

Abstract
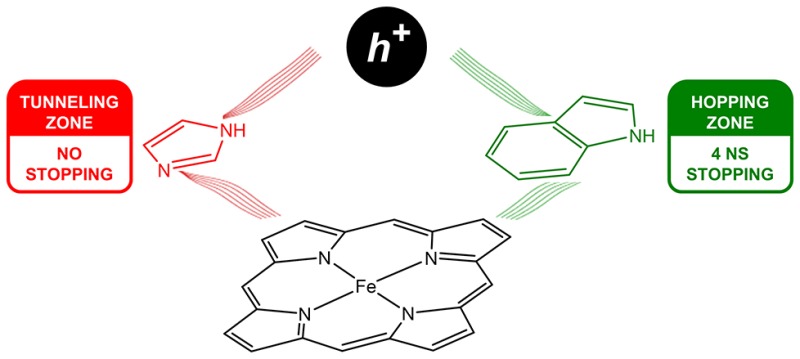
Electron-transfer kinetics have been measured in four conjugates of cytochrome P450 with surface-bound Ru-photosensitizers. The conjugates are constructed with enzymes from Bacillus megaterium (CYP102A1) and Sulfolobus acidocaldarius (CYP119). A W96 residue lies in the path between Ru and the heme in CYP102A1, whereas H76 is present at the analogous location in CYP119. Two additional conjugates have been prepared with (CYP102A1)W96H and (CYP119)H76W mutant enzymes. Heme oxidation by photochemically generated Ru3+ leads to P450 compound II formation when a tryptophan residue is in the path between Ru and the heme; no heme oxidation is observed when histidine occupies this position. The data indicate that heme oxidation proceeds via two-step tunneling through a tryptophan radical intermediate. In contrast, heme reduction by photochemically generated Ru+ proceeds in a single electron tunneling step with closely similar rate constants for all four conjugates.
Most biological redox transformations involve reagents with formal potentials in the ±1 V vs NHE range. At the periphery of this potential window proteins present a decidedly unsymmetrical medium for electron transfer (ET). Whereas reduction of peptides and small aromatic groups only proceeds at potentials more negative than −2.5 V vs NHE,1−3 one-electron oxidations of aromatic and sulfur-containing amino-acids, as well as the peptide backbone itself, can occur at potentials in the 1.0–1.5 V vs NHE range.4−11 We anticipate, then, that proteins are superexchange mediators of ET in reactions of low-potential redox couples. In contrast, oxidized amino acid radicals are known to be essential participants in many high-potential enzymatic redox reactions,12−21 and structural evidence suggests that they may play a far greater role than previously recognized.22
Elucidating the roles of protein radicals, particularly those of the aromatic amino acids tryptophan (W) and tyrosine (Y), in functional and protective pathways of high-potential enzymes continues to be an active area of research.22−25 The appearance of dioxygen in Earth’s atmosphere promoted the evolution of a vast array of O2-utilizing enzymes that generate high-potential reactive intermediates capable of oxidizing tryptophan and tyrosine. The cytochromes P450 (CYP) are prominent representatives of this enzyme class, responsible for a broad spectrum of vital metabolic functions.26 The accepted enzymatic reaction cycle of cytochrome P450 involves two high-potential reactive intermediates (compounds I and II) that participate directly in reactions with substrates.27−32 In prior work, we demonstrated that compound II can be prepared in the heme domain of Bacillus megaterium P450 (CYP102A1) by oxidation of the Fe3+-heme with a surface-attached Ru(diimine)33+ complex (E°(Ru3+/2+) ≈ 1.2 V vs NHE33,34).35 We have extended this work to the archaeal Sulfolobus acidocaldarius P450 (CYP119).30 A key structural difference between the two enzymes is an aromatic side chain hydrogen bonded to one of the heme proprionates (W96 in CYP102A1; H76 in CYP119) (Figure 1).36,37 We find that an intervening tryptophan residue (CYP102A1, W96; CYP119, H76W) is essential for promoting Fe3+(OH2)–heme oxidation by Ru(diimine)33+ in both CYP102A1 and CYP119 but that these residues appear to play no analogous role in the reduction of Fe3+(OH2)-heme by Ru(diimine)3+ (E°(Ru2+/+) ≈ −1.3 V vs NHE33,34).
Figure 1.
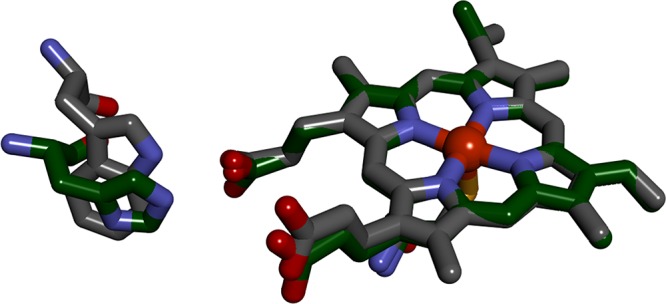
Overlay of the heme environments of CYP102A1 (gray carbon atoms) and CYP119 (green carbon atoms) illustrating the relative positions of W96 in CYP102A1 and H76 in CYP119. Atomic coordinates were taken from PDB IDs 2IJ2 (CYP102A1)34 and 1IO7 (CYP119).35
Experimental Procedures
Materials
Buffer salts were obtained from J.T. Baker. 5-Aminolevulenic acid and dithiothreitol were obtained from Sigma-Aldrich. [Ru(NH3)6]Cl3 was obtained from Strem Chemicals. All chemicals were used as received with no further purification. The ruthenium labeling reagent ([Ru(2,2′-bipyridine)2(5-iodoacetamido 1,10-phenanthroline)]2+, [Ru(bpy)2(IAphen)]2+),38 model complex ([Ru(2,2′-bipyridine)2(5-acetamido 1,10-phenanthroline)]2+, [Ru(bpy)2(Aphen)]2+),38 and p-methoxydimethylaniline (pMeODMA)39 were synthesized according to published procedures. Solutions were prepared using 18 MΩ-cm water unless otherwise noted. Mutagenesis primers were obtained from Operon.
Plasmid Preparation
The recombinant CYP102A1 (UniProt accession number P14779) heme domain, consisting of the first 463 residues with an N-terminal 6-histidine tag, was obtained courtesy of Professor Andrew Udit (Occidental College, Los Angeles California), within the pCWori+ vector. Recombinant CYP119 (UniProt accession number Q55080) with an N-terminal 6-histidine tag was obtained courtesy of Professor Paul Ortiz de Montellano (University of California, San Francisco), also within the pCWori+ vector. Qiagen Quik-Change site-directed mutagenesis was used to generate the desired P450 mutants. Mutagenesis primers (forward, 5′–3′) were CTAATTAAGAAGCAGCCGATGAATCACG (CYP102A1 C62A), CGATTGGTCTTAGCGGCTTTAAC (CYP102A1 C156S), GCTGGACGCATCAAAAAAATTGGTGCAAAGCGC (CYP102A1 K97C); GGACGCATGAAAAAAATCATTGCAAAGCGCATAATATC (CYP102A1 W96H); GATCCCCCTCTCCATTGTGAGTTAAGATCAATGTCAGC (CYP119 D77C), and CCTCAGATCCCCCTCTCTGGTGTGAGTTAAGATCAATGTC (CYP119 H76W).
Overexpression in E. coli
The P450 mutants CYP102A1-C62A/C156S/K97C (C97(CYP102A1)W96), CYP102A1-C62A/C156S/K97C/W96H (C97(CYP102A1)W96H), CYP119-D77C (C77(CYP119)H76), and CYP119-D77C/H76W (C77(CYP119)H76W) were overexpressed in the BL21-DE3 strain of E. coli. Overnight cultures of Luria–Bertani broth (25 mL) containing 100 μg/mL ampicillin and a single respective E. coli colony were incubated at 37 °C overnight, shaking at 180–200 rpm. Induction cultures of TB (1× TB for CYP102A1, 2× TB for CYP119) containing 200 μg/mL ampicillin, 1 μM thiamine, 0.4% glycerol, and 250 μL of mineral supplements (stock solution 100 mM FeCl3, 10 mM ZnCl2, 8.5 mM CoCl2, 8.5 mM Na2MoO4, 7 mM CaCl2, 7.5 mM CuCl2, 8 mM H3BO3) were inoculated with the overnight culture and incubated at 37 °C until reaching an optical density of ∼1 at 600 nm. Cultures were induced by addition of 1 mM IPTG, and 0.5 mM α-aminolevulenic acid, a heme precursor, was added. The temperature was lowered to 30 °C for 24 (CYP102A1) or 48 h (CYP119). Following expression, cells were harvested by centrifugation, and cell pellets were stored at −80 °C until needed.
Cell pellets were resuspended in cold wash buffer (50 mM Tris pH 8, 300 mM sodium chloride, 20 mM imidazole). A small spatula tip of each of two protease inhibitors (benzamidine hydrochloride and Pefabloc SC) was added, and cells were lysed by two to three cycles of probe-tip sonication (0.5 s on, 0.5 s off, for 5 min), cooled by an ice–water bath. After centrifugation (15 000 rpm, 1 h, 8 °C) to pellet cellular debris, the supernatant was loaded directly onto a Ni batch column. After thorough washing with wash buffer (1.5–2 L), protein was eluted (200 mM imidazole in wash buffer), and the colored (red/orange) fractions were collected and concentrated in 30 kDa centrifugal filters. Gel filtration chromatography was used to remove fragmented proteins, followed by buffer exchange into 20 mM Tris, pH 8, with 20 mM dithiothreitol (DTT) added to reduce intermolecular disulfide bonds. Purity was determined by UV–vis absorption (A420/A280), SDS-PAGE, and mass spectrometry. Protein not intended for immediate use was flash-frozen in liquid nitrogen (with 40% glycerol added to solution as cryoprotectant) and stored at −80 °C.
Conjugation to Ru-Photosensitizer
An approximately 3-fold excess of [Ru(bpy)2(IA-phen)]2+ was added to a ∼10 μM solution of P450 mutant in 20 mM Tris buffer (pH 8) and shaken in the dark at 4 °C. Labeling progress can be monitored by MALDI mass spectrometry; no further increase in the peak corresponding to the predicted mass of Ru2+–P450 was observed after 2 h. Excess [Ru(bpy)2(IA-phen)]2+ was removed during concentration in 30 kDa filters, followed by desalting on an FPLC HiPrep column.
To separate photosensitizer-labeled and unlabeled enzymes, protein samples were loaded onto an anion exchange MonoQ or HiPrep Q column equilibrated with 20 mM Tris buffer, pH 8 (Q wash buffer). The column was washed with Q wash buffer until UV–visible absorbance returned to baseline. The gradient was ramped quickly to 59% Q elution buffer (Q wash buffer + 250 mM sodium chloride), followed by a slow gradient of 59–65% Q elution buffer over 60 min. Successful conjugation and separation of Ru–P450 was verified by UV–vis, mass spectrometry, and fluorometry.
Laser Spectroscopy Sample Preparation
Laser samples were composed of ∼10 μM Ru–P450 conjugate, with and without oxidative quencher (17 mM [Ru(NH3)6]Cl3) or reductive quencher (10 mM pMeODMA) in buffered solution (pH 8, 50 mM sodium borate or 50 mM Tris); additionally, each buffer contained sodium chloride to prevent precipitation. pMeODMA is only sparingly soluble in water; aqueous stock solutions were prepared by dropwise addition of concentrated pMeODMA/DMSO solution into aqueous buffer (50 mM sodium borate, pH 8). Fresh pMeODMA solutions were prepared immediately prior to use and protected from light, as oxygenated solutions change from clear to pinkish/purple in ambient light. Laser samples were placed in a high-vacuum four-sided quartz fluorescence cuvette with high-vacuum Teflon valve, equipped with a small stir bar. Deoxygenation was achieved via 3 × 10–15 gentle pump-backfill cycles with argon on a Schlenk line, with 15 min of equilibration between each set of cycles. Additional details provided in Supporting Information.
For nanosecond-to-millisecond transient luminescence and absorption experiments, excitation was provided by 480 nm pulses from a tunable optical parametric oscillator (Spectra Physics, Quanta-Ray MOPO-700) pumped by the third harmonic from a Spectra Physics Q-switched Nd:YAG laser (Spectra-Physics, Quanta-Ray PRO-Series, 8 ns pulse width) operated at 10 Hz. Probe light was provided by a 75-W arc lamp (PTI model A 1010) that could be operated in continuous or pulsed mode and passed through the sample collinearly with the excitation pulse. After rejection of scattered light by appropriate long- and short-pass filters, and intensity modulation by a neutral density filter, probe wavelengths were selected by a double monochromator (Instruments SA DH-10) with 1 mm slits. Transmitted light was detected by a photomultiplier tube (PMT, Hamamatsu R928) and amplified by a 200 MHz wideband voltage amplifier DHPVA-200 (FEMTO).
Luminescence decays were monitored at 630 nm. Single wavelength transient absorption kinetics were monitored every 10 nm from 390 to 440 nm, averaging ∼500 shots per wavelength. Data from five separate time scales (2 μs, 40 μs, 400 μs, 10 ms, and 500 ms) were collected, log-compressed, and spliced together to produce full kinetics traces using Matlab software (Mathworks).
Results
Ru–P450 Conjugates
Electron-transfer kinetics measurements were performed on four Ru–P450 conjugates: two mutants of CYP102A1 and two of CYP119. The Ru–P450 from our previous work is a triple mutant of CYP102A1: C62A/C156S/K97C with [Ru(2,2′-bipyridine)2(5-acetamido-1,10-phenanthroline)]2+ ([Ru(bpy)2(APhen)]2+) covalently bound to C97 via a thioether linkage (RuC97(CYP102A1)W96). To investigate the importance of W96 in photochemical ET, we generated the analogous conjugate with a W96H mutation (RuC97(CYP102A1)W96H). The W96H mutation should preserve hydrogen-bonding with the heme propionates, which is thought to provide structural stability to the heme.40 But, unlike tryptophan, this side chain should not be susceptible to oxidation by photochemically generated Ru3+.11
We have prepared analogous Ru–P450 conjugates with an intervening histidine or tryptophan residue in thermophilic CYP119. The residue corresponding to CYP102A1-W96 is H76 in CYP119 (Figure 1); a cysteine mutation at residue 77 serves as the photosensitizer attachment point. The two Ru–CYP119 conjugates are referred to as RuC77(CYP119)H76 and RuC77(CYP119)H76W.
Luminescence Quenching Measurements
The Ru(diimine)3 photosensitizer luminesces upon 480 nm excitation. The luminescence decay of all four Ru–P450 conjugates is nonexponential, attributable to multiple photosensitizer conformations that do not exchange on the luminescence decay time scale. The nonexponential decay kinetics are most pronounced for mutants containing a tryptophan residue adjacent to the Ru-tethering point (RuC97(CYP102A1)W96, RuC77(CYP119)H76W). The mild oxidant [Ru(NH3)6]3+ is an efficient quencher of excited Ru(diimine)32+ complexes (*Ru2+),35 producing powerfully oxidizing Ru(diimine)33+ in high yield. In the presence of [Ru(NH3)6]3+ (17 mM), luminescence is strongly quenched, and the decay kinetics can be fit to a single exponential function (Table 1). There are small differences in quenched decay times; in particular, the decay time of quenched RuC77(CYP119)H76 is approximately two to three times longer than that found in the three other proteins (90 ns vs 30–50 ns). The origin of this difference is not readily apparent.
Table 1. Luminescence Decay Times of Four Ru–P450 Conjugates in the Absence and Presence of Quenchersa.
| enzyme | quencher | τmono, ns | τa, ns (ρa) | τb, ns (ρb) |
|---|---|---|---|---|
| RuC97(CYP102A1)W96 | none | 140 | 190 (0.65) | 52 (0.35) |
| [Ru(NH3)6]3+ | 30 | |||
| pMeODMA | 62 | |||
| RuC97(CYP102A1)W96H | none | 180 | 160 (0.80) | 310 (0.20) |
| [Ru(NH3)6]3+ | 33 | |||
| pMeODMA | 65 | |||
| RuC77(CYP119)H76 | none | 200 | 220 (0.85) | 45 (0.15) |
| [Ru(NH3)6]3+ | 91 | |||
| pMeODMA | 54 | |||
| RuC77(CYP119)H76W | none | 130 | 91 (0.75) | 320 (0.25) |
| [Ru(NH3)6]3+ | 48 | |||
| pMeODMA | 50 |
Quenchers: [Ru(NH3)6]3+, 17 mM; pMeODMA, 10 mM. The relative amplitudes of major (ρa) and minor (ρb) components in biexponential fits to the unquenched decays also are listed. Samples were excited at 480 nm, and luminescence was detected at 630 nm. Uncertainties in the decay times are ±10%, except for the single-exponential fits to the unquenched decays.
In prior work, we demonstrated that para-methoxy-N,N-dimethylaniline (pMeODMA) quenches *Ru2+ complexes to produce one-electron reduced forms (Ru+).39 In the presence of 10 mM pMeODMA, *Ru2+ luminescence is efficiently quenched, and the decay kinetics are monoexponential. In contrast to the oxidative quenching with [Ru(NH3)6]3+, there is very little difference in the quenched decay times among the four Ru–P450 conjugates (Table 1).
Transient Absorption Measurements
In the absence of electron-transfer quenchers, the transient absorption features of the Ru–P450 conjugates are characterized by a loss of Ru(diimine)32+ metal-to-ligand charge transfer (MLCT) absorbance in the 400–440 nm region due to depopulation of the ground-state photosensitizer. The spectral and temporal profiles of these transients are essentially identical for the free photosensitizer and all four Ru–P450 conjugates: the features return to baseline with the same time constants as the luminescence decay.
Oxidative Quenching
Transient absorption measurements demonstrate that [Ru(NH3)6]3+ quenches the *Ru2+ to produce electron-transfer products (Figure 2). The ligand field absorption bands of [Ru(NH3)6]3+/2+ are too weak to make any detectable contribution to the transient spectra. Photogenerated [Ru(bpy)2(Aphen)]3+ (Ru3+) is characterized by a bleach of [Ru(bpy)2(Aphen)]2+ MLCT absorption between 390 and 440 nm, and time-resolved absorption measurements reveal that it persists for several microseconds before reacting with [Ru(NH3)6]2+ to regenerate the Ru2+ sensitizer and oxidized quencher (Figure 2).
Figure 2.
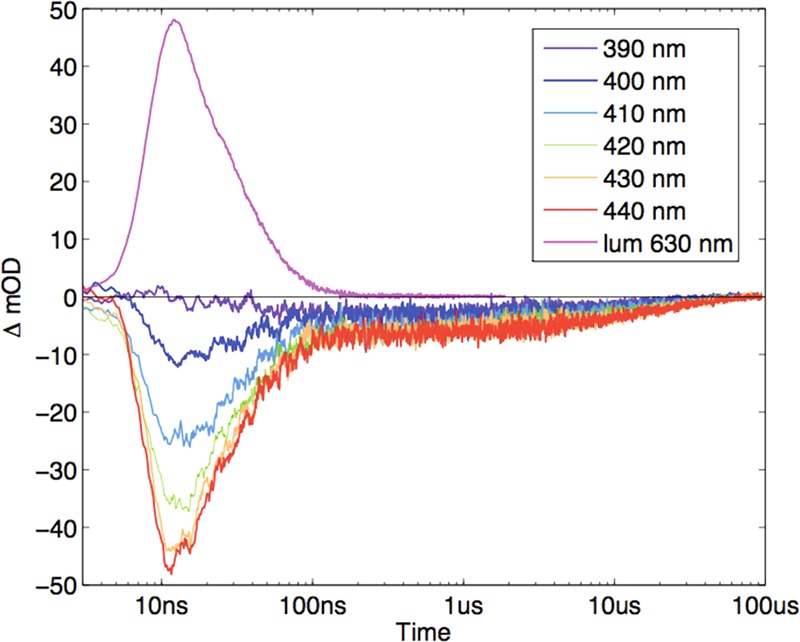
Transient absorption kinetics following 480 nm laser excitation of [Ru(bpy)2(Aphen)]2+ in the presence of [Ru(NH3)6]3+ (17 mM). The purple curve is a luminescence decay trace.
We reported previously that when [Ru(bpy)2(IAphen)]2+ is conjugated to C97 in CYP102A1, photogenerated RuC973+(CYP102A1)W96 sequentially generates ferric-porphyrin radical cation (Fe3+por•+) and Fe4+-hydroxide (Fe4+–OH, CII) intermediates. These oxidation processes are characterized by loss of absorbance at 420 nm (the peak of the Fe3+–OH2 heme Soret absorption) that persists for hundreds of milliseconds; formation of CII is characterized by the appearance of transient absorption at 440 nm on the millisecond time scale (Figure 3).
Figure 3.

Transient kinetics following oxidative quenching ([Ru(NH3)6]3+, 17 mM) in four Ru–P450 conjugates: λex = 480 nm; λobsd = 420 nm (green), 440 nm (red). Signals normalized to the magnitude of the 440 nm prompt bleach.
We subjected the other three Ru–P450 conjugates to identical oxidative flash-quench irradiation and recorded the transient kinetics (Figure 3). The CYP119 mutant with an intervening tryptophan, RuC77(CYP119)H76W, displays transient absorption spectra and kinetics similar to those of RuC97(CYP102A1)W96, albeit with smaller signal amplitudes (<5 mOD) (Figure 3). The lower ET yield in this variant may be a consequence of nonproductive oxidation of other nearby aromatic residues or less efficient competition with back electron transfer from reduced quencher. The results with RuC97(CYP102A1)W96 and RuC77(CYP119)H76W are in striking contrast to those of the Ru–P450 constructs with an intervening histidine residue (RuC97(CYP102A1)W96H and RuC77(CYP119)H76). Neither of the latter constructs reveals any evidence indicative of heme oxidation following photochemical generation of the Ru(diimine)33+ complex. In both cases, we observe only a bleach of the ground-state MLCT absorption features (390–440 nm) that returns to baseline within 200 μs. The spectral profiles and kinetics of these transients are nearly identical to that of the free photosensitizer ([Ru(bpy)2(Aphen)]2+) quenched with [Ru(NH3)6]3+ (Figure 2). These observations suggest that Ru(diimine)33+ oxidizes the P450 heme only in structures with an intervening tryptophan residue.
Reductive Quenching
Photochemical reduction of Ru–P450 conjugates was performed using pMeODMA as quencher. The resulting Ru+ species has nearly 1 eV of driving force for reduction of the ferric P450 heme: E°(Ru(bpy)32+/+ = −1.3 V vs NHE;33,34E°(P450 Fe3+/2+) = −0.43 V vs NHE.41 All four Ru–P450 conjugates exhibit similar transient absorption features under flash-quench conditions (Figure 4). Three distinct kinetics phases appear on the nanosecond, microsecond, and millisecond time scales following pulsed laser excitation of the photosensitizer. All transient absorption features decay back to baseline within a few hundred milliseconds, suggesting that the overall photochemical process is reversible. The fastest kinetics phase (τ ≈ 60 ns) is assigned to decay of *Ru(diimine)32+; transient absorption features associated with this state decay with the same time constant as the luminescence. Transient absorption at 510 nm, attributable to Ru(diimine)3+ and pMeODMA•+, develops with the same time constant. The subsequent microsecond kinetics phase is characterized by a bleach at 420 nm and increased absorbance at 440 nm, indicative of a red-shift in the heme Soret band. In the resting state of Fe3+-P450, the Fe center is axially ligated by cysteine thiolate and water ligands, producing a low-spin electronic configuration. Chemical reduction of Fe3+-P450 produces a five-coordinate high-spin ferrous heme with a blue-shifted Soret band.41 The red-shifted Soret observed following reductive quenching of the Ru–P450 conjugates is indicative of a low-spin ferrous heme.42 The microsecond heme reduction by Ru+ is likely faster than aquo ligand loss, resulting in a transient low-spin, six-coordinate ferrous species. This conclusion is consistent with the absorption profiles of Fe2+(CO)–CYP101 (P450cam from Pseudomonas putida), Fe2+(imidazole)–CYP101, and cryoreduced CYP101.43,44 The millisecond kinetics phase corresponds to reoxidation of the reduced heme by pMeODMA•+, resulting in a return to baseline of all transient absorption features and regeneration of Fe3+(OH2)–P450 and pMeODMA.
Figure 4.

Transient kinetics following reductive quenching (pMeODMA, 10 mM) of four Ru–P450 conjugates: λex = 480 nm; λobsd = 420 nm (green), 440 nm (red). Signals normalized to the magnitude of the 440 nm prompt bleach.
We performed a global least-squares analysis of the Ru–P450 reductive quenching kinetics recorded at 420 and 440 nm (and, for select mutants, 400, 410, 430 nm) to a three-exponential function with amplitude coefficients ρ1–3(λ) and rate constants γ1–3 (eq 1). We fixed the first observed rate constant to the value obtained from single-exponential fits to the quenched luminescence decay kinetics recorded at 630 nm. The remaining two rate constants, which were extracted from the global fitting, are listed in Table 2.
| 1 |
Table 2. Rate Constants for Ru-Sensitizer Quenching (γ1), Heme Reduction (γ2), and Heme Oxidation (γ3) Extracted from Global Fitting of Transient Absorption Data.
| enzyme | γ1 (s–1) | γ2 (s–1) | γ3 (s–1) |
|---|---|---|---|
| RuC97(CYP102A1)W96 | 1.6 × 107 | 3.6 × 104 | 1.1 × 102 |
| RuC97(CYP102A1)W96H | 1.6 × 107 | 6.0 × 104 | 1.9 × 102 |
| RuC77(CYP119)H76 | 1.9 × 107 | 5.7 × 104 | 1.4 × 102 |
| RuC77(CYP119)H76W | 1.9 × 107 | 8.1 × 104 | 1.3 × 102 |
The second rate constant corresponds to heme reduction. All four mutants exhibit very similar reduction rates, with maximum heme reduction complete at approximately 100 μs. Interestingly, after normalizing by the magnitude of the prompt *Ru2+ excited state bleach (440 nm), the magnitudes of the transient features associated with heme reduction (e.g., absorption at 440 nm) differ greatly among the four Ru–P450 conjugates. In particular, both CYP119 mutants exhibit absorption features that are greater by a factor of 2–3 than either of the CYP102A1 mutants (Figure 4).
All transient absorption features decay to baseline within 100 ms; this return rate is extremely sensitive to small amounts of oxygen. In our proposed flash-quench scheme, reoxidation of the ferrous center occurs via bimolecular recombination with pMeODMA•+. This recombination is expected to be a second-order process; however, the disappearance of the transient features is better modeled as a first-order process, possibly owing to reaction with oxygen or minor impurities.
Discussion
In a prior report on the kinetics of heme oxidation by Ru3+ in RuC97(CYP102A1)W96, we found spectroscopic evidence for stepwise oxidation of Fe3+(OH2)–heme to Fe4+(OH)–heme via an intermediate porphyrin radical cation. Electron transfer from the porphyrin to Ru3+C97 was remarkably rapid (8.5 × 105 s–1, pH 8), given the 20.8-Å distance from the Ru-center to the nearest aromatic carbon atom on the porphyrin ring (Figure 5).35 This result conflicts with the semiclassical ET theory prediction that the rate constant for this reaction should be 3 orders of magnitude smaller (4 × 102 s–1; −ΔG° = 0.2 eV; reorganization energy λ = 0.8 eV; distance decay factor β = 1.1 Å–1, see Supporting Information). Indeed, the failure to observe flash-quench induced heme oxidation in RuC77(CYP119)H76 is in better agreement with the slower predicted rate since porphyrin oxidation by Ru3+ apparently does not compete effectively with Ru3+ reduction by reduced quencher (complete in 100 μs).
Figure 5.
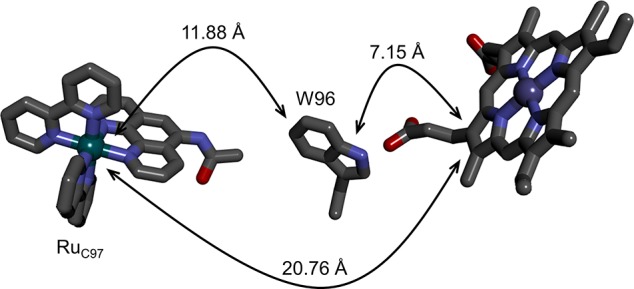
Structural model of RuC97–CYP102A1 (PDB ID 3NPL) highlighting the electron-transfer distances from RuC97 to the porphryin (20.76 Å), RuC97 to W96 (11.88 Å), and W96 to the porphyrin (7.15 Å).
The fact that we observe flash-quench heme oxidation only in Ru–P450s with intervening tryptophan residues strongly implicates the tryptophan radical cation as a reaction intermediate. Kinetics modeling of a stepwise hole-transfer reaction from Ru3+ to W96•+ to por•+, using distances taken from the RuC97(CYP102A1)W96 structure (PDB ID 3NPL)35 (Figure 5), predicts an apparent rate constant for porphyrin oxidation of 1.4 × 106 s–1 (Supporting Information), a value in remarkably good agreement with the experimentally derived quantity. The rate constant for hole transfer from W96•+ to por•+ is predicted to be more than 2 orders of magnitude greater, suggesting that a negligibly small concentration of W96•+ will build up during the porphyrin oxidation process (Figure 6).
Figure 6.

Photochemical ET reaction scheme in RuC97(CYP102A1) and RuC77(CYP119). Blue arrows indicate excitation processes, solid green arrows indicate bimolecular quenching reactions, dashed green arrows indicate bimolecular charge-recombination processes with quencher redox products, and red arrows indicate intraprotein ET reactions. With an intervening W residue (a, CYP102A, W96; CYP119 H76W), oxidative quenching of *Ru2+ by QO (left path) leads to heme oxidation via an intermediate Trp radical; reductive quenching by QR (right path) leads to heme reduction in a single-step tunneling reaction. With an intervening H residue (b, CYP102A, W96H; CYP119 H76), oxidative quenching of *Ru2+ by QO produces Ru3+ but not heme oxidation, whereas reductive quenching again leads to single-step electron transfer from Ru+ to the heme.
The kinetics of the high-potential heme oxidation reaction are in striking contrast to those of the low-potential heme reduction reaction. The estimated driving force for the Ru+ to Fe3+(OH2)–heme ET is 0.9 eV, close to the reorganization energy estimate of 0.8 eV. The ET distance is somewhat ambiguous for this reaction because the transferring electron could be localized on any one of the three diimine ligands. On the basis of the RuC97(CYP102A1) crystal structure,35 the shortest distances from any diimine ligand to the Fe center range from 19.5 to 23.8 Å. Semiclassical ET theory predicts that the rate constant for reactions over this distance range will be 103–105 s–1, in accord with the γ2 values listed in Table 2. Moreover, the rate constants for heme reduction vary by no more than a factor of 2 among the four conjugates. We conclude from this analysis that reduction of Fe3+(OH2)–heme by RuC97+ in CYP102A1 and by RuC77+ in CYP119 involves single step electron tunneling and that the intervening tryptophan (W96, W76) and histidine (H96, H76) residues serve only to mediate the superexchange coupling between the two redox sites (Figure 6).
The heme oxidation and reduction kinetics in RuC97(CYP102A1) and RuC77(CYP119) highlight the asymmetry between high and low potential ET reactions in proteins. The electron-tunneling timetables extracted from our studies of ET in Ru-modified proteins provide benchmarks for single-step electron tunneling in which the reduction potential of the oxidant is <1 V vs NHE. Our studies of Ru–P450 clearly demonstrate that multistep tunneling reactions via tryptophan (and tyrosine) radicals come into play when oxidants have potentials >1 V. In addition, the radicals of the sulfur-containing amino acids also might be participants in reactions with particularly high potential oxidants. For enzymes functioning with intermediates at potentials greater than 1 V, protein structure and composition are critically important factors that ensure oxidizing equivalents are delivered to intended targets rather than diffusing to low potential sinks via multistep tunneling. The obvious corollary is that strategic placement of tryptophan and tyrosine residues in enzymes can direct the flow of oxidizing equivalents over long distances with little loss of potential.
Glossary
Abbreviations
- ET
electron transfer
- MLCT
metal-to-ligand charge transfer
- [Ru(bpy)2(IAphen)]2+
[Ru(2,2′-bipyridine)2(5-iodoacetamido 1,10-phenanthroline)]2+
- [Ru(bpy)2(Aphen)]2+
[Ru(2,2′-bipyridine)2(5-acetamido-1,10-phenanthroline)]2+
- pMeODMA
p-methoxydimethylaniline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00432.
Additional experimental details, spectra of ferric and ferrous wild-type P450-BM3 and the ferrous–ferric difference spectrum, details of electron transfer rate calculations, and estimated electron transfer rate constants (PDF)
Author Present Address
† M.E.E.: Department of Chemistry, Yale University, 225 Prospect St., P.O. Box 208107, New Haven, CT 06520, U.S.A.
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK019038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was provided by the Arnold and Mabel Beckman Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Heinze J. (2016) Aliphatic and Aromatic Hydrocarbons - Reduction, in Organic Electrochemistry (Hammerich O., and Speiser B., Eds.), pp 861–890, Taylor and Francis Group, Boca Raton, FL. [Google Scholar]
- Holton D. M.; Edwards P. P.; Salmon G. A. (1984) Electrons, alkali metal-electron species and radical anions in substituted organic amides. J. Phys. Chem. 88, 3855–3859. 10.1021/j150661a034. [DOI] [Google Scholar]
- Knecht L. A.; Kolthoff I. M. (1962) N-Methylacetamide as a Polarographic Solvent. Inorg. Chem. 1, 195–203. 10.1021/ic50002a002. [DOI] [Google Scholar]
- Harriman A. (1987) Further Comments on the Redox Potentials of Tryptophan and Tyrosine. J. Phys. Chem. 91, 6102–6104. 10.1021/j100308a011. [DOI] [Google Scholar]
- Fourré I.; Bergès J.; Houée-Levin C. (2010) Structural and Topological Studies of Methionine Radical Cations in Dipeptides: Electron Sharing in Two-Center Three-Electron Bonds. J. Phys. Chem. A 114, 7359–7368. 10.1021/jp911983a. [DOI] [PubMed] [Google Scholar]
- Glass R. S.; Hug G. L.; Schoneich C.; Wilson G. S.; Kuznetsova L.; Lee T. M.; Ammam M.; Lorance E.; Nauser T.; Nichol G. S.; Yamamoto T. (2009) Neighboring Amide Participation in Thioether Oxidation: Relevance to Biological Oxidation. J. Am. Chem. Soc. 131, 13791–13805. 10.1021/ja904895u. [DOI] [PubMed] [Google Scholar]
- Glass R. S.; Petsom A.; Hojjatie M.; Coleman B. R.; Duchek J. R.; Klug J.; Wilson G. S. (1988) Facilitation of Electrochemical Oxidation of Dialkyl Sulfides Appended with Neighboring Carboxylate and Alcohol Groups. J. Am. Chem. Soc. 110, 4772–4778. 10.1021/ja00222a040. [DOI] [Google Scholar]
- Jovanovic S. V.; Harriman A.; Simic M. G. (1986) Electron-Transfer Reactions of Tryptophan and Tyrosine Derivatives. J. Phys. Chem. 90, 1935–1939. 10.1021/j100400a039. [DOI] [Google Scholar]
- Surdhar P. S.; Armstrong D. A. (1987) Reduction potentials and exchange reactions of thiyl radicals and disulfide anion radicals. J. Phys. Chem. 91, 6532–6537. 10.1021/j100310a022. [DOI] [Google Scholar]
- Yashiro H.; White R. C.; Yurkovskaya A. V.; Forbes M. D. E. (2005) Methionine radical cation: Structural studies as a function of pH using X- and Q-band time-resolved electron paramagnetic resonance spectroscopy. J. Phys. Chem. A 109, 5855–5864. 10.1021/jp051551k. [DOI] [PubMed] [Google Scholar]
- Navaratnam S.; Parsons B. J. (1998) Reduction Potential of Histidine Free Radicals: a Pulse Radiolysis Study. J. Chem. Soc., Faraday Trans. 94, 2577–2581. 10.1039/a803477j. [DOI] [Google Scholar]
- Stubbe J.; van der Donk W. A. (1998) Protein Radicals in Enzyme Catalysis. Chem. Rev. 98, 705–762. 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- Aubert C.; Mathis P.; Eker A. P. M.; Brettel K. (1999) Intraprotein Electron Transfer between Tyrosine and Tryptophan in DNA photolysase from Anacystis nidulans. Proc. Natl. Acad. Sci. U. S. A. 96, 5423–5427. 10.1073/pnas.96.10.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert C.; Vos M. H.; Mathis P.; Eker A. P. M.; Brettel K. (2000) Intraprotein Radical Transfer during Photoactivation of DNA Photolyase. Nature 405, 586–590. 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- DeGray J. A.; Lassmann G.; Curtis J. F.; Kennedy T. A.; Marnett L. J.; Eling T. E.; Mason R. P. (1992) Spectral-Analysis of the Protein-Derived Tyrosyl Radicals from Prostaglandin-H Synthase. J. Biol. Chem. 267, 23583–23588. [PubMed] [Google Scholar]
- Eklund H.; Eriksson M.; Uhlin U.; Nordlund P.; Logan D. (1997) Ribonucleotide Reductase - Structural Studies of a Radical Enzyme. Biol. Chem. 378, 821–825. [PubMed] [Google Scholar]
- Goodin D. B.; Mauk A. G.; Smith M. (1986) Studies of the Radical Species in Compound ES of Cytochrome c Peroxidase Altered by Site-Directed Mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 83, 1295–1299. 10.1073/pnas.83.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T. (1999) Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc. 121, 7939–7940. 10.1021/ja991541v. [DOI] [Google Scholar]
- Licht S.; Gerfen G. J.; Stubbe J. A. (1996) Thiyl radicals in ribonucleotide reductases. Science 271, 477–481. 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- Pogni R.; Baratto M. C.; Teutloff C.; Giansanti S.; Ruiz-Dueñas F. J.; Choinowski T.; Piontek K.; Martínez A. T.; Lendzian F.; Basosi R. (2006) A Tryptophan Neutral Radical in the Oxidized State of Versatile Peroxidase from Pleurotus eryngii: a Combined Multifrequency EPR and Density Functional Theory Study. J. Biol. Chem. 281, 9517–9526. 10.1074/jbc.M510424200. [DOI] [PubMed] [Google Scholar]
- Bernini C.; Pogni R.; Basosi R.; Sinicropi A. (2012) The nature of tryptophan radicals involved in the long-range electron transfer of lignin peroxidase and lignin peroxidase-like systems: Insights from quantum mechanical/molecular mechanics simulations. Proteins: Struct., Funct., Genet. 80, 1476–1483. 10.1002/prot.24046. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. (2015) Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U. S. A. 112, 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P.; Yamamoto J.; Martin R.; Iwai S.; Brettel K. (2015) Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6–4) photolyases. Chem. Commun. 51, 15502–15505. 10.1039/C5CC06276D. [DOI] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. (2015) Electron flow through biological molecules: does hole hopping protect proteins from oxidative damage?. Q. Rev. Biophys. 48, 411–420. 10.1017/S0033583515000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan M.; English A. M. (2017) LC-MS/MS suggests that hole hopping in cytochrome c peroxidase protects its heme from oxidative modification by excess H2O2. Chem. Sci. 8, 1152–1162. 10.1039/C6SC03125K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R., Ed. (2015) Cytochrome P450 - Structure, Mechanism, and Biochemistry, Springer, Switzerland. [Google Scholar]
- Green M. T.; Dawson J. H.; Gray H. B. (2004) Oxoiron(IV) in Chloroperoxidase Compound II is Basic: Implications for P450 Chemistry. Science 304, 1653–1656. 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- Behan R. K.; Hoffart L. M.; Stone K. L.; Krebs C.; Green M. T. (2006) Evidence for basic ferryls in cytochromes P450. J. Am. Chem. Soc. 128, 11471–11474. 10.1021/ja062428p. [DOI] [PubMed] [Google Scholar]
- Krest C. M.; Onderko E. L.; Yosca T. H.; Calixto J. C.; Karp R. F.; Livada J.; Rittle J.; Green M. T. (2013) Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem. 288, 17074–17081. 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. (2010) Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 330, 933–937. 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. (2013) Iron(IV)hydroxide pK(a) and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 342, 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi A. R.; Dawson J. H.; Hrycay E. G.; Bandiera S. M. (2015) Oxidizing Intermediates in P450 Catalysis: A Case for Multiple Oxidants. Adv. Exp. Med. Biol. 851, 63–81. 10.1007/978-3-319-16009-2_2. [DOI] [PubMed] [Google Scholar]
- Sutin N., and Creutz C. (1978) Properties and Reactivities of the Luminescent Excited States of Polypyridine Complexes of Ruthenium(II) and Osmium(II), in Inorganic and Organometallic Photochemistry (Wrighton M. S., Ed.), pp 1–27, American Chemical Society, Washington, DC. [Google Scholar]
- Creutz C.; Chou M.; Netzel T. L.; Okumura M.; Sutin N. (1980) Lifetimes, Spectra, and Quenching of the Excited-States of Polypyridine Complexes of Iron(II), Ruthenium(II), and Osmium(II). J. Am. Chem. Soc. 102, 1309–1319. 10.1021/ja00524a014. [DOI] [Google Scholar]
- Ener M. E.; Lee Y. T.; Winkler J. R.; Gray H. B.; Cheruzel L. (2010) Photooxidation of cytochrome P450-BM3. Proc. Natl. Acad. Sci. U. S. A. 107, 18783–18786. 10.1073/pnas.1012381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan H. M.; Seward H. E.; Toogood H. S.; Cheesman M. R.; Leys D.; Munro A. W. (2007) Structural and spectroscopic characterization of P450BM3 mutants with unprecedented P450 heme iron ligand sets - New heme ligation states influence conformational equilibria in P450BM3. J. Biol. Chem. 282, 564–572. 10.1074/jbc.M607949200. [DOI] [PubMed] [Google Scholar]
- Park S.-Y.; Yamane K.; Adachi S.-i.; Shiro Y.; Weiss K. E.; Maves S. A.; Sligar S. G. (2002) Thermophilic cytochrome P450 (CYP119) from Sulfolobus solfataricus: high resolution structure and functional properties. J. Inorg. Biochem. 91, 491–501. 10.1016/S0162-0134(02)00446-4. [DOI] [PubMed] [Google Scholar]
- Castellano F. N.; Dattelbaum J. D.; Lakowicz J. R. (1998) Long-lifetime Ru(II) complexes as labeling reagents for sulfhydryl groups. Anal. Biochem. 255, 165–170. 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- Mines G. A.; Bjerrum M. J.; Hill M. G.; Casimiro D. R.; Chang I.-J.; Winkler J. R.; Gray H. B. (1996) Rates of Heme Oxidation and Reduction in Ru(His33)cytochrome c at Very High Driving Forces. J. Am. Chem. Soc. 118, 1961–1965. 10.1021/ja9519243. [DOI] [Google Scholar]
- Munro A. W.; Malarkey K.; McKnight J.; Thomson A. J.; Kelly S. M.; Price N. C.; Lindsay J. G.; Coggins J. R.; Miles J. S. (1994) The Role of Tryptophan-97 of Cytochrome P450-BM3 from Bacillus megaterium in Catalytic Function - Evidence Against the Covalent Switching Hypothesis of P450 Electron Transfer. Biochem. J. 303, 423–428. 10.1042/bj3030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. (2013) A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whited C. A.; Belliston-Bittner W.; Dunn A. R.; Winkler J. R.; Gray H. B. (2009) Nanosecond photoreduction of inducible nitric oxide synthase by a Ru-diimine electron tunneling wire bound distant from the active site. J. Inorg. Biochem. 103, 906–911. 10.1016/j.jinorgbio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn A. R.; Dmochowski I. J.; Winkler J. R.; Gray H. B. (2003) Nanosecond Photoreduction of Cytochrome P450cam by Channel-Specific Ru-Diimine Electron Tunneling Wires. J. Am. Chem. Soc. 125, 12450–12456. 10.1021/ja0294111. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sugar S. G. (2002) Cryoradiolysis for the Study of P450 Reaction Intermediates. Methods Enzymol. 357, 103–115. 10.1016/S0076-6879(02)57670-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.