

Abstract
PURPOSE: Inflammatory breast cancer (IBC) is arguably the deadliest form of breast cancer due to its rapid onset and highly invasive nature. IBC carries 5- and 10-year disease-free survival rates of ~45% and <20%, respectively. Multiple studies demonstrate that in comparison with conventional breast cancer, IBC has a unique molecular identity. Here, we have identified platelet-derived growth factor receptor alpha (PDGFRA) as being uniquely expressed and active in IBC patient tumor cells. EXPERIMENTAL DESIGN: Here we focus on characterizing and targeting PDGFRA in IBC. Using gene expression, we analyzed IBC patient samples and compared them with non-IBC patient samples. Further, using IBC cells in culture, we determined the effect of small molecules inhibitors in both in vitro and in vivo assays. RESULTS: In IBC patients, we show more frequent PDGFRA activation signature than non-IBC samples. In addition, the PDGFRA activation signature is associated with shorter metastasis-free survival in both uni- and multivariate analyses. We also demonstrate that IBC cells express active PDGFRA. Finally, we show that PDGFRA targeting by crenolanib (CP-868-596), but not imatinib (STI571), two small molecule inhibitors, interferes with IBC cell growth and emboli formation in vitro and tumor growth in vivo. CONCLUSIONS: Our data suggest that PDGFRA may be a promising target for therapy in IBC.
Introduction
Inflammatory breast cancer (IBC) is arguably the most aggressive form of epithelial breast cancer. By the current Surveillance, Epidemiology and End Results Program estimates, IBC accounts for 1% to 3% of breast cancers in the United States, yet IBC is responsible for ~10% of breast cancer–related deaths [1], [2]. Most IBC physicians, researchers, and advocates estimate that the actual incidence of IBC is higher, potentially accounting for up to 10% of total breast cancers [3]. One main distinctive feature of IBC is the absence of a palpable mass in the breast [2]. As a result, IBC is diagnosed clinically by characteristic changes in the breast, which include “peau d'orange,” erythema, edema, swelling, and pain [1], [4]. Pathological findings indicate the presence of tumor emboli in the dermal and parenchymal lymphatic vessels of the skin overlying the breast [5]. Because of this, nearly 100% of IBC patients have axillary lymph node involvement and approximately one third of the patients show gross distant metastasis in organs such as lung, liver, and bone at first clinical presentation [6]. The lymphoangiogenic nature and tendency to invade dermal lymphatic vessels contribute significantly to the metastatic nature of this disease [7]. Undoubtedly, the metastatic behavior of IBC accounts for the poor clinical outcome with a current 5-year survival rate of less than 45%, much lower than that of conventional breast cancer [6].
Along with a distinctive clinical presentation, IBC has a molecular profile that is unique compared to other forms of breast cancer, which can be exploited to identify new therapeutic targets and improve patient survival [8], [9]. In a previous study, we observed that platelet-derived growth receptor alpha (PDGFRA), but not platelet-derived growth factor receptor beta (PDGFRB), is overexpressed in IBC compared to non-IBC patient samples [10]. In a separate study, we reported that a number of PDGFR-specific transcription factors were activated in IBC in a subtype-independent manner [11]. Like many receptor tyrosine kinases (RTKs), PDGFRA is involved in the progression of a variety of cancers, such as leukemias, gastrointestinal stromal tumors (GISTs), glioblastoma, and hepatocellular carcinoma, either by overexpression or by increased activity due, for example, to mutation or chromosomal rearrangement [12], [13], [14], [15].
Inhibition by small molecules directed to the ATP-binding site has been recognized as a way to therapeutically target these RTKs [16]. Imatinib (STI571) is routinely used as a treatment for several PDGFRA-expressing cancers such as GISTs and dermatofibrosarcoma protuberans by targeting the activated form of the receptor [17], [18]. However, tumors harboring a constitutively active mutation in PDGFRA are resistant to imatinib [19], [20]. Crenolanib (CP-868-596) is reported to be significantly more potent and effective against imatinib-resistant PDGFRA harboring activating D842I, D842V, D842Y, D1842-843IM, and deletion I843 mutations [21].
In the present study, we identified overexpressed and active PDGFRA pathway in IBC tumors and demonstrated sensitivity to crenolanib but not to imatinib. Crenolanib treatment induced a G2 cell cycle arrest of IBC cells and reduced emboli formation in a unique in vitro three-dimensional model system. Finally, in an orthotopic in vivo model, targeting PDGFRA with crenolanib significantly prevented IBC tumor growth. Our results suggest that PDGFRA is a viable target for treating IBC.
Materials and Methods
Cells and Materials
The SUM149 IBC cell line was grown in Ham's F12 medium (Mediatech Inc., Manassas, VA) supplemented with 5% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 1% penicillin-streptomycin (Mediatech Inc., Manassas VA), 1% antibiotic-antimycotic (Mediatech Inc., Manassas, VA), 1% L-glutamine (Mediatech Inc., Manassas, VA), 0.1% hydrocortisone (Sigma Aldrich, St. Louis, MO), 1% ITS cocktail (Mediatech Inc., Manassas VA), or 1% insulin (Sigma Aldrich, St. Louis, MO), 0.55% transferrin (Sigma Aldrich, St. Louis, MO), and 0.00067% selenium (Sigma Aldrich, St. Louis, MO) as described [10], [22], [23]. The KPL-4 cell line was kindly provided by Dr. Junichi Kurebayshi, and both the KPL-4 and MDA-MB-231 lines were grown in DMEM (Mediatech) supplemented with 10% FBS (Atlanta Biologicals). Human mammary epithelial cells (HMECs) were grown in 5% FBS (Atlanta Biologicals)-supplemented Ham's F-12 medium (Mediatech, Inc.) containing insulin, hydrocortisone, epidermal growth factor, and cholera toxin (Sigma Chemical Co.). Imatinib (STI571) was purchased from LC Laboratories (Woburn, MA), and crenolanib was provided under a materials transfer agreement by Arog Pharmaceuticals LLC (Dallas, TX).
Generation of a Predictive model for PDGFRA Activation
In order to construct a PDGFRA activation predictive model, public expression series from patients with GISTs with and without activating PDGFRA mutations were retrieved from the National Center for Biotechnology Information/Genbank GEO and ArrayExpress databases and authors' website [24], [25], [26], [27], [28], [29], [30], [31], [32]. An overview of the incorporated data sets is provided in Supplemental Table 1. The final pooled data set included 324 nonredundant GIST samples with PDGFRA mutational status available. Data preprocessing was performed as follows. The first step was to normalize each set separately: we used quantile normalization for the available processed data from non–Affymetrix-based sets, and Robust Multichip Average with the nonparametric quantile algorithm for the raw data from the Affymetrix-based data sets [33]. Normalization was done in R using Bioconductor and associated packages. Then, probe sets were mapped based on their EntrezGeneID. When multiple probes mapped to the same GeneID, we retained the one with the highest variance in a particular dataset.
Supplemental Figure 1.

Box-plot of mRNA expression of PDGFs according to the predicted PDGFRA activation status in breast cancers. (A) mRNA expression of PDGF-A in all 389 breast cancer samples according to the predicted PDGFRA activation status (“activated” versus “not activated”). (B-D) Similar to A, but for PDGF-B, PDGF-C, and PDGF-D, respectively. The P values (Student's t test) are .37, 1.3E-07, 2.6E-08, and .15, respectively.
The predictive model for PDGFRA activation was built using supervised analysis applied to the Ostrowski's set as learning set and the other pooled sets as validation set. In the learning set, we compared the expression profiles of 10,380 filtered genes between tumors with (N = 11) versus without (N = 18) activating PDGFRA mutation using Significance Analysis of Microarrays (SAM) with significance thresholds of P value inferior to 5% and FDR inferior to 25% [34]. The predictive model, based on the 1889 differentially expressed genes identified above, was constructed using the nearest shrunken centroid algorithm implemented in the pamr-package. The model incorporating all genes was trained using the Ostrowski's samples, and the training error rate was assessed using 10-fold cross-validation, allowing definition of the delta value corresponding to the minimal error rate [32]. The resulting model was then applied onto the pooled set of 213 GISTs (20 with and 193 without activating PDGFRA mutation) to assess its robustness in an independent validation set.
Once validated, the predictive model was applied onto an expression series of 137 IBC samples and 252 non-IBC samples collected through the International IBC Consortium. Collection criteria, sample characteristics (including the molecular subtype classification), and data preprocessing steps have been described previously [11]. Prior to applying the model, technical variation in gene expression between the Ostrowski's GIST series and the IBC/non-IBC series was removed using the limma-package to ensure optimal classification performance [32]. To investigate the molecular subtype independency of PDGFRA activation differences between IBC and non-IBC, a multivariate logistic regression model (logit link test) with the PDGFRA activation model as dependent variable and both the tumor phenotype (IBC/non-IBC) and the molecular subtypes as independent variables was performed. The molecular subtype of samples was defined using the PAM50 predictor [35].
RNA Extraction, Reverse Transcription, and PCR Analysis
Total RNA harvested from cells using Trizol reagent (Invitrogen, Carlsbad, CA) was converted to cDNA using the AMV-RT system (Promega Corp., Madison, WI) per the manufacturer's recommendations. PDGF-A, -B, -C, and -D transcripts were amplified using 10 μM forward and reverse primers (Integrated DNA Technologies, Inc., Coralville, IA) and GoTaq Green Master Mix (Promega Corp., Madison, WI) in a 25-μl reaction. The primers were:
PDGF-A: forward 5′-CACACCTCCTCGCTGTAGTATTTA-3′ and reverse 5′-GTTATCGGTGTAAATGTCATCCAA-3′
PDGF-B: forward 5′-TCCCGAGGAGCTTTATGAGA-3′ and reverse 5′-ACTGCACGTTGCGGTTGT-3′
PDGF-C: forward 5′-CATGCCATGGGGAGCCTCTTCGGGCTTCTC-3′; and reverse, 5′-CGGGATCCCTATCCTCCTGTGCTCCCTCT-3′
PDGF-D: forward 5′-GTGCAGAGTCCTAGATTCCC-3′ and reverse 5′-GAGGTGGTCTTGAGCTGCAG-3′
Control β-actin: forward 5′-TCGTGCGTGACATTAAGGAG-3′ and reverse 5′-AGCACTGTGTTGGCGTACAG-3′.
PCR products were separated on 1.2% TAE agarose gel and visualized by ethidium bromide staining.
Cell Viability Assay
Cell viability assays for drug treatment were performed by seeding 4000 cells/well in triplicates in a 96-well plate and treatment 16 hours later with imatinib (LC Laboratories, Woburn, MA), crenolanib (Arog Pharmaceuticals, Dallas, TX), or vehicle controls (phosphate-buffered saline [PBS] for imatinib and dimethyl sulfoxide [DMSO] for crenolanib) for 48 hours. Imatinib and crenolanib concentrations ranged from 50 nM to 400 μM and 50 nM to 100 μM, respectively. 3-(4, 5-Dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide (MTT; Invitrogen, Carlsbad, CA) solution, 5 mg/ml in PBS, was prepared fresh for each experiment; 50 μl of MTT was added in each well 48 hours posttreatment and incubated at 37°C in 5% CO2 for 3 hours to allow the reduction of MTT to formazan crystals. Formazan crystals were dissolved in 100 μl of DMSO, and absorbance was read at an optical density of 562 nm. Absorbance data are represented as percentage absorbance of untreated control cells. Data are expressed as the percentage difference between the vehicle and treated cells for each treatment and day.
Immunoblotting
For analysis of PDGFRA activation, cells were grown either in regular growth medium or in serum-free medium containing all supplements and 1% BSA (Sigma Aldrich) with or without the addition of 10 ng/ml PDGF-BB for 10 minutes. Proteins were harvested using radioimmunoprecipitation assay buffer (1× PBS, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS) with 5 μl/ml of protease inhibitor cocktail (Calbiochem, Gibbstown, NJ) and phosphatase inhibitor (Thermo Scientific, Waltham, MA). Cell lysates were sonicated and kept on ice in between sonication pulses, and protein concentrations were determined using a BCA protein assay (Pierce Scientific, Rockford, IL). Laemmli buffer was added to 30 μg protein, denatured for 5 minutes in boiling water, and separated by SDS-PAGE on precast Criterion 4% to 20% Tris-HCl gels (BioRad, Hercules, CA). Depending upon the experiment, proteins were transferred onto nitrocellulose membrane and probed for PDGFRA, phosphorylated PDGFRA (Cell Signaling, Beverly, MA), or GAPDH (Sigma Aldrich, St. Louis, MO) and detected using ECL chemiluminescent reagent (Millipore Co., Billerica, MA).
PDGF-AA and PDGF-BB Ligand Expression
Secretion of PDGF-B by breast cancer cell lines was measured using the R&D Systems Human Quantikine PDGF-AA and PDGF-BB enzyme-linked immunosorbent assays (ELISAs). Briefly, breast cancer cell lines were grown in 100-mm3 dishes to 75% confluence, and the medium was changed. Conditioned medium was harvested 48 hours later, and 100 μl sample assayed kit per manufacturer’s recommendations. Absorbance was read at 450 nm with wavelength correction at 540 nm.
FACS Analysis
SUM149 cells were grown in 100-mm dishes until 80% confluent and treated with either 2 μM or 5 μM crenolanib (or vehicle control) for 12 hours. Posttreatment cells were washed with PBS, released with 0.05% trypsin at 37°C for 3 to 5 minutes, transferred to 15-ml tube, centrifuged at 800 rpm, resuspended in 0.003% DNAse I in Ham's F12+ 5% FBS (Sigma Aldrich, St. Louis, MO), and incubated for 5 minutes on ice. Cells were centrifuged at 800 rpm, washed with PBS, and fixed with ice-cold 70% ethanol for 48 to 72 hours. Fixed cells were centrifuged at 800 rpm to remove ethanol, resuspended in PBS, washed, and incubated in DNA staining solution (200 μg/ml of DNAse free RNAse A; 5 Prime Inc., Gaithersburg, MD) and 20 μg/ml of propidium iodide (Invitrogen)) at room temperature for 30 minutes in the dark. Stained cells were transferred to FACS tubes and analyzed in a Becton Dickinson FACSCalibur flow cytometer. Data were processed using Cell Quest and ModFit software.
In Vitro Emboli Formation Assay
SUM149 and KPL-4 IBC cells were grown in the emboli culture system as previously described [36]. Briefly, monolayer cells were harvested by trypsinization, collected by brief centrifugation, and counted. A total of 5 ml of suspension of 100,000 single cells was placed in Greiner CELLSTAR suspension flasks (Greiner Bio-One, Monroe, NC) with normal growth medium +2.25% PEG8000 (Sigma Aldrich). Cells were treated immediately with imatinib or crenolanib along with appropriate vehicle controls. Suspension flasks were placed at 37°C with 5% CO2 on the platform of a Belly Button shaker (Stovall Life Science Inc., Greensboro, NC) and allowed to gently rock at approximately 40 rpm for 48 hours. Emboli formation was assessed by directly counting the number of emboli formed in control and treated flasks.
Therapeutic Efficiency of Crenolanib In Vivo
Ten-week-old female nude mice were kept in a controlled, sterile environment per a University of Delaware Institutional Animal Care and Use Committee–approved animal protocol. SUM149 cells were washed with PBS, harvested by trypsinization, and washed twice, and a suspension of 4 × 106 cells per mouse was made in 1:1 mixture of PBS and BD Matrigel matrix (BD Transduction Laboratories, San Jose, CA) on ice. Orthotopic mammary fat pad injections of cells were performed, and size and weight were measured daily. Tumor volume was measured using the formula mm3 = l × w2 × 0.5, where l is length and w is width of the tumor. Tumors were allowed to grow until 200 mm3 in size, mice were randomized into two groups, and Alzet model 1002 micro-osmotic pumps (DURECT Corporation, Cupertino, CA) containing crenolanib or DMSO were implanted subcutaneously. The micropumps deliver a steady dose of 15 mg/kg crenolanib per day at a rate of 0.25 μl/h for a total of 10 days. Mice were sacrificed at day 15 and necropsied. The experiment was performed in duplicate with n = 8 for the treatment group and n = 7 for the control group.
Statistical Analysis
Correlations between tumor groups and clinicopathological features were analyzed using the Fisher's exact test. Metastasis-free survival (MFS) was calculated from the date of diagnosis until the date of distant relapse. Follow-up was measured from the date of diagnosis to the date of last news for metastasis-free patients. Survivals were calculated using the Kaplan-Meier method and compared with the log-rank test. Univariate and multivariate MFS analyses were done using Cox regression analysis (Wald test). Variables tested in univariate analyses included patients' age; tumor grade; ER, PR, and ERBB2 status (positive versus negative); patients' age at diagnosis (≤50 vs > 50 years); pathological grade (3 vs 2); and predicted PDGFRA activation status (“activated” versus “not activated”).
Unless otherwise noted, all laboratory experiments were performed a minimum of three separate times with no less than three replicates per experiment, and statistical analysis of the combined experiments was performed using Graph Pad Prism and by the University of Delaware College of Agriculture and Natural Resources Statistics Laboratory. Power analysis was performed for the in vivo experiments. A one-way analysis of variance was used with Bonferroni's post hoc analysis for comparison between multiple groups. A Student's t test was used for comparison between two groups. Significance was defined as a P value < .05.
Results
Generation of a Predictive Model for PDGFRA Activation
Previously, we demonstrated that PDGFRA but not PDGFRB expression was increased in IBC patient samples and associated with increased phosphoinositide-3 kinase/Akt1 signaling [10]. To determine whether PDGFRA is active in IBC patients similar to other types of cancers, we set out to develop a predictive model for receptor activation. When comparing expression profiles from GIST samples of the learning set (GSE17743) with and without activating PDGFRA mutations, we identified 1889 unique and differentially expressed (P < 5%; FDR < 25%) genes. Twenty-nine percent and 71% of these genes were, respectively, up- and downregulated in PDGFRA-mutated samples. Using these genes, a predictive model for PDGFRA activation was constructed on the same series of expression profiles (GSE17743). Learning samples with and without activating PDGFRA mutations were correctly classified in 86% of the cases (OR = 40; P = 2.6E-04, Fisher's exact test; Figure 1A). The resulting model was applied onto a validation series of expression profiles of 213 GIST samples. An overall error rate of 86% was achieved (OR = 37; P = 3.0E-10, Fisher's exact test; Figure 1B), suggesting the robustness of our predictive model.
Figure 1.

Generation of a robust predictive model for PDGFRA activation in GIST and application to breast cancer samples. (A) Classification of 29 GIST samples from the learning set (Ostrowki's set). (Left) Box-plot showing the distribution of posterior probabilities, according to our predictive model, of presence of PDGFRA mutation in samples without (“WT”) and with (“mut.”) PDGFRA mutation. (Right) Cross-table between the observed PDGFRA status (“mut.” versus “WT”) and the predicted PDGFRA activation status according to our model. (B) Similar to A but applied to the validation series of 213 GIST samples. (C) Cross-table between the predicted “PDGFRA-activated” status and the clinical phenotype (IBC versus non-IBC) of 137 IBC samples and 252 non-IBC samples collected through the IBC International Consortium gene expression data set. (D) Kaplan-Meier MFS in patients with IBC according to the predicted PDGFRA activation status: “activated” (black curve) versus “not activated” (gray curve). The respective 5-year MFSs were 52% and 72%.
Evaluation of PDGFRA Activation in IBC Clinical Samples
When applying the predictive model for PDGFRA activation onto an expression series of breast cancer patients with IBC and with non-IBC, more patients with IBC (22%) had samples predicted to exhibit PDGFRA activation than patients with non-IBC (13%; OR = 1.79, P = .044, Fisher's exact test; Figure 1C). Interestingly, this difference of activation prediction between IBC and non-IBC samples was independent from the molecular subtype distribution between IBC and non-IBC (univariate: βIBC versus non-IBC = 1.8, P = .034, logit link test; multivariate: βIBC versus non-IBC = 2.0, P = .02, logit link test). Evaluation of PDGF ligands’ mRNA expression in function of this classification reveals elevated expression of PDGF-B in the predicted “PDGFRA-activated” samples (P = 1.3E-07) but not PDGF-A, -C, or -D (Supplemental Figure 1).
We then investigated whether the PDGFRA activation model had a prognostic value in IBC. MFS data were available for 121 nonmetastatic IBC patients of the International IBC Consortium series, including 59 patients who displayed metastatic relapse. With a median follow-up of 147 months (range, 5-419), the 5-year MFS rate was 67% (95% CI, 0.59-0.77). Twenty-five patients were predicted to be “PDGFRA-activated” and showed 52% 5-year MFS (95% CI, 0.36-0.76), whereas 96 patients were predicted to be “not PDGFRA-activated” and showed better survival with 72% 5-year MFS (95% CI, 0.63-0.81; P = .022, log-rank test; Figure 1D). In univariate analysis, the baseline clinicopathological variables (patients' age; tumor grade; and ER, PR, and ERBB2 status) were not associated with MFS. In multivariate analysis including these variables and the classification based on our “PDGFRA activation” predictive model, only the latter was significant (HR = 2.09, P = .015, Wald test; Table 1).
Table 1.
Uni- and Multivariate Analyses for MFS in Patients with IBC
| Characteristic | N | Univariate HR [95% CI] | P | N | Multivariate HR [95% CI] | P |
|---|---|---|---|---|---|---|
| Age, >50 vs <=50 years | 119 | 1.42 [0.85-2.38] | .177 | 115 | 1.09 [0.63-1.88] | .768 |
| Grade, 3 vs 2 | 115 | 1.16 [0.6-2.25] | .650 | 115 | 1.14 [0.58-2.25] | .701 |
| ER status (mRNA), positive vs negative | 121 | 1.19 [0.71-1.99] | .500 | 115 | 1.03 [0.57-1.86] | .934 |
| PR status (mRNA), positive vs negative | 121 | 1.11 [0.64-1.92] | .707 | 115 | 1.23 [0.68-2.23] | .502 |
| ERBB2 status (mRNA), positive vs negative | 121 | 1.82 [0.89-3.72] | .102 | 115 | 2.01 [0.93-4.34] | .074 |
| PDGFRA prediction, activated vs nonactivated | 121 | 1.91 [1.09-3.38] | 2.49E-02 | 115 | 2.09 [1.15-3.8] | 1.57E-02 |
Characterization of PDGFRA Expression and Activity in IBC Cells
Next, to further study PDGFRA, we chose the SUM149 and KPL-4 IBC cell lines and compared them to the non-IBC MDA-MB-231 cell line. The SUM149 IBC cell line, in particular, is shown to accurately represent IBC patient samples [11]. We set out to determine if PDGFRA is active in the breast cancer cells, potentially via a PDGF-PDGFRA autocrine loop. In the presence or absence of either serum or exogenous PDGF-BB, active PDGFRA is detected by immunoprecipitation using a phosphospecific antibody (Figure 2A). Quantitation of pPDGFRA levels demonstrates an average four-fold increase in active PDGFRA in unstimulated IBC cells versus the non-IBC MDA-MB-231 cell line (Figure 2B).
Figure 2.

Expression of PDGFRA and PDGF ligands in IBC cells. (A) Representative immunoblot for total and tyrosine phosphorylated PDGFRA in SUM149, KPL-4, and MDA-MB-231 cells grown in monolayer with and without 10% serum or 10-ng/ml exogenous PDGF-BB stimulation for 10 minutes. GAPDH was used as a loading control. (B) Quantitation of PDGFRA immunoblots. Densitometry was performed on nonsaturated immunoblots using ImageJ analysis. A ratio of pPDGFRA to total PDGFRA is reported. (C) Representative RT-PCR for PDGF ligands expressed by the SUM149 IBC cell line. Cells were grown under standard conditions. Control is β-actin. (D) Results of an ELISA assay for PDGF-AA and PDGF-BB produced by breast cancer cell lines and compared with HMECs. Cells were grown to 75% confluence; medium was changed and assayed 48 hours later. IBC cells were compared to the non-IBC MDA-MB-231 cells. Error bars represent SD, *P ≤ .001. All experiments were performed in at least triplicate.
Figure 2C is a representative RT-PCR image showing mRNA expression for all four PDGF ligands, PDGF-A, -B, -C, and -D, in the SUM149 cells. Homodimeric PDGF-A, -B, and -C and heterodimer PDGF-A and -B lead to several signaling cascades including the PI3K/Akt pathway, which has been shown to be important in IBC metastasis [10] [37]. As indicated above, we demonstrated PDGF-B expression in “PDGFRA-activated” IBC patient tumors.
To determine whether PDGF-BB was expressed and secreted by the cell lines, conditioned medium from the SUM149 and KPL-4 IBC cells was analyzed for expression of PDGF-BB and compared with PDGF-AA expression via ELISA. HMEC control cells produced very little PDGF ligands. In contrast, both IBC cell lines produced significantly higher levels (P < .001) of PDGF-BB compared to the MDA-MB-231 non-IBC cell line (Figure 2D). In contrast, PDGF-AA expression was similar for all three tumor cell lines analyzed but expressed at approximately half the concentration of PDGF-BB in the IBC cells. Taken together, these data suggest that IBC cells express active PDGFRA, with receptor activation potentially occurring through an autocrine mechanism due to expression of PDGF-BB.
Effect of PDGFRA Targeting on IBC Cell Viability
To determine if active PDGFRA can be exploited as a therapeutic target in IBC, we assessed the efficiency of RTK inhibitors. Imatinib (a.k.a. STI571 or Gleevec) is a commonly used RTK inhibitor for several PDGFRA-activated cancers. The effect of imatinib on IBC cell viability (based on the published t1/2 of imatinib) was evaluated after 48 hours. Figure 3A are results of treatment over a dose range of 0.05 to 200 μM imatinib, which failed to show any discernible dose response on SUM149 and KPL-4 IBC cell viability. Longer treatment times had no effect on IBC cell viability (data not shown). In contrast, MDA-MB-231 cells, which are reported to be sensitive to imatinib treatment [38], [39], were significantly affected (Supplemental Figure 2). Absorbance data are represented as percent viable cells, normalized to untreated cells.
Figure 3.

Pharmacologic inhibition of PDGFRA. (A) Dose response of imatinib and crenolanib treatment on SUM149 and KPL-4 cells grown in monolayer. Cells were treated with a dose range from 0 to 200 μM imatinib or 0 to 20 μM crenolanib for 48 hours and assessed by MTT. Effect on cell survival was compared in parallel with vehicle controls. Error bars represent SD, *P ≤ .001. ND = not done. (B) Cell cycle analysis by propidium iodide staining of SUM149 cells treated with 2 μM or 5 μM crenolanib for 12 hours. Untreated and DMSO-treated cells were used as controls. FACS analysis data were processed using CellQuest and Mod-Fit software. The bar graph is quantification of the data showing the percentage of cells in G1, G2/M, and S phases, Error bars represent SD,*P ≤ .05. (C) Dose response of imatinib and crenolanib treatment on SUM149 and KPL-4 cells grown as three-dimensional emboli. Cells were treated with a dose range from 0 to 200 μM imatinib or 0 to 5 μM crenolanib for 48 hours; emboli were harvested and assessed by direct counting, which was compared in parallel with vehicle controls. Error bars represent SD, *P ≤ .001. Each experiment was performed in at least triplicate.
Supplemental Figure 2.
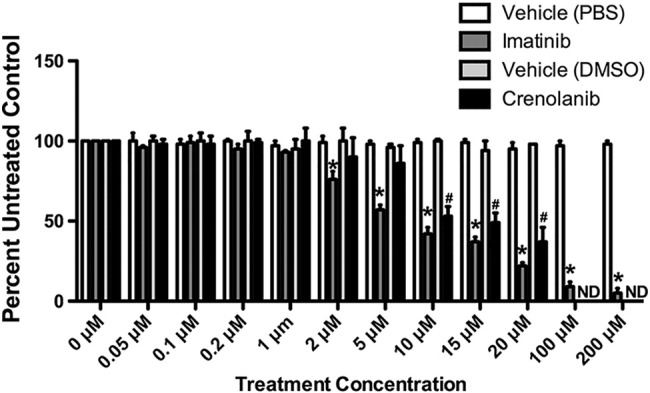
Pharmacologic inhibition of PDGFRA in MDA-MB-231 cells. Dose response of imatinib and crenolanib treatment on SUM149 and KPL-4 cells grown in monolayer. Cells were treated with a dose range from 0 to 200 μM imatinib or 0 to 20 μM crenolanib for 48 hours and assessed by MTT. Effect on cell survival was compared in parallel with vehicle controls. Error bars represent SD, *P ≤ .001. ND = not done.
Crenolanib (CP-868-596) has a mean reported terminal half-life of 14.0 ± 4.2 hours upon single-dose treatment with IC50 ranging from 1 ± 0.03 to 6.93 ± 0.03 μM for leukemic cell lines [40]. Based on the available pharmacokinetic data of crenolanib, a similar cell growth inhibition assay was performed over 48 hours on SUM149 cells. An effective crenolanib response for both the SUM149 and KPL-4 cell lines over a 0.05- to 20-μM range as compared to vehicle (DMSO)-treated control cells is shown in Figure 3A. MDA-MB-231 cells were also sensitive to crenolanib treatment but only in the higher 10- to 20-μM range (Supplemental Figure 2).
To determine if crenolanib affected the cell cycle or induced apoptosis of the SUM149 IBC cells, two higher concentrations, 2 μM and 5 μM, were chosen. Accumulation of cells in G2/M was observed after 12 hours of crenolanib treatment with both concentrations tested (Figure 3B). Quantitative analysis of the data demonstrated a significant difference between percentage of cells in G2/M phase with and without crenolanib treatment. No distinct apoptotic peaks were observed, nor were any other indicators that crenolanib-treated cells underwent apoptosis.
Effect of PDGFRA Targeting on IBC Emboli Formation
Because the main hallmark of IBC is the presence of florid tumor emboli in the dermal lymphatic vessels, we next determined if targeting PDGFRA affected emboli formation. Using an in vitro emboli culture system [36], we determined the effect of imatinib and crenolanib treatment on emboli formation. Because the MDA-MB-231 non-IBC cells do not form emboli in our assay, they were not tested [36]. Similar to the cell viability assay, a wide concentration (1-200 μM) of imatinib did not cause any detectable change in SUM149 or KPL-4 IBC emboli formation over a 48-hour treatment time (Figure 3C; again, longer treatment times had no effect; data not shown). In contrast, the effect of crenolanib on emboli formation over a 48-hour treatment resulted in the formation of significantly fewer emboli at all concentrations tested (Figure 3C).
Effect of PDGFRA Targeting on IBC Tumor Growth In Vivo
We next evaluated the efficiency of crenolanib treatment on IBC orthotopic tumor growth in female athymic nude mice. SUM149 IBC tumors were grown to 200 mm3, the mice were randomized, and treatment was started. Alzet micro-osmotic pumps containing either crenolanib or vehicle DMSO were implanted subcutaneously. Figure 4 demonstrates a significant difference in tumor growth between the vehicle control and crenolanib-treated mice. The average starting tumor sizes for control and crenolanib-treated mice were similar, 215.7 ± 5.1 mm3 and 210.9 ± 6.9 mm3, respectively, on day 1 of treatment. Tumor volume on the final day of treatment (day 10) was 753.9 ± 116 mm3 and 197.6 ± 50.7 mm3 for control and treated tumors, respectively (P < .0001). Tumor growth resumed once crenolanib treatment was halted (Supplemental Figure 3). Crenolanib treatment had no adverse effect on weight, activity, or general health (not shown). Thus, crenolanib appears to be cytostatic, effectively preventing IBC tumor growth in vivo.
Figure 4.

Crenolanib treatment of an orthotopic IBC tumor. Orthotopic SUM149 tumors were established in female athymic nude mice and grown to ~200 mm3 in size. Mice were randomized to control and treatment groups, and Alzet micro-osmotic pumps were implanted subcutaneously, represented as day 1. Micropumps contained either crenolanib or DMSO vehicle, and treatment lasted for 10 days. Tumors were measured daily. Error bars represent SD, *P ≤ .0001.
Supplemental Figure 3.

Crenolanib treatment of an orthotopic IBC tumor. Orthotopic SUM149 tumors were established in female athymic nude mice and grown to ~200 mm3 in size. Mice were randomized to control and treatment groups, and Alzet micro-osmotic pumps were implanted subcutaneously, represented as day 1. Micropumps contained either crenolanib or DMSO vehicle, and treatment lasted 10 days (arrow). Mice were kept for an additional 4 days, and tumors were measured daily.
Discussion
The current standard therapy for treating IBC is aggressive and pluridisciplinary, including systemic chemotherapy followed by radical mastectomy, radiotherapy, hormone therapy for hormone receptor–positive tumors, and anti-ERBB2 drug for ERBB2-positive tumors [1]. This treatment is relatively effective, although insufficient with long-term survival inferior to 50%; results in severe side effects; and offers poor life quality [3]. In the present study, we explored PDGFRA as a potential target for IBC treatment with a novel inhibitor, crenolanib.
Using a validated predictive model of PDGFRA activation developed from GIST expression data, we demonstrated, using the large International IBC Consortium series, that IBC patient samples showed more frequent PDGFRA activation than non-IBC samples independently from the molecular subtypes of samples. Then, we showed that patients with IBC samples predicted as “PDGFRA-activated” according to this predictive model showed shorter MFS than patients with IBC samples predicted as “not PDGFRA-activated.” Such prognostic value remained significant in multivariate analysis.
Previously published studies suggested that the non-IBC triple-negative cell line MDA-MB-231 responds to imatinib treatment [38], [39], [41]. However, in clinical studies, patients with PDGFR-expressing metastatic breast cancer showed no significant response to imatinib treatment [42], perhaps due to the presence of a resistant constitutively active PDGFR or perhaps because of absence of activation of PDGFR in the tumor. In this study, we find that the SUM149 and KPL-4 IBC cell lines are refractory to imatinib but not to crenolanib. This is likely due to the fact that imatinib prevents the activation of PDGFRA, whereas crenolanib targets the active form of the receptor [18], [19], [21]. Crenolanib was specifically designed to target imatinib-resistant GISTs [21].
Using a novel cell culture model to form IBC emboli, we show that crenolanib treatment leads to a significant decrease in IBC cells ability to form emboli. This result is similar to what we reported with TGFβ stimulation of IBC cells [36]. Previously, we reported low TGFβ expression and decreased TGFβ signaling in IBC patients and cell lines [11], [36]. As a consequence, TGFβ stimulation of IBC cells leads to the formation of fewer IBC emboli. Ingenuity pathway analysis networks demonstrated a merging of PDGFR and TGFβ signaling pathways in IBC [43]. In addition, TGFβ is shown to downregulate PDGFRA in lung fibroblasts [44]. Currently, we are studying the molecular interactions of these pathways in IBC.
IBC patients with “PDGFRA-activated” prediction were found in our study to overexpress PDGF-B ligand, which is produced and secreted at high levels by the IBC cell lines. Thus, our results suggest the possibility of autocrine PDGFRA signaling. Wang et al. demonstrated endosomal activation of PDGFRA by intracellular PDGF, which was sufficient to generate a physiological output including cell proliferation and survival of human hepatocellular cells [45]. Whether PDGFRA in IBC cells is being packaged with ligand resulting in autocrine stimulation or whether the receptor harbors similar activating mutations as observed in GISTs is currently under investigation.
The most striking and exciting finding is the significant inhibition of in vivo tumor growth by crenolanib treatment. Crenolanib-treated tumors remained roughly the same size during the course of treatment and increased in size only after treatment was halted. Like many chemotherapeutic compounds, crenolanib arrests cells at G2/M, suggesting that the efficiency of chemotherapeutic agents or radiotherapy may be improved by crenolanib treatment, a focus of current experiments. The effect of crenolanib treatment on tumor recurrences needs to be assessed. Most recurrences occur due to the outgrowth of tumor-initiating cells, and IBC has a very high proportion of tumor-initiating cells [46], [47], [48].
We acknowledge that, in this study, we tested only two IBC cell lines, one a triple-negative and the other HER2+; ideally, we would have liked to test other lines that cover the molecular heterogeneity of IBC. In our experience, we have found that the SUM149 cell line accurately represents IBC patient tumors and is an appropriate cell line to test in vitro observations made in patient samples.
Conclusions
Our current study suggests that expression of activated PDGFRA has a role in determining MFS in IBC patients. Our data suggest that crenolanib is a promising candidate to target PDGFRA.
The following are the supplementary data related to this article.
Description of the GIST and Breast Cancer Data Sets Used for mRNA Expression Analysis
Funding
This work was supported in part by the Congressionally Directed Medical Research Program, Breast Cancer Research Program, DAMB-17-03-1-0728 (K.L.v.G.), W81WXH-06-1-00495 (K.L.v.G.), W81XWH-08-1-0356 (K.L.v.G.), and W81XWH-10-1-0272 (M.J.J.).
Acknowledgements
This project was completed as a collaborative effort of the International Inflammatory Breast Cancer Consortium.
Footnotes
Conflicts of interest: Monique Carter, Justin Rains, and Abhijit Rama2chandran are employed by Arog Pharmaceuticals LLC. All other authors have no conflicts of interest.
References
- 1.Dawood S. Biology and management of inflammatory breast cancer. Expert Rev Anticancer Ther. 2010;10:209. doi: 10.1586/era.09.90. [DOI] [PubMed] [Google Scholar]
- 2.Dawood S, Valero V. Clinical Aspects of Inflammatory Breast Cancer: Diagnosis, Criteria, Controversy. In: Ueno NT, Cristofanilli M, editors. Inlfammatory Breast Cancer: An Update. Springer; New York, N.Y: 2012. pp. 11–20. [Google Scholar]
- 3.van Golen KL, Cristofanilli M. The Third International Inflammatory Breast Cancer Meeting. Breast Cancer Res. 2013;15:318. doi: 10.1186/bcr3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonnier P, Charpin C, Lejeune C, Romain S, Tubiana N, Beedassy B, Martin PM, Serment H, Piana L. Inflammatory carcinomas of the breast: a clinical, pathological, or a clinical and pathological definition? Int J Cancer. 1995;62:382–385. doi: 10.1002/ijc.2910620404. [DOI] [PubMed] [Google Scholar]
- 5.Kleer CG, van Golen KL, Merajver SD. Molecular biology of breast cancer metastasis. Inflammatory breast cancer: clinical syndrome and molecular determinants. Breast Cancer Res. 2000;2:423. doi: 10.1186/bcr89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodward WA, Cristofanilli M. Inflammatory breast cancer. Semin Radiat Oncol. 2009;19:256–265. doi: 10.1016/j.semradonc.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Vermeulen PB, van Golen KL, Dirix LY. Angiogenesis, lymphangiogenesis, growth pattern, and tumor emboli in inflammatory breast cancer: a review of the current knowledge. Cancer. 2010;116:2748–2754. doi: 10.1002/cncr.25169. [DOI] [PubMed] [Google Scholar]
- 8.Joglekar M, van Golen KL. Molecules That Drive the Invasion and Metastasis of Inflammatory Breast Cancer. In: Ueno NT, Cristofanilli M, editors. Inflammatory Breast Cancer: An Update. Springer; New York, NY USA: 2012. pp. 161–184. [Google Scholar]
- 9.Radunsky GS, van Golen KL. The current understanding of the molecular determinants of inflammatory breast cancer metastasis. Clin Exp Metastasis. 2005;22:615–620. doi: 10.1007/s10585-006-9000-7. [DOI] [PubMed] [Google Scholar]
- 10.Lehman HL, Van Laere SJ, van Golen CM, Vermeulen PB, Dirix LY, van Golen KL. Regulation of inflammatory breast cancer cell invasion through Akt1/PKBalpha phosphorylation of RhoC GTPase. Mol Cancer Res. 2012;10:1306–1318. doi: 10.1158/1541-7786.MCR-12-0173. [DOI] [PubMed] [Google Scholar]
- 11.Van Laere SJ, Ueno NT, Finetti P, Vermeulen PB, Lucci A, Robertson FM, Marsan M, Iwamoto T, Krishnamurthy S, Masuda H. Uncovering the molecular secrets of inflammatory breast cancer biology: an integrated analysis of three distinct Affymetrix gene expression data sets. Clin Cancer Res. 2013;19:4685–4696. doi: 10.1158/1078-0432.CCR-12-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oseini AM, Roberts LR. PDGFRalpha: a new therapeutic target in the treatment of hepatocellular carcinoma? Expert Opin Ther Targets. 2009;13:443. doi: 10.1517/14728220902719233. [DOI] [PubMed] [Google Scholar]
- 13.Ostman A, Heldin CH. PDGF receptors as targets in tumor treatment. Adv Cancer Res. 2007;97:247. doi: 10.1016/S0065-230X(06)97011-0. [DOI] [PubMed] [Google Scholar]
- 14.Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, Huse JT, Pedraza A, Utsuki S, Yasui Y. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010;24:2205. doi: 10.1101/gad.1972310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones AV, Cross NC. Oncogenic derivatives of platelet-derived growth factor receptors. Cell Mol Life Sci. 2004;61:2912. doi: 10.1007/s00018-004-4272-z. [DOI] [PubMed] [Google Scholar]
- 16.Fabbro D, Parkinson D, Matter A. Protein tyrosine kinase inhibitors: new treatment modalities? Curr Opin Pharmacol. 2002;2:374. doi: 10.1016/s1471-4892(02)00179-0. [DOI] [PubMed] [Google Scholar]
- 17.Ganjoo KN, Patel S. Current and emerging pharmacological treatments for gastrointestinal stromal tumour. Drugs. 2011;71:321. doi: 10.2165/11585370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Aleksandrov A, Simonson T. A molecular mechanics model for imatinib and imatinib:kinase binding. J Comput Chem. 2010;31:1550. doi: 10.1002/jcc.21442. [DOI] [PubMed] [Google Scholar]
- 19.Wang W-L, Conley A, Reynoso D, Nolden L, Lazar A, George S, Trent JC. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2011;67:15–24. doi: 10.1007/s00280-010-1513-8. [DOI] [PubMed] [Google Scholar]
- 20.Capdeville R, Silberman S, Dimitrijevic S. Imatinib: the first 3 years. Eur J Cancer. 2002;38(Suppl. 5):S77–S82. doi: 10.1016/s0959-8049(02)80607-4. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:4375–4384. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]
- 22.Lehman HL, Dashner EJ, Lucey M, Vermeulen P, Dirix L, Van Laere S, van Golen KL. Modeling and characterization of inflammatory breast cancer emboli grown in vitro. Int J Cancer. 2012;132:2283–2294. doi: 10.1002/ijc.27928. [DOI] [PubMed] [Google Scholar]
- 23.van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–2519. [PubMed] [Google Scholar]
- 24.Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, Neuville A, Wozniak A, Sciot R, Schoffski P. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:826–838. doi: 10.1158/1078-0432.CCR-11-1610. [DOI] [PubMed] [Google Scholar]
- 25.Astolfi A, Nannini M, Pantaleo MA, Di Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu A, Saponara M. A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number. Lab Invest. 2010;90:1285–1294. doi: 10.1038/labinvest.2010.110. [DOI] [PubMed] [Google Scholar]
- 26.Lee EJ, Kang G, Kang SW, Jang KT, Lee J, Park JO, Park CK, Sohn TS, Kim S, Kim KM. GSTT1 copy number gain and ZNF overexpression are predictors of poor response to imatinib in gastrointestinal stromal tumors. PLoS One. 2013;8:e77219. doi: 10.1371/journal.pone.0077219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Du X, Lazar AJ, Pollock R, Hunt K, Chen K, Hao X, Trent J, Zhang W. Genetic aberrations of gastrointestinal stromal tumors. Cancer. 2008;113:1532–1543. doi: 10.1002/cncr.23778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, Nilsson O. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129:1149–1161. doi: 10.1002/ijc.25755. [DOI] [PubMed] [Google Scholar]
- 29.Kang HJ, Nam SW, Kim H, Rhee H, Kim NG, Kim H, Hyung WJ, Noh SH, Kim JH, Yun CO. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene. 2005;24:1066–1074. doi: 10.1038/sj.onc.1208358. [DOI] [PubMed] [Google Scholar]
- 30.Rink L, Skorobogatko Y, Kossenkov AV, Belinsky MG, Pajak T, Heinrich MC, Blanke CD, von Mehren M, Ochs MF, Eisenberg B. Gene expression signatures and response to imatinib mesylate in gastrointestinal stromal tumor. Mol Cancer Ther. 2009;8:2172–2182. doi: 10.1158/1535-7163.MCT-09-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Hasegawa T, Shitashige M, Ono M, Shoji A, Sakuma T, Kuwabara H. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:4100–4108. doi: 10.1200/JCO.2007.14.2331. [DOI] [PubMed] [Google Scholar]
- 32.Ostrowski J, Polkowski M, Paziewska A, Skrzypczak M, Goryca K, Rubel T, Kokoszynska K, Rutkowski P, Nowecki ZI, Vel Dobosz AJ. Functional features of gene expression profiles differentiating gastrointestinal stromal tumours according to KIT mutations and expression. BMC Cancer. 2009;9:413. doi: 10.1186/1471-2407-9-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 34.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehman HL, Dashner EJ, Lucey M, Vermeulen P, Dirix L, Van Laere S, van Golen KL. Modeling and characterization of inflammatory breast cancer emboli grown in vitro. Int J Cancer. 2012;132:2283–2294. doi: 10.1002/ijc.27928. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwaiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiraga T, Nakamura H. Imatinib mesylate suppresses bone metastases of breast cancer by inhibiting osteoclasts through the blockade of c-Fms signals. Int J Cancer. 2009;124:215–222. doi: 10.1002/ijc.23903. [DOI] [PubMed] [Google Scholar]
- 39.Roussidis AE, Mitropoulou TN, Theocharis AD, Kiamouris C, Papadopoulos S, Kletsas D, Karamanos NK. STI571 as a potent inhibitor of growth and invasiveness of human epithelial breast cancer cells. Anticancer Res. 2004;24:1445–1447. [PubMed] [Google Scholar]
- 40.Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, Olszanski AJ, Cohen RB. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868, 596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol. 2009;27:5262–5269. doi: 10.1200/JCO.2009.21.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sims JT, Ganguly S, Fiore LS, Holler CJ, Park ES, Plattner R. STI571 sensitizes breast cancer cells to 5-fluorouracil, cisplatin and camptothecin in a cell type-specific manner. Biochem Pharmacol. 2009;78:249–260. doi: 10.1016/j.bcp.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cristofanilli M, Morandi P, Krishnamurthy S, Reuben JM, Lee BN, Francis D, Booser DJ, Green MA, Arun BK, Pusztais L. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann Oncol. 2008;19:1713–1719. doi: 10.1093/annonc/mdn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X, Wang X-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonner JC, Badgett A, Lindroos PM, Osornio-Vargas AR. Transforming growth factor beta 1 downregulates the platelet-derived growth factor alpha-receptor subtype on human lung fibroblasts in vitro. Am J Respir Cell Mol Biol. 1995;13:496–505. doi: 10.1165/ajrcmb.13.4.7546780. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Pennock SD, Chen X, Kazlauskas A, Wang Z. Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J Biol Chem. 2004;279:8038–8046. doi: 10.1074/jbc.M311494200. [DOI] [PubMed] [Google Scholar]
- 46.Griffiths CL, Olin JL. Triple negative breast cancer: a brief review of its characteristics and treatment options. J Pharm Pract. 2012;25:319–323. doi: 10.1177/0897190012442062. [DOI] [PubMed] [Google Scholar]
- 47.Stratford AL, Reipas K, Maxwell C, Dunn SE. Targeting tumour-initiating cells to improve the cure rates for triple-negative breast cancer. Expert RevMol Med. 2010;12 doi: 10.1017/S1462399410001535. [null-null] [DOI] [PubMed] [Google Scholar]
- 48.van Golen CM, van Golen KL. Inflammatory breast cancer stem cells: contributors to aggressiveness, metastatic spread and dormancy. Mol Biomarkers Diagn. 2012;S-8:1–4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of the GIST and Breast Cancer Data Sets Used for mRNA Expression Analysis