

Abstract
After DNA damage, the cell cycle is arrested to avoid propagation of mutations. Arrest in G2 phase is initiated by ATM‐/ATR‐dependent signaling that inhibits mitosis‐promoting kinases such as Plk1. At the same time, Plk1 can counteract ATR‐dependent signaling and is required for eventual resumption of the cell cycle. However, what determines when Plk1 activity can resume remains unclear. Here, we use FRET‐based reporters to show that a global spread of ATM activity on chromatin and phosphorylation of ATM targets including KAP1 control Plk1 re‐activation. These phosphorylations are rapidly counteracted by the chromatin‐bound phosphatase Wip1, allowing cell cycle restart despite persistent ATM activity present at DNA lesions. Combining experimental data and mathematical modeling, we propose a model for how the minimal duration of cell cycle arrest is controlled. Our model shows how cell cycle restart can occur before completion of DNA repair and suggests a mechanism for checkpoint adaptation in human cells.
Keywords: ATM, ATR, checkpoint recovery, G2, Plk1
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
DNA double‐strand breaks (DSBs) represent a serious threat to the genome integrity of a cell. Failure to recognize and repair these lesions can lead to mutations, genome instability, and cancer (Jackson & Bartek, 2009). To cope with DSBs cells launch a DNA damage response (DDR), involving a network of DNA damage sensors, signal transducers, and various effector pathways. Besides orchestrating DNA repair, a central component of DDR is activation of a checkpoint that blocks cell cycle progression (Bartek & Lukas, 2007; Medema & Macurek, 2011). This is particularly important in the G2 phase of the cell cycle, as cell division in the presence of DSBs may lead to aneuploidy and propagation of mutations to progeny. Surprisingly however, a growing body of evidence suggests that a cell cycle block is commonly reversed before all DNA lesions are repaired in both transformed and untransformed cells (Syljuasen et al, 2006; Deckbar et al, 2007; Tkacz‐Stachowska et al, 2011; Loewer et al, 2013). How cells correlate the checkpoint control with the completion of DNA repair and how they control the duration of a cell cycle arrest therefore remain unresolved issues.
Upon recruitment to DSBs, the ataxia telangiectasia mutated (ATM) kinase initiates a signaling cascade by phosphorylating more than 700 proteins, many of which are central proteins in various branches of the DDR (Matsuoka et al, 2007; Mu et al, 2007; Shiloh & Ziv, 2013). However, although crucial for initiating many of the responses, the role for ATM in maintaining a cell cycle arrest remains unclear, as acute inhibition of ATM after a checkpoint is initiated has limited effect on the efficiency of cell cycle resumption (Kousholt et al, 2012). Rather, ATM and Rad3‐related kinase (ATR), activated by the resected DNA ends, is a main controller of checkpoint duration (Sanchez et al, 1997; Shiotani & Zou, 2009).
To enforce a cell cycle arrest in G2, ATM‐ and ATR‐dependent signaling inhibits the activities of mitosis‐promoting kinases as cyclin‐dependent kinase 1 (Cdk1), Polo‐like kinase 1 (Plk1), and Aurora A (Lock & Ross, 1990; Smits et al, 2000; Krystyniak et al, 2006). In particular, ATM, ATR, and p38 activate Chk2, Chk1, and MK2, respectively, structurally distinct kinases that share similar consensus phosphorylation motifs (Reinhardt & Yaffe, 2009). Key targets of Chk1/Chk2/MK2 are Cdc25 phosphatases and their degradation or functional inactivation results in a rapid decrease in Cdk1 activity and suppression of cell cycle progression (Peng et al, 1997; Mailand et al, 2000; Reinhardt et al, 2007). In parallel, Plk1 is inactivated by ATM‐mediated dephosphorylation of Thr210 by phosphatase PP2A/B55α and by ATR‐mediated degradation of Bora, the co‐factor necessary for activation of Plk1 (Qin et al, 2013; Wang et al, 2015; Bruinsma et al, 2017). In addition to this rapid response, a checkpoint is reinforced by activation of p53 and expression of the Cdk inhibitor p21cip/waf and also by premature activation of APC/C‐Cdh1 that results in degradation of multiple proteins including Plk1 (Bunz et al, 1998; de Boer et al, 2016).
Interestingly, not only does ATM/ATR inhibit mitotic kinases, but the mitotic kinases have also been implicated in reversing the checkpoint. Both cyclin‐dependent kinase 1 (Cdk1) and Polo‐like kinase 1 (Plk1) phosphorylate multiple targets in the DDR (van Vugt et al, 2010). In particular, Plk1 promotes checkpoint recovery by inducing degradation of Claspin, a protein required for ATR‐dependent Chk1 activation (Mailand et al, 2006; Mamely et al, 2006; Peschiaroli et al, 2006). Plk1 activation is tightly linked to the central cell cycle engine, in which it in a feedback loop stimulates Cdk1 activity (Macurek et al, 2009). However, whereas Plk1 is redundant for unperturbed mitotic entry, it becomes essential for recovery from a DNA damage arrest in yeast and human cells, suggesting that Plk1‐mediated down‐regulation of the DDR is a key function to allow recovery from a cell cycle checkpoint (van Vugt et al, 2004). Identification of factors that control when Plk1 activity can start to accumulate during a DDR is therefore critical for understanding checkpoint recovery.
Here we show that DNA damage‐induced spread of ATM activity across chromatin prevents Plk1 activation. Activity of ATM is efficiently counteracted by the chromatin‐bound phosphatase Wip1, leading to Plk1 re‐activation despite the presence of active ATM at DNA break sites and active ATR. Based on a mathematical model, we suggest that the G2 checkpoint does not function by monitoring completion of repair but rather that the global ATM/Wip1 balance on chromatin controls the minimal duration of a checkpoint arrest.
Results
ATM kinase activity on chromatin is rapidly reversed
To monitor the DNA damage response pathway in live cells, we constructed ATM/ATR kinase activity reporter (ATKAR), a cellular biosensor that responds to phosphorylation by ATM and ATR. Depending on the type of DNA damage, ATM or ATR could phosphorylate ATKAR, which resulted in a change in Förster resonance energy transfer (FRET) efficiency in the nucleus, where presumably the predominant part of DNA damage signaling occurs (Figs 1A and EV1A–D). The kinetics of nuclear FRET‐ratio change differed between three DNA‐damaging agents in a manner that corresponded well with their different modes of action in causing DNA double‐strand breaks (DSBs). Whereas the topoisomerase II inhibitor etoposide and the nuclease cytolethal distending toxin (CDT) caused a slow accumulation of FRET‐ratio change, the radiomimetic neocarzinostatin (NCS) showed a rapid increase (Fig EV1B and C). Importantly, immunoprecipitation and live cell imaging experiments showed that ATKAR responded to ATM, but not to ATR or DNA‐PK activity after inducing double‐strand breaks with NCS (Figs 1B and EV1D–G). We therefore used ATKAR in combination with NCS to follow ATM activity in single live cells.
Figure 1. ATM kinase activity on chromatin is rapidly reversed.
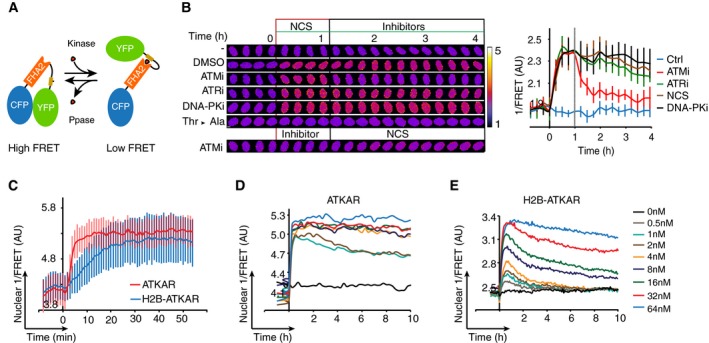
- Schematic outline of FRET‐based probe. Phosphorylation of designated sequence (beige) leads to interaction with the phospho‐binding FHA2 domain (red). This is considered to lead to a conformation change that separates the fluorophores (blue and green), resulting in a decrease in FRET.
- Change in FRET‐ratio of H2B‐ATKAR after neocarzinostatin (NCS) addition depends on ATM. Time‐lapse sequence (left) or quantification of 1/FRET (right) of U2OS cells expressing H2B‐ATKAR. Time point 0 indicates addition of 20 nM NCS, and time point 1 indicates addition of KU60019 (10 μM, ATMi), VE821 (1 μM, ATRi), or NU7026 (10 μM, DNAPKi). Thr‐Ala indicates alanine replacement of the designated phosphoacceptor in H2B‐ATKAR. Graph shows average and SD of at least 15 cells. Scale bars: 8 μm. Heat map indicates 1/FRET (AU).
- Quantification of 1/FRET of a mixed population of H2B‐ATKAR‐ and ATKAR‐expressing U2OS cells after addition of NCS (5 nM). H2B‐ATKAR and ATKAR expressing cells were identified by the localization pattern of the expressed constructs. Graph shows average and SD of median pixel value of at least seven cells. Time point 0 indicates addition of NCS.
- ATKAR signal reaches saturation at ˜4 nM NCS. Quantification of 1/FRET of U2OS cells expressing ATKAR, treated with the indicated NCS concentrations. Graph shows average 1/FRET of ≥ 10 U2OS cells/condition.
- H2B‐ATKAR signal responds in a dose‐dependent manner to NCS addition and is reversed over time. Graph shows average 1/FRET of ≥ 10 U2OS cells/condition.
Figure EV1. A FRET‐based biosensor to monitor the activity of ATM/ATR kinase in live cells.
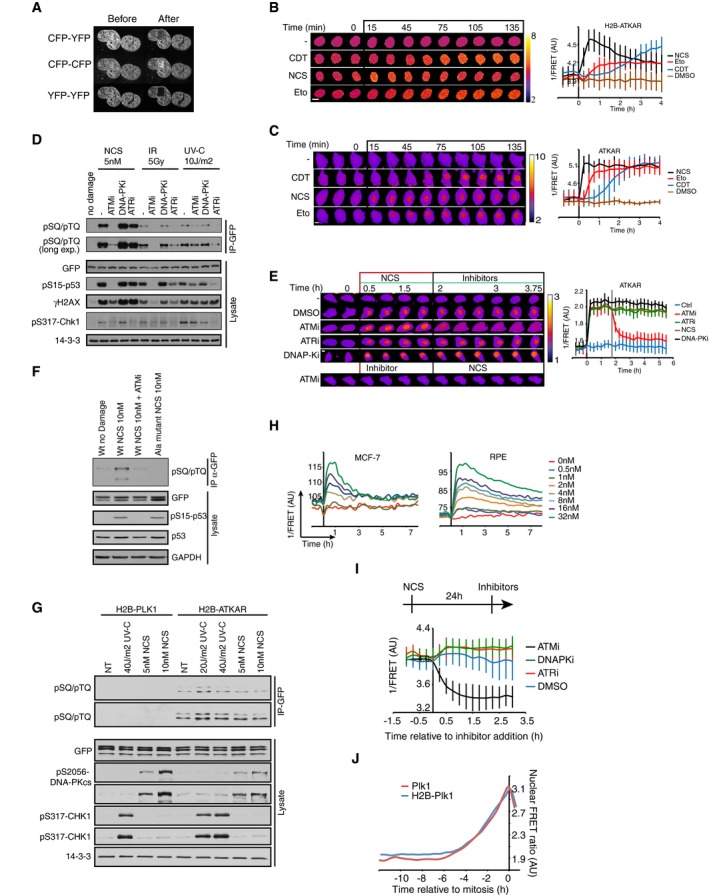
- Acceptor photobleaching of H2B‐ATKAR. U2OS cells expressing H2B‐ATKAR were photobleached using a 514 nm laser, and images were acquired by using CFP‐YFP, YFP‐YFP, and CFP‐CFP excitation‐emission before and after photobleaching. The bleached area is visible in the YFP‐YFP images.
- Kinetics of H2B‐ATKAR 1/FRET change after treatment with etoposide (Eto), neocarzinostatin (NCS) or cytolethal distending toxin (CDT). Time‐lapse sequence (left) or quantification of 1/FRET (right) of U2OS cells expressing H2B‐ATKAR. Graph shows average and SD of at least 15 cells. Time point 0 indicates addition of drugs. Scale bar: 15 μm. Heat map indicates 1/FRET (AU).
- Kinetics of ATKAR 1/FRET change after treatment with etoposide (Eto), neocarzinostatin (NCS), or cytolethal distending toxin (CDT). Time‐lapse sequence (left) or quantification of 1/FRET (right) of U2OS cells expressing H2B‐ATKAR. Graph shows average and SD of at least 15 cells. Time point 0 indicates addition of drugs. Scale bar: 15 μm. Heat map indicates 1/FRET (AU).
- H2B‐ATKAR phosphorylation after NCS addition depends on ATM. GFP pull‐down from U2OS cells expressing H2B‐ATKAR treated with NCS (5 nM) or exposed to IR (5 Gy) or UVC (10 J/m2). Immunoblots were probed with the indicated antibodies.
- Change in ATKAR FRET‐ratio after NCS addition depends on ATM. Time‐lapse sequence (left) or quantification of 1/FRET (right) of U2OS cells expressing ATKAR. Time point 0 indicates addition of 5 nM NCS, and time point 1 indicates addition of KU60019 (10 μM, ATMi), VE821 (1 μM, ATRi), or NU7026 (10 μM, DNAPKi). Graph shows average and SD of at least 7 cells. Scale bar: 10 μm. Heat map indicates 1/FRET (AU).
- GFP pull‐down from U2OS cells expressing ATKAR wild‐type (Wt) or alanine mutant (Ala) treated with NCS (10 nM) in presence or absence of ATM inhibitor. Immunoblot analysis shows the phosphorylation of ATKAR is on the expected target site residue and is ATM‐dependent upon NCS treatment.
- H2B‐ATKAR phosphorylation after NCS treatment depends on ATM. GFP pull‐down from AT cells expressing H2B‐PLK1 or H2B‐ATKAR FRET probe treated with 5–10 nM NCS or 20–40 J/m2 UV‐C. Immunoblots were probed with indicated antibodies. Note that phosphorylation of H2B‐ATKAR is not induced by DNA damage in cells lacking ATM.
- Quantification of 1/FRET of MCF‐7 and RPE cells expressing H2B‐ATKAR treated with the indicated concentrations of NCS. Graph shows average of at least 10 cells per condition.
- ATKAR phosphorylation depends on ATM activity long after NCS addition. U2OS cells expressing ATKAR were treated with 5 nM NCS. The indicated inhibitors were added 24 h later. Graph shows average and SD of at least 15 cells.
- Similar dynamics of phosphorylation of Plk1 sensor on chromatin or nucleoplasm. Quantification of nuclear 1/FRET of U2OS cells expressing either H2B‐Plk1 or Plk1 biosensor. Cells are synchronized upon mitotic entry in silico.
To study ATM activity at different subcellular locations, we either expressed ATKAR as a diffusible protein that does not shuttle rapidly across the nuclear membrane due to its size, or as a histone H2B fusion protein immobilized on chromatin. When comparing ATKAR and H2B‐ATKAR, we noted clear differences in their phosphorylation kinetics (Fig 1C–E). The nuclear response of ATKAR was detected within 2 min after addition of NCS and reached a steady state after 5 min, whereas H2B‐ATKAR rose at a slower pace and started to plateau after 20 min (Fig 1C). Similarly, although both ATKAR and H2B‐ATKAR showed a dose‐dependent response to NCS, ATKAR reached saturation already at 4 nM, whereas H2B‐ATKAR reached similar FRET‐ratio changes only at 32 nM (Fig 1D and E). Nuclear ATKAR signal remained high over time, whereas the H2B‐ATKAR signal started to revert within an hour after NCS addition at all concentrations in U2OS and MCF7 cancer cell lines or untransformed diploid RPE cells, suggesting that ATM‐mediated phosphorylation of an artificial substrate becomes less efficient when the substrate is targeted to chromatin (Figs 1D and E, and EV1H). Importantly, the FRET change of ATKAR and H2B‐ATKAR could be reverted by addition of an ATM inhibitor at various time points in the DDR, suggesting that the differential readout of both probes represent ATM activity (Figs 1B and EV1E and I). In contrast to ATKAR, a Plk1 biosensor showed similar nuclear kinetics whether or not coupled to H2B, indicating that fusion to histone H2B does not directly affect the FRET efficiency of this type of biosensor (Macurek et al, 2008) (Fig EV1J). Thus, the ATM‐dependent phosphorylation kinetics differs between nuclear‐soluble and chromatin‐bound ATKAR, where chromatin‐bound ATKAR is dephosphorylated rapidly after DSB induction.
Activation of Plk1 correlates with dephosphorylation of a chromatin‐bound ATM substrate
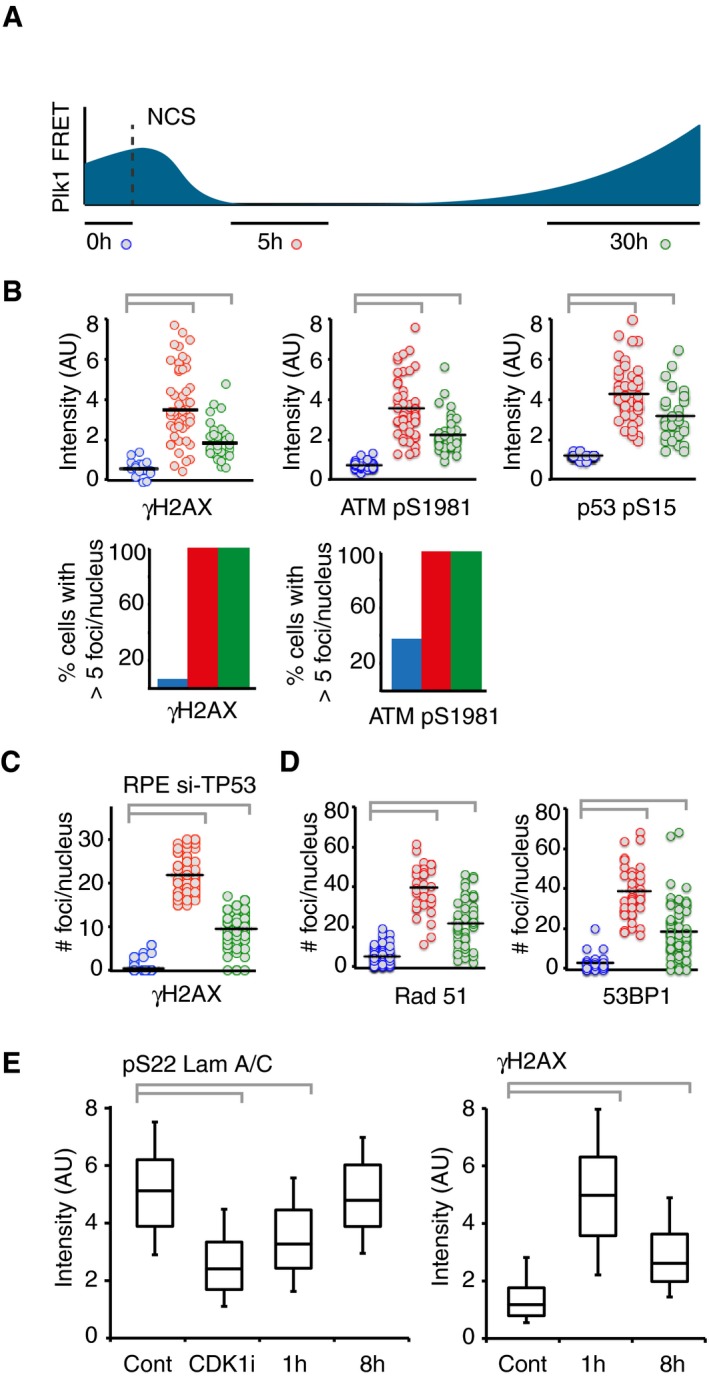
We next sought to follow ATM‐mediated phosphorylation throughout a DDR in single cells. We therefore monitored FRET‐ratios of single U2OS cells from before addition of NCS until recovery from a cell cycle arrest and subsequent mitotic entry. The NCS concentration was chosen to induce a clear cell cycle arrest in all cells, but at the same time allow for a subset of cells to eventually recover and resume cell division (Fig 2A). To focus on the cells that recovered from a cell cycle arrest, we monitored cells that resumed cell division within the first 24–33 h and synchronized them in silico upon mitotic entry. To detect when the cell cycle is restarted in this setup, we simultaneously followed cells expressing a Plk1 FRET probe. To minimize experimental variation, these cells were mixed with H2B‐ATKAR expressing cells and separated based on localization of the FRET probe. Strikingly, we find that Plk1 activity is detected around 15 h before mitotic entry, showing a clear correlation to when H2B‐ATKAR phosphorylation is reversed (Fig 2A). Similarly, in RPE cells depleted of p53 to allow recovery from a checkpoint, the appearance of Plk1 activity correlates with the disappearance of H2B‐ATKAR phosphorylation (Fig 2B). In contrast, ATKAR phosphorylation is sustained until mitotic entry, consistent with the large difference between ATKAR and H2B‐ATKAR also during initiation of a DDR (Figs 1C and 2C). Thus, Plk1 activity is observed once ATM‐dependent H2B‐ATKAR phosphorylation is reversed, consistent with a model in which ATM‐mediated phosphorylation blocks Plk1 activation.
Figure 2. Activation of Plk1 correlates with dephosphorylation of a chromatin‐bound ATM substrate.
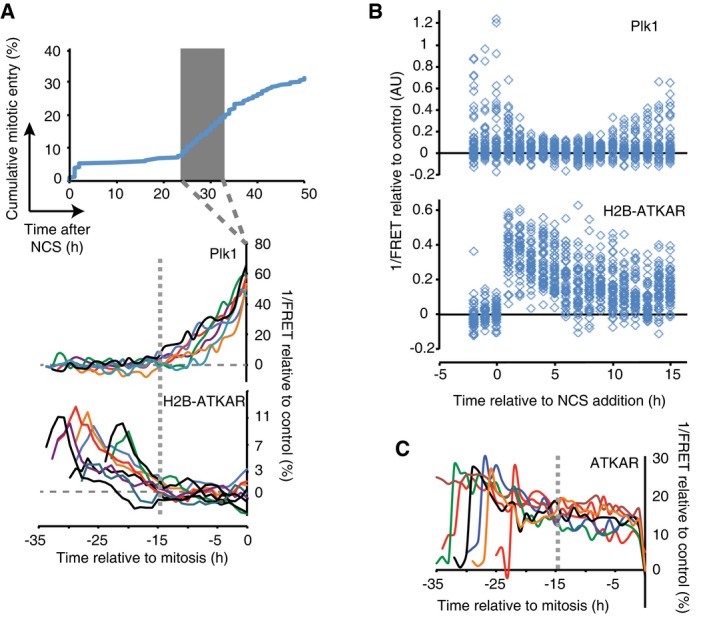
- Reversal of H2B‐ATKAR correlates with resumption of Plk1 activity during cell cycle restart. A mixed population of U2OS cells expressing H2B‐ATKAR or Plk1 FRET probe were treated with 2 nM NCS, and mitotic entry was followed over time (top). Cells entering mitosis 24 to 33 h after NCS addition (gray rectangle) were synchronized in silico on mitosis and 1/FRET of individual cells was quantified (bottom). Gray dotted vertical line indicates 15 h before mitosis.
- Resumption of Plk1 activity correlates with reversal of H2B‐ATKAR phosphorylation. A mixed population of RPE cells expressing H2B‐ATKAR or Plk1 FRET probe were transfected with p53 siRNA and treated with 8 nM NCS. 1/FRET was quantified of at least 41 cells per time point for each probe. H2B‐ATKAR or Plk1 FRET were recognized by their nuclear or whole‐cell localization. Each mark corresponds to one cell.
- ATKAR phosphorylation is sustained until mitotic entry during spontaneous checkpoint recovery. U2OS cells expressing ATKAR were followed during treatment with NCS (2 nM) and 1/FRET of cells spontaneously recovering 24–33 h later were plotted as in (A). Each line represents a single cell synchronized in silico upon mitotic entry. Gray dotted vertical line indicates 15 h before mitosis.
ATM and ATR control Plk1 activity at different time‐scales during a DDR
To test if and when ATM controls Plk1 activation, we added a small molecule inhibitor to ATM at different time points of a DDR. Whereas activity of Plk1 was rapidly reduced in control G2 cells treated with NCS, inhibition of ATM early after NCS addition allowed sustaining high Plk1 activity as determined by the level of pT210‐Plk1 modification (Fig 3A). Similarly, using high‐content imaging of cells expressing a Plk1 activity reporter, G2 cells show intermediate activity and mitotic cells high activity. Upon ATM inhibition early after NCS addition, many cells sustained Plk1 activity (Fig 3B). Interestingly, inhibition of ATR also affected the amount of cells showing Plk1 activity, indicating that both ATM and ATR can control Plk1 activity after NCS addition (Fig 3B).
Figure 3. ATM and ATR control Plk1 activity at different time‐scales during a DDR.
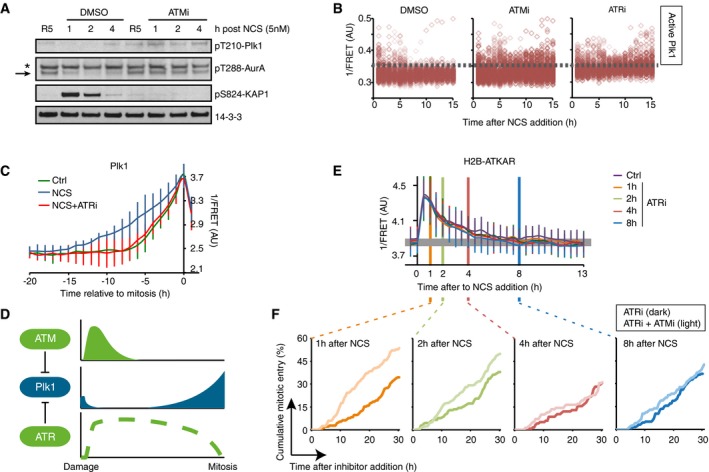
- ATM inhibits Plk1 activity after NCS treatment. RPE cells were synchronized by 2 mM HU for 16 and 5 h after release to fresh media treated with NCS (5 nM) and DMSO or ATMi (10 μM) for indicated times. Antibodies against pT210‐Plk1 and pT288‐Aurora A recognize active forms of Plk1 and Aurora A, respectively. Asterisk indicates a cross‐reacting band. Arrow indicates position of Aurora A.
- ATM activity contributes to Plk1 inhibition early after damage. U2OS cells expressing Plk1 FRET probe were treated with NCS (4 nM) and 15 min later ATMi, ATRi, or DMSO were added. Plots show 1/FRET of ˜500 cells/condition/time point. Dotted line shows approximate threshold below which Plk1 activity is not detected.
- ATR counteracts Plk1 activity after cell cycle restart. Plk1 FRET probe expressing U2OS cells were untreated (Ctrl) or treated with 2 nM NCS followed by DMSO or ATRi. 1/FRET of individual cells entering mitosis was quantified. Graph shows average and SD of 1/FRET from ≥ 10 cells, synchronized in silico on mitosis. Note that the duration of Plk1 activation is longer in cells recovering from DNA damage compared to unperturbed cells, and that the prolonged duration is reverted in the presence of ATR inhibitor.
- Schematic model. Whereas ATR inhibits Plk1 activity throughout a DDR, ATM determines when Plk1 can be activated to promote cell cycle resumption.
- ATR inhibition does not affect H2B‐ATKAR FRET‐ratio after NCS. ATR inhibitor was added to U2OS cells expressing H2B‐ATKAR at the indicated time points after 4 nM NCS addition. Graph shows average and SD of 1/FRET for ≥ 10 cells per condition.
- Synergistic effect of ATM and ATR inhibition early after NCS. Cumulative mitotic entry of U2OS cells after treatment with NCS (1 nM) followed by addition of ATRi (1 μM, dark lines) or ATRi and ATMi (1 and 10 μM, light lines) at different time points as indicated. For comparison, these time points are displayed as vertical lines in (D), where 4 nM NCS is used.
We next sought to investigate Plk1 activity once it restarted to promote recovery from a G2 checkpoint. As we previously reported, Plk1 activity is detected ~5 h before mitosis during both an unperturbed cell cycle and when cells containing damaged DNA are forced to enter mitosis by addition of caffeine (Macurek et al, 2008). However, in the absence of drugs that force a shutdown of the checkpoint, we detected a gradually increasing Plk1 activity from ~15 h before mitosis (Figs 2A and 3C). Thus, accumulation of Plk1 activity is slower during recovery from a DNA damage‐mediated cell cycle arrest compared to unperturbed growth (Liang et al, 2014). Interestingly, we find that inhibition of ATR completely reversed the slow Plk1 activation during spontaneous checkpoint recovery (Fig 3C and Appendix Fig S1A). This suggests a model in which both ATM and ATR control Plk1 activity, but function at different periods during a DDR (Fig 3D).
A prediction of such a model is that inhibition of ATM would only affect the cell cycle when a FRET change in H2B‐ATKAR is detected. In line with published reports, we find that inhibition of ATM simultaneously with induction of DNA damage overrides a checkpoint, whereas ATM inhibition after a checkpoint is instigated does not stimulate mitotic entry (Appendix Fig S1B), likely reflecting ATR activation due to ATM‐stimulated resection of DSBs (Kousholt et al, 2012). We therefore assessed the role of ATM in the presence of an ATR inhibitor. ATR inhibition did not markedly alter the dynamics of H2B‐ATKAR phosphorylation, and all cells that entered mitosis contained dephosphorylated H2B‐ATKAR several hours before recovery occurred (Fig 3E and Appendix Fig S1C). At NCS concentrations that allow spontaneous recovery, H2B‐ATKAR phosphorylation peaks 30 min after NCS addition and gradually declines until it is no longer detected 4–5 h later (Fig 1E). We therefore tested whether ATM inhibition had a synergistic effect on ATR inhibition at different time points after NCS addition. Interestingly, early after NCS addition, combined inhibition of ATM and ATR efficiently stimulated mitotic entry. In contrast, no synergistic effect was detected at later time points when H2B‐ATKAR phosphorylation was largely absent (Fig 3F and Appendix Fig S1B). Thus, our data are in support of a model in which ATM and ATR controls when Plk1 activity is initiated, whereas ATR counteracts Plk1 activity once initiated until cells reach mitosis (Fig 3D).
Cell cycle reactivation precedes complete termination of DDR during checkpoint recovery
ATM is activated at sites of double‐strand breaks, and we next sought to assess whether H2B‐ATKAR dephosphorylation and cell cycle restart corresponds to ATM inactivation at DNA damage foci. We therefore established a setup where we followed the appearance of Plk1 activity in live cells and subsequently fixed and quantified immunofluorescence from the same cells (Fig EV2A). Based on the level of Plk1 activity, we compared unperturbed G2 cells, cells in which Plk1 activity was reduced after NCS addition, and cells that had re‐initiated Plk1 activity after NCS addition (Figs 4A and EV2A). Interestingly, ATM‐dependent phosphorylation of H2AX and p53 as well as autophosphorylation of ATM remained after Plk1 was re‐activated (Fig 4B and C). Moreover, DNA repair proteins 53BP1 and Rad51 were present in nuclear foci after Plk1 re‐activation (Fig 4D). The notion that ATM activity is present at DNA damage foci after re‐activation of Plk1 is consistent with our observation that ATKAR, which is present in nucleoplasm but not targeted to chromatin, showed sustained nuclear ATM activity until mitosis after checkpoint recovery (Figs 2C and EV1I). Similarly, high‐content microscopy showed that Cdk1‐dependent phosphorylation of lamin A/C was present in G2 cells containing γH2AX foci (Figs 4E and EV2B) (Peter et al, 1990; Akopyan et al, 2014). Thus, Plk1 and Cdk1 activities that are both necessary for recovery from a G2 checkpoint can occur despite sustained ATM activity and despite repair proteins being assembled at DNA damage foci.
Figure EV2. As opposed to H2B‐ATKAR phosphorylation, ATM activity is sustained after Plk1 activation.
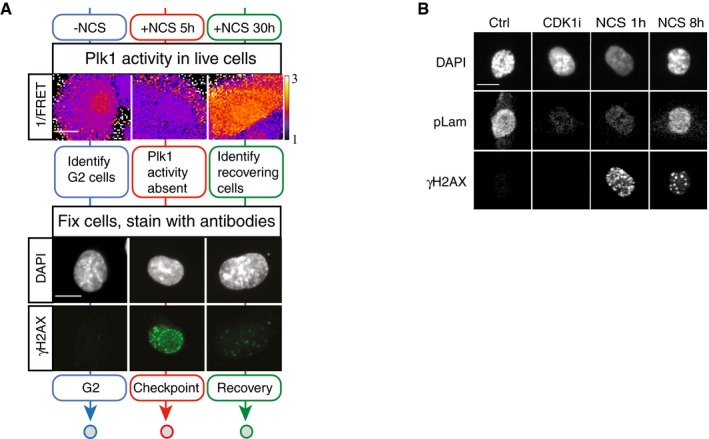
- Example of approach described in Fig 4A. Representative images of live cells depicting Plk1 activity and the same cells fixed and stained for γH2AX and DAPI are shown. Mock treated cells (blue), 5 h NCS (red), or 30 h NCS (green). Scale bar: 15 μm. Heat map indicates 1/FRET (AU).
- Example of images from experiment described and quantified in Fig 4E. Scale bar: 15 μm.
Figure 4. Cell cycle reactivation precedes complete termination of DDR during checkpoint recovery.
- Schematic of approach. U2OS cells expressing Plk1 FRET probe were treated with NCS (2 nM). Before fixation at indicated time points, 1/FRET was followed in individual live cells to detect undamaged G2 cells (0 h, blue), G2 arrested cells without detectable Plk1 activity (5 h, red), and recovering G2 cells with increasing Plk1 activity (30 h, green). After fixation, the corresponding cells were identified both based on position and morphology.
- Quantification of immunofluorescence of cells followed as in (A). Graphs show signal intensity or percentage of cells with nuclear foci detected by indicated antibodies.
- RPE cells expressing Plk1‐FRET were treated with siRNA for TP53 and followed as in (A and B). Times were adjusted to 4.5 h (red) and 17 h (green). Graph shows quantification of γH2AX foci.
- Quantification of immunofluorescence of cells followed as in (A and B). Graphs show amount of nuclear foci detected by indicated antibodies.
- Cdk1 re‐activation in presence of γH2AX phosphorylation. RPE p53−/− cells were pulsed with EdU and treated or not (Cont, Cdk1i) with 8 nM NCS; 1 or 8 h later, cells were fixed and EdU‐negative, 4n DNA content cells (assessed by DAPI intensity) were analyzed for pS22 Lam A/C or γH2AX using quantitative immunofluorescence. RO‐3306 (Cdk1i) was added for 2 h before fixation. The box plots represent the 90th, 75th, 50th, 25th, and 10th percentiles of at least 270 G2 cells per condition.
ATM activity is detected throughout chromatin upon localized DNA damage
Our results indicate that whereas ATM remains active throughout checkpoint recovery, H2B‐ATKAR responds to a subset of ATM activity that is silenced before Plk1 re‐activation. As H2B‐ATKAR is restricted to chromatin due to targeting by histone H2B, the detected ATM activity presumably occurs on chromatin. However, we did not detect enrichment of H2B‐ATKAR activity on DNA damage foci (not shown) and both γH2AX and pS1981 ATM staining persisted on foci after H2B‐ATKAR signal disappeared (Fig 4B and C), which indicates that ATM activity on DNA damage foci is poorly detected by H2B‐ATKAR. As DNA damage foci are restricted in size and histone H2B is present throughout chromatin, we therefore sought to test whether chromatin‐bound H2B‐ATKAR can be phosphorylated by ATM at sites distal to where DNA damage occurred. Upon localized DNA damage induced by laser microirradiation, H2B‐ATKAR detected a pan‐nuclear signal across chromatin, in contrast to γH2AX and BRCA1 that remained restricted in close proximity to DNA lesions (Fig 5A and B, and Appendix Fig S2A–C). During the time‐course of the experiment, there is little exchange of H2B‐ATKAR within chromatin, as YFP remained bleached in the microirradiated area (Fig 5A). However, due to this bleaching, caution is required when interpreting FRET changes within the damaged area. In addition, part of the global H2B‐ATKAR phosphorylation was sensitive to ATR inhibition, indicating that opposite to NCS, laser microirradiation can cause ATR‐dependent phosphorylation of H2B‐ATKAR. Nonetheless, inhibition of ATM abolished the FRET change of H2B‐ATKAR after laser microirradiation, indicating that ATM activity can reach chromatin far from the damaged area (Fig 5C). Taken together, this suggests that H2B‐ATKAR can detect phosphorylation across chromatin.
Figure 5. ATM activity is detected throughout chromatin upon localized DNA damage.
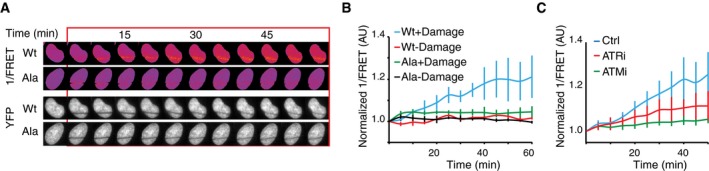
- H2B‐ATKAR 1/FRET spreads over chromatin after localized damage. U2OS cells expressing H2B‐ATKAR were laser‐microirradiated, and 1/FRET was followed over time. Note that bleaching of fluorophores precludes FRET analysis within the laser‐microirradiated area.
- Quantification of spread of H2B‐ATKAR (wt) or non‐phosphorylatable Ala‐H2B‐ATKAR (Ala) FRET change after laser microirradiation in U2OS cells. Measurements were performed distal to the laser‐microirradiated area. Graph shows average and SD of at least seven cells.
- H2B‐ATKAR detects both ATM and ATR activity after laser microirradiation. Quantification of spread of H2B‐ATKAR FRET change after laser microirradiation in U2OS cells in the presence of ATM or ATR inhibitors. Inhibitors were added 30 min before laser microirradiation. Graph shows average and SD of at least five cells. Measurements were performed as in (B).
Wip1 phosphatase counteracts ATM activity at chromatin
We next sought to identify why ATM‐dependent phosphorylation of H2B‐ATKAR on chromatin is more rapidly reverted compared to phosphorylation of diffusible ATKAR. We reasoned that a chromatin‐associated phosphatase could account for the difference in phosphorylation between ATKAR and H2B‐ATKAR and analyzed effects of expression or depletion of PPM1D (referred to as Wip1), which is known to counteract ATM‐mediated phosphorylations including γH2AX and ATM‐pS1981 (Shreeram et al, 2006; Yamaguchi et al, 2007; Macurek et al, 2010). Indeed, expression of mCherry‐Wip1 efficiently suppressed the H2B‐ATKAR signal after NCS treatment, but less efficiently counteracted the diffusible ATKAR (Fig 6A). Conversely, depletion of Wip1 by shRNA‐mediated RNAi increased the H2B‐ATKAR signal after addition of NCS and delayed its reversal during the recovery (Fig 6A and B). Similarly, overexpression or depletion of Wip1 blocked or potentiated, respectively, the spreading of ATM activity at chromatin after localized DNA damage caused by laser microirradiation (Fig 6C and D). Previously, we have shown that Wip1 counteracts a p53‐mediated cell cycle exit and allows cells to maintain recovery competence during the G2 checkpoint arrest (Lindqvist et al, 2009). Here, we noted that Wip1‐depleted cells failed to revert the H2B‐ATKAR signal and did not enter mitosis despite the presence of an ATR inhibitor (Fig 6E). This suggests that Wip1 controls when ATM‐mediated signaling on chromatin is reversed to allow initiation of a cell cycle restart (Fig 6F).
Figure 6. Wip1 counteracts ATM activity at chromatin.
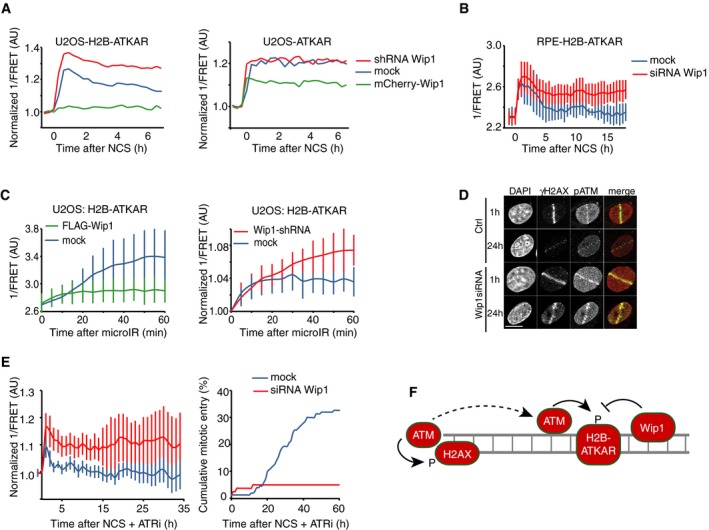
- Quantification of 1/FRET of mixed populations of U2OS cells expressing H2B‐ATKAR or ATKAR. Cells were mock transfected or transfected with Wip1 shRNA or mCherry‐Wip1 for 48 h and treated with NCS (8 nM). Graph shows average of ≥ 8 cells.
- Quantification of 1/FRET of RPE‐H2B‐ATKAR transfected with control or Wip1 siRNA treated with NCS (8 nM). Graph shows average of ≥ 8 cells; error bars indicate SD.
- Wip1 influences the spread of H2B‐ATKAR 1/FRET change. U2OS‐H2B‐ATKAR cells were transfected with mock (blue), FLAG‐Wip1 (green), or Wip1 shRNA (red) and microirradiated with 364 nm laser. 1/FRET distal to the damaged area was quantified. Graph shows average and SD of at least five cells.
- pS1981‐ATM is present throughout chromatin and counteracted by Wip1. U2OS cells were transfected with control or Wip1 siRNA, fixed after 1 or 24 h after microirradiation with 364 nm laser, and co‐stained for γH2AX and pS1981‐ATM. Scale bar: 15 μm.
- Wip1‐depleted cells do not enter mitosis in presence of ATRi. 1/FRET (left) and cumulative mitotic entry (right) were measured in U2OS cells expressing H2B‐ATKAR transfected with mock or Wip1 shRNA treated with NCS (4 nM) and ATRi. Graph shows average of 10 cells. Error bars indicate SD.
- Schematic model. Rather than DNA damage foci, H2B‐ATKAR signal detects ATM/Wip1 balance throughout chromatin.
ATM activity at chromatin determines the minimal duration of a checkpoint
To study possible implications of our findings, we assembled a simplified mathematical model in which ATM‐, Wip1‐, ATR‐, and Plk1‐dependent pathways are treated as functional entities (Fig 7A). The Plk1‐dependent pathways represent mitosis‐promoting cell cycle activities, and a high level of these activities is used as readout for mitosis (Fig 7B, top graph). The suppression of Plk1 by ATR represents multiple pathways, including ATR‐mediated phosphorylation of Bora and Chk1‐mediated phosphorylation of Wee1 (Lee et al, 2001; Qin et al, 2013). Similarly, the suppression of ATR by Plk1 represents many possible interactions, including phosphorylation of Claspin by Plk1 (Mailand et al, 2006; Mamely et al, 2006; Peschiaroli et al, 2006). Although we do not exclude that the cell cycle affects ATM‐dependent pathways, as for example through Chk2 or 53BP1 (van Vugt et al, 2010), we find no evidence for a major effect of Plk1 in regulating ATM activity (Fig EV3A).
Figure 7. ATM activity at chromatin determines the minimal duration of a checkpoint.
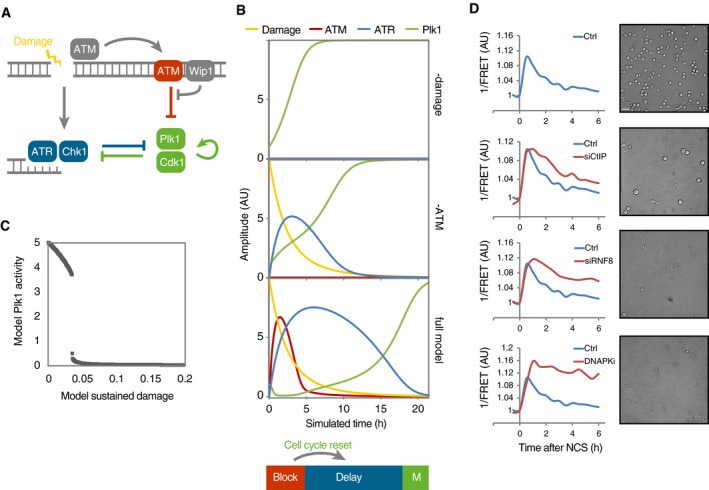
- Schematic outline of simplified mathematical model. Arrows represent differential equations.
- Simulation of model in the absence of damage (top) or ATM (middle) or containing all components (bottom). ATM activity on chromatin functions as a barrier that blocks Plk1 activity. Wip1 efficiently counteracts ATM‐mediated phosphorylations, which restricts ATM activity throughout chromatin to high damage levels. After reversal of the barrier, cell cycle signaling eventually overcomes ATR‐dependent activities, despite the presence of unrepaired DNA breaks. Due to the reset cell cycle activities (compare Plk1 and ATR in middle and lower graphs), a delay is introduced before ATR activities are overcome and mitosis occurs.
- Cell cycle progression depends on a threshold level of damaged DNA. Simulation of optimized model (Fig EV3C), set so that after the initial DNA damage 2, a proportion of damage is not repaired. Graph shows simulation to steady state for the indicated range of sustained DNA damage levels.
- Interference with DNA repair processes delays dephosphorylation of H2B‐ATKAR and mitotic entry. U2OS cells expressing H2B‐ATKAR were treated with indicated siRNAs or DNA‐PK inhibitor (NU7441, 2 μM), and 1/FRET was quantified in 10 cells after addition of 8 nM NCS (left). In parallel, mitotic cells were visualized by microscopy 24 h after treatment with 2 nM NCS and nocodazole (right). Scale bar: 50 μm.
Figure EV3. Checkpoint response with decreasing amounts of DNA damage.
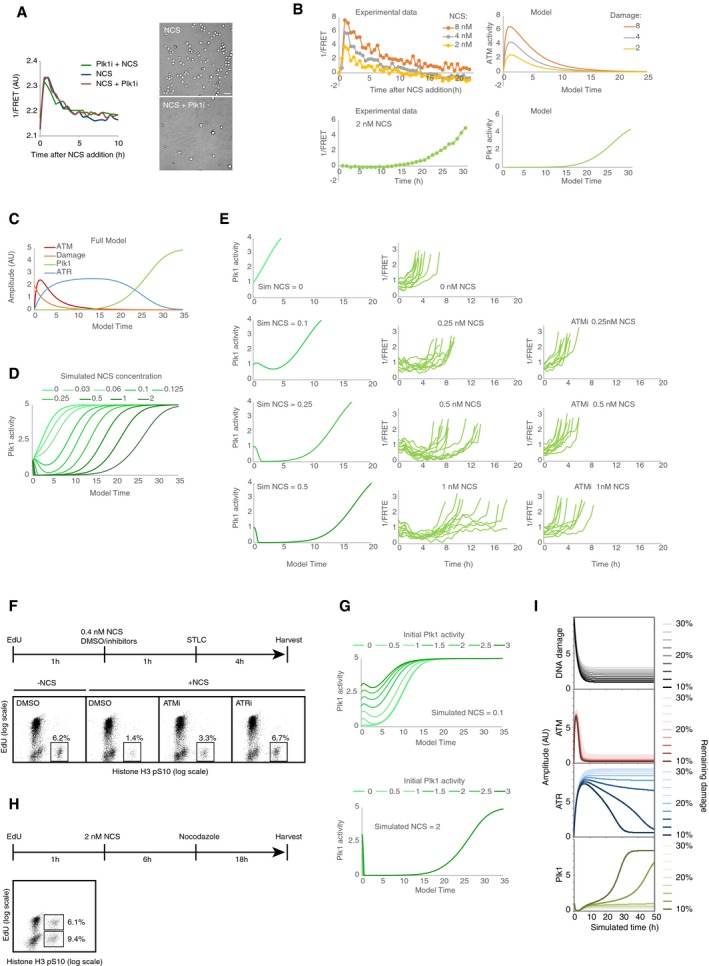
- Plk1 inhibition has no major effect on H2B‐ATKAR. Left, quantification of 1/FRET of at least 16 U2OS cells expressing H2B‐ATKAR after addition of 2 nM NCS. Plk1 inhibitor BI2536 was added 1 h before or immediately after NCS. Right, mitotic cells were visualized by microscopy 24 h after treatment with 2 nM NCS and nocodazole. Scale bar: 50 μm.
- Fitting of model parameters to experimental data of U2OS cells. Top, model ATM activity was fitted to H2B‐ATKAR signal after NCS addition. Left graph shows average signal of at least 100 cells per condition. Bottom, model Plk1 activity was fitted to Plk1 FRET signal during recovery from NCS treatment. Left graph shows average signal of nine cells entering mitosis 27–32 h after NCS addition, synchronized in silico upon mitotic entry.
- Model after fitting experimental data from (B).
- Simulation of model using decreasing levels of DNA damage (corresponding to 0–2 nM NCS). Graph shows Plk1 activity. Note delayed activation of Plk1 activity after lower levels of DNA damage and full reversal of Plk1 activity after higher doses of damage.
- Plk1 activity in recovering cells after NCS treatment. Cells were exposed to the indicated NCS concentrations, in the presence or absence of ATM inhibitor. Individual cells that showed a low level of Plk1 FRET signal upon NCS addition (indicating early G2 phase) and that subsequently entered mitosis were followed over time. Left, model predictions of Plk1 activity in early G2 cells after NCS addition. The model shows a factor 2 deviation from experimental data, possibly partly due to batch‐to‐batch variation in NCS potency.
- ATM and ATR controls recovery from a G2 checkpoint. U2OS cells were treated as indicated in schematic. Graphs show percentage of EdU‐negative, pHistone H3‐positive cells analyzed by flow cytometry.
- Simulation of model using low (0.1 nM NCS, top) or high (2 nM NCS, bottom) amounts of DNA damage at different initial activities of Plk1.
- Both S‐phase and G2 cells exposed to NCS recover from DNA damage. U2OS cells were treated as indicated in schematic and analyzed by flow cytometry. Graphs show percentage of EdU‐positive or EdU‐negative cells that are positive for pHistone H3.
- Cell cycle progression depends on a threshold level of damaged DNA. Simulation of model depicted in Fig 7A and B, but set so that 10–30% of initial damage is not repaired, as shown in top graph. Amount of remaining damage is color‐coded as indicated to the right. Above a threshold level of damage, ATM activity remains sufficiently high to ensure that ATR activity will be maintained and the cell cycle restart will not occur.
Simulating this model in the absence of ATM, we found that ATR prolonged the duration before high cell cycle activities developed. However, although the balance between ATR and cell cycle‐promoting activities determined the duration of a cell cycle arrest, the rising self‐amplifying cell cycle activities eventually forced inactivation of ATR and subsequent mitotic entry (Fig 7B, middle graph). In the full model, ATM activity was present when damage was high, and was rapidly suppressed by Wip1 when damage declined. Importantly, the resulting pulse of ATM activity heavily suppressed cell cycle activities, thereby ensuring that cell cycle activities started from a low level once ATM activities were countered by Wip1 (Fig 7B, lower graph). The lower cell cycle activities resulted in less efficient suppression of ATR‐dependent activities, which in turn resulted in a delay before these ATR‐dependent activities could be inactivated (Fig 7B, middle and lower graphs). Thus, in this simulation, a pulse of ATM activity resets cell cycle signaling, thereby ensuring an extended delay before mitosis occurs.
The duration of this delay is likely influenced by factors not included in the simplified model, as p53‐ and p38‐dependent activities may influence the self‐amplifying buildup of cell cycle regulators (Bunz et al, 1998; Reinhardt et al, 2010). Similarly, the spatial separation of ATM activation on DNA breaks and ATM function throughout chromatin, where Wip1 phosphatase efficiently dephosphorylates ATM targets, may result in more rapid dephosphorylation of ATM. Nonetheless, the model describes a framework for how ATM‐ and ATR‐dependent signaling determines the minimal duration of a checkpoint.
A threshold of ATM activity controls whether checkpoint recovery can occur
We next sought to test the validity of the model. To this end, we used time‐resolved data of H2B‐ATKAR and Plk1 reporter to estimate model constants in U2OS cells (Fig EV3B and C), and compared simulation of decreasing amount of double‐strand DNA breaks to experimental data. In both the model and analysis of Plk1 FRET, ATM did not completely suppress Plk1 activity at low levels of DNA damage, but rather imposed a delay before an increase in Plk1 activity resumed (Fig EV3D and E). Correspondingly, once resumed, the steepness of Plk1 activation increased, leading to a shorter arrest in G2 phase (Fig EV3D and E). Thus, similar mechanisms that govern the minimal duration of a G2 arrest are present after both high and low amounts of DNA damage (Fig EV3F). However, one key difference is that at low initial amounts of DNA damage Plk1 activity is not completely suppressed. As a consequence, Plk1 will affect ATR‐mediated signaling throughout the response and the initial Plk1 activity will influence the duration of an arrest (Fig EV3G). Due to the reset of cell cycle activities at higher damages, the initial Plk1 activity will have little impact on the duration of a delay (Fig EV3G). Indeed, after damages that impose a significant delay before Plk1 activity can resume, both cells receiving damage in S and G2 phases recover, presumably due to that S‐phase cells progressed to G2 before ATM activity decreased (Figs 2A and EV3H).
We next used the simplified model to test how sustained DNA damage affects a cell cycle arrest. In the model, damage that sustains ATM activity will lead to a steady state with intermediate ATM‐mediated phosphorylation, low cell cycle activity, and sustained ATR activation (Fig EV3I). Importantly, the appearance of a steady state with low cell cycle activities indicates the presence of a threshold of DNA damage. Above this threshold cells will not enter mitosis, whereas below this threshold, Plk1 activity can start to increase and eventually silence the checkpoint (Fig 7C). To test this prediction, we identified three treatments that prolonged H2B‐ATKAR signal after NCS addition: depletion of CtIP or RNF8 by siRNA, and inhibition of DNA‐PK (Fig 7D). We found that mitosis was delayed after these three treatments, indicating that sustained ATM activation can block mitosis (Fig 7D). However, we do detect phosphorylated ATM, H2AX, and p53 in cells that have restarted Plk1 and Cdk1 activity (Fig 4B and E), indicating that a low level of active ATM is not sufficient to block mitosis. These data are consistent with a threshold of ATM activity determining whether cells will recover from a checkpoint (Fig 7C).
The notion of a threshold is in line with our finding that repair factors are present at DNA damage foci after Plk1 activation (Fig 4D). Similarly, we do not detect more than 20 γH2AX foci in RPE cells that have restarted Plk1 activity (Fig 4C), in agreement with the report that after radiation, fibroblasts can enter mitosis with up to 20 γH2AX foci (Deckbar et al, 2007). We propose that spread of ATM activity on chromatin has two functions. First, it enforces a checkpoint arrest above a threshold of DSBs. Second, by re‐setting cell cycle activities it ensures that once DSB repair has decreased below this threshold, there will be a delay before cell cycle activities can rise to eventually override ATR‐mediated signaling. The ATM‐dependent barrier thereby ensures a minimal duration between its own silencing by Wip1 and mitotic entry. Thus, although a cell cannot efficiently sustain a G2 checkpoint in the presence of low amounts of damage, ATM activity ensures a considerable delay in the cell cycle during which DNA repair can occur.
Kap1 is an ATM/Wip1 target on chromatin
We next sought to assess through which endogenous substrates ATM enforces a barrier to cell cycle restart. We found that similar to H2B‐ATKAR, DNA damage‐induced phosphorylation of Kap1‐S824 and SMC3‐S1083 spread throughout chromatin in an ATM‐dependent manner and rapidly declined due to dephosphorylation by Wip1 (Figs 8A and B, and EV4A and B). This is in accordance with previously reported chromatin‐wide effects of Kap1 (also known as TRIM28 or TIF1B) and SMC3 after DNA damage (Ziv et al, 2006; Kim et al, 2010). Focusing on Kap1, we found that Wip1 interacted with Kap1 and efficiently dephosphorylated Kap1 S824 in vitro and in situ (Figs 8C and D, and EV4C and D). Moreover, combined inhibition of ATM and depletion of Wip1 sustained Kap1 S824 phosphorylation, indicating that the observed effect on S824 phosphorylation was not caused by increased ATM activity in the absence of Wip1 (Fig EV4E). In addition to Wip1, we found that PP4 to some extent contributed to Kap1 dephosphorylation in untransformed RPE cells (Fig EV4F and G) (Bulavin et al, 2002; Rauta et al, 2006; Lee et al, 2012). In contrast to other ATM targets including γH2AX and pS15‐p53, Kap1 S824 was dephosphorylated before cell cycle resumption was initiated (Figs 4B and 8E and F). Thus, phosphorylation of Kap1 S824 and H2B‐ATKAR is both regulated by ATM and Wip1 and follows similar spatiotemporal dynamics. Whereas overexpression of the wild‐type Kap1 did not impact on the recovery of cells from the checkpoint, overexpression of the Kap1‐S824A mutant stimulated cell cycle restart after DNA damage, showing that phosphorylation of Kap1 can influence the duration of a DDR (Fig 8G). This finding is in agreement with previously reported function of Kap1 phosphorylation in transcriptional repression of multiple genes including p21 and in chromatin relaxation (Li et al, 2007; Lee et al, 2012). Taken together, ATM‐ and Wip1‐dependent regulation of Kap1 contributes to determine the duration of a cell cycle arrest, most likely in concert with other ATM and Wip1 targets.
Figure 8. Kap1 is an ATM/Wip1 target on chromatin.
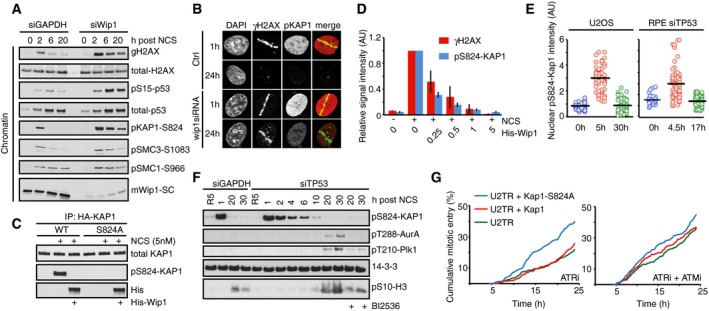
- U2OS cells transfected with GAPDH or Wip1 siRNA were treated with NCS (5 nM) and collected after 2, 6, and 20 h. Chromatin fractions were probed with indicated antibodies.
- RPE cells transfected with Wip1 siRNA were microirradiated, fixed 1 or 24 h later, and stained with the indicated antibodies.
- HA‐KAP1‐WT or HA‐KAP1‐S824A were immunopurified from cells exposed to NCS, incubated with His‐Wip1 and probed with pS824‐KAP1 or KAP1 antibody.
- U2OS cells were fixed 1 h after treatment with NCS, incubated with His‐Wip1 (0–5 ng/μl) and probed for γH2AX and pS824‐KAP1. Plot shows mean nuclear fluorescence intensity of at least 100 cells per condition; error bars indicate SD.
- Kap1 is dephosphorylated before Plk1 activation. RPE cells transfected with TP53 siRNA and U2OS cells were followed as in Fig 4A and stained for pS824‐Kap1. G2 cells (0 h, blue), G2‐arrested cells without detectable Plk1 activity (5 h, red), and recovering G2 cells with increasing Plk1 activity (30 h, green). For RPE cells, the times were modified as indicated. Black bars indicate median and circles correspond to individual cells.
- RPE cells transfected with GAPDH or TP53 siRNA were synchronized by HU, released to fresh media for 5 h (R5), and treated with NCS for indicated times. Nocodazole (NZ) was added 1 h after NCS. Where indicated, cells were incubated in the presence of BI2536. Whole‐cell lysates were probed with indicated antibodies. Antibodies against active forms of Plk1 and Aurora A kinase and to a mitotic marker pS10‐H3 were used to determine when cells recover from the checkpoint arrest.
- Overexpression of Kap1‐S824A phenocopies ATM inhibition. Cumulative mitotic entry of ≥ 300 U2OS cells expressing inducible HA‐tagged Kap1‐wt (red) or Kap1‐S824A (green) after treatment with NCS (4 nM) and subsequent treatment after 1 h with ATRi or ATRi + ATMi.
Figure EV4. Kap1 is an ATM/Wip1 target on chromatin.

- RPE cells were treated with NCS (8 nM) and collected after 1–8 h. Soluble and chromatin fractions were probed with indicated antibodies.
- U2OS or RPE cells were transfected with Wip1 siRNA, microirradiated, and stained after 1 or 24 h with the indicated antibodies. Scale bars: 8 μm or 15 μm (middle).
- U2OS and RPE cells were microirradiated in the presence of indicated inhibitors. After 1 h, cells were stained with the indicated antibodies. Scale bar: 10 μm.
- EGFP or EGFP‐Wip1 was immunoprecipitated from HEK293 cells using GFP‐Trap. Endogenous KAP1 and p53 were probed with antibodies. Coomassie Brilliant Blue (CBB).
- U2OS cells transfected with GAPDH or Wip1 siRNA were treated with NCS in combination with DMSO or ATMi for indicated times. Whole‐cell lysates were probed with indicated antibodies.
- U2OS cells transfected with GAPDH, Wip1, or PP4C siRNA were treated with NCS for indicated times. Soluble and chromatin fractions were probed with indicated antibodies.
- RPE cells transfected with GAPDH, Wip1, or PP4C siRNA were treated with NCS for indicated times. Whole‐cell lysates were probed with indicated antibodies.
Discussion
Our results suggest that the duration of a cell cycle arrest induced by DSBs in G2 phase is determined by three principal components. First, an ATM‐dependent signal that efficiently blocks cell cycle progression. This signal is not affected by cell cycle regulators, but is rapidly reversed by the phosphatase Wip1, likely complemented by other phosphatases. Second, an ATR‐dependent signal that counters cell cycle progression throughout the checkpoint. And third, the self‐amplifying mitotic entry network that counters the ATR‐mediated pathway in G2 phase, eventually leading to checkpoint recovery.
Plk1 and Cdk1 activity rise through G2 phase due to the self‐amplifying properties of the mitotic entry network (Macurek et al, 2009; Akopyan et al, 2014). Maintaining a self‐amplifying activity at a constant level is not a trivial task for a cell. Indeed, we recently found that during a DDR, cyclin B1 levels over time accumulate to levels far higher than is observed during an unperturbed cell cycle (Mullers et al, 2014). Thus, although ATR‐mediated activities slow down cell cycle progression, G2 activities may ultimately prevail and induce mitotic entry. In particular, Plk1 phosphorylates a large number of proteins involved in both DDR and cell cycle control and is essential for recovery from a cell cycle arrest in G2. By inhibiting Plk1 activity, ATM not only solidly enforces a cell cycle block, but also ensures that mitotic entry will be postponed to long after the ATM‐dependent signal is reversed.
The spatial regulation of the DDR on chromatin has recently attracted considerable attention (Altmeyer & Lukas, 2013). ATM activity is efficiently induced at damage sites, where it triggers establishment of DNA damage‐induced foci. A determinant of foci formation is ATM‐mediated phosphorylation of H2AX, which remains restricted to the damaged area, at least in part by insulation mediated by Brd4 (Floyd et al, 2013). Although DNA damage foci are likely to be crucial for repair and amplification of the DDR, the localized DNA damage needs to be communicated to the cell cycle machinery at a cellular scale. Here, we show that ATM activity is differentially maintained at distinct subnuclear compartments. Nucleoplasmic activity of ATM persists throughout the recovery, possibly because the counteracting Wip1 phosphatase is absent from the soluble nuclear fraction (Macurek et al, 2010) and ATM is inactivated by cytosolic phosphatases after nuclear envelope breakdown. Similarly, ATM in the foci remains active after the recovery has been initiated as documented by the rising Plk1 and Cdk1 activity. ATM present in the foci is likely continuously activated by persisting DNA lesions or possibly Wip1 may not be sufficient to abolish ATM activity in DNA damage foci until damage is repaired. In contrast, ATM activity present at undamaged chromatin distal to damage sites blocks cell cycle progression and is inactivated by Wip1 before the checkpoint recovery is initiated. In this sense, ATM's stimulatory effect on DNA repair may be sustained, while ATM's effect on the cell cycle is reversed. We suggest that the spatial separation of ATM activation and inactivation may be a powerful manner for simultaneously sustaining and terminating different aspects of ATM signaling.
In unicellular organisms, a checkpoint can eventually be overcome, despite that not all DNA lesions are repaired. In budding yeast, this process called adaptation depends heavily on the Plk1 homolog cdc5, which counteracts activating phosphorylation of the checkpoint kinase Rad53 (Toczyski et al, 1997; Donnianni et al, 2010). In contrast, multicellular organisms have evolved mechanisms that promote apoptosis or terminal cell cycle arrest, largely dependent on p53 (Bartek & Lukas, 2007; Belyi et al, 2010). We find that albeit the kinetics may differ, ATM‐Wip1‐Kap1 proteins function similarly in U2OS and RPE cells. However, untransformed RPE cells are more likely to terminally exit the cell cycle in G2, rather than to recover from DNA damage. Strikingly, the duration of the ATM activity present across chromatin overlaps with the time required for a DNA damage‐mediated cell cycle exit to become irreversible (Krenning et al, 2014; Mullers et al, 2014). We suggest that these mechanisms are inherent to all cells, but that they will only become apparent after inactivation of the p53 pathway. Of note, our model also elucidates the previously unexplained observations that cells can enter mitosis in the presence of low levels of damaged DNA (Lobrich & Jeggo, 2007). Such revival of adaptation may have deleterious consequences in multicellular organisms since segregation of unrepaired DNA during mitosis may cause aneuploidy and cancer.
Materials and Methods
Construction of ATM/ATR kinase activity reporter (ATKAR)
ATKAR is based on an established Plk1 biosensor (Fuller et al, 2008; Macurek et al, 2008), where the Plk1 target sequence from the parental biosensor was replaced with EPPLTQEI sequence derived from the residues 11–18 of human p53, a well‐established substrate of ATM and ATR kinases. To promote binding of the target site by FHA2 domain in the biosensor, the serine corresponding to Ser15 in p53 sequence was replaced by threonine and residue at +3 position was changed to isoleucine (Durocher et al, 2000). The DNA fragment corresponding to ATKAR was inserted in HindIII/XbaI sites of pcDNA4 plasmid. To generate H2B‐ATKAR, DNA fragment carrying H2B sequence was cloned in‐frame with ATKAR into the HindIII site. In contrast to a previously described ATM biosensor (Johnson et al, 2007), the FRET‐ratio change observed after NCS addition was largely reversed upon ATM inhibition, indicating that phosphorylation of ATKAR is reversible and depends on ATM activity also after initiation of a DDR.
Plasmids
Plasmid carrying human HA‐KAP1 was obtained from Addgene (ID: 45569; Liang et al, 2011). The S824A mutant of HA‐KAP1 was generated by Site‐directed mutagenesis kit (Agilent Technologies), and mutation was confirmed by sequencing. Fragments corresponding to HA‐KAP1‐WT and HA‐KAP1‐S824A were cloned into pcDNA4/TO plasmid. pRS‐Wip1 plasmid for shRNA‐mediated knockdown of Wip1 was described previously (Lindqvist et al, 2009).
Cell culture and transfections
U2OS, MCF‐7, and HEK293 cell lines and AT fibroblasts (GM05849, Coriell Institute) were cultured in Dulbecco's modified Eagle's medium (DMEM) + GlutaMAX (Life Technologies) supplemented with 6 or 10% heat‐inactivated fetal bovine serum, respectively (FBS, Hyclone), and 1% Penicillin/Streptomycin (Hyclone) at 37°C and 5% CO2. hTERT‐RPE1 cell lines were cultured in DMEM/Nutrient mixture‐F12 medium (DMEM/F12) + GlutaMAX (Life Technologies) supplemented with 10% heat‐inactivated fetal bovine serum and 1% Penicillin/Streptomycin (Hyclone) at 37°C and 5% CO2. To generate cells stably expressing ATKAR or H2B‐ATKAR, cells were transfected by linearized plasmids and selected by Zeocine for 3 weeks. For RNA interference experiments, cells were seeded at a density of 8,000 cells/well and transfected with SMARTpool ON‐TARGET plus siRNAs (20 nM, Dharmacon) targeting Wip1, ATR, CtIP, and p53, or RNF8 (20 nM, Thermo Scientific—GAGGGCCAAUGGACAAUUAUU) using HiPerFect (Qiagen) and OptiMEM (Life Technologies) at 48 h before analysis of phenotypes. Alternatively, cells were transfected with Silencer Select siRNA (5 nM, Life Technologies) targeting GAPDH (control), Wip1 (CGAAAUGGCUUAAGUCGAA), TP53 (GUAAUCUACUGGGACGGAA), or PP4C (UCAAGGCCCUGUGCGCUAA) using RNAiMAX (Life Technologies) and cells were analyzed 48 h after transfection. U2OS cells expressing KAP1 or KAP1‐S824A upon tetracycline induction were generated as described previously (Macurek et al, 2008). Where indicated, hRPE cells were synchronized in S‐phase by hydroxyurea (2 mM, 16 h), released to fresh media for 5 h to allow progression to G2 and treated with NCS for indicated times. To genetically inactivate TP53, hTERT‐RPE1 cells were transfected with a combination (1:1) of p53 CRISPR/Cas9 KO plasmid (coding for gRNA to the targeting sequences CCGGTTCATGCCGCCCATGC and CCCCGGACGATATTGAACAA) and a corresponding p53 HDR plasmid (Santa Cruz, sc‐416469) and stable clones were selected by treatment with puromycin (10 μg/ml) for 2 weeks. Correct targeting of both alleles of TP53 was confirmed by sequencing and loss of p53 expression by immunoblotting.
Live cell microscopy and Image processing
For live cell imaging, 8,000–10,000 cells were seeded in 96‐well imaging plates (BD Falcon) 24 h before imaging in CO2‐independent medium (Leibovitz's L15—Invitrogen) supplemented with 6 or 10% heat‐inactivated fetal bovine serum. Cells were followed on either a DeltaVision Spectris imaging system (Applied Precision) using a 20×, NA 0.7 objective, a Leica DMI6000 Imaging System using a 20×, NA 0.4 objective, or on an ImageXpress system (Molecular Devices) using a 20×, NA 0.45 objective. Images were processed and analyzed using ImageJ (http://rsb.info.nih.gov/ij/) or using custom written Matlab scripts. 1/FRET was quantified as the ratio of YFP emission–YFP excitation and CFP excitation–YFP emission as described previously (Hukasova et al, 2012). Unless stated otherwise, the median pixel value of the inversed nuclear FRET‐ratio was used. For spontaneous recovery of H2B‐ATKAR‐expressing cells, a moving average of 3 or 4 time points is shown.
Microirradiation
U2OS or RPE cells, grown on MatTek glass bottom dish (MatTek Corporation), were treated with 10 μM BrdU (Sigma) for 24 h. For microirradiation, the dish was mounted on stage of Leica DMI 6000B microscope stand (Leica) integrated with a pulsed nitrogen laser (20 Hz, 364 nm, Micropoint Ablation Laser System) that was directly coupled to the epifluorescence path of the microscope and focused through a Leica 40× HCX PL APO/1.25–0.75 oil‐immersion objective. Typically, 50 cells were microirradiated (150× 1pixel) in Leibovitz's L15 medium at 37°C, after which cells were either followed for 1 h or returned to incubator at 37°C to recover. Cells were fixed 1 or 24 h after microirradiation and analyzed on Zeiss LSM510 META confocal microscope equipped with a 63× Plan‐A (1.4 NA) oil‐immersion objective. Images were recorded using Zeiss LSM imaging software in multi‐track mode.
Immunofluorescence
For immunofluorescence, U2OS or RPE cells were seeded on 96‐well microscope plates (Falcon‐BD), or MatTek glass bottom dishes. Fixation was performed using 3.7% paraformaldehyde (Sigma) for 5 min at room temperature. Permeabilization was achieved by incubating cells with ice‐cold methanol for 2 min. Blocking, antibody and DAPI incubations were performed in TBS supplemented with 0.1% Tween‐20 (TBST) and 2% BSA (Sigma). Wash steps were performed in TBS supplemented with 0.1% Tween‐20. Images were acquired using either DeltaVision Spectris imaging system using a 20×, NA 0.7 objective, a Leica DMI6000 Imaging System using a 20×, NA 0.4 objective, or on an ImageXpress system using a 20×, NA 0.45 objective, and quantified as described (Akopyan et al, 2014).
Immunoprecipitation
U2OS or AT cells expressing ATKAR, H2B‐ATKAR, or H2B‐PLK1 FRET probe were collected 1 h after exposure to IR (5 Gy) or UVC (10–40 J/m2) or NCS (5–10 nM) and sonicated in cold IP buffer (50 mM HEPES pH 7.5, 250 mM NaCl, 0.25 NP‐40, 1% glycerol) supplemented with phosphatase inhibitor (PhosSTOP, Roche) and protease inhibitor (complete EDTA‐free, Roche). FRET probes were immunoprecipitated from cell extracts using GFP‐Trap (ChromoTek) for 2 h. Beads were washed three times with IP buffer and mixed with SDS sample buffer. Alternatively, HEK293 cells were transfected by empty EGFP or EGFP‐Wip1 plasmid using linear polyethylenimine MAX. After 48 h, cells were harvested to lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 3 mM MgCl2, 10% glycerol, 1% Tween‐20, 0.1% NP‐40) supplemented with benzonase (25 U/ml), protease, and phosphatase inhibitors (Roche), sonicated, and incubated on rotary shaker overnight at 4°C. Cell extract was centrifuged 15 min 20,000 g at 4°C, supernatant was incubated with GFP‐Trap beads for 1 h at 4°C, beads were washed four times with lysis buffer, and bound proteins were eluted with SDS sample buffer.
Subcellular fractionation
Cells were treated with DMSO or 5 nM NCS and collected after 2, 6, or 20 h. Subcellular fractionation was performed as described before (Macurek et al, 2010). Cells were incubated in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X‐100, and protease inhibitor cocktail) at 4°C for 10 min and centrifuged at 1,500 g for 2 min. The cytosolic fraction was collected, and sedimented cells were further incubated with buffer B (10 mM HEPES, pH 7.9, 3 mM EDTA, 0.2 mM EGTA, 1 mM DTT) and centrifuged at 2,000 g for 2 min. Nuclear soluble fraction was collected, pooled with cytosol (together forming a soluble fraction), and mixed with 4× SDS sample buffer. Remaining chromatin fraction was mixed with 1.25× volumes of 1× SDS sample buffer. All samples were boiled, sonicated, and separated on SDS–PAGE.
In vitro and in situ phosphatase assay
Cells expressing HA‐KAP1‐WT or HA‐KAP1‐S824A were treated with DMSO or NCS (5 nM) for 1 h and then extracted using IP buffer. Cell extracts were immunoprecipitated for 2 h using monoclonal anti‐HA tag antibody (Santa Cruz, sc7392) immobilized at pA/G beads (Pierce). Beads were stripped with 0.5 M NaCl to remove proteins interacting with KAP1. Beads were incubated with purified His‐Wip1 (100 ng) in phosphatase buffer (40 mM HEPES pH 7.4, 100 mM NaCl, 50 mM KCl, 1 mM EGTA, 50 mM MgCl2) for 20 min at 30°C. The reaction was stopped by addition of 4× SDS sample buffer. Alternatively, cells grown on coverslips were treated with NCS (5 nM) for 1 h, fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X‐100 and in situ phosphatase assay was performed as described (Munoz et al, 2013). Samples were blocked with 3% BSA in PBS and incubated at room temperature in ISB buffer (50 mM HEPES pH 7.0, 10 mM MgCl2, 25 mM NaCl, 2 mM CaCl2, 1 mM DTT) and His‐Wip1 (0.25–10 ng/μl) for 1 h. Reaction was stopped by addition of 20 mM NaF and 20 mM β‐glycerolphosphate in PBS. Samples were incubated with a mixture of anti‐γH2AX and anti‐pS824‐KAP1 antibodies for 1 h. After washing, samples were incubated with secondary antibodies and DAPI. Imaging was performed on ScanR microscope (Olympus), and 1,000 nuclei were counted per condition.
Reagents and antibodies
The following antibodies were from Cell Signaling: phospho‐histone H3 (#3377); pChk1‐S345 (133D3; #2348); pATM‐S1981 (10H11.E12; #4526); pH2AX (20E3; #9718), pp53‐s15 (#9284; #9286), Phospho‐(Ser/Thr) ATM/ATR Substrate (#2851), pT210‐Plk1 (#9062), pT288‐AurA (#3079), AurA (#3092), pS296‐Chk1 (#2349), pS317‐Chk1 (#12302). Antibodies against pSMC3‐S1083 (#IHC00070), pKap1‐S824 (#A300‐767A and GTX63711), Kap1 (#A300‐274A and GTX62973), pS966‐SMC1 (A300‐050A), and PP4C (GTX114659) were from Bethyl Lab and GeneTex. Additional antibodies included pH2AX (Clone JBW301; #05‐636, Millipore), 53BP1 (#NB100‐304, Novus Biologicals), pS10‐H3 (#05‐806, Millipore), Rad51 (FE#7946, Biogenes), and Alexa‐ and FITC‐coupled antibodies (Life Technologies). Antibodies against GFP (sc‐8334), 14‐3‐3, p53 (sc‐6243), p21 (sc‐397), Wip1 (sc‐130655), His (sc‐8036), and BRCA1 (sc‐6945) were obtained from Santa Cruz. Neocarzinostatin (NCS), etoposide, and DMSO were from Sigma. ATM inhibitor (#4176 and #3544) and DNA‐PK inhibitor (#2828) were from Tocris Bioscience and used at 10 μM. Alternatively, DNA‐PK inhibitor NU7441 was used at 2 μM. ATR inhibitor VE821 (Reaper et al, 2011) was obtained from (Tinib‐Tools) or was synthesized according to published procedure (Charrier et al, 2011) and used at 1 μM. Plk1 inhibitor BI2536 was from Boehringer Ingelheim Pharma and used at 100 nM. CDT was a generous gift from Teresa Frisan.
Simulations
We wrote ordinary differential equations assuming Michaelis–Menten kinetics to simulate the relation between the variables Damage, Cell Cycle, ATM, and ATR. Damage was added at the beginning of a simulation and assumed to decrease over time, simulating quick repair by non‐homologous end joining and slow repair by homologous recombination. Cell cycle contains a positive feedback loop to simulate the auto‐amplifying property of the mitotic entry network, of which Plk1 is a part. ATM is activated by damage, but is efficiently counteracted by a constant to simulate the balance of ATM and Wip1 on chromatin. ATR, simulating ATR‐Chk1 activity, is activated by Damage, but inactivated by cell cycle to simulate Plk1 and Cdk1 phosphorylation of DDR components. Both ATM and ATR inhibit cell cycle.
The equations were solved using Copasi 4.8, build 35 (www.copasi.org/). Parameterization was performed manually, restricting constants to 1, 2, or 10. The model is available at biomodels.net with the accession number MODEL1704030000.
Equations
Constants and initial values of variables:
| Constants | Value | Variables | Initial value |
|---|---|---|---|
| Kcc2a | 1 | CellCycleact | 1 |
| Kcc2e | 1 | CellCycletot | 10 |
| Kd2e | 1 | DHR | 3 |
| Kd2t | 2 | DNHEJ | 7 |
| Ke2cc | 1 | ATRact | 0 |
| Km1 | 1 | ATRtot | 10 |
| Km10 | 10 | ATMact | 0 |
| Kt2cc | 10 | ATMtot | 10 |
| Kti2t | 10 | ||
| Km10t | 10 |
Model optimization (Parameter estimation)
The model was optimized sequentially based on experimental data of ATM and Plk1 activity dynamics during DNA damage conditions (Fig EV3B). Parameter estimation was performed using the evolutionary programming built‐in optimization method in COPASI. First, the inverted FRET‐ratio of H2B‐ATKAR in U2OS cells after addition of three concentrations of NCS was set as model ATM activity and used to estimate the values for Kd2t, Kti2t, and Km1. Next, the inverted FRET‐ratio of Plk1‐FRET in U2OS cells entering mitosis after NCS treatment was set as model Plk1 activity and used to estimate the values for Kt2cc and Km10t.
Constants and initial values of variables after optimization:
| Constants | Value | Variables | Initial value |
|---|---|---|---|
| Kcc2a | 1 | CellCycleact | 1 |
| Kcc2e | 1 | CellCycletot | 5 |
| Kd2e | 1 | DHR | 0.6 |
| Kd2t | 10.1794 | DNHEJ | 1.4 |
| Ke2cc | 1 | ATRact | 0 |
| Km1 | 26.3699 | ATRtot | 10 |
| Km10 | 10 | ATMact | 0 |
| Kt2cc | 6.58694 | ATMtot | 10 |
| Kti2t | 31.054 | ||
| Km10t | 0.119845 |
Simulation of sustained damage
For dependence on sustained damage, the constant K damage was introduced such that
The initial values were set as DHR = 0.6 – K damage/2 and DNHEJ = 1.4 – K damage/2 (Optimized model, Fig 7C) and DHR = 3 – K damage/2 and DNHEJ = 7 – K damage/2 (Initial model, Fig EV3I).
Author contributions
HJ, JB, EM, KA, KB, LM, and AL performed experiments and analyzed data. RHM, LM, and AL supervised the study. TK and TH generated reagents. HJ, LM, and AL wrote the manuscript. All authors revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Marcel van Vugt, Camilla Sjögren, and Urban Lendahl for comments on the manuscript and Teresa Frisan and Nico Dantuma for kind gift of CDT and RNF8 siRNA. This work was supported by grants from the Swedish Research Council, the Swedish Foundation for Strategic Research, the Swedish Cancer Society and the Swedish Childhood Cancer Foundation (all to AL), Vinnova (TH), the Grant Agency of the Czech Republic (13‐18392S) and Norwegian Financial Mechanism 2009‐14 (Project Contract 7F14061, PHOSCAN). HJ was supported by the Wenner‐Gren Foundation and JB partly by the Grant Agency of the Charles University (project 836613).
The EMBO Journal (2017) 36: 2161–2176
Contributor Information
Libor Macurek, Email: libor.macurek@img.cas.cz.
Arne Lindqvist, Email: arne.lindqvist@ki.se.
References
- Akopyan K, Silva Cascales H, Hukasova E, Saurin AT, Mullers E, Jaiswal H, Hollman DA, Kops GJ, Medema RH, Lindqvist A (2014) Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol Cell 53: 843–853 [DOI] [PubMed] [Google Scholar]
- Altmeyer M, Lukas J (2013) To spread or not to spread–chromatin modifications in response to DNA damage. Curr Opin Genet Dev 23: 156–165 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2007) DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 19: 238–245 [DOI] [PubMed] [Google Scholar]
- Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio‐Kuter A, Levine AJ (2010) The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol 2: a001198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer HR, Guerrero Llobet S, van Vugt MA (2016) Controlling the response to DNA damage by the APC/C‐Cdh1. Cell Mol Life Sci 73: 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma W, Aprelia M, García‐Santisteban I, Kool J, Xu YJ, Medema RH (2017) Inhibition of Polo‐like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene 36: 1840–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace AJ, Appella E (2002) Amplification of PPM1D in human tumors abrogates p53 tumor‐suppressor activity. Nat Gen 31: 210–215 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, Mortimore M, O'Donnell ME, Pinder JL, Reaper PM, Rutherford AP, Wang PS, Young SC, Pollard JR (2011) Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem 54: 2320–2330 [DOI] [PubMed] [Google Scholar]
- Deckbar D, Birraux J, Krempler A, Tchouandong L, Beucher A, Walker S, Stiff T, Jeggo P, Lobrich M (2007) Chromosome breakage after G2 checkpoint release. J Cell Biol 176: 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Ferrari M, Lazzaro F, Clerici M, Tamilselvan Nachimuthu B, Plevani P, Muzi‐Falconi M, Pellicioli A (2010) Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet 6: e1000763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB (2000) The molecular basis of FHA domain: phosphopeptide binding specificity and implications for phospho‐dependent signaling mechanisms. Mol Cell 6: 1169–1182 [DOI] [PubMed] [Google Scholar]
- Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, Fydrych A, Ho R, Greenberger BA, Chen GC, Maffa A, Del Rosario AM, Root DE, Carpenter AE, Hahn WC, Sabatini DM et al (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller BG, Lampson MA, Foley EA, Rosasco‐Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM (2008) Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature 453: 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukasova E, Silva Cascales H, Kumar SR, Lindqvist A (2012) Monitoring kinase and phosphatase activities through the cell cycle by ratiometric FRET. J Vis Exp 59: 3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, You Z, Hunter T (2007) Monitoring ATM kinase activity in living cells. DNA Repair 6: 1277–1284 [DOI] [PubMed] [Google Scholar]
- Kim BJ, Li Y, Zhang J, Xi Y, Li Y, Yang T, Jung SY, Pan X, Chen R, Li W, Wang Y, Qin J (2010) Genome‐wide reinforcement of cohesin binding at pre‐existing cohesin sites in response to ionizing radiation in human cells. J Biol Chem 285: 22784–22792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousholt AN, Fugger K, Hoffmann S, Larsen BD, Menzel T, Sartori AA, Sorensen CS (2012) CtIP‐dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J Cell Biol 197: 869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenning L, Feringa FM, Shaltiel IA, van den Berg J, Medema RH (2014) Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol Cell 55: 59–72 [DOI] [PubMed] [Google Scholar]
- Krystyniak A, Garcia‐Echeverria C, Prigent C, Ferrari S (2006) Inhibition of Aurora A in response to DNA damage. Oncogene 25: 338–348 [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2001) Positive regulation of Wee1 by Chk1 and 14‐3‐3 proteins. Mol Biol Cell 12: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D (2012) Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP‐1 impacting the DNA damage response. EMBO J 31: 2403–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK (2007) Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1‐mediated transcriptional repression. J Biol Chem 282: 36177–36189 [DOI] [PubMed] [Google Scholar]
- Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ III, Ozato K, Zhu F (2011) Tripartite motif‐containing protein 28 is a small ubiquitin‐related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J Immunol 187: 4754–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Esposito A, De S, Ber S, Collin P, Surana U, Venkitaraman AR (2014) Homeostatic control of polo‐like kinase‐1 engenders non‐genetic heterogeneity in G2 checkpoint fidelity and timing. Nat Commun 5: 4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A, de Bruijn M, Macurek L, Bras A, Mensinga A, Bruinsma W, Voets O, Kranenburg O, Medema RH (2009) Wip1 confers G2 checkpoint recovery competence by counteracting p53‐dependent transcriptional repression. EMBO J 28: 3196–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobrich M, Jeggo PA (2007) The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7: 861–869 [DOI] [PubMed] [Google Scholar]
- Lock RB, Ross WE (1990) Inhibition of p34cdc2 kinase activity by etoposide or irradiation as a mechanism of G2 arrest in Chinese hamster ovary cells. Cancer Res 50: 3761–3766 [PubMed] [Google Scholar]
- Loewer A, Karanam K, Mock C, Lahav G (2013) The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol 11: 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH (2008) Polo‐like kinase‐1 is activated by aurora A to promote checkpoint recovery. Nature 455: 119–123 [DOI] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Medema RH (2009) Aurora‐A and hBora join the game of Polo. Cancer Res 69: 4555–4558 [DOI] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH (2010) Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 29: 2281–2291 [DOI] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288: 1425–1429 [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker‐Jensen S, Bartek J, Lukas J (2006) Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 23: 307–318 [DOI] [PubMed] [Google Scholar]
- Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R (2006) Polo‐like kinase‐1 controls proteasome‐dependent degradation of Claspin during checkpoint recovery. Curr Biol 16: 1950–1955 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- Medema RH, Macurek L (2011) Checkpoint control and cancer. Oncogene 31: 2601–2613 [DOI] [PubMed] [Google Scholar]
- Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J (2007) A proteomic analysis of ataxia telangiectasia‐mutated (ATM)/ATM‐Rad3‐related (ATR) substrates identifies the ubiquitin‐proteasome system as a regulator for DNA damage checkpoints. J Biol Chem 282: 17330–17334 [DOI] [PubMed] [Google Scholar]
- Mullers E, Silva Cascales H, Jaiswal H, Saurin AT, Lindqvist A (2014) Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 13: 2733–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DP, Kawahara M, Yannone SM (2013) An autonomous chromatin/DNA‐PK mechanism for localized DNA damage signaling in mammalian cells. Nucleic Acids Res 41: 2894–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H (1997) Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M (2006) SCFbetaTrCP‐mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 23: 319–329 [DOI] [PubMed] [Google Scholar]
- Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA (1990) In vitro disassembly of the nuclear lamina and M phase‐specific phosphorylation of lamins by cdc2 kinase. Cell 61: 591–602 [DOI] [PubMed] [Google Scholar]
- Qin B, Gao B, Yu J, Yuan J, Lou Z (2013) Ataxia telangiectasia‐mutated‐ and Rad3‐related protein regulates the DNA damage‐induced G2/M checkpoint through the Aurora A cofactor Bora protein. J Biol Chem 288: 16139–16144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, Kallioniemi A (2006) The serine‐threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat 95: 257–263 [DOI] [PubMed] [Google Scholar]
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR (2011) Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol 7: 428–430 [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB (2007) p53‐deficient cells rely on ATM‐ and ATR‐mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11: 175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Yaffe MB (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 21: 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, Yaffe MB (2010) DNA damage activates a spatially distinct late cytoplasmic cell‐cycle checkpoint network controlled by MK2‐mediated RNA stabilization. Mol Cell 40: 34–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica‐Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210 [PubMed] [Google Scholar]
- Shiotani B, Zou L (2009) Single‐stranded DNA orchestrates an ATM‐to‐ATR switch at DNA breaks. Mol Cell 33: 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, Bulavin DV (2006) Wip1 phosphatase modulates ATM‐dependent signaling pathways. Mol Cell 23: 757–764 [DOI] [PubMed] [Google Scholar]
- Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH (2000) Polo‐like kinase‐1 is a target of the DNA damage checkpoint. Nat Cell Biol 2: 672–676 [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Jensen S, Bartek J, Lukas J (2006) Adaptation to the ionizing radiation‐induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo‐like kinase 1 kinases. Cancer Res 66: 10253–10257 [DOI] [PubMed] [Google Scholar]
- Tkacz‐Stachowska K, Lund‐Andersen C, Velissarou A, Myklebust JH, Stokke T, Syljuasen RG (2011) The amount of DNA damage needed to activate the radiation‐induced G2 checkpoint varies between single cells. Radiother Oncol 101: 24–27 [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90: 1097–1106 [DOI] [PubMed] [Google Scholar]
- van Vugt MA, Bras A, Medema RH (2004) Polo‐like kinase‐1 controls recovery from a G2 DNA damage‐induced arrest in mammalian cells. Mol Cell 15: 799–811 [DOI] [PubMed] [Google Scholar]
- van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, Tan CS, Miao H, Keezer SM, Li J, Pawson T, Lewis TA, Carr SA, Smerdon SJ, Brummelkamp TR, Yaffe MB (2010) A mitotic phosphorylation feedback network connects Cdk1, Plk 1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 8: e1000287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Guo Q, Fisher LA, Liu D, Peng A (2015) Regulation of polo‐like kinase 1 by DNA damage and PP2A/B55alpha. Cell Cycle 14: 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Durell SR, Chatterjee DK, Anderson CW, Appella E (2007) The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K‐like kinases. Biochemistry 46: 12594–12603 [DOI] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker‐Jensen S, Bartek J, Shiloh Y (2006) Chromatin relaxation in response to DNA double‐strand breaks is modulated by a novel ATM‐ and KAP‐1 dependent pathway. Nat Cell Biol 8: 870–876 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File