

Abstract
The early steps of mammary tumorigenesis include loss of epithelial cell polarity, escape from anoikis, and acquisition of proliferative capacity. The genes responsible for these processes are predicted to be early diagnostic markers or new therapeutic targets. Here we tested 51 genes coamplified with ERBB2 in the 17q12–21 amplicon for these tumorigenic activities using an MCF10A 3D culture‐based screening system. We found that overexpression of retinoic acid receptor α (RARA) disrupted normal acinar structure and induced epithelial‐to‐mesenchymal transition (EMT). The mRNA levels of known EMT‐inducing factors, including SLUG, FOXC2, ZEB1, and ZEB2, were significantly increased upon RARA overexpression. Knockdown of ZEB1 suppressed the RARA‐mediated EMT phenotype. These results suggest that overexpression of RARA enhances malignant transformation during mammary tumorigenesis.
Keywords: 3D culture, Gene amplification, Breast cancer, EMT, ERBB2, RARA
Highlights
Retinoic acid receptor alpha (RARA) was identified as a transforming gene.
Enhanced expression of RARA triggered collapse of mammary luminal structures.
RARA overexpression induced epithelial‐to‐mesenchymal transition (EMT).
Knockdown of ZEB1 suppressed RARα‐induced EMT phenotypes.
Abbreviations
- Dox
doxycycline
- RARα
retinoic acid receptor alpha
- ERBB2
v-erbB-2 avian erythroblastic leukemia viral oncogene homolog B2
- ZEB
zinc finger E-box-binding homeobox
- FOXC2
forkhead box protein C2
- RXR
retinoid X receptor
- TGFB
transforming growth factor beta
- EMT
epithelial-to-mesenchymal transition
- DCIS
ductal carcinoma in situ
- IBC
invasive breast cancer
- MOI
multiplicity of infection
- TRE
tetracycline responsive element
- LBD
ligand-binding domain
- RARE
retinoic acid response element
- ER
estrogen receptor
- ATRA
all-trans retinoic acid
- BCSC
breast cancer stem cell
- MaSC
mammary stem cell
1. Introduction
The majority of breast cancers originate from the epithelial cell layers and progress through a continuum of changes to malignancy. Ductal carcinoma in situ (DCIS) is an early premalignant stage of breast cancer progression and is recognized as the proliferation of neoplastic epithelial cells within the duct, surrounded by myoepithelial cells and an intact basement membrane. DCIS is the most common type of non‐invasive breast cancer in women, yet has the potential to progress towards malignant invasive breast cancer (IBC) and subsequently metastatic cancer (Espina and Liotta, 2011).
Three‐dimensional (3D) culture of mammary epithelial cells embedded in Matrigel is an in vitro culture system for understanding the biological processes and signaling pathways that lead to the disruption of epithelial architecture at the early stages of mammary tumorigenesis in vivo (Debnath and Brugge, 2005; Vargo‐Gogola and Rosen, 2007; Yamada and Cukierman, 2007). Matrigel is rich in basement membrane proteins, such as type IV collagen, laminin, and heparan sulfate proteoglycan (Kleinman et al., 1982). When cultured in Matrigel, MCF10A, a spontaneously immortalized but non‐transformed human breast epithelial cell line, forms 3D acinar structures characterized by hollow lumens surrounded by polarized and growth‐arrested luminal epithelial cells. These structures arise through changes in many biological processes including proliferation, cell polarity, apoptosis, and cell cycle distribution, and resemble mammary acini in vivo, which constitute the multiple lobular units at the end of the mammary ducts (Vargo‐Gogola and Rosen, 2007; Debnath et al., 2003).
The amplification and overexpression of ERBB2 in breast cancer is correlated with poor prognosis due to an increased rate of metastasis and chemotherapy resistance. In general, this gene is overexpressed in 50–60% of DCIS (Lu et al., 2009). Muthuswamy et al. (2001) showed that activation of ERBB2 in 3D‐cultured MCF10A cells resulted in aberrant multi‐acinar structures with filled lumens, similar to ERBB2‐overexpressing DCIS in vivo. These cells, however, lacked an invasive phenotype, inconsistent with high metastatic rates of clinical ERBB2‐positive breast cancer, suggesting the existence of other cofactors necessary for malignant progression. Previous studies have shown coexpression of genes such as TGFB, RHOG, and FOS with ERBB2 induced invasive behaviors in the MCF10A 3D culture system (Seton‐Rogers et al., 2004; Witt et al., 2006). However, the clinical significance of these observations remains to be evaluated.
ERBB2 gene is not amplified alone, but is coamplified together with adjacent genes on the same chromosomal segment (Bieche et al., 1996; Kauraniemi et al., 2001). Given that breast tumors with gene amplification on the distal side of the chromosome 17q12–21 containing KRT20 and KRT19 are more aggressive than tumors with amplification restricted to the small region adjacent to ERBB2 (Lamy et al., 2011; Jacot et al., 2013), it is likely that the other genes localized in the same amplicon as ERBB2 play a significant role in breast cancer progression.
Here, we established an MCF10A 3D culture‐based screening system to identify genes that disrupt acini formation with or without oncogenic ERBB2 (ERBB2VE) expression. We found that overexpression of retinoic acid receptor alpha gene (RARA) in the 17q12–21 amplicon induces both the collapse of luminal morphology and an invasive phenotype. We also found that RARA upregulated EMT‐inducing transcription factors such as SLUG, FOXC2, ZEB1, and ZEB2, and TGF‐β–SMAD signaling‐activating factors including TGFBR1, TGFBR2, TGFB2, and SMAD3. Of these genes, ZEB1 in particular was identified as an essential gene in RARA‐induced EMT.
Our studies propose a model in which overexpression of RARA resulting from gene amplification contributes to the progression of mammary epithelial cell transformation towards more malignant invasive phenotypes through stimulation of the EMT interactome.
2. Materials and methods
2.1. Cell culture
MCF10A cells were purchased from the American Type Culture Collection (ATCC). Cells were maintained in growth medium consisting of DMEM/F12 (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/mL human epidermal growth factor (EGF) (BD), 0.5 μg/mL hydrocortisone (Sigma), 100 ng/mL cholera toxin (List Biological Laboratories), 10 μg/mL insulin (Sigma), 100 μg/mL streptomycin (Meiji‐Seika), and 100 U/mL penicillin (Meiji‐Seika).
2.2. Generation of MCF10A cell clones
A DNA fragment of rtTA‐Advanced (Clontech) was ligated into an EF1α promoter‐driven plasmid followed by the insertion of IRES‐neo. MCF10A cells were transfected with the resultant plasmid pEF‐rtTA‐Advanced‐IRES‐neo by calcium phosphate transfection and selected with 800 μg/mL G418 (MCF10A/Tet‐on). For generating clones inducibly‐expressing ERBB2 V659E, the MCF10A/Tet‐on clones were cotransfected with two vectors, a puromycin‐resistant gene expression vector and a Tet‐responsive (Clontech) ERBB2VE expression vector (pTRE‐Tight‐ERBB2VE) (1:25 ratio), and selected with 0.5 μg/mL puromycin (MCF10A/Tet‐on/TRE‐ERBB2VE). The MCF10A/Tet‐on and MCF10A/Tet‐on/TRE‐ERBB2VE clones were then transfected with a vector encoding a murine ecotropic retroviral receptor (pLenti6/Ubc/mSlc7a1, Addgene) and selected with 15 μg/mL blasticidin (MCF10A/Tet‐on/Eco and MCF10A/Tet‐on/TRE‐ERBB2VE/Eco, respectively). During serial cloning steps, we selected MCF10A clones with the ability to form hollow acini‐like structures as most suitable for 3D culture‐based screening.
2.3. Viral infection
The construction of the pMXs retroviral vectors and retroviral packaging was performed as previously described (Saito et al., 2012). Lentiviral packaging was performed by cotransfection of 293T cells with HIV‐based vectors including an expression construct and the pPACK packaging plasmids (System Biosciences). MCF10A cells were seeded at a density of 2.0 × 105 cells in each well of a 12‐well plate, followed by infection with a 1:1 mixture of diluted viruses and growth medium the next day. After 24 h for retroviruses, or 16–20 h for lentiviruses, the viral supernatant was replaced with growth medium for an additional 24 h period following which the cells were moved to new dishes.
2.4. Three‐dimensional morphogenesis assay
Forty microliters of Matrigel (growth factor reduced, BD) was added to each well of 48‐well plates and then the plates were incubated at 37 °C to allow solidification of the Matrigel. MCF10A cells were trypsinized and resuspended in assay medium (DMEM/F12 supplemented with 2% horse serum, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, 100 μg/mL streptomycin, and 100 U/mL penicillin). Five thousand cells were seeded onto the surface of the solidified Matrigel in a mixture of 400 μL of assay medium containing 5 ng/mL EGF and 2% Matrigel. On day 4, the assay medium supplemented with 5 ng/mL EGF and 2% Matrigel was replaced, and from day 7 the liquid medium was replaced with fresh assay medium containing 1 ng/mL EGF and 2% Matrigel every 4 days. For assays with type I collagen, Matrigel was mixed with a bovine collagen solution (PureCol, Advanced BioMatrix) at a final concentration of 50% Matrigel and 1.2 mg/mL collagen I, and 50 μl of the mixture was used to coat 48‐well plates. Before mixing, the collagen I‐PureCol was neutralized by addition of 10 × PBS and 100 mM NaOH (10×), and the pH was adjusted to 7.2–7.6. Cells were seeded on the underlayer using the same procedure described above.
2.5. Antibodies
For immunoblotting, the following antibodies were used. α‐Tubulin (DM1A), Sigma–Aldrich (T9026); FLAG M2, Sigma; Neu (ERBB2), Santa Cruz (sc‐284); E‐cadherin, BD (610182); N‐cadherin, BD (610921); Vimentin (D21H3), Cell Signaling (#5741S); RARα, Cell Signaling (#2554S); Akt1 (2H10), Cell Signaling (#2967); phospho‐Akt (Ser473), Cell Signaling (#9271); Smad2, Cell Signaling (#5339S); phospho‐Smad2, Cell Signaling (#3108S); HRP‐linked sheep anti‐mouse IgG, GE healthcare (NA931); HRP‐linked donkey anti‐rabbit IgG, GE healthcare (NA934).
2.6. Dual‐luciferase reporter assay
MCF10A cells were co‐transfected with pRL‐SV40 (Promega), 2 × DR5‐tk/pGL3, which has two retinoic acid response elements, 2 × DR5 (kindly provided by Dr. Shigeaki Kato), and pMXs‐RARA or pMXs‐raraΔ408‐416 by calcium phosphate transfection; total DNA was equalized by the addition of empty vector. The day after transfection, extracts were harvested and assayed with the dual‐luciferase reporter kit (Promega).
2.7. Quantitative real‐time PCR
Total RNA was extracted using Chomczynski's method (Chomczynski and Sacchi, 1987) or ISOGEN (NIPPON GENE) and incubated with reverse transcriptase SuperScript III (Invitrogen) using random hexamers to obtain cDNA. Real‐time PCR was performed by the StepOnePlus real‐time PCR system (Applied Biosystems) using Taqman gene expression assays (Applied Biosystems) and Taqman gene expression master mix (Applied Biosystems). Quantification of relative mRNA expression levels was normalized to 18S ribosomal RNA. The following Taqman gene expression assays were used. SNAIL, Hs00195591_m1; SLUG, Hs00950344_m1; FOXC2, Hs00270951_s1; ZEB1, Hs00232783_m1; ZEB2, Hs00207691_m1; TGFBR1, Hs00610320_m1; TGFBR2, Hs00234253_m1; TGFB1, Hs00998133_m1; TGFB2, Hs00234244_m1; SMAD3, Hs00969210_m; 18S, Hs99999901_s1.
2.8. Knockdown viral vectors
H1 promoter‐driven ZEB1 knockdown vectors were constructed from an HIV‐based SIN lentiviral vector (System biosciences), into which a puromycin resistance marker was introduced. An H1 promoter‐driven ZEB2 knockdown vector was constructed from the pMXs retroviral vector bearing blasticidin resistance marker. Pairs of oligonucleotide sequences encoding shRNA are listed in Table S1.
3. Results
3.1. Overexpression of RARA gene disrupted hollow lumen structures of 3D‐cultured MCF10A mammary epithelial cells
To identify genes cooperating with ERBB2 in the early stages of breast cancer tumorigenesis under microenvironments mimicking that of the mammary gland, we first established an MCF10A clone suitable for 3D culture screening. To evaluate the capability of the clone to induce disruption of 3D structures, and to enable screening of genes cooperating with ERBB2, we introduced an oncogenic ERBB2 expression unit under the control of a tetracycline‐responsive DNA element (MCF10A/Tet‐on/TRE‐ERBB2VE). We used a full‐length ERBB2 mutant, ERBB2 V659E, which forms homo‐ and heterodimers with other ERBB receptor family members and whose tyrosine kinase activity is constitutively activated (Yarden and Sliwkowski, 2001; Baselga and Swain, 2009). During the serial cloning steps, we chose the MCF10A clones most suitable for screening. When embedded in Matrigel, the MCF10A clone was required to show normal development of acinus structures without ERBB2VE induction (Figure S1B, Dox.(−) panel). Normal acinus had well‐ordered spheroid structures and its inner area seemed to have a lower cell density. Then, the induction of ERBB2VE (Figure S1B, Dox.(+) panel) had to induce clear phenotypic changes including distorted luminal structures with high density of inner cells, “multi‐acinar” morphology. To efficiently introduce target genes into human cells, we established an MCF10A clone expressing an ecotropic retroviral receptor (mSlc7a1) (MCF10A/Tet‐on/TRE‐ERBB2VE/Eco, Figure S1A).
We selected the 52 genes (51 genes plus wild type ERBB2) in the 17q12–21 amplicon and divided them into 10 subgroups consisting of five to six genes (Table S2). Each subgroup was introduced into the MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells and cultured with or without doxycycline to examine the morphological changes induced by genes cooperating with ERBB2VE (Figure 1A, lower panels) or acting alone (upper panels) in the same cells, respectively. Albeit in the absence of ERBB2VE (Dox(−)), subgroup #3 showed remarkable filled luminal structures with larger spherical bodies (approximately 200 μm in diameter) compared with the mock control (less than approximately 120 μm) and #10 showed aggregation (Figure 1B, S2). Subgroup #6 enhanced the aggregation by day 15 when ERBB2VE expression was induced at day 4 (Figure 1B, S2). We subsequently tested each of the 16 individual genes from these three subgroups, and found that the RARA gene from subgroup #3 caused similar aberrant structures in both the absence and presence of ERBB2VE (Figure 1C), as seen in the subgroup #3‐infected cells. We did not detect any remarkable transforming activity in candidate genes included in both subgroup #6 and #10 using the method of evaluating the function of those alone (Supplementary file 2).
Figure 1.
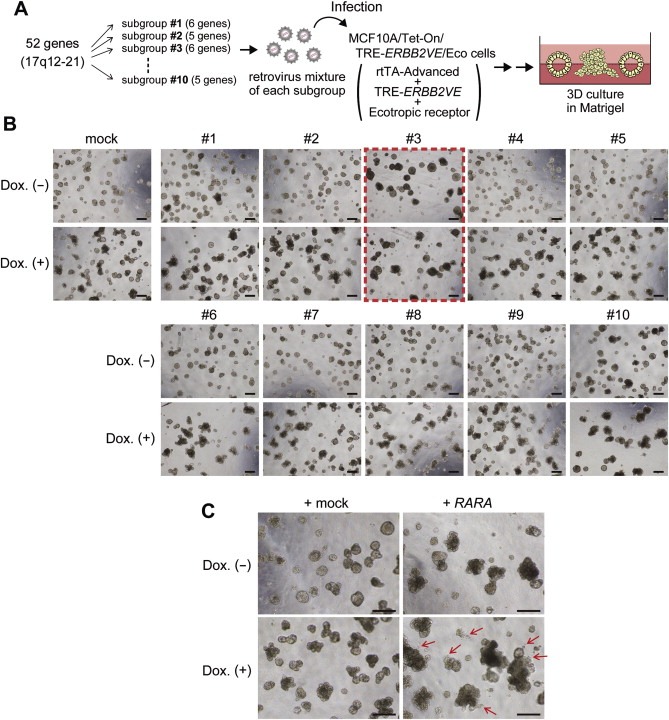
MCF10A 3D culture‐based screening for genes coamplified with ERBB2 and identification of RARA as a gene inducing filled‐lumen structures. (A) Overview of the screening system. Fifty‐two genes on the 17q12–21 amplicon including wild type ERBB2 were selected as screening targets. We divided full‐length cDNAs corresponding to those genes into ten subgroups and introduced them into MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells using retroviruses. The cells were embedded in Matrigel and allowed to proliferate. (B) Selection of transformed gene subgroups using a 3D culture system. Cells were infected with retrovirus mixture for each subgroup, embedded in Matrigel and cultured for 15 days. In the bottom panels, ERBB2VE was induced with Dox at day 4. Scale bars represent 200 μm. Magnified pictures are provided in Figure S2. (C) Activity of RARA in both ERBB2VE (−) and (+) backgrounds. MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells were transfected with mock (left panels) or RARA (right panels) retroviruses and were cultured on Matrigel for 14 days. In the right panels, ERBB2VE was induced by the addition of Dox at day 4. Arrows indicate raptured structures. Scale bars represent 200 μm.
3.2. Overexpression of RARA induced invasive transformation in Matrigel‐containing collagen I cultures
Coexpression of RARA and ERBB2VE led to the formation of larger cell clusters with a slightly protrusive outgrowth (Figure 1C), raising the possibility that RARA conferred migratory or invasive activities to MCF10A cells. To test this hypothesis, we cultured RARA‐expressing cells in collagen I‐containing Matrigel, as previously performed, to assess the invasiveness of MCF10A cells (Seton‐Rogers et al., 2004). Intriguingly, we noticed that the MCF10A cells overexpressing RARA displayed a lattice‐like network of invasive projections in the Matrigel‐collagen I mixture, while those expressing ERBB2VE alone did not (Figure 2A). This response of RARA‐expressing cells was dependent on the multiplicity of infection (MOI) of retroviruses, whereas control virus did not induce such response (Figure 2B and C). In addition, coexpression of both RARA and ERBB2VE resulted in thicker cords containing multiple acini‐like structures (Figure 2A). These results suggest that RARA is a potent invasion‐associated gene that is qualitatively different from ERRBB2VE, which is an inducer of acinar expansion without invasion into the surrounding matrix.
Figure 2.
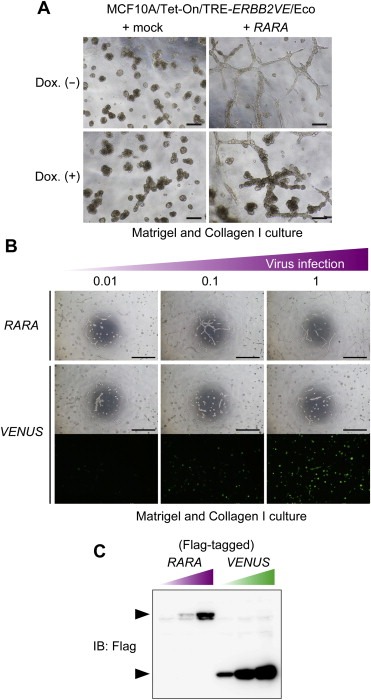
RARA‐induced invasive transformation in Matrigel containing collagen I in a manner dependent on its expression levels. (A) Identification of an invasive phenotype induced by RARA in Matrigel and collagen I culture. MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells overexpressing RARA were cultured in a 1:1 mixture of Matrigel and collagen I for 11 days. ERBB2VE was induced by the addition of Dox at day 4. Scale bars represent 200 μm. (B) Detection of dose‐dependent RARA activity. MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells were infected with retroviruses containing RARA or Venus with Flag epitope tag at the indicated MOIs. Cells were grown in a 1:2 mixture of Matrigel and Collagen I for 4 days in the absence of Dox. Scale bars represent 1 mm. (C) MOIs‐related expression levels. Protein levels in the cells used in (B) were analyzed by immunoblotting using the antibody against Flag. The upper band indicates the expression of RARα, and the lower band indicates that of Venus.
RARα, which is encoded by RARA, is a nuclear receptor for retinoic acid and functions as a transcriptional regulator for genes involving multicellular development, differentiation, and apoptosis. In the absence of ligand or in the presence of antagonists, target genes are silenced due to the binding of corepressors (CoRs) to RARα. Binding of RARα agonists to the ligand binding pocket induces allosteric changes in the ligand binding domain (LBD), leading to the movement of the C‐terminal helix H12, which then generates a novel interaction surface for coactivators (CoAs) (Bastien and Rochette‐Egly, 2004; Altucci and Gronemeyer, 2001). In a previous study, it was demonstrated that deletion of H12 increases CoR‐binding and results in the repression of gene expression (Farboud et al., 2003). To examine the role of the transcriptional activity of RARα for the invasive protrusion phenotype, we constructed a RARα deletion mutant (RARαΔ408–416) lacking the H12 motif. In a retinoic acid response element (RARE)‐Luc reporter assay, the wild type RARα enhanced luciferase activity in a dose‐dependent manner whereas the RARαΔ408–416 mutant rather suppressed it (Figure S3A). When we introduced retrovirus for the mutant at the same MOI as the wild type, MCF10A cells expressing RARαΔ408–416 did not form protrusions in the Matrigel and collagen I culture (Figure S3B). Thus, it is likely that the transcriptional activation by RARα, not the transcriptional repression in cooperation with CoR, is required for the invasive phenotype in cells. The dominant negative activity of RARαΔ408–416 likely resulted from an antagonistic role against intrinsically activated endogenous RAR proteins, which is insufficient to cause transformation.
3.3. RARA induced EMT‐related gene signatures
In 2D cultures, we observed that MCF10A cells overexpressing RARA displayed a fibroblast‐like morphology, whereas control mock‐infected cells grew in well‐ordered epithelial clusters (Figure 3A), suggesting that RARA acts as an EMT‐inducing factor. To address this hypothesis, we examined the expression of epithelial and mesenchymal markers by immunoblotting. In MCF10A cells expressing RARA, the epithelial marker E‐cadherin was downregulated, whereas mesenchymal markers N‐cadherin and vimentin were upregulated (Figure 3B). These changes in E‐cadherin and N‐cadherin expression were totally abrogated by the mutant raraΔ408–416 (Figure S3C), suggesting that RARA induces EMT in MCF10A cells via RARα‐mediated transcriptional activation. In contrast, ERBB2VE did not affect the epithelial morphology in monolayer cultures nor the expression of EMT markers (Figure 3B). N‐cadherin upregulation by RARA was also seen in MDA‐MB‐361 breast cancer cells (Figure S3D).
Figure 3.
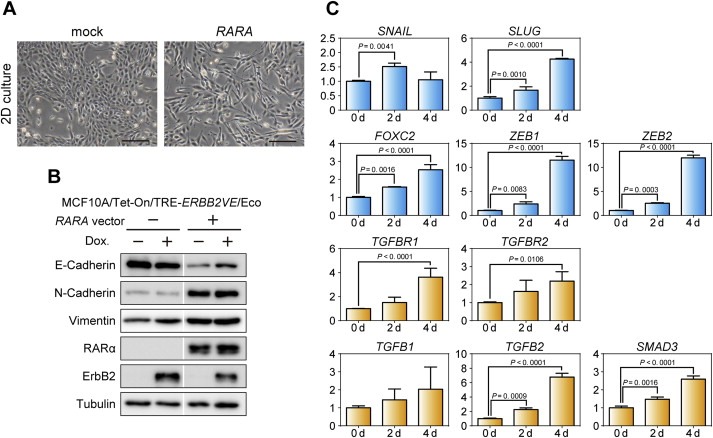
Induction of EMT‐like morphological changes and upregulation of EMT‐related genes by RARA overexpression. (A) Mesenchymal‐like transition induced by RARA. MCF10A/Tet‐on/TRE‐ERBB2VE/Eco cells overexpressing the indicated genes exhibited different morphologies in 2D culture. Scale bars represent 200 μm. (B) Expression analyses for EMT markers. The expression levels of the indicated proteins were analyzed by immunoblotting. Tubulin was used as a loading control. To evaluate effects of ERBB2VE, Dox‐treated samples for 2 days (Dox (+) lanes) were simultaneously evaluated, but revealed to be negligible compared with the effects by RARA. White line, trimmed margin of unrelated sample lanes. (C) Quantitative analyses of mRNA changes induced by RARA. The mRNA expression levels of the indicated genes were measured by real‐time PCR. MCF10A/Tet‐on cells were infected with lentiviruses for TRE‐RARA (MCF10A/Tet‐on/TRE‐RARA), and RNA was extracted at the indicated days after Dox addition. Real‐time PCR values were normalized to the internal control, 18S ribosomal RNA. The y‐axis depicts the fold‐change in each normalized mRNA compared with Dox (−) cells (0 d). The represented data are shown as mean ± s.d.. Dunnett's multiple comparisons test were performed to assess statistical significance.
To examine whether the expression levels of EMT‐related genes were regulated by RARA, we performed real‐time PCR analysis. RARA‐overexpressing MCF10A cells exhibited approximately 4‐, 2.5‐, and 12‐fold increases at day 4 in the mRNA levels of the SLUG, FOXC2, and ZEB gene families, respectively, compared with control MCF10A cells. Also, the mRNA levels of TGFBR1, TGFBR2, TGFB2, and SMAD3 increased by 4‐, 2.5‐, 7‐, and 2.5‐fold, respectively, at day 4 in RARA‐overexpressing MCF10A cells (Figure 3C). Time‐course experiments revealed that changes in E‐cadherin and N‐cadherin occurred after 24 h of RARA induction (Figure S4), suggesting that transcriptional cascades involving EMT‐inducing factors are activated during this time period. These results suggest that RARα activates the EMT signaling program before inducing a protrusive behavior in 3D cultures with collagen I.
Because RARA induced upregulation of key factors involved in the TGF‐β/SMAD signaling pathway (Figure 3C), we hypothesized that RARA activated this pathway and then induced invasion in 3D cultures as well as EMT. Therefore, we examined whether the inhibition of the TGF‐β/SMAD pathway suppressed phenotypic changes in RARA‐expressing MCF10A cells. Optimal concentration of an inhibitor of the TGF‐β receptor type I, SB‐431542 (Figure S5A), partially suppressed changes in the expression levels of EMT markers (Figure S5B) and protrusion (Figure S5C). These results suggest that the TGF‐β signaling pathway is partially responsible for EMT and invasion caused by RARA. We subsequently examined whether the activation of the TGF‐β pathway was sufficient to induce such changes. Stimulation of parental MCF10A cells with TGF‐β1 led to both slight downregulation of E‐cadherin and upregulation of N‐cadherin (Figure S5D). Also, modest effects on the phenotype were observed in 3D cultures using Matrigel and collagen I (Figure S5E), consistent with the previous result showing that the expression of TGF‐β1 without the activation of ERBB2 hardly displayed any invasive activity (Seton‐Rogers et al., 2004). These results suggest that TGF‐β1 alone is not sufficient to induce an invasive phenotype in MCF10A cells.
3.4. ZEB1 knockdown in RARA‐expressing MCF10A cells inhibited EMT and protrusion
To further investigate the mechanism of EMT induced by RARA, we established MCF10A cells that express RARA under the control of a tetracycline‐responsive DNA element. We examined the effects of silencing of ZEB1 and ZEB2 using short hairpin RNAs (shRNAs) in the cells because these genes exhibited the most marked increase in expression levels among the known key EMT‐inducing transcription factors (Figure 3C). We obtained two shRNAs (Figure 4A) and one shRNA (Figure 4B) that efficiently suppressed ZEB1 and ZEB2 mRNAs induced by RARA, respectively. Western blot analysis of EMT markers revealed that reduction of ZEB1 inhibited RARA‐induced EMT, whereas that of ZEB2 did not (Figure 4C). Furthermore, we observed ZEB1 knockdown inhibited the protrusive phenotype in Matrigel and collagen I cultures caused by RARA (Figure 4D). These results indicate that ZEB1 is required for the RARA‐mediated EMT and invasive phenotype.
Figure 4.
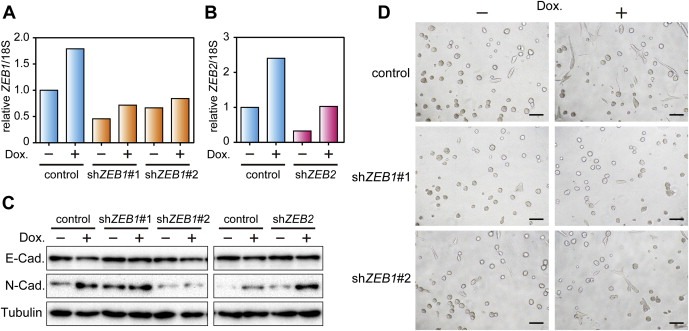
Essential roles of ZEB1 in RARA‐induced invasive phenotype. (A, B) Assessment of knockdown effects of shRNAs targeting ZEB1 and ZEB2. MCF10A/Tet‐on/TRE‐RARA cells infected with indicated shRNA viral vectors were cultured in the absence or presence of Dox for 3 days. ZEB1 and ZEB2 mRNA was analyzed by real‐time PCR analysis. The data were normalized by the amount of 18S rRNA and shown as a relative value compared with the control shRNA‐infected cells without Dox. Representative results of one of two independent experiments are shown. (C) Inhibition of RARA‐induced typical EMT marker transition by ZEB1 knockdown. Protein extracts from the ZEB1‐ or ZEB2‐knockdown cells were analyzed by immunoblotting. These cells were prepared as shown in (A) and (B), and cultured in Dox for 4 days with one passage. Tubulin was used as a loading control. (D) Inhibition of an RARA‐induced protrusive phenotype in Matrigel and collagen I cultures by ZEB1 knockdown. The ZEB1‐knockdown cells were prepared as shown in (A), and cultured in Dox for 2 days in 2D cultures and for 4 additional days in 3D cultures. Invasive protrusions of these cells in Matrigel and collagen I cultures were observed on day 4.
4. Discussion
ERBB2 amplification is a so‐called “driver” mutation (Stratton et al., 2009) that confers growth advantages on cancer cells. However, accumulating evidence suggests that breast cancer development probably results from the involvement of some other genes coamplified with ERBB2 in the 17q12–21 amplicon, not through ERBB2 acting alone. For example, previous studies identified that STARD3 and C35 localized within the amplicon expressed with ERBB2, and contributed to upregulated proliferation, decreased cell death, cell cycle progression (Kao and Pollack, 2006), and malignancy of breast cancer cells (Katz et al., 2010). We recently reported that GRB7, which is also located in the 17q12–21 amplicon, transforms NIH3T3 cells due to enhancement of ERBB2‐mediated signaling pathways (Saito et al., 2012). In this report, we examined the genes in the amplicon for their ability to transform 3D‐cultured MCF10A, and identified RARA, which disrupted acinar structures (Figure 1C) and induced EMT (Figure 3A and B). In contrast to the synergetic transforming activity of ERBB2 and GRB7 (Saito et al., 2012), RARA independently induced an invasive phenotype (Figure 2A). These studies indicate that the 17q12–21 amplicon harbors a surprising number of genes that play functional roles in the development and progression of breast cancers.
RARA amplification in parallel with ERBB2 in breast tumors was first reported by Keith et al. (1993) and is observed in 26.7% of ERBB2‐amplified breast cancer patients. Such simultaneous amplification is statistically associated with malignant lesions, such as lymph node invasion (Lamy et al., 2011), suggesting that RARA could have the potential to efficiently promote progression to malignancy. Furthermore, Peng et al. (2004) described that RARα expression was enhanced in premalignant MCF10AT and malignant MCF10CA1a cells at the protein level, and suggested the association between upregulation of RARα and malignant transformation of MCF10A cells. Our results that RARA overexpression contributes to the phenotypic change of non‐malignant cells to invasiveness support these hypotheses for the first time. We propose that RARA expression levels could be an important indicator to evaluate breast cancer malignancy.
Classically, RAR family members form heterodimers with retinoid X receptor (RXR) family members. Their ligand, retinoic acid (RA), activates these heterodimers, consequently leading to differentiation, apoptosis, and cell cycle arrest through transcriptional activation (Bastien and Rochette‐Egly, 2004; Altucci and Gronemeyer, 2001). Consistent with these observations, a recent study revealed that ERBB2 +/RARA + breast cancer cells undergo apoptosis by the combination of a specific ERBB2 inhibitor, trastuzumab and a pan‐RAR ligand, all‐trans retinoic acid (ATRA) (Paroni et al., 2012). Here we demonstrated that overexpression of RARα accompanied by transcriptional activation induced large acinar structures presumably filled with surviving inner cells (Figure 1C and S3A). These results are apparently opposed to the effect of ATRA. It is known that ATRA differentially regulates all α, β and γ members of RARs. A previous report has shown that ATRA suppresses RARα expression in premalignant MCF10AT and malignant MCF10CA1a cells, while it suppresses RARγ expression only in MCF10CA1a cells (Peng et al., 2004). Also, it is known that distinct RXR‐RAR isotype combinations lead to different expression patterns of RA‐responsive genes (Chiba et al., 1997). Thus, we assume that the individual role of each RAR subtype is different and that phenotypes of breast cancer cells caused by treatment with ATRA represent effects on not only RARα but also RARβ and γ. Further analyses are needed to contribute to the understanding of the whole regulatory role of RARs.
In addition to typical heterodimer formation, another mechanism of RARα‐induced cell‐fate decisions could be considered. Previous studies implicated that RARα is an essential component of the estrogen receptor (ER) transcription complex and regulates the expression of estrogen‐target genes, leading to cell proliferation in ER‐positive cell lines (Ross‐Innes et al., 2010; Toma et al., 1998). Activation of the classical RARα pathways could deplete RARα from the ER complex (Ross‐Innes et al., 2010). Thus, any shift between the classical and non‐classical RARα‐pathways is assumed to influence cell fate. Previous evidence showed that intracellular lipid binding proteins, CRABP‐II and FABP5, delivered RA from the cytosol to each particular nuclear receptor RARα and PPARβ/γ, respectively (Schug et al., 2007). It can be considered that dominant expression of FABP5 abolishes CRABP‐II‐mediated import of RA to RARα‐RXR heterodimers, activating the non‐classical pathway, while that of CRABP‐II stimulates the classical pathway. Thus, the relative expression levels between those intracellular lipid binding proteins might be important to determine which classical or non‐classical pathway RARα preferentially contributes to. Taken together, whether RARα promotes or suppresses tumor progression relies on several cellular contexts including the expression levels of ligands, nuclear hormone receptors and intracellular lipid‐binding proteins. Efforts to identify RARα interaction targets in MCF10A cells will provide a molecular basis for the positive transforming activity observed in this study.
We observed that overexpression of RARA in MCF10A cells induced EMT (Figure 3A and B). The mechanisms inducing EMT are quite complex because a variety of signal transduction cascades such as the TGF‐β, Wnt, Notch, EGF, FGF, and HIF pathways are involved (Polyak and Weinberg, 2009; Yang and Weinberg, 2008; Thiery et al., 2009). Which pathway is employed is dependent on the tissue, and even in the same tissue, on the particular cellular context. Our study suggests that MCF10A cells utilize, in part, the TGF‐β pathway in the context of RARA overexpression (Figure S5). Repression of E‐cadherin is an established hallmark of EMT, which is directly or indirectly mediated by EMT‐inducing transcription factors including SNAIL, TWIST, SLUG, ZEB1, and ZEB2 (reviewed in Polyak and Weinberg (2009); Yang and Weinberg (2008); Thiery et al. (2009)). There is also extensive crosstalk among the factors that make up the “EMT interactome”. In this network, a single EMT‐inducing transcription factor affects the expression of other EMT‐inducing transcription factors (Taube et al., 2010). It is likely that the EMT‐interactome in mammary epithelial MCF10A cell lines is activated within 24 h after induction of RARA expression, as shown in Figure S4. During this period, RARα may activate the transcription of several EMT‐related genes and subsequently regulate other EMT inducers, coordinately contributing to execution of the EMT program. Our results that ZEB1‐depleted cells did not induce EMT phenotypes (Figure 4) strongly suggest that ZEB1 is a key factor for the RARα‐activated EMT interactome. We also found that RARA overexpression upregulates TGFB2 (Figure 3C), raising the possibility that RARα is involved in autocrine TGF‐β/ZEB/miR‐200 signaling (Burk et al., 2008; Gregory et al., 2011).
Accumulating evidence suggests that cells undergoing EMT acquire stem cell‐like properties (Mani et al., 2008). RARα may also contribute to these changes. A recent report showed that mammary epithelium obtained from RARα1 knockout mice contained a greater percentage of progenitors but fewer mammary stem cells (MaSCs) compared with wild type, suggesting that RARα plays an important role in the maintenance of MaSCs, and affects the mammary epithelial hierarchy (Cohn et al., 2010). Furthermore, another report has shown that ZEB1 is required for the initial acquisition of both CD44hi expression, which is a marker of breast cancer stem cells (BCSCs), and the stem‐like activity of CD44hi cells (Chaffer et al., 2013). In pancreatic cancer cells, ZEB1 maintains stem‐like properties via downregulation of miR‐183, 200c, and 203, which suppress the expression of stem cell factors such as BMI1, SOX2, and KLF4 (Wellner et al., 2009). Future experiments are required to investigate the potential role of the RARα–ZEB1 axis as a promoting factor of MaSCs and BCSCs.
5. Conclusion
We established an MCF10A 3D culture‐based screening system and performed a screen of genes coamplified with ERBB2 in the 17q12–21 amplicon. As a result, we revealed that RARA induced the malformation of acinar structures in Matrigel and facilitated an invasive phenotype in Matrigel/collagen I 3D culture. Furthermore, RARA upregulated the transcription of EMT‐related genes including ZEB1, which was a key mediator of EMT in RARA‐expressing MCF10A cells. Our results suggest that overexpression of RARA is a potentially important factor for breast cancer malignancy.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgments
We would like to acknowledge the secretarial assistance of Kumiko Semba. This research was partially supported by Japan Society for the Promotion of Science KAKENHI 23241064 and a grant for Translational Research Program: Achievement of personalized medicine and acceleration of anti‐cancer drug development by using gene expression analysis technology from the New Energy and Industrial Technology Development Organization (NEDO).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.09.005.
Doi Ayano, Ishikawa Kosuke, Shibata Nao, Ito Emi, Fujimoto Jiro, Yamamoto Mizuki, Shiga Hatsuki, Mochizuki Hiromi, Kawamura Yoshifumi, Goshima Naoki, Semba Kentaro, Watanabe Shinya, (2015), Enhanced expression of retinoic acid receptor alpha (RARA) induces epithelial‐to‐mesenchymal transition and disruption of mammary acinar structures, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.09.005.
Contributor Information
Ayano Doi, Email: ad0i_i0da@yahoo.co.jp.
Kosuke Ishikawa, Email: IshikawaKosuke@gmail.com.
Kentaro Semba, Email: ksemba@waseda.jp.
References
- Altucci, L. , Gronemeyer, H. , 2001. The promise of retinoids to fight against cancer. Nat. Rev. Cancer. 1, 181–193. [DOI] [PubMed] [Google Scholar]
- Baselga, J. , Swain, S.M. , 2009. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer. 9, 463–475. [DOI] [PubMed] [Google Scholar]
- Bastien, J. , Rochette-Egly, C. , 2004. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 328, 1–16. [DOI] [PubMed] [Google Scholar]
- Bieche, I. , Tomasetto, C. , Regnier, C.H. , Moog-Lutz, C. , Rio, M.C. , Lidereau, R. , 1996. Two distinct amplified regions at 17q11-q21 involved in human primary breast cancer. Cancer Res. 56, 3886–3890. [PubMed] [Google Scholar]
- Burk, U. , Schubert, J. , Wellner, U. , Schmalhofer, O. , Vincan, E. , Spaderna, S. , Brabletz, T. , 2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer, C.L. , Marjanovic, N.D. , Lee, T. , Bell, G. , Kleer, C.G. , Reinhardt, F. , D'Alessio, A.C. , Young, R.A. , Weinberg, R.A. , 2013. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba, H. , Clifford, J. , Metzger, D. , Chambon, P. , 1997. Distinct retinoid X receptor-retinoic acid receptor heterodimers are differentially involved in the control of expression of retinoid target genes in F9 embryonal carcinoma cells. Mol. Cell. Biol. 17, 3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski, P. , Sacchi, N. , 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Cohn, E. , Ossowski, L. , Bertran, S. , Marzan, C. , Farias, E.F. , 2010. RARalpha1 control of mammary gland ductal morphogenesis and wnt1-tumorigenesis. Breast Cancer Res. 12, R79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath, J. , Brugge, J.S. , 2005. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer. 5, 675–688. [DOI] [PubMed] [Google Scholar]
- Debnath, J. , Muthuswamy, S.K. , Brugge, J.S. , 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Espina, V. , Liotta, L.A. , 2011. What is the malignant nature of human ductal carcinoma in situ?. Nat. Rev. Cancer. 11, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud, B. , Hauksdottir, H. , Wu, Y. , Privalsky, M.L. , 2003. Isotype-restricted corepressor recruitment: a constitutively closed helix 12 conformation in retinoic acid receptors beta and gamma interferes with corepressor recruitment and prevents transcriptional repression. Mol. Cell. Biol. 23, 2844–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, P.A. , Bracken, C.P. , Smith, E. , Bert, A.G. , Wright, J.A. , Roslan, S. , Morris, M. , Wyatt, L. , Farshid, G. , Lim, Y.Y. , Lindeman, G.J. , Shannon, M.F. , Drew, P.A. , Khew-Goodall, Y. , Goodall, G.J. , 2011. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell. 22, 1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacot, W. , Fiche, M. , Zaman, K. , Wolfer, A. , Lamy, P.J. , 2013. The HER2 amplicon in breast cancer: topoisomerase IIA and beyond. Biochim. Biophys. Acta. 1836, 146–157. [DOI] [PubMed] [Google Scholar]
- Kao, J. , Pollack, J.R. , 2006. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer. 45, 761–769. [DOI] [PubMed] [Google Scholar]
- Katz, E. , Dubois-Marshall, S. , Sims, A.H. , Faratian, D. , Li, J. , Smith, E.S. , Quinn, J.A. , Edward, M. , Meehan, R.R. , Evans, E.E. , Langdon, S.P. , Harrison, D.J. , 2010. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of Syk. Br. J. Cancer. 103, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauraniemi, P. , Barlund, M. , Monni, O. , Kallioniemi, A. , 2001. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 61, 8235–8240. [PubMed] [Google Scholar]
- Keith, W.N. , Douglas, F. , Wishart, G.C. , McCallum, H.M. , George, W.D. , Kaye, S.B. , Brown, R. , 1993. Co-amplification of erbB2, topoisomerase II alpha and retinoic acid receptor alpha genes in breast cancer and allelic loss at topoisomerase I on chromosome 20. Eur. J. Cancer. 29A, 1469–1475. [DOI] [PubMed] [Google Scholar]
- Kleinman, H.K. , McGarvey, M.L. , Liotta, L.A. , Robey, P.G. , Tryggvason, K. , Martin, G.R. , 1982. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 21, 6188–6193. [DOI] [PubMed] [Google Scholar]
- Lamy, P.J. , Fina, F. , Bascoul-Mollevi, C. , Laberenne, A.C. , Martin, P.M. , Ouafik, L. , Jacot, W. , 2011. Quantification and clinical relevance of gene amplification at chromosome 17q12-q21 in human epidermal growth factor receptor 2-amplified breast cancers. Breast Cancer Res. 13, R15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Guo, H. , Treekitkarnmongkol, W. , Li, P. , Zhang, J. , Shi, B. , Ling, C. , Zhou, X. , Chen, T. , Chiao, P.J. , Feng, X. , Seewaldt, V.L. , Muller, W.J. , Sahin, A. , Hung, M.C. , Yu, D. , 2009. 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell. 16, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, S.A. , Guo, W. , Liao, M.J. , Eaton, E.N. , Ayyanan, A. , Zhou, A.Y. , Brooks, M. , Reinhard, F. , Zhang, C.C. , Shipitsin, M. , Campbell, L.L. , Polyak, K. , Brisken, C. , Yang, J. , Weinberg, R.A. , 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy, S.K. , Li, D. , Lelievre, S. , Bissell, M.J. , Brugge, J.S. , 2001. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroni, G. , Fratelli, M. , Gardini, G. , Bassano, C. , Flora, M. , Zanetti, A. , Guarnaccia, V. , Ubezio, P. , Centritto, F. , Terao, M. , Garattini, E. , 2012. Synergistic antitumor activity of lapatinib and retinoids on a novel subtype of breast cancer with coamplification of ERBB2 and RARA. Oncogene. 31, 3431–3443. [DOI] [PubMed] [Google Scholar]
- Peng, X. , Yun, D. , Christov, K. , 2004. Breast cancer progression in MCF10A series of cell lines is associated with alterations in retinoic acid and retinoid X receptors and with differential response to retinoids. Int. J. Oncol. 25, 961–971. [PubMed] [Google Scholar]
- Polyak, K. , Weinberg, R.A. , 2009. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 9, 265–273. [DOI] [PubMed] [Google Scholar]
- Ross-Innes, C.S. , Stark, R. , Holmes, K.A. , Schmidt, D. , Spyrou, C. , Russell, R. , Massie, C.E. , Vowler, S.L. , Eldridge, M. , Carroll, J.S. , 2010. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 24, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, M. , Kato, Y. , Ito, E. , Fujimoto, J. , Ishikawa, K. , Doi, A. , Kumazawa, K. , Matsui, A. , Takebe, S. , Ishida, T. , Azuma, S. , Mochizuki, H. , Kawamura, Y. , Yanagisawa, Y. , Honma, R. , Imai, J. , Ohbayashi, H. , Goshima, N. , Semba, K. , Watanabe, S. , 2012. Expression screening of 17q12-21 amplicon reveals GRB7 as an ERBB2-dependent oncogene. FEBS Lett. 586, 1708–1714. [DOI] [PubMed] [Google Scholar]
- Schug, T.T. , Berry, D.C. , Shaw, N.S. , Travis, S.N. , Noy, N. , 2007. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 129, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seton-Rogers, S.E. , Lu, Y. , Hines, L.M. , Koundinya, M. , LaBaer, J. , Muthuswamy, S.K. , Brugge, J.S. , 2004. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. U S A. 101, 1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton, M.R. , Campbell, P.J. , Futreal, P.A. , 2009. The cancer genome. Nature. 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube, J.H. , Herschkowitz, J.I. , Komurov, K. , Zhou, A.Y. , Gupta, S. , Yang, J. , Hartwell, K. , Onder, T.T. , Gupta, P.B. , Evans, K.W. , Hollier, B.G. , Ram, P.T. , Lander, E.S. , Rosen, J.M. , Weinberg, R.A. , Mani, S.A. , 2010. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. U S A. 107, 15449–15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery, J.P. , Acloque, H. , Huang, R.Y. , Nieto, M.A. , 2009. Epithelial-mesenchymal transitions in development and disease. Cell. 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Toma, S. , Isnardi, L. , Raffo, P. , Riccardi, L. , Dastoli, G. , Apfel, C. , LeMotte, P. , Bollag, W. , 1998. RARalpha antagonist Ro 41-5253 inhibits proliferation and induces apoptosis in breast-cancer cell lines. Int. J. Cancer. 78, 86–94. [DOI] [PubMed] [Google Scholar]
- Vargo-Gogola, T. , Rosen, J.M. , 2007. Modelling breast cancer: one size does not fit all. Nat. Rev. Cancer. 7, 659–672. [DOI] [PubMed] [Google Scholar]
- Wellner, U. , Schubert, J. , Burk, U.C. , Schmalhofer, O. , Zhu, F. , Sonntag, A. , Waldvogel, B. , Vannier, C. , Darling, D. , zur Hausen, A. , Brunton, V.G. , Morton, J. , Sansom, O. , Schuler, J. , Stemmler, M.P. , Herzberger, C. , Hopt, U. , Keck, T. , Brabletz, S. , Brabletz, T. , 2009. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495. [DOI] [PubMed] [Google Scholar]
- Witt, A.E. , Hines, L.M. , Collins, N.L. , Hu, Y. , Gunawardane, R.N. , Moreira, D. , Raphael, J. , Jepson, D. , Koundinya, M. , Rolfs, A. , Taron, B. , Isakoff, S.J. , Brugge, J.S. , LaBaer, J. , 2006. Functional proteomics approach to investigate the biological activities of cDNAs implicated in breast cancer. J. Proteome Res. 5, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K.M. , Cukierman, E. , 2007. Modeling tissue morphogenesis and cancer in 3D. Cell. 130, 601–610. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Weinberg, R.A. , 2008. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell. 14, 818–829. [DOI] [PubMed] [Google Scholar]
- Yarden, Y. , Sliwkowski, M.X. , 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data