

Abstract
Myotis rufoniger is a vesper bat in the genus Myotis. Here we report the whole genome sequence and analyses of the M. rufoniger. We generated 124 Gb of short-read DNA sequences with an estimated genome size of 1.88 Gb at a sequencing depth of 66× fold. The sequences were aligned to M. brandtii bat reference genome at a mapping rate of 96.50% covering 95.71% coding sequence region at 10× coverage. The divergence time of Myotis bat family is estimated to be 11.5 million years, and the divergence time between M. rufoniger and its closest species M. davidii is estimated to be 10.4 million years. We found 1,239 function-altering M. rufoniger specific amino acid sequences from 929 genes compared to other Myotis bat and mammalian genomes. The functional enrichment test of the 929 genes detected amino acid changes in melanin associated DCT, SLC45A2, TYRP1, and OCA2 genes possibly responsible for the M. rufoniger’s red fur color and a general coloration in Myotis. N6AMT1 gene, associated with arsenic resistance, showed a high degree of function alteration in M. rufoniger. We further confirmed that the M. rufoniger also has bat-specific sequences within FSHB, GHR, IGF1R, TP53, MDM2, SLC45A2, RGS7BP, RHO, OPN1SW, and CNGB3 genes that have already been published to be related to bat’s reproduction, lifespan, flight, low vision, and echolocation. Additionally, our demographic history analysis found that the effective population size of Myotis clade has been consistently decreasing since ~30k years ago. M. rufoniger’s effective population size was the lowest in Myotis bats, confirming its relatively low genetic diversity.
Introduction
M. rufoniger is a species of vesper bat in the family Vespertilionidae [1]. It can be distinguished from other bats by its rusty orange fur (S1 Picture) and is therefore called ‘golden bat’ or ‘red bat’ in South Korea (Republic of Korea). Recently, its scientific name has been re-assigned to M. rufoniger from Myotis formosus tsuensis based on a molecular phylogeny and morphological study of bats [2]. Although its population is not assessed systematically, it is apparently a rare species that has only been collected in a handful of localities [2]. In South Korea, M. rufoniger is protected and designated as a natural monument. Being one of the most well-known and iconic protected wild animals in South Korea, even an M. rufoniger exhibition center is also in operation (golden bat exhibition center in Hampyeong County, South Korea).
In 2012, a fruit bat Pteropus alecto and an insectivorous Myotis davidii genomes were published, reporting bat-specific amino acid sequences on TP53 (Tumor Protein P53) and MDM2 (MDM2 Proto-Oncogene) genes that are associated with DNA damage checkpoint and DNA repair pathways. They provided insights into bats’ evolution of high metabolic rate and an increased amount of free radicals that are associated with flight [3]. In 2013, the Brandt’s bat (Myotis brandtii) genome analysis further identified unique amino acid sequence changes in GHR (Growth Hormone Receptor), IGF1R (Insulin Like Growth Factor 1 Receptor), FSHB (Follicle Stimulating Hormone Beta Subunit), SLC45A2 (Solute carrier family 45, member 2), RGS7BP (Regulator of G-protein signaling 7 binding protein), RHO (Rhodopsin), OPN1SW (Opsin 1 [Cone Pigments], Short-Wave-Sensitive), and CNGB3 (cyclic nucleotide gated channel beta 3) genes, providing new insights into bats’ delayed ovulation during hibernation, long lifespan, small body size, low vision, and echolocation [4].
As such, a set of close but distinct species genomes enable prediction of variants that may contain significant geno-phenotype association information [5–8]. This close species comparative genomics approach has been applied to M. rufoniger genome analyses to identify the species-specific variants that can confer functional and hence evolutionary adaptation of the M. rufoniger. To confirm and further identify such species-specific or bat-specific sequence changes, it is critical to evaluate such amino acid changes with as many genomes as possible at different levels of background comparison species. Thus, it is important to build biological resource with continuous sequencing of various species genomes.
A complete mitochondrial genome of M. rufoniger has been published already [9], while no whole-genome sequence has been reported yet, thus limiting the investigation of autosomal genetic signatures for environmental adaptations of M. rufoniger. Furthermore, the demographic history of a species can only be reconstructed accurately from deeply sequenced whole genomes [10,11]. Here, we provide a whole genome analysis of M. rufoniger by producing massively parallel short DNA sequences (approximately 124 Gb) with its genomic features and unique amino acid sequences, accompanied by its demographic history and genetic diversity.
Results
Whole genome sequences of M. rufoniger
The genomic DNA from the wild carcass of M. rufoniger found in Gosudonggul cave, Danyang, in South Korea, was sequenced using Illumina HiSeq2000 platform. A total of 124 Gb of paired-end short DNA sequences were produced with a read length of 100 bp, and target insert sizes of 566 bp and 574 bp from two genomic libraries. After reducing low sequencing quality reads and possible microbial contaminated reads, we acquired a total of 115 Gb of DNA sequences (Table 1; S1 Table). To confirm the species identification of the sample, a phylogenetic analysis was performed using a multiple sequence alignment of mitochondrial cytochrome b sequences, and our sample was verified to be the closest to M. rufoniger (S1 Fig). We performed a K-mer analysis (K = 17) using the M. rufoniger whole genome sequences, and its genome size was predicted to be approximately 1.88 Gb (S2 Fig; S2 Table). This is similar to those of other bat species and smaller than those of other mammals [3,4,12]. Since there is no de novo assembled M. rufoniger genome, the DNA reads were aligned to all three available Myotis bat reference genomes (M. brandtii, M. davidii, and M. lucifugus; Table 1; S3 Table). The consensus DNA sequences of M. rufoniger were generated by substituting M. rufoniger single nucleotide variants (SNVs) detected from the whole genome sequencing data against the three reference genomes. The genome coverage (≥10×, 87.63%) was the highest when the M. lucifugus assembly was the reference, while the coding sequence (CDS) coverage (≥10×, 96.60%) was the highest when the M. davidii genome was the reference. The number of SNVs (58,660,193) was the lowest when the M. brandtii was the reference, while the number of small insertions and deletions was the lowest when the M. daividii was the reference. However, when the M. brandtii assembly was the reference, the mapping rate (96.50%) and the number of consensus genes (17,247) was the highest. Therefore, we used M. brandtii reference-guided M. rufoniger consensus sequences for following analyses.
Table 1. Sequencing and mapping statistics of M. rufoniger genome.
| Myotis bat references | |||
|---|---|---|---|
| M. brandtii | M. davidii | M. lucifugus | |
| Sequenced reads | 1,240,507,648 | ||
| Low quality filtered reads | 1,152,154,630 | ||
| Contamination filtered reads | 1,149,260,980 | ||
| Mapped reads | 1,109,045,915 | 1,101,733,443 | 1,095,435,354 |
| Mapping rate | 96.50% | 95.86% | 95.32% |
| Deduplicated reads | 1,023,540,144 | 1,017,982,320 | 1,010,388,456 |
| Depth coverage | 49.5 × | 51.5 × | 49.1 × |
| 1× genome coverage | 90.17% | 87.65% | 92.29% |
| 5× genome coverage | 87.83% | 85.68% | 89.88% |
| 8× genome coverage | 86.47% | 84.60% | 88.52% |
| 10× genome coverage | 85.58% | 83.88% | 87.63% |
| 1× CDS coverage | 98.86% | 99.10% | 98.13% |
| 5× CDS coverage | 97.68% | 98.19% | 96.29% |
| 8× CDS coverage | 96.60% | 97.33% | 94.88% |
| 10× CDS coverage | 95.71% | 96.60% | 93.81% |
| The number of homozygous SNVs | 50,615,560 | 56,635,858 | 50,441,069 |
| The number of heterozygous SNVs | 8,044,633 | 8,008,552 | 8,306,592 |
| The number of homozygous indels | 4,197,521 | 4,231,431 | 4,313,780 |
| The number of heterozygous indels | 517,253 | 457,868 | 512,018 |
| The number of consensus genes | 17,274 | 16,419 | 17,141 |
| The number of consensus genes without gap | 12,841 | 12,618 | 12,941 |
We examined the base composition of M. rufoniger genome (S4 Table) and found that the ratio of GC content was higher in the CDS region (52.15 ~ 52.95%) than that of the whole genome (41.49 ~ 42.28%), and this is concordant with the other mammalian species (the GC content ratios of the other species were 48.98 ~ 54.35% in the CDS region and 37.82 ~ 42.93% in the whole genome; S5 Table). The proportion of known transposon-derived repeats in the M. rufoniger genome was 19.05%, which is comparable with those of other bats (19.34 ~ 33.32%), but significantly lower than those of other mammalian genomes (25.27 ~ 51.62%; S6 Table).
The body of the M. rufoniger carcass used in this study was damaged and its morphological sex determination was not possible. We therefore compared its heterozygosity in X-chromosomal scaffolds with that of the other male Myotis bat (S7 Table). Since mammalian males have both X and Y sex chromosomes while females have two X chromosomes, a male individual has to show a lower heterozygosity in X chromosome than that of a female individual. Our sample showed an even lower heterozygosity in X-chromosomal scaffolds than that of the male M. davidii individual, indicating that it is clearly a male.
Relationship of M. rufoniger to other mammalian species
We identified orthologous gene clusters from 13 mammalian genomes (seven bat genomes: Myotis brandtii, Myotis lucifugus, Myotis davidii, Eptesicus fuscus, Pteropus alecto, Pteropus vampyrus, and Rousettus aegyptiacus, and six other mammalian genomes: Homo sapiens, Mus musculus, Bos taurus, Equus caballus, Heterocephalus glaber, and Monodelphis domestica; S2 Table) using OrthoMCL software [13]. The M. rufoniger genes were matched and added to the orthologous clusters of M. brandtii’s genes, and 6,782 single-copy gene families among the 14 species were identified [7].
The divergence times of M. rufoniger and the 13 mammals were estimated using 1,258,141 four-fold degenerate sites from the 6,782 single-copy genes (Fig 1). The closest clade to bats was E. caballus (horse) and B. taurus (cow) clade (diverged at 79.45 million years ago [MYA]). The Myotis bats diverged into its current clade approximately 11.52 MYA. M. davidii was estimated to be the closest to the M. rufoniger among the three Myotis bats used in this study. The divergence time between M. rufoniger and M. davidii was estimated to be 10.41 million years, indicating a fairly recent divergence which is suitable for our close species comparative genomics analyses.
Fig 1. Phylogenetic relationships and divergence times in bats and mammalian species.

The estimated divergence time (million years ago; MYA) is given at the nodes, with the 95% confidence intervals in parentheses. The calibration times of M. brandtii—H. sapiens (97.5 MYA), and M. brandtii—P. alecto (62.6 MYA) were derived from the TimeTree database. Colored branches and circles represent bat groups (blue: insect-eating microbat, red: fruits-eating mega bat). M. domestica, used as an outgroup species, was excluded in this figure.
M. rufoniger specific amino acid sequences
From previous studies [3,4], bat-specific amino acid sequences within FSHB, GHR, IGF1R, TP53, MDM2, SLC45A2, RGS7BP, RHO, OPN1SW, and CNGB3 genes were reported. They represent some general characteristics of Myotis bats: delayed ovulation (FHSB), long lifespan (GHR and IGF1R), powered flight (TP53 and MDM2), echolocation (SLC45A2 and RGS7B), and low vision (RHO, OPN1SW, and CNGB3). The same amino acid sequences within the above listed genes were also identified in the M. rufoniger genome (S3 Fig).
To further identify bat-specific amino acid sequences linked to environmental adaptations and unique evolutionary features, we investigated M. rufoniger specific amino acid changes compared to other Myotis bats. A total of 3,366 unique amino acid changes (uAACs) from 2,125 genes were identified in M. rufoniger. Among them, 1,696 uAACs from 1,228 genes were predicted to be function altering by the protein variation effect analyzer (PROVEAN) software (variant score ≤ -2.5; S8 Table) [14]. These function-altering variants and associated genes can provide insights onto M. rufoniger specific evolutionary adaptation. When gap-containing genes were excluded, 1,239 uAACs from 929 genes were predicted to be function altering (S9 Table). A functional enrichment analysis of the 929 genes having function-altering uAACs was performed using DAVID (Database for Annotation Visualization and Integrated Discovery) tool [15]. The probably function-altered genes were significantly enriched in reproduction related terms (Gene Ontology [GO] analysis with a P–value ≤ 0.05 by EASE scores [modified Fisher’s exact test] and with a 10% of false discovery rate [FDR]) including reproductive processes in a multicellular organism (P-value: 0.00026, 39 genes, GO: 0048609), ovulation cycle (P-value: 0.00052, 11 genes, GO: 0042698), and gamete generation (P-value: 0.0017, 31 genes, GO: 0007276; S10 Table). In M. rufoniger, pigment related terms were significantly enriched in the function-altered genes as in the melanin biosynthetic process (P-value: 0.0037, 4 genes, GO: 0042438; S10 Table). The genes are DCT (Dopachrome tautomerase), SLC45A2 (Solute carrier family 45), TYRP1 (Tyrosinase-related protein 1), and OCA2 (Oculocutaneous albinism II). We could identify some uAACs in DCT, SLC45A2, TYRP1, and OCA2 genes in the other Myotis bats as function-altering using PROVEAN software (Table 2; S11 Table) [14] indicating those genes are not only specific to M. rufoniger’s red fur color. The multiple sequence alignments and specific amino acid sequences are presented in S4 Fig.
Table 2. Myotis bats’ uAACs within melanin associated genes.
| Gene | Description | The number of uAACs (The number of uAACs with PROVEAN score ≤ -2.5) | |||
|---|---|---|---|---|---|
| M. rufoniger | M. brandtii | M. davidii | M. lucifugus | ||
| DCT | Dopachrome tautomerase | 2 (2) | 0 (0) | 0 (0) | 1 (0) |
| SLC45A2 | Solute carrier family 45 | 3 (1) | 0 (0) | 0 (0) | 0 (0) |
| TYRP1 | Tyrosinase-related protein 1 | 2 (1) | 0 (0) | 1 (1) | 0 (0) |
| OCA2 | Oculocutaneous albinism II | 3 (3) | 1 (0) | 0 (0) | 2 (2) |
As we were interested in the variants’ whole gene function, all the PROVEAN variant scores of each gene were summed up. We then ranked the sums of the function-altered gene candidates (S12 Table). For the top 20 genes, the same analysis was carried out in the other Myotis bat species for comparison (Table 3; S13 and S14 Tables). We ranked M. rufoniger’s genes according to the numbers of variants and summed variant scores, and compared them to those of other Myotis bat genes. From the result, N6AMT1 (N-6 Adenine-Specific DNA Methyltransferase 1) showed the fourth-lowest sum of variants scores (sum of PROVEAN variant scores: -40.285 and number of variants: six in M. rufoniger; no uAAC in the other Myotis bats), indicating a high degree of function alteration of N6AMT1 in M. rufoniger. The multiple sequence alignments and specific amino acid sequences are presented in S5 Fig.
Table 3. The summed PROVEAN variant score for top 20 ranked genes. The lower the more significant.
| Gene | Description | Sum of the PROVEAN variant scores | |
|---|---|---|---|
| M. rufoniger | Other Myotis bats | ||
| BCO1 | Beta-Carotene Oxygenase 1 | -51.29 | -8.13 ~ 0 |
| CCDC184 | Coiled-coil Domain Containing 184 | -47.77 | -2.88 ~ 0 |
| PCM1 | Pericentriolar material 1 protein | -40.61 | -19.04 ~ 0 |
| N6AMT1 | N-6 adenine-specific DNA methyltransferase 1 | -40.29 | 0 ~ 0 |
| MDN1 | Midasin AAA ATPase 1 | -37.90 | -32.07 ~ -5.80 |
| CEP350 | Centrosomal protein 350 | -37.30 | -15.16 ~ -4.92 |
| WNK2 | WNK Lysine Deficient Protein Kinase 2 | -34.67 | -17.12 ~ -4.79 |
| SPG11 | Spastic Paraplegia 11 | -29.39 | -18.19 ~ 0 |
| C10orf12 | Uncharacterized protein | -29.01 | -18.97 ~ 0 |
| C1orf228 | Uncharacterized protein | -28.71 | -8.22 ~ 0 |
| NOC2L | NOC2 Like Nucleolar Associated Transcriptional Repressor | -28.61 | 0 ~ 0 |
| CPLX3 | Complexin 3 | -27.85 | -7.67 ~ 0 |
| KIAA1462 | Junctional Protein Associated With Coronary Artery Disease | -27.73 | -3.22 ~ 0 |
| ALPK2 | Alpha Kinase 2 | -26.00 | -9.09 ~ 0 |
| PTRH1 | peptidyl-tRNA hydrolase 1 | -25.30 | 0 ~ 0 |
| MYO16 | Myosin XVI | -24.71 | -14.57 ~ 0 |
| KIAA0922 | Transmembrane protein 131-like | -24.59 | -4.33 ~ 0 |
| OAT | Ornithine aminotransferase | -24.44 | -11.60 ~ 0 |
| SPAG17 | sperm associated antigen 17 | -23.71 | -16.24 ~ -2.18 |
| IPMK | Inositol Polyphosphate Multikinase | -23.47 | 0 ~ 0 |
Demographic history and the genetic diversity of Myotis bats
Deeply sequenced genomes allow the estimation of population structure history [10,11]. To investigate the demographic history of Myotis bats, a pairwise sequentially Markovian coalescent (PSMC) model inference analysis was conducted [11]. M. rufoniger was estimated as the most flourished species around 30 ~ 300 k years ago compared to the other Myotis bats, and its demographic history showed that its population peak was 50 k years ago (Fig 2). However, its effective population size was dramatically decreased during the last glacial period (10 ~ 50 k years ago), and it was estimated to be the lowest in present. Also, a consistent decline in the effective population size of Myotis bats since ~30 k years ago was found (Fig 2). We further investigated the genomic diversity (which can be affected by population size) of the M. rufoniger and compared it to those of the other Myotis bats. The M. rufoniger genetic diversity (0.00391), based on the heterozygous SNV rate, was lower than those of the M. davidii (0.00471) and the M. brandtii (0.00614), confirming the M. rufoniger’s low effective population size (S13 Table).
Fig 2. Demographic history of Myotis bats.

Tsurf, atmospheric surface air temperature (T indicates temperature, surf indicates surface); RSL, relative sea level; 10 m.s.l.e., 10 m sea level equivalent; g, generation time (years); μ, mutation rate per base pair per year.
Discussion
The function-altering variants containing genes of the M. rufoniger were concentrated in reproduction associated pathways. Prolonged sperm storage is a common behavior in Vespertilionidae (including genus Myotis, Pipistrellus, Nyctalus, Eptesicus, Vesoertilio, Chalinolobus, and Plecotus) and Rhinolophidae family species [16–18], and thus it is probably not M. rufoniger specific as reproduction related genes are always under strong natural selection. Therefore, such hits can be regarded as general or even a kind of artefact. Therefore, further detailed functional verification is necessary to understand the roles of each uAAC in functional categories such as reproduction.
The previous bat studies reported unique variants representing some general characteristics of Myotis bats: delayed ovulation (FHSB), long lifespan (GHR and IGF1R), powered flight (TP53 and MDM2), echolocation (SLC45A2 and RGS7B), and low vision (RHO, OPN1SW, and CNGB3) [3, 4]. We confirmed the reported amino acid sequences within the above listed genes were conserved in the M. rufoniger as in other bat genomes (S3 Fig), supporting that those variants are Myotis bat-wide not one species-specific nor individual-specific variants.
We also found that the function-altering unique variants containing the genes of the M. rufoniger were concentrated in a melanin associated pathway including DCT, SLC45A2, TYRP1, and OCA2 genes. TYRP1 and DCT genes are known to influence the quantity and quality of melanin [19]. Mutations on the TYRP1 gene are known to be associated with “rufous/red albinism”, causing reddish-brown skin or red hair [20–22]. The protein (melanocyte-specific transporter protein) encoded by OCA2 gene has been reported as a transporter of tyrosine, the precursor to melanin synthesis [23]. Variants within the SLC45A2 gene have shown association with hair color, affecting red/yellow pheomelanin pathway [24–25]. In this context, we suggest that the variants within the DCT, SLC45A2, TYRP1, and OCA2 genes are likely responsible for the M. rufoniger’s rusty orange fur color, which distinguish it from the other bats. However, fewer but certain uAACs within the DCT, SLC45A2, TYRP1, and OCA2 genes were also found in the other Myotis bats, suggesting a possible general role of those genes in bat-wide coloration. Therefore, a set of bat species with color diversity will be required for further analyses to understand the precise geno-phenotype associations in coloration.
In the M. rufoniger genome, N6AMT1 gene showed a high degree of function alteration. The protein (HemK methyltransferase family member 2) encoded by N6AMT1 gene can transform monomethylarsonous acid to dimethylarsinic acid, conferring resistance of mammalian cells to arsenic-induced toxicity [26]. An elemental analysis in the tissues from the M. rufoniger individual analyzed in the present study showed a very high concentration of arsenic in its intestinal tissue [27], suggesting that the M. rufoniger individual was exposed to food or water highly contaminated with arsenic. In this context, uAACs in N6AMT1 can be considered as a possible chemical adaptation signature. It can be a generally stressful condition for the bats living in caves, because caves are hazardous due to various harmful elements and chemicals such as heavy metals [28]. However, until a further experimental validation is carried out, it is a very speculative hypothesis of rapid adaptation to harmful cave elements. Furthermore, this study used genomic data from only one individual for each species and the variant comparison analysis can be biased by individual-specific variants. More genomes are necessary to be sequenced to confirm that our finding’s general applicability in both M. rufoniger and Myotis bats.
The M. rufoniger showed the lowest effective population size compared to other Myotis bats from the PSMC analysis. This does not necessarily indicate that they are the most endangered as they are fairly widely spread. It is perhaps more reasonable to attribute it to very recent human encroachment. It is also shown that the population size of the M. rufoniger was dramatically decreased during the latter part of the last glacial period. There was a consistent decline of Myotis bat family’s effective population size since ~30 k years ago. However, we cannot determine whether it is a bat-wide or Myotis bat specific phenomenon either, because we conducted the PSMC analysis using a small number of species. The dramatic decrease in the population size of the M. rufoniger could be a Korean M. rufoniger specific phenomenon. Therefore, a set of diverse species will be required to accurately model the bats’ effective population size history.
In conclusion, we report the first whole genome sequences of the M. rufoniger with bioinformatics analyses such as multiple sequence alignment, function altering uAAC prediction, functional enrichment, and population size estimation using PSMC. These provided us with some speculative insights on how bats adapted to environment sustaining the current behavioral, physiological, and demographic features such as bat’s prolonged sperm storage during reproduction, long lifespan, powered flight, low vision, echolocation, possible arsenic resistance, fur coloration, and consistently decreasing effective population sizes in their demographic history.
Materials and method
Sample and genome sequencing
The partially decomposed carcass of M. rufoniger was found in August 2012 at Gosudonggul cave in Danyang, South Korea (coordinates: 36° 59' 18.67'' N, 128° 22' 53.26'' E; elevation: 180.65 m) by Gosudonggul cave management office staff. The M. rufoniger was moved to the Natural Heritage Center of the Cultural Heritage Administration (South Korea) and kept frozen (Keeping temperature: -70°C). We acquired the M. rufoniger sample from the Natural Heritage Center under the Cultural Heritage Administration permit in June 2013. The wing membrane tissue, which seemed not decayed, was acquired for genomic DNA sequencing (S1 Picture).
Its genomic DNA was sequenced using Illumina HiSeq2000 platform, with a read length of 100 bp and insert sizes of 566 and 574 bp from two genomic libraries. The DNA was extracted using a Tiangen Micro DNA Kit. The amount of DNA was quantified by fluorometry using a UV spectrophotometer (Tecan F200). A partial DNA degradation was identified by electrophoresis (S2 Picture), but it was judged good resulting in no significant sequencing quality problems. The genomic DNA was sheared to approximately 566 bp and 574 bp using a Covaris S2 Ultrasonicator, and then used in the preparation of whole-genome shotgun libraries according to Illumina’s library preparation protocols. The efficacy of each step of the library construction was ascertained using a 2100 Bioanalyzer (S6 Fig). The final dilutions of the two libraries were then sequenced using a HiSeq2000 sequencer with the TruSeq PE Cluster kit v3-cbot HS and TruSeq SBS kit v3-HS for 200 cycles.
Sequencing read filtering
A DNA read was filtered out when the read’ Q20 base content was lower than 70%, using IlluQCPRLL.pl script of NGSQCToolkit (ver 2.3.3; S1 Table) [29]. We additionally downloaded genome data of the other bats (Myotis brandtii, Myotis davidii, Myotis lucifugus, Pteropus Alecto, and Pteropus vampyrus) from the NCBI database. Low quality DNA reads from the other bats were also filtered out using the same method (S1 Table). The possible microbial contaminated DNA reads were filtered out when the reads’ alignment scores to microbial (bacteria and fungi) genomes were higher than that to the Myotis bat genomes (M. brandtii, M. davidii, and M. lucifugus; S3 Table). We used microbial genomes that were downloaded from Ensembl database and the three Myotis bat genome assemblies were from NCBI. The DNA reads were mapped to the microbial and Myotis bat genomes using BWA-MEM algorithm of BWA (version 0.7.15) with Mark shorter split hits as a secondary option (-M) [30]. The rmdup command of SAMtools (ver 0.1.19) was used to remove PCR duplicates [31]. The possible microbial contaminated reads in the other Myotis bats were also filtered out by using the same method but mapping was to their own genome assemblies (S1 Table).
Species identification
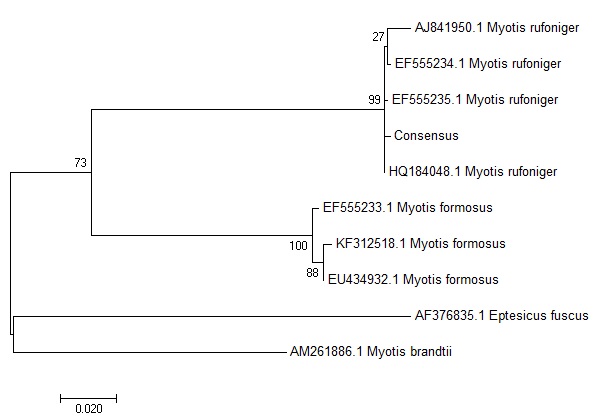
To construct the phylogenetic tree and identify the sample’s species, the mitochondrial cytochrome b consensus sequence was generated by mapping the sequencing data to the M. rufoniger mitochondrial cytochrome b sequence (GenBank accession number: HQ184048.1) using BWA-MEM algorithm of BWA (version 0.7.15) with Mark shorter split hits as a secondary option (–M) [30]. The reads were realigned using the GATK (version 2.5–2) RealignerTargetCreator and IndelRealigner algorithms to minimize the read alignment mismatches [32]. The rmdup command of SAMtools (ver 0.1.19) was used to remove PCR duplicates [31]. The variants from the whole genome sequencing data against the reference were called using mpileup command of SAMtools (version 0.1.19) with–E option to minimize the noise, -A option to use regardless of insert size constraint, -q 20 to consider only high quality mapping reads, and–Q 30 to consider only high quality bases [31]. The called variants were filtered using vcfutils.pl varFilter command of SAMtools (ver 0.1.19) with -d 8 option (minimal depth of 8) [31]. SNVs detected from the whole genome sequencing data against the reference were substituted to construct mitochondrial cytochrome b consensus sequence of the M. rufoniger using vcfutils.pl vcf2fq command of SAMtools (ver 0.1.19) with -l 5 option (no indel present within a 5 bp window) [31]. A multiple sequence alignment of the mitochondrial cytochrome b sequences was conducted using MUSCLE (version 3.8.31) program with default options [2,33]. A phylogenetic analysis was conducted using MEGA 7.0 program [34]. The phylogenetic tree was inferred by using a Maximum Likelihood method based on the Tamura-Nei model with 1,000 replicates bootstrapping [35,36]
Genome size estimation
For a K-mer analysis, KmerFreq_HA command of SOAPec program in SOAPdenovo2 package was used with a K = 17 option [37]. The genome size was estimated from the number of K-mers (depth > 3) divided by the peak depth of the K-mer graph.
Mapping sequence data
The filtered sequenced reads were mapped to the three Myotis bat genome assemblies (M. brandtii, M. davidii, and M. lucifugus) using BWA-MEM algorithm of BWA (version 0.7.15) with Mark shorter split hits as a secondary option (-M) [30]. The reads were realigned using the GATK (version 2.5–2) RealignerTargetCreator and IndelRealigner algorithms to minimize the read alignment mismatches [32]. The rmdup command of SAMtools (ver 0.1.19) was used to remove PCR duplicates [31]. The variants of M. rufoniger were called using mpileup command of SAMtools with–E, -q 20, -A, and–Q 30 options [31]. The called variants were filtered using vcfutils.pl varFilter command of SAMtools (ver 0.1.19) with -d 8 and -D 250 options (minimal depth of 8 and maximal depth of 250) [31]. The DNA consensus sequences of M. rufoniger were generated by substituting the M. rufoniger SNVs detected from the whole genome sequencing data to each reference bat genome using the vcfutils.pl vcf2fq command of SAMtools (ver 0.1.19) with -l 5 option [31].
Repeat annotation
RepeatMasker (version 4.0.5) was used to identify transposable elements by aligning the M. rufoniger consensus genome sequence against a library of M. brandtii with default option [38]. For the comparison of the M. rufoniger’s repeat annotation with the other species, the same method was used for other genome references.
Sex determination
Male M. davidii reads were mapped to the M. brandtii (a male sample) genome assembly using BWA-MEM algorithm of BWA 0.7.15 version [30] with–M option. The reads were realigned using the GATK (version 2.5–2) RealignerTargetCreator and IndelRealigner algorithms to minimize the read mismatches [32]. The rmdup command of SAMtools (ver 0.1.19) was used to remove PCR duplicates [31]. Variants were called using mpileup command of SAMtools with–E, -q 20, -A, and–Q 30 options [31]. The called variants were filtered using vcfutils.pl varFilter command of SAMtools (ver 0.1.19) with -d 8 and -D 250 options [31]. To identify X-chromosomal scaffolds from the M. brandtii genome assembly, we conducted BLASTn (Blast 2.2.26) with an E-value cutoff of 1E-6 using X-chromosomal CDS sequences of human as queries [39]. Scaffolds having greater than or equal to 10 top hits were considered as X-chromosomal scaffolds.
Comparative evolutionary analysis
Orthologous gene families between M. rufoniger’s sequence and 13 mammalian genomes (Seven bat genomes: M. brandtii, M. lucifugus, M. davidii, E. fuscus, P. alecto, P. vampyrus, and R. aegyptiacus, and six other mammals: H. sapiens, M. musculus, B. taurus, E. caballus, H. glaber, and M. domestica; S2 Table) were identified using OrthoMCL software (version 2.0.9) by matching and adding the M. rufoniger genes to orthologous clusters of M. brandtii’s genes, and 6,782 single-copy gene families among the 14 species were collected [7,13]. M. brandtii, M. lucifugus, M. davidii, E. fuscus, P. alecto, P. vampyrus, R. aegyptiacus, H. sapiens, M. musculus, B. taurus, E. caballus, H. glaber, and M. domestica genomes and gene sets were downloaded from the NCBI database (S3 Table).
To construct a phylogenetic tree and estimate the divergence time of bats and other mammals, 1,258,141 four-fold degenerate sites from the 6,782 single-copy gene families were used to conduct the multiple sequence alignment (MSA) using MUSCLE (version 3.8.31) program [33]. The phylogenetic tree was constructed among the 14 species using a Randomized Axelerated Maximum Likelihood (RAxML) program (version 8.2) [40]. In RAxML program, GTR gamma model was used as the nucleotide substitution model. In order to check the branch reliability, 1,000 rapid bootstrapping was used. The M. domestica was used as an outgroup species. Based on this phylogenetic tree topology, the divergence time was estimated using Reltime-ML in MEGA 7.0 program [34]. In this process, the divergence time at the node between M. brandtii–H. sapiens was constrained to be 97.5 MYA, and M. brandtii–P. alecto was constrained to be 62.6 MYA based on the TimeTree database [41].
Amino acid changes were identified by constructing MSAs among the 6,782 single copy gene families using Clustal Omega (version 1.2.4) [42]. Function-altering amino acid changes were predicted using PROVEAN (version 1.1.5) [14]. The MSAs of the DCT, SLC45A2, TYRP1, OCA2, and N6AMT1 genes were manually checked, and uAACs within misaligned regions were excluded in the variant score analysis. Only homozygous amino acid variants were considered in the amino acid sequence comparison analysis to reduce bias from individual-specific variants.
Demographic history and genetic diversity
The demographic history of the Myotis bats was estimated using a pairwise sequential Markovian coalescent (PSMC) program [11]. We mapped the downloaded genome sequencing data of Myotis bats to the M. lucifugus genome assembly using BWA-MEM algorithm of BWA 0.7.15 version [30] with–M option. The reads were realigned using the GATK (version 2.5–2) RealignerTargetCreator and IndelRealigner algorithms to minimize the read mismatches [32]. The rmdup command of SAMtools (ver 0.1.19) was used to remove PCR duplicates [31]. SAMtools was used to extract diploid genome information from the BAM files [31]. Options of -N25 -t15 -r -p “4+25*2+4+6” were used for the PSMC analysis. The generation time for the Myotis bats was estimated as sum of average maturation and gestation time (S16 Table) [43,44]. The mutation rate of Myotis bats was estimated by multiplying reported neutral mutation rate for mammals (2.2 × 10−9 per base pair per year) by the generation time (1.2 year) [45]. Atmospheric surface air temperature and global relative sea level data of the past three million years were obtained and used for this analysis [46]. The genomic diversity was calculated by dividing the number of heterozygous SNVs by its genome size (bp) [8]. For the genomic diversity calculation, variants were called using mpileup command of SAMtools with–E, -q 20, -A, and–Q 30 options [31]. The called variants were filtered using vcfutils.pl varFilter command of SAMtools (ver 0.1.19) with -d 8 and -D 250 options [31].
Supporting information
(JPG)
{kind=link}
(JPG)
{kind=link}
The phylogenetic relationship of Myotis bats was inferred from the alignment of mitochondrial cytochrome b coding sequences. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Each node has its species name and GenBank accession number.
(JPG)
{kind=link}
The x-axis represents depth, and the y-axis represents proportion, as calculated by the frequency at a given depth divided by the total frequency at all depths.
(PDF)
Previously reported bats’ unique amino acid sequence changes within FSHB, GHR, IGF1R, TP53, and MDM2 are highlighted in yellow; (A), Alignment of FSHB encoded peptide sequences; (B), Alignment of GHR-encoded peptide sequences; (C), Alignment of IGF1R encoded peptide sequences; (D), Alignment of TP53-encoded peptide sequences; (E), Alignment of MDM2 encoded peptide sequences; (F), Alignment of SLC45A2-encoded peptide sequences; (G), Alignment of RGS7BP-encoded peptide sequences; (H), Alignment of RHO-encoded peptide sequences; (I), Alignment of OPN1SW-encoded peptide sequences; (J), Alignment of CNGB3-encoded peptide sequences.
(PDF)
Myotis bats’ uAACs within DCT, SLC45A2, TYRP1, and OCA2 genes are highlighted (yellow if PROVEAN score of variant ≤ -2.5, unless green). The domain region of human sequences are shaded in gray; (A) Alignment of DCT encoded peptide sequences; (B) Alignment of SLC45A2 encoded peptide sequences; (C) Alignment of TYRP1 encoded peptide sequences; (D) Alignment of OCA2 encoded peptide sequences.
(PDF)
M. rufoniger specific amino acid sequence changes within N6AMT1 gene are highlighted in yellow.
(PDF)
(A) Sequencing library 574bp QC; (B) Sequencing library 566bp QC.
(PDF)
(XLSX)
(XLSX)
(XLSX)
The GC contents were calculated by dividing the number of [(C+G+S)–(M+K+R+Y)] ~ [(C+G+S) + (M+K+R+Y)] by the number of (sum—gap); The proportion of A, C, G, and T, and the proportion of W, S, M, K, and Y were calculated by dividing each number by the number of (sum-gap)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
Korea Institute of Science and Technology Information (KISTI) provided us with Korea Research Environment Open NETwork (KREONET), which is an academic internet connection service.
Data Availability
All sequencing files are available from the National Center for Biotechnology Information (NCBI) database (566bp: SRX2755014, 574bp: SRX2755088).
Funding Statement
This work was supported by ‘the bioinformatics marker discovery analysis system using genomic big data’ Research Fund (1.150014.01) of Ulsan National Institute of Science & Technology (UNIST). It was also supported by ‘Software Convergence Technology Development Program’ through the Ministry of Science, ICT and Future Planning (S0503-17-1007), PGI of Genome Research Foundation internal research fund, a grant (NIBR201603103) from the National Institute of Biological Resources funded by the Ministry of Environment, Republic of Korea, and Ulsan city's Genome Korea Project. Geromics Inc. provided support in the form of salaries for OC, JHJ, and JB, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
References
- 1.Tomes RF, editor ON THE CHARACTERS OF FOUR SPECIES OF BATS INHABITING EUROPE AND ASIA, AND THE DESCRIPTION OF A NEW SPECIES OF VESPERTILIO INHABITING MADAGASCAR. Proceedings of the Zoological Society of London; 1858: Wiley Online Library.
- 2.Ruedi M, Csorba G, Lin L-K, Chou C-H. Molecular phylogeny and morphological revision of Myotis bats (Chiroptera: Vespertilionidae) from Taiwan and adjacent China. Zootaxa. 2015;3920(1):301–42. [DOI] [PubMed] [Google Scholar]
- 3.Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2012:1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seim I, Fang X, Xiong Z, Lobanov AV, Huang Z, Ma S, et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nature communications. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, et al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nature communications. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yim H-S, Cho YS, Guang X, Kang SG, Jeong J-Y, Cha S-S, et al. Minke whale genome and aquatic adaptation in cetaceans. Nature genetics. 2014;46(1):88–92. doi: 10.1038/ng.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung O, Jin S, Cho YS, Lim J, Kim H, Jho S, et al. The first whole genome and transcriptome of the cinereous vulture reveals adaptation in the gastric and immune defense systems and possible convergent evolution between the Old and New World vultures. Genome biology. 2015;16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Cho YS, Kim H-M, Chung O, Kim H, Jho S, et al. Comparison of carnivore, omnivore, and herbivore mammalian genomes with a new leopard assembly. Genome Biology. 2016;17(1):211 doi: 10.1186/s13059-016-1071-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YM, Choi EH, Kim SK, Jang KH, Ryu SH, Hwang UW. Complete mitochondrial genome of the Hodgson's bat Myotis formosus (Mammalia, Chiroptera, Vespertilionidae). Mitochondrial DNA. 2011;22(4):71–3. doi: 10.3109/19401736.2011.624598 [DOI] [PubMed] [Google Scholar]
- 10.Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences. 2012;109(36):E2382–E90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–6. doi: 10.1038/nature10231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith JD, Gregory TR. The genome sizes of megabats (Chiroptera: Pteropodidae) are remarkably constrained. Biology letters. 2009;5(3):347–51. doi: 10.1098/rsbl.2009.0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome research. 2003;13(9):2178–89. doi: 10.1101/gr.1224503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one. 2012;7(10):e46688 doi: 10.1371/journal.pone.0046688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 16.Racey P. The prolonged storage and survival of spermatozoa in Chiroptera. Journal of Reproduction and Fertility. 1979;56(1):391–402. [DOI] [PubMed] [Google Scholar]
- 17.Hartman CG. On the survival of spermatozoa in the female genital tract of the bat. The Quarterly Review of Biology. 1933;8(2):185–93. [Google Scholar]
- 18.Kitchener D. Reproduction in female Gould's wattled bat, Chalinolobus gouldii (Gray)(Vespertilionidae), in Western Australia. Australian journal of zoology. 1975;23(1):29–42. [DOI] [PubMed] [Google Scholar]
- 19.Sturm RA, Duffy DL. Human pigmentation genes under environmental selection. Genome biology. 2012;13(9):248 doi: 10.1186/gb-2012-13-9-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newton J, Cohen-Barak O, Hagiwara N, Gardner JM, Davisson MT, King RA, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. The American Journal of Human Genetics. 2001;69(5):981–8. doi: 10.1086/324340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boissy RE, Zhao H, Oetting WS, Austin LM, Wildenberg SC, Boissy YL, et al. Mutation in and lack of expression of tyrosinase-related protein-1 (TRP-1) in melanocytes from an individual with brown oculocutaneous albinism: a new subtype of albinism classified as" OCA3". American journal of human genetics. 1996;58(6):1145 [PMC free article] [PubMed] [Google Scholar]
- 22.Manga P, Kromberg J, Box N, Sturm R, Jenkins T, Ramsay M. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. The American Journal of Human Genetics. 1997;61(5):1095–101. doi: 10.1086/301603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S-T, Nicholls RD, Jong MT, Fukai K, Spritz RA. Organization and sequence of the human P gene and identification of a new family of transport proteins. Genomics. 1995;26(2):354–63. [DOI] [PubMed] [Google Scholar]
- 24.Han J, Kraft P, Nan H, Guo Q, Chen C, Qureshi A, et al. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 2008;4(5):e1000074 doi: 10.1371/journal.pgen.1000074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X, Dong G-X, Hu X-S, Miao L, Zhang X-L, Zhang D-L, et al. The genetic basis of white tigers. Current biology. 2013;23(11):1031–5. doi: 10.1016/j.cub.2013.04.054 [DOI] [PubMed] [Google Scholar]
- 26.Ren X, Aleshin M, Jo WJ, Dills R, Kalman DA, Vulpe CD, et al. Involvement of N-6 adenine-specific DNA methyltransferase 1 (N6AMT1) in arsenic biomethylation and its role in arsenic-induced toxicity. Environmental health perspectives. 2011;119(6):771 doi: 10.1289/ehp.1002733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu HJ, Kang JH, Lee S, Choi YJ, Oh D, Lim JD, et al. Elemental analysis of the liver, kidney, and intestine tissues from a Hodgson's bat (Myotis formosus tsuensis). Korean Journal of Veterinary Research. 2016;56(1):51–2. [Google Scholar]
- 28.Monge G, Jimenez-Espejo FJ, García-Alix A, Martínez-Ruiz F, Mattielli N, Finlayson C, et al. Earliest evidence of pollution by heavy metals in archaeological sites. Scientific reports. 2014;5:14252–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one. 2012;7(2):e30619 doi: 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular biology and evolution. 2016:msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular biology and evolution. 1993;10(3):512–26. [DOI] [PubMed] [Google Scholar]
- 36.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985:783–91. doi: 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 37.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarailo‐Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 2009:410 1–4. [DOI] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 40.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. doi: 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22(23):2971–2. doi: 10.1093/bioinformatics/btl505 [DOI] [PubMed] [Google Scholar]
- 42.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology. 2011;7(1):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, et al. Human ageing genomic resources: integrated databases and tools for the biology and genetics of ageing. Nucleic acids research. 2012:gks1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iannelli M. Mathematics of Biology: Lectures given at a Summer School of the Centro Internazionale Matematico Estivo (CIME) held in Cortona (Arezzo), Italy, June 18–30, 1979: Springer Science & Business Media; 2011.
- 45.Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proceedings of the National Academy of Sciences. 2002;99(2):803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bintanja R, Van de Wal R. North American ice-sheet dynamics and the onset of 100,000-year glacial cycles. Nature. 2008;454(7206):869–72. doi: 10.1038/nature07158 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(JPG)
(JPG)
The phylogenetic relationship of Myotis bats was inferred from the alignment of mitochondrial cytochrome b coding sequences. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Each node has its species name and GenBank accession number.
(JPG)
The x-axis represents depth, and the y-axis represents proportion, as calculated by the frequency at a given depth divided by the total frequency at all depths.
(PDF)
Previously reported bats’ unique amino acid sequence changes within FSHB, GHR, IGF1R, TP53, and MDM2 are highlighted in yellow; (A), Alignment of FSHB encoded peptide sequences; (B), Alignment of GHR-encoded peptide sequences; (C), Alignment of IGF1R encoded peptide sequences; (D), Alignment of TP53-encoded peptide sequences; (E), Alignment of MDM2 encoded peptide sequences; (F), Alignment of SLC45A2-encoded peptide sequences; (G), Alignment of RGS7BP-encoded peptide sequences; (H), Alignment of RHO-encoded peptide sequences; (I), Alignment of OPN1SW-encoded peptide sequences; (J), Alignment of CNGB3-encoded peptide sequences.
(PDF)
Myotis bats’ uAACs within DCT, SLC45A2, TYRP1, and OCA2 genes are highlighted (yellow if PROVEAN score of variant ≤ -2.5, unless green). The domain region of human sequences are shaded in gray; (A) Alignment of DCT encoded peptide sequences; (B) Alignment of SLC45A2 encoded peptide sequences; (C) Alignment of TYRP1 encoded peptide sequences; (D) Alignment of OCA2 encoded peptide sequences.
(PDF)
M. rufoniger specific amino acid sequence changes within N6AMT1 gene are highlighted in yellow.
(PDF)
(A) Sequencing library 574bp QC; (B) Sequencing library 566bp QC.
(PDF)
(XLSX)
(XLSX)
(XLSX)
The GC contents were calculated by dividing the number of [(C+G+S)–(M+K+R+Y)] ~ [(C+G+S) + (M+K+R+Y)] by the number of (sum—gap); The proportion of A, C, G, and T, and the proportion of W, S, M, K, and Y were calculated by dividing each number by the number of (sum-gap)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All sequencing files are available from the National Center for Biotechnology Information (NCBI) database (566bp: SRX2755014, 574bp: SRX2755088).