

Abstract
The cytopathic effects of Zika virus (ZIKV) are poorly characterized. Innate immunity controls ZIKV infection and disease in most infected patients through mechanisms that remain to be understood. Here, we studied the morphological cellular changes induced by ZIKV and addressed the role of interferon‐induced transmembrane proteins (IFITM), a family of broad‐spectrum antiviral factors, during viral replication. We report that ZIKV induces massive vacuolization followed by “implosive” cell death in human epithelial cells, primary skin fibroblasts and astrocytes, a phenomenon which is exacerbated when IFITM3 levels are low. It is reminiscent of paraptosis, a caspase‐independent, non‐apoptotic form of cell death associated with the formation of large cytoplasmic vacuoles. We further show that ZIKV‐induced vacuoles are derived from the endoplasmic reticulum (ER) and dependent on the PI3K/Akt signaling axis. Inhibiting the Sec61 ER translocon in ZIKV‐infected cells blocked vacuole formation and viral production. Our results provide mechanistic insight behind the ZIKV‐induced cytopathic effect and indicate that IFITM3, by acting as a gatekeeper for incoming virus, restricts virus takeover of the ER and subsequent cell death.
Keywords: cell death, cytopathic effect, IFITM, paraptosis, ZIKA virus
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Introduction
The recent Zika virus (ZIKV) epidemics in South East Asia, French Polynesia, the Caribbean islands, and the Americas, and its association with neurological disorders including Guillain‐Barré syndrome and microcephaly (Cauchemez et al, 2016; Lessler et al, 2016; Weaver et al, 2016) have triggered a global public health emergency. ZIKV infection mainly occurs after a bite by infected Aedes mosquitoes, through maternal–fetal transmission, and less frequently by sexual transmission (Musso & Gubler, 2016; Petersen et al, 2016). The frequency of Guillain‐Barré syndrome is estimated to be 1/5,000 cases (Cao‐Lormeau et al, 2016; Lessler et al, 2016). Congenital malformations and adverse outcomes associated with ZIKV acquisition during pregnancy are likely due to placental infection and viral transmission to the fetus, with the virus displaying a tropism for neuronal cells (Garcez et al, 2016; Miner et al, 2016; Nowakowski et al, 2016; Quicke et al, 2016; Tang et al, 2016). However, ZIKV infection is generally asymptomatic or, in about 20% of the cases, associated with mild and non‐specific symptoms (Lessler et al, 2016; Petersen et al, 2016). The infection is thus self‐limiting in most cases. Mice lacking the interferon α/β receptor or transcription factors implicated in interferon production develop neurological disease and succumb to ZIKV infection (Dowall et al, 2016; Lazear et al, 2016; Rossi et al, 2016). This suggests that the innate immune response controls viral spread and disease development (Xie et al, 2016), through mechanisms that are not clearly understood.
Zika virus is a member of the Flavivirus genus of the Flaviviridae family, which includes dengue virus (DENV), yellow fever virus (YFV), West Nile virus (WNV), Japanese encephalitis virus (JEV), and tick‐borne encephalitis virus (TBEV). Despite differences in cell tropism and pathogenesis, flaviviruses employ similar replication strategies. The ZIKV RNA genome encodes a single polyprotein that is cleaved post‐translationally into three structural proteins [capsid/core (C), premembrane (prM), and envelope (E)] and seven nonstructural proteins. In cell culture or animal models, ZIKV infects a variety of cell types, such as skin cells (dermal fibroblasts and epidermal keratinocytes), epithelial cells, trophoblasts, neuronal cells, dendritic cells, and macrophages (Hamel et al, 2015; Garcez et al, 2016; Miner et al, 2016; Nowakowski et al, 2016; Quicke et al, 2016; Tabata et al, 2016; Tang et al, 2016). Several receptors, including DC‐SIGN, Axl, Tyro3, and TIM‐1, promote ZIKV binding or entry into target cells, with a major role for the TAM receptors Axl and TIM‐1 (Hamel et al, 2015; Tabata et al, 2016; Meertens et al, 2017). Flaviviral entry occurs through endocytosis and pH‐dependent fusion, while subsequent steps including synthesis of viral proteins, RNA replication, and particle assembly take place in the endoplasmic reticulum (ER) (Blázquez et al, 2014; Ravindran et al, 2016). Flaviviruses rearrange ER membranes to build novel organelle‐like structures (also termed viral factories) used for viral production (Blázquez et al, 2014; Blanchard & Roingeard, 2015; Paul & Bartenschlager, 2015; Ravindran et al, 2016). Recent genetic CRISPR‐based screens identified that ER protein complexes involved in signal sequence recognition, N‐linked glycosylation, and ER‐associated degradation (ERAD) are required for flavivirus replication (Marceau et al, 2016; Savidis et al, 2016a; Zhang et al, 2016). Membrane remodeling and accumulation of viral proteins trigger a stress pathway in the ER and activate the unfolded protein response (UPR). This response induces ERAD (Blázquez et al, 2014). The UPR and autophagy pathways are intimately intertwined. Formation of autophagosomes in skin fibroblasts may facilitate ZIKV replication (Hamel et al, 2015), whereas autophagy assists DENV in part by regulating lipid droplet formation and lipid metabolism (Heaton & Randall, 2010).
In addition to serving as the site for virus replication and production, the ER is central to the cytopathic effects observed in flavivirus‐infected cells. The ERAD pathway and the ER membrane complex (EMC) are primary drivers of WNV‐induced cell death (Ma et al, 2015). Cell death resulting from flavivirus infection may occur via different mechanisms, with apoptosis being an important pathway, as demonstrated with WNV‐, JEV‐, ZIKV‐, and DENV‐infected cells (Ghosh Roy et al, 2014; Frumence et al, 2016). Other reported mechanisms of flavivirus‐induced cell death include necrosis, pyroptosis, and necroptosis (Ghosh Roy et al, 2014; Danthi, 2015; Frumence et al, 2016; Suwanmanee & Luplertlop, 2016). How the UPR and cell death are linked in ZIKV infection is unknown. Furthermore, the relative contribution of various cell death pathways to ZIKV‐induced cytopathy has not been explored.
The immune‐related interferon‐induced transmembrane proteins IFITM1, IFITM2, and IFITM3 are restriction factors that protect uninfected cells from viral infection. They block the entry of diverse viruses by inhibiting virus–cell fusion (Brass et al, 2009; Everitt et al, 2012; Diamond & Farzan, 2013; Smith et al, 2014). The mechanisms involved include altering the biophysical properties or cholesterol content of the cellular membranes in which they are found (Amini‐Bavil‐Olyaee et al, 2013; Li et al, 2013; Desai et al, 2014). These proteins inhibit many enveloped viruses, including influenza A virus (IAV), the flaviviruses WNV and DENV, severe acute respiratory syndrome coronavirus, hepatitis C virus, and Ebola virus (Brass et al, 2009; Zhu et al, 2015; Gorman et al, 2016) as well as HIV‐1 and SIV (Lu et al, 2011). We and others identified new antiviral functions of IFITM proteins during HIV‐1 infection: IFITM3 in the virus‐producing cell is incorporated into nascent virions and decreases virus–cell fusion (Compton et al, 2014; Tartour et al, 2014; Yu et al, 2015).
It has been recently reported that IFITM proteins inhibit ZIKV replication and can prevent ZIKV‐induced cell death (Savidis et al, 2016b). Here we confirm and extend these initial observations using transformed and primary human cells. We show that IFITM3 is an important component of the type‐I interferon response conferring protection against ZIKV infection. Combining time‐lapse microscopy, electron microscopy, and other techniques, we describe in detail the fate of ZIKV‐infected cells. We report a striking succession of events including cytoplasmic vacuole formation, cell shrinkage, and “implosion”, which is exacerbated when levels of IFITM3 are low. We further demonstrate that the large vacuoles are derived from the ER, require the Sec61 translocon for their formation, and are associated with paraptosis‐like cell death of ZIKV‐infected cells. These findings reinforce the crucial importance of intrinsic virus restriction, mediated largely by IFITM proteins, in the control of ZIKV infection and avoidance of cytopathy. Furthermore, these findings provide functional insight into the mechanisms driving cell death during ZIKV infection, which likely contribute to ZIKV‐associated disease in vivo.
Results
IFITM3 inhibits ZIKV replication in epithelial cell lines and primary fibroblasts
To confirm the effect of IFITM protein expression on ZIKV replication, we generated 293T cells expressing IFITM1, IFITM2, or IFITM3 (Fig EV1A) and challenged them with an African isolate of ZIKV (HD78788, referred to as HD78), at a multiplicity of infection (MOI) of 1. In control cells, the virus establishes a productive infection and spreads rapidly, as scored by the appearance of cells expressing viral envelope (E) protein (stained with the pan‐flavivirus anti‐E antibody 4G2) (Fig EV1B). IFITM3 severely impaired productive ZIKV infection, whereas IFITM1 and IFITM2 exerted a lower antiviral effect (Fig EV1B and C). IFITM3 is primarily localized in the endosomal/lysosomal compartment. To determine whether the location of IFITM3 is important for the restriction, we used 293T cells expressing a variant of IFITM3 lacking the amino‐terminus (IFITM3 ∆1‐21), which accumulates at the cell surface (Chesarino et al, 2014; Jia et al, 2014; Compton et al, 2016). IFITM3 ∆1‐21 restricted ZIKV replication less efficiently than the full‐length protein (Fig EV1C). Murine IFITM3 also potently inhibited ZIKV replication (Fig EV1C). Two other ZIKV strains, isolated from individuals from French Polynesia and New Caledonia (PF13 and NC14, respectively; Hamel et al, 2015; Barba‐Spaeth et al, 2016; Chouin‐Carneiro et al, 2016), exhibited a similar sensitivity to IFITM3 (Fig EV1D). In human newborn foreskin fibroblasts (HFF) and adult dermal fibroblasts (HDFa), which both display relatively low levels of basal IFITM3, transduction with a lentivector resulted in about 60–80% of the cells expressing IFITM3 (Fig EV1E). ZIKV infection was reduced by fourfold to fivefold in these cells, with 4G2+ cells appearing predominantly in the IFITM3‐low population (Fig EV1E).
Figure EV1. Expression of IFITMs in 293T cells and human primary fibroblasts restricts ZIKV replication.
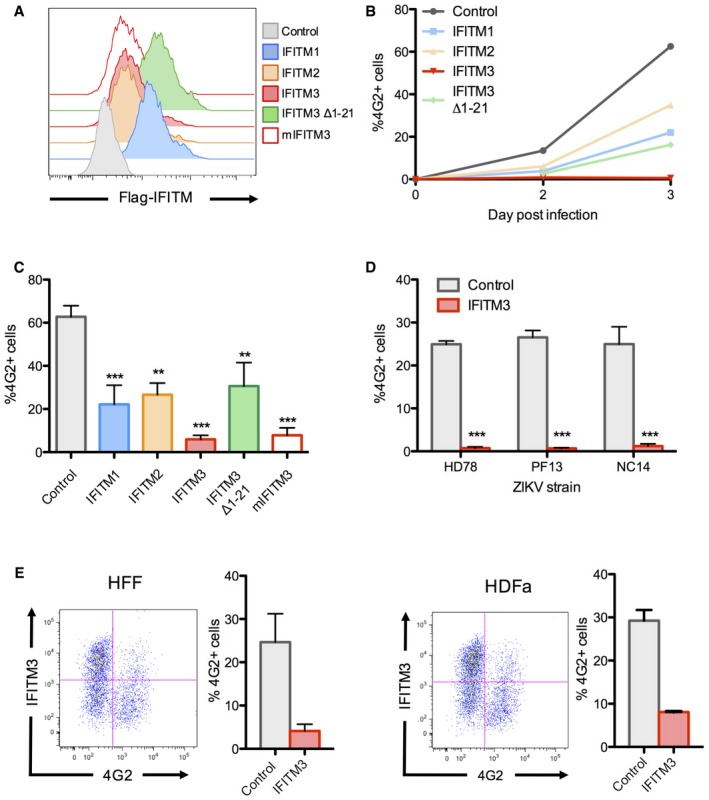
- Stable IFITM‐positive cell lines were produced in 293T cells, and the expression of each protein was measured by flow cytometry.
- 293T cells were infected with ZIKV HD78 (MOI 1), and viral replication was measured at days 2 and 3 post‐infection (pi) by flow cytometry using the anti‐E protein antibody 4G2, as illustrated in one representative experiment.
- Results from four independent experiments as in (B) were quantified at day 3 pi. Mean ± SEM.
- 293T cells expressing or not IFITM3 were infected with the indicated ZIKV strains. Viral replication was measured at day 2 or 3 post‐infection (pi) by flow cytometry using the anti‐E protein antibody 4G2. Results are mean ± SEM of three independent experiments.
- Human foreskin fibroblasts (HFF) (left panels) or human adult dermal fibroblasts (HDFa) (right panels) overexpressing or not IFITM3 were infected with ZIKV, and the % of E‐positive cells was measured by flow cytometry at day 2 pi. For each cell type, one representative experiment is shown on the left and the mean ± SEM of three independent experiments on the right.
HeLa cells express basal levels of IFITM3, which are upregulated by type‐1 interferon (IFN; Fig 1A; Compton et al, 2014). To measure the impact of endogenous IFITM3 on ZIKV infection, we transduced HeLa cells with a lentivector expressing shRNA targeting IFITM3 (sh‐IFITM3). Protein levels of IFITM3 were reduced by > 80%, in the presence or absence of IFN, when compared to control cells treated with a scrambled shRNA (sh‐SCR; Fig 1A). Knockdown of IFITM3 led to a 10‐fold enhancement of ZIKV infection, whereas pre‐treatment of control HeLa cells with IFN strongly inhibited infection (Fig 1B). However, IFN exerted only a modest antiviral effect in sh‐IFITM3 cells when compared to sh‐SCR cells (Fig 1B), demonstrating that IFITM3 is a major contributor to the anti‐ZIKV response mediated by IFN (Hamel et al, 2015; Bayer et al, 2016; Lazear et al, 2016). Silencing of IFITM3 via siRNA in HDFa cells (Fig EV2A) also resulted in enhanced sensitivity to ZIKV, confirming an antiviral role for endogenous IFITM3 in primary cells.
Figure 1. ZIKV‐induced cytopathic effects and cytoplasmic vacuolization.
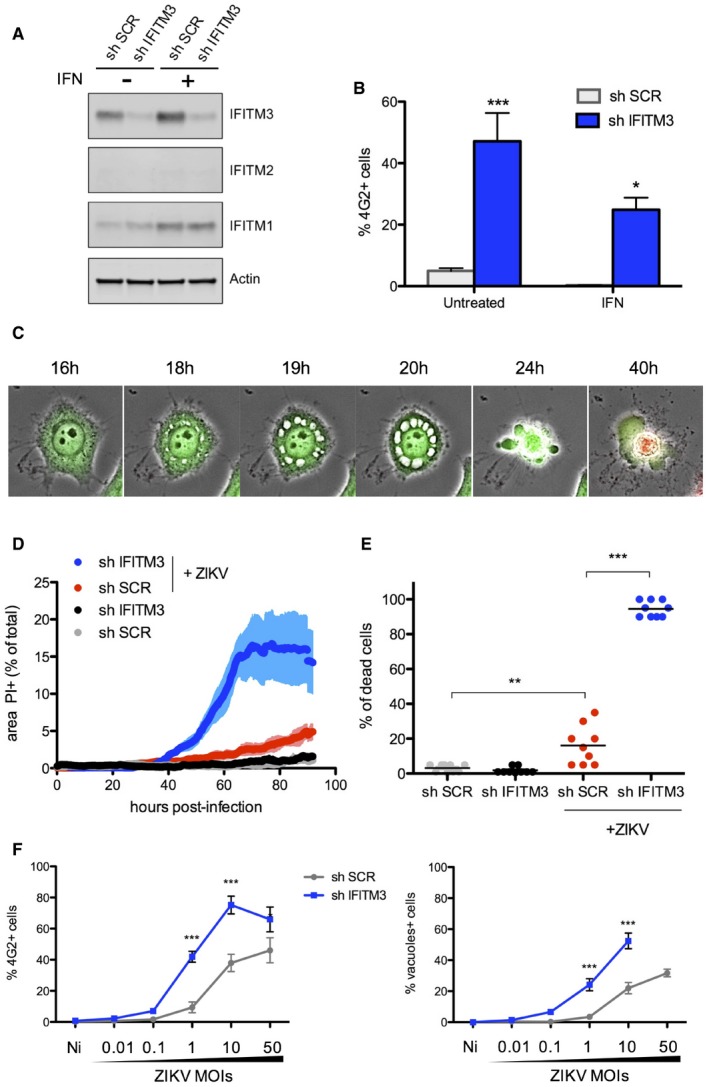
- HeLa cells were transduced to stably express a control scrambled shRNA (sh‐SCR) or shRNA targeting IFITM3 (sh‐IFITM3). The level of endogenous IFITMs in the presence or absence of IFN‐α (1,000 IU/ml for 48 h) was assessed by Western blot.
- HeLa sh‐SCR or sh‐IFITM3 treated or not with IFN‐α were infected with ZIKV HD78 (MOI 1), and the % of E‐positive cells was determined by flow cytometry at day 2 pi. Mean ± SEM of three independent experiments.
- HeLa sh‐SCR or sh‐IFITM3 cells were infected with ZIKV (MOI 1) for 2–4 days in the presence of propidium iodide (PI) for time‐lapse microscopy. The cells also expressed GFP to facilitate visualization of the cytoplasm and the vacuoles. Still images were extracted from Movie EV1 (from HeLa sh‐IFITM3 infected cells) at the indicated time points.
- The area of the PI+ signal was quantified for each condition. Nine fields from three independent experiments were analyzed and plotted as mean ± SEM. Movie EV3 is a representative example of data used for quantification.
- The % of dead cells from 9 fields was visually scored at day 3 pi and plotted as mean.
- ZIKV E expression determined by flow cytometry and the proportion of cells displaying vacuoles was scored by visual examination of at least 200 cells at 24 h pi in HeLa sh‐SCR or sh‐IFITM3 cells infected with the indicated MOIs. Mean ± SEM of three independent experiments is shown.
Figure EV2. Endogenous IFITM3 protein inhibits ZIKV replication at an early stage.
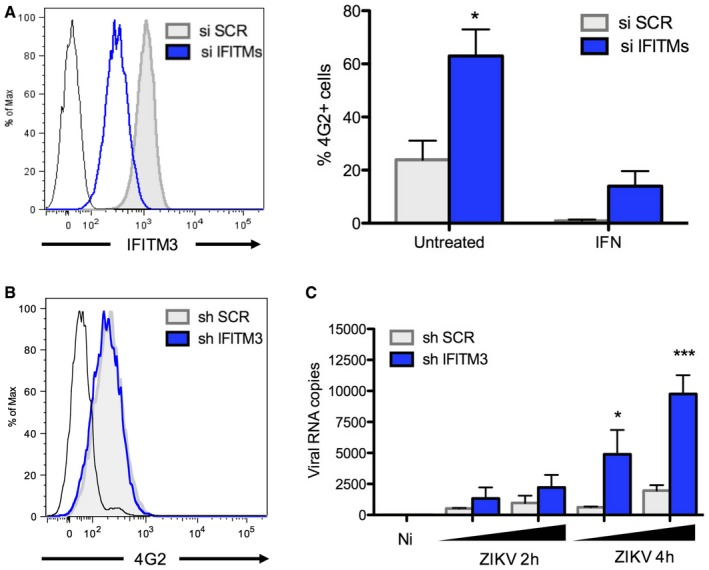
- Effect of IFITM3 in HDFa cells. Left panel: The levels of endogenous IFITM3 were assessed by flow cytometry in HDFa transfected with a control siRNA or siRNA targeting IFITM3. Black curve: isotype control. Right panel: The cells were pretreated or not with IFN‐α (1,000 IU/ml for 48 h) and infected with ZIKV HD78 (MOI 1) for 24 h. Infected cells were scored by 4G2 staining and flow cytometry (right panel). Mean ± SEM from three independent experiments is shown.
- IFITM3 does not affect virus attachment to target cells. HeLa sh‐SCR or sh‐IFITM3 were incubated with ZIKV for 1 h at 4°C. The mean fluorescence intensity (MFI) of 4G2 staining was determined by flow cytometry. Black curve: isotype control.
- IFITM3 prevents intracellular accumulation of viral RNA. HeLa sh‐SCR or sh‐IFITM3 were infected with ZIKV for 2 or 4 h at 37°C. The black triangle indicates increasing amounts of virus (MOI 3 and 10). Virus remaining at the cell surface was removed by trypsin treatment. The cellular RNA was extracted, and RT‐qPCR was performed to quantify viral RNA. Mean ± SEM of three independent experiments.
IFITM3 inhibits an early step of ZIKV replication
To elucidate the step of the ZIKV life cycle inhibited by IFITM3, we examined virus binding to target cells. HeLa sh‐SCR or sh‐IFITM3 cells were incubated with ZIKV on ice for one hour in order to measure levels of bound viral material using flow cytometry (Fig EV2B). No significant difference in virus attachment was observed in the presence or absence of IFITM3. To assess virus entry, we incubated target cells with ZIKV for 2 h at 37°C, followed by trypsin treatment (to eliminate surface‐bound virus) and cell lysis to measure intracellular viral RNA by RT–qPCR (Hamel et al, 2015; Meertens et al, 2017). We observed a strong augmentation of viral RNA (vRNA) in IFITM3‐silenced cells at two and four hours post‐viral exposure (Fig EV2C). vRNA was barely detectable when cells were incubated on ice to block endocytosis (not shown), suggesting that trypsinization removes most of the virions that remained bound to cell surfaces. Thus, the antiviral effect of IFITM3 occurs at an early stage of the ZIKV life cycle following virus attachment.
Analysis of ZIKV‐induced cytopathy
Flaviviruses are cytopathic in cell culture, and previous studies have shown that IFITM3 restricts virus cytopathicity (Ghosh Roy et al, 2014; Savidis et al, 2016b). However, a detailed and mechanistic description of these events has not been performed for ZIKV. Using HeLa cells in which IFITM3 was silenced or not, we visualized cells using time‐lapse fluorescence microscopy following incubation with virus, with images acquired every 10 min for up to 96 h. The cells also expressed GFP to facilitate visualization. The addition of propidium iodide (PI), a membrane‐impermeable dye which stains cellular DNA, allowed us to monitor the terminal death event. Representative examples are shown in Fig 1C and Movies EV1, EV2 and EV3. ZIKV (HD78 strain) induces a mild cytopathic effect in control cells at a multiplicity of infection (MOI) of 1, driving the appearance of few PI+ cells and allowing for ongoing cell proliferation (Fig 1D and Movies EV1 and EV3). In the absence of IFITM3, however, PI+ labeling occurred in virtually all cells. A visual scoring of cells showed that 15% of IFITM3‐positive cells and > 95% of IFITM3‐silenced cells die at 72 h pi, respectively (Fig 1E). Longitudinal analysis demonstrated that death of ZIKV‐infected cells is preceded by the formation of massive cytoplasmic vacuoles (Fig 1C) and cytoplasmic contraction typically occurred 2–5 h post‐vacuolization (see a quantification of the duration of events in Fig EV3A). Approximately 6 h later, membrane integrity was lost and death was confirmed by PI staining. This final phase is characterized by bubble‐like membrane extensions and an “implosive” destruction of the cell (Movies EV1, EV2 and EV3). Similar cell death phenomena can also be observed in control HeLa cells, albeit to a lower frequency (Fig 1D and E, and Movies EV1, EV2 and EV3). Single‐cell analysis showed that, once initiated, vacuolization and death proceeded similarly in both IFITM3− and IFITM3+ cells (Fig EV3A). A similar cytopathic effect was observed in cells infected with ZIKV strains NC14 and PF13 (not shown). Of note, exposure of HeLa sh‐IFITM3 cells to dengue virus DENV2 16681 strain, derived from a dengue hemorrhagic fever patient (Medina et al, 2015), led to productive infection without massive vacuolization of cells (Fig EV4).
Figure EV3. ZIKV‐induced cytoplasmic vacuolization and cytopathic effects.
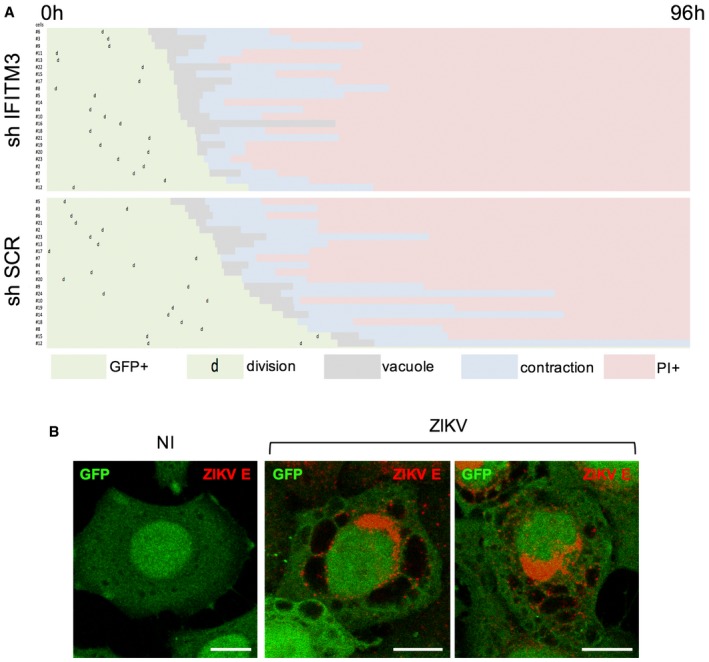
- Time‐lapse analysis of the morphological changes observed in ZIKV‐infected cells. HeLa sh‐SCR and HeLa sh‐IFITM3 cells were infected with ZIKV HD78 (MOI 1) and recorded using time‐lapse microscopy. For each condition, about 20 individual cells that end up dying upon ZIKV infection were selected for further analysis. Morphological changes were visually scored for each cell. Results are representative of three independent experiments.
- HeLa sh‐SCR cells were infected or not with ZIKV HD78 (MOI 1) for 24 h, fixed, and stained to detect ZIKV E protein (red). One single Z slice representative of three independent experiments is shown. Scale bars: 5 μm.
Figure EV4. Morphological changes induced by ZIKV HD78 and DENV2.
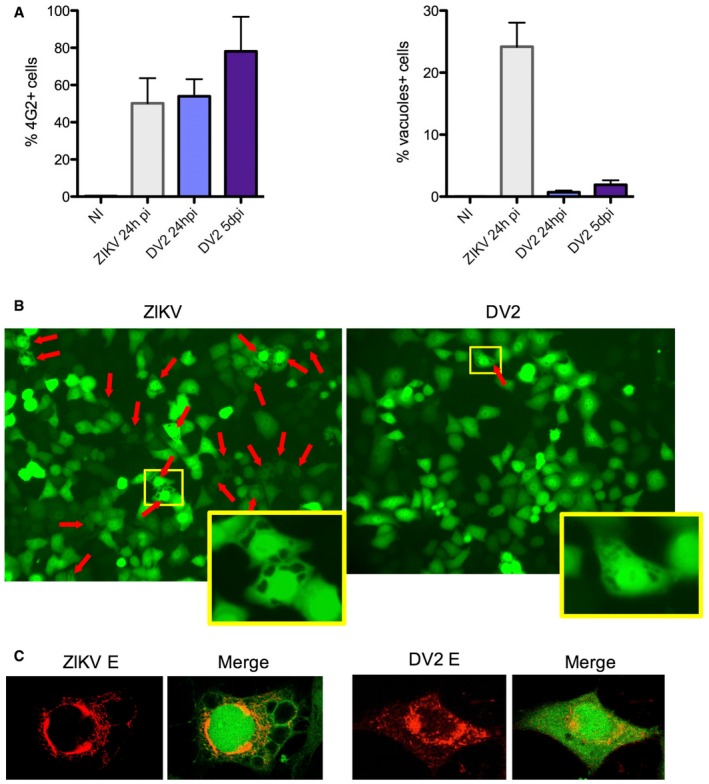
- HeLa sh‐IFITM3 were infected with ZIKV HD78 or DV2 for 24 h or 5 days pi. Left panel: The levels of infection were determined by 4G2 staining and flow cytometry. Right panel: The proportion of cells displaying vacuoles was scored by visual examination of at least 200 cells at 24 h pi (right panel). Mean ± SEM of three independent experiments.
- One example of a representative field from (A) is shown for ZIKV (left) and DENV2 (right) infected cells at 24 h pi. Red arrows point to vacuole+ cells. For each condition, an enlarged view of a vacuolated cell is shown.
- Examples of ZIKV‐ and DENV‐infected cells, with 4G2 staining and confocal microscopy analysis.
We then quantified more precisely the impact of IFITM3 on ZIKV infection and subsequent cytopathic effects. We performed a dose–response analysis of viral inocula in HeLa sh‐SCR and sh‐IFITM3 cells. We then measured the proportion of cells expressing E protein or displaying vacuoles at 24 h pi (Fig 1F). The proportion of infected cells and vacuolated cells increased in parallel with the MOI. The viral inoculum required to obtain similar levels of infection and cytopathy was at least 10 times higher when IFITM3 was present.
Large cytoplasmic vacuoles were also visible in ZIKV‐infected primary human astrocytes (Fig 2A) and skin fibroblasts (HDFa cells; Fig 2B and Movie EV4). We silenced IFITM3 in HDFa cells with siRNA and observed that small vesicles form and accumulate in the vicinity of the nucleus following ZIKV infection, which then increase in size and toward the cell periphery and fuse together (Fig 2B and Movie EV4). As observed in HeLa cells, this vacuolization preceded shrinkage and death. This cytopathic effect was visible but less frequent when IFITM3 was not silenced (not shown).
Figure 2. ZIKV induces cytoplasmic vacuolization in primary cells.
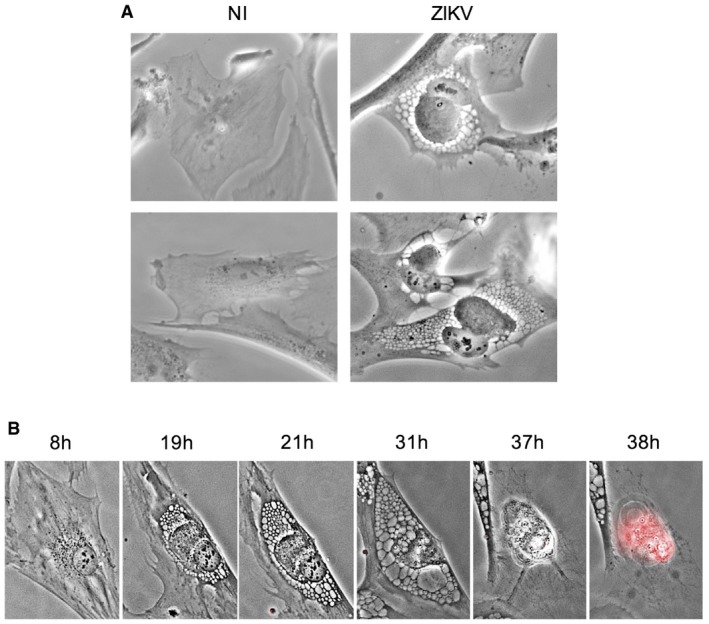
- Primary human astrocytes were infected with ZIKV HD78 (MOI 1) and observed 24 h pi. Non‐infected (NI) astrocytes are shown as control. Images were taken with a bright‐field objective. Representative panels from three independent experiments are shown.
- Primary adult human dermal fibroblasts (HDFa) were infected ZIKV HD78 (MOI 1) for 2–4 days in the presence of propidium iodide (PI) for time‐lapse microscopy. Still images were extracted from Movie EV4 at the indicated time points.
Altogether, these results show that ZIKV induces massive cytoplasmic vacuolization in various cell types, which is associated with cell death. When IFITM3 levels are low, sensitivity of target cells to productive infection is augmented, and the proportion of cells displaying vacuolization and death is also elevated.
Characterization of cytoplasmic vacuoles in ZIKV‐infected cells
To assess the causes and consequences of cellular vacuolization, we first used immunofluorescence microscopy to study ZIKV‐infected cells. At 24 h pi, little to no E protein staining was apparent in control HeLa cells incubated with ZIKV at a MOI of 1 (Fig 3A). However, in the absence of IFITM3, infected cells displayed a typical perinuclear accumulation of ZIKV E protein (Fig 3A and B). All cells displaying vacuolization were positive for E protein, indicating that this process is associated with productive infection. Close examination at a higher magnification revealed that E protein is predominantly found in a dense perinuclear region, and to a lesser extent, at the limiting edges and lumens of the large vacuoles (Fig 3B). In the presence of IFITM3, HeLa cells exhibiting large vacuoles were much less frequent, but when present, they displayed a pattern of E protein staining similar to cells in which IFITM3 is silenced (Fig EV3B). We also performed fluorescence in situ hybridization (FISH) imaging at 24 h pi (Savidis et al, 2016b; Meertens et al, 2017) to localize vRNA. vRNA accumulated throughout the cytoplasm in infected cells, including the perinuclear region and in the vicinity of the large cytoplasmic vacuoles (Fig 3C).
Figure 3. Large cytoplasmic vacuoles are the direct result of productive ZIKV infection.
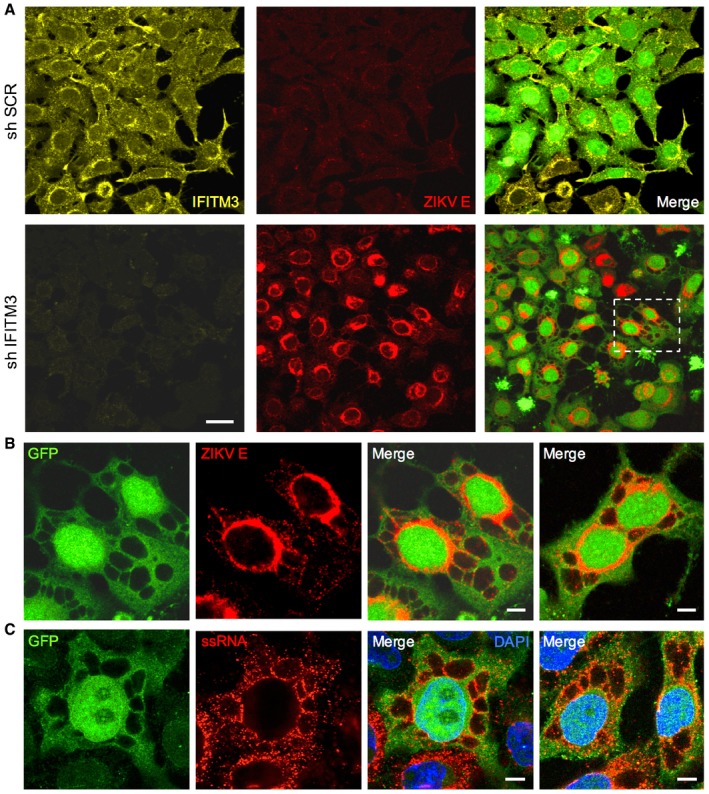
- HeLa sh‐SCR or sh‐IFITM3 cells were infected with ZIKV HD78 (MOI 1) for 24 h, fixed, and stained to detect E (red) and IFITM3 (yellow) proteins. The cells also expressed GFP to facilitate visualization of the cytoplasm and the vacuoles. One field representative of three independent experiments is shown.
- Detail of ZIKV‐infected cell in (A) showing large vacuoles and 4G2 staining (red). Images are average Z‐stacks of three consecutive slices. An additional example from a different field is also shown in the right panel.
- HeLa sh‐IFITM3 cells were infected with ZIKV HD78 (MOI 10) for 24 h, fixed, and subjected to FISH for the detection of viral ssRNA. Two examples from two fields are shown. A single Z slice is shown.
To provide ultrastructural detail, we performed transmission electron microscopy of ZIKV‐infected cells at 24 h pi (Fig 4). Non‐infected HeLa cells displayed normal morphology and nuclear membrane integrity (Fig 4A). In sharp contrast, ZIKV‐infected cells presented striking disturbances. An electron dense perinuclear region with numerous convoluted membranes contained abundant virus‐like particles (Fig 4B). This region resembles the “membranous web” induced by DENV infection and likely corresponds to viral factories, the primary sites of viral production (Blázquez et al, 2014; Blanchard & Roingeard, 2015; Paul & Bartenschlager, 2015; Ravindran et al, 2016). The significantly larger, cytoplasmic vacuoles were also observed throughout the cell cytoplasm (Fig 4B and C). They appeared virtually empty and only occasionally contained virus‐like particles within their lumen, indicating that they are not major sites of virion production or accumulation. In addition, the outer nuclear membrane of infected cells was deformed, swollen, and in some cases, contiguous with large vacuoles in the cytoplasm, suggesting that these large cytoplasmic vacuoles may originate from ER‐related membranes (Fig 4D).
Figure 4. Ultrastructural analysis of ZIKV‐infected HeLa cells.
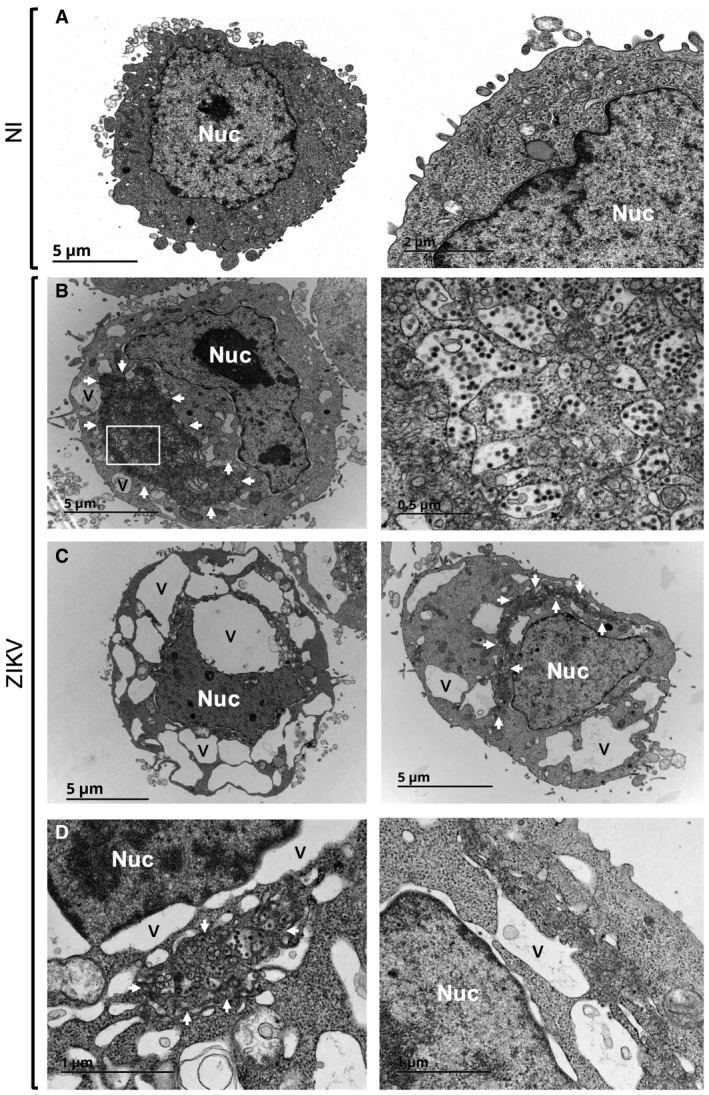
- Transmission electron microscopy (TEM) of non‐infected HeLa sh‐IFITM3 cells.
- TEM of ZIKV‐infected HeLa sh‐IFITM3 cells at 24 h pi displays cytoplasmic vacuolization (indicated with the letter V) and active virion production (enlarged on the right).
- Examples of ZIKV‐infected cells exhibiting large vacuoles.
- Enlarged views of dilated nuclear membranes in ZIKV‐infected cells.
Role of the ER in the massive vacuolization of ZIKV‐infected cells
To learn more about the origins of virus‐induced vacuoles, we stained cells with anti‐protein disulfide isomerase (PDI), an ER‐resident enzyme. In non‐infected cells, PDI was mostly associated with the nuclear envelope and in dispersed structures in the cell periphery (Fig 5A). However, in ZIKV‐infected cells displaying vacuolization, the pattern of PDI staining was altered: a large fraction of vacuoles was decorated with PDI, while nuclear envelope staining was less evident (Fig 5A). These findings strongly suggest that ZIKV induces large‐scale ER rearrangements and that virus‐induced vacuoles originate from ER‐derived membranes. Consistent with their “empty” nature, these vacuoles did not stain positive with the lipid droplet reagent Bodipy 493/503, nor were they positive for the lysosomal protein LAMP1 (not shown).
Figure 5. ZIKV infection triggers ER‐derived vacuoles and ER stress.
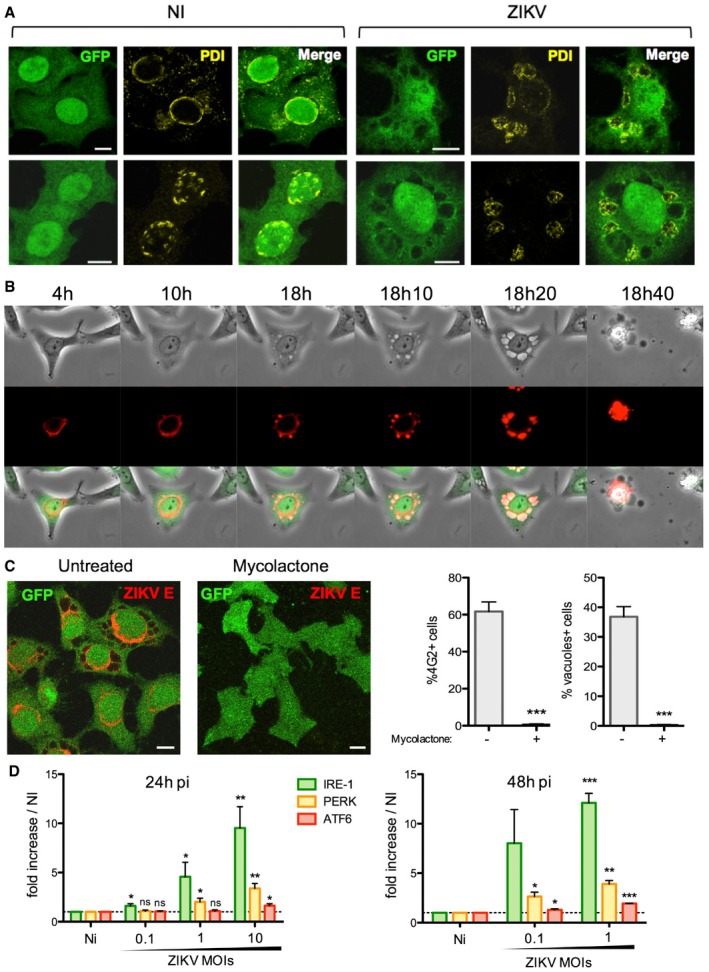
- HeLa sh‐IFITM3 cells were infected with ZIKV HD78 (MOI 1) for 24 h, fixed, and stained for protein disulfide isomerase (PDI) (yellow). Two representative cells for each condition over three independent experiments are shown. Images are average Z‐stacks of three consecutive slices. Scale bars, 10 μm.
- HeLa sh‐IFITM3 cells stably expressing a fluorescent ER‐tracker (DsRed2‐ER) shown in red were infected with ZIKV HD78 (MOI 1) for 2 days for time‐lapse microscopy. Still images were extracted from Movie EV5 at the indicated time points.
- HeLa sh‐IFITM3 were infected with ZIKV HD78 (MOI 0.1 or 1) for 2 h and then treated with or without mycolactone (20 nM), an inhibitor of the Sec61 translocon. After 24 h, cells were stained for viral E protein and analyzed by confocal microscopy (left panels). ZIKV E expression was determined by flow cytometry, and the proportion of cells displaying vacuoles was scored by visual examination of at least 200 cells (right). Results are mean ± SEM of three independent experiments. Statistical significance was determined by unpaired t‐tests. ***P < 0.001. Scale bars, 10 μm.
- ZIKV‐induced ER stress. Levels of IRE‐I, PERK, and ATF6 mRNA were determined by RT–PCR at 24 and 48 h pi. Results are mean ± SEM for three independent experiments. Statistical significance was determined by unpaired t‐tests. ***P < 0.001; **P < 0.01; *P < 0.05; ns, P > 0.05.
To confirm that the vacuoles originated from the ER, we generated HeLa cells transfected with a fluorescent ER marker (dsRed–calreticulin–KDEL fusion protein known as dsRed‐ER). Real‐time imaging indicated that in non‐infected cells, dsRed‐ER was present mainly around the nucleus (data not shown). Upon ZIKV infection, the red signal expanded and colocalized with vacuoles that were made visible by phase contrast (Fig 5B and Movie EV5). At later time points following cell contraction, membranous red blebs were observed in dying cells, suggesting that the blebs and ER‐derived vacuoles are one and the same (Fig 5B).
We further analyzed the involvement of the ER in ZIKV replication and in the formation of massive vacuoles by using a mycobacterial virulence factor, mycolactone. Mycolactone is a diffusible macrolide that blocks the biogenesis of secretory and integral membrane proteins by inhibiting the Sec61 translocon, the channel guiding nascent polypeptides into the ER lumen (Hall et al, 2014; Guenin‐Mace et al, 2015; Kalies & Romisch, 2015; Baron et al, 2016; McKenna et al, 2016; Ravindran et al, 2016). Recent screen‐based studies revealed that Sec61 is required for DENV and ZIKV replication (Heaton et al, 2016; Marceau et al, 2016; Zhang et al, 2016). In accordance with these findings, cells treated with mycolactone at 2 h following ZIKV exposure were completely refractory to infection and vacuolization (Fig 5C), while partial inhibition occurred when mycolactone was added at later time points (Fig EV5A). Mycolactone directly targets the α subunit of the Sec61 translocon (Baron et al, 2016), so we used cells expressing a mutant Sec61α resistant to the effects of mycolactone (Sec61 mut) (Baron et al, 2016). ZIKV infection was not inhibited by mycolactone in cells expressing mutant Sec61 (Fig EV5B), implying that Sec61 blockade protects against ZIKV infection. Of note, Brefeldin A, an inhibitor of protein transport from the ER to the Golgi apparatus, also inhibited ZIKV infection (Fig EV5A). Together, these results indicate that viral production requires Sec61 activity. Moreover, when viral protein synthesis is blocked by mycolactone, no vacuolization is observed.
Figure EV5. Effect of mycolactone on ZIKV replication.
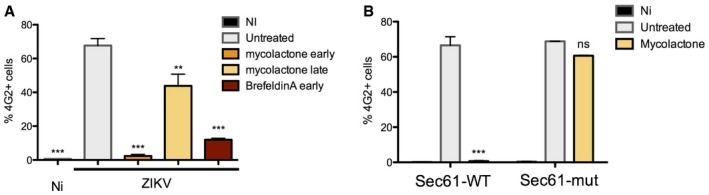
- HeLa sh‐IFITM3 were infected with ZIKV HD 78 (MOI 1) for 24 h in the presence or absence of mycolactone (20 nM) added at 2 h (mycolactone early) or 15 h pi (mycolactone late). Brefeldin A (0.5 μg/ml) was added during the 24 h of infection. The levels of infection were determined by 4G2 staining and flow cytometry. Results are mean ± SEM of three independent infections.
- 293T cells expressing WT Sec61 or a mutated Sec61 resistant to mycolactone (Sec61‐mut) were infected with ZIKV HD 78 (MOI 1) for 3 days in the presence or absence of mycolactone (20 nM). The levels of infection were determined by 4G2 staining and flow cytometry. Results are mean ± SEM of three independent infections.
Various RNA viruses, including flaviviruses, trigger ER stress, likely through the influx of nascent unfolded proteins, and as a result, infections activate the unfolded protein response (UPR) (Blázquez et al, 2014; Jheng et al, 2014). The UPR leads to upregulation of genes encoding ER chaperones, attenuation of translation, initiation of the ER quality control system, and ER‐associated degradation (ERAD) to restore ER homeostasis (Fu & Gao, 2014). RNA‐activated protein kinase‐like ER kinase (PERK), inositol‐requiring enzyme‐1 (IRE1), and activating transcription factor 6 (ATF6) make up three effector pathways of the UPR (Blázquez et al, 2014; Fu & Gao, 2014; Jheng et al, 2014). While little to no effect on PERK and ATF6 mRNA levels was detected in ZIKV‐infected cells (Fig 4D), we observed a 10‐fold increase in IRE1 mRNA levels (Fig 5D). Overall, our data demonstrate that ZIKV infection leads to massive, Sec61‐dependent rearrangements of the ER, including an activation of the UPR.
ZIKV replication and paraptosis‐like cell death
During viral infections and under other cellular stresses, autophagy and cell death programs are triggered when the UPR is insufficient to reestablish the ER steady state (Blázquez et al, 2014; Fu & Gao, 2014; Jheng et al, 2014). While flavivirus infection is often associated with apoptosis, one of the most characterized mechanisms of programmed cell death, other possibilities exist. Our attention was drawn to “paraptosis”, a form of cell death characterized by swelling of the ER and mitochondria as well as cytoplasmic vacuolization (Sperandio et al, 2000, 2004; Ranjan & Iwakumazr, 2016). Paraptosis is naturally observed during development and neurodegeneration and can be induced by expression of insulin‐like growth factor I receptor (IGF‐IR; Sperandio et al, 2000, 2004; Ranjan & Iwakumazr, 2016). Furthermore, paraptosis can be triggered in cancer cells through treatment with various anti‐tumor agents or cyclosporin A (CsA; Ram & Ramakrishna, 2014; Lee et al, 2016). These studies have indicated that paraptosis is dependent on the activation of phosphoinositide 3‐kinase (PI3K) and MAP kinases (Sperandio et al, 2004; Ram & Ramakrishna, 2014).
The cytoplasmic vacuoles observed in ZIKV‐infected cells and in cells treated with the paraptosis‐inducer CsA displayed similar morphology and in both cases were positively stained by a fluorescent ER marker (Fig 6A). To better understand the cellular pathways important for virus‐induced vacuole formation and cell death, we examined the effects of various kinase inhibitors. HeLa cells were treated with inhibitors 2 h following ZIKV exposure to rule out any effect on virus attachment and entry (Fig 6B). Two pan‐PI3K inhibitors, 3MA and wortmannin, the latter being reported to block paraptotic vacuoles (Ram & Ramakrishna, 2014), efficiently blocked vacuole formation following ZIKV infection. However, 3MA and wortmannin block both class‐1 and class‐3 PI3K (also known as Vps34), and the latter class has a well‐described role in autophagosome formation during the process of autophagy. Therefore, we tested other inhibitors exhibiting greater specificity: the class‐1‐specific PI3K inhibitor ZSTK474 (Kong & Yamori, 2007), the class‐3‐specific PI3K inhibitor SAR405 (Ronan et al, 2014), and the Akt‐specific inhibitor Triciribine (Akt V) (Yang et al, 2004). None of these inhibitors blocked ZIKV infection (Fig 6B). However, our results strongly suggest that the class‐1 PI3K/Akt signaling axis is involved in cytopathic vacuole formation in ZIKV‐infected cells (Fig 6C). In contrast, while inhibition of class‐3 PI3K diminished clustering of the autophagosome marker LC3 (Fig EV6A), it had no effect on vacuole formation in ZIKV‐infected cells (Fig 6C). Comparable levels of infection and vacuole formation are observed in cells in which LC3 is silenced, further excluding a role for autophagosomes in ZIKV‐induced cytopathy (Fig EV6B).
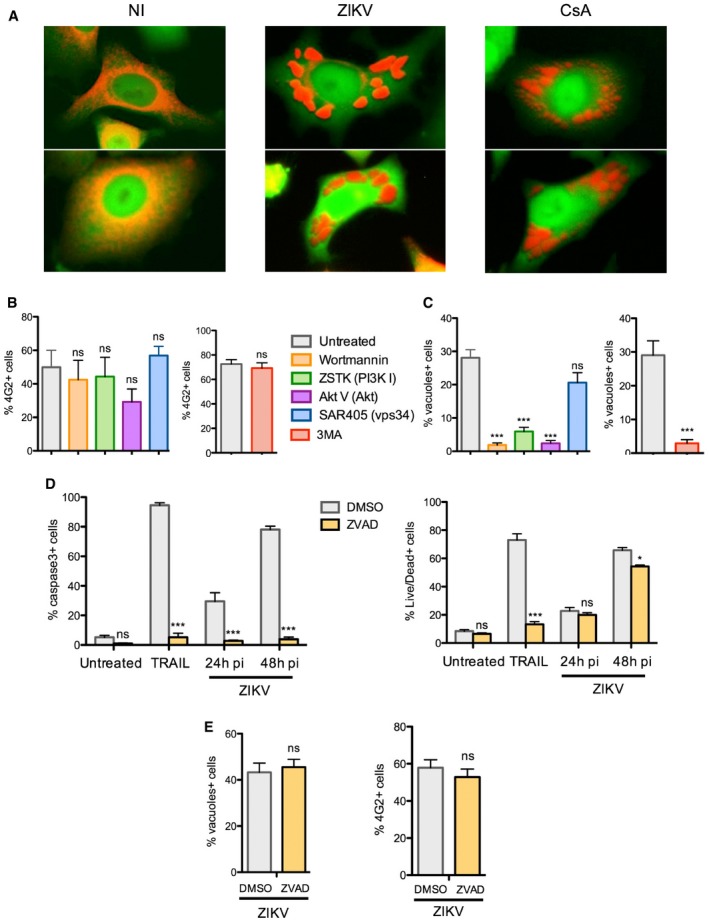
Figure 6. ZIKV triggers paraptosis‐like cell death.
-
AComparison of large cytoplasmic vacuoles induced by ZIKV or cyclosporine A. HeLa sh‐IFITM3 expressing the fluorescent ER‐tracker were infected with ZIKV HD 78 (MOI 1) or treated with cyclosporin A (10 μM) for 2 days and analyzed by time‐lapse microscopy. Two representative still images for each condition are shown.
-
B, CHeLa sh‐IFITM3 were infected with ZIKV HD 78 (MOI 1) for 24 h in the presence or absence of the pan PI3K inhibitor Wortmannin, the class 1‐specific PI3K inhibitor ZSTK474 (1 μM), the Akt inhibitor Triciribine (Akt V) (20 μM), the class 3‐specific PI3K (vps34) inhibitor SAR405 (1 μM), or the pan‐PI3K inhibitor 3MA (5 mM). (B) The percentage of E+ cells was quantified with 4G2 staining and flow cytometry. (C) In parallel, the proportion of cells displaying vacuoles was scored by visual examination of at least 200 cells. Results are mean ± SEM for three independent experiments. Statistical significance was determined using unpaired t‐tests. ***P < 0.001; ns, P > 0.05.
-
D, EHeLa sh‐IFITM3 were infected with ZIKV HD 78 (MOI 1) for 24 or 48 h in the presence or absence of the pan‐caspase inhibitor ZVAD‐FMK. (D) The percentage of cells expressing activated caspase‐3 (left panel) and the percentage of dying cells quantified with live/dead staining (right panel) were measured by flow cytometry. The apoptosis inducer TRAIL was used as a control. (E) The percentage of E+ cells was quantified with 4G2 staining and flow cytometry (left panel). In parallel, the proportion of cells displaying vacuoles was scored by visual examination of at least 200 cells (right panel). Results are mean ± SEM for three independent experiments. Statistical significance was determined using ANOVA and Bonferroni post‐tests (D) or unpaired t‐tests (E).***P < 0.001; *P < 0.05; ns, P > 0.05.
Figure EV6. Impact of LC3 on ZIKV‐induced vacuole formation.
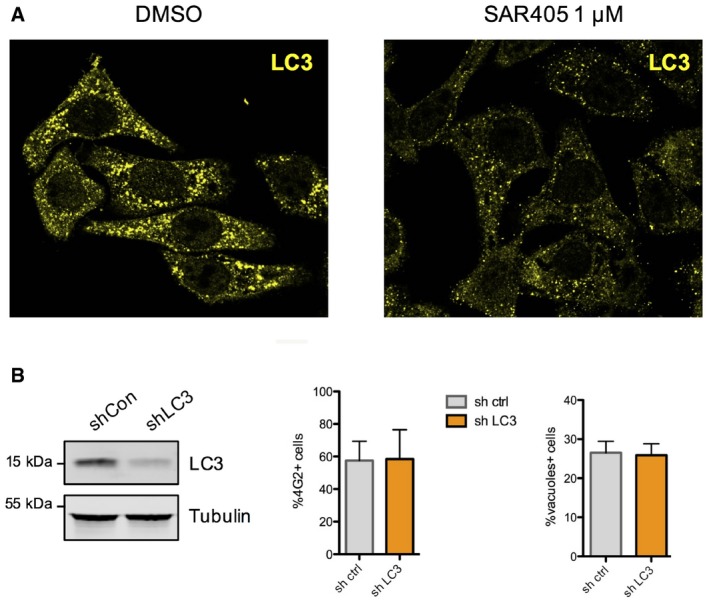
- The class 1 PI3K inhibitor blocks autophagy assessed by LC3 clustering in HeLa sh‐IFITM3 treated with rapamycin (5 mM) and concanamycin A (1 μM), which prevents fusion of phagosomes with lysosomes.
- HeLa cells expressing a shRNA control or a shRNA targeting LC3 were infected with ZIKV HD78 (moi 1) for 24 h, and the % of E+ cells as well as the % of vacuole‐containing cells were quantified by flow cytometry and microscopy, respectively. The efficiency of silencing was assessed by Western blot (left panel). Results are shown as mean ± SEM from three independent experiments.
Unlike apoptosis, paraptosis is unaffected by caspase inhibitors or Bcl‐2‐like anti‐apoptotic proteins (Sperandio et al, 2000, 2004; Ranjan & Iwakumazr, 2016). We thus tested the effect of a pan‐caspase inhibitor (ZVAD‐FMK) on ZIKV‐induced cytopathy. We observed an elevation of caspase‐3 activation in infected cells, which was inhibited by treatment with ZVAD‐FMK (Fig 6D). Nonetheless, the appearance of dead cells was not inhibited by this compound, as detected using a LIVE/DEAD staining reagent at 24 and 48 h pi (Fig 6D). We confirmed that ZVAD‐FMK inhibited caspase‐3 activation and cell death triggered by apoptosis inducer TRAIL (Fig 6D). In addition to the negligible effect of ZVAD‐FMK on ZIKV‐induced cell death, ZVAD‐FMK did not prevent productive viral infection or vacuolization following ZIKV exposure (Fig 6E).
Altogether, these results indicate that ZIKV induces a caspase‐independent, paraptosis‐like cell death, which can be prevented through inhibition of class‐I PI3K/Akt signaling.
Discussion
Interferon‐induced transmembrane proteins inhibit the replication and pathogenesis of a wide array of viruses, including flaviviruses. We confirm that IFITM3 overexpression blocks the replication of ZIKV (Savidis et al, 2016b). We further show that, in epithelial cells and primary dermal fibroblasts, IFITM3 is a key IFN‐induced host protein that prevents virus infection and subsequent cytopathic effects. IFITM3 inhibits an early step of the ZIKV cycle, as reflected by a reduced number of viral genomes in target cells following virus exposure. The role of IFITM proteins as inhibitors of virus entry is well established, yet further work is needed to determine whether IFITM3 prevents ZIKV–cell fusion events in endomembranes.
Removing endogenous IFITM3 unveiled a series of cytopathic events associated with ZIKV replication, including the formation of giant vacuoles in the cytoplasm. Infected cells tolerate these virus‐induced structures for a few hours before undergoing cytoplasmic collapse and succumbing to cell death. The formation of vacuolar structures has been reported in some cell types infected with WNV or other viruses such as chikungunya virus and poliovirus (Suhy et al, 2000; Chu & Ng, 2003; Hussain et al, 2016), but their origins have not been fully characterized. The results presented here suggest that cytopathic phenotypes induced by other viruses deserve more detailed descriptions.
The ZIKV‐induced vacuoles we describe stain positive for a calreticulin‐based fluorescent ER marker and for the ER‐resident enzyme PDI. Our ultrastructural analysis further indicates that they may, in part, originate from the nuclear membrane, which is contiguous with the ER. Genetic screens recently identified components of the ER as being required for flavivirus replication and cell death (Ma et al, 2015; Heaton et al, 2016; Marceau et al, 2016; Zhang et al, 2016). We also show that the Sec61 blocker mycolactone abrogates ZIKV replication, the formation of intracellular vacuoles, and cell death, indicating that events taking place in the ER drive this destructive pathway. Since ER membranes serve as scaffolds for viral protein synthesis and virion assembly (Paul & Bartenschlager, 2015; Ravindran et al, 2016), it is possible that the giant, ER‐derived vacuoles facilitate ZIKV production. While we did not observe large quantities of virus‐like particles inside the vacuoles, we show that ZIKV RNA and E protein concentrate at their borders, raising the possibility that they are important for an intermediate step of virus replication. Alternatively, vacuole formation and cell death following ZIKV infection may promote virus release and spread from infected cells.
Similar ER‐derived vacuoles have been observed in the absence of viral infection, revealing that ZIKV infection sets off a cell death program hard‐wired into cells (Ram & Ramakrishna, 2014; Lee et al, 2016). The virus‐induced structures highlighted here are highly reminiscent of those generated during so‐called paraptosis, a non‐apoptotic form of cell death (Sperandio et al, 2000, 2004; Ram & Ramakrishna, 2014; Lee et al, 2016; Ranjan & Iwakumazr, 2016). A previous report found that paraptosis could be prevented through inhibition of PI3K activity (Ram & Ramakrishna, 2014). Here we extend these findings by showing that ZIKV‐induced paraptosis‐like death requires class 1 but not class 3 PI3K activity, implicating an involvement of the PI3K/Akt pathway and ruling out a major role for autophagy. Using models of mice susceptible to ZIKV infection (Lazear et al, 2016), administration of class‐I PI3K inhibitors will help determine whether paraptosis‐like cytopathy is responsible for morbidity in vivo. Interestingly, we observe that caspase‐3 activation is detected among cells exposed to ZIKV. However, inhibition of caspase activity does not prevent cell death, strongly suggesting that apoptosis is not the major driving force for the cytopathic effects observed in ZIKV‐infected cells. Other questions that deserve further attention are whether paraptotic signaling suppresses or modulates apoptosis in ZIKV‐infected cells and whether apoptosis occurs among non‐infected bystander cells.
Paraptosis has been reported to occur in fish cells infected with iridovirus (Huang et al, 2015). To our knowledge, our findings represent the first description of this type of cell death being induced by a mammalian virus, not to mention a virus pathogenic to humans. Paraptosis‐like vacuoles were observed in several cell types (HeLa, primary fibroblasts, and astrocytes) infected with various ZIKV strains. However, infection with one DENV strain did not trigger a similar cytopathic effect. It will be worth testing other DENV strains as well as other flavivirus types, in order to determine whether paraptosis‐like cytopathy is a selective feature of ZIKV. It will also be of interest to determine whether individual viral proteins play a role in initiating paraptosis.
It has been reported that a program resembling autophagy is triggered in ZIKV‐infected cells (Blázquez et al, 2014; Hamel et al, 2015; Souza et al, 2016). Importantly, the virus‐induced vacuoles described here do not stain positive for LC3 or LAMP1, suggesting that they do not correspond to autophagosomes or autophagolysosome fusions. Furthermore, they are not loaded with neutral lipids, excluding their role as lipid droplets observed in some DENV‐ or HCV‐infected cells (Paul & Bartenschlager, 2015). Nonetheless, future work will shed light on how genuine autophagy may impact paraptosis and if these processes are cooperative or antagonistic.
While the cytopathic events described here were detectable in cells containing IFITM3, these phenomena were much more apparent upon IFITM3 silencing. This suggests that once incoming ZIKV virions have surmounted the intracellular barrier mediated by IFITM3, a pathway leading to ER stress and cell death is triggered. As such, the proportion of dying cells following exposure to ZIKV was much lower when IFITM3 was present. Thus, our results suggest that IFITM3 is a major component of host innate control of ZIKV and is likely implicated in the low pathogenicity observed in most infected individuals (Lessler et al, 2016). IFITM3 is required to prevent pathogenesis of IAV or WNV infections in mice (Everitt et al, 2012; Gorman et al, 2016). A single nucleotide polymorphism (SNP) (rs12252‐C) in human IFITM3 is predicted to produce a truncated form of IFITM3, which is confined to the plasma membrane (Everitt et al, 2012). Since this polymorphism may be associated with severe outcomes following IAV infection (Everitt et al, 2012; Zhang et al, 2013), it will be worth examining whether the severity of ZIKV infection is associated with polymorphisms in IFITM genes.
Materials and Methods
Cells, lentivectors, and viruses
293T cells (ATCC) stably expressing IFITM proteins were established via transfection with pQCXIP plasmids containing amino‐terminal FLAG‐tagged IFITM sequences and puromycin selection. HeLa cells (ATCC) stably expressing shRNA were established via transduction with a pGIPZ‐GFP‐based lentivector expressing shRNA clones (scrambled and IFITM3 (V3LHS_325106), Thermo Scientific). HeLa cells stably expressing a fluorescent, calreticulin‐based ER marker were established via transfection with pDsRed2‐ER Vector (Clontech 632409) and G418 selection. Primary human dermal fibroblasts‐adult (HDFa) and human foreskin fibroblasts (HFF) were purchased from ATCC. Normal human astrocytes were purchased from Lonza and grown in AGM™ Astrocyte Growth Medium (AGM BulletKit CC‐3187 & CC‐4123). HeLa cells stably expressing a control shRNA or shRNA for LC3 were previous described (Coulon et al, 2016).
ZIKV strains HD78788, PF13, and NC14 were previously described (Hamel et al, 2015; Barba‐Spaeth et al, 2016; Chouin‐Carneiro et al, 2016). The dengue virus strain DENV2 was previously described (Medina et al, 2015). Virus stocks were produced on Vero cells or C6/36 cells and tittered on Vero cells using a TCID50 assay. Cells were seeded 1 day prior to infection and overlaid with ZIKV or DENV2 at the desired multiplicity of infection (MOI) in a final volume of 200 μl. After 2 h at 37°C, the inoculum was removed and replaced with fresh medium.
Flow cytometry analysis
The following antibodies were used: anti‐IFITM3 (Proteintech, 11714‐1‐AP), anti‐FLAG M2 (Sigma), anti‐E protein (4G2, purified from the ATCC hybridoma), anti‐caspase‐3 (BD Pharmingen 559341). For stainings, cells were fixed with 4% PFA and permeabilized with 0.1% Triton X‐100, followed by 30‐min incubations with primary and secondary antibodies. For cell death staining, LIVE/DEAD® Fixable Aqua Dead Cell Stain kit (ThermoFisher L34965) was used. Cells were stained for 30 min in PBS prior to fixation with 4% PFA. Samples were acquired with a FACS Canto II (Becton Dickinson), and data were analyzed in FlowJo.
Western blotting
Cells were lysed in 1% Triton X‐100/PBS in the presence of protease inhibitor cocktail, and 10–20 μg of protein lysate was loaded into 12% Bis‐Tris SDS–PAGE gels. The following antibodies were used: IFITM1 (Proteintech, 60074‐1‐Ig), IFITM2 (Proteintech, 66137‐1‐Ig), IFITM3 (Abcam, EPR5242/ab109429), actin (Santa Cruz Biotechnology), tubulin (Santa Cruz Biotechnology), and LC3 (MBL International PM036). DyLight‐coupled secondary antibodies (Thermo Fisher) were used for protein detection on a Li‐Cor Odyssey imaging system.
Time‐lapse microscopy
HeLa or HDFa cells were plated in Hi‐Q4 microdishes (10,000 cells per chamber) (Ibidi). The following day, cells were infected with ZIKV at the indicated MOI in the presence of propidium iodide (10 μg/ml, Molecular Probes, Life Technologies, P3566). Transmission and fluorescence images were taken every 5 or 10 min for up to 96 h using a Nikon Biostation IMQ, with three fields captured simultaneously for each condition. Images were analyzed in FIJI.
Confocal immunofluorescence microscopy
HeLa cells stably expressing pGIPZ‐shRNA‐GFP were plated and infected with ZIKV at MOI 1 for 24 h. The cells were then washed, fixed, permeabilized with Triton X‐100 0.1%, and stained with 4G2, anti‐IFITM3 (Proteintech, 11714‐1‐AP), anti‐LC3 antibody (MBL International PM036), and anti‐PDI (Abcam, RL90/ab2792). Confocal microscopy was carried out on a Zeiss LSM700 using a 63× objective. Representative medial sections or combined Z‐stacks are shown as indicated. Images were analyzed in FIJI.
Fluorescent in situ hybridization
HeLa sh‐IFITM3 (10,000 cells per well) were seeded into 96‐well glass bottom plates (Eppendorf) and infected with ZIKV HD78 at an MOI of 1 for 24 h. Cells were washed with PBS, fixed with 4% PFA for 30 min at room temperature, and permeabilized with 0.5% Triton X‐100 in PBS. FISH was performed using the QuantiGene ViewRNA ISH Assay Kit (Affymetrix) according to the manufacturer's instructions. Viral plus strand RNA was detected using a probe (Affymetrix) designed to specifically hybridize ZIKV HD78 RNA. Cellular DNA was stained with Nucblue (Thermo Fisher). Images were acquired with an inverted confocal microscope (Zeiss LSM700) using a 63× magnification and analyzed in FIJI.
Transmission electron microscopy
HeLa sh‐IFITM3 cells were fixed for 24 h in 4% PFA and 1% glutaraldehyde (Sigma) in 0.1 M phosphate buffer (pH 7.2). Cells were washed in PBS and post‐fixed with 2% osmium tetroxide (Agar Scientific) for 1 h. Cells were fully dehydrated in a graded series of ethanol solutions and propylene oxide. The impregnation step was performed with a mixture of (1:1) propylene oxide/Epon resin (Sigma) and left overnight in pure resin. Cells were then embedded in resin blocks, which were allowed to polymerize for 48 h at 60°C. Ultra‐thin sections (70 nm) of blocks were obtained with a Leica EM UC7 ultramicrotome (Wetzlar). Sections were stained with 5% uranyl acetate (Agar Scientific) and 5% lead citrate (Sigma), and observations were made with a JEOL 1011 transmission electron microscope.
Real‐time qRT–PCR
Total RNA was extracted from cells using Qiamp RNeasy extraction kit (Qiagen). 500 ng of RNA was used for cDNA synthesis using SuperScript II reverse transcriptase (Life Technologies) in an Eppendorf EP Mastercycler Gradient S thermocycler. The following primers were used to amplify viral cDNA as described (Meertens et al, 2017): forward‐AARTACACATACCARAACAAAGTGGT; reverse‐ TCCRCTCCCYCTYTGGTCTTG. cDNAs corresponding to cellular transcripts were amplified using the following primers: IRE‐1, forward‐AGAGAAGCAGCAGACTTTGTC, reverse‐GTTTTGGTGTCGTACATGGTGA; ATF6, forward‐GACAGTACCAACGCTTATGCC, reverse‐CTGGCCTTTAGTGGGTGCAG; PERK, forward‐GGAAACGAGAGCCGGATTTATT, reverse‐ACTATGTCCATTATGGCAGCTTC. cDNA amplification was performed by qPCR using 500 nM of each primer, 25 ng of cDNA, and 10 μl of SYBR Green. An activation step of 15 min at 95°C was followed by 40 amplification cycles of 95°C for 15 s, 60°C for 20 s, and 72°C for 30 s. Viral RNA was quantified by comparing each sample's threshold cycle (C T) value with a ZIKV RNA standard curve, obtained by limiting dilution of a vector encoding the target sequence of the primers. Levels of cellular transcripts were normalized to GAPDH. Results are expressed as fold induction relative to the non‐infected condition.
Kinase inhibitor treatment
HeLa sh‐IFITM3 cells were incubated with ZIKV at an MOI of 1. After 2 h at 37°C, the inoculum was removed and fresh medium containing or not 250 nM of wortmannin (SIGMA W1628), 1 μM of ZSTK474 (SelleckChem S1072), 20 μM of Akt Inhibitor V Triciribine (Calbiochem 124012), 1 μM of SAR405 (Clinisciences A8883), or 5 mM of 3‐methyladenine (3MA) (Sigma M9281) was added. After 24 h, cells were collected and analyzed by flow cytometry and microscopy.
Cell death analysis
HeLa sh‐IFITM3 cells were infected with ZIKV at an MOI of 1. After 2 h at 37°C, the inoculum was removed and replaced with fresh medium containing or not 20 μM of the pan‐caspase inhibitor ZVAD‐FMK (Sigma). Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) (Sigma) was used at 0.2 μg/ml as a positive control. After 24 h and 48 h, cells were collected and analyzed by flow cytometry for activated caspase‐3 expression, LIVE/DEAD staining, and viral protein E expression. Vacuole formation was quantified by microscopy.
RNAi in primary HFDa cells
HDFa cells were seeded in 24‐well plates (30,000 cells per well) and transfected with 100 nM siRNA targeting IFITM3 (Silencer Select, Ambion; s195035) using Lipofectamine RNAiMAX (Thermo Fisher). After 72 h, IFITM3 protein levels were assessed by flow cytometry and cells were infected with ZIKV.
ZIKV–cell attachment assay
HeLa cells (sh‐SCR and sh‐IFITM3) were incubated for 60 min at 4°C with different MOI of ZIKV. Cells were washed twice with PBS‐BSA 1% and stained for 1 h with 4G2 antibody at 4°C followed by a goat anti‐mouse Alexa Fluor 647 antibody and LIVE/DEAD aqua (Life Technologies). Cells were then fixed and analyzed by flow cytometry.
Blockade of Sec61 translocon with mycolactone
HeLa cells (sh‐IFITM3) were infected with ZIKV at an MOI of 1. After 2 h at 37°C, the inoculum was removed and replaced with fresh medium containing or not mycolactone (20 nM; Guenin‐Mace et al, 2015) or 0.5 μg/ml of Brefeldin A (Sigma). At 24 h, cells were fixed, stained, and analyzed by flow cytometry and confocal microscopy. 293T cells expressing Dox‐inducible wild‐type or mutant Sec61 (Baron et al, 2016) were treated with 500 ng/ml doxycycline for 24 h and infected with ZIKV in the presence or absence of mycolactone for an additional 24 h. Cells were then fixed, stained, and analyzed by flow cytometry.
Induction and inhibition of autophagy
To induce autophagy and allow visualization of LC3 clustering, cells were incubated with medium containing rapamycin (20 μM, InSolution, CalBiochem) and concanamycin A (1 μM C9705, Sigma) for 4 h at 37°C. To inhibit the induction of autophagy, cells were pretreated with SAR405 (1 μM) for 24 h prior to treatment with rapamycin/concanamycin A. LC3 proteins levels were assessed by Western blot and confocal microscopy.
Statistical analysis
Statistical analysis was performed using Prism, with tests indicated in figure legends.
Author contributions
BM, AAC, and OS conceived the study. All authors performed experiments and/or participated in the experimental design. BM, AAC, and OS wrote and edited the manuscript. All authors approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Review Process File
Acknowledgements
We thank members of the Virus & Immunity Unit for discussions. AC was supported from postdoctoral fellowships from the Pasteur Foundation and ANRS. Work was supported by grants from the “CHIKV‐Viro‐Immuno” ANR‐14‐CE14‐0015‐01 and “TIMTAMDEN” ANR‐14‐CE14‐0029 projects, the Labex IBEID program ANR‐10‐IHUB‐0002, the Labex VRI program ANR‐10‐LABX‐77 and Institut Pasteur1. We thank Van‐Mai Cao‐Lormeau, Institut Louis Malardé (French Polynesia) and Myrielle Dupont‐Rouzeyrol (Institut Pasteur Nouvelle Calédonie) for the gift of reagents. We thank Stephane Petres (Institut Pasteur) for preparation of the 4G2 antibody and the Imagopole (Institut Pasteur) for help in image acquisition.
The EMBO Journal (2017) 36: 1653–1668
Footnotes
Correction added on 14 June 2017, after first online publication: the Acknowledgements section has been updated.
Contributor Information
Alex A Compton, Email: alex.compton@pasteur.fr.
Olivier Schwartz, Email: schwartz@pasteur.fr.
References
- Amini‐Bavil‐Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU (2013) The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13: 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba‐Spaeth G, Dejnirattisai W, Rouvinski A, Vaney MC, Medits I, Sharma A, Simon‐Loriere E, Sakuntabhai A, Cao‐Lormeau VM, Haouz A, England P, Stiasny K, Mongkolsapaya J, Heinz FX, Screaton GR, Rey FA (2016) Structural basis of potent Zika‐dengue virus antibody cross‐neutralization. Nature 536: 48–53 [DOI] [PubMed] [Google Scholar]
- Baron L, Paatero AO, Morel JD, Impens F, Guenin‐Mace L, Saint‐Auret S, Blanchard N, Dillmann R, Niang F, Pellegrini S, Taunton J, Paavilainen VO, Demangel C (2016) Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med 213: 2885–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr, Cherry S, Sadovsky Y, Coyne CB (2016) Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 19: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard E, Roingeard P (2015) Virus‐induced double‐membrane vesicles. Cell Microbiol 17: 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blázquez A‐BB, Escribano‐Romero E, Merino‐Ramos T, Saiz J‐CC, Martín‐Acebes MA (2014) Stress responses in flavivirus‐infected cells: activation of unfolded protein response and autophagy. Front Microbiol 5: 266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139: 1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao‐Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, Vial AL, Decam C, Choumet V, Halstead SK, Willison HJ, Musset L, Manuguerra JC, Despres P, Fournier E, Mallet HP (2016) Guillain‐barre syndrome outbreak associated with Zika virus infection in French Polynesia: a case‐control study. Lancet 387: 1531–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette‐Artur P, Eyrolle‐Guignot D, Salje H, Van Kerkhove MD, Abadie V, Garel C, Fontanet A, Mallet HP (2016) Association between Zika virus and microcephaly in French Polynesia, 2013‐15: a retrospective study. Lancet 387: 2125–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarino NM, McMichael TM, Yount JS (2014) Regulation of the trafficking and antiviral activity of IFITM3 by post‐translational modifications. Future Microbiol 9: 1151–1163 25405885 [Google Scholar]
- Chouin‐Carneiro T, Vega‐Rua A, Vazeille M, Yebakima A, Girod R, Goindin D, Dupont‐Rouzeyrol M, Lourenco‐de‐Oliveira R, Failloux AB (2016) Differential Susceptibilities of Aedes aegypti and Aedes albopictus from the Americas to Zika Virus. PLoS Negl Trop Dis 10: e0004543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JJH, Ng ML (2003) The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol 84: 3305–3314 [DOI] [PubMed] [Google Scholar]
- Compton AA, Bruel T, Porrot F, Mallet A, Sachse M, Euvrard M, Liang C, Casartelli N, Schwartz O (2014) IFITM proteins incorporated into HIV‐1 virions impair viral fusion and spread. Cell Host Microbe 16: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton AA, Roy N, Porrot F, Billet A, Casartelli N, Yount JS, Liang C, Schwartz O (2016) Natural mutations in IFITM3 modulate post‐translational regulation and toggle antiviral specificity. EMBO Rep 17: 1657–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon PG, Richetta C, Rouers A, Blanchet FP, Urrutia A, Guerbois M, Piguet V, Theodorou I, Bet A, Schwartz O, Tangy F, Graff‐Dubois S, Cardinaud S, Moris A (2016) HIV‐infected dendritic cells present endogenous MHC class II‐restricted antigens to HIV‐specific CD4+ T cells. J Immunol 197: 517–532 [DOI] [PubMed] [Google Scholar]
- Danthi P (2015) Viruses and the diversity of cell death. Annu Rev Virol 3: 1–21 [DOI] [PubMed] [Google Scholar]
- Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB (2014) IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus‐endosome hemifusion. PLoS Pathog 10: e1004048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Farzan M (2013) The broad‐spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13: 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowall SD, Graham VA, Rayner E, Atkinson B, Hall G, Watson RJ, Bosworth A, Bonney LC, Kitchen S, Hewson R (2016) A susceptible mouse model for zika virus infection. PLoS Negl Trop Dis 10: e0004658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt A, Clare S, Pertel T, John S, Wash R, Smith S, Chin C, Feeley E, Sims J, Adams D (2012) IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumence E, Roche M, Krejbich‐Trotot P, El‐Kalamouni C, Nativel B, Rondeau P, Misse D, Gadea G, Viranaicken W, Despres P (2016) The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN‐beta production and apoptosis induction. Virology 493: 217–226 [DOI] [PubMed] [Google Scholar]
- Fu XL, Gao DS (2014) Endoplasmic reticulum proteins quality control and the unfolded protein response: the regulative mechanism of organisms against stress injuries. BioFactors 40: 569–585 [DOI] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK (2016) Zika virus impairs growth in human neurospheres and brain organoids. Science 352: 816–818 [DOI] [PubMed] [Google Scholar]
- Ghosh Roy S, Sadigh B, Datan E, Lockshin RA, Zakeri Z (2014) Regulation of cell survival and death during Flavivirus infections. World J Biol Chem 5: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman MJ, Poddar S, Farzan M, Diamond MS (2016) The interferon‐stimulated gene IFITM3 restricts West Nile virus infection and pathogenesis. J Virol 90: 8212–8225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenin‐Mace L, Baron L, Chany AC, Tresse C, Saint‐Auret S, Jonsson F, Le Chevalier F, Bruhns P, Bismuth G, Hidalgo‐Lucas S, Bisson JF, Blanchard N, Demangel C (2015) Shaping mycolactone for therapeutic use against inflammatory disorders. Sci Transl Med 7: 289ra285 [DOI] [PubMed] [Google Scholar]
- Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, Simmonds RE (2014) The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 10: e1004061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera‐Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao‐Lormeau VM, Choumet V, Briant L, Desprès P, Amara A, Yssel H, Missé D (2015) Biology of Zika virus infection in human skin cells. J Virol 89: 8880–8896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Randall G (2010) Dengue virus‐induced autophagy regulates lipid metabolism. Cell Host Microbe 8: 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Moshkina N, Fenouil R, Gardner TJ, Aguirre S, Shah PS, Zhao N, Manganaro L, Hultquist JF, Noel J, Sachs D, Hamilton J, Leon PE, Chawdury A, Tripathi S, Melegari C, Campisi L, Hai R, Metreveli G, Gamarnik AV et al (2016) Targeting viral proteostasis limits influenza virus, HIV, and Dengue virus infection. Immunity 44: 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Huang X, Yang Y, Wang W, Yu Y, Qin Q (2015) Involvement of fish signal transducer and activator of transcription 3 (STAT3) in nodavirus infection induced cell death. Fish Shellfish Immunol 43: 241–248 [DOI] [PubMed] [Google Scholar]
- Hussain KM, Lee RC, Ng MM, Chu JJ (2016) Establishment of a novel primary human skeletal myoblast cellular model for chikungunya virus infection and pathogenesis. Sci Rep 6: 21406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jheng J‐R, Ho J‐Y, Horng J‐T (2014) ER stress, autophagy, and RNA viruses. Front Microbiol 5: 388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R, Xu F, Qian J, Yao Y, Miao C, Zheng YM, Liu SL, Guo F, Geng Y, Qiao W, Liang C (2014) Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell Microbiol 16: 1080–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalies KU, Romisch K (2015) Inhibitors of protein translocation across the ER membrane. Traffic 16: 1027–1038 [DOI] [PubMed] [Google Scholar]
- Kong D, Yamori T (2007) ZSTK474 is an ATP‐competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci 98: 1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS (2016) A mouse model of zika virus pathogenesis. Cell Host Microbe 19: 720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Kim IY, Saha S, Choi KS (2016) Paraptosis in the anti‐cancer arsenal of natural products. Pharmacol Ther 162: 120–133 [DOI] [PubMed] [Google Scholar]
- Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, Carcelen AC, Ott CT, Sheffield JS, Ferguson NM, Cummings DA, Metcalf CJ, Rodriguez‐Barraquer I (2016) Assessing the global threat from Zika virus. Science 353: aaf8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Markosyan RM, Zheng YM, Golfetto O, Bungart B, Li M, Ding S, He Y, Liang C, Lee JC, Gratton E, Cohen FS, Liu SL (2013) IFITM proteins restrict viral membrane hemifusion. PLoS Pathog 9: e1003124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Pan Q, Rong L, He W, Liu SL, Liang C (2011) The IFITM proteins inhibit HIV‐1 infection. J Virol 85: 2126–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi J‐GG, Shi G, Qi L, Manjunath N, Wu H (2015) A CRISPR‐based screen identifies genes essential for west‐nile‐virus‐induced cell death. Cell Rep 12: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi Y, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE (2016) Genetic dissection of Flaviviridae host factors through genome‐scale CRISPR screens. Nature 535: 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M, Simmonds RE, High S (2016) Mechanistic insights into the inhibition of Sec61‐dependent co‐ and post‐translational translocation by mycolactone. J Cell Sci 129: 1404–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina FA, Torres‐Malavé G, Chase AJ, Santiago GA, Medina JF, Santiago LM, Muñoz‐Jordán JL (2015) Differences in type I interferon signaling antagonism by dengue viruses in human and non‐human primate cell lines. PLoS Negl Trop Dis 9: e0003468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet‐Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao‐Lormeau V‐M, Coulpier M, Missé D, Jouvenet N, Tabibiazar R, Gressens P, Schwartz O, Amara A (2017) Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep 18: 324–333 [DOI] [PubMed] [Google Scholar]
- Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS (2016) Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165: 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D, Gubler DJ (2016) Zika virus. Clin Microbiol Rev 29: 487–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval‐Espinosa C, Bershteyn M, Kriegstein AR (2016) Expression analysis highlights AXL as a candidate zika virus entry receptor in neural stem cells. Cell Stem Cell 18: 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D, Bartenschlager R (2015) Flaviviridae replication organelles: Oh, what a tangled web we weave. Annu Rev Virol 2: 289–310 [DOI] [PubMed] [Google Scholar]
- Petersen LR, Jamieson DJ, Powers AM, Honein MA (2016) Zika virus. N Engl J Med 374: 1552–1563 [DOI] [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O'Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS (2016) Zika virus infects human placental macrophages. Cell Host Microbe 20: 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram BM, Ramakrishna G (2014) Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim Biophys Acta 1843: 2497–2512 [DOI] [PubMed] [Google Scholar]
- Ranjan A, Iwakumazr T (2016) Non‐canonical cell death induced by p53. Int J Mol Sci 17: 2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran MS, Bagchi P, Cunningham CN, Tsai B (2016) Opportunistic intruders: how viruses orchestrate ER functions to infect cells. Nat Rev Microbiol 14: 407–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, Marquette JP, El‐Ahmad Y, Filoche‐Romme B, Schio L, Garcia‐Echeverria C, Goulaouic H, Pasquier B (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC (2016) Characterization of a novel murine model to study zika virus. Am J Trop Med Hyg 94: 1362–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, Guo Z, Green S, Kowalik TF, Brass AL (2016a) Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16: 232–246 [DOI] [PubMed] [Google Scholar]
- Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL (2016b) The IFITMs inhibit zika virus replication. Cell Rep 15: 2323–2330 [DOI] [PubMed] [Google Scholar]
- Smith S, Weston S, Kellam P, Marsh M (2014) IFITM proteins‐cellular inhibitors of viral entry. Curr Opin Virol 4: 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, Figueira CP, Paredes BD, Nonaka CK, Azevedo CM, Rocha VP, Bandeira AC, Mendez‐Otero R, Dos Santos RR, Soares MB (2016) Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep 6: 39775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio S, de Belle I, Bredesen DE (2000) An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci 97: 14376–14381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, Bredesen DE (2004) Paraptosis: mediation by MAP kinases and inhibition by AIP‐1/Alix. Cell Death Differ 11: 1066–1075 [DOI] [PubMed] [Google Scholar]
- Suhy DA, Giddings TH Jr, Kirkegaard K (2000) Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy‐like origin for virus‐induced vesicles. J Virol 74: 8953–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanmanee S, Luplertlop N (2016) Immunopathogenesis of dengue virus‐induced redundant cell death: apoptosis and pyroptosis. Viral Immunol 30: 13–19 [DOI] [PubMed] [Google Scholar]
- Tabata T, Petitt M, Puerta‐Guardo H, Michlmayr D, Wang C, Fang‐Hoover J, Harris E, Pereira L (2016) Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 20: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, Christian KM, Didier RA, Jin P, Song H, Ming GL (2016) Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18: 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartour K, Appourchaux R, Gaillard J, Nguyen XN, Durand S, Turpin J, Beaumont E, Roch E, Berger G, Mahieux R, Brand D, Roingeard P, Cimarelli A (2014) IFITM proteins are incorporated onto HIV‐1 virion particles and negatively imprint their infectivity. Retrovirology 11: 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Costa F, Garcia‐Blanco MA, Ko AI, Ribeiro GS, Saade G, Shi PY, Vasilakis N (2016) Zika virus: history, emergence, biology, and prospects for control. Antiviral Res 130: 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Shan C, Shi PY (2016) Restriction of zika virus by host innate immunity. Cell Host Microbe 19: 566–567 [DOI] [PubMed] [Google Scholar]
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ (2004) Akt/protein kinase B signaling inhibitor‐2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 64: 4394–4399 [DOI] [PubMed] [Google Scholar]
- Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL (2015) IFITM proteins restrict HIV‐1 infection by antagonizing the envelope glycoprotein. Cell Rep 13: 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Zhao Y, Li N, Peng YC, Giannoulatou E, Jin RH, Yan HP, Wu H, Liu JH, Liu N, Wang DY, Shu YL, Ho LP, Kellam P, McMichael A, Dong T (2013) Interferon‐induced transmembrane protein‐3 genetic variant rs12252‐C is associated with severe influenza in Chinese individuals. Nat Commun 4: 1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS (2016) A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535: 164–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, He Z, Yuan J, Wen W, Huang X, Hu Y, Lin C, Pan J, Li R, Deng H, Liao S, Zhou R, Wu J, Li J, Li M (2015) IFITM3‐containing exosome as a novel mediator for anti‐viral response in dengue virus infection. Cell Microbiol 17: 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Review Process File