

Summary
Nicotinamide adenine dinucleotide (NAD+) biosynthetic pathway, mediated by nicotinamide phosphoribosyltransferase (NAMPT), a key NAD+ biosynthetic enzyme, plays a pivotal role in controlling many biological processes, such as metabolism, circadian rhythm, inflammation, and aging. Over the past decade, NAMPT-mediated NAD+ biosynthesis, together with its key downstream mediator, namely the NAD+-dependent protein deacetylase SIRT1, has been demonstrated to regulate glucose and lipid metabolism in a tissue-dependent manner. These discoveries have provided novel mechanistic and therapeutic insights into obesity and its metabolic complications, such as insulin resistance, an important risk factor for developing type 2 diabetes and cardiovascular disease. This review will focus on the importance of adipose tissue NAMPT-mediated NAD+ biosynthesis and SIRT1 in the pathophysiology of obesity and insulin resistance. We will also critically explore translational and clinical aspects of adipose tissue NAD+ biology.
Keywords: NAD+, NAMPT, SIRT1, PPARγ, adipose tissue, obesity, insulin resistance
Introduction
The current epidemic of obesity, which is largely driven by our modern lifestyle, crucial features of which are excess calorie intake and insufficient physical activity, has already imposed a substantial economic burden on health systems worldwide, and this situation is projected worsen [1,2]. Obesity is characterized by the pathological expansion of adipose tissue, and adipose tissue dysfunction that involves dysregulated production of adipose tissue-secreted proteins (adipokines) and lipids, low-grade inflammation, and accumulation of extracellular matrix (ECM) [3–6]. Insulin resistance is an important systemic metabolic abnormality tightly associated with obesity. Insulin-resistant individuals demonstrate impaired ability of insulin to: i) stimulate glucose uptake in skeletal muscle; ii) suppress glucose production in the liver; and iii) suppress hydrolysis of triglycerides into fatty acids in adipose tissue. Thus, insulin resistance is critically involved in the pathogenesis of many cardiometabolic diseases, such as type 2 diabetes, atherogenic dyslipidemia, metabolic syndrome, nonalcoholic fatty liver disease (NAFLD), and coronary heart disease [4,7–9]. Intriguingly, accumulating evidence has demonstrated that insulin sensitivity can be dynamically modulated by the alterations in nutritional input and these metabolic adaptations are accompanied by dynamic response of adipose tissue that involves not only quantity but also quality alterations. For example, a very recent and elegant study conducted in people with obesity and insulin resistance shows that diet-induced moderate 5% weight loss is sufficient to reduce intra-abdominal adipose tissue mass as well as intrahepatic triglyceride contents, and improve insulin sensitivity in liver, skeletal muscle, and adipose tissue [10]. Additional weight loss of 11–16% induces stepwise changes in adipose tissue mass as well as adipose tissue expression of genes involved in lipid metabolism, ECM remodeling, and oxidative stress, and further increases skeletal muscle insulin sensitivity. In contrast, short-term overfeeding increases adipose tissue mass and deteriorates multi-organ insulin sensitivity with concomitant increase in adipose tissue markers of oxidative stress and ECM remodeling [11–13]. Although it is very difficult to address causality of these fascinating associations, these findings lend support to the hypothesis that adipose tissue plays a pivotal role in the pathophysiology of obesity and multi-organ insulin resistance and thereby it could be an important therapeutic target. Supporting these hypotheses, recent data obtained from the studies conducted in genetically engineered animals have shed light on the extraordinary capability of adipose tissue to critically regulate whole-body glucose and lipid metabolism, particularly insulin sensitivity [3–6]. As discussed below, we have recently identified classical coenzyme nicotinamide adenine dinucleotide (NAD+) as a new physiological regulator of adipose tissue function and multi-organ insulin sensitivity, providing important mechanistic and therapeutic insights into obesity-associated multi-organ insulin resistance [14]. The overall purpose of this review is to explore the pathophysiological significance and therapeutic potential of adipose tissue NAD+ biology in obesity and insulin resistance. First, we will summarize recent progress in NAD+ biology research and highlight our recent work regarding the novel role of adipose NAD+ biosynthesis in regulating whole body glucose metabolism and multi-organ insulin sensitivity. Second, we will discuss the importance of a key downstream mediator of the NAD+ biosynthetic pathway, namely the NAD+ dependent protein deacetylase SIRT1, in adipocyte biology and obesity. Finally, we will explore clinical aspects and translational potential of these experimental findings.
Impaired NAMPT-mediated NAD+ biosynthesis in adipocytes: A novel mechanism for obesity-associated multi-organ insulin resistance
How is NAD+ biosynthesized in mammals?
NAD+ is a universal and essential coenzyme found in all species. Intracellular NAD+ homeostasis is dynamically regulated by a balance between synthesis and degradation of NAD+ mediated by multiple enzymatic reactions [15–18]. NAD+ biosynthetic pathways involve four major precursors, nicotinamide and nicotinic acid (two distinctive forms of vitamin B3), tryptophan, and nicotinamide riboside (NR) (Figure 1). Among them, mammals predominantly use nicotinamide and nicotinamide phosphoribosyltransferase (NAMPT), a dimeric type II phosphoribosyltransferase, functions as the rate-limiting enzyme in this NAD+ salvage pathway starting from nicotinamide [19,20]. NAMPT generates a key NAD+ intermediate, nicotinamide mononucleotide (NMN), from nicotinamide and 5-phosphoribosyl 1-pyrophosphate (PRPP). The second enzyme nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) catalyzes the conversion of NMN to NAD+ in the presence of ATP. Mammals have three NMNAT isoforms (NMNAT-1, NMNAT-2 and NMNAT-3), located in the nucleus, cytosol, and mitochondria, respectively. NR, a newly identified NAD+ precursor, is phosphorylated and converted into NMN by nicotinamide riboside kinase. Nicotinic acid, a key NAD+ precursor in the classical Preiss-Handler pathway, is used by nicotinic acid phosphoribosyltransferase (NAPRT) to produce nicotinic acid mononucleotide (NaMN). NaMN is converted into nicotinic acid dinucleotide (NaAD+) by NMNAT and NaAD+ is then amidated to NAD+ by NAD+ synthase. NAD+ is also synthesized de novo by multiple enzymatic steps starting from tryptophan. As discussed below, emerging evidence has demonstrated that NAD+ acts not only as a classical redox cofactor but also as a pleiotropic regulator controlling various important biological processes together with these NAD+-consuming enzymes [15–18,21–23]. In this section, we will primarily focus on the recent progress in the understanding of the pathophysiological significance of the key NAD+ biosynthetic enzyme, NAMPT, in metabolic regulation.
Figure 1. Mammalian NAD+ biosynthetic pathways.
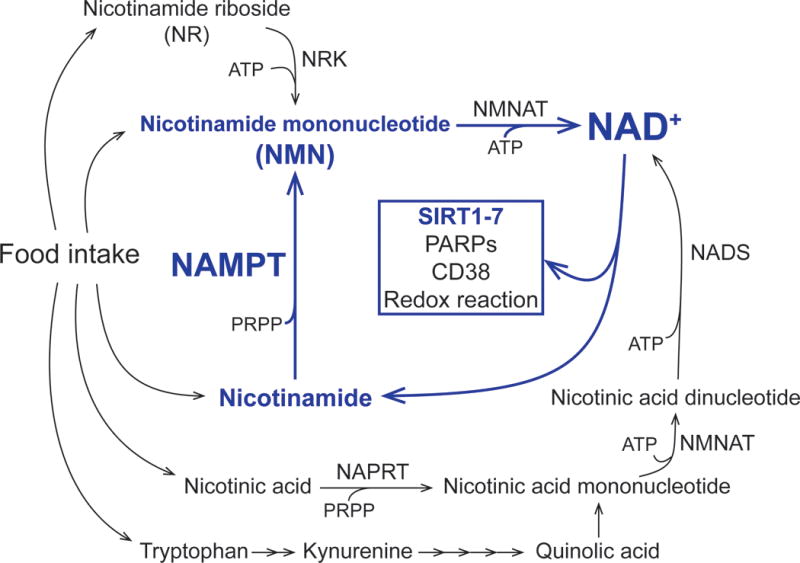
NAD+ biosynthesis is mediated by multiple enzymatic reactions. In mammals, nicotinamide phosphoribosyltransferase (NAMPT) functions as the rate-limiting enzyme in mammalian NAD+ biosynthetic pathway from the point of nicotinamide. NAMPT-mediated NAD+ biosynthesis critically regulates metabolic function through key NAD+-consuming enzymes, such as poly ADP ribose polymerases (PARPs), sirtuins, and CD38, in a tissue-dependent manner. See the text for the details and references. Abbreviations: NMNAT, nicotinamide/nicotinic acid mononucleotide adenylyltransferase; NRK, nicotinamide riboside kinase; NAPRT, nicotinic acid phosphoribosyltransferase; NADS, NAD+ synthase; PRPP; 5-phosphoribosyl 1-pyrophosphate.
NAMPT-mediated NAD+ biosynthesis plays diverse roles in metabolic regulation
Global knockout of Nampt in mice results in lethality in homozygotes, but, interestingly, heterozygotes have impaired glucose tolerance [24]. Recent studies have revealed that the NAD+ biosynthetic pathway mediated by NAMPT is critically involved in regulating a number of important metabolic pathways in a tissue-dependent manner. In the liver, some [25–27], but not all, studies [17,28,29] have found that Nampt mRNA/protein expression levels decrease in high-fat diet-fed obese mice. Interestingly, fasting increases hepatic NAMPT levels [30] and enhanced NAD+ biosynthesis improves hepatic metabolic function by decreasing triglyceride (TG) accumulation [26,27,31]. In contrast, dominant-negative, enzymatically-inactive Nampt transgenic mice increase TG accumulation and display insulin resistance and NAFLD phenotype [31]. In skeletal muscle, NAMPT is induced by energy deprivation or exercise through an AMPK-dependent mechanism [32,33]. Importantly, although overexpression of Nampt does not affect mitochondrial oxidative metabolism or whole-body glucose metabolism [34], loss of Nampt severely impairs mitochondrial oxidative metabolism and causes progressive muscle degeneration, which is reversed by systemic administration of NR [35]. In the heart, Nampt gene expression is reduced by various stresses such as ischemia, ischemia/reperfusion, and pressure overload [36]. Overexpression of Nampt in the heart decreases the number of apoptotic cells and reduces the size of myocardial infarction after ischemia reperfusion injury [36]. In addition, it was also reported that cardiac-specific Nampt overexpression suppresses mitochondrial protein hyperacetylation and protects mice from cardiac dysfunction and hypertrophy induced by isoproterenol infusion [37]. Interestingly, a very recent study demonstrated that rod or cone photoreceptor-specific Nampt deletion impairs retinal mitochondrial energy metabolism and causes severe retinal degeneration [38].
Although we will not focus on this aspect in this review, it should be noted that mammals have an extracellular form of NAMPT (eNAMPT) actively secreted by many cell types, including adipocytes, hepatocytes, immune cells, and cardiomyocytes [17,24]. In fact, the pioneering studies by Shin-ichiro Imai’s group have suggested that adipose tissue-derived eNAMPT is enzymatically active and it is critically involved in regulating glucose-stimulated insulin secretion and physical activity by modulating NAD+ biosynthesis in pancreatic islets and hypothalamus respectively [24,39,40]. Given that efferent sympathetic nerves to adipose tissue is involved in regulating whole-body insulin sensitivity [41], it will be of great importance to dissect out this potential complex neuron-adipocyte metabolic crosstalk composed of two distinct forms of NAMPT. Taken together, these results demonstrate the pathophysiological importance of NAMPT-mediated NAD+ biosynthesis in tissue-specific and whole-body metabolic function.
NAMPT-mediated NAD+ biosynthesis in adipocytes regulates multi-organ insulin sensitivity
In adipose tissue, NAD+ biosynthesis largely relies on NAMPT [14,39]. Intriguingly, previous studies have suggested that adipose tissue NAMPT-mediated NAD+ biosynthesis is very responsive to the changes in nutritional input and these alterations are associated with the pathophysiological alterations in whole-body glucose metabolism and insulin sensitivity. For example, high-fat diet-induced obesity and aging, two major risk factors of insulin resistance, decrease Nampt mRNA and protein expression and NAD+ contents in adipose tissue [25,42]. Although the precise molecular mechanisms remain poorly understood, inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα), could trigger to the reduction in adipose tissue Nampt expression in obese mice [43,44]. In contrast, calorie restriction, which improves insulin sensitivity, markedly increases NAMPT protein levels and NAD+ concentrations in adipose tissue [45,46].
These findings led us to hypothesize that adipose NAMPT-mediated NAD+ biosynthesis plays a critical role in regulating whole-body glucose metabolism, particularly insulin sensitivity. To test these key hypotheses, we recently generated and analyzed adipocyte-specific Nampt knockout (ANKO) mice using adiponectin Cre transgenic mice [14]. Surprisingly, data obtained from the hyperinsulinemic euglycemic clamp procedures demonstrated that ANKO mice have severe insulin resistance in liver, skeletal muscle, heart, and adipose tissue, even under a regular chow-fed condition. Interestingly, such severe metabolic deterioration is not accompanied by the alterations in body weight or whole-body adiposity. Loss of Nampt causes severe adipose tissue dysfunction, manifested by local adipose tissue inflammation, increased plasma FFA concentrations, and decreased production of a key insulin-sensitizing adipokine, adiponectin. Remarkably, oral administration of NMN, a key NAD+ intermediate (Figure 1), in the drinking water, is able to largely restore adipose tissue NAD+ biosynthesis and to completely normalize insulin resistance and the alterations in plasma adiponectin and FFA concentrations in ANKO mice. Data obtained from the studies conducted in rodents have demonstrated that adiponectin knockout mice have severe diet-induced insulin resistance [47], whereas administration of adiponectin recombinant protein improves multi-organ insulin resistance in obese rodents [48,49]. In addition, it has long been recognized that elevated plasma FFA availability causes insulin resistance in skeletal muscle and liver by increasing the production of lipotoxic fatty acid metabolites, such as diacylglycerol and ceramide [9]. It is therefore likely that hypoadiponectinemia and excess FFA availability contribute to the pathogenesis of multi-organ insulin resistance induced by adipocyte-specific NAD+ depletion. Taken together, our new results have strongly suggested that impaired NAMPT-mediated NAD+ biosynthesis in adipocytes could constitute a novel mechanism underlying the pathogenesis of obesity-associated multi-organ insulin resistance (Figure 2). Given that adipose tissue Nampt gene expression is regulated in a circadian manner [50–52], peaking during the dark period when mice are active and relatively insulin-sensitive [53], NAMPT-mediated NAD+ biosynthesis in adipocytes could have a significant impact on physiological changes in whole-body insulin sensitivity by affecting adiponectin and FFA production. Future studies are warranted to further investigate the normal physiological functions of adipose NAMPT-mediated NAD+ biosynthesis in the regulation of glucose metabolism and insulin sensitivity.
Figure 2. Impaired NAMPT-mediated NAD+ biosynthesis in adipocytes causes insulin resistance in liver and skeletal muscle.
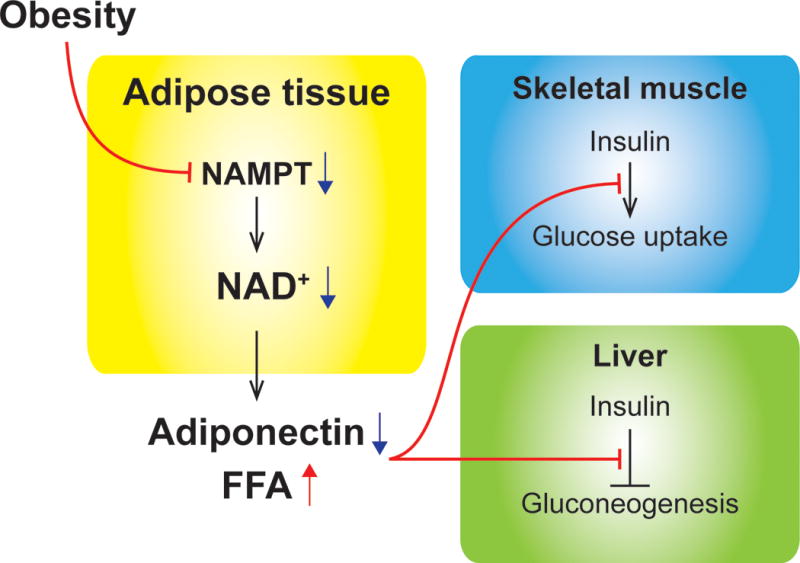
Obesity is known to decrease adipose tissue NAMPT expression and NAD+ contents in rodents and humans. Reduction of NAMPT-mediated NAD+ biosynthesis in adipocytes severely impairs insulin action in liver and skeletal muscle by decreasing adiponectin and increasing free fatty acids (FFA) production [14].
SIRT1: A key molecular link between NAMPT-mediated NAD+ biosynthesis and adipocyte biology
SIRT1 acts a key downstream mediator of NAMPT-mediated NAD+ biosynthesis
What is the downstream mechanism that links NAD+ and metabolic function? In mammals, NAD+ is utilized in various enzymatic reactions mediated by the key NAD+-consuming enzymes, such as poly ADP ribose polymerases (PARPs), cyclic ADP ribose hydrolase (e.g., CD38), and seven sirtuins (SIRT1-SIRT7) [15–18,21–23]. Among them, SIRT1 has been well studied and most characterized over the past decade [21,54]. SIRT1 is a NAD+-dependent protein deacetylase that plays a pivotal role in regulating cellular metabolism through lysine (Lys) residue deacetylation on both histone and non-histone proteins. Emerging evidence has suggested that SIRT1 functions as a key downstream mediator of NAMPT-mediated NAD+ biosynthesis in many biological processes, including metabolism, circadian rhythm, aging, and inflammation [21,55,56]. For example, it was recently reported that NAMPT-mediated NAD+ biosynthesis and SIRT1 regulate the circadian core clock mechanism [50,51]. More specifically, both NAMPT protein levels and NAD+ contents display circadian oscillation patterns that are driven by the core clock machinery. NAMPT-mediated NAD+ biosynthesis suppresses the CLOCK: BMAL1 complex-mediated transcription through SIRT1 activity. Finally, CLOCK binds to the canonical E-boxes on Nampt gene and up-regulates Nampt gene expression. These findings have revealed a fascinating negative feedback loop composed of NAMPT-mediated NAD+ biosynthesis, SIRT1, and CLOCK: BMAL1 complex, providing a novel molecular link between metabolism and circadian rhythm.
SIRT1 deacetylates and regulates key metabolic regulators in adipocytes
Interestingly, data obtained from the recent acetylome studies identified numerous SIRT1-reuglated, acetylated proteins in matured adipocytes [57]. Provided that acetylation is a pivotal posttranslational protein modification that controls protein functions and many biological processes [58], it is postulated that the alteration in SIRT1 activity has profound impact on adipocyte biology and functions. Indeed, recent results obtained from in vitro and in vivo studies have firmly established the significance of SIRT1 in adipocyte biology and whole-body glucose metabolism (Figure 3). For example, SIRT1 deacetlyates FoxO1 at multiple residues (i.e. Lys242, Lys245, and Lys262) and enhances production of adiponectin, a key insulin-sensitizing adipokine, in a FoxO1-dependent manner [59–61]. Thus, genetic or pharmacological activation of SIRT1 by resveratrol administration increases plasma adiponectin concentrations and insulin sensitivity in vivo [61–63]. SIRT1 has been reported to interact physically with the RelA/p65 subunit of the transcription factor nuclear factor B (NF-κB) and suppress transcriptional activity by deacetylating RelA/p65 at Lys310 [64]. SIRT1 inhibition results in hyperacetylation of NF-κB and increased gene expression of NF-κB targets, such as IL-6, TNFα, and monocyte chemoattractant protein 1 (MCP1), in 3T3L1 matured adipocytes [65,66]. Similarly, SIRT1 deacetylates Lys136 of cyclic AMP response element binding protein (CREB) and Lys136 acetylation stimulates CREB activity and increases the expression of key inflammatory genes in adipocytes [67]. Consistent with these in vitro results, systemic delivery of antisense oligonucleotides or genetic deletion of SIRT1 induces adipose tissue inflammation and macrophage activation [42,68–70]. Indeed, adipocyte-specific Nampt deletion also causes macrophage infiltration in adipose tissue [14], suggesting the important role of NAMPT-NAD+-SIRT1 as a negative regulator of macrophage activation, a key feature that contributes to the development of obesity-associated insulin resistance. Recently, it was also reported that SIRT1 inhibits adipocyte hyperplasia though the deacetylation of Lys323 on c-Myc [71]. Finally, it should be noted that SIRT1 enhances the enzymatic activity and secretion of eNAMPT by deacetylating Lys53 on NAMPT, thus affecting hypothalamic function and physical activity [39].
Figure 3. SIRT1 deacetylates and regulates key metabolic regulators in adipocytes.
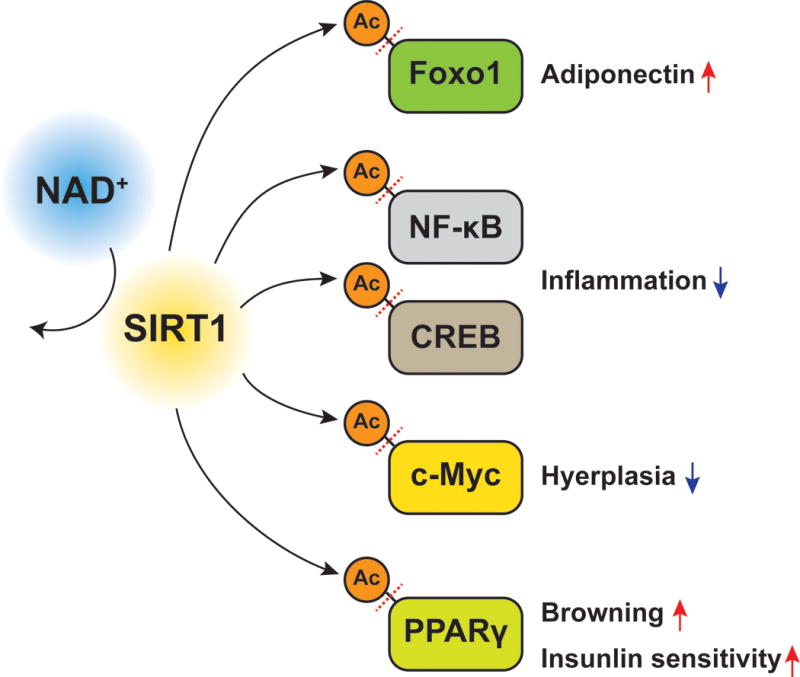
SIRT1 is a NAD+-dependent protein deacetylase that regulates many biological processes. In adipocytes, SIRT1 deacetylates lysine residues on key metabolic regulators, including the forkhead transcriptional factor (Foxo1), nuclear factor kappa B (NF-κB), cyclic AMP response element binding protein (CREB), c-Myc, and PPARγ. SIRT1-mediated lysine deacetylation of these proteins affects key metabolic functions in adipocytes. See the text for the details and references.
NAMPT-NAD+-SIRT1 controls PPARγ function by post-translational protein modifications
The nuclear receptor peroxisome proliferator activated receptor gamma (PPARγ) is a master regulator of adipocyte biology and a molecular target for the treatment of obesity-associated insulin resistance [72]. Adiponectin PPARγ knockout mice exhibit severe insulin resistance and hepatosteatosis [73], whereas adipocyte-specific deletion of the Nuclear Receptor Corepressor (NCoR), a repressor of PPARγ, enhances multi-organ insulin sensitivity in obese mice [74], demonstrating the importance of adipocyte PPARγ function in the regulation of whole-body insulin sensitivity. SIRT1 is classically known to inhibit adipogenesis by repressing the transcriptional activity of PPARγ [75] and thus adipocyte-specific inactivation of SIRT1 results in increased whole-body adiposity in vivo [42]. More recently, SIRT1 has been reported to be critically involved in regulating adipocyte PPARγ function through post-translational protein modifications. SIRT1 deacetylates PPARγ on Lys268 and Lys293, leading to browning of white adipose tissue and repressing insulin resistance genes [76]. Indeed, inhibition of SIRT1 also affects another important post-translational protein modification of PPARγ in adipocytes, namely serine-273 (Ser273) phosphorylation. Phosphorylation of this particular residue on PPARγ has been reported to result in the selective deactivation of PPARγ towards a subset of its obesity-linked target genes involved in regulating whole-body glucose metabolism and insulin sensitivity [77–80]. Interestingly, ANKO mice have increased acetylation and Ser273 phosphorylation of PPARγ and decreased gene expression in a majority of obesity-linked PPARγ targets including adiponectin and adipsin in adipose tissue, and these alterations are nearly completely reversed by administration of NMN or the PPARγ ligand rosiglitazone [14]. Adipocyte-specific Sirt1 knockout (ATKO) mice also have insulin resistance and very similar molecular phenotypes in adipose tissue on short-term, but not chronic, high fat diet feeding [69]. In addition, high-fat diet feeding increases both acetylation and Ser273 phosphorylation of PPARγ in adipose tissue and these protein modifications are reversed by administration of PPARγ agonists [76,77]. Although the mechanistic link between these two distinct post-translational protein modifications of PPARγ is not entirely clear, one can speculate that SIRT1 inactivation-induced PPARγ hyperacetylation leads to Ser273 phosphorylation in adipocytes. Supporting this concept, it was recently reported that hyperacetylation of Lys293, but not of Lys268, could increase Ser273 phosphorylation of PPARγ in human 293 fibroblasts [76]. In addition, recent studies have suggested that SIRT1 mediates such a molecular interplay between acetylation and phosphorylation in proteins involved in key metabolic signaling pathways, such as insulin receptor substrate 2 (IRS-2) [81], CREB [67], signal transducer and activator of transcription 3 (STAT3) [82], mitogen-activated protein kinase kinase-1 (MEK1) [83], and p70 ribosomal S6 kinase (S6K1) [84]. Alternatively or additionally, it is possible that SIRT1 regulates Ser273 phosphorylation of PPARγ through post-translational protein modifications of its key upstream kinases, such as cyclin-dependent kinase 5 (CDK5) and extracellular signal-regulated kinase (ERK) [77,78,80]. We found that ANKO mice have increased phosphorylation of CDK5 in adipose tissue and that pharmacological inhibition of SIRT1 increases CDK5 phosphorylation in matured adipocytes [14]. Similarly, ATKO mice increase adipose tissue CDK5 phosphorylation under a high fat diet [69]. Indeed, it was recently reported that CDK5 is also acetylated at Lys33 residue of ATP binding domain [85], which could be deacetylated by SIRT1. It will certainly be of great interest to determine whether CDK5 acetylation affects its phosphorylation status and thus Ser273 phosphorylation of PPARγ in adipocytes. Dissecting the complex molecular mechanisms responsible for multi-layer SIRT1-mediated post-translational protein modifications of PPARγ will be one important research direction in this field.
A NAMPT-NAD+-SIRT1-PPARγ axis in adipocytes: A novel therapeutic target in obesity and insulin resistance?
High fat diet-induced obesity leads to a decrease in NAMPT-mediated NAD+ biosynthesis and SIRT1 activity [25,42,60] and an increase in acetylation and Ser273 phosphorylation of PPARγ [76,77] in adipose tissue. Therefore, the summation of data obtained from animal and cell culture models outlined above has strongly suggested that impaired adipose NAMPT-NAD+-SIRT1-PPARγ axis contributes to the development of obesity and its metabolic complications and thus it could be a promising therapeutic target for obesity treatment. Indeed, adipocyte-specific overexpression of human SIRT1 was recently reported to ameliorate systemic metabolic complications, such as insulin resistance, glucose intolerance, and dyslipidemia, in aged mice [86]. In addition, adipocyte-specific inactivation of nicotinamide N-methyltransferase prevents diet-induced insulin resistance by increasing NAD+ contents and SIRT1 activity in adipose tissue [87]. Numerous studies conducted in obese rodents have demonstrated that systemic administration of key NAD+ intermediates, such as NMN and NR, exerts remarkable beneficial effects on obesity and its metabolic complications, such as insulin resistance and NAFLD [16,18,25,56,88]. However, it is still unclear whether enhancing NAD+ biosynthesis in adipocytes is sufficient to counteract obesity-associated pathologies. Further studies are awaited to determine the metabolic benefits induced by adipocyte-specific Nampt overexpression in obese rodents and to carefully evaluate the efficacy of NAD+ intermediates in adipose tissue function in vivo. Interestingly, our data suggest that loss of NAMPT results in a decrease in PPAR γ function through Ser273 phosphorylation without affecting classical PPARγ lipogenic targets or whole-body adiposity [14]. Therefore, it is hoped that enhancing NAD+ biosynthesis in adipocytes alone could be novel mechanism-based, insulin sensitizing therapy that can lead to the selective activation of PPARγ through post-translational protein modifications, without the unfavorable side effect profile of the PPARγ agonists [72].
Exploring the translational potential of adipose tissue NAD+ biology in obesity and insulin resistance
As demonstrated above, considerable progress has been made in understanding the importance of adipose NAMPT-mediated NAD+ biosynthesis and SIRT1 in the pathophysiology of obesity and its metabolic complications. The next question is whether we could translate these experimental findings into practical medicine and provide new mechanism-based intervention for obesity and insulin resistance in people. Promisingly, numerous clinical studies have firmly demonstrated the strong association between obesity-associated metabolic complications and NAMPT-NAD+-SIRT1-PPARγ axis in adipose tissue in people. Accumulating evidence suggests that subcutaneous adipose tissue expression of both NAMPT and SIRT1 is negatively correlated with markers of adiposity (i.e. body mass index [BMI]), is positively associated with whole-body insulin sensitivity, and thus it significantly decreases in people with obesity, compared to healthy lean people [68,89–94]. Interestingly, a recent study conducted in BMI-discordant monozygotic twins demonstrated that subcutaneous adipose tissue gene expression of NAMPT and SIRT1 is reduced in the heavier twins, compared to their leaner co-twins, suggesting the relationship between impaired adipose NAMPT-NAD+-SIRT1 axis and acquired obesity [92]. Contrary to the alterations associated with obesity, adipose tissue gene expression of NAMPT and SIRT1 increases after the insulin-sensitizing treatments with lifestyle interventions (i.e. hypocaloric diet and exercise) and bariatric surgery [94–97] in people with obesity. However, adipose tissue NAD+ concentrations in people have not yet been evaluated or reported, possibly due to the technical difficulty in accurate NAD+ measurement [55]. Additional clinical studies are needed to fully understand the pathophysiological importance of adipose NAMPT-mediated NAD+ biosynthesis in obesity and insulin resistance. Furthermore, it was recently reported that rosiglitazone treatment decreases adipose tissue Ser273 phosphorylation of PPARγ in patients with early diagnosed type 2 diabetes, and that these alterations are associated with the improvement in insulin sensitivity [78]. Taken together, these results are highly consistent with the experimental findings obtained from studies conducted in rodents discussed above, suggesting the high translational potential of adipose tissue NAD+ and SIRT1 biology in obesity and insulin resistance.
Remarkably, these promising clinical and experimental data have fueled increasing enthusiasm to investigate the effects of resveratrol and other synthetic sirtuin-activating compounds (STACs) [88] on metabolic health in randomized-controlled trials in the last decade (Table 1). Resveratrol supplementation has been reported to improve markers of insulin resistance, such as homeostasis model assessment of insulin resistance (HOMA-IR), in people who have metabolic abnormalities, including obesity [98], NAFLD [99], and type 2 diabetes [100,101]. However, one limitation of these studies is that they did not use the hyperinsulinemic euglycemic clamp, a gold-standard method to evaluate insulin sensitivity. Indeed, conflicting data have also been reported: resveratrol supplementation does not affect whole-body or tissue-specific insulin sensitivity in different (e.g. healthy non-obese people [102]) or even similar study populations (e.g. obese [103] and diabetic people [104,105]). In addition, administration of SRT2104, one of chemical STACs, does not improve insulin sensitivity in people with type 2 diabetes [106,107]. One possible explanation for these apparently inconsistent data is that the beneficial effects of STACs supplementation on insulin sensitivity could depend on very specific pre-existing metabolic health conditions, dosage and duration of supplementation, bioavailability of STACs, or a combination of these factors. Additional studies are needed to reach a definitive conclusion about the effect of STACs on insulin sensitivity in people. Provided that NMN and NR are natural endogenous compounds found in daily foods and are well-tolerated in rodents [18,108], their translational and therapeutic potential could be highly promising, and effects of supplementation of these NAD+ intermediates have started to be evaluated in people [109].
Table 1.
Randomized controlled trials evaluating the effects of STACs on insulin sensitivity
| Dose (day) | Duration | Study subjects | Measure of insulin sensitivity | Reference |
|---|---|---|---|---|
| Resveratrol | ||||
| 10 mg | 4 weeks | T2D men | HOMA-IR ↓ | [101] |
| 1 g | 45 days | T2D | HOMA-IR ↓ | [100] |
| 150 mg | 30 days | Obese men | HOMA-IR ↓ | [98] |
| 300 mg | Single dose | Overweight/obese men | Postprandial insulin levels ↓ | [112] |
| 300 mg | 12 weeks | NAFLD | HOMA-IR ↓ | [99] |
| 100 mg | 60 days | T2D | HOMA-IR → | [105] |
| 1 - 2 g | 2 weeks | Overweight/obese men | HOMA-IR → | [113] |
| 1.5 g | 4 weeks | Obese men | HECP → | [103] |
| 75 mg | 12 weeks | Nonobese women | HECP → | [102] |
| 3 g | 8 weeks | NAFLD men | HECP → | [114] |
| 500 mg | 12 weeks | NAFLD | HOMA-IR → | [115] |
| 150 mg | 30 days | T2D | HECP → | [104] |
| SRT2104 | ||||
| 0.5 - 2g | 28 days | Nonobese elderly | Oral glucose tolerance → | [107] |
| 0.25 - 2g | 28 days | T2D | Postprandial glucose/insulin levels → | [106] |
Abbreviations: T2D, type 2 diabetes; NAFLD, non-alcoholic fatty liver disease; HOMA-IR, homeostasis model assessment of insulin resistance; HECP, hyperinsulinemic euglycemic clamp procedure.
Conclusions and prospects
In the past decade, we have witnessed conceptual breakthrough and significant progress in the research field of NAD+ and SIRT1 biology. As discussed in this review, the summation of studies conducted in adipocyte-specific knockout/transgenic mouse models has revealed an extraordinary capacity for the adipose NAMPT-NAD+-SIRT1-PPARγ axis to regulate whole-body glucose metabolism and insulin sensitivity, thereby providing important mechanistic and therapeutic insights into obesity and its metabolic complications, particularly insulin resistance. Nevertheless, several fascinating questions remain to be addressed in the future. First, it will be of great interest to elucidate the molecular mechanism responsible for the regulation of Nampt expression in adipocytes. In other cell types, Nampt gene transcription has been reported to be controlled by AMP-activated protein kinase (AMPK) [32], CLOCK: BMAL1 complex [50,51], FoxO transcriptional factors [110], c-Myc [111], or microRNA (miR-34a) [26]. However, Nampt regulation in adipose tissue or adipocytes is not clear. A deeper understanding of this regulatory mechanism will potentially lead to the development of drugs that can specifically target adipose NAMPT-mediated NAD+ biosynthesis. Second, the precise mechanisms underlying regulation of NAD+ homeostasis in adipocytes remain to be investigated. Indeed, we found that both adipose tissue and adipocytes express NARPT protein and administration of nicotinic acid improves insulin resistance in ANKO mice [14]. The contribution of other NAD+ biosynthetic enzymes (e.g. NMNATs, NAPRT), NAD+ degrading enzymes (e.g. CD38), and NAD+ intermediates (e.g. NR) in adipocyte NAD+ biology and glucose metabolism should be further explored. Third, NAD+ is required for the enzymatic activities of other sirtuins (SIRT2-SIRT7): it will be of great interest to investigate the functional role of other sirtuins in adipocyte biology, PPARγ post-translational protein modifications, and whole-body glucose metabolism. Lastly, it is to be hoped that the effects of NAD+ intermediates on adipose tissue function and multi-organ insulin sensitivity will be comprehensively tested in people with obesity and insulin resistance in randomized, controlled trials. The results from these studies will further improve our understanding of the pathophysiological significance and therapeutic potential of adipose tissue NAD+ biology in obesity and insulin resistance.
Acknowledgments
The authors apologize to those whose work is not cited due to space limitations. We would like to thank members in the Yoshino lab for excellent discussion and suggestion in this manuscript. JY is supported by grants from the National Institute of Diabetes and Kidney Diseases DK104995, DK 56341 (Nutrition and Obesity Research Center), DK 37948 and DK 20579 (Diabetes Research Center), KL2 Career Developmental Awards (UL1 TR00450), and the Longer Life Foundation. S.Y. is supported by the Sumitomo Life Welfare and Culture Foundation.
Footnotes
The authors declare that there is no conflict of interest.
References
- 1.Caballero B. The global epidemic of obesity: an overview. Epidemiol Rev. 2007;29:1–5. doi: 10.1093/epirev/mxm012. [DOI] [PubMed] [Google Scholar]
- 2.Swinburn BA, Sacks G, Hall KD, McPherson K, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378:804–14. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- 3.Scherer PE. The Multifaceted Roles of Adipose Tissue-Therapeutic Targets for Diabetes and Beyond: The 2015 Banting Lecture. Diabetes. 2016;65:1452–61. doi: 10.2337/db16-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–81. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115:911–9. doi: 10.1016/j.jaci.2005.02.023. quiz 20. [DOI] [PubMed] [Google Scholar]
- 6.Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34:2–11. doi: 10.1016/j.diabet.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 8.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–94. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 9.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–6. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magkos F, Fraterrigo G, Yoshino J, Luecking C, et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016;23:591–601. doi: 10.1016/j.cmet.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franck N, Gummesson A, Jernas M, Glad C, et al. Identification of adipocyte genes regulated by caloric intake. J Clin Endocrinol Metab. 2011;96:E413–8. doi: 10.1210/jc.2009-2534. [DOI] [PubMed] [Google Scholar]
- 12.Fabbrini E, Yoshino J, Yoshino M, Magkos F, et al. Metabolically normal obese people are protected from adverse effects following weight gain. J Clin Invest. 2015;125:787–95. doi: 10.1172/JCI78425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boden G, Homko C, Barrero CA, Stein TP, et al. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7:304re7. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stromsdorfer KL, Yamaguchi S, Yoon MJ, Moseley AC, et al. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016;16:1851–60. doi: 10.1016/j.celrep.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–9. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garten A, Schuster S, Penke M, Gorski T, et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–46. doi: 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Sauve AA. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbapap.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–63. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, Zhang X, Bheda P, Revollo JR, et al. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat Struct Mol Biol. 2006;13:661–2. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- 21.Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15(Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verdin E. The many faces of sirtuins: Coupling of NAD metabolism, sirtuins and lifespan. Nat Med. 2014;20:25–7. doi: 10.1038/nm.3447. [DOI] [PubMed] [Google Scholar]
- 23.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Revollo JR, Korner A, Mills KF, Satoh A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–75. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi SE, Fu T, Seok S, Kim DH, et al. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. 2013;12:1062–72. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gariani K, Menzies KJ, Ryu D, Wegner CJ, et al. Eliciting the mitochondrial unfolded protein response via NAD repletion reverses fatty liver disease. Hepatology. 2015 doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penke M, Larsen PS, Schuster S, Dall M, et al. Hepatic NAD salvage pathway is enhanced in mice on a high-fat diet. Mol Cell Endocrinol. 2015;412:65–72. doi: 10.1016/j.mce.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 29.Chang WC, Jia H, Aw W, Saito K, et al. Beneficial effects of soluble dietary Jerusalem artichoke (Helianthus tuberosus) in the prevention of the onset of type 2 diabetes and non-alcoholic fatty liver disease in high-fructose diet-fed rats. Br J Nutr. 2014;112:709–17. doi: 10.1017/S0007114514001421. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Yang T, Baur JA, Perez E, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou CC, Yang X, Hua X, Liu J, et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. 2016;173:2352–68. doi: 10.1111/bph.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulco M, Cen Y, Zhao P, Hoffman EP, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–73. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canto C, Jiang LQ, Deshmukh AS, Mataki C, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–9. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frederick DW, Davis JG, Davila A, Jr, Agarwal B, et al. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J Biol Chem. 2015;290:1546–58. doi: 10.1074/jbc.M114.579565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frederick DW, Loro E, Liu L, Davila A, Jr, et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016;24:269–82. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu CP, Oka S, Shao D, Hariharan N, et al. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–91. doi: 10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin JB, Kubota S, Ban N, Yoshida M, et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016;17:69–85. doi: 10.1016/j.celrep.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon MJ, Yoshida M, Johnson S, Takikawa A, et al. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD(+) and Function in Mice. Cell Metab. 2015;21:706–17. doi: 10.1016/j.cmet.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai S-i. The NAD World 2.0: the importance of the inter-tissue communication mediated by NAMPT/NAD+/SIRT1 in mammalian aging and longevity control. npj Systems Biology and Applications. 2016;2:16018. doi: 10.1038/npjsba.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uno K, Katagiri H, Yamada T, Ishigaki Y, et al. Neuronal pathway from the liver modulates energy expenditure and systemic insulin sensitivity. Science. 2006;312:1656–9. doi: 10.1126/science.1126010. [DOI] [PubMed] [Google Scholar]
- 42.Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012;16:180–8. doi: 10.1016/j.cmet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kralisch S, Klein J, Lossner U, Bluher M, et al. Interleukin-6 is a negative regulator of visfatin gene expression in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2005;289:E586–90. doi: 10.1152/ajpendo.00090.2005. [DOI] [PubMed] [Google Scholar]
- 44.Gouranton E, Romier B, Marcotorchino J, Tourniaire F, et al. Visfatin is involved in TNFalpha-mediated insulin resistance via an NAD(+)/Sirt1/PTP1B pathway in 3T3-L1 adipocytes. Adipocyte. 2014;3:180–9. doi: 10.4161/adip.28729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, Bruno J, Easlon E, Lin SJ, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–7. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song J, Ke SF, Zhou CC, Zhang SL, et al. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J Gerontol A Biol Sci Med Sci. 2014;69:44–57. doi: 10.1093/gerona/glt122. [DOI] [PubMed] [Google Scholar]
- 47.Maeda N, Shimomura I, Kishida K, Nishizawa H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–7. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 48.Yamauchi T, Kamon J, Waki H, Terauchi Y, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–6. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 49.Berg AH, Combs TP, Du X, Brownlee M, et al. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–53. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 50.Ramsey KM, Yoshino J, Brace CS, Abrassart D, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–4. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakahata Y, Sahar S, Astarita G, Kaluzova M, et al. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–7. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ando H, Yanagihara H, Hayashi Y, Obi Y, et al. Rhythmic messenger ribonucleic acid expression of clock genes and adipocytokines in mouse visceral adipose tissue. Endocrinology. 2005;146:5631–6. doi: 10.1210/en.2005-0771. [DOI] [PubMed] [Google Scholar]
- 53.Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, et al. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23:372–81. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–45. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD(+)) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–15. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–71. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim SY, Sim CK, Tang H, Han W, et al. Acetylome study in mouse adipocytes identifies targets of SIRT1 deacetylation in chromatin organization and RNA processing. Arch Biochem Biophys. 2016;598:1–10. doi: 10.1016/j.abb.2016.03.025. [DOI] [PubMed] [Google Scholar]
- 58.Choudhary C, Weinert BT, Nishida Y, Verdin E, et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–50. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 59.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–95. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 60.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem. 2006;281:39915–24. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 61.Banks AS, Kon N, Knight C, Matsumoto M, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–41. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ajmo JM, Liang X, Rogers CQ, Pennock B, et al. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–42. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beaudoin MS, Snook LA, Arkell AM, Simpson JA, et al. Resveratrol supplementation improves white adipose tissue function in a depot-specific manner in Zucker diabetic fatty rats. Am J Physiol Regul Integr Comp Physiol. 2013;305:R542–51. doi: 10.1152/ajpregu.00200.2013. [DOI] [PubMed] [Google Scholar]
- 64.Yeung F, Hoberg JE, Ramsey CS, Keller MD, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoshizaki T, Milne JC, Imamura T, Schenk S, et al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. 2009;29:1363–74. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin QQ, Yan CF, Lin R, Zhang JY, et al. SIRT1 regulates TNF-alpha-induced expression of CD40 in 3T3-L1 adipocytes via NF-kappaB pathway. Cytokine. 2012;60:447–55. doi: 10.1016/j.cyto.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 67.Paz JC, Park S, Phillips N, Matsumura S, et al. Combinatorial regulation of a signal-dependent activator by phosphorylation and acetylation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17116–21. doi: 10.1073/pnas.1420389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillum MP, Kotas ME, Erion DM, Kursawe R, et al. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–45. doi: 10.2337/db11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mayoral R, Osborn O, McNelis J, Johnson AM, et al. Adipocyte SIRT1 knockout promotes PPARgamma activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol Metab. 2015;4:378–91. doi: 10.1016/j.molmet.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu F, Zheng X, Lin B, Liang H, et al. Diet-induced obesity and insulin resistance are associated with brown fat degeneration in SIRT1-deficient mice. Obesity (Silver Spring) 2016;24:634–42. doi: 10.1002/oby.21393. [DOI] [PubMed] [Google Scholar]
- 71.Abdesselem H, Madani A, Hani A, Al-Noubi M, et al. SIRT1 Limits Adipocyte Hyperplasia through c-Myc Inhibition. J Biol Chem. 2016;291:2119–35. doi: 10.1074/jbc.M115.675645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahmadian M, Suh JM, Hah N, Liddle C, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–66. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang F, Mullican SE, DiSpirito JR, Peed LC, et al. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18656–61. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li P, Fan W, Xu J, Lu M, et al. Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell. 2011;147:815–26. doi: 10.1016/j.cell.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Picard F, Kurtev M, Chung N, Topark-Ngarm A, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–6. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qiang L, Wang L, Kon N, Zhao W, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150:620–32. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi JH, Banks AS, Estall JL, Kajimura S, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–6. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi JH, Banks AS, Kamenecka TM, Busby SA, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–81. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choi SS, Kim ES, Koh M, Lee SJ, et al. A novel non-agonist peroxisome proliferator-activated receptor gamma (PPARgamma) ligand UHC1 blocks PPARgamma phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J Biol Chem. 2014;289:26618–29. doi: 10.1074/jbc.M114.566794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Banks AS, McAllister FE, Camporez JP, Zushin PJ, et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517:391–5. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282:34356–64. doi: 10.1074/jbc.M706644200. [DOI] [PubMed] [Google Scholar]
- 82.Nie Y, Erion DM, Yuan Z, Dietrich M, et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yeung F, Ramsey CS, Popko-Scibor AE, Allison DF, et al. Regulation of the mitogen-activated protein kinase kinase (MEK)-1 by NAD(+)-dependent deacetylases. Oncogene. 2015;34:798–804. doi: 10.1038/onc.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hong S, Zhao B, Lombard DB, Fingar DC, et al. Cross-talk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J Biol Chem. 2014;289:13132–41. doi: 10.1074/jbc.M113.520734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee J, Yun N, Kim C, Song MY, et al. Acetylation of cyclin-dependent kinase 5 is mediated by GCN5. Biochem Biophys Res Commun. 2014;447:121–7. doi: 10.1016/j.bbrc.2014.03.118. [DOI] [PubMed] [Google Scholar]
- 86.Xu C, Bai B, Fan P, Cai Y, et al. Selective overexpression of human SIRT1 in adipose tissue enhances energy homeostasis and prevents the deterioration of insulin sensitivity with ageing in mice. Am J Transl Res. 2013;5:412–26. [PMC free article] [PubMed] [Google Scholar]
- 87.Kraus D, Yang Q, Kong D, Banks AS, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–62. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nature Reviews Molecular Cell Biology. 2016 doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pedersen SB, Olholm J, Paulsen SK, Bennetzen MF, et al. Low Sirt1 expression, which is upregulated by fasting, in human adipose tissue from obese women. Int J Obes (Lond) 2008;32:1250–5. doi: 10.1038/ijo.2008.78. [DOI] [PubMed] [Google Scholar]
- 90.Rutanen J, Yaluri N, Modi S, Pihlajamaki J, et al. SIRT1 mRNA expression may be associated with energy expenditure and insulin sensitivity. Diabetes. 2010;59:829–35. doi: 10.2337/db09-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song YS, Lee SK, Jang YJ, Park HS, et al. Association between low SIRT1 expression in visceral and subcutaneous adipose tissues and metabolic abnormalities in women with obesity and type 2 diabetes. Diabetes Res Clin Pract. 2013;101:341–8. doi: 10.1016/j.diabres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 92.Jukarainen S, Heinonen S, Rämö JT, Rinnankoski-Tuikka R, et al. Obesity Is Associated With Low NAD+/SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. The Journal of Clinical Endocrinology & Metabolism. 2015;101:275–83. doi: 10.1210/jc.2015-3095. [DOI] [PubMed] [Google Scholar]
- 93.Barth S, Klein P, Horbach T, Dotsch J, et al. Expression of neuropeptide Y, omentin and visfatin in visceral and subcutaneous adipose tissues in humans: relation to endocrine and clinical parameters. Obes Facts. 2010;3:245–51. doi: 10.1159/000319508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kovacikova M, Vitkova M, Klimcakova E, Polak J, et al. Visfatin expression in subcutaneous adipose tissue of pre-menopausal women: relation to hormones and weight reduction. Eur J Clin Invest. 2008;38:516–22. doi: 10.1111/j.1365-2362.2008.01964.x. [DOI] [PubMed] [Google Scholar]
- 95.Pivovarova O, Gogebakan O, Sucher S, Groth J, et al. Regulation of the clock gene expression in human adipose tissue by weight loss. Int J Obes (Lond) 2016;40:899–906. doi: 10.1038/ijo.2016.34. [DOI] [PubMed] [Google Scholar]
- 96.Rappou E, Jukarainen S, Rinnankoski-Tuikka R, Kaye S, et al. Weight Loss Is Associated With Increased NAD(+)/SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue. J Clin Endocrinol Metab. 2016;101:1263–73. doi: 10.1210/jc.2015-3054. [DOI] [PubMed] [Google Scholar]
- 97.Moschen AR, Wieser V, Gerner RR, Bichler A, et al. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J Hepatol. 2013;59:1315–22. doi: 10.1016/j.jhep.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 98.Timmers S, Konings E, Bilet L, Houtkooper RH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–22. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen S, Zhao X, Ran L, Wan J, et al. Resveratrol improves insulin resistance, glucose and lipid metabolism in patients with non-alcoholic fatty liver disease: a randomized controlled trial. Dig Liver Dis. 2015;47:226–32. doi: 10.1016/j.dld.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Movahed A, Nabipour I, Lieben Louis X, Thandapilly SJ, et al. Antihyperglycemic effects of short term resveratrol supplementation in type 2 diabetic patients. Evid Based Complement Alternat Med. 2013;2013:851267. doi: 10.1155/2013/851267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brasnyo P, Molnar GA, Mohas M, Marko L, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106:383–9. doi: 10.1017/S0007114511000316. [DOI] [PubMed] [Google Scholar]
- 102.Yoshino J, Conte C, Fontana L, Mittendorfer B, et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012;16:658–64. doi: 10.1016/j.cmet.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poulsen MM, Vestergaard PF, Clasen BF, Radko Y, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes. 2013;62:1186–95. doi: 10.2337/db12-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Timmers S, de Ligt M, Phielix E, van de Weijer T, et al. Resveratrol as Add-on Therapy in Subjects With Well-Controlled Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care. 2016;39:2211–7. doi: 10.2337/dc16-0499. [DOI] [PubMed] [Google Scholar]
- 105.Bashmakov YK, Assaad-Khalil SH, Abou Seif M, Udumyan R, et al. Resveratrol promotes foot ulcer size reduction in type 2 diabetes patients. ISRN Endocrinol. 2014;2014:816307. doi: 10.1155/2014/816307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baksi A, Kraydashenko O, Zalevkaya A, Stets R, et al. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br J Clin Pharmacol. 2014;78:69–77. doi: 10.1111/bcp.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Libri V, Brown AP, Gambarota G, Haddad J, et al. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS One. 2012;7:e51395. doi: 10.1371/journal.pone.0051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mills KF, Yoshida S, Stein LR, Grozio A, et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24:795–806. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tao R, Wei D, Gao H, Liu Y, et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem. 2011;286:14681–90. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Menssen A, Hydbring P, Kapelle K, Vervoorts J, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E187–96. doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Williams CB, Hughes MC, Edgett BA, Scribbans TD, et al. An examination of resveratrol’s mechanisms of action in human tissue: impact of a single dose in vivo and dose responses in skeletal muscle ex vivo. PLoS One. 2014;9:e102406. doi: 10.1371/journal.pone.0102406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dash S, Xiao C, Morgantini C, Szeto L, et al. High-dose resveratrol treatment for 2 weeks inhibits intestinal and hepatic lipoprotein production in overweight/obese men. Arterioscler Thromb Vasc Biol. 2013;33:2895–901. doi: 10.1161/ATVBAHA.113.302342. [DOI] [PubMed] [Google Scholar]
- 114.Chachay VS, Macdonald GA, Martin JH, Whitehead JP, et al. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2092–103. e1–6. doi: 10.1016/j.cgh.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 115.Faghihzadeh F, Adibi P, Hekmatdoost A. The effects of resveratrol supplementation on cardiovascular risk factors in patients with non-alcoholic fatty liver disease: a randomised, double-blind, placebo-controlled study. Br J Nutr. 2015;114:796–803. doi: 10.1017/S0007114515002433. [DOI] [PubMed] [Google Scholar]