, *p< 0.05, NS: not significant.
Discussion
Genetics and environmental factors play an important role in CRC development, with increasing attention directed toward the intestinal microbiota as a key environmental component (26). In general, the microbiota is thought to play a pro-carcinogenic role in CRC with numerous CRC mouse models demonstrating tumor reduction in antibiotic treated or GF mice (27). Here we utilized gnotobiotic ApcMin/+ and ApcMin/+;Il10−/− mice to define the relationship between inflammation, microbial status and tumorigenesis. We observed that despite genetic susceptibility in both ApcMin/+ and ApcMin/+;Il10−/− mice, colonic inflammation in the latter mice fosters development of colon tumors, which was associated with an altered luminal microbiota composition. Gnotobiotic experiments revealed that E. coli colibactin but not F. nucleatum FadA and Fap2 adhesins promote colon tumorigenesis, suggesting an intricate interaction between host genetics and bacteria.
We observed an inflammation dependent increase in colon tumorigenesis in 129 SvEv ApcMin/+;Il10−/− mice, which is in line with previous reports on Il10 deficient C57BL/6 ApcMin/+ and ApcΔ468 mice (13,14). However, in the small intestine compartment, tumors developed at a comparable rate regardless of Il10 status in ApcMin/+ mice, which is in contrast to previous findings showing a delay in small intestinal polyp formation in Il10 deficient Apc Δ468 mice (15). Possible explanations for the differences in small intestine tumor formation may be due to genetic background differences which have been shown to strongly modulate tumor multiplicity, particularly in the small intestine of ApcMin/+ mice (28). Nevertheless, our findings that ApcMin/+;Il10−/− and ApcMin/+ small intestine tumor development significantly correlated with age but not inflammation suggests that age-related factors are a primary driver of small intestinal tumorigenesis.
We observed changes in the abundance of 26 genera that correlated with colon inflammation and/or tumor number. In contrast to previous sequencing results with ApcMin/+ and T cell specific ApcMin/+;Il10−/− mice, no significant increases in the Bacteroides or Porphyromonas genera were observed (14). These differences are likely due to a combination of factors including sampling location (stool vs tissue), mouse genetic background, husbandry and 16S sequencing methods (V1–V3 region vs V3–V4). Importantly, Akkermansia, Blautia, Dorea, Enterococcus, and Escherichia/Shigella, which positively correlated with tumor number, have been previously associated as increased in mucosal tissue (17,29,30) or stools from either human CRC patients (31–33) or AOM/DSS mice (34,35). Conversely, some of the genera that negatively correlated with tumors and inflammation or just inflammation included Clostridium XIVa, Lachnospiracea and Roseburia, which have been implicated as butyrate producers and were decreased in CRC stool (31,33) or stools from AOM/DSS mice transplanted with stools from human patient samples (34). Thus, our sequencing results suggest there are changes in the microbiota that are associated with colon inflammation and tumor status.
The interaction between bacteria and the host in the context of intestinal carcinogenesis is complex. One study, using the chemical AOM/DSS regimen, reported that GF mice developed more colonic tumors than mice colonized with a complex biota, suggesting certain bacteria can have a beneficial role in CRC (36). Since microbial composition is a key determinant of colon tumor burden in AOM/DSS mice (35,37), this chemical model may better capture the protective functions of bacteria than the genetic ApcMin/+ mouse model.
Nevertheless, the role of bacteria in ApcMin/+ intestinal tumorigenesis is complex, with one report showing fewer tumors in the middle region of the small intestine in GF ApcMin/+ mice (9) while another report showed reduced tumors throughout the intestine in GF ApcMin/+ mice (10). The difference in tumor distribution is not clear. Our finding that bacteria promote colon tumors in ApcMin/+;Il10−/− mice is in line with a study showing reduced colon polyp numbers in ApcΔ468;CD4CreIl10f/f mice following broad-spectrum antibiotic treatment (14).
Numerous studies have implicated Fusobacterium spp., in particular F. nucleatum, as carcinogenic, based on associative studies showing the presence of the bacterium in the luminal and mucosal compartment of human CRC patients using genomic analyses (3,4,24,33). In addition, daily gavage of F. nucleatum (strain EAVG_002; 7/1 or ATCC 25586) for 8–24 weeks was shown to promote intestinal tumorigenesis in C57BL/6 ApcMin/+ mice (23–25). Surprisingly, ApcMin/+ and ApcMin/+;Il10−/− mice colonized with fadA+, fap2+/− F. nucleatum isolates from CRC patients failed to promote intestinal tumorigenesis, in the presence (SPF) or absence of complex biota (gnotobiotic). The absence of tumorigenesis in mono-associated ApcMin/+ mice was not due to poor colonization, since a high load of F. nucleatum (107 CFU/g) was recovered from these mice. The discrepancy between our study and previous F. nucleatum ApcMin/+ studies (23–25) is unclear but could be the result of strain specific properties (EAVG_002 (19,38) and ATCC 25586 (39,40) vs strains tested here), mouse genetic background differences and different microbial environments, as microbial communities are notoriously different between institutions. Nevertheless, our gnotobiotic approach clearly showed that presence of FadA and Fap2 adhesins in F. nucleatum is not sufficient to induce either inflammation or cancer, as opposed to E. coli pks+ mono-associated ApcMin/+;Il10−/− mice. Thus, it is possible that only a select group of F. nucleatum strains possess carcinogenic abilities, which require interactions with other specific members of the microbial community. It will be important to define these interactions and test a larger set of F. nucleatum strains to determine the role of these bacteria in CRC pathogenesis.
Several studies have found an association between pks+ E. coli and human CRC patients patients (17,29). Furthermore, pks+ E. coli isolates from mice or human CRC patients have a pro-tumorigenic effect in GF AOM/Il10−/−, SPF ApcMin/+, and SPF AOM/DSS mice (17,29,41). However, because the pks-associated clbA gene is implicated in the production of siderophores located in the enterobactin (ent) and yersiniabactin (HPI) loci (42), and our previous observation was based on removal of the entire pks island, it was unclear whether the decreased tumorigenesis observed in AOM/Il10−/− mice was the consequence of dual siderophore/colibactin impairment, or solely due to abolished pks activity. Using a mutant with defective ClbP, the key enzyme implicated in pre-colibactin cleavage and generation of the active form (43), we demonstrated the colibactin-producing E. coli murine isolate NC101 is responsible for the pro-tumorigenic effect of the bacterium in ApcMin/+;Il10−/− mice. Whether clbA contributes to colibactin-mediated tumorigenesis is still unclear and would need to be investigated, especially since a recent in vitro study showed that iron levels and E. coli iron sensors regulate clbA transcription and colibactin production (44). Since our studies were performed using a mono-association approach, and therefore without competitive pressure from other microorganisms, the full extent of iron acquisition on E. coli pks+ induced carcinogenesis remains unclear.
Recent studies have attempted to dissect the contributions of intrinsic (organ specific stem cell division rates, aging) and extrinsic factors (hereditary mutations, lifestyle, environmental exposure, etc.), to overall cancer risk in humans (45–47). However, the interplay between all these factors makes it difficult to tease out the various contributions using epidemiological data. Nevertheless, these studies suggest that small intestine cancers with a relatively low lifetime risk are driven by intrinsic risk factors, while 82.9% of the mutation signatures in CRCs are from extrinsic factors, correlating with a much higher lifetime risk (46). We postulate that one of the extrinsic factors contributing to CRC risk is the microbiota, which not coincidentally is also affected by lifestyle and environmental factors (48,49). Interestingly, the concentration of bacteria increases along the gastrointestinal tract with 103–104 bacteria/mL in the small intestine to 1011 bacteria/mL in the colon, mirroring the distribution of cancer risk along the human intestinal tract (50). Similarly, in the ApcMin/+;Il10−/− model, age strongly correlates with small intestine tumor numbers while inflammation and bacteria composition play a strong role in colon tumorigenesis. Elucidating the mechanisms by which specific bacteria interact with other microbiota members to promote carcinogenesis will generate important insights into the pathophysiology of CRC.
Supplementary Material
Acknowledgments
The authors would like to thank the University of Florida Animal Care Services, particularly the Germ Free Services division for assistance with SPF and gnotobiotic mouse experiments and the staff at the Molecular Pathology Core for assistance with histology. We also thank Dr. Emma Allen-Vercoe for sharing the F. nucleatum strains.
Data Availability: All sequences have been uploaded to the NCBI SRA (National Center for Biotechnology Information Sequence Read Archive) under BioProject PRJNA350319. See Supplementary Table S3 for individual accession numbers.
Financial support: This research was supported by National Institutes of Health grants R01DK047700, R01DK073338 and R21 CA195226 to C. Jobin. Y. Yang was supported by the Crohn’s & Colitis Foundation of America (CCFA) research fellowship award (CCFA Ref. #409472). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borges-Canha M, Portela-Cidade JP, Dinis-Ribeiro M, Leite-Moreira AF, Pimentel-Nunes P. Role of colonic microbiota in colorectal carcinogenesis: a systematic review. Rev Esp Enferm Dig. 2015;107:659–71. doi: 10.17235/reed.2015.3830/2015. [DOI] [PubMed] [Google Scholar]
- 4.Brennan CA, Garrett WS. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol. 2016;70:395–411. doi: 10.1146/annurev-micro-102215-095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol. 2016;17:230–40. doi: 10.1038/ni.3384. [DOI] [PubMed] [Google Scholar]
- 6.Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015;372:1441–52. doi: 10.1056/NEJMra1403718. [DOI] [PubMed] [Google Scholar]
- 7.Jackstadt R, Sansom OJ. Mouse models of intestinal cancer. J Pathol. 2016;238:141–51. doi: 10.1002/path.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 9.Dove WF, Clipson L, Gould KA, Luongo C, Marshall DJ, Moser AR, et al. Intestinal neoplasia in the ApcMin mouse: independence from the microbial and natural killer (beige locus) status. Cancer Res. 1997;57:812–4. [PubMed] [Google Scholar]
- 10.Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, et al. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33:1231–8. doi: 10.1093/carcin/bgs137. [DOI] [PubMed] [Google Scholar]
- 11.Cooper HS, Everley L, Chang WC, Pfeiffer G, Lee B, Murthy S, et al. The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodium-induced colitis. Gastroenterology. 2001;121:1407–16. doi: 10.1053/gast.2001.29609. [DOI] [PubMed] [Google Scholar]
- 12.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–8. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang EH, Park JC, Appelman H, Weinberg AD, Banerjee M, Logsdon CD, et al. Induction of inflammatory bowel disease accelerates adenoma formation in Min +/− mice. Surgery. 2006;139:782–8. doi: 10.1016/j.surg.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Dennis KL, Wang Y, Blatner NR, Wang S, Saadalla A, Trudeau E, et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013;73:5905–13. doi: 10.1158/0008-5472.CAN-13-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dennis KL, Saadalla A, Blatner NR, Wang S, Venkateswaran V, Gounari F, et al. T-cell Expression of IL10 Is Essential for Tumor Immune Surveillance in the Small Intestine. Cancer Immunol Res. 2015;3:806–14. doi: 10.1158/2326-6066.CIR-14-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe. 2016;20:215–25. doi: 10.1016/j.chom.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344–55. doi: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nougayrède J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 23.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–15. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y-N, Yu T-C, Zhao H-J, Sun T-T, Chen H-M, Chen H-Y, et al. Berberine may rescue Fusobacterium nucleatum-induced colorectal tumorigenesis by modulating the tumor microenvironment. Oncotarget. 2015;6:32013–26. doi: 10.18632/oncotarget.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor−κ, Up-Regulating Expression of MicroRNA-21. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pope JL, Tomkovich S, Yang Y, Jobin C. Microbiota as a mediator of cancer progression and therapy. Transl Res. 2017;179:139–54. doi: 10.1016/j.trsl.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–12. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009;656:85–106. doi: 10.1007/978-1-4419-1145-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859–67. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 30.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–47. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–9. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE. 2013;8:e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66:462–70. doi: 10.1007/s00248-013-0245-9. [DOI] [PubMed] [Google Scholar]
- 34.Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2:20. doi: 10.1186/2049-2618-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, et al. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692–13. doi: 10.1128/mBio.00692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhan Y, Chen P-J, Sadler WD, Wang F, Poe S, Núñez G, et al. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 2013;73:7199–210. doi: 10.1158/0008-5472.CAN-13-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zackular JP, Baxter NT, Chen GY, Schloss PD. Manipulation of the Gut Microbiota Reveals Role in Colon Tumorigenesis. 2016;1 doi: 10.1128/mSphere.00001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cochrane KLS. Doctoral dissertation. The University of Guelph; 2016. Elucidating potential virulence factors in Fusobacterium nucleatum [Internet] Available from: https://atrium.lib.uoguelph.ca/xmlui/bitstream/handle/10214/9623/Cochrane_Kyla_201605_PhD.pdf?sequence=3. [Google Scholar]
- 39.Kaplan CW, Ma X, Paranjpe A, Jewett A, Lux R, Kinder-Haake S, et al. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect Immun. 2010;78:4773–8. doi: 10.1128/IAI.00567-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han YW, Ikegami A, Rajanna C, Kawsar HI, Zhou Y, Li M, et al. Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol. 2005;187:5330–40. doi: 10.1128/JB.187.15.5330-5340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932–42. doi: 10.1136/gutjnl-2013-305257. [DOI] [PubMed] [Google Scholar]
- 42.Martin P, Marcq I, Magistro G, Penary M, Garcie C, Payros D, et al. Interplay between siderophores and colibactin genotoxin biosynthetic pathways in Escherichia coli. PLoS Pathog. 2013;9:e1003437. doi: 10.1371/journal.ppat.1003437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, et al. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J Biol Chem. 2011;286:35562–70. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tronnet S, Garcie C, Rehm N, Dobrindt U, Oswald E, Martin P. Iron Homeostasis Regulates the Genotoxicity of Escherichia coli That Produces Colibactin. Infect Immun. 2016;84:3358–68. doi: 10.1128/IAI.00659-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu S, Powers S, Zhu W, Hannun YA. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016;529:43–7. doi: 10.1038/nature16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Podolskiy DI, Gladyshev VN. Intrinsic Versus Extrinsic Cancer Risk Factors and Aging. Trends Mol Med. 2016 doi: 10.1016/j.molmed.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Sullivan O, Cronin O, Clarke SF, Murphy EF, Molloy MG, Shanahan F, et al. Exercise and the microbiota. Gut Microbes. 2015;6:131–6. doi: 10.1080/19490976.2015.1011875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2015;7:17–44. doi: 10.3390/nu7010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.