

Abstract
Vitepyrroloids A–D (1–4), four new 2-cyano-substituted pyrrole-ring-containing labdane diterpenoids, were isolated from the leaves of Vitex trifolia. Their structures were elucidated based on spectroscopic data analysis. The absolute configuration of compound 1 was determined by X-ray diffraction. Compounds 1–4 are unprecedented labdane diterpenoids featuring a 2-cyano-substituted pyrrole ring. Compound 1 showed cytotoxic activity against a human nasopharyngeal carcinoma cell line (CNE1) with an IC50 value of 8.7 μM.
Graphical abstract
Vitex trifolia L. (Verbenaceae), a small deciduous shrub, is widely distributed in Fujian, Guangdong, Guangxi, and Yunnan provinces in mainland China. The fruits of V. trifolia have been traditionally used for the treatment of colds, migraine, headache, and rheumatism.1 The leaf extract has been reported to show cytotoxic activity.2 Previous studies on this plant have focused on the isolation of various diterpenoids, especially the labdane diterpenoids.3–5 Labdane-type diterpenes possess significant biological properties, such as cytotoxic,6 antibacterial,7 and anti-inflammatory activities.8 Herein, four novel labdane diterpenoid alkaloids, vitepyrroloids A–D (1–4), were isolated from the leaves of V. trifolia. Compounds 1–4 are unprecedented labdane diterpenoid alkaloids containing a cyano-substituted pyrrole cyclic system. In compounds 2–4, the pyrrole amino proton is replaced by ethyl butanoate, butanoic acid, and hydroxyethyl groups, respectively. The structures of compounds 1–4 were elucidated via spectroscopic data analysis, and the absolute configuration of 1 was determined by X-ray crystallographic analysis. Compound 1 was evaluated for cytotoxic activity against a well-differentiated human nasopharyngeal carcinoma cell line (CNE1) and a poorly differentiated human nasopharyngeal carcinoma (CNE2) cell line.
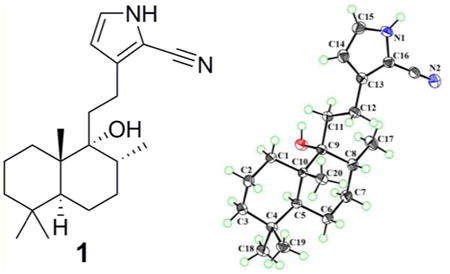
Compound 1 was obtained as colorless crystals with a molecular formula of C21H32N2O based on the HREIMS ion at m/z 328.2507 [M]+ (calcd for C21H32N2O, 328.2509), suggesting seven degrees of unsaturation. The IR spectrum showed absorptions typical for hydroxy (3348 cm−1), amino (3601 cm−1), and C≡N (2210 cm−1) groups. The 1H NMR spectrum indicated the presence of three methyl singlets [δH 0.82 (3H, s), 0.86 (3H, s), 0.93 (3H, s)] and one methyl doublet [δH 0.94 (3H, d, J = 6.6 Hz)]. A secondary amide group was revealed by the broad singlet at δH 8.74 (1H, br s). The 13C NMR spectrum (Table 1) of 1 exhibited 21 carbon signals, including four methyls (δC 16.6, 34.0, 22.2, 16.4), seven sp3 methylenes (δC 32.1, 18.9, 42.0, 21.9, 31.5, 36.0, 23.5), two sp3 methines (δC 46.6, 36.8), two sp2 methines (δC 110.2, 123.5), an oxygenated tertiary carbon (δC 77.3), two sp3 quaternary carbons (δC 33.6, 43.5), and three carbons without hydrogens at low field (δC 138.1, 99.3, 114.6). The aforementioned data further confirmed the presence of a cyano group, which was implied by the IR spectrum (2210 cm−1). 1H–1H COSY correlations of NH/H-15/H-14 (Figure 1) indicated a –NH–CH–CH– spin system. The small coupling constants for H-14 (J = 2.8 Hz) and H-15 (J = 2.8 Hz) suggested a pyrrole cyclic system. Four olefinic carbons, one C≡N bond, and one pyrrole cyclic system accounted for five out of the seven degrees of unsaturation, suggesting a dicyclic diterpenoid skeleton for 1. Detailed analysis of the 2D NMR spectrum suggested compound 1 is a labdane-type diterpenoid alkaloid.10 The HMBC cross-peaks of H-14/C-12, C-16; H-12/C-13, C-16; and H-15/C-13, C-16 and the weak cross-peak from H-15 to the cyano carbon indicated the presence of a 2-cyano-substituted pyrrole ring, which was confirmed by the base peak m/z 105 in the EIMS (Figure S2, Supporting Information). The HMBC cross-peaks of H-12/C-9 and H-11/C-13 (Figure 1) revealed the 2D structure of 1 as shown. Key NOESY correlations of H3-19/H3-20, H3-20/H-8, H3-20/H-11, and H3-18/H-5 suggested that CH3-19, CH3-20, and H-8 are cofacial, and these were assigned as β-oriented, with CH3-17 and CH3-18 having an α-orientation. Crystals of 1 were obtained from MeOH and subjected to X-ray diffraction using Cu Kα radiation (Figure 2). Thus, the absolute configuration of compound 1, given the trivial name vitepyrroloid A, was defined as 5S, 8R, 9R, and 10S.
Table 1. 1H and 13C NMR Chemical Shifts (δ) of Compounds 1–4a.
| 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| position | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC |
| 1a | 1.50, overlap | 32.1 | 1.48, overlap | 32.0 | 1.67, overlap | 29.9 | 1.49, overlap | 33.8 |
| 1b | 1.50, overlap | 1.48, overlap | 1.51, overlap | 1.49, overlap | ||||
| 2a | 1.56, overlap | 18.9 | 1.54, overlap | 18.8 | 1.66, overlap | 23.8 | 1.61, m | 18.9 |
| 2b | 1.48, overlap | 1.47, overlap | 1.66, overlap | 1.49, m | ||||
| 3a | 1.33, overlap | 42.0 | 1.32, overlap | 41.9 | 4.44, dd (10.8, 4.0) | 81.0 | 1.30, m | 43.9 |
| 3b | 1.14, td (12.9, 3.7) | 1.13, td (12.9, 3.4) | 1.15, td (13.3, 3.2) | |||||
| 4 | 33.6 | 33.5 | 37.9 | 34.2 | ||||
| 5 | 1.40, m | 46.6 | 1.40, m | 46.6 | 1.55, m | 45.8 | 1.63, d (1.8) | 47.7 |
| 6a | 1.52, overlap | 21.9 | 1.51, overlap | 21.8 | 1.51, overlap | 21.3 | 5.37, q-like (2.4) | 70.4 |
| 6b | 1.29, overlap | 1.28, overlap | 1.28, overlap | |||||
| 7a | 1.48, overlap | 31.5 | 1.48, overlap | 31.5 | 1.48, overlap | 31.2 | 1.60, overlap | 36.3 |
| 7b | 1.28, overlap | 1.27, overlap | 1.32, overlap | 1.55, overlap | ||||
| 8 | 1.80, m | 36.8 | 1.78, m | 36.8 | 1.76, m | 36.8 | 2.12, m | 31.9 |
| 9 | 77.3 | 77.2 | 77.2 | 77.2 | ||||
| 10 | 43.5 | 43.4 | 43.0 | 43.9 | ||||
| 11a | 1.92, ddd (14.4, 11.7, 6.3) | 36.0 | 1.89, ddd (14.4, 11.8, 6.2) | 35.9 | 1.90, ddd (14.6, 9.9, 7.7) | 35.8 | 1.92, ddd (14.4, 11.9, 6.1) | 35.8 |
| 11b | 1.69, ddd (14.4, 11.3, 5.9) | 1.67, ddd (14.4, 11.5, 5.9) | 1.69, overlap | 1.79, ddd (14.4, 11.5, 5.6) | ||||
| 12 | 2.66, m | 23.5 | 2.61, m | 23.8 | 2.62, m | 23.8 | 2.65, m | 23.7 |
| 13 | 138.1 | 138.3 | 138.1 | 138.0 | ||||
| 14 | 6.11, t (2.8) | 110.2 | 6.00, d (2.6) | 109.4 | 6.00, d (2.6) | 109.6 | 6.03, d (2.6) | 109.4 |
| 15 | 6.82, t (2.8) | 123.5 | 6.70, d (2.6) | 126.5 | 6.71, d (2.6) | 126.7 | 6.83, d (2.6) | 127.5 |
| 16 | 99.3 | 102.0 | 102.1 | 102.1 | ||||
| 17 | 0.94, d (6.6) | 16.6 | 0.93, d (6.8) | 16.6 | 0.94, d (6.8) | 16.4 | 0.96, d (6.8) | 16.3 |
| 18 | 0.86, s | 34.0 | 0.85, s | 33.9 | 0.85, s | 28.5 | 0.94, s | 33.8 |
| 19 | 0.82, s | 22.2 | 0.81, s | 22.2 | 0.84, s | 16.7 | 0.98, s | 23.9 |
| 20 | 0.93, s | 16.4 | 0.92, s | 16.4 | 0.93, s | 16.3 | 1.24, s | 19.3 |
| CN | 114.6 | 114.0 | 114.0 | 114.1 | ||||
| 1′ | 4.01, t (6.9) | 48.0 | 4.04, t (7.0) | 47.9 | 4.11, t (5.2) | 51.3 | ||
| 2′ | 2.09, quintet (6.9) | 26.4 | 2.10, m | 26.2 | 3.91, t (5.2) | 62.3 | ||
| 3′ | 2.26, t (6.9) | 30.9 | 2.30, m | 30.3 | ||||
| 4′ | 172.6 | 176.0 | ||||||
| 5′ | 4.12, q (7.1) | 60.9 | ||||||
| 6′ | 1.24, t (7.1) | 14.4 | ||||||
| OAc-3 | 171.4 | |||||||
| 2.03, s | 21.6 | |||||||
| OAc-6 | 170.8 | |||||||
| 2.03, s | 22.2 | |||||||
| NH | 8.74, br s | |||||||
1H NMR measured at 400 MHz, 13C NMR measured at 100 MHz, and spectra obtained in CDCl3 with TMS as internal standard. Assignments were supported with HSQC and HMBC NMR spectra.
Figure 1.
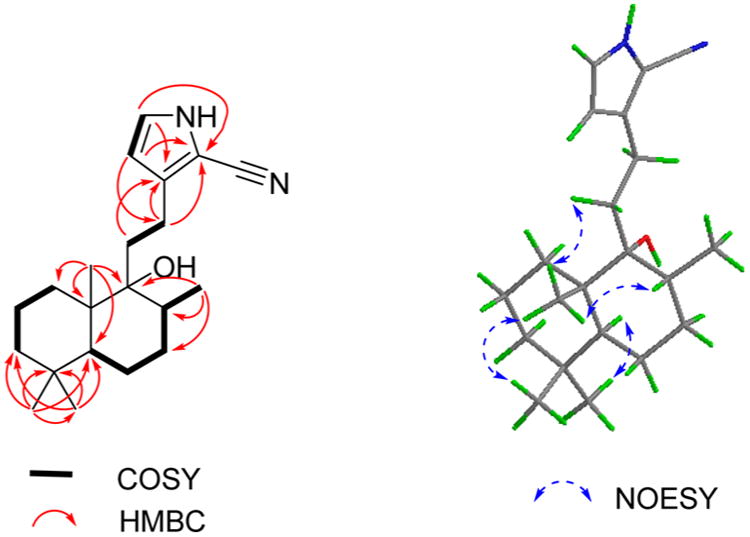
1H–1H COSY (bold), selected HMBC (red arrows), and key NOESY (blue arrows) correlations for compound 1.
Figure 2.
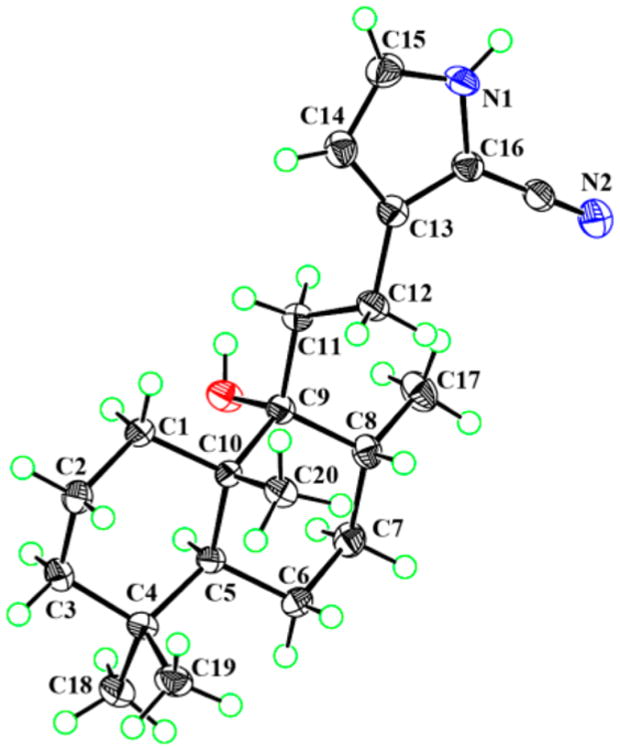
PLATON drawing of compound 1.
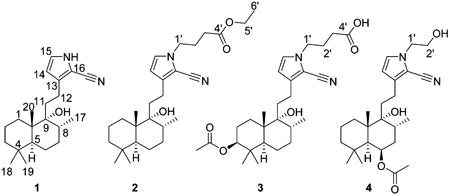
Compound 2 exhibited a molecular formula of C27H42N2O3 as determined by the HREIMS ion at m/z 442.3188 [M]+ (calcd for C27H42N2O3, 442.3190). Its IR spectrum showed absorptions for hydroxy (3394 cm−1) and cyano (2210 cm−1) groups. The NMR data of 2 were closely similar to those of 1, except for the presence of three methylenes, an ethoxy group, and an ester carbonyl group. Moreover, the amino proton observed in the 1H NMR spectrum of 1 was absent from the spectrum 2, indicating the occurrence of a substituent on the pyrrole nitrogen of 2. The COSY cross-peaks of H-1′/H-2′/H-3′ and H-5′/H-6′ revealed the presence of –CH2–CH2–CH2– and –OCH2CH3 moieties (Figure S14, Supporting Information), with the correlations of H-3′ and H-5′ with C-4′ in the HMBC spectrum indicating a –(CH2)3COOC2H5 moiety. Furthermore, the observation of HMBC cross-peaks of H-1′/C-15, C-16 and of H-15/C-1′ suggested that the ethyl butyrate moiety is connected to the nitrogen atom (Figure S16, Supporting Information). The relative configuration of 2 was assigned as being the same as that of 1 based on a detailed NOESY analysis (Figure S17, Supporting Information). Biogenetically, the structure of compound 2 should have the same absolute configuration as that of 1. Thus, compound 2 (vitepyrroloid B) was determined as 5S, 8R, 9R, and 10S.
Compound 3 was obtained as an amorphous powder and gave the molecular formula C27H40N2O5 based on its sodiated molecular ion peak in the HRESIMS at m/z 495.2824 [M + Na]+ (calcd 495.2829). Its IR spectrum exhibited characteristic absorptions for hydroxy (3398 cm−1) and cyano (2210 cm−1) groups. In comparison to 2, the 1H and 13C NMR data of 3 (Table 1) did not show signals of an ethoxy group, but it did contain those for an acetoxy group. The HMBC cross-peaks from the oxygenated methine proton (δH 4.44) to C-18 (δC 28.5), C-19 (δC 16.7), and the acetoxy carbon (δC 171.4) suggested that the acetoxy group is located at C-3 (Figure 3). The COSY cross-peaks of H-1′/H-2′/H-3′ in combination with the correlations from H-1′ to C-15 and C-16 in the HMBC spectrum indicated a butyric acid moiety to be connected to the pyrrole nitrogen. Key NOESY correlations of H3-19/H3-20, H3-20/H-8, H3-20/H-11, H3-18/H-5, H3-18/H-3, and H-3/H-5 (Figure 3) suggested that CH3-19, CH3-20, and H-8 are cofacial, and they were assigned as β-oriented, with H-3, CH3-17, and CH3-18 α-oriented. On the basis of comparison with 1, compound 3 (vitepyrroloid C) was established as 3S, 5S, 8R, 9R, and 10S. Moreover, it is worth mentioning that the protonated molecular ion peak at m/z 473.3009 [M + H]+ for compound 3 was observed in an LC-HRESIMS experiment on the original MeOH extract of V. trifolia (Supporting Information, Figure S34). This result clearly indicated that compounds featuring a 2-cyano-substituted pyrrole ring exist in the crude product of the plant and, thus, are not artifact products.
Figure 3.
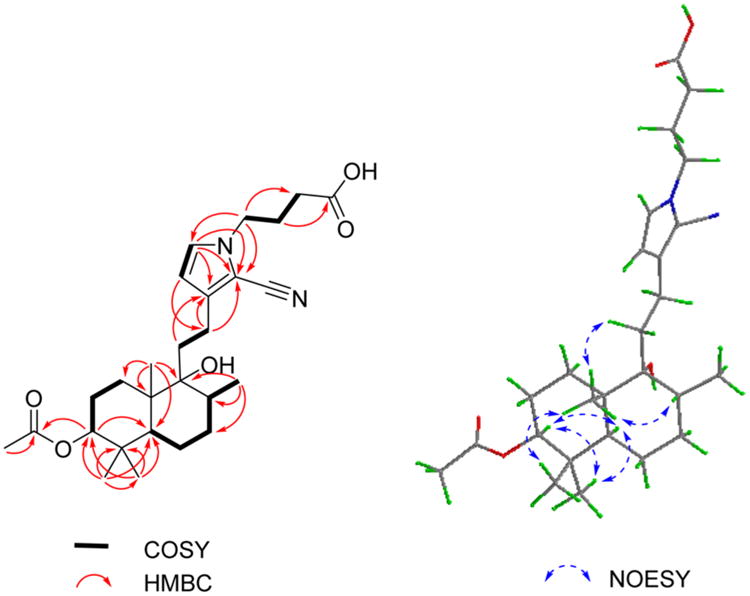
1H–H COSY (bold), selected HMBC (red arrows), and key NOESY (blue arrows) correlations for compound 3.
The molecular formula of compound 4 was determined as C25H38N2O4 from the sodiated HRESIMS ion at m/z 453.2717 [M + Na]+ (calcd for C25H38N2O4Na, 453.2724). The 1H and 13C NMR data (Table 1) indicated 4 to be closely similar to 3 structurally, except for the absence of two sp3 CH2 units and a carbonyl carbon and the presence of an oxygenated methylene group. The COSY cross-peaks from H-5 (δH 1.63) to the oxygenated methine proton H-6 (δH 5.37) suggested that the acetoxy group in 4 is connected to C-6 (Figure S30, Supporting Information). The COSY correlations of H-1′/H-2′ and the HMBC cross-peaks of H-1′/C-15 and H-1′/C-16 indicated that a –CH2CH2OH moiety is located at the nitrogen atom of the pyrrole ring (Figures S30 and S32, Supporting Information). The NOESY correlations of H3-19/H3-20, H3-20/H-8, H3-20/H-11, H3-18/H-5, H3-18/H-6, and H-6/H-5 were consistent with CH3-19, CH3-20, and H-8 being cofacial with β-orientations, with H-6, CH3-17, and CH3-18 assigned as α-oriented (Figure S33, Supporting Information). Accordingly, the structure of compound 4 (vitepyrroloid D) was determined as 5S, 6R, 8R, 9R, and 10S.
Compounds 1–4 were evaluated for in vitro cytotoxicity against the CNE1 and CNE2 cell lines. As shown in Table S1 in the Supporting Information, compound 1 was the most active isolated compound against these two cell lines, with IC50 values of 8.7 and >10 μM, respectively. Compound 1 is an unprecedented labdane diterpenoid alkaloid containing a cyano moiety and a pyrrole ring. The N–H group may be necessary for manifestation of cytotoxic activity, because the less active compounds 2–4 are derivatives of 1, but are substituted at the pyrrole ring nitrogen atom. Compounds 1–4 may be derived from the precursors of geranylgeranyl pyrophosphate (GGPP), ammonia, and an amino acid as shown in Figure 4. Protonation of GGPP can initiate a concerted cyclization sequence, terminated by loss of a proton from a methyl, yielding (+)-copayl PP. Next, sequential oxidation and reduction could produce intermediate i. A subsequent attack on intermediate i by ammonia (NH3) or an amino acid (L-glutamic acid or L-serine) followed by decarboxylation would give intermediate ii, which could be converted to intermediate iii via Schiff base and Mannich reactions. Finally, oxidation of the intermediate iii followed by the loss of CO2 and H2O leads to intermediate v or compound 1. Oxidation and acetylation of the intermediate v gave compounds 3 and 4. Compound 2 could be yielded from the intermediated v via estenification using ethanol (Figure 4).11−14
Figure 4.
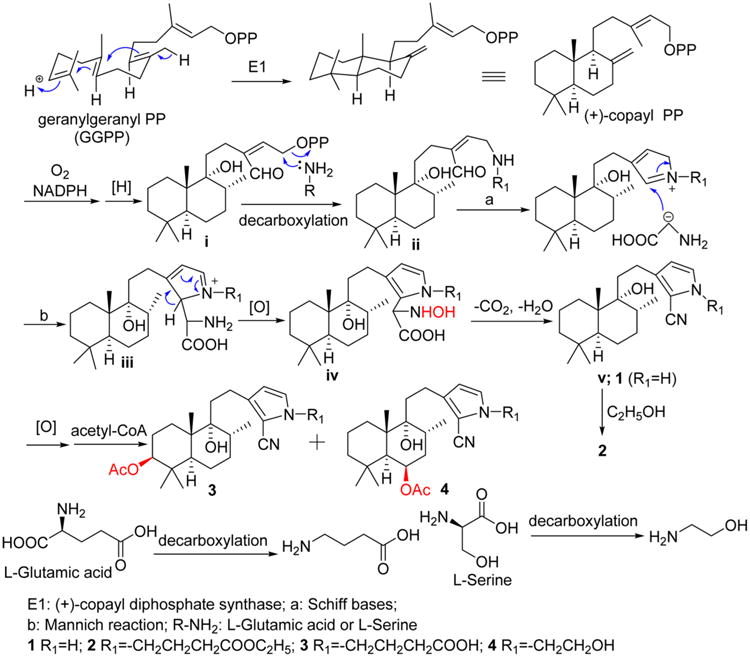
Proposed biosynthetic origin of compounds 1–4.
Experimental Section
General Experimental Procedures
The melting point was measured on an X-5 melting point instrument and was uncorrected. Optical rotations were obtained with a PerkinElmer 341 automatic polarimeter. UV spectra were recorded with MeOH as the solvent using a Shimadzu UV-2450 spectrophotometer. IR spectra were determined on a Bruker Tensor 37 infrared spectrophotometer with KBr pellets. The 1D and 2D NMR spectra were obtained on a Bruker AM-400 NMR spectrometer with tetramethylsilane (TMS) as an internal reference. HREIMS were measured on a Thermo MAT95XP high-resolution mass spectrometer, and EIMS on a Thermo DSQ EIMS spectrometer. HRESIMS were acquired on a Shimadzu LCMS-IT-TOF instrument, and the ESIMS on an Agilent 1200 series LC-MS/MS system. RP-C18 silica gel (Fuji, 40–75 μm), MCI gel (CHP20P, 75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan), silica gel (200–300 Mesh Marine Chemical Ltd., Qingdao, People’s Republic of China), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden) were used for column chromatography (CC). Semipreparative HPLC separations were carried out on a LC-20AT Shimadzu liquid chromatography system with a YMC-Pack ODS-A column and an Agilent SB-C18 column (250 × 9.4 mm, 5 μm) connected with an SPD-M20A diode array detector.
Plant Material
The leaves of Vitex trifolia were collected in September 2014 from Xishuangbanna in Yunnan Province, People’s Republic of China, and were authenticated by Prof. Xinxin Zhou of the School of Chinese Materia Medica, Guangzhou University of Chinese Medicine. A voucher specimen (XG-2014002) has been deposited at the School of Pharmacy Sciences, Sun Yat-sen University.
Extraction and Isolation
The air-dried, powdered leaves of V. trifolia (30 kg) were percolated four times with 95% EtOH at room temperature for 3 days. After removing the organic solvent, the residue was partitioned with EtOAc–H2O. The EtOAc extract (1.5 kg) was subjected to silica gel CC using petroleum ether–EtOAc mixtures (1:0, 10:1, 3:1, 1:1, 1:4, 0:1 v/v) to obtain fractions A–F. Fraction C (100 g) was subjected to silica gel CC eluting with CH2Cl2–EtOAc (from 200:1 to 1:1) to provide six fractions, C1–C6. C3 (23 g) was loaded onto a Sephadex LH-20 column and eluted with CH2Cl2–MeOH (1:1) to yield two subfractions, C3a and C3b. Fraction C3a was further subjected to silica gel CC (CH2Cl2–MeOH, 300:1 to 95:5) to give four fractions, C3a1–C3a4. Subfraction C3a2 was purified by semipreparative HPLC (80% CH3CN in H2O, 1.5 mL/min) to yield 3 (5 mg) and 4 (4.6 mg). Fraction B (234 g) was further subjected to silica gel CC, by eluting with CH2Cl2–MeOH (from 300:1 to 30:1), to obtain three fractions, B1–B3. Fraction B1 (100 g) was separated using silica gel CC to give fraction B1a, which was further fractionated over a RP-C18 column (MeOH–H2O, 40% to 90%, v/v) to yield three fractions, B1a1–B1a3. Fraction B1a2 was subjected to silica gel CC (cyclohexane–EtOAc, 10:1 to 1:1) to give three subfractions, B1a2-1–B1a2-3. Subfraction B1a2-1 was separated by semipreparative HPLC (80% MeOH–H2O, 1.5 mL/min) to yield compounds 1 (8.5 mg) and 2 (7.4 mg).
Vitepyrroloid A (1): colorless crystals (MeOH); mp 145–146°C; [α]25D +10 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 247 (4.09) nm; IR (KBr) νmax 3601, 3348, 2922, 2848, 2210, 1645, 1462, 1410, 1385, 1255, 1119, 901, 758, 665, 579, 499 cm−1; 1H and 13C NMR data, see Table 1; HREIMS m/z 328.2507 [M]+ (calcd for C21H32N2O, 328.2509).
Vitepyrroloid B (2): amorphous powder; [α]25D +9 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 249 (3.96) nm; IR (KBr) νmax 3394, 2920, 2850, 2210, 1731, 1643, 1460, 1414, 1375, 1254, 1188, 1026, 752, 648, 499 cm−1; 1H and 13C NMR data, see Table 1; HREIMS m/z 442.3188 [M]+ (calcd for C27H42N2O3, 442.3190).
Vitepyrroloid C (3): amorphous powder; [α]25D +18 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 249 (3.61) nm; IR (KBr) νmax 3398, 3192, 2926, 2854, 2210, 1732, 1643, 1468, 1419, 1373, 1255, 1028, 970, 800, 648, 496 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 495.2824 [M + Na]+ (calcd for C27H40N2O5Na, 495.2829).
Vitepyrroloid D (4): amorphous powder; [α]25D –2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 250 (3.87) nm; IR (KBr) νmax 3396, 3192, 2924, 2848, 2212, 1728, 1645, 1466, 1414, 1375, 1057, 1020, 955, 750, 613, 499 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 453.2717 [M + Na]+ (calcd for C25H38N2O4Na, 453.2724).
X-ray Crystallographic Analysis of Compound 1
The crystal structure and absolute configuration of 1 were determined by single-crystal X-ray diffraction analysis. A suitable crystal was selected and tested on an Xcalibur, Onyx, Nova diffractometer with the temperature kept at 103(2) K during data collection. The structure was solved and refined, using the programs XS and olex2.refine, respectively. The program X-Seed was used as an interface to the SHELX programs and to prepare the figures. Crystal data for compound 1: C21H32N2O (M = 328.50); orthorhombic, space group P212121 (no. 19), a = 7.45094(6) Å, b = 9.21543(8) Å, c = 26.7264(2) Å, V = 1835.14(3) Å3; Z = 4, T = 103(2) K, μ(Cu Kα) = 0.558 mm−1, Dcalc = 1.1889 g/cm3, 13 373 reflections measured, 3577 unique (Rint = 0.0278, Rsigma = 0.0238), which were used in all calculations. The final R1 was 0.0298 and wR2 was 0.0738 (all data). Flack parameter = 0.2(2). Crystallographic data for 1 have been deposited at the Cambridge Crystallographic Data Centre under the reference number CCDC 1519552.
Cytotoxicity Assay
The well-differentiated human nasopharyngeal carcinoma (CNE1) and the poorly differentiated human nasopharyngeal carcinoma (CNE2) cell lines were obtained from Sun Yat-sen University Cancer Center and cultured on RPMI-1640 medium at 37°C in a humidified atmosphere with 5% CO2. The cells were seeded at 2000–4000 cells/well in 96-well plates. For the test compound treatment experiments, the cells were treated in triplicate with graded concentrations of each substance (predissolved in DMSO) for a period of 3 days. At the end of the compound treatment period, MTT solution (20 μL, 5 mg/mL) in PBS (PBS without MTT as the blank) was fed to each well of the culture plate (containing 100 μL of medium). After 4 h of incubation, the formazan crystals that formed in the well were dissolved with 100 μL of DMSO for optical density reading at 492 nm.15 All IC50 values were calculated by nonlinear regression analysis (GraphPad Prism).
Supplementary Material
Acknowledgments
This study was supported in part by the Guangdong Province Frontier and Key Technology Innovation Program (2015B010109004), the National Natural Science Foundation of China (Nos. 81471138, 81573310, 81371793), and the Medical Scientific Research Foundation of Guangdong Province (No. A2014212). Partial support was also provided by NIH CA177584 awarded to K.H.L.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.6b01195.
IR and NMR data for compounds 1–4, EIMS of compound 1, HREIMS for compounds 1 and 2, HRESIMS for 3 and 4, LC-HRESIMS for methanol extract of V. trifolia, and cytotoxicity for 1–4 (PDF) Crystallographic data (CIF)
Notes: The authors declare no competing financial interest.
References
- 1.Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China. Vol. 1. Beijing Chemical Industry Press; Beijing: 2010. pp. 340–341. [Google Scholar]
- 2.Ghani A. Medicinal Plants of Bangladesh: Chemical Constituents and Uses. Asiatic Society of Bangladesh; Dhaka: 1998. p. 320. [Google Scholar]
- 3.Tiwari N, Thakur J, Saikia D, Gupta MM. Phytomedicine. 2013;20:605–610. doi: 10.1016/j.phymed.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Zhou T, Zhang SW, Zhang XH, Xuan LJ. Planta Med. 2009;75:367–370. doi: 10.1055/s-0028-1112211. [DOI] [PubMed] [Google Scholar]
- 5.Zheng CJ, Zhu JY, Yu W, Ma XQ, Rahman K, Qin LP. J Nat Prod. 2013;76:287–291. doi: 10.1021/np300679x. [DOI] [PubMed] [Google Scholar]
- 6.Itokawa H, Morita H, Katou I, Takeya K, Cavalheiro AJ, de Oliveira RC, Ishige M, Motidome M. Planta Med. 1988;54:311–315. doi: 10.1055/s-2006-962442. [DOI] [PubMed] [Google Scholar]
- 7.Corlay N, Lecso-Bornet M, Leborgne E, Blanchard F, Cachet X, Bignon J, Roussi F, Butel MJ, Awang K, Litaudon M. J Nat Prod. 2015;78:1348–1356. doi: 10.1021/acs.jnatprod.5b00206. [DOI] [PubMed] [Google Scholar]
- 8.Zheng CJ, Huang BK, Wang Y, Ye Q, Han T, Zhang QY, Zhang H, Qin LP. Bioorg Med Chem. 2010;18:175–181. doi: 10.1016/j.bmc.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Bisset NG, Choudhury AK, Waleker MD. Phytochemistry. 1974;13:255–258. [Google Scholar]
- 10.Pal M, Li SH, Tewari SK, Sun HD. Chem Nat Compd. 2013;49:635–638. [Google Scholar]
- 11.Olafsdottir ES, Jorgensen LB, Jaroszewski JW. Phytochemistry. 1992;31:4129–4134. [Google Scholar]
- 12.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 3. John Wiley & Sons; U.K.: 2009. pp. 228–223.pp. 362–365. [Google Scholar]
- 13.Zagrobelny M, Bak S, Moller BL. Phytochemistry. 2008;69:1457–1468. doi: 10.1016/j.phytochem.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Jaroszewski JW, Jensen PS, Cornett C, Byberg JR. Biochem Syst Ecol. 1988;16:23–28. [Google Scholar]
- 15.Carmichael J, Degraff WG, Gazdar AF, Minna JD, Mitchell JB. Cancer Res. 1987;47:943–946. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.