

Abstract
Despite advances in the treatment of asthma, optimization of symptom control remains an unmet need in many patients. These patients, labeled severe asthma, are responsible for a substantial fraction of the disease burden. In these patients research is needed to define the cellular and molecular pathways contributing to disease which in large part are refractory to corticosteroid treatment. The causes of steroid-resistant asthma are multifactorial and result from complex interactions of genetics, environmental factors, and innate and adaptive immunity. Adaptive immunity, addressed here, integrates the activities of distinct T cell subsets and by definition is dynamic and responsive to an ever-changing environment and the influences of epigenetic modifications. These T-cell subsets exhibit different susceptibilities to the actions of corticosteroids and, in some, corticosteroids enhance their functional activation. Moreover, these subsets are not fixed in lineage differentiation but can undergo transcriptional reprogramming in a bidirectional manner between protective and pathogenic effector states. Together, these factors contribute to asthma heterogeneity between patients but also in the same patient at different stages of their disease. Only by carefully defining mechanistic pathways, delineating their sensitivity to corticosteroids, and determining the balance between regulatory and effector pathways, will precision medicine become a reality with selective and effective application of targeted therapies.
Keywords: Asthma, T cell subsets, steroid sensitivity, plasticity
1.0 Introduction
Asthma is a complex syndrome characterized by altered airway function and airway inflammation. Over the last several decades, there has been significant progress in both the understanding and treatment of asthma. The introduction of inhaled corticosteroids, avoiding the many adverse events of systemic corticosteroids, significantly impacted the quality of life of asthmatics with significant reductions in asthma mortality and morbidity, as well as the need for hospitalization and urgent care visits. Despite these advances, a number of issues remain unresolved and unmet needs for asthmatics persist. It is estimated that 10–15% of asthmatics remain uncontrolled on what is considered optimum therapy and upwards of 40% of asthmatics may show little to no improvement (even deterioration) of lung function on high doses of inhaled corticosteroids (1–3). The efficacy of corticosteroids in asthma likely occurs at several levels, primarily reducing inflammation and edema. Effects on smooth muscle contractility or airway remodeling are less apparent. The anti-inflammatory effects are mediated by targeting steroid-sensitive (SS) cells, cells which undergo apoptosis or alter functional programs when exposed to corticosteroids. Since corticosteroids may not be equally effective in all patients, it is likely that other cell types and pathways contributing to asthma are not steroid-sensitive and are major contributors to steroid-resistant (SR) asthma. Examples of corticosteroid-sensitive cells that are implicated in asthma pathogenesis are CD4+ T cells and eosinophils while neutrophils and CD8+ T cells have been shown to be insensitive to corticosteroids. Given the efficacy of corticosteroids in a large proportion of asthmatics, the development of novel therapeutics has been to address the needs of patients whose asthma is uncontrolled or requires high doses of corticosteroids. Additionally, since corticosteroids have little disease-modifying activity and are only effective when taken (4), targeting pathways or mediators contributing to disease progression are important objectives. Corticosteroids may also have a limited role in preventing asthma exacerbations, further suggesting that the triggers of exacerbation may involve cells and pathways that are less responsive to corticosteroids. Different triggers of asthma exacerbation result in diverse immune-inflammatory outcomes, including accumulation of different inflammatory cells (5–10) and pathways that exhibit differential steroid responsiveness (Table 1).
Table 1.
Asthma Pathogenesis.
| Trigger | Allergen Exposure | Virus Infection | Diesel Exhaust/Ozone | Smoking |
|---|---|---|---|---|
| Predominant pathway | Th2, IgE | Th1, LT | Thl, LT | Thl, LT |
| Predominant cell type | Eosinophils CD4 T cells | Neutrophils CD8 T cells | Neutrophils CD8 T cells | Neutrophils CD8 T cells |
LT: leukotrienes
Central to these issues is the large variety of T-lymphocyte subsets implicated in asthma pathobiology and the integration of adaptive and innate immunity leading to airway dysfunction and inflammation. In this review, we will consider the individual contributions of several distinct T-cell subsets, their integration into pathology-driving circuits, the pathways activated by different asthma triggers, and the identification of pathways that are SS and those which remain unaffected by corticosteroids contributing to severe asthma. In addition to infiltrating and resident T lymphocytes, airway structural cells participate in the inflammatory responses releasing important mediators that regulate T-cell subsets and innate lymphocyte (ILC) responses. Oxidative stress is an important regulator of airway inflammatory responses, especially in severe asthma associated with increased levels of reactive oxygen species (ROS) and reduced anti-oxidant responses (11, 12). The increased levels of ROS contribute in different ways to the chronic inflammatory responses, airway hypercontractility, and corticosteroid insensitivity (13–15).
This immuno-inflammatory complexity positions asthma as a syndrome with many overlapping phenotypes when clinical and physiological parameters are considered. Clustering of asthma phenotypes based on these parameters has had some benefit in furthering our understanding of the significant heterogeneity of asthma. However, only by understanding the underlying mechanisms and pathways (endotypes) involved will we advance personalized treatment. Defining asthma clusters based on differentially-expressed genes appears more promising to capture the diverse pathways of asthma pathobiology that could lead to identification of important targets for therapy and offer insights towards achieving personalized medicine (16–18). This approach has been used to identify genes from sputum cells that associated pathway-based transcriptomic clusters to clinically-important features of asthma (19).
A major challenge faced in a clustering approach is the heterogeneity of the disease, not only between patients but also in the same patient at different stages of their disease. Based on clinical and physiological parameters, the stability of a patient remaining in the same asthma cluster over time is controversial (20, 21). This may not be surprising given the number of triggers resulting in an asthma exacerbation. These individual triggers have the capacity to activate distinct pathways characterized by unique patterns of inflammatory cell accumulation and the response to corticosteroids (Table 1). Notwithstanding the important gene-environment interactions that govern these outcomes, epigenetic mechanisms link gene regulation to these environmental triggers. Important epigenetic marks that govern the distinct stages of asthma from inception to maintenance, to exacerbation, and to resolution, are in large part translated to T-cell subset function and cytokine release. Understanding these intricate and dynamic interactions between functionally activated T-cell subsets and asthma pathophysiology requires well-characterized experimental models and longitudinal cellular, molecular, and genetic clinical investigations, with the ultimate goal of identifying novel therapeutic approaches.
2.0 The Role of CD4+ T Cells in Patients with Asthma
For several decades, asthma has been considered a T helper 2 (Th2)-driven inflammatory disease, characterized by eosinophilic inflammation, Th2 cytokine production [i.e., interleukin-4 (IL-4), IL-5, and IL-13], and airway hyperresponsiveness (AHR) (22, 23). In the earliest studies, type 2 cytokine-producing CD4+ T cells were identified in the bronchoalveolar lavage (BAL) fluid and airway tissue of asthmatics (24). In mice, the transfer of Th2 cells was sufficient to induce airway eosinophilia and AHR (25, 26). Naive T cells can, under different conditions, differentiate into Th1, Th2, Th9, Th17, or T regulatory (Treg) cells, each characterized by the release of distinct cytokine profiles and effector functions. Each of these T-cell subsets has been associated with allergic inflammation indicating the complex fate decisions and multifaceted involvement in disease pathology (27). During differentiation, each of the T-cell subsets is subject to the cytokine milieu and the epigenetic environment for the activities they are programmed to exhibit. As is increasingly evident, the fate decisions may not be unidirectional or terminal but the cells can undergo transcriptional reprogramming exhibiting different effector functions.
In this section, we focus on the contribution of type 2 cytokine-producing CD4+ T cells in the development of asthma. Differentiation into Th2 cells is dependent on the cytokine IL-4 followed by expression of the key transcription factor GATA3. Th2 cells release IL-4 and IL-13 which promote IgE production in human B cells, and growth and differentiation of human B cells and monocytes (28). Th2 cells protect against nematodes but also play a central role in allergic immune responses (29, 30). Based on in vitro and in vivo studies in mice as well as in humans, the processes of Th1/Th2 polarization in CD4+ T cells is reciprocally regulated (31–33). The expression of the key type 1 transcription factor T-BET and production of IFNγ does not only lead to the differentiation of Th1 cells but also exerts an inhibitory function on the maturation of Th2 cells (28). Likewise, GATA3 expression and the presence of IL-4 favors Th2 polarization, but additionally inhibit the differentiation of Th1 cells (28).
The differentiation of Th2 cells is highlighted by molecular events leading to the activation of the IL-4/IL-13 pathway (34–37). IL-4 binds to a receptor composed of an IL-4Rα chain and the common γ chain, inducing oligomerization. IL-13 binds to its own specific receptor subunit IL-13Rα1 chain to which IL-4 cannot bind, and additionally to the IL-4Rα chain (IL-4 receptor α) (38). IL-4 activates the Janus tyrosine kinases (JAK1 and JAK3), while IL-13 transmits its signal through JAK1 and the Tyk2 kinase. The activated kinases initiate the phosphorylation of the intracellular molecule signal transducer and activator of transcription 6 (STAT6). Once phosphorylated, STAT6 forms homodimers which translocate to the nucleus and bind to IL-4/IL-13 responsive regulatory gene regions.
The pathophysiological importance of type 2 cytokine production has been demonstrated in several studies as increased levels of IL-4, IL-5, and IL-13 were observed in asthmatic patients (39–44). GATA3 gene expression in BAL cells and bronchial biopsies from asthmatics significantly correlated with IL-5 mRNA levels and AHR (45). In cells from induced sputum of asthmatics compared to healthy controls, elevated type 2 cytokine receptor expression of IL-4Rα and IL-5Rα were present which correlated with increased expression of GATA3 and STAT6 (46). Genetic studies have associated single nucleotide polymorphisms (SNP) within the IL-4/IL-13 pathway with susceptibility to asthma (39, 47–49).
Atopic patients, regardless of asthma status, exhibited increased allergen-specific CD4+ T-cell activation and IL-5 production after house dust mite (HDM) stimulation of peripheral blood mononuclear cells (PBMC) (50). IL-5 production was significantly elevated in PBMC and BAL cells from asthmatics (50, 51). The assessment of SS and SR asthmatics and healthy controls revealed lower IL-13 levels in CD4+ vs. CD8+ T cells while levels of the anti-inflammatory cytokine IL-10 were higher in CD4+ T cells from controls and SS asthmatics compared to SR asthmatics (52). A decrease in IL-10 production by CD4+CD45RO+ T cells has previously been correlated with severe asthma (53, 54).
2.1 The Role of CD4+ T Cells in Experimental Asthma
The roles of IL-4 and IL-13 in the induction of Th2 responses and lung allergic responses in experimental models of asthma were initially demonstrated in genetically-manipulated mice deficient in IL-4 or IL-13 (37, 55–57). IL-4-deficient mice in contrast to wild-type (WT) mice showed substantial attenuation of IL-5 secretion, IgE production, eosinophilic inflammation, and AHR (56, 58). IL-13-deficient mice were unable to develop AHR even though airway inflammation and high levels of IL-4 and IL-5 were detectable (57). AHR was restored after administration of recombinant IL-13, indicating that IL-13 was also necessary for AHR. In contrast to IL-4-deficient mice, IL-13-deficient mice failed to generate goblet cells responsible for mucus overproduction (59), a characteristic feature of asthma. Hence, IL-13 may be more relevant for mucus hypersecretion and subepithelial fibrosis. The strongest effects were seen in mice deficient in both IL-4 and IL-13 (60). Here, complete abrogation of eosinophil infiltration, IgE production, and IL-5 secretion were seen, and differentiation to a Th1-like phenotype was demonstrated. Thus, the expression of either IL-4 or IL-13 may be sufficient to induce a Th2 response but their concomitant release seems to exert an additive effect on the immune response to allergen. As IL-4 and IL-13 mediate their signaling through IL-4Rα, this receptor plays a central role in allergic inflammation. In IL-4Rα-deficient mice, IgE production was diminished and Th2 differentiation was impaired (61). Mice lacking IL-4Rα were also unable to develop AHR, induce mucus production, or airway inflammation (62).
The JAK-STAT signaling pathway is involved in many of the physiologic events that are dysregulated in asthma which is why this pathway has been targeted in various immune disorders (63). Only a few studies have investigated the effects on asthma pathogenesis after inhibition of the JAK pathway. Using the JAK1 and JAK3 inhibitor R256, the in vitro differentiation of Th2 cells was prevented by blocking the phosphorylation of STAT6 and STAT5 without affecting Th1 and Th17 differentiation (64). Lung allergic responses including AHR, eosinophilia, airway inflammation, and Th2 cytokine production in the BAL fluid were prevented in a model of experimental asthma when R256 was administered during the sensitization phase (64). As shown in vitro, cytokine release by Th1, Th17, and Treg cells in the BAL fluid remained unaltered in R256-treated mice in response to allergen sensitization and challenge (64). When mice were treated with R256 during the challenge phase in an acute model or a secondary allergen challenge model, AHR, eosinophilia, and goblet cell metaplasia were significantly attenuated (64). However, Th2 cytokine levels including IL-4, IL-5, and IL-13 in the BAL fluid were not reduced in response to R256 treatment during the challenge phase in both models suggesting that the inhibitory effects were limited to an early stage of Th2 polarization and potentially downstream of the Th2 cytokine receptor signaling pathway (64). Other drugs (tofacinitinib, pyridine 6) targeting the JAK family members have been reported to reduce lung allergic responses in vivo (65, 66). BAL cells from asthmatics and healthy controls cultured in the presence of tofacinitinib alone or in combination with the corticosteroid dexamethasone (DEX) decreased the production of IFNγ, IL-13, and IL-17 (67). Unlike R256, these pan-JAK inhibitors also altered Treg, Th1, and Th17 responses.
Naive cells from mice lacking STAT6 were not capable of differentiating into Th2 cells (68–70). Following allergen sensitization and challenge, STAT6-deficient mice failed to develop AHR and airway eosinophilia (71, 72). Consistent with these observations, lower levels of the Th2 cytokines IL-4 and IL-5, but increases in the Th1 cytokine IFNγ were detected (72). Moreover, total serum and ovalbumin (OVA)-specific IgE production were completely abolished accompanied by significantly increased IgG2a responses in the serum and increased IFNγ levels in the BAL fluid. Following reconstitution with IL-5, AHR and airway eosinophilia were similar to WT mice (72). Of note, STAT6-deficient mice receiving IL-5 still developed a Th1 response after sensitization and challenge as reflected by high IFNγ and IgG2a production paralleled by low levels of IL-4 and the absence of goblet cell metaplasia, total IgE, and OVA-specific IgE (72).
Another gene regulated through the JAK/STAT pathway is the serine/threonine kinase Pim1 whose activity can be induced by type 2 cytokines. The role of PIM1 kinase has mainly been studied in tumor pathogenesis (73–77) but expression of Pim1 was critical to the IL-5-induced survival of eosinophils (78, 79) and promoted cell survival in T cells (80). Further, PIM1 expression was elevated in BAL eosinophils compared to blood cells from asthmatics in response to in vitro allergen exposure (81). Levels of PIM1 kinase were increased in the lungs of sensitized and challenged mice (82). To determine the role of PIM1 kinase in experimental asthma, mice were treated with a PIM1 kinase inhibitor during the challenge phase resulting in dose-response outcomes with significantly lower BAL eosinophil numbers, goblet cell metaplasia, AHR, and BAL cytokine levels including IL-4, IL-5, IL-13, and IFNγ in comparison to sham-treated mice (82). Interestingly, CD4+ as well as CD8+ T cell numbers in the lungs were significantly diminished in response to in vivo treatment with the inhibitor during the challenge phase (82). In vitro cultures confirmed these findings with reduced proliferative responses of both T-cell subsets in response to T-cell receptor (TCR) stimulation (anti-CD3/CD28) combined with the PIM1 kinase inhibitor, leading to lower cytokine production of IL-4, IL-5, IL-13, and IFNγ in the supernatants of cultured cells (82). Based on findings on the pharmacological inhibition of PIM1 kinase activity in an allergen-induced model of experimental asthma, de Vries and colleagues investigated whether PIM1 kinase activity protects against the development of HDM-induced allergic asthma in vivo by preserving airway epithelial integrity (83). Pim1-deficient mice demonstrated a similar increase in lung allergic responses including eosinophilia, mucus production, and AHR in comparison to WT mice exposed to HDM while the Th2 cytokines IL-5 and IL-10 were significantly increased in HDM-treated Pim1-deficient mice (83). Albeit in a relatively small population, primary bronchial epithelial cells (PBEC) from healthy controls and moderate/severe asthmatics cultured by air-liquid interface suggested that the inhibition of the PIM1 kinase reduced the viral load of cells infected with human rhinovirus (HRV) in both the healthy controls and asthmatics (84). Analyses of the expression of IFNβ and IL-29 showed strongly enhanced mRNA levels upon HRV infection compared to mock-infected control PBEC in healthy controls accompanied by enhanced STAT1 phosphorylation which was further enhanced in the presence of the PIM1 kinase inhibitor (84).
An important relationship exists between PIM1 kinase and the Runt-related transcription factor (Runx). PIM1 kinase regulates RUNX expression in vitro (85). Runx3 is required for epigenetic silencing in cytotoxic lineage thymocytes (86). Importantly in the current context, Runx3 cooperates with T-bet to repress the production of IL-4 by binding to the Il-4 silencer element in the Th2 cytokine locus and promotes the production of IFN-γ in Th1 cells (87–89). Loss of RUNX3 results in the spontaneous development of inflammatory bowel disease and allergic asthma (90, 91). The contribution of PIM1 in the pathogenesis of atopic diseases was confirmed in an experimental model of peanut allergy (92). The inhibition of PIM1 kinase attenuated peanut-induced allergic responses in vivo and was associated with reduced expression of Gata3 as well as levels of Il-17a and Rorγt. Inhibition of PIM1 kinase was associated with increased expression of Runx3, identifying an important inverse relationship between PIM1 kinase and Runx3; the higher the levels of PIM1 kinase, the lower the levels of Runx3 and greater Th2 levels; the higher the levels of Runx3, a greater repression of IL-4 was observed paralleled by reduced allergic disease.
3.0 Pathogenic Role of CD8+ T Cells in Asthma
Among the different T cell subsets implicated in asthma pathogenesis, the role of CD8+ T cells has received limited attention. CD8+ T cells have been associated with recognition and destruction of cells that are infected with viruses and other intracellular pathogens. There are now increasing reports of the presence of CD8+ T cells in the BAL fluid and sputum of asthmatic patients as well as increased numbers of CD8+ T-cell numbers in patients with status asthmaticus (93–95). In experimental models of asthma in mice, CD8+ T cells were shown to contribute to the development of methacholine-induced AHR, airway eosinophilia, and mucus hyperproduction. The role of CD8+ T cells has gained support because of the important properties they exhibit in some subsets of asthmatics (93–98). Unlike CD4+ T cells, CD8+ T cells appear to be steroid-insensitive (52, 99). CD8+ T cells, similar to other subsets, have the capacity to undergo transcriptional reprogramming and release pro-allergic cytokines (100). Many environmental triggers selectively lead to the accumulation of CD8+ T cells in the lung including viruses, changes in air quality (ozone and diesel exhaust), and cigarette smoke (5–10) (Table 1).
3.1 Role of CD8+ T Effector (Teff) Cells in Experimental Asthma
A number of studies reported that CD8+ T cells play a protective role in allergic disease (101–105). In a rat model of experimental asthma, depletion of CD8+ T cells upregulated late airway responses, AHR, and airway inflammation (101, 102) while administration of antigen-primed CD8+ T cells suppressed these responses (103). Regulatory or suppressive effects on lung allergic responses have been associated with secretion of type 1 cytokines such as IL-12 and IFNγ (104, 105). Immunization of mice with a vaccine comprised of antigen and cationic liposome-DNA complexes generated antigen-specific CD8+ T cells which, when adoptively transferred into sensitized and challenged mice, suppressed development of AHR, airway eosinophilia, and Th2 cytokine production. Suppression by the transferred CD8+ T cells was dependent on IFNγ production (106).
The plasticity of CD8+ T cells and their pro-asthmatic activities have been demonstrated in many ways. We initially showed that the depletion of CD8+ T cells attenuated AHR and airway inflammation in mice exposed to allergen exclusively via the airways, in the absence of systemic sensitization (107). The responses were restored following reconstitution of CD8+ T cells. Using CD8-deficient mice, lacking expression of CD8α, allergen-induced AHR and airway inflammation as well as IL-13 levels were significantly reduced compared to WT mice. This failure to fully develop AHR and lung inflammation was not the result of depletion of CD8α on other cell types as reconstitution of the mice with antigen-primed CD8+ T cells restored development of lung allergic responses and AHR through the release of IL-13 (108). IL-13-deficient CD8+ T cells were unable to restore these responses. Thus, in this study and others CD8+ T cells were shown to be prime sources of type 2 cytokines (109–113).
Two distinct populations of antigen-experienced CD8+ T cells, labeled effector memory (Teff) and central memory (Tcm) CD8+ T cells have been described (114–116). They differed significantly in their functional and migratory properties and were distinguished by the relative expression of CD62 ligand (CD62L) and CCR7. Antigen-experienced CD62Lhigh and CCR7high CD8+ Tcm homed preferentially to lymph nodes whereas antigen-experienced CD62LlowCCR7low CD8+ Teff cells trafficked more efficiently to non-lymphoid tissues and sites of inflammation and acquired effector function more rapidly. In vitro, CD8+ T cells can be differentiated in culture to either of these subtypes. When cultured in the presence of IL-15, antigen-specific CD8+ T cells developed the phenotypic and functional characteristics of CD8+ Tcm, whereas CD8+ T cells cultured in the presence of IL-2 showed characteristics of CD8+ Teff (116, 117).
To examine the function of CD8+ Teff and Tcm in the development of experimental asthma, CD8-deficient mice received either of these subsets after sensitization and prior to challenge (103). Transfer of Teff but not Tcm fully enabled development of allergen-induced lung allergic responses; the transferred Teff were recovered from the lung whereas Tcm were recovered from the local lymph nodes. Moreover, the Teff in the lung were shown to have expanded significantly and to produce IL-13. Even in WT mice, transfer of Teff further enhanced the development of AHR, airway eosinophilia, and mucus production as well as increased IL-13 levels in the BAL fluid. These studies established the role of a distinct subset of CD8+ Teff cells in the development of allergen-induced AHR and airway inflammation (Figure 1).
Figure 1. Recruitment of CD8+ Teff to the lung.
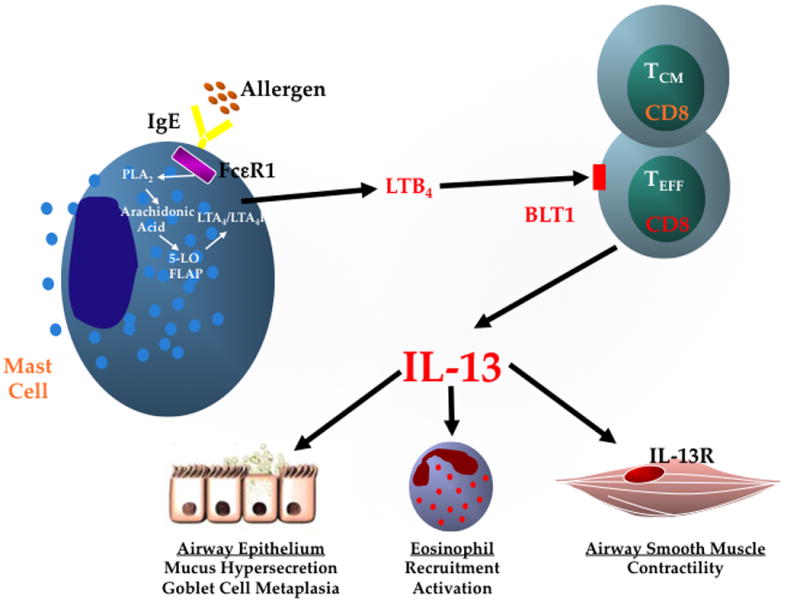
Schematic of the CD8+/BLT1+/IL-13+ axis in asthma pathogenesis. Lung mast cells, activated through the high affinity IgE receptor (FcεRI) after cross-linking with allergen, initiate a cascade resulting in the rapid synthesis and release of LTB4. Activated CD8+ Teff upregulate expression of BLT1, the high-affinity receptor for LTB4 and binding of LTB4 to BLT1 on CD8+ Teff results in their recruitment and accumulation in the lung. In the lung, in the presence of IL-4, CD8+ Teff undergo transcriptional reprogramming and differentiate into Tc2 cells releasing IL-13, which affects many of the cell types involved in asthma pathogenesis. PLA2, Phospholipase A2; FLAP, 5-LO activating protein; LTA4H, LTA4 hydrolase.
3.2 NOTCH Signaling Regulates CD8+ Teff Differentiation
Examining transcription factor expression in activated cells, strong induction of Notch1 was demonstrated specific to Teff in contrast to Tcm (118). The NOTCH signaling pathway is a highly conserved program for cell fate decisions in all organisms (119, 120). The signal induced by ligand binding is transmitted intracellularly by a process involving proteolytic cleavage of the NOTCH receptor by γ-secretase and nuclear translocation of the intracellular domain of the NOTCH family protein. γ-secretase inhibitors (GSI) suppress NOTCH activation. Some studies have suggested that NOTCH activation suppresses CD8+ T cell effector functions (121, 122) while inhibition of NOTCH signaling reduced the numbers of IFNγ-producing CD8+ T cells suggesting that NOTCH activation may be required for CD8+ T cell function (123). Treatment of CD8+ Teff with a GSI strongly inhibited NOTCH signaling in these cells and after adoptive transfer, GSI-treated Teff failed to restore AHR and airway inflammation in sensitized and challenged CD8-deficient recipients or enhance these responses in WT recipients (118). The effects of GSI were associated with increased expression of the NOTCH ligand DELTA1 in Teff and treatment with DELTA1-Fc resulted in decreased lung allergic responses associated with increased IFNγ in the BAL fluid. Thus, the NOTCH signaling pathway was an important regulator of CD8+ Teff differentiation to a pro-allergic effector pathway.
3.3 ERK1/2 Signaling Regulates CD8+ Teff Activation
Mitogen-activated protein kinases (MAPKs) play important roles in intracellular signal transduction in many cells. Extracellular signal-regulated kinase 1/2 (ERK1/2) was activated in the lungs of allergen sensitized and challenged mice when compared to normal, healthy lungs (124); activation was associated with many asthma-related events including goblet cell metaplasia and MUC5AC production (125), Th2 differentiation (126), B cell proliferation (127), and rolling and migration of eosinophils (128). Activation of ERK1/2 appeared important in the development of allergic inflammation (129–131). Treatment of CD8+ Teff with an ERK1/2 inhibitor prior to the secondary allergen challenge of sensitized WT mice resulted in significant decreases in AHR, airway eosinophilia, mucus production, and IL-5 and IL-13 levels in BAL fluid compared to recipients receiving untreated CD8+ Teff (132).
3.4 Importance of CD4+ T Cell-Derived IL-4 in CD8+ T Cell Activation
The relative contributions of CD4+ and CD8+ T cells in the development of experimental asthma appear dependent on the model used, likely accounting for the differences reported by different groups. In light of the capacity of CD8+ T cells to produce type 2 cytokines such as IL-4, IL-5, and IL-13 in mice and even in atopic asthmatics (133), the requirement for interactions between CD4+ and CD8+ T cells was investigated. Such interactions have been shown for CD4+ T cell priming of CD8+ T cells against bacterial or viral challenges (134–136). In the context of acute infection, CD4+ T cells were required during the maintenance phase of long-lived memory CD8+ T cell development (137). The indication of a requirement for interaction between the two subsets in a setting of asthma emerged in studies of MHC class-I restricted OVA peptide-specific TCR transgenic OT-1 mice (109). Following sensitization and challenge to OVA, because of the absence of functional CD4+ T cells, these mice failed to develop lung allergic responses. In contrast, transfer of CD4+ T cells prior to sensitization restored responsiveness leading to AHR, airway eosinophilia, goblet cell metaplasia, production of type 2 cytokines, and an increased number of lung CD8+IL-13+ T cells. Transfer of naive CD4+ T cells from Il-4-deficient mice before sensitization failed to trigger development of lung allergic responses or increases in CD8+IL-13+ T cells in the recipients (109). To more directly demonstrate the requirement for IL-4, recombinant IL-4 was administered to OT-1 mice during sensitization, which effectively restored the full spectrum of lung allergic responses including increases in CD8+IL-13+ T cells. To further establish the role of IL-4 in the activation of IL-13-producing CD8+ T cells, anti-IL-4 was administered to CD8-deficient mice prior to transfer of antigen-primed CD8+ T cells. Recipients of anti-IL-4 failed to develop AHR, airway inflammation, goblet cell metaplasia, or increases in IL-13-producing CD8+ T cells (100). IL-4 is known to stimulate a number of intracellular signaling cascades in CD8+ T cells (138). IFNγ and IL-4 reciprocally regulate type 2 polarization of CD8+ T cells. IL-4 activated effector CD8+ T cells to express type 2 as well as type 1 cytokines. Endogenously-produced IL-4 was a direct stimulator of CD8+ T-cell proliferation and these responses were enhanced in a model of airway disease (139). In a tumor model, tumor-derived IL-4 induced the expression of type 2 cytokines and Gata3 by responding CD8+ T cells (140). Taken together, the data indicated that CD4+ T cells and IL-4 during the allergen sensitization phase were central to the full activation of CD8+ T cells to convert to IL-13 producers, which was essential to development of the full spectrum of lung allergic responses.
3.5 LTB4-BLT1 Pathway in CD8+ T Cell Activation
BLT1 is a high affinity receptor specific for LTB4, whereas BLT2 is a low affinity receptor which also binds other eicosanoids. These two receptors differ in their expression pattern: BLT1 is expressed primarily on leukocytes, whereas BLT2 is expressed more ubiquitously (141). LTB4 was described as a chemoattractant for myeloid leukocytes (142, 143) while BLT1 was shown to be expressed on granulocytes, macrophages, monocytes, and eosinophils, and to a lesser extent on naive lymphocytes (141, 144–148). BLT1 also acts as an important attractant for differentiated T cells. BLT1 expression on mouse CD4+ T cells that have been differentiated in vitro to effector phenotypes (149). In vitro, activated CD4+ T cells cultured under non- (Th0), Th1-, or Th2-polarizing conditions all had increased levels of mRNA encoding Blt1 compared with naive cells, which expressed little Blt1 (149). In contrast, expression of BLT2 on naive T cells or on Th0, Th1, or Th2 effector cells was not detected. BLT1 expression was induced in CD4+ T cells leaving the lymph node and entering the tissue after activation by antigen (149). Similarly, BLT1 expression on mouse CD8+ T cells following in vitro differentiation to effector phenotypes has been shown (150, 151).
The LTB4-BLT1 pathway appears to play an important role in the recruitment and activation of IL-13-producing CD8+ Teff in the lung (Figure 1). In vitro-generated CD8+ Teff express higher mRNA levels of the LTB4 high affinity receptor, Blt1, than CD8+ Tcm (150, 151). To investigate whether BLT1 expression was essential for the development of CD8+ T-cell-mediated allergen-induced AHR and inflammation, BLT1+/+CD8+ or BLT1−/−CD8+ T cells were transferred into CD8-deficient mice (152). A similar approach was taken by adoptively transferring in vitro-generated CD8+ Teff. Only CD8+ T cells or Teff expressing BLT1 were capable of fully reconstituting all of the lung allergic responses, including increases in BAL IL-13 levels. These data established the essential and functional role of the LTB4 receptor on CD8+ T cells for accumulation in the lungs of sensitized and challenged mice. Further, numbers of BLT1+CD8+IL-13+ T cells were significantly increased in the lungs or BAL fluid of sensitized and challenged recipients (152). Initially, it was reported that trafficking of Teff into the airway did not differ in the presence or absence of this receptor following adoptive transfer and airway challenge of naive (non-sensitized) recipients (149). However, using sensitized and challenged mice as opposed to naive recipient mice, migration of transferred BLT1−/− Teff into the lung as well as BAL fluid was significantly impaired compared to transferred BLT1+/+ Teff (152). As recipient mice were sensitized to OVA prior to OVA challenge, LTB4 production in the lungs was shown to be significantly higher than levels in recipients that were non-sensitized and challenged only (153). The increased levels of LTB4 played a pivotal role in enhancing the recruitment of transferred BLT1+/+ Teff into the lung. In a different lung disease model, BLT1 was shown to contribute to the development of lung rejection and obliterative bronchiolitis by mediating effector CD8+ T cell trafficking into the lung (154). Different members of the chemokine family are known to be subset-selective chemoattractants for T cells. CCL2 and CCL5 may be important in the recruitment of CD8+ T cells (155). CD8+ Teff were reported to migrate in response to CCL5 as well as LTB4 in vitro (156). It appears that the LTB4-dependent pathway and signals contribute at least one essential link to a chain of molecular events that lead to efficient recruitment of CD8+ Teff to the allergic airways (Fig. 1).
3.6 The IgE-FcεRI-Mast Cell-CD8-BLT1-IL-13 Connection
Many of the studies carried out in mice utilized systemic sensitization and challenge. This experimental model was shown to be (relatively) IgE- and mast cell-independent as mast cell-deficient mice and B cell-deficient mice developed comparable levels of airway allergic responses as control littermates (157, 158). To compare different approaches, responses in a well-characterized mast cell-dependent system were examined. In this model, CD8+ T cells were shown to be required for development of altered airway responsiveness (159). Briefly, following 10-day allergen exposure via the airways (in the absence of systemic sensitization with alum), FcεRI-deficient mice failed to develop altered airway function, less airway inflammation, and lower IL-13 levels (160). Transfer of WT bone marrow-derived mast cells (BMMC) into FcεRI-deficient recipients fully restored these responses. Transferred GFP-labeled mast cells were detected in the tracheal preparations, confirming that IgE-FcεRI-mast cells were involved in the responses after 10 days of airway allergen exposure (160).
IL-13 was shown to be critical as these responses did not develop in Il-13-deficient mice and were attenuated in WT mice treated with an IL-13 receptor antagonist. Transfer of WT BMMC into Il-13-deficient mice similarly failed to restore lung allergic responses, suggesting that IL-13 was derived from another cell type other than mast cells (160). One explanation for the failure to develop altered airway function and airway inflammation in these experiments could have been the absence of an allergen-specific IgE response. To address this possibility, different strains of mice were passively sensitized with allergen-specific (OVA) IgE (161) which was administered prior to airway challenge. Mast cell-, FcεRI-, IL-13-, BLT11, and CD8-deficient mice all failed to develop alterations in airway responsiveness following passive sensitization with OVA-specific IgE and limited airway allergen challenge (159, 160). Recipient CD8-deficient mice, reconstituted with CD8+ Teff, restored all of the responses. Moreover, only Teff expressing BLT1 but not BLT1−/− Teff were capable of doing so. The numbers of BLT1+ Teff cells were significantly higher in the lungs of recipients than recipients of BLT1−/− Teff. In addition, the induction of increased airway responsiveness and their accumulation in the lung following transfer of BLT1+CD8+ Teff could be blocked by administration of an LTB4 receptor antagonist. In parallel, BAL levels of LTB4 were monitored. LTB4 levels in mast cell- and FcεRI-deficient mice were significantly lower compared to control littermates following passive sensitization and challenge, suggesting that mast cells were a primary source of LTB4. Thus, in both mast cell-independent (systemic sensitization) and mast cell-dependent systems, support for an LTB4-BLT1-CD8+ Teff-IL-13 module was demonstrated.
These results were further confirmed using leukotriene A4 hydrolase (LTA4H)-deficient mice which failed to synthesize LTB4 (162). The role of this lipid mediator was investigated in allergen-induced airway alterations using two approaches. First, LTA4H-deficient and WT mice were systemically sensitized and challenged to allergen via the airways (mast cell-independent). Second, the animals were initially passively sensitized with OVA-specific IgE, followed by challenge with OVA via the airways (mast cell- dependent). In both approaches, LTA4H-deficient mice developed little AHR and eosinophilia, and IL-13 levels in the BAL fluid were significantly lower compared to WT mice. Transfer of LTA4H+/+ but not LTA4H−/− BMMC into LTA4H-deficient mice reconstituted AHR and lung allergic responses in passively sensitized recipients.
Although the importance of LTB4 in allergic airway disease and T cell activation was suggested over two decades ago (145), the role of LTB4 in the recruitment of antigen-specific effector CD8+ T cells in both mast cell-dependent and -independent allergic airway responses underscores how control of this LTB4-BLT1 pathway may provide novel therapeutic opportunities for the treatment of asthma.
3.7 Transcriptional Reprogramming of CD8+ T Cells from a Tc1 to a Tc2 Phenotype
T helper cell lineage commitment and differentiation fate have often been viewed in a unidirectional non-reversible manner, with endpoints exhibiting distinct effector functions. It now appears that the different T-cell subsets exhibit plasticity and can undergo transcriptional reprogramming, redirecting their functional roles. In large part, these differentiation decisions are dictated by specific cytokines in the microenvironment. Most studies have focused on CD4+ T cells, with examples of reciprocal regulation of polarization by IL-4 and IL-12/IFNγ (163, 164). In contrast, little is known about the mechanisms underlying the functional plasticity of CD8+ T cells (100, 165) or the pathways involved. Epigenetic mechanisms regulate CD4+ T cell lineage differentiation from Th0 to Th1, Th2, regulatory T cells, and Th17 subsets (166–168). Th2 polarization is regulated by association of the permissive histone modification trimethylation of lysine 4 of histone 3 (H3K4me3) with the IL-4 promoter and repressive trimethylation of lysine 27 of histone 3 (H3K27me3) with the IFNγ promoter (167). Alterations in H3K4me3, H3K27me3, and histone acetylation regulate CD8+ T cell memory development (168–171), and permissive histone modifications poise cytokine gene promoters for rapid transcription after stimulation. Little is known about the interrelationships of histone regulation, asthma pathophysiology, and CD8+ T cell plasticity as opposed to fixed lineage commitment.
We proposed that antigen-specific CD8+ T cells committed to IFNγ production, when exposed to IL-4 in vitro or an atopic environment, transit through distinct differentiation stages characterized by changes in transcription and translation resulting in IL-13-producing CD8+ T cells (100). In a different system, Hayashi et al. (172) showed that Th1 cells, committed to IFNγ production, can also convert to Th2 cytokine production such as IL-13 under specific conditions. As reviewed by O’Shea and Paul (164), it is still unclear what is meant by the terms lineage stability and plasticity/conversion, and how they can be distinguished.
Based on our findings of the contributions of CD4+IL-4+ T cells in the initial activation of CD8+ T cells (109), we examined the role of IL-4 during the functional differentiation of CD8+ T cells in a carefully regulated and staged in vitro system (100). This system was used to identify mechanisms controlling the plasticity of CD8+ T cells, focusing initially on the functional development of CD8+ T cells into IFNγ or IL-13 producers in vitro. To examine the mechanisms involved in this “stepwise” conversion, we monitored expression of the lineage-specific master regulatory transcription factors that control T cell differentiation. Before TCR re-stimulation, but after IL-4 treatment, Gata3 expression levels significantly increased in CD8+ T cells, whereas T-bet levels (and repressor of GATA3) decreased. Differences in Runx3 and Eomes expression were also observed. Interestingly, changes in lineage-specific transcription factors required only IL-4 and were not dependent on TCR re-stimulation. In contrast, IL-4-mediated induction of IL-13 mRNA and protein expression required TCR re-stimulation. Thus, IL-4 alone was sufficient to alter lineage-specific transcription factor levels during CD8+ T cell differentiation, poising these cells to produce cytokines, but only after TCR engagement. In addition, we observed IL-4-induced epigenetic programming of CD8+ T cells that poised the Il-13 locus for transcription after TCR re-stimulation in vitro or after adoptive transfer and allergen challenge in vivo. H3K4me3 binding to the Gata3 and Il-13 promoters was dependent on IL-4 treatment and occurred even in the absence of TCR re-stimulation, whereas H3K4me3 histone modifications at the T-bet and Ifnγ promoters were not regulated by IL-4. The in vitro and in vivo data demonstrated that IL-4 alone, in the absence of TCR re-stimulation, resulted in epigenetic poising of the Il-13 locus through the gain of permissive and loss of repressive histone modifications, which were co-regulated with recruitment of RNA polymerase II. IL-4 was also required for Gata3 expression in CD8+ T cells and IL-4-dependent recruitment of GATA3 protein to the Il-13 promoter. IL-4-mediated Gata3 promoter recruitment coincided with histone modifications and RNA polymerase II recruitment, but was temporally separated from the enhancement of IL-13 transcription and protein production. TCR ligation was required for IL-13 protein production, but interestingly, GATA3 was not associated with the Il-13 promoter during the TCR re-stimulation stage during in vitro differentiation. Similar to CD4+ T cells, the binding of GATA3 at the Il-13 locus in CD8+ T cells corresponded with changes in histone methylation. Binding of GATA3 was associated with significantly enhanced permissive H3K4me3 and significantly decreased repressive H3K27me3 histone marks to the Il-13 locus in CD8+ T cells treated with IL-4. Enhanced IL-13 transcription and protein production in CD8+ T cells were temporally associated with sustained permissive H3K4me3. The data support a model in which IL-4-mediated GATA3 recruitment regulates histone modifications at the promoter, which then poise the Il-13 locus for transcription after a TCR stimulus. In the absence of IL-4, GATA3 was not recruited to the Il-13 promoter, and consequently, TCR re-stimulation did not result in cytokine production.
As a result, an allergic inflammatory lung microenvironment containing IL-4 supports asthma pathogenesis through epigenetically poising CD8+ T cells for Tc2 conversion via differential histone modifications at lineage-specific promoter regions (100). These observations indicate that the Tc2 pathway requires an extrinsic stimulus, IL-4, whereas the Tc1 pathway may represent a default pathway, because addition of IL-4 did not greatly affect the histone modifications at the T-bet or Ifnγ locus. Further understanding of the molecular mechanisms by which IL-4 and GATA3 regulate the plasticity and/or stable conversion of CD8+ T cells may reveal novel therapeutic strategies and new targets for the treatment of asthma.
3.8 Steroid Insensitivity of CD8+ T Cells
Corticosteroids effectively suppress inflammatory responses through repression of many immune genes by means of interaction with the glucocorticoid receptor. To some extent, this explains the benefits in many patients with asthma, but not in those asthmatic patients who suffer a decrease in lung function despite high doses of inhaled or oral corticosteroid treatment (173–175). Paradoxic effects of corticosteroids on neutrophils, such as increased LTB4-induced intracellular Ca2+ mobilization, chemotaxis, and enhanced survival have been reported (176, 177). These effects were mediated through upregulation of BLT1 expression, a major neutrophil chemoattractant receptor (176, 177). Susceptibility to corticosteroids differs among T-cell subpopulations and states of maturity (178). Because administration of corticosteroids to asthmatic patients results in significant decreases in numbers of CD4+ but not CD8+ T cells in peripheral blood (179), activated CD8+ T cells are likely more resistant to corticosteroids than CD4+ T cells. Therefore, CD8+ Teff are proposed to play a more important role in the pathophysiology of inflammatory diseases than CD4+ T cells after initiation of corticosteroid treatment.
Studies in mice showed that activated CD8+ T cells were more resistant to DEX than CD4+ T cells in the presence of IL-2; DEX treatment upregulated BLT1 expression on Teff in a dose-dependent manner likely through increased IL-2R expression (99). The consequences of increased BLT1 expression were enhanced by LTB4-induced intracellular Ca2+ influx, phosphorylation of ERK1/2, in vitro chemotaxis, CD8+ Teff-mediated AHR and allergic airway inflammation. The effects of corticosteroids on the upregulation of BLT1 on CD8+ Teff and their ability to potentiate AHR and allergic airway inflammation is proposed to represent an example of a “steroid-insensitive” pathway. Enhancement or amplification of the LTB4/BLT1 pathway by corticosteroids represents a potentially important negative factor in asthmatic patients receiving high doses of inhaled or oral corticosteroids.
Glucocorticoid-insensitivity of lymphocytes has also been described in a number of human diseases (180–184). DEX treatment of human CD4+ and CD8+ T cells revealed significant differences: in normal CD4+ T cells but not CD8+ T cells, DEX induced histone H4K5 acetylation (185). DEX responses were impaired in CD8+ T cells compared to CD4+ T cells when IL-10 and MAPK phosphatase induction were monitored. Further, the findings suggested that activating transcription factor-2, via acetylation of the histone H4K5 residue, controls steroid-mediated gene transactivation and accounts for the differences in corticosteroid responsiveness between CD4+ and CD8+ T cells (185). When peripheral blood CD4+ and CD8+ T cells were examined from SS and SR asthmatics, the expansion of anti-CD3/CD28 activated cells was markedly different in the presence of corticosteroid (52). In both SS- and SR- activated CD4+ T cells, expansion was significantly reduced in the presence of corticosteroid. In contrast, SR asthmatic CD8+ T cell expansion was maintained after corticosteroid treatment. The data demonstrated that human CD8+ T cells, similar to mouse CD8+ T cells, are relatively steroid-resistant compared to CD4+ T cells.
3.9 Role of CD8+ T Cells in Human Asthma
Whereas the studies in experimental asthma enable manipulation and direct evaluation of the role of CD8+ T cells in the development of airway inflammation and altered airway function, studies in asthmatics are necessarily associative. CD8+ T cells may be triggered by allergens as a result of cross-presentation of exogenous allergen by MHC I on airway dendritic cells to CD8+ T cells. Increasing evidence further supports the contribution of CD8+ T cells in asthma pathogenesis. CD8+ T cells have been demonstrated in the BAL fluid and sputum of patients with asthma (186, 187) and in lung tissue of subjects with fatal asthma (188). Further, an association between CD8+ T cells in sputum and asthma severity, as measured by bronchial hyperresponsiveness to histamine, has been demonstrated (133). In addition to production of type 2 cytokines by CD4+ T cells, CD8+ T cells were also found to produce IL-4 and IL-5 in the airways of asthmatics (94, 133). In patients with severe asthma, transcriptome analyses showed large changes in circulating CD8+ but not CD4+ T cells (189). In non-smokers with fatal asthma, CD8+ but not CD4+ T cells were significantly increased in the lung (190). A unique population of IL-6Rα high effector memory CD8+ T cells was found in peripheral blood of asthmatics with an increased frequency compared to healthy control subjects (191). These cells proliferated, survived, and produced high levels of the type 2 cytokines IL-5 and IL-13 and expressed increased levels of GATA3 and decreased levels of T-BET and BLIMP-1 in comparison with other effector memory CD8+ T cells. GATA3 was shown to be required for IL-6Rα expression. Further, IL-6Rα high effector memory CD8+ T cells exclusively produced IL-5 and IL-13 in response to asthma-associated respiratory syncytial virus and bacterial superantigens (191).
In a 7.5-year follow-up study including 32 patients with asthma, the annual fall in post-bronchodilator forced expiratory volume in 1 second (FEV1) was not related to the thickness of the reticular basement membrane layer or to eosinophil counts in bronchial biopsies, but was correlated with an increase in CD8+ T cell numbers (192). These data prompted the conclusion that the annual decline in lung function could be predicted by the bronchial CD8+ T cell infiltrate. This also suggested that the inflammatory phenotype in asthmatics has prognostic significance, and because of CD8+ T cell insensitivity to corticosteroid, requires novel therapeutic strategies. This same group also carried out a 14-year follow-up study of asthmatics to determine which inflammatory markers in the bronchial mucosa were associated with lung function decline (193). Lung function decline was associated with both CD4+ and CD8+ bronchial T cells at baseline, but at follow-up, lung function decline was marked by high CD8+ and CD3+ T cell numbers and granzyme counts. In asthmatics, numbers of CD8+ human bronchial intraepithelial lymphocytes were significantly higher than CD4+ T cells, further supporting the notion that CD8+ T cells in the mucosa contribute to asthma pathogenesis (194). Given the association of severe asthma with airway neutrophilia, a greater than 3-fold increase in the expression of MAPK3, shown to be critical for airway neutrophilia but not eosinophilia, was demonstrated in CD8+ T cells from asthmatics compared to healthy controls; no increases were seen in CD4+ T cells (195).
In a study comparing BAL fluid from mild-moderate asthmatics to healthy controls, an association of CD8+BLT1+IL-13+ T cells with asthma was shown (196): a subset of CD8+ T cells expressed the high-affinity receptor for LTB4 (BLT1) and produced IL-13. Numbers of CD8+ T cells in the airways of patients with asthma were significantly higher compared to healthy subjects. The frequency of these CD8+ T cells was inversely related to airflow obstruction defined by measurements of FEV1 and FEF[25–75] percent predicted values. The results, albeit indirect, supported a pathogenic role for CD8+BLT1+IL-13+ T cells in airway obstruction. These findings are in agreement with the results described in experimental asthma in mice. As in sensitized and challenged mice (103), the data identified a subset of CD8+ T cells in asthmatic airways expressing BLT1 and producing IL-13 similar to CD8+ Teff in the mouse model. In asthmatics, numbers of CD8+BLT1+IL-13+ T cells correlated with lower lung function and reticular basement membrane thickening.
The phenotype and consequences of activation of human CD4+ and CD8+ T cells were further investigated in two subsets of asthmatics defined as SS or SR (52). The percentages of T cells that expressed BLT1 were higher in patients with asthma than in those without asthma; in patients with SR asthma, the largest percentages of BLT1-expressing cells were detected in activated CD8+ T cells. This increase in BLT1-expressing CD8+ T cells from SR asthmatics was maintained in the presence of corticosteroid. The increases in BLT1 expression were functional as determined by the increased intracellular Ca2+ flux following ligation of the receptor by LTB4.
As discussed, CD8+ T cell expansion, especially in SR asthma, was relatively resistant to corticosteroids compared to CD4+ T cells. The pathway leading to the generation of lipid mediators, including LTB4, is activated in many inflammatory diseases but is resistant to corticosteroid treatment (197). The generation of LTB4 appears to be an important contributor to SR asthma as confirmed in this study. In patients with SS and SR asthma, significant differences between CD4+ and CD8+ T cells were demonstrated (52). Activation with anti-CD3/CD28 resulted in the expansion of BLT1-expressing CD8+ T cells in SS and SR asthmatics, in contrast to CD4+ T cells. The numbers of BLT1+CD8+ T cells were higher in patients with asthma compared to controls. Within the asthmatic group, increases in CD8+ T cell numbers were greater in patients with SR asthma compared to SS asthmatics. Expansion of activated CD4+ T cells with anti-CD3/CD28 and IL-2 was significantly decreased in the presence of DEX, whereas the effects of DEX on expansion of activated CD8+ T cells was lower; the smallest effects were seen in CD8+ T cells from patients with SR compared to SS asthma. When cytokine levels in cultures of CD4+ and CD8+ T cells activated with anti-CD3/CD28 were assessed, levels of IL-13 were higher in CD8+ T cells than in CD4+ T cells. In contrast, levels of the anti-inflammatory cytokine IL-10 were elevated in CD4+ T cells from controls and patients with SS asthma compared with CD4+ T cells from patients with SR asthma. This inverse relation between IL-13 and IL-10 (increased IL-13 and decreased IL-10 levels in the BAL fluid) has previously correlated with disease severity (53, 54). Hence, corticosteroids may exert some of their beneficial effects through the induction of IL-10 production (53, 198). Levels of IFNγ, which may down-regulate Th2 cytokine production and asthmatic responses, were also shown to be lowest in CD4+ and CD8+ T cells from patients with SR asthma (52).
Thus, a pathway, similar to that in the mouse, is activated in asthmatics, leads to the generation of lipid mediators, is insensitive to corticosteroids, and results in the activation and accumulation of steroid-insensitive, IL-13-producing CD8+ T cells in the lung. This positions the LTB4-BLT1-CD8+ T cell axis as a major contributor to severe asthma in humans.
4.0 CD4+ Tregs Modulate Lung Allergic Responses
In humans and mice, CD4+ T cells represent a large subpopulation of lymphocytes that play key roles in developing and controlling immune responses to self and non-self antigens. Any imbalances may result in disease (199). A small subset of CD4+ T cells has been identified as Tregs based on their capacity to suppress in vitro proliferation of target T cells and regulate host immune responses through cell-to-cell contact and production of immunomodulatory cytokines, including IL-10 and TFG-β (200–202). These regulatory T cells express a lineage-specific transcription master regulator, forkhead box P3 (FOXP3), a member of the forkhead/winged-helix protein family, and a distinctive phenotype including constitutive, high expression of the alpha chain of the interleukin 2 receptor (CD25), cytotoxic T lymphocyte antigen 4 (CTLA-4), glucocorticoid-induced tumor necrosis factor receptor (GITR) and other surface molecules. Among the CD4+ Tregs, two important subsets have been defined, naturally occurring Foxp3+CD4+CD25+ Tregs (nTregs), which are thymus-derived, and secondary lymphoid-derived inducible Tregs (iTregs) which develop from peripheral naive T cells, often in the context of antigen-specific inflammation, referred to as adaptive Tregs. Despite their differences in origin and development, there are no current consistent or reliable markers to distinguish nTregs from iTregs (203, 204). A number of non-CD4+ Tregs have also been described (205), but CD4+ Tregs have dominated research and interest, in part because defects in their function and/or numbers resulted in identifiable immunological imbalances and autoimmunity in both humans and mice. Males with mutations in FOXP3, encoded on the X chromosome (IPEX syndrome), suffer from immune dysregulation, polyendocrinopathy, and enteropathy (206). This is a rare disorder characterized by multiorgan autoimmunity in early life often resulting in death without treatment. In Scurfy mice, an X-linked frameshift mutation results in scurfin protein lacking the forkhead domain is responsible for multiorgan autoimmunity in hemizygous males with similar manifestations to the human counterparts with IPEX (207).
Significant progress has been made in our understanding of the development and function of Tregs as it relates to their indispensable role in the maintenance of immunological tolerance and immune homeostasis. In studies of multiple strains of mice in which Foxp3 in CD4+ T cells has been attenuated or overexpressed, expression of this gene has been shown to play a pivotal role in determining the outcomes of disease (208, 209). We have focused on the role of nTregs in the development of experimental models of asthma in mice with various allergens including OVA, whole extracts of ragweed (RW), HDM, as well as various OVA peptides including SINFEKL and OVA323-339 (132, 210–213).
4.1 Regulation of Lung Allergic Responses by nTregs
To assess the importance of nTregs in the development of lung allergic responses, the effects of antibody depletion with anti-CD25 and anti-CD4 antibodies in sensitized and challenged Balb/c mice were investigated. We compared the effects of treating sensitized mice with anti-CD25 (a combination of 7D4 and PC61 antibodies which was shown to deplete Foxp3+ cells) or anti-CD4 (GK1.5) after sensitization but prior to challenge. Depletion of CD4+ cells, presumably removing both CD4+ nTregs and CD4+ T effector cells, prevented the development of AHR and airway eosinophilia, associated with lower levels of the type 2 cytokines, IL-4, IL-5, and IL-13 and increased levels of IL-10 and IFNγ in the BAL fluid. In contrast, treatment of sensitized and challenged mice with anti-CD25, which depleted nTregs, resulted in increased AHR, increased numbers of eosinophils and lymphocytes, higher levels of IL-4, IL-5, and IL-13 and lower levels of IL-10 and IFNγ in the BAL fluid. Depletion of CD25+ cells prior to allergen challenge of sensitized mice showed a more extensive alteration of lung histopathology, including increases in the influx of inflammatory cells, mucus hyperproduction, and goblet cell metaplasia. This was in contrast to depletion of CD4+ T cells following sensitization and challenge, where mice showed little lung pathology. Similar results were obtained in C57BL/6 mice indicating that the effects of CD4 and CD25 antibody treatment were not strain-dependent (214, 215). Taken together, the results of depletion experiments identified the regulatory control that nTregs exhibited on the development of the full spectrum of lung allergic responses. The differential effects of administering anti-CD4 and anti-CD25 on the development of experimental asthma highlighted how the balance between lung regulatory and effector CD4+ T cells may impact clinical outcomes.
The suppressive activities of nTregs on the development of lung allergic responses were further investigated by transferring these cells into sensitized and challenged recipient mice (216). Of particular interests were nTregs residing in the lungs of naive donor mice, albeit in low numbers. Isolated nTregs from the lungs of naive Balb/c and C57BL/6 (WT) mice were characterized by flow cytometry for expression of surface markers and intracellular molecules. High constitutive expression of CD25, CTLA-4, and GITR on the cell surface and intracellular IL-10, TGF-β, and Foxp3 were detected in nTregs compared to naive CD4+CD25− T cells. Further, TCR-induced proliferation of activated T cells was significantly reduced in the presence of nTregs. Several mechanisms of suppression of proliferation have been suggested including the release of the immunoregulatory cytokines IL-10 and TGF-β and cell-to-cell contact via surface molecules such as CTLA-4 (216–218). The consequences of intratracheal transfer of isolated, nTregs and naive Foxp3−CD4+CD25− conventional T (Tconv) cells from lungs of naive donors on the development of lung allergic responses prior to allergen challenge of sensitized recipients were also determined (216). Sensitized and challenged C57BL/6 mice given Tconv cells developed significant increases in lung resistance (RL) compared to mice challenged alone. In contrast, AHR in recipients of nTregs was significantly decreased. Increased AHR in mice which received Tconv cells was associated with increased numbers of both eosinophils and lymphocytes, and higher levels of IL-4, IL-5, and IL-13 and lower levels of IL-10 and IFNγ in BAL fluid. In contrast, recipients of nTregs had reduced airway inflammation, lower levels of IL-4, IL-5, and IL-13 and higher levels of IL-10 and IFNγ in BAL fluid. Significantly, suppression of lung allergic responses was dependent on both IL-10 and TGF-β (216, 217); nTregs from IL-10-deficient mice failed to suppress the development of lung allergic responses in sensitized and challenged recipient mice (216). We and others demonstrated that nTregs regulated immune responses in an antigen-independent manner (211, 219–221); however, antigen-specific regulation by nTregs has also been reported (222). Given the development of nTregs in the context of the thymus microenvironment and self-antigen (223, 224) and limited TCR repertoire (225–227), it is unclear whether nTregs can become antigen-specific as there are no reliable marker(s) distinguishing nTregs and iTregs.
4.2 nTregs Attenuate Mast Cell-Dependent Lung Allergic Responses
Given that changes in function and structural thickening of airway smooth muscle have been suggested as contributing factors to the severity of asthma (228–230), the effects of nTregs on contractility of tracheal smooth muscle (TSM) induced by electrical field stimulation (EFS) were examined. EFS is an in vitro technique developed for monitoring TSM tone and is critically dependent on antigen-specific IgE, B cells, expression of the high affinity IgE receptor FcεRI, mast cells, and mediators released from cellular granules (159, 231, 232). Following sensitization through aerosolized allergen challenge in the absence of systemic sensitization, a significantly lower electrical current was required to elicit 50% of maximum contraction (EFS50) when compared to EFS50 of TSM from control mice (231). Intratracheal transfer of nTregs into mice on day 8 of aerosolized allergen challenges but not on the first day of aerosolized allergen sensitization prevented increases in TSM contractility. To identify the mechanism involved in nTreg regulation of TSM contractility, we determined the effects of nTregs on mast cell degranulation. Following crosslinking surface-bound IgE with antigen, mast cells, derived in vitro from bone marrow, were co-cultured with nTregs and degranulation was monitored by β-hexosaminidase release. Degranulation of mast cells was decreased in cells co-cultured with nTregs but not with naive CD4+CD25− T cells. Similar effects of Tregs on mast cells have been shown in other experimental models of anaphylaxis (233, 234) and contact hypersensitivity (235).
4.3 Activation of nTregs
Functional activation of nTregs is usually through TCR ligation (200–203). Results of in vitro suppression assays have demonstrated TCR-activated nTregs were more effective in preventing proliferation of target cells (236–238) and observations in TCR transgenic mice that co-express an agonist ligand demonstrated increased numbers of nTregs (239). Expression of agonist ligand in peripheral, secondary lymphoid tissues resulted in the activation of nTregs (240). Somewhat in contrast, we did not observe requirements for TCR activation of nTregs to suppress proliferation of target cells (241). Moreover, suppression was seen in an antigen-independent fashion (211). A number of environmental factors and compounds including farm environments (242, 243), retinoic acid (244), and rapamycin (245) have been reported to maintain and/or increase numbers of nTregs and their regulatory function. Some of these effects may be mediated through epigenetic modifications including methylation and histone modifications of the FOXP3 locus as histone deacetylase inhibitors have been shown to increase numbers and suppressive activities of Tregs in vitro and in vivo (246–249).
Although interactions between CD4+ and CD8+ T cells are often necessary for the generation of optimal immune responses (199, 250), few, if any, studies have examined such interactions in the functional activation of Tregs. We demonstrated a critical requirement for engagement of MHC I on nTregs by CD8 for the functional activation of nTregs (251) (Figure 2). As described elsewhere in this chapter, a critical role for CD8+ T cells in the development of lung allergic responses was initially shown by the failure of sensitized and challenged CD8-deficient mice to develop the full spectrum of lung allergic responses. In contrast to the suppression of lung allergic responses seen following transfer of WT nTregs into sensitized and challenged WT recipients, nTregs from WT mice were shown to enhance lung allergic responses, including AHR, airway inflammation, and increases in BAL fluid levels of IL-5 and IL-13 in sensitized and challenged CD8-deficient mice. To address the differences in the responses of WT and CD8-deficient recipients, CD8+ T cells were isolated by negative and positive selection from spleens of naive and sensitized WT donors, and injected into sensitized CD8-deficient recipients prior to intratracheal instillation of nTregs and allergen challenge (251). Reconstitution with primed (previously sensitized) but not naive CD8+ T cells restored the full spectrum of lung allergic responses, comparable to those seen in sensitized and challenged WT mice. Both negatively- and positively-selected CD8+ T cells from sensitized donors were equally effective in restoring AHR, inflammation, Th2 cytokines, and lung histopathology. Surprisingly, the suppressive effects of nTregs on CD8+ T cell-mediated airway responses was only seen in CD8-deficient recipients reconstituted with negatively-selected, but not positively-selected CD8+ T cells. The interpretation was that the bound antibodies on CD8 following positive selection prevented the engagement with MHC I on nTregs. The importance of CD8-MHC I engagement on in vivo regulatory function was also demonstrated by investigating activities of nTregs from β2-microglobulin-deficient (β2m−/−) mice or following blocking of MHC I on WT nTregs with anti-MHC I antibody. Both anti-MHC I-treated nTregs from WT donors and nTregs from β2m-deficient mice, which do not express MHC I, failed to suppress the development of lung allergic responses in sensitized and challenged WT recipients. In parallel, in vitro co-culture of WT nTregs with negatively-selected naive CD8+ T cells isolated from naive WT mice produced significantly higher amounts of IL-10 and TGF-β than co-cultures of these cells in the presence of either anti-CD8, anti-MHC I, or nTregs alone. Taken together, both in vitro and in vivo data demonstrate a previously unreported mechanism for activating nTreg activity, one which involves the interaction of MHC I on lung nTregs and CD8 on T cells (or possibly other CD8-expressing cells in the lung, e.g., DC). Activation of these lung nTregs effectively reduced AHR, eosinophilic lung inflammation, Th2 cytokine production, and lung histopathology (mucus hypersecretion/production and goblet cell metaplasia), likely through the production and secretion of IL-10 and TGF-β (216).
Figure 2. Pathways signaling the suppressive or enhancing activities of nTregs.
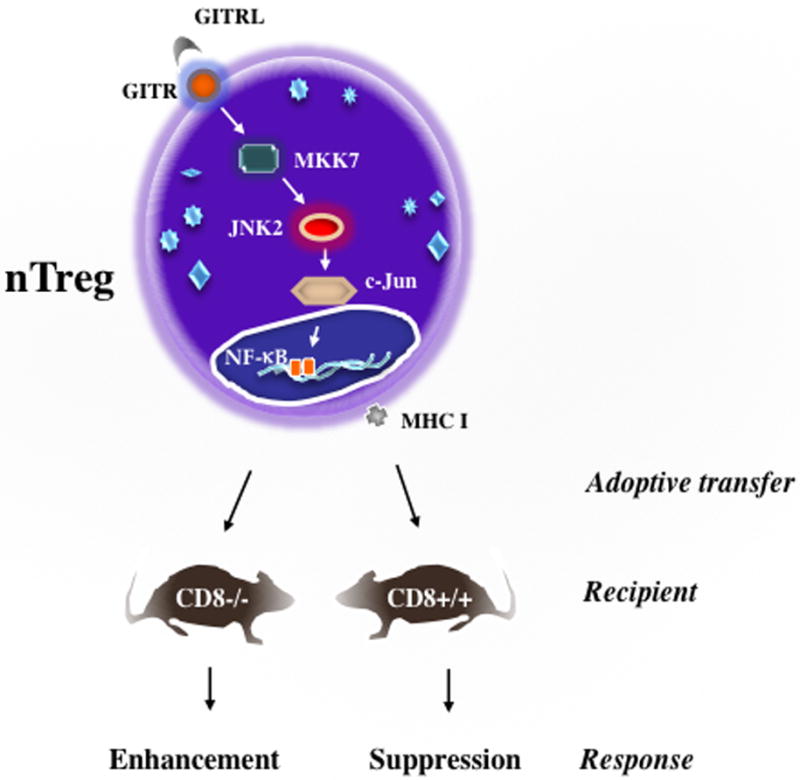
nTregs obtained from WT (CD8+) donors when transferred into sensitized and challenged WT recipients suppress the development of lung allergic responses. Interactions between CD8 and MHC I are essential. In the absence of CD8 (CD8-deficient mice), nTregs from these mice when transferred into sensitized and challenged WT recipients, enhance development of lung allergic responses. Enhancement is triggered by transcriptional reprogramming of the nTregs, mediated through ligation of GITR and activation of the GITRL-GITR-JNK pathway. In CD8+/+ mice signaling through CD8-MHC I dominates and is associated with the maintenance of the regulatory phenotype and suppressive activities of nTregs even in face of GITR ligation.
Interactions between CD8 and nTregs via MHC I expression appeared to play an additional important role in the retention of regulatory function (241). In the absence of this cell-to-cell interaction, nTregs from CD8-deficient mice were functionally altered, having a mixed phenotype of suppressor and effector cells. These cells had similar levels of expression of CD25, CD39, IL-6Rα but higher GITR and lower CTLA-4, IL-10, TGF-β, and Foxp3 levels when compared to WT nTregs. The levels of IL-6 were significantly increased. As a consequence, Foxp3+CD4+CD25+ T cells from lungs of CD8-deficient mice, in contrast to WT mice, failed to suppress the development of lung allergic responses in sensitized and challenged WT recipients. The absence of suppression was associated not only with high levels of IL-5 and IL-13, but also significant levels of IL-6 in the BAL fluid of the recipients. IL-6 is a proinflammatory cytokine that plays a prominent role in host defense and response to injury (252). The combination of IL-6 and TFG-β is central to the differentiation of Th17 cells (253). Similar to nTregs from CD8-deficient mice, IL-6-treated nTregs from WT donors failed to decrease lung responses in recipients which was consistent with other studies (254–256). In contrast, interference with IL-6 activity by administering anti-IL-6 or genetic depletion of IL-6 restored suppression of lung responses in recipients given nTregs from CD8-deficient or IL-6-deficient mice (241). Collectively, these data demonstrated the inhibitory effects of endogenous or exogenous IL-6 on nTreg function leading to reduced Foxp3 expression and suppressive activities.
4.4 Functional Reprogramming of nTregs
The variable effector functions of nTregs, dependent on environmental cues, suggested an ability to undergo transcriptional reprogramming, the underpinning of plasticity. Plasticity of nTregs was first described in the mid 2000s, including conversion of Tregs into IL-4+ (257), IL-13+ (214), IL-17+ (258), or IL-17+/IFNγ+ T cells (259). Prior to these reports, nTregs were thought to be terminally-differentiated, consistent with the concept of unidirectional lineage commitment. In the absence of CD8, the functional development of nTregs was altered in that they were unable to prevent the development of lung responses in sensitized and challenged WT recipients (241). This failure was associated with decreased expression of Foxp3, IL-10, TGF-β, CTLA-4, and increased production of endogenous IL-6. However, the loss of suppression was not terminally fixed as restoration of regulatory function in CD8-deficient nTregs could be achieved. CD8-deficient newborn mice received negatively-isolated CD8+ T cells from naive WT mice by intraperitoneal injection over several weeks. When mice reached 6–8 weeks of age, analyses of nTregs from these mice demonstrated recovery of regulatory phenotype and function. Levels of IL-6, IL-10, TGF-β, and Foxp3 were now comparable to those seen in WT nTregs. When transferred into sensitized and challenged WT recipients, these nTregs prevented the development of AHR, airway inflammation, and increased type 2 cytokines. The restoration of suppression in CD8-deficient nTregs following CD8+ T-cell reconstitution suggested that reprogramming remained possible, potentially after intrathymic development. The functional program exhibited by nTregs was not only dependent on CD8-MHC I interactions (214, 241, 251) but on a number of pathways including overexpression of IL-4Rα variant (260), cytokines (254, 261), OX40 stimulation (262), or GITR agonist antibody (214, 263).
In the presence of intact interactions between CD8 and MHC I, the suppressive activities of nTregs were maintained despite the presence of elevated levels of IL-6 in the BAL fluid of sensitized and challenged WT recipients (241, 261). However, preventing this interaction resulted in functional instability and loss of suppressive activity could easily be triggered by IL-6 or GITR ligation. Exposure to endogenous IL-6 disrupted the regulatory function of CD8-deficient nTregs by decreasing Foxp3 expression. In the absence of CD8-MHC I interactions, ligation of GITR on nTregs with host GITR ligand (GITRL) in the lungs was shown to be critical in the conversion of nTregs to pathogenic IL-13+CD4+ T effector cells. This GITR-GITRL-triggered conversion was mediated through a MAPK signaling pathway that involved c-Jun N-terminal kinase 2 (JNK2). Disruption of this signaling pathway, including administering anti-GITRL antibody, GITR- and JNK2-deficient nTregs, or incubation with the JNK inhibitor, SP600125, prevented the conversion of nTregs to IL-13+CD4+ T effector cells and enhancement of lung allergic responses (214, 264, 265). This signaling cascade was shown ultimately to involve the activation of NF-κB in the nucleus, resulting in effector cell conversion (Figure 2).
4.5 Role of iTregs Cell Activities in Experimental Asthma
In contrast to nTregs, iTregs appear to be antigen-induced and derived differently, but share many features. iTregs were generated in vitro by culturing activated (anti-CD3/CD28) Foxp3−CD4+CD25− T cells from spleens of naive mice with TGF-β (261). Virus transduction of Foxp3 in CD4+ T cells has also been reported to endow these cells with a regulatory phenotype and function (209). iTregs shared many of characteristic phenotypes of nTregs with high expression of CD25, CTLA-4, GITR, OX40, and intracellular IL-10 and Foxp3 (261). Similar to nTregs, iTregs produced significant amounts of IL-10, but unlike nTregs, moderate amounts of IFNγ also were detected in culture supernatants by ELISA. Adoptive transfer of iTregs into sensitized and challenged WT recipient mice decreased development of the spectrum of lung allergic responses (AHR, inflammation, Th2 cytokines). At this level, these changes were comparable to those of nTregs, consistent with previous reports (266, 267).
4.6 Functional Reprogramming of iTregs
Similar to nTregs, iTregs exhibited functional instability under different conditions, but differences in transcriptional reprogramming of iTregs and nTregs were demonstrated (261). Enhancement of lung allergic responses mediated by nTregs was shown to depend on increases in airway eosinophilia and IL-13, as recipients treated with anti-IL-13 antibody prior to cell transfer or given nTregs from Il-13-deficient mice, developed no additional increases, with similar lung responses as seen in sensitized and challenged mice given CD4+ Tconv cells. In contrast, transfer of iTregs resulted in increases in AHR, airway neutrophilia and IL-17 in the BAL fluid, and these changes were prevented by treatment with anti-IL-17 antibody or transfer of iTregs generated from cells isolated from Il-17-deficient mice. Divergence in the signal transduction pathways promoting the conversion of iTregs and nTregs into effector cells was also demonstrated in sensitized and challenged CD8-deficient recipients (261). Conversion of nTregs into IL-13+CD4+ T effector cells was shown to depend on signaling through GITR and any interference with the cascade of GITR-MKK7-JNK2-cJun prevented the enhancement in recipient mice (214, 264, 265). Interestingly, despite being expressed by iTregs, neither GITR nor OX40 signaling appeared to play a role in the conversion of iTregs into effector cells in vitro or in vivo (261). In contrast to nTregs, treatment of recipients with anti-IL-6, but not anti-GITRL, anti-OX40, or control antibody, prevented the enhancement and conversion of iTregs into IL-17+CD4+ T effector cells, as transferred cells recovered from anti-IL-6-treated mice had decreased levels of intracellular RORγt and IL-17 compared to cells recovered from control antibody-treated recipients. Collectively, the reprogramming pathways and enhancement appeared distinct and cytokine-specific in iTregs and nTregs.
4.7 Role of Tregs in Human Asthma
The importance of Tregs in man is underscored by their absence and consequences in the IPEX syndrome (206). However, except for a few reports (268–270), most studies on human CD4+CD25+ Tregs have been carried out on cells enriched from peripheral blood and found within the population of CD4+CD25high (an arbitrary delineation). Even in this subset heterogeneity was present because of the absence of specific markers to distinguish Foxp3+CD4+CD25high from the significant presence of activated Foxp3−CD4+CD25hi cells (271). In contrast to mouse, activated human CD4+ T cells also transiently express FOXP3, but have little or no suppressive activities in vitro (272–274), further contributing to the difficult and sometimes controversial descriptions of subsets of human Tregs. Expression of Treg-associated markers including CTLA-4, HLA-DR and GITR was also found on activated T cells, indistinguishable from bona fide Tregs. Several different classifications include CD127low Tregs, CD45RA+Foxp3low naïve Tregs (nTregs), CD45RA−Foxp3high effector Tregs (eTregs), CD45RA−Foxp3low cytokine secreting, non-suppressive cells, and memory CD45RO+ Tregs (mTregs), which display differences not only in phenotype but also in function and behavior (275). In contrast to CD45RA+ Tregs, CD45RO+ Tregs were reported to be more suppressive (276), but were prone to acquire effector function, secreting IL-17 in vitro (271). The eTregs were capable of greater expansion than nTregs, and fewer genes were detected by microarray gene expression analysis of nTregs when compared to eTregs (276). ICOS+eTregs preferentially expressed IL-10 (277) and suppressor function of BAL fluid-derived Tregs was dependent on TGF-β (268). Similar to mouse, regulatory functions and phenotypes of human Tregs were directly correlated to the expression and activity levels of Foxp3. The numbers of Foxp3+CD4+ T cells also appeared to correlate with the clinical outcomes of many disease states including asthma (268–270). Various therapeutic approaches with some success promoting expression of Foxp3 and/or numbers of Foxp3+CD4+ Tregs have been investigated, including allergen-specific immunotherapy (278), vitamin D3 (279), rapamycin (280, 281), and infusion of Tregs as cell-based advances (both unmodified, expanded and genetically-modified cells) are becoming safer and more feasible (275, 282).
Although human and mouse iTregs can be generated in vitro by TCR stimulation in the presence of IL-2 and TGF-β (283, 284), mouse iTregs are functionally similar to nTregs as both can prevent the development of lung allergic responses in recipient mice (261, 267); human iTregs do not consistently exhibit suppressive activities in vitro (284). Lack of suppression was also reported in activated TCR-induced expression of FOXP3 in human cells (272–274), suggesting other molecular mechanism(s) may be required for endowing regulatory function. Although both mouse iTregs and nTregs retained the capacity to undergo transcriptional reprogramming and conversion into pathogenic effector T cells, it is unclear whether human Tregs possess a similar plasticity in vivo and is an important consideration in the development of Treg-based therapy.
5.0 Gamma-Delta (γδ) T Cells in Experimental and Human Asthma
γδ T cells are a small subset which develop in the fetal thymus and primarily localize in mucosal tissues such as lung, skin, uterus, and intestine in mice (285). Studies on the role of γδ T cells in allergic inflammation, including asthma, are limited given their small numbers. Experiments using δ-deficient mice identified their potential role in the development of allergen-induced AHR and airway inflammation in an experimental model of asthma (286). Deficiency or deletion of the entire γδ T cell population resulted in enhanced AHR (287). This function of γδ T cells was independent of αβ T cells, eosinophilic airway inflammation and cytokine responses. The activity of γδ T cells in the development of AHR was dependent on activation of the TNFα/TNFα-receptor 2 (TNFR2) pathway, as demonstrated in TNF-α transgenic, Tnfα-deficient, Tnfr1-, or Tnfr2- deficient mice (287, 288). γδ T cells were also shown to play a key role in the suppressive effects of Mycobacterium leprae-derived heat shock protein on allergen-induced AHR and eosinophilic airway inflammation, as deletion of the entire γδ T cell population abolished the effects of this molecule (289).
Subsets of γδ T cells appeared to play distinct roles in allergic airway inflammation. The small subpopulation of pulmonary Vγ4+ T cells played a central role in the suppression of AHR without modulating airway eosinophilia and goblet cell metaplasia and the effects were dependent on expression of IFNγ and MHC class I (290, 291). Long-term or repeated allergen exposure has been shown to induce resolution of AHR and airway inflammation in allergen sensitized mice (292). In this setting, Vγ4+ T cells were shown to play a key role in the resolution of AHR, which was independent of induction of antigen-specific tolerance of αβ T cells (292). Functional differentiation of these suppressive Vγ4+ T cells required antigen stimulation, whereas they still suppressed AHR without antigen stimulation once suppressive activities of the cells were induced (293). Induction of suppressive Vγ4+ T cells required CD8+ DCs in the lymphoid tissues and the effects were triggered through direct cell-cell contact (294).
In contrast to suppressive Vγ4+ T cells, Vγ1+ T cells increased allergen-induced AHR, eosinophilic airway inflammation, and Th2 cytokines in the airways through enhancing sensitization to allergen (295). Induction of enhancing Vγ1+ T cells required CD8+ DCs in the lymphoid tissues and the effects were also triggered through direct cell-cell contact (294). Vγ1+ T cells were further shown to play a critical role in the development of AHR following acute ozone exposure through TNα-induced activation (296). Vγ1+ T cells synergized with another small subset of T cells in the lung, invariant natural killer T (iNKT) cells, and induced AHR without eosinophilic airway inflammation (297). Regardless of these enhancing effects of Vγ1+ T cells on lung responses, they did not produce IL-4 or IL-13 which was in contrast to iNKT cells. IFNγ, TNFα or IL-4 were required for the differentiation of Vγ1+ T cells (298). γδ T cell numbers were increased during rhinovirus-induced asthma exacerbations in asthmatics and animal models of asthma (299); the increases in γδ T cell numbers were negatively associated with asthma exacerbations. Recently, γδ T cells have been shown to produce IL-17 and were proposed to play a pathogenic role in asthma (300, 301). However, in the airways of asthmatics, an association of IL-17-producing γδ T cells with asthma was not observed (302).
5.1 Natural Killer T (NKT) Cells in Experimental and Human Asthma
iNKT cells, also present in small numbers, have been linked to the pathogenesis of asthma, in large part because they can rapidly release large amounts of cytokines and chemokines following activation by glycolipids, many of which are found in microorganisms (303). To date, controversy remains about the role and magnitude of the contributions of iNKT cells within the lungs of asthmatics (304–306). We suggested this variability could be due to a functional plasticity within the iNKT subset (307). We demonstrated that IFNγ played a critical role in dictating the consequences of iNKT cell activation to become either IFNγ or IL-13 producers in the initiation phase of the development of AHR and airway inflammation (307). In experimental asthma, iNKT cells were shown to enhance Th2 responses through adjuvant effects and obesity-induced functional upregulation of iNKT cells, which augmented Th2 cytokine responses (308, 309). Whether iNKT cells are activated in asthmatics and whether the pathogenic effects of activated iNKT cells that were observed in mice are reproducible in human asthma still remain to be determined (310).
6.0 Transcriptomic Profiling of Immunologically-Relevant Specimens Contributing to Asthma
To determine whether the clinical heterogeneity of asthma is also reflected in underlying molecular mechanisms, global gene expression profiles have been monitored in human asthmatics including isolated leukocyte populations (311–314), bronchial and epithelial biopsies (315–319), and nasal lavage samples (320). Comparing asthmatics and healthy controls, two distinct subgroups of patients were identified with a “Th2 high” or a “Th2 low” transcriptomic profile with key markers of asthma including levels of IL-5 and IL-13, AHR, serum IgE, blood and airway eosinophilia, subepithelial fibrosis, airway MUCIN gene expression, and improvement with inhaled corticosteroids (5). A signature of IL-13-induced genes including PERIOSTIN, CLCA1, and SERPINB2 correlated with the “Th2 high” group (5). These studies provided mechanistic insights into mild-to-moderate and atopic asthma. Further clustering based on transcriptomic endotypes of asthma (TEA) was performed in the sputum and blood of patients with asthma identifying three major TEA clusters (321): TEA cluster 1 included asthmatics with a history of intubation, lower pre-bronchodilator FEV1, a higher bronchodilator response, and higher exhaled nitric oxide levels; TEA cluster 2 contained individuals that were hospitalized for asthma; asthmatics grouped within the TEA cluster 3 had normal lung function, low exhaled nitric oxide levels, and lower inhaled steroid requirements.
Few studies have investigated molecular pathways on a global level with a focus on severe (189, 322) or SR asthma (323, 324). Measuring mRNA and noncoding RNA expression by microarray in circulating CD4+ and CD8+ T cells revealed expression changes specific to CD8+ T cells in patients with severe asthma but not with CD4+ T cells (189). Differentially-expressed genes in these CD8+ T cells were attributable to multiple pathways involved in T-cell activation and additionally with expression changes of noncoding RNA species. Surprisingly, no differences were detectable in both T-cell subsets in non-severe asthmatics compared to healthy controls.
Supervised clustering of 42 gene sets associated with asthma and immune and/or inflammatory pathways was performed using transcriptomic data derived from bronchial biopsies and epithelial brushings from the same patients with moderate to severe asthma to examine if transcriptomic signatures may help distinguish between Th2 high eosinophilic and non-Th2 asthma (322). Nine gene set signatures expressed in bronchial biopsies and airway epithelial brushings distinguished these two asthma subtypes and corticosteroid responsiveness. In two independent cohorts of SS and SR asthmatics, gene expression profiles in PBMC were identified which predicted the clinical response to inhaled corticosteroid treatment with a suggested 84% accuracy (323). In a smaller cohort of children with SR asthma, with controlled persistent asthma, and healthy controls, molecular endotyping identified gene networks related to decreased glucocorticoid receptor signaling and increased activity of the MAPK and Jun kinase cascades in children with SR asthma (324). In a large cohort including 1,170 asthmatic adults from the Asthma BioRepository for Integrative Genomic Exploration (Asthma BRIDGE) and the Childhood Asthma Management Program (CAMP) study transcriptomic profiles were analyzed in whole blood and CD4+ T cells to assess the relationship to chronic and acute asthma control (325). In the initial discovery cohort using whole blood samples, 1,106 genes were found to be significantly overrepresented and associated with asthma control; 53% of these were replicated in at least one of the three replication cohorts determining gene expression signatures in either whole blood or CD4+ T cells. These studies identified eosinophilic and granulocytic inflammatory signals to be enriched in poorly-controlled asthmatics while immature lymphocytic patterns were associated with positive asthma control.
These analyses demonstrated both the usefulness and limitations of transcriptomic-driven classifications of asthmatics based on endotyping in addition to clinical parameters. This approach may improve the outcomes specifically in asthmatics that fail to respond to corticosteroid therapy and benefit the most from treatments prone to target type-2-mediated inflammation. However, asthma heterogeneity exists not only between patients but in the same patient at different time-points during disease progression. In addition, asthma exacerbations are triggered by numerous external stimuli that activate different pathways regulated by complex and dynamic gene-environment interactions (Table 1). In order to address the multifactorial entities of asthma, future investigations need to be developed in well-phenotyped populations with cohort-specific definitions of optimal asthma control and steroid-responsiveness.
In addition, this must be coupled with sampling of different immunologically-relevant specimens from the same patient at multiple time-points to delineate the longitudinal contributions to disease pathogenesis. Adding to the complexity is that regulatory regions of genes, in spite of being epigenetically-poised for expression, may not be actively transcribed until an appropriate environmental signal is received. Therefore, coupling transcriptomic analyses at baseline and after activation may be necessary to identify regulatory regions that control expression of genes relevant to severe asthma. Further, integration of different “omics” datasets including genetics, genomic, proteomic, and metabolomics will provide a unique opportunity to identify true causal genes and endotypes responsible for the disease.
7.0 Role of the Steroidogenic Enzyme CYP11A1 in Asthma Pathobiology
The cholesterol side-chain cleavage P450 enzyme (CYP11A1) is a key regulator of steroid biogenesis as the first and rate-limiting enzyme in the steroidogenic pathway converting cholesterol to pregnenolone (326). CYP11A1 is expressed primarily in the cortex of the adrenal gland, testis, ovary, placenta, and thymus (326–328). CYP11A1 is also expressed in various nonendocrine tissues, including the lung, spleen, intestine, stomach, liver, brain, and kidney (326–328). Activation of CYP11A1 results in a spectrum of steroid hormones, including glucocorticoids that are known to play a role in T-cell function (328, 329). In humans, mutations in the CYP11A1 gene result in steroid hormone deficiency and can cause the rare and potentially fatal condition, lipoid congenital adrenal hyperplasia (330–332). Patients with a heterozygous or homozygous mutation of CYP11A1 exhibit adrenal insufficiency and sex reversal (333, 334). Depletion of Cyp11a1 in mice or rabbits results in steroid deficiency, female external genitalia, and death (329, 335, 336).
During the differentiation of CD8+ T lymphocytes to a Tc2 phenotype, transcriptional profiling identified a marked upregulation of Cyp11a1 transcripts (337). Differentiation of CD8+ T cells in the presence of IL-2 plus IL-4, compared with IL-2 alone, resulted in significant increases in CYP11A1 gene expression, protein levels, and enzymatic activity (337). Inhibition of the enzymatic activation of CYP11A1 with aminoglutethimide (AMG) or silencing of Cyp11a1 using shRNA blocked the functional conversion of CD8+ T cells from IFNγ- to IL-13-producing cells (337). Expression of the lineage-specific transcription factors T-bet or Gata3 was not affected by inhibition of CYP11A1 activity indicating that Cyp11a1 was downstream of these master regulatory transcription factors. Silencing of Gata3 prevented the activation of CYP11A1 (337). Adoptive transfer of AMG-treated CD8+ T cells failed to restore AHR and airway inflammation in sensitized and challenged CD8-deficient mice. These studies identified CYP11A1 as a key regulator of CD8+ Tc2-cell transdifferentiation (337).
Trans- and cis-regulatory elements have been identified in the Cyp11a1 promoter region to regulate tissue-specific expression of CYP11A1 (328, 329, 338). In particular, transcription factors of the GATA protein family (GATA3, GATA4) play an important role in the regulation of CYP11A1 (328, 329, 338) and Cyp11a1 expression was decreased in Gata3-deficient mice (339). Levels of histone H3K27 trimethylation predominantly associated with repression of genes, were increased in Th1 cells at Th2 gene loci (Gata3, Cyp11a1, Il-4, and Il-13) when compared to levels in Th2 cells (340). In differentiated and polarized T-cell subsets, Cyp11a1 gene expression was 100–300-fold higher in Th2 cells compared with Th1 (341, 342) or Th17 cells (342), and 30-fold higher in Tc2 cells compared with Tc1 cells (341). As shown in CD8+ Tc2 cells (337), AMG decreased IL-13 cytokine production in polarized CD4+ Th2 cells without impairing IFNγ production in polarized Th1 cells (342). Silencing of Cyp11a1 by shRNA in cultured Th2 cells significantly lowered levels of IL-4 and IL-13 mRNA and protein. Similar to Tc2 CD8+ T cells (337), Gata3 mRNA levels remained unaffected after silencing of Cyp11a1 (342). These data indicate that CYP11A1 critically regulates Th2 and Tc2 cell differentiation and type 2 cytokine production in both T-cell subsets downstream of the essential lineage-specific transcription factor Gata3.
Recently, we identified a novel regulatory mechanism mediated through CYP11A1 which promoted IL-4-induced CD8+ T cell conversion to IL-13 production (343). Adoptive transfer of 1,25D3 (the active form of vitamin D3)-treated CD8+ T cells into sensitized and challenged CD8-deficient recipients prevented development of lung allergic immune responses. In CD8+ T cells in an IL-4-rich environment, 1,25D3 altered recruitment of the transcription factor vitamin D receptor (VDR) to the Cyp11a1 promoter in vitro and in vivo. These data confirmed the role of CYP11A1 in the molecular programming of CD8+ T cell conversion from an IFNγ- to an IL-13-secreting phenotype. In addition to the importance of CYP11A1 in the regulation of experimental asthma in mice, our data in two large cohorts of asthmatic children, uniquely linked genetic variants in CYP11A1 to the susceptibility to childhood asthma (343). CYP11A1 SNPs (rs4886595, rs4432229, and rs11632698) revealed significant protective effects in asthma, further diminished the risk to develop childhood asthma in combination with asthma-modifier SNPs in the VDR, and correlated with allele-specific CYP11A1 gene expression (343). Translating findings from the mouse model of experimental asthma to human studies provided a unique tool to identify similar mechanistic links between the steroidogenic enzyme CYP11A1 as a key regulator of CD8+ Tc2 and CD4+ Th2 cell transcriptional programming and plasticity and susceptibility to childhood asthma.
8.0 Conclusions
Asthma is a common chronic disease of the airways and continues to challenge both clinicians and researchers. The search for common genetic factors is stymied by the number of triggers, epigenetic confounders, cells and mediators involved in dictating pathobiological consequences for development, maintenance, and exacerbation of the disease. Asthma is no longer considered a single disease but a syndrome, one not simply mediated by CD4+ T cell responses to allergen. The effector pathways are not likely stable but responsive to an ever-changing environment and numerous asthma triggers. A considerable degree of the heterogeneity in asthma between patients, and in the same patient at different stages of their disease, is reflected in the balance between different effector T-cell subsets and regulatory T cells. Each cell type is not fixed in lineage commitment but can undergo transcriptional reprogramming to a protective or enhancing effector phenotype. As a consequence, no single drug can be effective in all patients at all times. As the pathways or endotypes of the disease continue to be unraveled and characterized as steroid-sensitive or steroid-insensitive, more precise therapeutic targeting will become a reality for individual patients.
Acknowledgments
We thank Diana Nabighian for her assistance in preparation of the manuscript and the many members of the Gelfand laboratory who through the years contributed to the work. This work was supported in part by NIH grants HL-36577 and AI-77609, the Colorado Bioscience Discovery Evaluation Grant Program, the Eugene F. and Easton M. Crawford Charitable Lead Unitrust, and the Joanne Siegel Fund.
References
- 1.Martin RJ, et al. The Predicting Response to Inhaled Corticosteroid Efficacy (PRICE) trial. J Allergy Clin Immunol. 2007;119:73e80. doi: 10.1016/j.jaci.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Expert Panel Report 3 (EPR-3) Guidelines for the diagnosis and management of asthma – summary report. J Allergy Clin Immunol. 2007;120:S94–S138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 3.Bateman ED, Boushey HA, Bousquet J, et al. Can guideline-defined asthma control be achieved? The Gaining Optimal Asthma Control study. Am J Respir Crit Care Med. 2004;170:836–844. doi: 10.1164/rccm.200401-033OC. [DOI] [PubMed] [Google Scholar]
- 4.Guilbert TW, et al. Long-term inhaled corticosteroids in preschool children at high risk for asthma. N Engl J Med. 2006;354:1985–1997. doi: 10.1056/NEJMoa051378. [DOI] [PubMed] [Google Scholar]
- 5.Woodruff PG, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coyle AJ, Erard F, Bertrand C, Walti S, Pircher H, Le Gros G. Virus-specific CD8+ cells can switch to interleukin 5 production and induce airway eosinophilia. J Exp Med. 1995;181:1229–1233. doi: 10.1084/jem.181.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravensberg AJ, et al. CD8(+) T cells characterize early smoking-related airway pathology in patients with asthma. Respir Med. 2013;107:959–966. doi: 10.1016/j.rmed.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 8.Lan Q, et al. Occupational exposure to diesel engine exhaust and alterations in lymphocyte subsets. Occup Environ Med. 2015;72:354–359. doi: 10.1136/oemed-2014-102556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujimaki H, Ui N, Endo T. Induction of inflammatory response of mice exposed to diesel exhaust is modulated by CD4(+) and CD8(+) T cells. Am J Respir Crit Care Med. 2001;164:1867–1873. doi: 10.1164/ajrccm.164.10.2009095. [DOI] [PubMed] [Google Scholar]
- 10.Larsen ST, Matsubara S, McConville G, Poulsen SS, Gelfand EW. Ozone increases airway hyperreactivity and mucus hyperproduction in mice previously exposed to allergen. J Toxicol Environ Health A. 2010;73:738–747. doi: 10.1080/15287391003614034. [DOI] [PubMed] [Google Scholar]
- 11.Comhair SA, et al. Correlation of systemic superoxide dismutase deficiency to airflow obstruction in asthma. Am J Respir Crit Care Med. 2005;172:306–313. doi: 10.1164/rccm.200502-180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkham P, Rahman I. Oxidative stress in asthma and COPD: antioxidants as a therapeutic strategy. Pharmacol Ther. 2006;111:476–494. doi: 10.1016/j.pharmthera.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Extracellular glutathione peroxidase induction in asthmatic lungs: evidence for redox regulation of expression in human airway epithelial cells. FASEB J. 2001;15:70–78. doi: 10.1096/fj.00-0085com. [DOI] [PubMed] [Google Scholar]
- 14.Dweik RA, et al. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc Natl Acad Sci USA. 2001;98:2622–2627. doi: 10.1073/pnas.051629498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 16.Chung KF. New treatments for severe treatment-resistant asthma: targeting the right patient. Lancet Respir Med. 2013;1:639–652. doi: 10.1016/S2213-2600(13)70128-0. [DOI] [PubMed] [Google Scholar]
- 17.Pavord ID, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–659. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- 18.Chung KF. Defining phenotypes in asthma: a step towards personalized medicine. Drugs. 2014;74:719–728. doi: 10.1007/s40265-014-0213-9. [DOI] [PubMed] [Google Scholar]
- 19.Yan X, et al. Noninvasive analysis of the sputum transcriptome discriminates clinical phenotypes of asthma. Am J Respir Crit Care Med. 2015;191:1116–1125. doi: 10.1164/rccm.201408-1440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kupczyk M, et al. Stability of phenotypes defined by physiological variables and biomarkers in adults with asthma. Allergy. 2014;69:1198–1204. doi: 10.1111/all.12445. [DOI] [PubMed] [Google Scholar]
- 21.Shin Bomi, et al. The transition of sputum inflammatory cell profiles is variable in stable asthma patients. Asia Pac Allergy. 2017;7:19–28. doi: 10.5415/apallergy.2017.7.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shalaby KH, Martin JG. Overview of asthma; the place of the T cell. Curr Opin Pharmacol. 2010;10:218–225. doi: 10.1016/j.coph.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Robinson DS, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 25.Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of airway mucus production By T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. J Exp Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG, Foster PS. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol. 1998;161:1501–1509. [PubMed] [Google Scholar]
- 27.Huber M, Lohoff M. Change of paradigm: CD8+ T cells as important helper for CD4+ T cells during asthma and autoimmune encephalomyelitis. Allergo J Int. 2015;24:8–15. doi: 10.1007/s40629-015-0038-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rengarajan J, Szabo SJ, Glimcher LH. Transcriptional regulation of Th1/Th2 polarization. Immunol Today. 2000;21:479–483. doi: 10.1016/s0167-5699(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 29.Romagnani S. Lymphokine production by human T cells in disease states. Annu Rev Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 30.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 31.Lack G, et al. Nebulized IFN-gamma inhibits the development of secondary allergic responses in mice. J Immunol. 1996;157:1432–1439. [PubMed] [Google Scholar]
- 32.Parronchi P, et al. IL-4 and IFN (alpha and gamma) exert opposite regulatory effects on the development of cytolytic potential by Th1 or Th2 human T cell clones. J Immunol. 1992;149:2977–2983. [PubMed] [Google Scholar]
- 33.Skapenko A, Niedobitek GU, Kalden JR, Lipsky PE, Schulze-Koops H. Generation and regulation of human Th1-biased immune responses in vivo: a critical role for IL-4 and IL-10. J Immunol. 2004;172:6427–6434. doi: 10.4049/jimmunol.172.10.6427. [DOI] [PubMed] [Google Scholar]
- 34.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 35.Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402:B18–B23. doi: 10.1038/35037014. [DOI] [PubMed] [Google Scholar]
- 36.Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, Tumes DJ, Okamoto Y. Th2 cells in health and disease. Annu Rev Immunol. 2016 doi: 10.1146/annurev-immunol-051116-052350. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 37.McKenzie GJ, et al. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–432. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 38.Hilton DJ, Zhang JG, Metcalf D, Alexander WS, Nicola NA, Willson TA. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci USA. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 40.Umetsu DT, DeKruyff RH. The regulation of allergy and asthma. Immunol Rev. 2006;212:238–255. doi: 10.1111/j.0105-2896.2006.00413.x. [DOI] [PubMed] [Google Scholar]
- 41.Finkelman FD, Hogan SP, Hershey GK, Rothenberg ME, Wills-Karp M. Importance of cytokines in murine allergic airway disease and human asthma. J Immunol. 2010;184:1663–1674. doi: 10.4049/jimmunol.0902185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corren J. Anti-interleukin-13 antibody therapy for asthma: one step closer. Eur Respir J. 2013;41:255–256. doi: 10.1183/09031936.00124212. [DOI] [PubMed] [Google Scholar]
- 43.Maes T, Joos GF, Brusselle GG. Targeting interleukin-4 in asthma: lost in translation? Am J Respir Cell Mol Biol. 2012;47:261–270. doi: 10.1165/rcmb.2012-0080TR. [DOI] [PubMed] [Google Scholar]
- 44.Saha SK, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–691. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura Y, et al. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol. 1999;103:215–222. doi: 10.1016/s0091-6749(99)70493-8. [DOI] [PubMed] [Google Scholar]
- 46.Taha R, Hamid Q, Cameron L, Olivenstein R. T helper type 2 cytokine receptors and associated transcription factors GATA-3, c-MAF, and signal transducer and activator of transcription factor-6 in induced sputum of atopic asthmatic patients. Chest. 2003;123:2074–2082. doi: 10.1378/chest.123.6.2074. [DOI] [PubMed] [Google Scholar]
- 47.Cameron L, et al. Th2 cell-selective enhancement of human IL13 transcription by IL13-1112C>T, a polymorphism associated with allergic inflammation. J Immunol. 2006;177:8633–8642. doi: 10.4049/jimmunol.177.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabesch M, et al. IL-4/IL-13 pathway genetics strongly influence serum IgE levels and childhood asthma. J Allergy Clin Immunol. 2006;117:269–274. doi: 10.1016/j.jaci.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 49.Michel S, et al. Unifying candidate gene and GWAS Approaches in Asthma. PLoS One. 2010;5:e13894. doi: 10.1371/journal.pone.0013894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang C, Rolland JM, Ward C, Quan B, Walters EH. IL-5 production by bronchoalveolar lavage and peripheral blood mononuclear cells in asthma and atopy. Eur Respir J. 1997;10:624–632. [PubMed] [Google Scholar]
- 51.Kenyon NJ, Kelly EA, Jarjour NN. Enhanced cytokine generation by peripheral blood mononuclear cells in allergic and asthma subjects. Ann Allergy Asthma Immunol. 2000;85:115–120. doi: 10.1016/S1081-1206(10)62450-7. [DOI] [PubMed] [Google Scholar]
- 52.Chung EH, et al. Leukotriene B4 receptor 1 is differentially expressed on peripheral T cells of steroid-sensitive and -resistant asthmatics. Ann Allergy Asthma Immunol. 2014;112:211–216. doi: 10.1016/j.anai.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects and patients with asthma. J Allergy Clin Immunol. 1996;97:1288–1296. doi: 10.1016/s0091-6749(96)70197-5. [DOI] [PubMed] [Google Scholar]
- 54.Matsumoto K, et al. Decrease of interleukin-10-producing T cells in the peripheral blood of severe unstable atopic asthmatics. Int Arch Allergy Immunol. 2004;134:295–302. doi: 10.1159/000079167. [DOI] [PubMed] [Google Scholar]
- 55.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 56.Brusselle G, Kips J, Joos G, Bluethmann H, Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. Am J Respir Cell Mol Biol. 1995;12:254–259. doi: 10.1165/ajrcmb.12.3.7873190. [DOI] [PubMed] [Google Scholar]
- 57.Walter DM, et al. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J Immunol. 2001;167:4668–4675. doi: 10.4049/jimmunol.167.8.4668. [DOI] [PubMed] [Google Scholar]
- 58.Coyle AJ, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- 59.Wills-Karp M, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 60.McKenzie GJ, Fallon PG, Emson CL, Grencis RK, McKenzie AN. Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J Exp Med. 1999;189:1565–1572. doi: 10.1084/jem.189.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noben-Trauth N, Shultz LD, Brombacher F, Urban JF, Jr, Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci USA. 1997;94:10838–10843. doi: 10.1073/pnas.94.20.10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cohn L, Homer RJ, MacLeod H, Mohrs M, Brombacher F, Bottomly K. Th2-induced airway mucus production is dependent on IL-4Ralpha, but not on eosinophils. J Immunol. 1999;162:6178–6183. [PubMed] [Google Scholar]
- 63.O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashino S, et al. Janus kinase 1/3 signaling pathways are key initiators of TH2 differentiation and lung allergic responses. J Allergy Clin Immunol. 2014;133:1162–1174. doi: 10.1016/j.jaci.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ghoreschi K, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsunaga Y, et al. Effects of a Janus kinase inhibitor, pyridone 6, on airway responses in a murine model of asthma. Biochem Biophys Res Commun. 2011;404:261–267. doi: 10.1016/j.bbrc.2010.11.104. [DOI] [PubMed] [Google Scholar]
- 67.Southworth T, et al. Anti-inflammatory potential of PI3Kdelta and JAK inhibitors in asthma patients. Respir Res. 2016;17:124. doi: 10.1186/s12931-016-0436-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 69.Shimoda K, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 70.Takeda K, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 71.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–948. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomkinson A, Kanehiro A, Rabinovitch N, Joetham A, Cieslewicz G, Gelfand EW. The failure of STAT6-deficient mice to develop airway eosinophilia and airway hyperresponsiveness is overcome by interleukin-5. Am J Respir Crit Care Med. 1999;160:1283–1291. doi: 10.1164/ajrccm.160.4.9809065. [DOI] [PubMed] [Google Scholar]
- 73.Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D, Telerman A. The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc Natl Acad Sci U S A. 1989;86:8857–8861. doi: 10.1073/pnas.86.22.8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cibull TL, et al. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J Clin Pathol. 2006;59:285–288. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11:23–34. doi: 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 76.Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA, Skorski T. Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood. 2002;99:4531–4539. doi: 10.1182/blood.v99.12.4531. [DOI] [PubMed] [Google Scholar]
- 77.Valdman A, Fang X, Pang ST, Ekman P, Egevad L. Pim-1 expression in prostatic intraepithelial neoplasia and human prostate cancer. Prostate. 2004;60:367–371. doi: 10.1002/pros.20064. [DOI] [PubMed] [Google Scholar]
- 78.Andina N, Didichenko S, Schmidt-Mende J, Dahinden CA, Simon HU. Proviral integration site for Moloney murine leukemia virus 1, but not phosphatidylinositol-3 kinase, is essential in the antiapoptotic signaling cascade initiated by IL-5 in eosinophils. J Allergy Clin Immunol. 2009;123:603–611. doi: 10.1016/j.jaci.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 79.Temple R, et al. Microarray analysis of eosinophils reveals a number of candidate survival and apoptosis genes. Am J Respir Cell Mol Biol. 2001;25:425–433. doi: 10.1165/ajrcmb.25.4.4456. [DOI] [PubMed] [Google Scholar]
- 80.Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med. 2005;201:259–266. doi: 10.1084/jem.20042020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stout BA, Bates ME, Liu LY, Farrington NN, Bertics PJ. IL-5 and granulocyte-macrophage colony-stimulating factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3 protein expression in human eosinophils. J Immunol. 2004;173:6409–6417. doi: 10.4049/jimmunol.173.10.6409. [DOI] [PubMed] [Google Scholar]
- 82.Shin YS, et al. Inhibition of Pim1 kinase activation attenuates allergen-induced airway hyperresponsiveness and inflammation. Am J Respir Cell Mol Biol. 2012;46:488–497. doi: 10.1165/rcmb.2011-0190OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Vries M, et al. Pim1 kinase activity preserves airway epithelial integrity upon house dust mite exposure. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1344–L1353. doi: 10.1152/ajplung.00043.2015. [DOI] [PubMed] [Google Scholar]
- 84.Vries M, et al. Inhibition of Pim1 kinase, new therapeutic approach in virus-induced asthma exacerbations. Eur Respir J. 2016;47:783–791. doi: 10.1183/13993003.00171-2015. [DOI] [PubMed] [Google Scholar]
- 85.Aho TL, Sandholm J, Peltola KJ, Ito Y, Koskinen PJ. Pim-1 kinase phosphorylates RUNX family transcription factors and enhances their activity. BMC Cell Biol. 2006;7:21–29. doi: 10.1186/1471-2121-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taniuchi I, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 87.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate IFNγ and silence IL-4 in T helper type 1 cells. Nature Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 88.Kohu K, et al. The Runx3 transcription factor augments Th1 and down-modulates Th2 phenotypes by interacting with and attenuating GATA3. J Immunol. 2009;183:7817–7824. doi: 10.4049/jimmunol.0802527. [DOI] [PubMed] [Google Scholar]
- 89.Lee SH, et al. Runx3 inhibits IL-4 production in T cells via physical interaction with NFAT. Biochem Biophys Res Commun. 2009;381:214–217. doi: 10.1016/j.bbrc.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 90.Brenner O, et al. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proc Natl Acad Sci USA. 2004;101:16016–16021. doi: 10.1073/pnas.0407180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol. 2009;9:106–115. doi: 10.1038/nri2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang M, et al. Inhibition of Pim1 kinase prevents peanut allergy by enhancing Runx3 expression and suppressing T(H)2 and T(H)17 T-cell differentiation. J Allergy Clin Immunol. 2012;130:932–944. doi: 10.1016/j.jaci.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Sullivan S, et al. Activated, cytotoxic CD8+ T lymphocytes contribute to the pathology of asthma death. Am J Respir Crit Care Med. 2001;164:560–564. doi: 10.1164/ajrccm.164.4.2102018. [DOI] [PubMed] [Google Scholar]
- 94.Ying S, et al. Expression of IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asthmatics. J Immunol. 1997;158:3539–3544. [PubMed] [Google Scholar]
- 95.Hamzaoui A, Chaouch N, Grairi H, Ammar J, Hamzaoui K. Inflammatory process of CD8+ CD28− T cells in induced sputum from asthmatic patients. Mediators Inflamm. 2005;3:160–166. doi: 10.1155/MI.2005.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Durrant DM, Metzger DW. Emerging roles of T helper subsets in the pathogenesis of asthma. Immunol Invest. 2010;39:526–549. doi: 10.3109/08820131003615498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Robinson DS. The role of the T cell in asthma. J Allergy Clin Immunol. 2010;126:1081–1093. doi: 10.1016/j.jaci.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 98.Schaller MA, Lundy SK, Huffnagle GB, Lukacs NW. CD8+ T cell contributions to allergen induced pulmonary inflammation and airway hyperreactivity. Eur J Immunol. 2005;35:2061–2070. doi: 10.1002/eji.200425715. [DOI] [PubMed] [Google Scholar]
- 99.Ohnishi H, et al. Corticosteroids enhance CD8+ T cell-mediated airway hyperresponsiveness and allergic inflammation by upregulating leukotriene B4 receptor 1. J Allergy Clin Immunol. 2008;121:864–871. doi: 10.1016/j.jaci.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 100.Jia Y, et al. Step-wise epigenetic and phenotypic alterations poise CD8+ T cells to mediate airway hyperresponsiveness and inflammation. J Immunol. 2013;190:4056–4065. doi: 10.4049/jimmunol.1202640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kallinich T, Beier KC, Wahn U, Stock P, Hamelmann H. T-cell co-stimulatory molecules: their role in allergic immune reactions. Eur Resp J. 2007;29:1246–1255. doi: 10.1183/09031936.00094306. [DOI] [PubMed] [Google Scholar]
- 102.O’Sullivan SM. Asthma death, CD8+ T cells, and viruses. Proc Am Thorac Soc. 2005;2:162–165. doi: 10.1513/pats.200502-016AW. [DOI] [PubMed] [Google Scholar]
- 103.Miyahara N, et al. Effector CD8+ T cells mediate inflammation and airway hyperresponsiveness. Nature Med. 2004;10:865–869. doi: 10.1038/nm1081. [DOI] [PubMed] [Google Scholar]
- 104.Wells JW, Cowled CJ, Giorgini A, Kemeny DM, Noble A. Regulation of allergic airway inflammation by class I-restricted allergen presentation and CD8 T-cell infiltration. J Allergy Clin Immunol. 2007;119:226–234. doi: 10.1016/j.jaci.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 105.Sarantopoulos S, Lu L, Cantor H. Qa-1 restriction of CD8+ suppressor T cells. J Clin Invest. 2004;114:1218–1221. doi: 10.1172/JCI23152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takeda K, et al. Vaccine-induced CD8+ T cell dependent suppression of airway hyperresponsiveness and inflammation. J Immunol. 2009;183:181–190. doi: 10.4049/jimmunol.0803967. [DOI] [PubMed] [Google Scholar]
- 107.Hamelmann E, et al. Requirement for CD8+ T cells in the development of airway hyperresponsiveness in a murine model of airway sensitization. J Exp Med. 1996;183:1719–1729. doi: 10.1084/jem.183.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miyahara N, et al. Contribution of antigen-primed CD8+ T cells to the development of airway hyperresponsiveness and inflammation is associated with IL-13. J Immunol. 2004;172:2549–2558. doi: 10.4049/jimmunol.172.4.2549. [DOI] [PubMed] [Google Scholar]
- 109.Koya T, et al. CD8+ T cell-mediated airway hyperresponsiveness and inflammation is dependent on CD4+IL-4+ T cells. J Immunol. 2007;179:2787–2796. doi: 10.4049/jimmunol.179.5.2787. [DOI] [PubMed] [Google Scholar]
- 110.Erard F, Wild MT, Garcia-Sanz JA, LeGros G. Switch of CD8+ lymphocytes can develop into non-cytolytic CD8−CD4− cells which produce IL-4, IL-5, IL-10, and help B cells. Science. 1993;260:1802–1805. doi: 10.1126/science.8511588. [DOI] [PubMed] [Google Scholar]
- 111.Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271–279. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 112.Cerwenka A, Carter LL, Reome JB, Swain SL, Dutton RW. In vivo persistence of CD8 polarized T cell subsets producing type 1 or type 2 cytokines. J Immunol. 1998;161:97–105. [PubMed] [Google Scholar]
- 113.Seneviratne SL, et al. Allergen-specific CD8+ T cells and atopic disease. J Clin Invest. 2002;10:1283–1291. doi: 10.1172/JCI15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 115.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;91:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 116.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 117.Manjunath N, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J Clin Invest. 2001;108:871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Okamoto M, et al. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Exp Med. 2008;205:1087–1097. doi: 10.1084/jem.20072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deftos ML, He YW, Ojala EW, Bevan MJ. Correlating notch signaling with thymocyte maturation. Immunity. 1998;9:777–786. doi: 10.1016/s1074-7613(00)80643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morimura T, Goitsuka R, Zhang Y, Saito I, Reth M, Kitamura D. Cell cycle arrest and apoptosis induced by Notch1 in B cells. J Biol Chem. 2000;275:36523–36531. doi: 10.1074/jbc.M006415200. [DOI] [PubMed] [Google Scholar]
- 121.Wong KK, et al. Notch ligation by Delta1 inhibits peripheral immune responses to transplantation antigens by a CD8+ cell-dependent mechanism. J Clin Invest. 2003;112:1741–1750. doi: 10.1172/JCI18020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vigouroux S, et al. Induction of antigen-specific regulatory T cells following overexpression of a Notch ligand by human B lymphocytes. J Virol. 2003;77:10872–10880. doi: 10.1128/JVI.77.20.10872-10880.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Palaga T, Miele L, Golde TE, Osborne BA. TCR-mediated notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J Immunol. 2003;171:3019–3024. doi: 10.4049/jimmunol.171.6.3019. [DOI] [PubMed] [Google Scholar]
- 124.Kumar A, et al. Mechanical stretch activates nuclear factor-kappaB, activator protein-1, and mitogen-activated protein kinases in lung parenchyma: implications in asthma. FASEB J. 2003;17:1800–1811. doi: 10.1096/fj.02-1148com. [DOI] [PubMed] [Google Scholar]
- 125.Atherton HC, Jones G, Danahay H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am J Physiol Lung Cell Mol Physiol. 2003;285:L730–L739. doi: 10.1152/ajplung.00089.2003. [DOI] [PubMed] [Google Scholar]
- 126.Amashita M, et al. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc Natl Acad Sci USA. 1999;96:1024–1029. doi: 10.1073/pnas.96.3.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Richards JD, Davé SH, Chou CH, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J Immunol. 2001;166:3855–3864. doi: 10.4049/jimmunol.166.6.3855. [DOI] [PubMed] [Google Scholar]
- 128.Boehme SA, et al. Activation of mitogen-activated protein kinase regulates eotaxin-induced eosinophil migration. J Immunol. 1999;163:1611–1618. [PubMed] [Google Scholar]
- 129.Pahl A, Zhang M, Kuss H, Szelenyi I, Brune K. Regulation of IL-13 synthesis in human lymphocytes: implications for asthma therapy. Br J Pharmacol. 2002;135:1915–1926. doi: 10.1038/sj.bjp.0704656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Duan W, Chan JH, Wong CH, Leung BP, Wong WS. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J Immunol. 2004;172:7053–7059. doi: 10.4049/jimmunol.172.11.7053. [DOI] [PubMed] [Google Scholar]
- 131.Chialda L, Zhang M, Brune K, Pahl A. Inhibitors of mitogen-activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophilia. Respir Res. 2005;6:36. doi: 10.1186/1465-9921-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ohnishi H, et al. Mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2-dependent pathways are essential for CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Allergy Clin Immunol. 2009;123:249–257. doi: 10.1016/j.jaci.2008.10.054. [DOI] [PubMed] [Google Scholar]
- 133.Cho SH, Stanciu LA, Holgate ST, Johnston SL. Increased interleukin-4, interleukin-5, and interferon-γ in airway CD4+ and CD8+ T cells in atopic asthma. Am J Resp Crit Care Med. 2005;171:224–230. doi: 10.1164/rccm.200310-1416OC. [DOI] [PubMed] [Google Scholar]
- 134.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 136.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 137.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Acacia de Sa Pinheiro A, et al. IL-4 induces a wide-spectrum intracellular signaling cascade in CD8+ T cells. J Leukoc Biol. 2007;81:1102–1110. doi: 10.1189/jlb.0906583. [DOI] [PubMed] [Google Scholar]
- 139.Morris SC, et al. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cell proliferation. J Immunol. 2009;182:1429–1438. doi: 10.4049/jimmunol.182.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Apte SH, et al. IFN-gamma inhibits IL-4-induced type 2 cytokine expression by CD8 T cells in vivo and modulates the anti-tumor response. J Immunol. 2010;185:998–1004. doi: 10.4049/jimmunol.0903372. [DOI] [PubMed] [Google Scholar]
- 141.Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B (4) receptors. Prostaglandins Leuko Essent Fatty Acids. 2003;69:123–134. doi: 10.1016/s0952-3278(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 142.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 143.Smith MJ, Ford-Hutchinson AW, Bray MA. Leukotriene B: a potential mediator of inflammation. J Pharm Pharmacol. 1980;32:517–518. doi: 10.1111/j.2042-7158.1980.tb12985.x. [DOI] [PubMed] [Google Scholar]
- 144.Huang WW, Garcia-Zepeda EA, Sauty A, Oettgen HC, Rothenberg ME, Luster AD. Molecular and biological characterization of the murine leukotriene B4 receptor expressed on eosinophils. J Exp Med. 1998;188:1063–1074. doi: 10.1084/jem.188.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Payan DG, Missirian-Bastian A, Goetzl EJ. Human T-lymphocyte subset specificity of the regulatory effects of leukotriene B4. Proc Natl Acad Sci USA. 1984;81:3501–3505. doi: 10.1073/pnas.81.11.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Leppert D, Hauser SL, Kishiyama JL, An S, Zeng L, Goetzl EJ. Stimulation of matrix metalloproteinase-dependent migration of T cells by eicosanoids. FASEB J. 1995;9:1473–1481. doi: 10.1096/fasebj.9.14.7589989. [DOI] [PubMed] [Google Scholar]
- 147.Bacon KB, Camp RD, Cunningham FM, Woollard PM. Contrasting in vitro lymphocyte chemotactic activity of the hydroxyl enantiomers of 12-hydroxy-5, 8, 10, 14-eicosatetraenoic acid. Br J Pharmacol. 1988;95:966–974. doi: 10.1111/j.1476-5381.1988.tb11727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang WW, Garcia-Zepeda EA, Sauty A, Oettgen HC, Rothenberg ME, Luster AD. Molecular and biological characterization of the murine leukotriene B4 receptor expressed on eosinophils. J Exp Med. 1998;188:1063–1074. doi: 10.1084/jem.188.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tager AM, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 150.Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol. 2003;4:965–973. doi: 10.1038/ni972. [DOI] [PubMed] [Google Scholar]
- 151.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol. 2003;4:974–981. doi: 10.1038/ni971. [DOI] [PubMed] [Google Scholar]
- 152.Miyahara N, et al. Leukotriene B4 receptor-1 is essential for allergen-mediated recruitment of CD8+ T cells and airway hyperresponsiveness. J Immunol. 2005;174:4979–4984. doi: 10.4049/jimmunol.174.8.4979. [DOI] [PubMed] [Google Scholar]
- 153.Henderson WR, Jr, et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. J Exp Med. 1996;184:1483–1494. doi: 10.1084/jem.184.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Medoff BD, et al. BLT1-mediated T cell trafficking is critical for rejection and obliterative bronchiolitis after lung transplantation. J Exp Med. 2005;202:97–110. doi: 10.1084/jem.20042481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 156.Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLOS Pathogens. 2011;7:1–17. doi: 10.1371/journal.ppat.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takeda K, et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. 1997;186:449–454. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885–892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Taube C, et al. The leukotriene B4 receptor (BLT1) is rezuired for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J Immunol. 2006;176:3157–3164. doi: 10.4049/jimmunol.176.5.3157. [DOI] [PubMed] [Google Scholar]
- 160.Taube C, et al. Mast cells, FcepsilonRI, and IL-13 are required for development of airway hyperresponsiveness after aerosolized allergen exposure in the absence of adjuvant. J Immunol. 2004;172:6398–6406. doi: 10.4049/jimmunol.172.10.6398. [DOI] [PubMed] [Google Scholar]
- 161.Oshiba A, et al. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig) E and IgG1 in mice. J Clin Invest. 1996;97:1398–1408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Miyahara N, et al. Leukotriene B4 release from mast cells in IgE-mediated airway hyperresponsiveness and inflammation. Am J Respir Cell Mol Biol. 2009;40:672–682. doi: 10.1165/rcmb.2008-0095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 164.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Erard F, Wild MT, Garcia-Sanz JA, Le Gros G. Switch of CD8 T cells to noncytolytic CD8-CD4- cells that make TH2 cytokines and help B cells. Science. 1993;260:1802–1805. doi: 10.1126/science.8511588. [DOI] [PubMed] [Google Scholar]
- 166.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci USA. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Araki Y, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181:865–868. doi: 10.4049/jimmunol.181.2.865. [DOI] [PubMed] [Google Scholar]
- 171.Denton AE, Russ BE, Doherty PC, Rao S, Turner SJ. Differentiation-dependent functional and epigenetic landscapes for cytokine genes in virus-specific CD8+ T cells. Proc Natl Acad Sci USA. 2011;108:15306–15311. doi: 10.1073/pnas.1112520108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci USA. 2007;104:14765–14770. doi: 10.1073/pnas.0706378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Backman KS, Greenberger PA, Patterson R. Airways obstruction in patients with long-term asthma consistent with “irreversible asthma”. Chest. 1997;112:1234–1240. doi: 10.1378/chest.112.5.1234. [DOI] [PubMed] [Google Scholar]
- 174.Ulrik CS, Backer V. Nonreversible airflow obstruction in life-long nonsmokers with moderate to severe asthma. Eur Respir J. 1999;14:892–896. doi: 10.1034/j.1399-3003.1999.14d27.x. [DOI] [PubMed] [Google Scholar]
- 175.Taniguchi H, Hoshino K, Kobayashi M. The decline of pulmonary function among patients with chronic asthma treated with inhaled corticosteroid. J Asthma. 2002;39:217–225. doi: 10.1081/jas-120002471. [DOI] [PubMed] [Google Scholar]
- 176.Stankova J, Turcotte S, Harris J, Rola-Pleszczynski M. Modulation of leukotriene B4 receptor-1 expression by dexamethasone: potential mechanism for enhanced neutrophil survival. J Immunol. 2002;168:3570–3576. doi: 10.4049/jimmunol.168.7.3570. [DOI] [PubMed] [Google Scholar]
- 177.Obinata H, Yokomizo T, Shimizu T, Izumi T. Glucocorticoids up-regulate leukotriene B4 receptor-1 expression during neutrophilic differentiation of HL-60 cells. Biochem Biophys Res Commun. 2033;309:114–119. doi: 10.1016/s0006-291x(03)01554-7. [DOI] [PubMed] [Google Scholar]
- 178.Fernández-Ruiz E, et al. IL-2 protects T cell hybrids from the cytolytic effect of glucocorticoids. Synergistic effect of IL-2 and dexamethasone in the induction of high-affinity IL-2 receptors. J Immunol. 1989;143:4146–4151. [PubMed] [Google Scholar]
- 179.Corrigan CJ, et al. CD4 T-lymphocyte activation in asthma is accompanied by increased serum concentrations of interleukin-5. Effect of glucocorticoid therapy. Am Rev Respir Dis. 1993;147:540–547. doi: 10.1164/ajrccm/147.3.540. [DOI] [PubMed] [Google Scholar]
- 180.Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: pathologenic features and decreased susceptibility to immunoregulation. J Immunol. 2007;179:6514–6523. doi: 10.4049/jimmunol.179.10.6514. [DOI] [PubMed] [Google Scholar]
- 181.Fasth AE, et al. Skewed distribution of pro-inflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res Ther. 2007;9:R87. doi: 10.1186/ar2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yokoyama Y, et al. The CD4CD28null and the regulatory CD4+CD25high T-cell phenotypes in patients with ulcerative colitis during active and quiescent disease, following colectomy. Cytokine. 2011;56:466–470. doi: 10.1016/j.cyto.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 183.Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005;205:158–169. doi: 10.1111/j.0105-2896.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- 184.Yao H, Rahman I. Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol. 2012;303:557–566. doi: 10.1152/ajplung.00175.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li L-B, Leung DYM, Strand MJ, Goleva E. ATF2 impairs glucocorticoid receptor-mediated transactivation in human CD8+ T cells. Blood. 2007;110:1570–1577. doi: 10.1182/blood-2007-01-070755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Walker C, et al. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med. 1994;150:1038–1048. doi: 10.1164/ajrccm.150.4.7921434. [DOI] [PubMed] [Google Scholar]
- 187.Lee SY, et al. Distribution and cytokine production of CD4 and CD8 T-lymphocyte subsets in patients with acute asthma attacks. Ann Allergy Asthma Immunol. 2001;86:659–664. doi: 10.1016/S1081-1206(10)62295-8. [DOI] [PubMed] [Google Scholar]
- 188.Faul JL, et al. Lung immunopathology in cases of sudden asthma death. Eur Respir J. 1997;10:301–307. doi: 10.1183/09031936.97.10020301. [DOI] [PubMed] [Google Scholar]
- 189.Tsitsiou E, et al. Transcriptome analysis shows activation of circulating CD8+ T cells in patients with severe asthma. J Allergy Clin Immunol. 2012;129:95–103. doi: 10.1016/j.jaci.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 190.Oda H, et al. Interleukin-18 expression, CD8+ T cells, and eosinophils in lungs of nonsmokers with fatal asthma. Ann Allergy Immunol. 2014;112:23–28. doi: 10.1016/j.anai.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 191.Lee N, et al. IL-6 receptor □ defines effector memory CD8+ T cells producing Th2 cytokines and expanding in asthma. Am J Respir Crit Care Med. 2014;190:1383–1394. doi: 10.1164/rccm.201403-0601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.van Rensen ELJ, et al. Bronchial CD8 cell infiltrate and lung function decline in asthma. Am J Respir Crit Care Med. 2005;172:837–841. doi: 10.1164/rccm.200504-619OC. [DOI] [PubMed] [Google Scholar]
- 193.den Otter I, et al. Lung function decline in asthma patients with elevated bronchial CD8, CD4 and CD3 cells. Eur Respir J. 2016;48:393–402. doi: 10.1183/13993003.01525-2015. [DOI] [PubMed] [Google Scholar]
- 194.Hirosako S, et al. CD8 and CD103 are highly expressed in asthmatic bronchial intraepithelial lymphocytes. Int Arch Allergy Immunol. 2010;153:157–165. doi: 10.1159/000312633. [DOI] [PubMed] [Google Scholar]
- 195.Holand T, et al. A role for mitogen kinase kinase 3 in pulmonary inflammation validated from a proteomic approach. Pulm Pharmacol Ther. 2014;27:156–163. doi: 10.1016/j.pupt.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 196.Dakhama A, et al. IL-13-producing BLT1-positive CD8 cells are increased in asthma and are associated with airway obstruction. Allergy. 2013;68:666–673. doi: 10.1111/all.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 198.John M, et al. Inhaled corticosteroids increase interleukin-10 but reduce macrophage inflammatory protein-1-alpha, granulocyte-macrophage colony-stimulating factor, and interferon-gamma release from alveolar macrophages in asthma. Amer J Resp Crit Care Med. 1998;157:256–262. doi: 10.1164/ajrccm.157.1.9703079. [DOI] [PubMed] [Google Scholar]
- 199.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 201.Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. N Immunol. 2009;10:689–695. doi: 10.1038/ni.1760. [DOI] [PubMed] [Google Scholar]
- 202.von Boehmner Harald. Mechanisms of suppression by suppressor T cells. N Immunol. 2005;6:338–444. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 203.Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3+CD4+CD25+ regulatory T cells are not mirror images of each other. Trends in Immunol. 2008;29:429–435. doi: 10.1016/j.it.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 204.Lin X, et al. Advances in distinguishing natural from induced Foxp3+ regulatory cells. Int J Clin Exp Pathol. 2013;6:116–123. [PMC free article] [PubMed] [Google Scholar]
- 205.Ligocki AJ, Niederkorn JY. Advances on non-CD4+Foxp3+ T regulatory cells: CD8+, type 1, and double negative T regulatory cells in organ transplantation. Transplantation. 2015;99:11553–11559. doi: 10.1097/TP.0000000000000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bennet CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused mutations of Foxp3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 207.Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramshell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162:2546–2554. [PubMed] [Google Scholar]
- 208.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 209.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 210.Joetham A, et al. Airway hyperresponsiveness in the absence of CD4+ T cells after primary but not secondary challenge. Am J Respir Cell Mol Biol. 2005;33:89–96. doi: 10.1165/rcmb.2004-0414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Joetham A, et al. Antigen specificity is not required for modulation of lung allergic responses by naturally occurring regulatory T cells. J Immunol. 2009;183:1821–1827. doi: 10.4049/jimmunol.0900303. [DOI] [PubMed] [Google Scholar]
- 212.Herz U, Lumpp U, Daser A, Gelfand EW, Renz H. Murine animal models to study the central role of T cells in immediate-type hypersensitivity responses. Adv Exp Med Biol. 1996;409:25–32. doi: 10.1007/978-1-4615-5855-2_4. [DOI] [PubMed] [Google Scholar]
- 213.Renz H, Bradley K, Larsen GL, McCall C, Gelfand EW. Comparison of the allergenicity of ovalbumin and ovalbumin peptide 323–339. Differential expansion of V beta-expressing T cell populations. J Immunol. 1993;151:7206–7213. [PubMed] [Google Scholar]
- 214.Joetham A, et al. Plasticity of regulatory T cells: subversion of suppressive function and conversion to enhancement of lung allergic responses. J Immunol. 2008;180:7117–7124. doi: 10.4049/jimmunol.180.11.7117. [DOI] [PubMed] [Google Scholar]
- 215.Leech MD, Benson RA, DeVries A, Fitch PM, Howie SE. Resolution of Der p1-induced allergic airway inflammation is dependent on CD4+CD25+Foxp3+ regulatory cells. J Immunol. 2007;179:7050–7058. doi: 10.4049/jimmunol.179.10.7050. [DOI] [PubMed] [Google Scholar]
- 216.Joetham A, et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-β. J Immunol. 2007;178:1433–1442. doi: 10.4049/jimmunol.178.3.1433. [DOI] [PubMed] [Google Scholar]
- 217.Presser K, et al. Coexpression of TGF-beta1 and IL-10 enables regulatory T cells to completely suppress airway hyperreactivity. J Immunol. 2008;181:7751–7758. doi: 10.4049/jimmunol.181.11.7751. [DOI] [PubMed] [Google Scholar]
- 218.Taylor PA, Lees CJ, Fournier S, Allison JP, Sharpe AH, Blazar BR. B7 expression on T cells down-regulates immune responses through CTLA-4 ligation via T-T interactions. J Immunol. 2004;172:34–39. doi: 10.4049/jimmunol.172.1.34. [DOI] [PubMed] [Google Scholar]
- 219.Li J, et al. Ex vivo activated OVA specific and non-specific CD4+CD25+ regulatory T cells exhibit comparable suppression to OVA mediated T cell responses. Cell Immunol. 2006;241:75–84. doi: 10.1016/j.cellimm.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 220.Szymczak-Workman AL, Workman CJ, Vignali DA. Cutting edge: regulatory T cells do not require stimulation through their TCR to suppress. J Immunol. 2009;182:5188–92. doi: 10.4049/jimmunol.0803123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T-cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 222.Akbari O, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOSS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nature Med. 2002;8:1024–1032. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 223.Salaün J, et al. Thymic epithelium tolerizes for histocompatibility antigens. Science. 1990;247:1471–1474. doi: 10.1126/science.247.4949.1471. [DOI] [PubMed] [Google Scholar]
- 224.Modigliani Y, Coutinho A, Pereira P, Le Douarin N, Thomas-Vaslin V, Burlen-Defranoux O, Salaün J, Bandeira A. Establishment of tissue-specific tolerance is driven by regulatory T cells selected by thymic epithelium. Euro J Immunol. 1996;26:1807–15. doi: 10.1002/eji.1830260822. [DOI] [PubMed] [Google Scholar]
- 225.Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol. 2010;184:56–66. doi: 10.4049/jimmunol.0902379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Relland LM, et al. The TCR repertoires of regulatory and conventional T cells specific for the same foreign antigen are distinct. J Immunol. 2012;189:3566–3574. doi: 10.4049/jimmunol.1102646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fujishima M, Hirokawa, Fujishima N, Sawada K. TCRalphabeta repertoire diversity of human naturally occurring CD4+CD25+ regulatory T cells. Immunol Lett. 2005;99:193–197. doi: 10.1016/j.imlet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 228.Cutz E, Levison H, Cooper DM. Ultrastructure of airways in children with asthma. Histopathology. 1978;2:407–421. doi: 10.1111/j.1365-2559.1978.tb01735.x. [DOI] [PubMed] [Google Scholar]
- 229.Jenkins HA, et al. Histopathology of severe childhood asthma: a case series. Chest. 2003;124:32–41. doi: 10.1378/chest.124.1.32. [DOI] [PubMed] [Google Scholar]
- 230.Masu K, Ohno I, Suzuki K, Okada S, Hattori T, Shirato K. Proliferative effects of eosinophil lysates on cultured human airway smooth muscle cells. Clin Exp Allergy. 2002:32595–601. doi: 10.1046/j.0954-7894.2002.01243.x. [DOI] [PubMed] [Google Scholar]
- 231.Larsen GL, et al. Mice that overexpress Cu/Zn superoxide dismutase are resistant to allergen-induced changes in airway control. Am J Physiol Lung Cell Mol Physiol. 2000;279:L350–L359. doi: 10.1152/ajplung.2000.279.2.L350. [DOI] [PubMed] [Google Scholar]
- 232.Larsen GL, et al. Increased acetylcholine release in tracheas from allergen-exposed IgE-immune mice. Am J Physiol. 1994;266:L263–L270. doi: 10.1152/ajplung.1994.266.3.L263. [DOI] [PubMed] [Google Scholar]
- 233.Kanjarawi R, et al. Regulatory CD4+Foxp+T cells control the severity of anaphylaxis. PLOS One. 2013;8:e69183. doi: 10.1371/journal.pone.0069183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gri G, et al. CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity. 2008;29:771–781. doi: 10.1016/j.immuni.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Su W, et al. Induced CD4+ forkhead box protein-positive T cells inhibit mast cell function and established contact hypersensitivity through TGF-β. J Allergy Clin Immunol. 2012;130:444–452. doi: 10.1016/j.jaci.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 236.Takahashoi T, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 237.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Read S, Mauze S, Asseman C, Bean A, Coffman R, Powrie F. CD38+CD45RlowCD4+ T cells: a population of T cells with immune regulatory activities in vitro. Eur J Immunol. 1998;28:3435–3447. doi: 10.1002/(SICI)1521-4141(199811)28:11<3435::AID-IMMU3435>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 239.Jordan MS, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 240.Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198:249–258. doi: 10.1084/jem.20030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Joetham A, et al. CD8 regulates the endogenous production of IL-6 in naturally occurring T regulatory cells and maintains their suppressive phenotype in allergic lung disease. J Immunol. 2011;186:113–120. doi: 10.4049/jimmunol.1001663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lluis A, et al. Increased regulatory T-cell numbers are associated with farm milk exposure and lower atopic sensitization and asthma in childhood. J Allergy Clin Immunol. 2014;133:551–559. doi: 10.1016/j.jaci.2013.06.034. [DOI] [PubMed] [Google Scholar]
- 243.Martin H, Taube C. Regulatory T cells and regulation of allergic airway disease. Am J Clin Exp Immunol. 2012;1:166–178. [PMC free article] [PubMed] [Google Scholar]
- 244.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Imnunol. 2008;181:2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+ Foxp3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 246.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLOS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lal G, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Imnunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tao R, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 249.Reilly CM, et al. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/F1 mice. J Autoimmun. 2008;31:123–130. doi: 10.1016/j.jaut.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 250.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. 2013;37:273–283. doi: 10.1152/advan.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Joetham A, et al. Functional activation of naturally occurring lung CD4+CD25+ regulatory cells on lung allergic responses requires CD8 and MHC I interaction. Proc Natl Acad Sci. 2007;104:15057–15062. doi: 10.1073/pnas.0706765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8:S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL–17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 254.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 255.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25++ T cell regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 256.Hunter CA, Jones SA. IL-6 as a key cytokine in health and disease. Nat Immunol. 2015;16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 257.Wan YY, Flavell RA. Regulatory T cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 258.Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- 259.Zhou X, et al. Instability of the transcription factor Foxp3 leads to generation of pathogenic memory T cells in vivo. Nature Immunol. 2009;10:1000–1008. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Massoud AH, Charbonnier LM, Lopez D, Pellegrini M, Phipatanakul W, Chatila TA. An asthma-associated IL4R variant exacerbates airway inflammation by promoting conversion of regulatory T cells to Th17-like cells. Nature Med. 2016;22:1013–1022. doi: 10.1038/nm.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Joetham A, et al. Inducible and naturally occurring T regulatory cells mediate effector functions via divergent transcriptional pathways. J Allergy Clin Immunol. 2017 doi: 10.1016/j.jaci.2016.06.051. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4+CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 263.McHugh RS, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 264.Joetham A, et al. Loss of T regulatory cell suppression following signaling through the GITR is dependent on c-Jun N-terminal kinase activation. J Biol Chem. 2012;287:17100–17108. doi: 10.1074/jbc.M111.316943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Joetham A, et al. JNK2 regulates the functional plasticity of naturally occurring T regulatory cells and the enhancement of lung allergic responses. J Immunol. 2014;193:2238–2247. doi: 10.4049/jimmunol.1400604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.McGee HS, Agrawal DK. Naturally occurring and inducible T-regulatory cells modulating immune response in allergic asthma. Am J Respir Crit Care Med. 2009;180:211–225. doi: 10.1164/rccm.200809-1505OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Xu W, et al. Adoptive transfer of inducible-Treg cells effectively attenuates murine airway allergic inflammation. PloS One. 2012;7:e40314. doi: 10.1371/journal.pone.0040314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Hartl D, et al. Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J Allergy Clin Immunol. 2007;119:1258–1266. doi: 10.1016/j.jaci.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 269.Hamzaoui A, Ammar J, Hamzaoui K. Regulatory T cell in induced sputum of asthmatic children: association with inflammatory cytokines. Multi Resp Med. 2010;5:22–30. doi: 10.1186/2049-6958-5-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Malovrh MM, Rozman A, Skrgat S, Sila M, Selb J, Korosec P. Bronchial thermoplasty resulted in an increase of CD4+CD25+ regulatory T cells in severe asthma patients. Eur Resp J. 2016;48:A4906. [Google Scholar]
- 271.d’Hennezel E, Yurchenko E, Sgouroudis E, Hay V, Piccirillo CA. Single-cell analysis of the human T regulatory population uncovers functional heterogeneity and instability within Foxp3 cells. J Immunol. 2011;186:6788–6797. doi: 10.4049/jimmunol.1100269. [DOI] [PubMed] [Google Scholar]
- 272.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 273.Hoffmann P, et al. Isolation of CD4+CD25+ regulatory T cells for clinical trials. Biol Blood Marrow Transplant. 2006;12:267–274. doi: 10.1016/j.bbmt.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 274.Allan SE, et al. Activation-induced Foxp3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 275.Miyara M, Sakaguchi S. Human Foxp3+CD4+ regulatory T cells: their knowns and unknowns. Immunol and Cell Biol. 2011;89:346–351. doi: 10.1038/icb.2010.137. [DOI] [PubMed] [Google Scholar]
- 276.Miyara M, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the Foxp3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 277.Ito T, et al. Two functional subsets of Foxp3+ regulatory T cells in human thymus and periphery. Immunity. 2008;28:870–880. doi: 10.1016/j.immuni.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Swamy RS, et al. Epigentic modifications and improved regulatory T-cell function in subjects undergoing dual sublingual immunotherapy. J Allergy Clin Immunol. 2012;130:215–224. doi: 10.1016/j.jaci.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Urry Z, et al. The role of 1α,25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur J Immunol. 2012;42:2697–2708. doi: 10.1002/eji.201242370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten J. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–1023. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 281.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3 regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 282.Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P. Regulatory T cells in many flavors control asthma. Mucosal Immunol. 2010;3:216–229. doi: 10.1038/mi.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–27. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 284.Tran DQ, Ramsey H, Shevach EM. Induction of Foxp3 expression in naive human CD4FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–90. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Shibata K. Close link between development and function of gamma-delta T cells. Microbiol Immunol. 2012;56:217–227. doi: 10.1111/j.1348-0421.2012.00435.x. [DOI] [PubMed] [Google Scholar]
- 286.Lahn M, et al. Negative regulation of airway responsiveness that is dependent on γδ T cells and independent of αβ T cells. Nat Med. 1999;5:1150–1156. doi: 10.1038/13476. [DOI] [PubMed] [Google Scholar]
- 287.Kanehiro A, et al. Tumor necrosis factor-alpha negatively regulates airway hyperresponsiveness through gamma-delta T cells. Am J Respir Crit Care Med. 2001;164:2229–2238. doi: 10.1164/ajrccm.164.12.2012059. [DOI] [PubMed] [Google Scholar]
- 288.Kanehiro A, et al. Requirement for the p75 TNF-alpha receptor 2 in the regulation of airway hyperresponsiveness by gamma delta T cells. J Immunol. 2002;169:4190–4197. doi: 10.4049/jimmunol.169.8.4190. [DOI] [PubMed] [Google Scholar]
- 289.Rha YH, et al. Effect of microbial heat shock proteins on airway inflammation and hyperresponsiveness. J Immunol. 2002;169:5300–5307. doi: 10.4049/jimmunol.169.9.5300. [DOI] [PubMed] [Google Scholar]
- 290.Lahn M, et al. MHC class I-dependent Vgamma4+ pulmonary T cells regulate alpha beta T cell-independent airway responsiveness. Proc Natl Acad Sci USA. 2002;99:8850–8855. doi: 10.1073/pnas.132519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hahn YS, et al. V gamma 4+ gamma delta T cells regulate airway hyperreactivity to methacholine in ovalbumin-sensitized and challenged mice. J Immunol. 2003;171:3170–3178. doi: 10.4049/jimmunol.171.6.3170. [DOI] [PubMed] [Google Scholar]
- 292.Cui ZH, Joetham A, Aydintug MK, Hahn YS, Born WK, Gelfand EW. Reversal of allergic airway hyperreactivity after long-term allergen challenge depends on gammadelta T cells. Am J Respir Crit Care Med. 2003;168:1324–1332. doi: 10.1164/rccm.200305-634OC. [DOI] [PubMed] [Google Scholar]
- 293.Jin N, et al. Mismatched antigen prepares gamma delta T cells for suppression of airway hyperresponsiveness. J Immunol. 2005;174:2671–2679. doi: 10.4049/jimmunol.174.5.2671. [DOI] [PubMed] [Google Scholar]
- 294.Cook L, et al. Evidence that CD8+ dendritic cells enable the development of gammadelta T cells that modulate airway hyperresponsiveness. J Immunol. 2008;181:309–319. doi: 10.4049/jimmunol.181.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Hahn YS, et al. Different potentials of gamma delta T cell subsets in regulating airway responsiveness: V gamma 1+ cells, but not V gamma 4+ cells, promote airway hyperreactivity, Th2 cytokines, and airway inflammation. J Immunol. 2004;172:2894–2902. doi: 10.4049/jimmunol.172.5.2894. [DOI] [PubMed] [Google Scholar]
- 296.Matsubara S, et al. Vgamma1+ T cells and tumor necrosis factor-alpha in ozone-induced airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2009;40:454–463. doi: 10.1165/rcmb.2008-0346OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Jin N, et al. Airway hyperresponsiveness through synergy of γδ T cells and NKT cells. J Immunol. 2007;179:2961–2968. doi: 10.4049/jimmunol.179.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Jin N, et al. Allergic airway hyperresponsiveness-enhancing gammadelta T cells develop in normal untreated mice and fail to produce IL-4/13, unlike Th2 and NKT cells. J Immunol. 2009;182:2002–2010. doi: 10.4049/jimmunol.0803280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Glanville N, et al. γδ T cells suppress inflammation and disease during rhinovirus-induced asthma exacerbations. Mucosal Immunol. 2013;6:1091–1100. doi: 10.1038/mi.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Roark CL, Simonian PL, Fontenot AP, Born WK, O’Brien RL. gammadelta T cells: an important source of IL-17. Curr Opin Immunol. 2008;20:353–357. doi: 10.1016/j.coi.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Nakada EM, Shan J, Kinyanjui MW, Fixman ED. Adjuvant-dependent regulation of interleukin-17 expressing γδ T cells and inhibition of Th2 responses in allergic airways disease. Respir Res. 2014;15:90. doi: 10.1186/s12931-014-0090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hinks TS, et al. Innate and adaptive T cells in asthmatic patients: Relationship to severity and disease mechanisms. J Allergy Clin Immunol. 2015;136:323–333. doi: 10.1016/j.jaci.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Gapin L. iNKT cell autoreactivity: what is 'self' and how is it recognized? Nat Rev Immunol. 2010;10:272–277. doi: 10.1038/nri2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Akbari O, et al. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med. 2006;354:1117–1129. doi: 10.1056/NEJMoa053614. [DOI] [PubMed] [Google Scholar]
- 305.Vijayanand P, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. 2007;356:1410–1422. doi: 10.1056/NEJMoa064691. [DOI] [PubMed] [Google Scholar]
- 306.Thomas SY, Chyung YH, Luster AD. Natural killer T cells are not the predominant T cell in asthma and likely modulate, not cause, asthma. J Allergy Clin Immunol. 2010;125:980–984. doi: 10.1016/j.jaci.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Matsuda H, et al. Plasticity of invariant NKT cell regulation of allergic airway disease is dependent on IFN-γ production. J Immunol. 2010;185:253–262. doi: 10.4049/jimmunol.0902301. [DOI] [PubMed] [Google Scholar]
- 308.Chen YP, Zhang JH, Li CQ, Sun QX, Jiang XH. Obesity enhances Th2 inflammatory response via natural killer T cells in a murine model of allergic asthma. Int J Clin Exp Med. 2015;8:15403–15412. [PMC free article] [PubMed] [Google Scholar]
- 309.Nie H, et al. Invariant NKT cells act as an adjuvant to enhance Th2 inflammatory response in an OVA-induced mouse model of asthma. PLoS One. 2015;10:e0119901. doi: 10.1371/journal.pone.0119901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Berzins SP, Ritchie DS. Natural killer T cells: drivers or passengers in preventing human disease? Nat Rev Immunol. 2014;14:640–666. doi: 10.1038/nri3725. [DOI] [PubMed] [Google Scholar]
- 311.Aoki T, et al. Expression profiling of genes related to asthma exacerbations. Clin Exp Allergy. 2009;39:213–221. doi: 10.1111/j.1365-2222.2008.03186.x. [DOI] [PubMed] [Google Scholar]
- 312.Baines KJ, Simpson JL, Bowden NA, Scott RJ, Gibson PG. Differential gene expression and cytokine production from neutrophils in asthma phenotypes. Eur Respir J. 2010;35:522–531. doi: 10.1183/09031936.00027409. [DOI] [PubMed] [Google Scholar]
- 313.Bjornsdottir US, et al. Pathways activated during human asthma exacerbation as revealed by gene expression patterns in blood. PLoS One. 2011;6:e21902. doi: 10.1371/journal.pone.0021902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Chen E, Miller GE, Walker HA, Arevalo JM, Sung CY, Cole SW. Genome-wide transcriptional profiling linked to social class in asthma. Thorax. 2009;64:38–43. doi: 10.1136/thx.2007.095091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Chamberland A, Madore AM, Tremblay K, Laviolette M, Laprise C. A comparison of two sets of microarray experiments to define allergic asthma expression pattern. Exp Lung Res. 2009;35:399–410. doi: 10.1080/01902140902745174. [DOI] [PubMed] [Google Scholar]
- 317.Choy DF, et al. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol. 2011;186:1861–1869. doi: 10.4049/jimmunol.1002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, Taplin C, Pare PD, et al. Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am J Respir Crit Care Med. 2010;181:889–898. doi: 10.1164/rccm.200907-1071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Laprise C, Sladek R, Ponton A, Bernier MC, Hudson TJ, Laviolette M. Functional classes of bronchial mucosa genes that are differentially expressed in asthma. BMC Genomics. 2004;5:21. doi: 10.1186/1471-2164-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Bosco A, Ehteshami S, Panyala S, Martinez FD. Interferon regulatory factor 7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J Allergy Clin Immunol. 2012;129:88–94. doi: 10.1016/j.jaci.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Yan X, et al. Noninvasive analysis of the sputum transcriptome discriminates clinical phenotypes of asthma. Ann Am Thorac Soc. 2016;13:S104–S105. doi: 10.1513/AnnalsATS.201510-681MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kuo CS, et al. A transcriptome-driven analysis of epithelial brushings and bronchial biopsies to define asthma phenotypes in U-BIOPRED. Am J Respir Crit Care Med. 2017;195:443–455. doi: 10.1164/rccm.201512-2452OC. [DOI] [PubMed] [Google Scholar]
- 323.Hakonarson H, et al. Profiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patients. Proc Natl Acad Sci USA. 2005;102:14789–14794. doi: 10.1073/pnas.0409904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Persson H, et al. Transcriptome analysis of controlled and therapy-resistant childhood asthma reveals distinct gene expression profiles. J Allergy Clin Immunol. 2015;136:638–648. doi: 10.1016/j.jaci.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 325.Croteau-Chonka DC, et al. Gene expression profiling in blood provides reproducible molecular insights into asthma control. Am J Respir Crit Care Med. 2017;195:179–188. doi: 10.1164/rccm.201601-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Pazirandeh A, Xue Y, Rafter I, Sjövall J, Jondal M, Okret S. Paracrine glucocorticoid activity produced by mouse thymic epithelial cells. FASEB J. 1999;13:893–901. doi: 10.1096/fasebj.13.8.893. [DOI] [PubMed] [Google Scholar]
- 327.Cima I, et al. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med. 2004;200:1635–1646. doi: 10.1084/jem.20031958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lavoie HA, King SR. Transcriptional regulation of steroidogenic genes: STARD1, CYP11A1 and HSD3B. Exp Biol Med (Maywood) 2009;234:880–907. doi: 10.3181/0903-MR-97. [DOI] [PubMed] [Google Scholar]
- 329.Shih MC, Chiu YN, Hu MC, Guo IC, Chung BC. Regulation of steroid production: analysis of Cyp11a1 promoter. Mol Cell Endocrinol. 2011;336:80–84. doi: 10.1016/j.mce.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 330.Kim CJ, et al. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J Clin Endocrinol Metab. 2008;93:696–702. doi: 10.1210/jc.2007-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Al Kandari H, Katsumata N, Alexander S, Rasoul MA. Homozygous mutation of P450 side-chain cleavage enzyme gene (CYP11A1) in 46,XY patient with adrenal insufficiency, complete sex reversal, and agenesis of corpus callosum. J Clin Endocrinol Metab. 2006;91:2821–2826. doi: 10.1210/jc.2005-2230. [DOI] [PubMed] [Google Scholar]
- 332.Bhangoo A, Anhalt H, Ten S, King SR. Phenotypic variations in lipoid congenital adrenal hyperplasia. Pediatric Endocrinology Reviews. 2006;3:258–271. [PubMed] [Google Scholar]
- 333.Tajima T, Fujieda K, Kouda N, Nakae J, Miller WL. Heterozygous mutation in the cholesterol side chain cleavage enzyme (p450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocrinol Metab. 2001;86:3820–3825. doi: 10.1210/jcem.86.8.7748. [DOI] [PubMed] [Google Scholar]
- 334.Parajes S, et al. A novel entity of clinically isolated adrenal insufficiency caused by a partially inactivating mutation of the gene encoding for P450 side chain cleavage enzyme (CYP11A1) J Clin Endocrinol Metab. 2011;96:E1798–E1806. doi: 10.1210/jc.2011-1277. [DOI] [PubMed] [Google Scholar]
- 335.Pang S, et al. Inherited congenital adrenal hyperplasia in the rabbit: absent cholesterol side-chain cleavage cytochrome P450 gene expression. Endocrinology. 1992;131:181–186. doi: 10.1210/endo.131.1.1611996. [DOI] [PubMed] [Google Scholar]
- 336.Yang X, Iwamoto K, Wang M, Artwohl J, Mason JI, Pang S. Inherited congenital adrenal hyperplasia in the rabbit is caused by a deletion in the gene encoding cytochrome P450 cholesterol side-chain cleavage enzyme. Endocrinology. 1993;132:1977–1982. doi: 10.1210/endo.132.5.7682938. [DOI] [PubMed] [Google Scholar]
- 337.Jia Y, et al. Steroidogenic enzyme Cyp11a1 regulates Type 2 CD8+ T cell skewing in allergic lung disease. Proc Natl Acad Sci USA. 2013;110:8152–8157. doi: 10.1073/pnas.1216671110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Sher N, Yivgi-Ohana N, Orly J. Transcriptional regulation of the cholesterol side chain cleavage cytochrome P450 gene (CYP11A1) revisited: binding of GATA, cyclic adenosine 3′,5′-monophosphate response element-binding protein and activating protein (AP)-1 proteins to a distal novel cluster of cis-regulatory elements potentiates AP-2 and steroidogenic factor-1-dependent gene expression in the rodent placenta and ovary. Mol Endocrinol. 2007;21:948–962. doi: 10.1210/me.2006-0226. [DOI] [PubMed] [Google Scholar]
- 339.Wei G, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35:299–311. doi: 10.1016/j.immuni.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Horiuchi S, et al. Genome-wide analysis reveals unique regulation of transcription of Th2-specific genes by GATA3. J Immunol. 2011;186:6378–6389. doi: 10.4049/jimmunol.1100179. [DOI] [PubMed] [Google Scholar]
- 341.Chtanova T, Kemp RA, Sutherland AP, Ronchese F, Mackay CR. Gene microarrays reveal extensive differential gene expression in both CD4(+) and CD8(+) type 1 and type 2 T cells. J Immunol. 2001;167:3057–3063. doi: 10.4049/jimmunol.167.6.3057. [DOI] [PubMed] [Google Scholar]
- 342.Wang M, et al. The steroidogenic enzyme Cyp11a1 is essential for development of peanut-induced intestinal anaphylaxis. J Allergy Clin Immunol. 2013;132:1174–1183. doi: 10.1016/j.jaci.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Schedel M, et al. 1,25D3 prevents CD8(+)Tc2 skewing and asthma development through VDR binding changes to the Cyp11a1 promoter. Nat Commun. 2016;7:10213. doi: 10.1038/ncomms10213. [DOI] [PMC free article] [PubMed] [Google Scholar]