

We present a mechanism by which oxygen is reduced to water in living organisms.
Keywords: electron transfer, proton transfer, cytochrome aa3, membrane protein, ligand, kinetics, mechanism
Abstract
Heme-copper oxidases catalyze the four-electron reduction of O2 to H2O at a catalytic site that is composed of a heme group, a copper ion (CuB), and a tyrosine residue. Results from earlier experimental studies have shown that the O–O bond is cleaved simultaneously with electron transfer from a low-spin heme (heme a/b), forming a ferryl state (PR; Fe4+=O2−, CuB2+–OH−). We show that with the Thermus thermophilus ba3 oxidase, at low temperature (10°C, pH 7), electron transfer from the low-spin heme b to the catalytic site is faster by a factor of ~10 (τ ≅ 11 μs) than the formation of the PR ferryl (τ ≅110 μs), which indicates that O2 is reduced before the splitting of the O–O bond. Application of density functional theory indicates that the electron acceptor at the catalytic site is a high-energy peroxy state [Fe3+–O−–O−(H+)], which is formed before the PR ferryl. The rates of heme b oxidation and PR ferryl formation were more similar at pH 10, indicating that the formation of the high-energy peroxy state involves proton transfer within the catalytic site, consistent with theory. The combined experimental and theoretical data suggest a general mechanism for O2 reduction by heme-copper oxidases.
INTRODUCTION
Cytochrome c oxidase (CytcO) catalyzes the reduction of O2 to H2O in the respiratory chain of aerobic organisms using reduced cytochrome (cyt.) c as an electron donor. The enzyme is a transmembrane protein that is composed of two or more subunits, which harbor four redox-active metal sites. During turnover, electrons from cyt. c are transferred first to the primary electron acceptor CuA, then to the low-spin heme intermediate electron acceptor, and finally to the catalytic site, which consists of a heme group and a copper ion, CuB, in close proximity, as well as a redox-active Tyr residue (Fig. 1) [for a review of the structure and function of oxidases, see related studies (1–14)]. The heme-copper oxidases typically pump, on average, 0.5 to 1 proton across the membrane per electron transferred from cyt. c to the catalytic site.
Fig. 1. Structure of the catalytic site.
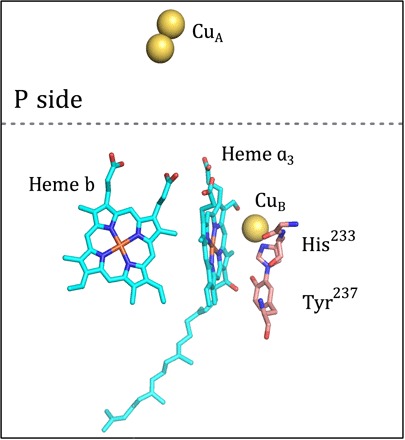
The redox-active cofactors of cyt. ba3. The low-spin heme b and heme a3 are depicted in cyan; CuB and the dinuclear CuA are indicated by yellow spheres. The cross-linked residues His233 and Tyr237, located in proximity of the catalytic site, are shown. The image was prepared using the software PyMOL [Protein Data Bank code 3S8F (20)]. The dashed line is the approximate position of the membrane surface on the positive (p) side of the membrane.
The oxidases are classified based on their structural details, particularly the architecture of the proton pathways, as members of one of three classes denoted by letters A, B, and C (15–17). The type of heme groups varies in different oxidases, and it is not strictly related to the class of the CytcO. In the B-type CytcO from Thermus thermophilus, the low-spin intermediate electron acceptor is heme b, whereas heme a3 resides in the catalytic site; that is, the CytcO is a cyt. ba3. The well-studied A-type CytcOs from Rhodobacter sphaeroides, Paracoccus denitrificans, and mitochondria are all aa3 CytcOs.
The A-family bacterial oxidases use two proton pathways starting at the surface on the negative (n) side of the membrane, namely, the K and D pathways. The ba3 CytcO from T. thermophilus harbors only one functional proton pathway (18), which approximately overlaps in space with the K pathway in the A-type oxidases, and it is therefore referred to as the K pathway analog. Structures of the ba3 CytcO have been determined at atomic resolution using x-ray crystallography (19–21).
Detailed kinetic and mechanistic information for the T. thermophilus ba3 CytcO has been obtained, for example, from studies of the reaction of the reduced CytcO with O2 and the comparison of this reaction sequence to that in the well-studied A-type oxidases. With the A-type oxidases, dioxygen binds first to heme a3 at the catalytic site, forming the oxo-ferrous intermediate called A, first described by Chance et al. (22). It was assumed originally that after O2 binding to CytcO, a peroxy intermediate (Eq. 1) is formed at heme a3 [see related studies (23–26)]
| (1) |
Hence, this state was referred to as P. However, on the basis of an analysis of the optical absorption spectra of the P state, Weng and Baker (27) suggested that the O–O bond is broken in the P state, indicating that a ferryl state (=O2−) is formed (Eq. 2). Further support for this suggestion was obtained from resonance Raman experiments (28–31). In 1999, Fabian et al. (32) demonstrated that one of the oxygen atoms of the O2 molecule bound to the two-electron reduced CytcO was released to solvent, which was interpreted to show that the O–O bond is broken upon formation of P
| (2) |
The source of the electron and proton at the left-hand side of the reaction varies depending on the initial state of the CytcO. If only the catalytic site is reduced, then the electron and proton are transferred from a Tyr residue (Tyr288 or Tyr237 in R. sphaeroides aa3 CytcO or T. thermophilus ba3 CytcO, respectively) (26, 29, 33–35), which forms a tyrosyl radical, TyrO•. The state that is formed is denoted PM (Eq. 3)
| (3) |
The time constant of this reaction is 200 to 300 μs (36, 37), and the PM state is stable over a time scale of minutes (38).
When the P state is formed in the fully reduced CytcO, the electron is transferred from the low-spin heme a iron (), and the Tyr residue donates only a proton. The state is referred to as PR (Eq. 4)
| (4) |
The PR formation time constants were found in the range 30 to 70 μs (37, 39–44), depending on the species from which the CytcO is isolated.
The summary above shows that the PM and PR states have the same structure; the only difference is the donor of the “fourth” electron, which is Tyr288 or the low-spin heme, respectively [reviewed by Kaila et al. (9)]. Note that the formation of PM/PR is not associated with any proton uptake from the solution to the catalytic site (45–47); the Tyr residue is the hydrogen or proton donor.
Although a peroxy state [Fe3+–O−–O−(H+)] has not been previously observed upon reaction of the reduced CytcO with O2, on the basis of theoretical studies, it has been predicted that this state (called IP) is formed transiently on the reaction path leading from state A to state PM (48–50). IP has not been observed because it is higher in energy than state A. Here, we investigated the formation of the PR state in the T. thermophilus ba3 CytcO as a function of temperature at neutral and high pH (10). The data indicate that at neutral pH and low temperature (10°C), electron transfer from heme b to the catalytic site is about 10 times faster than the formation of the PR state, which suggests that a true peroxy state is formed transiently before the O–O bond is broken in PR. This difference in rate constants was not observed at higher temperatures (~45°C), where the electron transfer and PR formation displayed the same rate constants, just as in the A-type CytcOs at room temperature. Furthermore, at high pH (10), the difference in rate constants (at low temperature) was significantly smaller than at neutral pH, which suggests that formation of a transient peroxy state [Fe3+–O−–O−(H+)] involves internal proton transfer within the catalytic site.
RESULTS
Experimental results
The reduced cyt. ba3 CytcO, with CO bound to heme a3, was mixed with an O2-saturated solution after which the CO ligand was removed by means of a laser flash to allow O2 to bind to heme a3. Absorbance changes were monitored at 560 and 610 nm. At 560 nm, there is a maximum in the contribution of the redox changes of heme b, whereas at 610 nm, the binding of O2 and the formation of the “peroxy” (P) intermediate are observed [a broad trough near 615 nm and a peak at 610 to 612 nm (51, 52)]. Figure 2 shows absorbance changes as a function of time after CO dissociation at t = 0. The starting level of the traces just after the flash (adjusted to zero) reflects the absorbance of the reduced CytcO relative to that of the CytcO-CO complex.
Fig. 2. Absorbance changes during reaction of the fully reduced ba3 CytcO with O2.
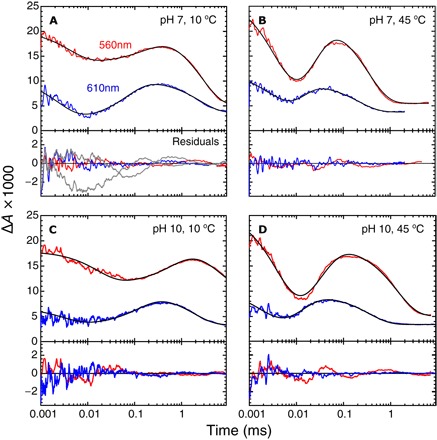
(A and B) pH 7 at 10° and 45°C, respectively. (C and D) pH 10 at 10° and 45°C, respectively. At 610 and 560 nm, the absorbance changes are mainly attributed to redox changes at heme a3 and heme b, respectively. The black lines are fits of the data with a model that is described by a sum of three exponential functions. The rate constants obtained from the fit are given in the text and in Table 1. The difference between the fit and the data (that is, the residuals) is shown below each panel. In addition, in (A), we show the residuals of a fit with a single rate constant (5 × 104 s−1) for electron transfer from heme b to the catalytic site and PR formation (gray lines). Experimental conditions after mixing: 0.6 to 0.8 μM CytcO (scaled to 1 μM), 0.05% DDM, 90 mM Hepes (pH 7) or 90 mM CAPS (pH 10), and ~1 mM O2. The cuvette path length was 1.00 cm. The 560-nm traces are shifted up by 1.7 × 10−3 units for clarity.
We first discuss data at 10°C and pH 7. At 610 nm (Fig. 2A), the first decrease in absorbance is associated with the binding of O2 to heme a3 with a rate constant of 2.8 × 105 ± 0.8 × 105 s−1 (τ ≅ 3.6 μs). The increase in absorbance is associated with the formation of the PR state with a rate constant of 9 × 103 ± 1 × 103 s−1 (τ ≅ 110 μs), whereas the slowest decrease in absorbance is associated with oxidation of the CytcO with a rate constant of 400 ± 60 s−1 (τ ≅ 2.5 ms). At 560 nm (Fig. 2A), the initial decrease in absorbance is associated with oxidation of heme b with a rate constant of 9 × 104 ± 4 × 104 s−1 (τ ≅11 μs), which is followed in time by re-reduction of heme b (τ = 180 ± 30 μs) and oxidation (τ = 2.6 ± 0.2 ms). The data indicate that at 10°C and pH 7, oxidation of heme b (decrease in absorbance at 560 nm, τ ≅ 11 μs) was ~10 times faster than the formation of state PR (increase in absorbance at 610 nm, τ ≅ 110 μs).
At high temperature (45°C) and pH 7 (Fig. 2B), the decrease in absorbance at 560 nm (oxidation of heme b) and the increase in absorbance at 610 nm (formation of PR) displayed rate constants of 1.9 × 105 ± 0.2 × 105 s−1 (τ ≅ 5.3 μs) and 1.5 × 105 ± 0.5 × 105 s−1 (τ ≅ 6.7 μs), respectively, that is, the same within the error. Figure 2 (C and D) shows absorbance changes at pH 10 and at 10° and 45°C, respectively. At higher pH, the ratios of the rate constants of heme b oxidation and PR formation were ~3 and ~1.3 at 10° and 45°C (the rate constant values are given in Table 1), respectively; that is, the difference in rates at a low temperature was smaller than at neutral pH.
Table 1. Rate constants for the early steps of O2 reduction at low and high temperature for pH 7 and 10, respectively.
Errors are the SDs for n = 3 (pH 10 data) or n = 4 (pH 7 data) measurements.
| Rate constant (s−1) (Time constant) (μs) | |||
| T (°C) | pH 7 | pH 10 | |
| O2 binding (610-nm decay) | 10 | (2.8 ± 0.8) × 105 (3.6) | (2.9 ± 0.1) × 105 (3.4) |
| 45 | (2.7 ± 0.8) × 105 (3.7) | (4 ± 1) × 105 (2.5) | |
| Heme b oxidation (kb) (560-nm decay) | 10 | (9 ± 4) × 104 (11) | (2.6 ± 0.6) × 104 (38) |
| 45 | (1.9 ± 0.2) × 105 (5.3) | (1.9 ± 0.2) × 105 (5.3) | |
| PR formation (kP) (610-nm increase) | 10 | (9 ± 1) × 103 (110) | (8.7 ± 0.3) × 103 (120) |
| 45 | (1.8 ± 0.5) × 105 (5.5) | (1.5 ± 0.5) × 105 (6.7) | |
| Ratio (kb/kP) | 10 | 10 | 3 |
| 45 | 1.1 | 1.3 | |
Figure 3A shows the temperature dependence of the rate constant for the absorbance change attributed to PR formation at 610 nm (black trace) and those attributed to heme b oxidation at 560 nm (red trace) at pH 7. As seen in the figure, the difference between these rate constants was significantly larger at low than at high temperature. Figure 3 also shows the temperature dependence of the kinetic component associated with the binding of O2 (blue trace, τ = 2 to 4 μs). This time constant was essentially temperature-independent, presumably because the slower O2 binding at lower temperatures is compensated for by the increased solubility of O2.
Fig. 3. Temperature dependence of the rate constants.
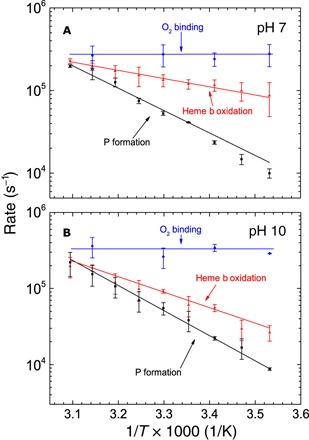
The rates of O2 binding (Arrhenius plots), heme b oxidation, and PR formation are shown as indicated in the graphs for (A) pH 7 and (B) pH 10. Conditions were the same as in Fig. 2.
Figure 4 shows absorbance changes at 610 nm associated with the reaction of the mixed-valence ba3 CytcO (that is, reduced heme a3 and CuB and oxidized heme b and CuA) with O2 at ~22°C and neutral pH. The increase in absorbance is associated with the formation of the PM state with a rate constant of 7.1 × 103 ± 0.7 × 103 s−1 (τ ≅ 140 μs), consistent with results from earlier studies (36, 37). The rate constant of this reaction is pH-independent in the pH range 6.1 to 9.2 (53), and it is not associated with proton uptake from the solution (46).
Fig. 4. Kinetics of the reaction of mixed-valence CytcO with O2.
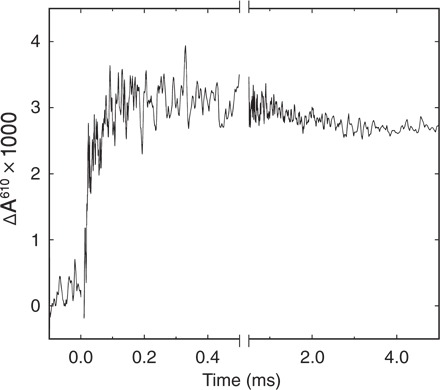
The reaction was monitored at 610 nm. Experimental conditions after mixing: 0.6 to 0.8 μM CytcO (scaled to 1 μM), 0.05% DDM, and 90 mM Hepes (pH 7.4). A laser artifact at t = 0 has been truncated for clarity.
Computation results
Previous computational studies (see Introduction), have suggested a two-step mechanism for the O–O bond cleavage upon reaction of the two-electron reduced CytcO with O2 (Fig. 5) (48, 50). Starting from state A, in the first step, a proton is transferred from Tyr237 (see Fig. 1) to the oxygen molecule, together with two electrons (one from the heme iron and one from either CuB or Tyr), resulting in the formation of a Fe3+–O−–O−(H+) peroxide intermediate (IP). In the second step (dashed line), the O–O bond is cleaved, forming state PM, with a hydroxyl group on CuB and a ferryl.
Fig. 5. Mechanism of the initial steps of O2 reduction catalyzed by the ba3 CytcO.
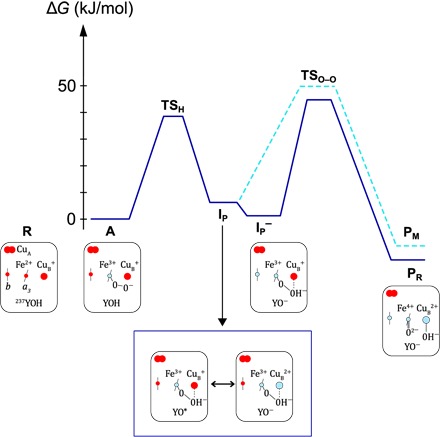
Energy profiles for the initial reaction steps after binding of O2 to heme a3, reflecting the approximate rate constants observed at pH 7 and 10°C. The reduced CytcO (state R) binds O2 to state A. In state IP, a peroxy state is formed upon oxidation of the heme a3 iron and either CuB or Tyr237. The peroxy state is stabilized by a proton from Tyr237. The reaction sequence below the diagram illustrates the specific electron transfers within the ba3 CytcO. Two possible configurations that are in resonance are shown for state IP. It is a geometric single minimum with an electronic structure that is a mixture (resonance) of two main electronic configurations.
Considering the reaction of the four-electron reduced CytcO with O2, when is the electron transferred from the low-spin heme b to the catalytic site? The present calculations show that the electron affinity of intermediate A is significantly lower than that of the electron donor heme b [also compare the study of Blomberg (54)], which is expected because there is no strong electron acceptor at the catalytic site. On the other hand, in the peroxide intermediate IP, an electron acceptor, either Cu2+ or the tyrosyl radical, is created, resulting in an increase of the calculated electron affinity by about ~50 kJ/mol (not shown in Fig. 5). This result indicates that the product of the electron transfer is a reduced peroxide intermediate, IP− (Fig. 5), that relaxes to PR upon cleaving the O–O bond.
The transition state for O–O bond cleavage (TSO–O) was determined for both IP and IP− intermediates. The calculated barrier heights are quite similar, 38.5 and 36.8 kJ/mol, respectively (from IP and IP−, respectively, to TSO–O in Fig. 5). The rate constants for PR and PM formation are determined by the energy difference between intermediate A and the highest point on the energy profiles. A transition state was determined for the initial proton transfer step (TSH), but because the calculated barrier height depends on the number of water molecules included in the model, a definite value could not be determined. However, it is estimated that TSH is similar to or lower than TSO–O, and therefore, the position of TSO–O (that is, the highest point) determines the formation rate constant of both PR and PM. The height of this rate-limiting barrier is the sum of the endergonicity for the formation of the peroxide intermediate (IP or IP−) and the O–O bond cleavage barrier relative to the peroxide.
Using the present model without considering any charges in the K pathway, we found the peroxide intermediate IP to be 30.5 kJ/mol above intermediate A (this level is not shown in Fig. 5), which would lead to a total barrier for PM formation of almost 70 kJ/mol. This is much higher than the activation free energy of about 50 kJ/mol, corresponding to the experimentally observed rate constant. Therefore, in previous computational studies, we investigated factors that could influence the barrier height. In particular, it was shown that a positive charge, for example, at Lys362 of the R. sphaeroides CytcO (55), in the vicinity of the negatively charged tyrosinate, significantly stabilizes the peroxide intermediate IP relative to A (48, 50). Therefore, it can be suggested that the positive charge would yield an estimated difference between IP and A, as shown in Fig. 5, yielding an overall barrier of about 50 kJ/mol for PM formation (highest barrier from A to PM; see Fig. 5). Note as well that with the smaller energy difference between A and IP, the latter would remain unobservable (populated to ~5%), in agreement with the experiments. Furthermore, the calculated electron affinity of intermediate IP is similar to the calculated electron affinities of other intermediates of the catalytic cycle, which precede electron transfer to the catalytic site (54). Hence, it is reasonable to assume that the electron transfer step to form state IP− is somewhat exergonic, putting this state close enough in energy to intermediate A to become observable, in accordance with the present experiments (see Fig. 5). Because the O–O bond cleavage barrier relative to the preceding peroxide state is rather similar for IP and IP− (see above), the difference in rate constants for PM and PR formation is mainly the result of the exergonicity of the electron transfer from heme b to IP, forming IP−. In summary, the qualitative picture of the O–O bond cleavage step obtained from the quantum chemical calculations is used to construct the energy profiles presented in Fig. 5, wherein the detailed relative energies are adapted to fit the present experimental data at low temperatures and neutral pH.
DISCUSSION
As described in detail in the Introduction, early models assumed that immediately after O2 binding to heme a3, a peroxy state is formed, which is the first reaction intermediate that is experimentally observed. Accordingly, this state was called P. Results from studies done after 1990 indicate that the O–O bond is already broken in the P state.
In an earlier study with the T. thermophilus ba3 CytcO, we noted that oxidation of heme b (absorbance decrease at 560 nm) was slightly faster than formation of PR (absorbance increase at 610 nm) at ambient temperatures (56), but the difference was not sufficiently large to be discussed in depth at that time. Furthermore, on the basis of results obtained earlier with the A-type CytcO, we assumed that heme b oxidation and PR formation should display the same rate constants, and therefore, in the model, both processes were fitted to the same transition rate constant. Here, we observed that the difference in rate constants is significantly more pronounced at low temperatures, where, at 10°C (pH 7), oxidation of the low-spin heme b occurs faster by a factor of ~10 than formation of the PR state; that is, it was not possible to fit the two processes to the same rate constant (see residuals in Fig. 2A). This relatively large difference in rate constants also allowed us to separately determine the temperature dependence of each process (Fig. 3).
The O2 binding is slightly faster than that previously observed (51) (5.3 μs) and falls between the values obtained by Szundi et al. (57) (~1 or ~10 μs), depending on experimental conditions. Using a slightly different experimental approach, Szundi et al. (57) suggested that the CO ligand may interfere with O2 binding, thereby slowing, for example, heme b oxidation by a factor of ~10, from 5 to 60 μs. As discussed previously (56), we did not observe this interference from CO dissociation.
The Arrhenius activation energy for PR formation in the R. sphaeroides CytcO is ~20 kJ/mol (37), that is, lower than that for PR formation with the ba3 CytcO but about the same as that for heme b oxidation (see Table 2). In earlier studies, the value for another transition (F→O) was determined [~42 kJ/mol (58)]. This reaction involves both electron transfer from heme b to the catalytic site and proton transfer from the solution. Previous experience has shown that the type of computational studies used in this study to interpret the data yields better agreement with the experiment for free energies than for the partitioning into enthalpy and entropy contributions, which may be related to the fact that the source for this partitioning is not always located in the active site (49, 59). Therefore, we present the parameters in Table 2 without further discussion.
Table 2. Thermodynamic parameters.
The parameters were determined by fitting the data in Fig. 3 with the expression . The range of A values was 30 to 40% of the values in the table (when taking into account the error bars in Fig. 3).
| pH 7 | pH 10 | |||
| Ea (kJ/mol) | ln A | Ea (kJ/mol) | ln A | |
|
Heme b oxidation (560-nm first decay) |
17 ± 2 | 18 | 40 ± 4 | 27 |
|
PR formation (610-nm increase) |
60 ± 5 | 34 | 60 ± 5 | 34 |
When heme b becomes oxidized before state PR is formed, a question regarding the identity of the electron acceptor arises. Upon binding of O2, that is, forming of state A, in the fully reduced CytcO, none of the redox sites can accept any additional electrons. State PR is formed over a time scale of 110 μs at 10°C (Fig. 2A and Table 1). Consequently, if PR was the first intermediate to be formed at the catalytic site after binding of O2 (intermediate A), there would be no available electron acceptor over the time scale wherein we observed oxidation of heme b (τ ≅ 11 μs at 10°C; see Fig. 2A). The PM state was formed with a time constant of ~140 μs at ~20°C (Fig. 4); that is, this state was not formed before electron transfer from heme b. To explain the data, we must therefore consider mechanisms to create an electron acceptor after binding of O2 but before breaking of the O–O bond. One plausible possibility is to form a transient peroxy intermediate over the same time scale as electron transfer from heme b. This intermediate was suggested by Blomberg et al. (48–50), and a reaction scheme including this state (IP) is presented in Fig. 5. The absorbance change associated with the A→IP− reaction would then correspond to the difference between states heme b2+ Fe2+–O2 and heme b3+ Fe3+–O−–O−(H+), which would be dominated by the absorbance change associated with oxidation of heme b. This is because the O–O bond length in state Fe2+–O2 is in between that of dioxygen and peroxide, similar to a superoxide coordinated to a ferric iron (Fe3+–O2−) (60), and we assume that no major absorbance changes occur at heme a3 during the transition from a superoxide-like state to a peroxy state.
According to the theoretical model (48–50), in state IP, the heme a3 iron is oxidized and an electron is transferred to O2 from either CuB or Tyr237, wherein the latter in both cases is also the proton donor (Fig. 5). In the absence of an electron at heme b, state IP would relax to PM. However, if heme b is reduced, as in our experiments, then the formation of IP would be accompanied by electron transfer from heme b to the catalytic site, forming state IP−, which is lower in energy than IP. In this model, observation of IP− is possible only if the energy level of this state is sufficiently close to that of state A. As outlined in Results, the time constant for the overall A→PR/M reaction is dependent on the energy difference between state A and the highest transition state TSO–O (Fig. 5). Because IP− is lower in energy than IP and the energy difference between IP and TSO–O is approximately the same as that between IP− and TSO–O, the highest energy barrier along the trajectory from A to PR is lower than that leading to PM. This difference explains why the formation of PR occurs over a shorter time scale than the formation of PM. Furthermore, according to the model, the IP state would not be observed upon the reaction of the mixed-valence CytcO with O2 because state IP is higher in energy than IP−; that is, with the mixed-valence CytcO, state IP would not be populated.
Next, we discuss differences in the data obtained at pH 7 and 10, respectively. We note that at high pH, the rate constants of heme b oxidation and PR formation displayed more similar slopes in the Arrhenius plots than at pH 7 (Fig. 3). The proposed model involves proton transfer from the Tyr residue to stabilize the peroxy state in IP (Fig. 5) (48), which would also take place at high pH because the Tyr237 residue in the ba3 CytcO is presumably protonated [for example, see the study of Koutsoupakis et al. (52)]. To explain the larger slope in the temperature dependence of heme b oxidation at high pH, we assume that the K pathway “below” Tyr237 becomes more negative (or less positive) upon increasing the pH. This change in the charge would stabilize the proton at Tyr237 such that the height of the transition state TSH would increase slightly at high pH. This change in the barrier height would act to slow the initial reaction step A→IP. In the T. thermophilus ba3 CytcO, which lacks a residue equivalent to Lys362, the overall charge may be determined by the protonation state of several groups in the K pathway, including Tyr244, Tyr248 (SU I), and Glu15 (SU II) (18, 52, 61, 62).
Assuming that the suggested scenario is generally applicable for respiratory oxidases, the data from this study would point to a mechanism for the initial steps of O2 reduction at the catalytic site: Electron transfer from the low-spin heme to the catalytic site would coincide with formation of a peroxy state, as originally suggested (23, 25), but the peroxy state Fe3+–O−–O−(H+) would only be formed transiently, and it would not always be observed. This is because the intermediate would be observed (that is, populated to a sufficient concentration) only if the energy level of IP− is sufficiently close to that of intermediate A, which may not be the case in all CytcOs.
A question then arises, why is the formation of the “true” peroxy state [Fe3+–O−–O−(H+)] observed in the ba3 CytcO but has not been observed in earlier studies with the A-type CytcOs? One possible explanation is that structural differences in the K pathway, as discussed above, would lead to differences in the transition state TSH (Fig. 5). Another possibility is that the different CytcOs are optimized to operate at different temperatures. With the ba3 CytcO, electron transfer from heme b to the catalytic site and PR formation are presumably synchronized at temperatures >45°C, but the different slopes in the temperature dependencies of the rate constants lead to a clear separation in the rate constants at 10°C. Other A-type CytcOs are presumably optimized to operate at lower temperatures, and a separation of the two rate constants would be observed only at temperatures significantly lower than 10°C. In other words, we assume the same mechanism but slightly shifted relative energy levels in the diagram in Fig. 5 for the different CytcOs.
SUMMARY
Earlier theoretical studies predicted a high-energy peroxy state, IP [Fe3+–O−–O−(H+)], formed along the reaction pathway leading to O–O bond breaking at the catalytic site of the heme-copper oxidases. Here, we showed that at low temperatures, with the ba3 T. thermophilus CytcO, an electron acceptor is formed at the catalytic site after ~11 μs, before the O–O bond breaking (τ ≅ 110 μs). This electron acceptor is suggested to be the predicted IP state, which, after electron transfer to the catalytic site (state IP−), becomes significantly populated in the T. thermophilus CytcO at low temperature.
MATERIALS AND METHODS
Purification of ba3 CytcO
T. thermophilus HB8 strain YC 1001 (with a deletion of the cba gene) was transformed with a plasmid encoding the ba3 CytcO gene with a 7-His tag at the N terminus of subunit I and cultivated at 60°C with mild shaking (125 rpm), as described by Chen et al. (63) [see also the study of Keightley et al. (64)]. The harvested cells were suspended in 100 mM Hepes (pH 8) at a ratio of about 1:3, together with a small amount of deoxyribonuclease. The cells were broken using a constant flow cell disrupter at 190 MPa (Constant Systems). The protease inhibitor phenylmethylsulfonyl fluoride was added but after breaking the cells to avoid excessive foaming during the breaking procedure. The solution was centrifuged for 1 hour at 205,000g at 16°C. The pellet was homogenized in 50 ml of 50 mM Hepes (pH 8) and 2.5% Triton X-100 for each 10 g of cell membrane, and the sample was incubated overnight at 4°C. The sample was centrifuged for 1 hour at 170,000g, and the obtained supernatant was supplemented with 10 mM imidazole and loaded onto a prepacked 5-ml Ni–nitrilotriacetic acid affinity chromatography column equilibrated with 10 mM Hepes (pH 8), 150 mM NaCl, 10 mM imidazole, and 1% Triton X-100. After binding of the CytcO, the column was washed with 10 mM Hepes (pH 8), 150 mM NaCl, 40 mM imidazole, and 1% Triton X-100, until the solution passing through the column was clear. Elution of the enzyme was obtained with 10 mM Hepes (pH 8), 150 mM NaCl, 250 mM imidazole, and 1% Triton X-100. The brown elution fractions were pooled and dialyzed overnight at 4°C in 1 liter of 5 mM Hepes (pH 8) and 0.05% Triton X-100 first and then in 1 liter of 5 mM Hepes (pH 8) and 0.05% dodecyl-β-d-maltoside (DDM). The enzyme was stored in this last buffer at 4°C until use.
The concentration of heme a3 was determined from the fully reduced minus oxidized absorption difference spectrum using the absorption coefficient ε(613–658) = 6.3 mM−1 cm−1. The concentration of heme b was calculated from the fully reduced spectrum using ε(560–590) = 26 mM−1 cm−1.
Flow-flash measurements
Purified ba3 CytcO was diluted to a final concentration of ~10 μM with 5 mM Hepes and 0.05% DDM (pH 7.4) and placed in a Thunberg cuvette. The sample was made anaerobic on a vacuum line, and air was replaced for N2. The CytcO was then reduced by 2 mM sodium ascorbate using 0.5 μM phenazine methosulfate as a mediator. Incubation under 150 kPa N2 for approximately 1 hour led to the formation of the fully reduced state. Last, the atmosphere was exchanged for CO, and complete binding to the catalytic site was observed after ~20 min of incubation under 100 kPa CO.
The flow-flash measurements were performed using a modified stopped-flow setup (Applied Photophysics) (65, 66). Briefly, the ba3 CytcO was mixed at a ratio of 1:5 with oxygen-saturated buffer [100 mM Hepes and 0.05% DDM (pH 7) or 100 mM 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) and 0.05% DDM (pH 10)]. After ~30 ms, a 10-ns laser flash (200 mJ, 532 nm; Nd-YAG laser Quantel) was used to dissociate the CO ligand, which initiated the catalytic reaction. Absorbance changes were monitored at specific wavelengths (see text and figure legends). The temperature in the optical chamber was varied using a thermostated water bath.
To prepare the mixed-valence CytcO, the oxidized CytcO [~10 μM in 5 mM Hepes (pH 7.4) and 0.05% DDM] was transferred to a Thunberg cuvette, after which air was replaced by N2. Next, the gas was exchanged for CO, after which the sample was incubated for 2 to 3 hours. Formation of the mixed valence state was confirmed from the optical absorption spectrum (oxidized heme b and reduced heme a3).
Data handling and analysis
The data were collected using a digital oscilloscope in which 107 sampling points were averaged to ~103 points using a pseudo-logarithmic oversampling function available in the LKS software (Applied Photophysics). If necessary, the traces were smoothed by averaging nine points in a moving time window. Data points in the time window 0 to 1 μs were removed to facilitate the analysis by eliminating a laser artifact (because of incomplete shielding of the detector from the laser flash). The signals were fitted to a model describing irreversible, sequential reactions using KinTek software (KinTek Corp.).
Computational details
Density functional theory was used to study the O–O bond cleavage in CytcO. The same model and methodology were used as those described by Blomberg and Siegbahn (67). The model of the catalytic site was based on the crystal structure of CytcO from R. sphaeroides (1). It contained heme a3 and the CuB complex, including the cross-linked tyrosine. This part of the catalytic site is very similar to that of the ba3 CytcOs, which means that the same model could be used to describe the details of the O–O bond cleavage step in both systems. The hybrid density functional B3LYP*-D3 [with 15% exact exchange and dispersion correction (68–70)] was used together with a polarized double zeta basis set (lacvp*) for the geometry optimizations and together with the large cc-pvtz(−f) basis plus lacv3p+ for the metals for the energy calculations (71). Polarizing effects from the surrounding protein were included using the self-consistent reaction field approach, and zero-point corrections to the energies were obtained from Hessian calculations. Entropy effects on the relative energies were found to be negligible for the investigated part of the reaction. For details on the model and the methods, see the study of Blomberg and Siegbahn (67). As previously noted (54), the computational results yield a qualitative picture of the reaction mechanism. In particular, spin states and redox properties of heme groups may not be correctly reproduced by density functional theory. Another difficulty concerns the description of water molecules in the catalytic site. The number and positions of water molecules seen in the crystal structures of the catalytic site vary for the different CytcOs. Because this situation is difficult to handle computationally, the most accurate relative energies are obtained when no water molecules are included in the model. An obvious exception is the investigation of proton transfer reactions, where water molecules need to be included in the model. To obtain a more quantitative picture of the energetics of a reaction mechanism, a careful combination of computational and experimental results has to be used (54, 72).
Acknowledgments
The wild-type T. thermophilus strain was obtained from R. B. Gennis at the University of Illinois at Urbana-Champaign. Funding: These studies were supported by grants from the Knut and Alice Wallenberg Foundation (KAW 2013.0006) and the Swedish Research Council (2014-4306 and 2015-04512). Author contributions: F.P. and C.v.B. (N.G. initially supervised F.P.) performed experiments and evaluated experimental data. M.R.A.B. performed calculations. P.B., P.Ä., and C.v.B. interpreted the data. P.B. and M.R.A.B. (the part on calculations) wrote the manuscript. All authors commented on the text, format of presentation, and data evaluation and interpretation. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Hosler J. P., Ferguson-Miller S., Mills D. A., Energy transduction: Proton transfer through the respiratory complexes. Annu. Rev. Biochem. 75, 165–187 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshikawa S., Muramoto K., Shinzawa-Itoh K., Aoyama H., Tsukihara T., Shimokata K., Katayama Y., Shimada H., Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 1757, 1110–1116 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Namslauer A., Brzezinski P., Structural elements involved in electron-coupled proton transfer in cytochrome c oxidase. FEBS Lett. 567, 103–110 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Brzezinski P., Gennis R. B., Cytochrome c oxidase: Exciting progress and remaining mysteries. J. Bioenerg. Biomembr. 40, 521–531 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brzezinski P., Ädelroth P., Design principles of proton-pumping haem-copper oxidases. Curr. Opin. Struct. Biol. 16, 465–472 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Richter O.-M. H., Ludwig B., Electron transfer and energy transduction in the terminal part of the respiratory chain—Lessons from bacterial model systems. Biochim. Biophys. Acta 1787, 626–634 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Ferguson-Miller S., Hiser C., Liu J., Gating and regulation of the cytochrome c oxidase proton pump. Biochim. Biophys. Acta 1817, 489–494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rich P. R., Maréchal A., Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. J. R. Soc. Interface 10, 183–196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaila V. R. I., Verkhovsky M. I., Wikström M., Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 110, 7062–7081 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Konstantinov A. A., Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 586, 630–639 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Blomberg M. R. A., Siegbahn P. E. M., Proton pumping in cytochrome c oxidase: Energetic requirements and the role of two proton channels. Biochim. Biophys. Acta 1837, 1165–1177 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Popović D. M., Leontyev I. V., Beech D. G., Stuchebrukhov A. A., Similarity of cytochrome c oxidases in different organisms. Proteins 78, 2691–2698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Ballmoos C., Ädelroth P., Gennis R. B., Brzezinski P., Proton transfer in ba3 cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta 1817, 650–657 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Wikström M., Sharma V., Kaila V. R. I., Hosler J. P., Hummer G., New perspectives on proton pumping in cellular respiration. Chem. Rev. 115, 2196–2221 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Pereira M. M., Santana M., Teixeira M., A novel scenario for the evolution of haem–copper oxygen reductases. Biochim. Biophys. Acta 1505, 185–208 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Hemp J., Gennis R. B., Diversity of the heme–copper superfamily in archaea: Insights from genomics and structural modeling. Results Probl. Cell Differ. 45, 1–31 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Lee H. J., Reimann J., Huang Y., Ädelroth P., Functional proton transfer pathways in the heme–copper oxidase superfamily. Biochim. Biophys. Acta 1817, 537–544 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Chang H. Y., Hemp J., Chen Y., Fee J. A., Gennis R. B., The cytochrome ba3 oxygen reductase from Thermus thermophilus uses a single input channel for proton delivery to the active site and for proton pumping. Proc. Natl. Acad. Sci. U.S.A. 106, 16169–16173 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soulimane T., Buse G., Bourenkov G. P., Bartunik H. D., Huber R., Than M. E., Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 19, 1766–1776 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiefenbrunn T., Liu W., Chen Y., Katritch V., Stout C. D., Fee J. A., Cherezov V., High resolution structure of the ba3 cytochrome c oxidase from Thermus thermophilus in a lipidic environment. PLOS ONE 6, e22348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luna V. M., Chen Y., Fee J. A., Stout C. D., Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: Structural analysis and role of oxygen transport channels in the heme–Cu oxidases. Biochemistry 47, 4657–4665 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Chance B., Saronio C., Leigh J. S. Jr., Functional intermediates in the reaction of membrane-bound cytochrome oxidase with oxygen. J. Biol. Chem. 250, 9226–9237 (1975). [PubMed] [Google Scholar]
- 23.Wikström M., Energy-dependent reversal of the cytochrome oxidase reaction. Proc. Natl. Acad. Sci. U.S.A. 78, 4051–4054 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babcock G. T., Varotsis C., Zhang Y., O2 activation in cytochrome oxidase and in other heme proteins. Biochim. Biophys. Acta 1101, 192–194 (1992). [PubMed] [Google Scholar]
- 25.Babcock G. T., Wikström M., Oxygen activation and the conservation of energy in cell respiration. Nature 356, 301–309 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Babcock G. T., How oxygen is activated and reduced in respiration. Proc. Natl. Acad. Sci. U.S.A. 96, 12971–12973 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng L., Baker G. M., Reaction of hydrogen peroxide with the rapid form of resting cytochrome oxidase. Biochemistry 30, 5727–5733 (1991). [DOI] [PubMed] [Google Scholar]
- 28.Proshlyakov D. A., Ogura T., Shinzawa-Itoh K., Yoshikawa S., Appelman E. H., Kitagawa T., Selective resonance Raman observation of the “607 nm” form generated in the reaction of oxidized cytochrome c oxidase with hydrogen peroxide. J. Biol. Chem. 269, 29385–29388 (1994). [PubMed] [Google Scholar]
- 29.Proshlyakov D. A., Pressler M. A., Babcock G. T., Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 95, 8020–8025 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proshlyakov D. A., Ogura T., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T., Microcirculating system for simultaneous determination of Raman and absorption spectra of enzymatic reaction intermediates and its application to the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry 35, 76–82 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa T., Ogura T., Time-resolved resonance Raman investigation of oxygen reduction mechanism of bovine cytochrome c oxidase. J. Bioenerg. Biomembr. 30, 71–79 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Fabian M., Wong W. W., Gennis R. B., Palmer G., Mass spectrometric determination of dioxygen bond splitting in the “peroxy” intermediate of cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 96, 13114–13117 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gennis R. B., Multiple proton-conducting pathways in cytochrome oxidase and a proposed role for the active-site tyrosine. Biochim. Biophys. Acta 1365, 241–248 (1998). [Google Scholar]
- 34.Blomberg M. R. A., Siegbahn P. E. M., Babcock G. T., Wikström M., O–O bond splitting mechanism in cytochrome oxidase. J. Inorg. Biochem. 80, 261–269 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Blomberg M. R. A., Siegbahn P. E. M., Babcock G. T., Wikström M., Modeling cytochrome oxidase: A quantum chemical study of the O–O bond cleavage mechanism. J. Am. Chem. Soc. 122, 12848–12858 (2000). [Google Scholar]
- 36.Hill B. C., Greenwood C., Spectroscopic evidence for the participation of compound A (Fea32+-O2) in the reaction of mixed-valence cytochrome c oxidase with oxygen at room temperature. Biochem. J. 215, 659–667 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karpefors M., Ädelroth P., Namslauer A., Zhen Y., Brzezinski P., Formation of the “peroxy” intermediate in cytochrome c oxidase is associated with internal proton/hydrogen transfer. Biochemistry 39, 14664–14669 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Oda K., Ogura T., Appelman E. H., Yoshikawa S., The intrinsic stability of the second intermediate following the dioxygen-bound form in the O2 reduction by cytochrome c oxidase. FEBS Lett. 570, 161–165 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Hill B. C., Greenwood C., The reaction of fully reduced cytochrome c oxidase with oxygen studied by flow-flash spectrophotometry at room temperature. Evidence for new pathways of electron transfer. Biochem. J. 218, 913–921 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han S., Ching Y.-C., Rousseau D. L., Cytochrome c oxidase: Decay of the primary oxygen intermediate involves direct electron transfer from cytochrome a. Proc. Natl. Acad. Sci. U.S.A. 87, 8408–8412 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verkhovsky M. I., Morgan J. E., Wikström M., Oxygen binding and activation: Early steps in the reaction of oxygen with cytochrome c oxidase. Biochemistry 33, 3079–3086 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Ädelroth P., Ek M., Brzezinski P., Factors determining electron-transfer rates in cytochrome c oxidase: Investigation of the oxygen reaction in the R. sphaeroides enzyme. Biochim. Biophys. Acta 1367, 107–117 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Sucheta A., Szundi I., Einarsdóttir Ó., Intermediates in the reaction of fully reduced cytochrome c oxidase with dioxygen. Biochemistry 37, 17905–17914 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Graf S., Fedotovskaya O., Kao W.-C., Hunte C., Ädelroth P., Bott M., von Ballmoos C., Brzezinski P., Rapid electron transfer within the III-IV supercomplex in Corynebacterium glutamicum. Sci. Rep. 6, 34098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salomonsson L., Faxén K., Ädelroth P., Brzezinski P., The timing of proton migration in membrane-reconstituted cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 102, 17624–17629 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karpefors M., Ädelroth P., Aagaard A., Smirnova I. A., Brzezinski P., The deuterium isotope effect as a tool to investigate enzyme catalysis: Proton-transfer control mechanisms in cytochrome c oxidase. Isr. J. Chem. 39, 427–437 (1999). [Google Scholar]
- 47.von Ballmoos C., Gennis R. B., Ädelroth P., Brzezinski P., Kinetic design of the respiratory oxidases. Proc. Natl. Acad. Sci. U.S.A. 108, 11057–11062 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blomberg M. R. A., Siegbahn P. E. M., Quantum chemistry applied to the mechanisms of transition metal containing enzymes—Cytochrome c oxidase, a particularly challenging case. J. Comput. Chem. 27, 1373–1384 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Blomberg M. R. A., Borowski T., Himo F., Liao R.-Z., Siegbahn P. E. M., Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 114, 3601–3658 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Blomberg M. R. A., Siegbahn P. E. M., Quantum chemistry as a tool in bioenergetics. Biochim. Biophys. Acta 1797, 129–142 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Siletsky S. A., Belevich I., Jasaitis A., Konstantinov A. A., Wikström M., Soulimane T., Verkhovsky M. I., Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta 1767, 1383–1392 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Koutsoupakis C., Kolaj-Robin O., Soulimane T., Varotsis C., Probing protonation/deprotonation of tyrosine residues in cytochrome ba3 oxidase from Thermus thermophilus by time-resolved step-scan Fourier transform infrared spectroscopy. J. Biol. Chem. 286, 30600–30605 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oliveberg M., Brzezinski P., Malmström B. G., The effect of pH and temperature on the reaction of fully reduced and mixed-valence cytochrome c oxidase with dioxygen. Biochim. Biophys. Acta 977, 322–328 (1989). [DOI] [PubMed] [Google Scholar]
- 54.Blomberg M. R. A., Mechanism of oxygen reduction in cytochrome c oxidase and the role of the active site tyrosine. Biochemistry 55, 489–500 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Brändén M., Sigurdson H., Namslauer A., Gennis R. B., Ädelroth P., Brzezinski P., On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 98, 5013–5018 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Ballmoos C., Lachmann P., Gennis R. B., Ädelroth P., Brzezinski P., Timing of electron and proton transfer in the ba3 cytochrome c oxidase from Thermus thermophilus. Biochemistry 51, 4507–4517 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Szundi I., Funatogawa C., Fee J. A., Soulimane T., Einarsdóttir Ó., CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus. Proc. Natl. Acad. Sci. U.S.A. 107, 21010–21015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giuffrè A., Forte E., Antonini G., D’Itri E., Brunori M., Soulimane T., Buse G., Kinetic properties of ba3 oxidase from Thermus thermophilus: Effect of temperature. Biochemistry 38, 1057–1065 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Isaksen G. V., Åqvist J., Brandsdal B. O., Protein surface softness is the origin of enzyme cold-adaptation of trypsin. PLOS Comput. Biol. 10, e1003813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blomberg L. M., Blomberg M. R. A., Siegbahn P. E. M., A theoretical study on the binding of O2, NO and CO to heme proteins. J. Inorg. Biochem. 99, 949–958 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Smirnova I., Chang H.-Y., von Ballmoos C., Ädelroth P., Gennis R. B., Brzezinski P., Single mutations that redirect internal proton transfer in the ba3 oxidase from Thermus thermophilus. Biochemistry 52, 7022–7030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smirnova I., Reimann J., von Ballmoos C., Chang H.-Y., Gennis R. B., Fee J. A., Brzezinski P., Ädelroth P., Functional role of Thr-312 and Thr-315 in the proton-transfer pathway in ba3 cytochrome c oxidase from Thermus thermophilus. Biochemistry 49, 7033–7039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Y., Hunsicker-Wang L., Pacoma R. L., Luna E., Fee J. A., A homologous expression system for obtaining engineered cytochrome ba3 from Thermus thermophilus HB8. Protein Exp. Purif. 40, 299–318 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Keightley J. A., Zimmermann B. H., Mather M. W., Springer P., Pastuszyn A., Lawrence D. M., Fee J. A., Molecular genetic and protein chemical characterization of the cytochrome ba3 from Thermus thermophilus HB8. J. Biol. Chem. 270, 20345–20358 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Rydström Lundin C., von Ballmoos C., Ott M., Ädelroth P., Brzezinski P., Regulatory role of the respiratory supercomplex factors in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 113, E4476–E4485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Björck M. L., Zhou S., Rydström Lundin C., Ott M., Ädelroth P., Brzezinski P., Reaction of S. cerevisiae mitochondria with ligands: Kinetics of CO and O2 binding to flavohemoglobin and cytochrome c oxidase. Biochim. Biophys. Acta 1858, 182–188 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Blomberg M. R. A., Siegbahn P. E. M., How cytochrome c oxidase can pump four protons per oxygen molecule at high electrochemical gradient. Biochim. Biophys. Acta 1847, 364–376 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Becke A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- 69.Reiher M., Salomon O., Hess B. A., Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 107, 48–55 (2001). [Google Scholar]
- 70.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Jaguar 7.6 (Schrödinger-LLC, New York, 2009).
- 72.Blomberg M. R. A., Siegbahn P. E. M., Improved free energy profile for reduction of NO in cytochrome c dependent nitric oxide reductase (cNOR). J. Comput. Chem. 37, 1810–1818 (2016). [DOI] [PubMed] [Google Scholar]