

Abstract
A theoretical model of haemoglobin is presented to explain an anomalous cationic Hofmeister effect observed in protein aggregation. The model quantifies competing proposed mechanisms of non-electrostatic physisorption and chemisorption. Non-electrostatic physisorption is stronger for larger, more polarizable ions with a Hofmeister series Li+< K+< Cs+. Chemisorption at carboxylate groups is stronger for smaller kosmotropic ions, with the reverse series Li+ > K+ > Cs+. We assess aggregation using second virial coefficients calculated from theoretical protein–protein interaction energies. Taking Cs+ to not chemisorb, comparison with experiment yields mildly repulsive cation–carboxylate binding energies of 0.48 kBT for Li+ and 3.0 kBT for K+. Aggregation behaviour is predominantly controlled by short-range protein interactions. Overall, adsorption of the K+ ion in the middle of the Hofmeister series is stronger than ions at either extreme since it includes contributions from both physisorption and chemisorption. This results in stronger attractive forces and greater aggregation with K+, leading to the non-conventional Hofmeister series K+ > Cs+ ≈ Li+.
Keywords: non-electrostatic cation interaction, Hofmeister effects, haemoglobin aggregation, chemisorption model, physisorption, second virial coefficient
1. Introduction
Hofmeister effects refer to differences in the behaviour of a solution arising from the specific identity of the electrolyte. Hofmeister originally observed the effect of salts on protein solubility [1]. He reported a series in the ability of cations (in sulphate and other salts) to precipitate egg-white protein in the order Li+< Na+< K+< NH4+, alongside a similar series in anions for which the effect was stronger. Specific ion effects have subsequently been observed in a wide range of systems, animal [2–5], mineral [6–9] and otherwise [10–13] in nature. Hofmeister's series was found widely across these systems, even in ionic liquids [14]. An observation could be made that smaller, strongly hydrated ions ‘kosmotropes’ such as Li+, Na+ are found at one end of the series, while larger, weakly hydrated ions like K+, NH4+ are found at the other. So the Hofmeister effect could be understood to be controlled by a single property of the ions, whether size, polarizability or ion–water interaction [15].
Depending on the specific conditions (pH, surface hydrophobicity), the Hofmeister series can be found in both the forward and reverse series [3,16,17]. Unsurprisingly, minor permutations between ions frequently appear. But a more intriguing ‘violation’ of the conventional Hofmeister series was observed by Medda et al. [18] in the aggregation of haemoglobin molecules. Turbidity measurements showed a cation series in aggregation of Li+< Cs+< Na+< K+ ≈ Rb+. The lighter cations appeared in the conventional order, Li+< Na+< K+. The anomaly here was Cs+. As a large chaotropic ion, we would conventionally expect Cs+ to appear to the right of K+. Instead, it had an effect similar to that of Li+. The same anomalous cation sequence was found in an independent experiment devoted to measuring the Brownian motion of BSA protein [19].
Medda et al. [18] interpreted the Cs+ anomaly to be a consequence of the highly heterogeneous nature of protein surfaces. Proteins have both charged and uncharged surface residues, thus ions specifically compete for these sites through two different types of ion–surface interaction. The process is illustrated in figure 1. On the one hand, ions adsorb to the protein surface or backbone through physisorption (interaction at a distance from the surface). Electrostatic physisorption is not ion-specific, but non-electrostatic physisorption is ion-specific. Depending on the nature of the surface, whether hydrophobic or hydrophilic, non-electrostatic physisorption alone may lead to a standard or reversed Hofmeister series [17,20]. In this work, we quantify non-electrostatic physisorption through ionic dispersion forces [21,22] using a standard dielectric model of the protein surface [23]. This model gives the surface (the protein backbone) a somewhat hydrophobic character with non-electrostatic physisorption following the series Cs+> K+> Li+.
Figure 1.
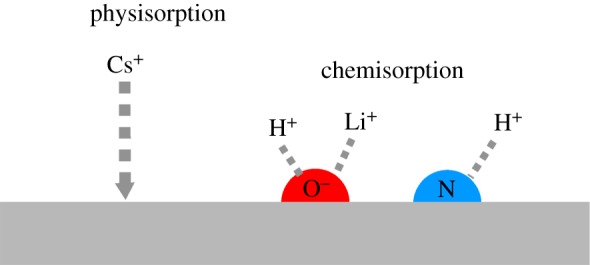
Illustration of physisorption and chemisorption processes.
Alongside non-electrostatic physisorption, however, ions may also interact directly with the surface through chemisorption, binding to specific sites scattered across the protein surface; see figure 2. Non-electrostatic physisorption is expected to be greater for the more polarizable Cs+ ion, with an effect on aggregation in the series Li+< K+< Cs+. But we would expect the kosmotropic Li+ to bind more strongly to kosmotropic carboxylate sites, leading to a chemisorption series in the reverse order, Cs+< K+< Li+. The observed behaviour is a combination of the two effects of physisorption and chemisorption. Chemisorption of Li+ and physisorption of Cs+ are both relatively (and equally) strong compared with K+. It is the ion giving a higher effective surface charge that inhibits aggregation [18,19].
Figure 2.
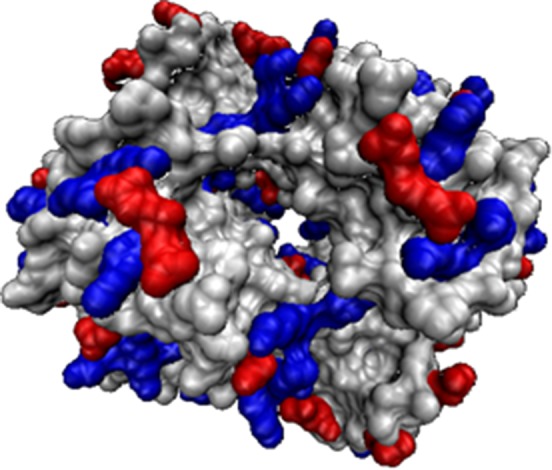
Structural image of the haemoglobin tetramer indicating surface heterogeneity. Negatively charged residues (carboxylates) are red and positively charged residues (amines) are blue; grey regions are uncharged.
Understanding specific cation interactions with proteins is of fundamental importance in life sciences. It is indeed well established that most cells have developed an energy-consuming mechanism to keep low concentrations of sodium and high concentrations of potassium in the cytosol. This is done against concentration gradients by means of ATP energy and an enzyme machine. In this paper, we show how the notions of competing physisorption and chemisorption may be quantified in a theoretical model that provides an estimate of the chemisorption binding energy of kosmotropic cations.
2. Physisorption model
The physisorption profile of ions between two protein surfaces separated by distance L may be estimated using a Poisson–Boltzmann description of the electrolyte. The concentration profile of ion i at a distance z from the surface is given by the Boltzmann relation
| 2.1 |
where ci0 is the bulk concentration of the ion and μi(z) is its interaction energy with the surface (i.e. excess chemical potential) which contains two parts, μi(z) = qiψ(z) + μNESi(z). The first is an electrostatic component qiψ(z) determined by the charge qi of the ion and the electrostatic potential ψ(z) at that point. The second is a non-electrostatic interaction μNESi(z). For this component, we apply Ninham's ion dispersion interaction [25] with each protein surface, μNESi(z) = μdispi(z) + μdispi(L − z), where
| 2.2 |
with 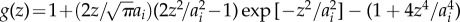
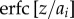 . Here, ion specificity is determined by two parameters, the ion radius [24] ai and the dispersion parameter Bi. Bi is predominantly determined by ion polarizability [25], although also modulated by ion size. Calculation of the Bi coefficient uses the dielectric function of water [26] and a model dielectric function for the protein molecules [23]. Dispersion coefficients and ion radii are given in table 1. The dispersion coefficient of Cs+ is augmented by a factor of 1.5 to enable non-electrostatic physisorption of Cs+ stronger than that of K+ (see μdispi(0) in the table). Ion–protein dispersion interactions of this kind have proved useful in anticipating reversal in the anionic Hofmeister series at pH above and below the isoelectric point of lysozyme [16,27], attributed to charge reversal due to non-electrostatic physisorption [28].
. Here, ion specificity is determined by two parameters, the ion radius [24] ai and the dispersion parameter Bi. Bi is predominantly determined by ion polarizability [25], although also modulated by ion size. Calculation of the Bi coefficient uses the dielectric function of water [26] and a model dielectric function for the protein molecules [23]. Dispersion coefficients and ion radii are given in table 1. The dispersion coefficient of Cs+ is augmented by a factor of 1.5 to enable non-electrostatic physisorption of Cs+ stronger than that of K+ (see μdispi(0) in the table). Ion–protein dispersion interactions of this kind have proved useful in anticipating reversal in the anionic Hofmeister series at pH above and below the isoelectric point of lysozyme [16,27], attributed to charge reversal due to non-electrostatic physisorption [28].
Table 1.
Gaussian radii ai of ions, static polarizability α0 (in vacuum), dispersion coefficients Bi and non-electrostatic physisorption energy (ion dispersion energy at contact) for ions in water interacting with a model protein surface.
| ion | ai (Å) | α0 (Å3) | Bi (10−50 J m3) | μdispi(0) (kBT) |
|---|---|---|---|---|
| Li+·5H2O | 2.56 | 6.648 | −0.700 | −20.754 |
| K+ | 0.96 | 0.814 | −0.322 | −26.591 |
| Cs+ | 0.96 | 2.402 | −1.371 | −27.805 |
| Cl− | 1.86 | 4.861 | −1.258 | −23.538 |
| H3O+ | 0.97 | 0.963 | −0.382 | −27.496 |
| OH−·3H2O | 2.39 | 7.305 | −1.498 | −21.983 |
The non-electrostatic interaction energy μNESi(z) (or μdispi(z)) falls with distance from the surface. But the non-electrostatic physisorption energy is its value when the ion is located at the surface, μpsi = μdispi(z = 0), and is a balance between polarizability (through Bi) and ion size, μpsi = 16Bi/[3(π)1/2a3].
The electrostatic potential ψ(z) is determined by solving Poisson's equation for the given charge distributions qici(z) with surface charge determined by chemisorption.
3. Chemisorption model
We apply a competitive site-binding model of chemisorption [29], involving ion binding to surface sites with a finite surface density. This model quantifies the notion of ion site binding which has been proposed as a mechanism that disrupts standard Hofmeister series [18,30]. We take haemoglobin in the form of a tetramer, spherical with diameter 5.5 nm. The surface charge of the haemoglobin tetramer is pH-dependent through charge regulation [31]. We consider H+ binding to both carboxylate and amine sites,
| 3.1 |
and
| 3.2 |
The pK binding constants here are intrinsic equilibrium constants, meaning they are evaluated using ion concentrations ci(z = 0) at the site rather than bulk concentrations ci0. We adapt acid constants and site densities for haemoglobin from Matthew et al. [32], averaging over values measured for individual amino acids. Their acid constants are pKMHGc = 5.46 averaged over acidic (carboxylate) residues and pKMHGa = 8.97 for basic (amine) residues. Matthew, Hanania and Gurd presented ‘intrinsic’ acid constants, already adjusted for the electrostatic physisorption energy of H+ (adjusting acid constants is discussed further in [33]). We adjust the acid constants further in order to match the IEP. We take pKc = pKMHGc + ΔGK/(kT ln 10), where the adjustment ΔGK =−5.32 kBT, is fitted to ensure that the surface potential in 100 mM CsCl (including ion dispersion interactions) is zero at the IEP, pH 7.1. We interpret this fitted adjustment as a correction for non-electrostatic physisorption, and as compensation for averaging the various pK values of the different amino acid residues. The final acid constants used are pKc = 3.15 and pKa = 6.66. Site densities are Nc = 0.800 nm−2 and Na = 0.863 nm−2 for carboxylate and amine sites, respectively. Site densities are determined from the total number of unmasked (titratable) residues (76 carboxylate sites and 82 amine sites in a haemoglobin tetramer), smeared over the surface of the spherical tetramer. Surface potentials and total protein charge are shown below as a function of pH in figure 4.
Figure 4.

(a) Surface potential and (b) total haemoglobin tetramer charge as a function of pH in 100 mM solutions of LiCl, KCl or CsCl.
Ion-specific chemisorption is modelled through competitive cation binding [29,34] at carboxylate sites,
| 3.3 |
We assume here that ion binding (apart from H+) at amine sites is weak [35]. Likewise, we assume that anion binding at carboxylate sites is negligible. Cations compete with hydrogen ions to bind to the carboxylate sites. In this work, we treat pKM as a fitting parameter to align theory with experiment.
The total surface charge is the sum of charges at carboxylate and amine sites:
| 3.4 |
The charge at each site is determined by the amount Γ of ion (H+ and M+) chemisorbed [29]
| 3.5 |
and
| 3.6 |
Here, qe refers to the elementary charge, qe = 1.6022 × 10−19 C. Chemisorption quantities Γ are determined from physisorbed concentrations cpsi = ci(z = 0) and binding constants,
| 3.7 |
| 3.8 |
| 3.9 |
The A parameters here are a measure of the total degree of binding at each site,
| 3.10 |
and
| 3.11 |
4. Protein interaction free energy
Protein aggregation is determined by the interaction free energy G(L) between two protein tetramers. We estimate the interaction free energy of the spherical haemoglobin tetramers from the interaction free energy Gflat (taken as a surface energy, the free energy per unit area) of an idealized flat haemoglobin surface. That is, the Poisson–Boltzman model-determining ion concentration profiles and electrostatic potential is solved for the flat surface. The relationship between the spherical and flat interaction energies is given by the proximity force approximation of Derjaguin [36], 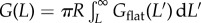 , where R is the radius of a tetramer.
, where R is the radius of a tetramer.
The free energy Gflat(L) is composed of various contributions [33,37],
| 4.1 |
Here, GvdW refers to the protein–water–protein van der Waals interaction GvdW =−A/12πL2. We use a Hamaker constant A = 3.522 kT (1.450 × 10−20 J). Gen accounts for the configuration entropy of the ion concentration profiles [38]. Gel describes the electrostatic energy of the electric field generated by surface and ion charges [37,38]. For brevity, we omit details of these terms.
GNES accounts for non-electrostatic physisorption, through the non-electrostatic interaction energy μNESi(z),
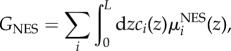 |
4.2 |
Gchem accounts for chemisorption, the charge transfer energy required to form the surface site charges σc and σa. Using the framework for describing competitive site binding [29], the chemisorption energy is Gchem = Gcchem + Gachem, with
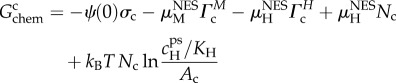 |
4.3 |
and
| 4.4 |
Three qualitatively distinct parts of the chemisorption free energy can be noted. The first is an electrostatic energy, −ψ(0)σ, which partially cancels [39] against Gel. The middle term involves ‘μNESΓ’. This is significant since it means that the magnitude of the chemisorption energy is determined in part by the non-electrostatic physisorption energy μNES of potential-determining ions. The third part involves ‘kBT N lnA’ which describes the entropy of surface sites.
5. Matching theory to experiment
The experiment we are modelling in this paper is based on turbidimetric pH titrations of haemoglobin [18]. This technique provided us information on the effect of pH on the aggregation/disaggregation of haemoglobin molecules in the presence of different chloride salts. In figure 3, the extent of aggregation is estimated by the ratio of transmittance 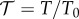 at different pH values (T0 is the transmittance of the optically clear haemoglobin solution). The protein suspension is optically clear below pH 5 (T/T0 = 1); then the transmittance falls as aggregation of haemoglobin proceeds as a consequence of the increase in pH. T/T0 reaches a minimum at about pH 7.4 (IEP); then it increases due to the re-dissolution of protein aggregates, reaching a T/T0 of about 1 at pH > 9. Optically clear protein solutions are obtained at pH values far from the IEP due to repulsive forces as a consequence of ion adsorption due to the electric charges carried by the protein surface at a pH far from the IEP. Figure 3 shows how haemoglobin solubility is modulated by the presence of different 50 mM chloride salts in a cation-specific way. At the starting pH 4.5 haemoglobin carries a positive net charge, so that cations behave as coions. In figure 3, if we consider the trends at
at different pH values (T0 is the transmittance of the optically clear haemoglobin solution). The protein suspension is optically clear below pH 5 (T/T0 = 1); then the transmittance falls as aggregation of haemoglobin proceeds as a consequence of the increase in pH. T/T0 reaches a minimum at about pH 7.4 (IEP); then it increases due to the re-dissolution of protein aggregates, reaching a T/T0 of about 1 at pH > 9. Optically clear protein solutions are obtained at pH values far from the IEP due to repulsive forces as a consequence of ion adsorption due to the electric charges carried by the protein surface at a pH far from the IEP. Figure 3 shows how haemoglobin solubility is modulated by the presence of different 50 mM chloride salts in a cation-specific way. At the starting pH 4.5 haemoglobin carries a positive net charge, so that cations behave as coions. In figure 3, if we consider the trends at 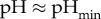 , cations promote haemoglobin aggregation in the order Rb+ > K+ ≈ Na+ > Cs+ > Li+ (50 mM salts) or K+ ≈ Rb+ > Na+ > Cs+ > Li+ (100 mM).
, cations promote haemoglobin aggregation in the order Rb+ > K+ ≈ Na+ > Cs+ > Li+ (50 mM salts) or K+ ≈ Rb+ > Na+ > Cs+ > Li+ (100 mM).
Figure 3.
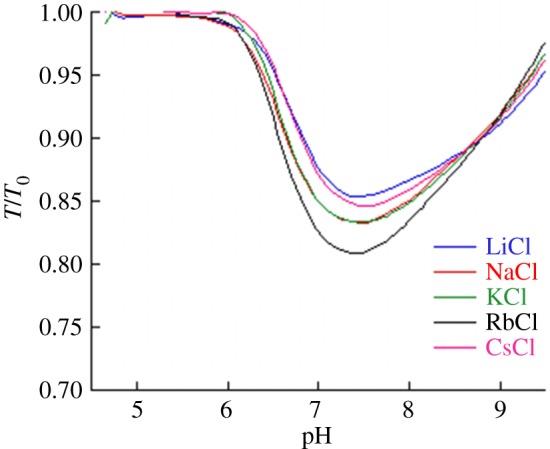
Experimental turbidity measurements of haemoglobin in 50 mM alkali chloride salts. Adapted from [18].
The transmittance measured in turbidity experiments falls as aggregation of haemoglobin tetramers proceeds. That is, the turbidance (absorbance) 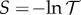 rises, proportional to the concentration cagg of aggregates. If aggregation is dominated by association of two tetramers, then cagg is proportional [40,41] to the second virial coefficient [42–44]
rises, proportional to the concentration cagg of aggregates. If aggregation is dominated by association of two tetramers, then cagg is proportional [40,41] to the second virial coefficient [42–44]
| 5.1 |
b is evaluated here with respect to the separation distance L between protein surfaces, rather than the distance between protein centres r = L + 2R, assuming hard sphere contact between tetramers. Hence, the concentration of aggregates cagg is determined by the protein–protein interaction G(L). Experimental second virial coefficients have often been used to characterize protein aggregation [45–48]. If particles are non-interacting (G = 0), then b = 0, corresponding to an ideal transmittance of 100%. If particles aggregate (G < 0), then b becomes negative and transmittance is reduced. We use the absorbance relationship 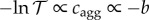 to deduce plausible estimates for cation binding constants pKM. Algebraic details are given in the electronic supplementary material.
to deduce plausible estimates for cation binding constants pKM. Algebraic details are given in the electronic supplementary material.
The chaotropic ion Cs+ is likely to bind weakly to the kosmotropic-like carboxylate site. We remove ambiguity by assuming it does not bind at all, with pKCs =−∞.
The experimental transmittance 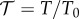 of 1 mg ml−1 haemoglobin in 50 mM chloride solution is shown [18] in figure 3. We focus on Li+, K+ and Cs+ representatives of the opposing ends and the middle of the cation Hofmeister series. Transmittances at pH 7 are 0.8541, 0.8324 and 0.8465 for Li+, K+ and Cs+, respectively. With the second virial coefficient b proportional to
of 1 mg ml−1 haemoglobin in 50 mM chloride solution is shown [18] in figure 3. We focus on Li+, K+ and Cs+ representatives of the opposing ends and the middle of the cation Hofmeister series. Transmittances at pH 7 are 0.8541, 0.8324 and 0.8465 for Li+, K+ and Cs+, respectively. With the second virial coefficient b proportional to 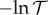 , we adjust cation binding constants to achieve the least error in
, we adjust cation binding constants to achieve the least error in
| 5.2 |
(see the electronic supplementary material for the derivation).
The resulting binding constants are pKLi =−0.21 and pKK =−1.31, equivalent to a binding energy of 0.484 kBT for Li+ and 3.02 kBT for K+, positive values indicating that binding is relatively unfavourable. Curiously, even for Li+ the binding energy is positive (unfavourable), indicating that cation binding to carboxylate sites is relatively weak. By comparison, the carboxylate binding energy of H+ (with pKc = 3.15) is −7.25 kBT. Chemisorption of Li+ is 12.6 times stronger than K+, which, combined with different non-electrostatic physisorption, is sufficient to drive the difference in transmittance observed experimentally.
The 2.5 kBT difference in Li+ and K+ binding energies found here is in reasonable agreement with the difference of 0.9 kcal mol−1 (1.5 kBT) found by molecular dynamics simulations [49], although the absolute binding energies from MD are more attractive. The discrepancy may be tentatively attributed to the mean-field smeared surface charge applied here, neglecting the discrete nature of binding sites as well as the specific amino acid residues hosting the carboxylate and amine sites, which is included in molecular dynamics simulations. Discrete surface charges tend to increase counterion physisorption, increasing repulsion (or diminishing attraction) between surfaces [50]. Anions are counterions for the mildly positive haemoglobin surface charge found in our model. To balance against this effect in order to maintain the same interaction under conditions of discrete surface charge, we would expect the slightly stronger cation binding which was found in MD simulation.
The calculated surface potential and total protein (haemoglobin) charge are shown in figure 4 as a function of pH in 100 mM salt solution. The anomalous Hofmeister series K+>Li+ > Cs+ is found in the surface potential. Cs+ and Li+ swap above pH 9 but remain anomalous with respect to K+. Interestingly, the Hofmeister effect is much stronger in the total protein charge, and follows the conventional series Li+ > K+ > Cs+. This observation in the total charge supports our interpretation that the overall Hofmeister effect is a competition between the two mechanisms of chemisorption and non-electrostatic physisorption, each with opposing Hofmeister series.
Calculated short-range interaction free energies G(L) for haemoglobin in 50 mM and 100 mM LiCl, KCl, CsCl at pH 7 are shown in figure 5. We find that at small separations the interactions follow the anomalous Hofmeister series, K+<Cs+≤Li+. At 50 mM salt concentrations we can additionally observe that, above a separation of 4.5 nm, the interaction for K+ crosses that of Li+, with the interaction for Li+ becoming the more attractive. A crossover point (not significant on the scale shown in figure 5) also occurs in 100 mM salts at 5 nm. It is apparent that the experimentally observed anomaly in the Hofmeister series for aggregation behaviour is controlled by short-range protein interactions.
Figure 5.

Theoretical short-range interaction energies between two haemoglobin tetramers (diameter 5.5 nm) at pH 7 in (a) 50 mM and (b) 100 mM LiCl, KCl and CsCl.
The reason why K+ induces stronger attraction at short separations can be attributed in part to the hydration model used for the ions. Li+ is taken as strongly hydrated, hence carrying a larger hydrated ion radius. Although lithium's dispersion B coefficient is larger, its dispersion interaction is weakened over the larger radius and so the short-range non-electrostatic physisorption of K+ is stronger. Consequently, the amount of K+ at the protein surface is greater than Li+, leading to a larger positive electrostatic potential (stronger attractive electrostatic energy Gel) and a stronger attractive chemisorption energy Gchem, due to the component −μNESKΓKc in equation (4.3).
In the case of Cs+, the attractive non-electrostatic chemisorption term −μNESCsΓCsc is not present, so the overall interaction in Cs+ is relatively less attractive than K+, leading to less aggregation. In the case of Li+, as discussed above, non-electrostatic physisorption is weaker, leading to less aggregation. K+ has a sufficient amount of both mechanisms, physisorption and chemisorption, that leads to a more attractive short-range interaction and therefore stronger aggregation.
6. Conclusion
Physisorption at the protein backbone and chemisorption at protein charge sites present as two competing mechanisms, each with a cationic Hofmeister series reverse to the other. The K+ ion, lying in the middle of a standard Hofmeister sequence between Li+ and Cs+, experiences a sufficient amount of both mechanisms that results in greater K+ adsorption than the other cations. The consequence is a more attractive short-range interaction leading to higher protein aggregation. Overall, the specific ion effect in any given salt is governed by the relative weights of the two mechanisms.
The model was tested against haemoglobin turbidity measurements, fitting chemisorption binding energies to carboxylate sites (assuming no chemisorption of Cs+) with the help of calculated second virial coefficients. Fitted binding energies are realistic with weakly unfavourable (positive) binding energies of 0.484 kBT for Li+ and 3.02 kBT for K+. This indicates that Li+ binds 12.6 times more strongly than K+, consistent with the notion that the kosmotropic Li+ should have greater affinity with the kosmotropic carboxylate site [49,51].
Chemisorption binding energies were fitted in this study. Improvements can be made by evaluating binding energies from first principles rather than fitting. A model of ion–ion interactions, already found to be useful in bulk solutions [44,52], may help achieve this.
In the model applied here, accessible chemisorption sites (carboxylates, amines) were located only at the surface of a protein, taken to be impenetrable to ions. In some situations, particularly for denatured proteins, it may be worthwhile exploring a model which allows ions to penetrate into the volume of the protein molecule, with chemisorption taking place at sites throughout the protein volume [53]. Anisotropy in the distribution of ions bound at charge sites is also an important factor in protein aggregation, requiring a more sophisticated three-dimensional modelling [54]. The methodology presented here can be extended to these kinds of three-dimensional models.
Supplementary Material
Supplementary Material
Competing interests
We declare we have no competing interests.
Funding
We acknowledge the grant of resources from the National Computational Infrastructure (NCI), which is supported by the Australian Government. A.S. thanks FIR 2016 and Fondazione di Sardegna (L.R. 7/2007 year 2016—DGR 28/21 17.05.2015) for support.
References
- 1.Hofmeister F. 1888. Zur Lehre von der Wirkung der Salze. Arch. Exp. Pathol. Pharmakol. 24, 247–260. ( 10.1007/BF01918191) [DOI] [Google Scholar]
- 2.LoNostro P, Ninham BW. 2012. Hofmeister phenomena: an update on ion specificity in biology. Chem. Rev. 112, 2286–2322. ( 10.1021/cr200271j) [DOI] [PubMed] [Google Scholar]
- 3.Paterová J, Rembert KB, Heyda J, Kurra Y, Okur HI, Liu WR, Hilty C, Cremer PS, Jungwirth P. 2013. Reversal of the Hofmeister series: specific ion effects on peptides. J. Phys. Chem. B 117, 8150–8158. ( 10.1021/jp405683s) [DOI] [PubMed] [Google Scholar]
- 4.Nostro PL, Ninham BW, Carretti E, Dei L, Baglioni P. 2015. Specific anion effects in Artemia salina. Chemosphere 135, 335–340. ( 10.1016/j.chemosphere.2015.04.080) [DOI] [PubMed] [Google Scholar]
- 5.Salis A, Monduzzi M. 2016. Not only pH. Specific buffer effects in biological systems. Curr. Opin. Colloid Interface Sci. 23, 1–9. ( 10.1016/j.cocis.2016.04.004) [DOI] [Google Scholar]
- 6.Vakarelski IU, Teramoto N, McNamee CE, Marston JO, Higashitani K. 2012. Ionic enhancement of silica surface nanowear in electrolyte solutions. Langmuir 28, 16072–16079. ( 10.1021/la303223q) [DOI] [PubMed] [Google Scholar]
- 7.Lützenkirchen J. 2013. Specific ion effects at two single-crystal planes of sapphire. Langmuir 29, 7726–7734. ( 10.1021/la401509y) [DOI] [PubMed] [Google Scholar]
- 8.Kumar E, Bhatnagar A, Hogland W, Marques M, Sillanpää M. 2014. Interaction of inorganic anions with iron-mineral adsorbents in aqueous media: a review. Adv. Colloid Interface Sci. 203, 11–21. ( 10.1016/j.cis.2013.10.026) [DOI] [PubMed] [Google Scholar]
- 9.Chang WZ, Leong YK. 2014. Ageing and collapse of bentonite gels—effects of Li, Na, K and Cs ions. Rheol. Acta 53, 109–122. ( 10.1007/s00397-013-0744-0) [DOI] [Google Scholar]
- 10.Kou R, Zhang J, Wang T, Liu G. 2015. Interactions between polyelectrolyte brushes and Hofmeister ions: chaotropes versus kosmotropes. Langmuir 31, 10461–10468. ( 10.1021/acs.langmuir.5b02698) [DOI] [PubMed] [Google Scholar]
- 11.Oncsik T, Trefalt G, Borkovec M, Szilagyi I. 2015. Specific ion effects on particle aggregation induced by monovalent salts within the Hofmeister Series. Langmuir 31, 3799–3807. ( 10.1021/acs.langmuir.5b00225) [DOI] [PubMed] [Google Scholar]
- 12.Jaspers M, Rowan AE, Kouwer PHJ. 2015. Tuning hydrogel mechanics using the Hofmeister effect. Adv. Funct. Mater. 25, 6503–6510. ( 10.1002/adfm.201502241) [DOI] [Google Scholar]
- 13.Pavlovic M, Huber R, Adok-Sipiczki M, Nardin C, Szilagyi I. 2016. Ion specific effects on the stability of layered double hydroxide colloids. Soft. Matter 12, 4024–4033. ( 10.1039/C5SM03023D) [DOI] [PubMed] [Google Scholar]
- 14.Oncsik T, Desert A, Trefalt G, Borkovec M, Szilagyi I. 2016. Charging and aggregation of latex particles in aqueous solutions of ionic liquids: towards an extended Hofmeister series. Phys. Chem. Chem. Phys. 18, 7511–7520. ( 10.1039/C5CP07238G) [DOI] [PubMed] [Google Scholar]
- 15.Collins KD. 2012. Why continuum electrostatics theories cannot explain biological structure, polyelectrolytes or ionic strength effects in ion–protein interactions. Biophys. Chem. 167, 43–59. ( 10.1016/j.bpc.2012.04.002) [DOI] [PubMed] [Google Scholar]
- 16.Boström M, Parsons DF, Salis A, Ninham BW, Monduzzi M. 2011. Possible origin of the inverse and direct Hofmeister series for lysozyme at low and high salt concentrations. Langmuir 27, 9504–9511. ( 10.1021/la202023r) [DOI] [PubMed] [Google Scholar]
- 17.Schwierz N, Horinek D, Sivan U, Netz RR. 2016. Reversed Hofmeister series—the rule rather than the exception. Curr. Opin. Colloid Interface Sci. 23, 10–18. ( 10.1016/j.cocis.2016.04.003) [DOI] [Google Scholar]
- 18.Medda L, Carucci C, Parsons DF, Ninham BW, Monduzzi M, Salis A. 2013. Specific cation effects on hemoglobin aggregation below and at physiological salt concentration. Langmuir 29, 15 350–15 358. ( 10.1021/la404249n) [DOI] [PubMed] [Google Scholar]
- 19.Medda L, Monduzzi M, Salis A. 2015. The molecular motion of bovine serum albumin under physiological conditions is ion specific. Chem. Commun. 51, 6663–6666. ( 10.1039/C5CC01538C) [DOI] [PubMed] [Google Scholar]
- 20.Schwierz N, Horinek D, Netz RR. 2010. Reversed anionic Hofmeister series: the interplay of surface charge and surface polarity. Langmuir 26, 7370–7379. ( 10.1021/la904397v) [DOI] [PubMed] [Google Scholar]
- 21.Parsons DF, Boström M, LoNostro P, Ninham BW. 2011. Hofmeister effects: interplay of hydration, nonelectrostatic potentials, and ion size. Phys. Chem. Chem. Phys. 13, 12352–12367. ( 10.1039/C1CP20538B) [DOI] [PubMed] [Google Scholar]
- 22.Salis A, Ninham BW. 2014. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 43, 7358–7377. ( 10.1039/C4CS00144C) [DOI] [PubMed] [Google Scholar]
- 23.Boström M, Tavares FW, Bratko D, Ninham BW. 2005. Specific ion effects in solutions of globular proteins: comparison between analytical models and simulation. J. Phys. Chem. B 109, 24 489–24 494. ( 10.1021/jp0551869) [DOI] [PubMed] [Google Scholar]
- 24.Parsons DF, Ninham BW. 2009. Ab initio molar volumes and Gaussian radii. J. Phys. Chem. A 113, 1141–1150. ( 10.1021/jp802984b) [DOI] [PubMed] [Google Scholar]
- 25.Parsons DF, Ninham BW. 2010. Importance of accurate dynamic polarizabilities for the ionic dispersion interactions of alkali halides. Langmuir 26, 1816–1823. ( 10.1021/la902533x) [DOI] [PubMed] [Google Scholar]
- 26.Dagastine RR, Prieve DC, White LR. 2000. The dielectric function for water and its application to van der Waals forces. J. Colloid Interface Sci. 231, 351–358. ( 10.1006/jcis.2000.7164) [DOI] [PubMed] [Google Scholar]
- 27.Salis A, Cugia F, Parsons DF, Ninham BW, Monduzzi M. 2012. Hofmeister series reversal for lysozyme by change in pH and salt concentration: insights from electrophoretic mobility measurements. Phys. Chem. Chem. Phys. 14, 4343–4346. ( 10.1039/C2CP40150A) [DOI] [PubMed] [Google Scholar]
- 28.Parsons DF, Ninham BW. 2011. Surface charge reversal and hydration forces explained by ionic dispersion forces and surface hydration. Colloids Surf, A 383, 2–9. ( 10.1016/j.colsurfa.2010.12.025) [DOI] [Google Scholar]
- 29.Parsons DF, Salis A. 2015. The impact of the competitive adsorption of ions at surface sites on surface free energies and surface forces. J. Chem. Phys. 142, 134 707 ( 10.1063/1.4916519) [DOI] [PubMed] [Google Scholar]
- 30.Okur HI, Hladílková J, Rembert KB, Cho Y, Heyda J, Dzubiella J, Cremer PS, Jungwirth P. 2017. Beyond the Hofmeister series: ion-specific effects on proteins and their biological functions. J. Phys. Chem. B 121, 1997–2014. ( 10.1021/acs.jpcb.6b10797) [DOI] [PubMed] [Google Scholar]
- 31.Ninham BW, Parsegian VA. 1971. Electrostatic potential between surfaces bearing ionizable groups in ionic equilibrium with physiologic saline solution. J. Theor. Biol. 31, 405–428. ( 10.1016/0022-5193(71)90019-1) [DOI] [PubMed] [Google Scholar]
- 32.Matthew JB, Hanania GIH, Gurd FRN. 1979. Electrostatic effects in hemoglobin: hydrogen ion equilibriums in human deoxy- and oxyhemoglobin A. Biochemistry 18, 1919–1928. ( 10.1021/bi00577a011) [DOI] [PubMed] [Google Scholar]
- 33.Parsons DF, Salis A. 2016. Hofmeister effects at low salt concentration due to surface charge transfer. Curr. Opin. Colloid Interface Sci. 23, 41–49. ( 10.1016/j.cocis.2016.05.005) [DOI] [Google Scholar]
- 34.Miklavic SJ, Ninham BW. 1990. Competition for adsorption sites by hydrated ions. J. Colloid Interface Sci. 134, 305–311. ( 10.1016/0021-9797(90)90140-J) [DOI] [Google Scholar]
- 35.Balos V, Kim H, Bonn M, Hunger J. 2016. Dissecting Hofmeister effects: direct anion–amide interactions are weaker than cation–amide binding. Angew. Chem. 55, 8125–8128. ( 10.1002/anie.201602769) [DOI] [PubMed] [Google Scholar]
- 36.Chan DYC, Pashley RM, White LR. 1980. A simple algorithm for the calculation of the electrostatic repulsion between identical charged surfaces in electrolyte. J. Colloid Interface Sci. 77, 283–285. ( 10.1016/0021-9797(80)90445-2) [DOI] [Google Scholar]
- 37.Parsons DF, Ninham BW. 2012. Nonelectrostatic ionic forces between dissimilar surfaces: a mechanism for colloid separation. J. Phys. Chem. C 116, 7782–7792. ( 10.1021/jp212154c) [DOI] [Google Scholar]
- 38.Overbeek JTG. 1990. The role of energy and entropy in the electrical double layer. Colloids Surf. 51, 61–75. ( 10.1016/0166-6622(90)80132-N) [DOI] [Google Scholar]
- 39.Deniz V, Parsons DF. 2013. Effect of nonelectrostatic ion interactions on surface forces involving ion adsorption equilibria. J. Phys. Chem. C 117, 16416–16428. ( 10.1021/jp404086u) [DOI] [Google Scholar]
- 40.Hirschfelder JO, McClure FT, Weeks IF. 1942. Second virial coefficients and the forces between complex molecules. J. Chem. Phys. 10, 201–214. ( 10.1063/1.1723708) [DOI] [Google Scholar]
- 41.Swope WC, Andersen HC, Berens PH, Wilson KR. 1982. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: application to small water clusters. J. Chem. Phys. 76, 637–649. ( 10.1063/1.442716) [DOI] [Google Scholar]
- 42.Lund M, Jönsson B. 2003. A mesoscopic model for protein–protein interactions in solution. Biophys. J. 85, 2940–2947. ( 10.1016/S0006-3495(03)74714-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tessier PM, Sandler SI, Lenhoff AM. 2004. Direct measurement of protein osmotic second virial cross coefficients by cross-interaction chromatography. Protein Sci. 13, 1379–1390. ( 10.1110/ps.03419204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duignan TT, Baer MD, Mundy CJ. 2016. Ions interacting in solution: moving from intrinsic to collective properties. Curr. Opin. Colloid Interface Sci. 23, 58–65. ( 10.1016/j.cocis.2016.05.009) [DOI] [Google Scholar]
- 45.Neal BL, Asthagiri D, Velev OD, Lenhoff AM, Kaler EW. 1999. Why is the osmotic second virial coefficient related to protein crystallization? J. Cryst. Growth 196, 377–387. ( 10.1016/S0022-0248(98)00855-0) [DOI] [Google Scholar]
- 46.Ho JGS, Middelberg APJ, Ramage P, Kocher HP. 2003. The likelihood of aggregation during protein renaturation can be assessed using the second virial coefficient. Protein Sci. 12, 708–716. ( 10.1110/ps.0233703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Brun V, Friess W, Schultz-Fademrecht T, Muehlau S, Garidel P. 2009. Lysozyme–lysozyme self-interactions as assessed by the osmotic second virial coefficient: impact for physical protein stabilization. Biotechnol. J. 4, 1305–1319. ( 10.1002/biot.200800274) [DOI] [PubMed] [Google Scholar]
- 48.Quigley A, Williams DR. 2015. The second virial coefficient as a predictor of protein aggregation propensity: a self-interaction chromatography study. Eur. J. Pharm. Biopharm. 96, 282–290. ( 10.1016/j.ejpb.2015.07.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Becconi O, Ahlstrand E, Salis A, Friedman R. 2017. Protein–ion interactions: simulations of bovine serum albumin in physiological solutions of NaCl, KCl and LiCl. Isr. J. Chem. 57, 403–412. ( 10.1002/ijch.201600119) [DOI] [Google Scholar]
- 50.Khan MO, Petris S, Chan DYC. 2005. The influence of discrete surface charges on the force between charged surfaces. J. Chem. Phys. 122, 104 705 ( 10.1063/1.1856925) [DOI] [PubMed] [Google Scholar]
- 51.Collins KD. 2004. Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process. Methods 34, 300–311. ( 10.1016/j.ymeth.2004.03.021) [DOI] [PubMed] [Google Scholar]
- 52.Duignan TT, Parsons DF, Ninham BW. 2014. A continuum solvent model of ion–ion interactions in water. Phys. Chem. Chem. Phys. 16, 22014–22027. ( 10.1039/C4CP02822H) [DOI] [PubMed] [Google Scholar]
- 53.Barbosa NSV, Lima ERA, Tavares FW. 2015. The electrostatic behavior of the bacterial cell wall using a smoothing function to describe the charge-regulated volume charge density profile. Colloids Surf. B 134, 447–452. ( 10.1016/j.colsurfb.2015.06.066) [DOI] [PubMed] [Google Scholar]
- 54.Lund M. 2016. Anisotropic protein–protein interactions due to ion binding. Colloids Surf. B 137, 17–21. ( 10.1016/j.colsurfb.2015.05.054) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.