

Abstract
Familial amyotrophic lateral sclerosis (FALS) is a fatal motor neuron disease that is caused by mutations in the gene encoding superoxide dismutase-type 1 (SOD1). The affected regions of the FALS brain are characterized by aggregated SOD1, and the mutations that destabilize SOD1 appear to promote its aggregation in vitro. Because dissociation of the native SOD1 dimer is required for its in vitro aggregation, we initiated an in silico screening program to find drug-like molecules that would stabilize the SOD1 dimer. A potential binding site for such molecules at the SOD1 dimer interface was identified, and its importance was validated by mutagenesis. About 1.5 million molecules from commercial databases were docked at the dimer interface. Of the 100 molecules with the highest predicted binding affinity, 15 significantly inhibited in vitro aggregation and denaturation of A4V, a FALS-linked variant of SOD1. In the presence of several of these molecules, A4V and other FALS-linked SOD1 mutants such as G93A and G85R behaved similarly to wild-type SOD1, suggesting that these compounds could be leads toward effective therapeutics against FALS.
Keywords: drug discovery, in silico screening, docking, thermopeutics, thermodynamics
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig's disease, is a fatal motor neuron disease that affects > 35,000 Americans and many more individuals worldwide. About 20% of ALS cases are monogenic and autosomal dominant [familial ALS (FALS)]. The most common cause of FALS are point mutations in the gene-encoding superoxide dismutase 1 (SOD1), a β-sheet-rich dimeric metalloenzyme that is normally responsible for scavenging superoxide ions (1, 2). FALS does not appear to arise from a loss of this important activity. Studies with transgenic mice suggest that FALS may result from a “gain of toxic function” due to aggregation of the mutant form of SOD1 (3). Because the 114 known SOD1 FALS mutations are distributed throughout the primary sequence and tertiary structure, it is proposed that the mutations affect, in different ways, the structural stability of SOD1 (1, 2). One or more FALS SOD1 mutations have been linked to decreased metal binding (4-6), decreased formation of a stabilizing intramolecular disulfide (7), decreased structural stability, and increased propensity to monomerize (8) and aggregate (7, 9-14). Occupancy of the zinc and copper binding sites (one each per subunit) may prevent SOD1 aggregation (10). Thus, the prevention of SOD1 demetallation could slow the onset and progression of FALS, but a practical solution to doing so in vivo is elusive.
We have pursued an alternative strategy to inhibit SOD1 aggregation: stabilization of the SOD1 native dimer with small, drug-like molecules (15). This strategy was based on the notion that SOD1 monomerization is required for aggregation, which is supported by the observation that insertion of an engineered intersubunit disulfide bond into the FALS SOD1 mutant A4V prevented its aggregation (16). Furthermore, a detailed analysis of the aggregation of SOD1 (10) supports the proposal that monomerization of the protein is required for in vivo aggregation.
The precedent for the discovery of small-molecule stabilizers of a native protein oligomer involves a protein aggregation disease that is analogous to FALS: familial amyloid polyneuropathy (FAP). FAP is caused by mutations in the gene encoding transthyretin (TTR) (17, 18). Many FAP mutations destabilize the native TTR tetramer, facilitating its dissociation, partial unfolding, and aggregation (17, 19). The natural ligand of TTR, thyroxine, stabilizes the tetramer and prevents its aggregation in vitro. Drug-like molecules that are thyroxine analogs bind and stabilize the native TTR tetramer, preventing its aggregation in vitro (20-24). These compounds could potentially be used for the treatment of FAP (25).
Unlike the example of TTR, there are no natural ligands of SOD1 to serve as a molecular scaffold for the design of small-molecule stabilizers. Therefore, we decided to take an in silico screening approach (docking) by using a library of ≈1.5 million drug-like molecules to select for compounds that could potentially bind at the dimer interface. We report here that 15 compounds identified by this method have the ability to significantly stabilize A4V (and other FALS variants) and prevent its aggregation in vitro. These compounds could lead to candidate therapeutics for FALS.
Materials and Methods
Cloning and Purification. Cloning, expression, and purification of human SOD1, WT, and the various FALS and other mutants described in the investigation were carried out as described in ref. 16.
Database Preparation and Docking. All computations were carried out on an 18-node Beowulf Linux Cluster (each node = 2.0 GHz Pentium processor, Intel, Santa Clara, CA). Raw structure data files obtained from vendors (Table 1, which is published as supporting information on the PNAS web site) were filtered to remove wrong structures. Database preparation and docking were carried out by using a trial version of the firstdiscovery suite (Schrödinger, Portland, OR), which included glide v2.5, the primary tool for docking (26). A detailed description of the docking methodology is described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Purification of Recombinant SOD1 Dimer and Metal Analysis. SOD1 dimer was purified on a Superdex 75 (16/60) gel filtration column (Amersham Pharmacia) to produce starting material for each aggregation experiment. Metal analyses were carried by inductive coupled plasma mass spectrometry (see Supporting Materials and Methods). WT and G93A were nearly fully metalated, and G85R and A4V were deficient in zinc and copper, respectively (see Table 2, which is published as supporting information on the PNAS web site). Possible effects of the heterogeneity of G85R and A4V will be discussed below.
Preparation of Apo-SOD1 and Variants. The procedure of Fridovich and coworkers (27) was followed, with minor modifications (see Supporting Materials and Methods). The loss of Cu and Zn were confirmed by using inductive coupled plasma mass spectrometry analysis; all variants prepared in this way contained <0.2% of Cu and Zn (see Table 2).
Aggregation of SOD1 and Mutants. Aggregation assays were prepared by adding a stock solution of compound to a protein solution (final concentrations: 100 μM compound and 50 μM protein). After a 15-min preincubation period at 37°C, 5 mM EDTA was added to initiate aggregation. Aliquots were periodically removed and analyzed (for amount of SOD1 dimer present; this value correlated in all cases with the appearance of oligomers) by gel filtration on a Superdex 200 (3.2/30) gel filtration column (Amersham Pharmacia). All chromatography was performed in Tris-buffered saline, pH 7.4 (20 mM Tris/150 mM NaCl), on a Waters 2690 Alliance HPLC and monitored at 220 and 276 nm. The assays were repeated in triplicate and showed <5% variation between individual experiments. Assays in the absence of EDTA were carried as described in ref. 16. For Apo-A4V experiments, buffers were treated with Chelex 100 (except those containing EDTA), and experiments were performed in plastic tubes to avoid introduction of contaminating zinc into the apoprotein.
Guanidinium Chloride Unfolding. Equilibrium unfolding transition, as a function of GdnCl concentration, was monitored by fluorescence spectroscopy. The fluorescence measurements were done on a f-4500 spectrofluorometer (Hitachi, Tokyo) in a 1-cm cell connected to a circulation water bath. The excitation and emission wavelengths were fixed at 278 and 348 nm, respectively, after making appropriate corrections for buffer and GdnCl. The slit width was 5 nm for both monochromators. Each measurement was an average of five readings. Protein concentration used for fluorescence experiment was 5 μM. The data were analyzed directly for a two-state (N → U) transition as follows: the raw data for the GdnCl-induced denaturation studies were converted to fractions of the protein in the unfolded state (fu) as a function of GdnCl concentration by using the equation:
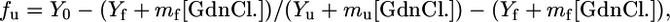 |
where Y0 is the observed spectroscopic property, Yf and mf are the slope and intercept of the folded-state baseline and Yu and mu represent the respective values of the unfolded baseline. The folded fraction was calculated as (fn = 1 - fu), and the equilibrium constant was determined by Keq = fu/fn. The free energy of unfolding was determined by using the equation ΔG = -RT·ln(Keq), where T is the temperature in Kelvin, and R is the universal gas constant (1.987 cal·mol-1·K-1).
Aggregation of α-Synuclein. Samples of α-synuclein were dissolved in PBS (pH 7.4) and filtered through a Millipore Microcon 100K MWCO filter. Samples were incubated at 37°C without agitation. A 100 μM aqueous solution of Thioflavin T (Thio T, Sigma) was prepared and filtered through a 0.2-μm polyether sulfone filter. At various time points, aliquots of the α-synuclein incubations were diluted to 10 μM in water. Fluorescence measurements for the 300 μM α-synuclein incubations were performed in a 384-well microplate as described in ref. 28. Fluorescence at 490 nm was measured by using the LJL Biosystems (Sunnyvale, CA) plate reader (excitation: 450 nm, bandwidth 30 nm; emission: 490 nm, bandwidth 10 nm).
Results and Discussion
Filling a Hydrophobic Cavity at the A4V SOD1 Dimer Interface Stabilizes It Against Unfolding and Aggregation. To look for suitable binding sites for small molecules at the SOD1 dimer interface, we used the program voidoo (Uppsala Software Factory, Uppsala), which detects cavities in protein (29). Five cavities were detected by the program, one of which was at the dimer interface of both WT and A4V. The cavity (shown as a Gaussian surface representation in Fig. 1a Right) is centered with the Cβ carbon of residue 148 as the point of origin (Fig. 1a Left). The site is predominantly hydrophobic in nature with a few hydrogen bond donors and acceptors.
Fig. 1.

When the cavity at the SOD1 dimer interface is partially filled by mutagenesis, the resultant protein is more stable and aggregates more slowly. (a Left) shows a surface representation of A4V mutant SOD1 dimer colored to show the two subunits. A deep cavity at the dimer interface is highlighted by the blue box. The surface was generated by using a water molecule as a probe. (a Right) A model of the SOD1 cavity in the variant A4V/V7F/V148F. Note the quartet of phenylalanine sidechains at the dimer interface. (b Left) GdnCl unfolding of A4V (black), WT (green) and the A4V/V7F/V148C triple mutant (red) plotted as fraction unfolded vs. GdnCl concentration. (b Right) Loss of SOD1 dimers over time parallels the formation of aggregates (data not shown). The rate of aggregation is inversely correlated to the stability toward denaturation: A4V (black), WT (green), and A4V/V7F/L148C (red).
To investigate the effect of partially capping the cavity with hydrophobic moieties, residues V148 and V7, the sidechains of which protrude into the cavity, were mutated to phenylalanine (Fig. 1a Right). Molecular modeling suggested that the four Phe residues at the interface could be easily accommodated with no steric clashes (Supporting Materials and Methods and Fig. 1b). Filling cavities in protein structures with hydrophobic side chains often stabilizes the protein structure (30, 31), lysozyme being a classic example (32).
Three variants of SOD1, in which the V7F and V148F mutations were introduced into WT, A4V, and G93A, were cloned, expressed in Escherichia coli and purified as described in ref. 15. Each protein was subjected to guanidine chloride (GdnCl) unfolding (Fig. 1b), and fluorescence intensity (348 nm) was monitored at 25°C. WT was completely unfolded at 3.5 M GdnCl (Cm = 3.2 M, where Cm is the midpoint of transition), whereas A4V was completely unfolded at 1.9 M GdnCl (Cm = 1.51 M) (Fig. 1b). A4V/V7F/V148F was found to be more stable compared with A4V but less stable than WT (unfolded at 2.1 M, Cm = 1.8 M) (Fig. 1b). G93A/V7F/V148F was slightly more resistant to denaturation than G93A (Fig. 6 c and d, which is published as supporting information on the PNAS web site). No significant effect of the two V → F mutations on the denaturation of WT (i.e., WT vs. V7F/V148F) could be measured (data not shown). This unexpected observation has motivated a comparison of the crystal structures of all of the V → F variants.
The V → F mutations stabilized both A4V and G93A against EDTA-induced aggregation. A4V/V7F/V148F aggregated more slowly than A4V but significantly faster than WT (Fig. 1c). Similarly, G93A/V7F/V148F aggregated slightly more slowly than G93A (Fig. 6).
Preparation of a Compound Database and High-Throughput Docking of Compounds to the Cavity at the A4V Interface. An in silico screening approach was undertaken to identify compounds from commercially available databases (Table 1) with a potential to bind at the SOD1 dimer interface and stabilize the dimer. Prefilters were used to select a subset of compounds that are more suited toward a particular target (see Supporting Materials and Methods for an overview; see also Fig. 2b). Structure data files for 15 commercially available libraries were gathered (Supporting Materials and Methods and Table 1). Preparation of a suitable database for docking requires several steps (see Supporting Materials and Methods). The distributions of various physicochemical properties (relaxed set of Lipinski rules in this case) (33) of the database used in the docking calculation are shown in Fig. 2a (also see Supporting Materials and Methods).
Fig. 2.

The protein and small-molecule portions of the docked complexes occupy overlapping space. (a) The grid box used for our docking calculation. The green box represents the conformational search space for the ligand; the pink box represents the boundary conditions used for the docking calculation. One hundred of the 2,000 final poses obtained from docking are shown docked at the dimer interface. (b) Superposition of five SOD1 x-ray structures WT, apo-WT, S134N, H46R, and A4V (Protein Data Bank codes: 1SPD, 1HL4, 1N19, 1OEZ, and 1UXL), showing critical residues of the dimer interface cavity used in docking calculations. The mean rms deviation (Cα)is <0.6 Å, indicating that the dimer interface is rigid in nature, which suggests that docked molecules should bind to all mutants. A superposition of 20 of the top 100 docked molecules is also shown.
Docking calculations was carried out by using a trial version of Schrödinger software, glide v2.5 (26). The docking calculation has two distinct steps: docking of ligands and scoring of hits.
The protein structural data file for A4V (Protein Data Bank ID code 1UXM) was used for all calculations described below. A primary grid box of 7 Å (purple) and a secondary ligand containment box (green) were generated around the Cβ carbon of residue 148 for the docking calculation of the protein after the removal of water and the addition of hydrogen atoms as the center of mass, as shown in Fig. 2a.
A detailed description of the glide methodology has been published in refs. 26 and 34 and is beyond the scope of discussion here. The molecules obtained after docking were analyzed and sorted by glidescores (26, 34). The top 100 binders were examined; the superimposed docked structures are shown in Fig. 2a. It is noteworthy that approved drugs such as baclofen, dapsone, and triamcinolone were among the top 100 hits. Superposition of x-ray structures of WT, apo-WT, S134N, H46R, and A4V (Protein Data Bank ID codes 1SPD, 1HL4, 1OZU, 1OEZ and 1UXM) reveal very low rms deviation (<0.6 Å for Cα) between residues that make up the binding pocket in these variants as compared with A4V (Fig. 2b), suggesting that compounds are likely to bind to several mutants.
Fifteen of the Top 100 in Silico “Hits” Significantly Inhibited A4V Aggregation. A4V aggregation assays (with EDTA; see Materials and Methods) were carried out in presence of the top 100 hits obtained as described above. The effect of each compound was compared with A4V (thick black line, Fig. 3a) and with WT (thick red line) in the absence of added compounds. About 15 of the top 100 compounds significantly slowed A4V aggregation; that is, in the presence of these compounds, ≤25% of the dimer had disappeared after 12 h, whereas 50% was lost in their absence (these compounds, arbitrarily numbered 1-15, are shown in Fig. 7, which is published as supporting information on the PNAS web site). In the presence of several of these compounds, A4V aggregation closely resembled WT (≈5% dimer loss after 12 h). The shape of the A4V aggregation curve may reflect the heterogeneity of the protein with respect to metallation: the initial rapid phase may represent the population lacking copper (apo-A4V does not show this “biphasic” behavior, see below).
Fig. 3.

Fifteen compounds significantly slowed the loss A4V, G85R, and G93A SOD1 dimer under aggregating conditions. (a) EDTA-induced loss of A4V dimer in the absence (solid black line) and presence of the top 100 compounds, as predicted by the docking calculation. The average of three trials is plotted for each compound; variation was <5%. The loss of WT was very slow under these conditions (dashed red line). (b) EDTA-induced loss of G85R dimer in the absence (solid black line) and presence of the 15 best inhibitors of A4V aggregation (see a). (c) EDTA-induced loss of G93A dimer in the absence (black line) in presence of the 15 best inhibitors of A4V aggregation. (d) EDTA-induced loss of WT dimer in the absence (black line) and presence of the 15 best inhibitors of A4V aggregation. Note the difference in the scale of the y axes.
The Inhibitory Effect Was Independent of Metal Binding Site Occupancy. Because the aggregation assay described above used EDTA to promote metal loss and accelerate aggregation (8), the observed inhibitory effect of a given compound could have been due to inhibition of demetallation rather than inhibition of dimer dissociation. To rule out the former possibility, the effects of compounds 1-15 (Fig. 8a, which is published as supporting information on the PNAS web site) on the aggregation of A4V in the absence of EDTA were measured (Fig. 8a). All 15 compounds slowed aggregation of A4V under these conditions. All 15 compounds also inhibited the aggregation of the completely demetallated apo-A4 (also in the absence of EDTA; Fig. 8c). This effect suggests that these molecules can bind and stabilize the apo-A4V dimer (crystalline apo-WT and metallated WT are indistinguishable with respect to the cavity that is the focus of our screen, Fig. 9b, which is published as supporting information on the PNAS web site).
The Inhibitory Effect of These Compounds Is Likely to Be Due to Their Affinity for the Cavity at the A4V Interface. To validate the rationale behind our in silico screening approach, we tested four of the most potent A4V aggregation inhibitors (2, 3, 4, and 7) to determine whether they were capable of inhibiting the aggregation of A4V/V7F/V148F, where the putative binding site had been disturbed. There was no appreciable change in the aggregation rate of A4V/V7F/V148F in presence of these compounds (Fig. 8c). Furthermore, a set of 20 arbitrarily chosen compounds from the initial database were found to have no effect on the aggregation of A4V (Fig. 8d). Finally, none of the top 15 A4V inhibitors affected the aggregation of α-synuclein (Fig. 8e).
The A4V Aggregation Inhibitors also Inhibited Aggregation of Other FALS-Linked SOD1 Mutants. Because the cavity at the A4V dimer interface was conserved in other FALS-linked SOD1 mutants (see above), we expected that the A4V inhibitors may also inhibit the aggregation of these proteins. Aggregation (EDTA-induced) of both G93A and G85R were significantly inhibited by several of the A4V inhibitors (Fig. 3 b and c). Interestingly, compounds 2, 3, 4, and 7 were among the best inhibitors in each case, as was the case with A4V. WT SOD1 was also subjected to aggregation in the presence of these compounds under the same conditions as described above. The aggregation of WT SOD1 under these conditions was too slow to observe a significant inhibition (Fig. 3d).
All 15 A4V Aggregation Inhibitors also Stabilized A4V Against Denaturation. If the A4V aggregation inhibitors act by binding A4V dimer and inhibiting its dissociation, they should also stabilize the native dimer against chaotrope-induced unfolding (see Fig. 4b). Although the completely unfolded state is probably not relevant to the aggregation pathway (10), these experiments provided a convenient and well precedented method to measure the relative stability of the native A4V dimer in the presence and absence of small molecules. All 15 of the aggregation inhibitors significantly protected A4V from GdnCl-induced unfolding (Fig. 4a; black dashed line is A4V, and red dashed line is WT). As controls, 10 of the 85 compounds that did not show significant aggregation inhibition were tested, and none of these compounds had a significant effect on unfolding (data not shown). The unfolding curves were analyzed directly, assuming a two-state (N → U) transition, and thermodynamic properties were measured by fitting the data to a linear extrapolation model (35). The stabilization of A4V in presence of the compounds was expressed as ΔG values, which are listed in Table 3, which is published as supporting information on the PNAS web site. These values reflect the binding energy of the compounds, presumably to the cavity at the dimer interface (see Fig. 4b). Four compounds (2, 3, 4, and 7) stabilized A4V nearly to WT levels. These four compounds were among the most potent aggregation inhibitors.
Fig. 4.

Chemical denaturation of SOD1 was inhibited by compounds that inhibited aggregation and were calculated to bind to the cavity at the interface. (a) GdnCl-induced unfolding of A4V in the presence of the 15 best aggregation inhibitors; plotted as fraction folded vs. [GdnCl]. The data has been fitted to a two-state model to allow estimation of thermodynamic parameters (Table 3). (b) Schematic and simplified representation of the reaction analyzed here (complete unfolding is a convenient experimental system but is not required for aggregation): SOD1 dimer can be stabilized relative to the unfolded state by binding to drug-like molecules. The A4V dimer (Center, red circles denote residue Val4) is much less stable than the WT dimer (Left). However, the binding of the drug molecule (purple square) to the unstable A4V dimer can decrease its free energy (ΔG of binding in green; Table 3). Binding results in depopulation of the aggregation-prone metal-free A4V monomer, decreasing the rate of aggregation.
Discussion
ALS, or Lou Gehrig's disease, is a degenerative and invariably deadly motor neuron disease that affects >35,000 Americans. Typically, one to five years elapses between the diagnosis of ALS and death by asphyxiation. There are no effective treatments. The linkage of the gene encoding SOD1 to a subset of cases of FALS has provided hope that this situation may change, and the work reported here represents our initial efforts to stimulate this change. It is unclear whether SOD1-linked FALS is similar in etiology to sporadic ALS, which constitutes the great majority of cases, but if a similarity exists, the strategy introduced here may represent a general approach to ALS.
The aggregation of mutant forms of SOD1 may be pathogenic in FALS. This process is very complex, even under controlled in vitro conditions, because it may require the loss of copper and zinc, reduction of an intrasubunit disulfide, monomerization, and partial unfolding (7, 9-14). The work here was based on the premise that stabilization of the SOD1 native dimer will inhibit its aggregation regardless of the exact pathway because it will deplete the population of the aggregating species, which may be a partially unfolded apomonomer. It is very important to note that one product of this simple screen (compound 2), even without optimization by medicinal chemistry, stabilizes the A4V dimer to an extent comparable with the difference in stability between the invariably lethal A4V and WT SOD1 (see Table 3).
The strategy of stabilizing a native oligomer to prevent its pathogenic aggregation has been successfully exploited by Kelly et al. (18) in the analogous case of transthyretin. In the case of SOD1, because there are no known endogenous ligands (unlike thyroxine in the case of transthyretin) to serve as a starting point for the identification of stabilizing compounds, we embarked on an in silico screening program. This approach is practical for academic laboratories where medicinal chemistry resources are limited. The objective of the in silico screen, which was to select an easily screenable compound set that would have a high likelihood of binding to the A4V dimer, was met 15 of our top 100 hits had significant activity in experimental assays for A4V aggregation and A4V unfolding. Several control experiments support the proposal that these compounds are binding to the cavity at the A4V interface, the intended mechanism of action. Of course, some of the compounds that were not in the top 100 may have activity (although 20 randomly chosen compounds from the original library had no activity).
The group of drug-like compounds that are reported here are chemically (Fig. 7) and structurally similar (Fig. 5) and, therefore, represent a good starting point for optimization by iterative medicinal chemistry. Modeling of the interaction of these compounds with the A4V dimer interface show that the shared aromatic moiety may occupy the space between the two Val 148 residues of the SOD1 subunits (introduction of an intersubunit disulfide at this position, by mutagenesis, was shown to stabilize the dimer of A4V against aggregation (16). After some improvement of potency and optimization of other critical biological properties (ADME, reduced toxicity), we believe that a compound that can be administered to SOD1 transgenic mouse models of FALS to test the central hypothesis of this work: that SOD1 dimer stabilization should slow the onset and progression of FALS.
Fig. 5.

Aggregation inhibitors have similar structural features and are predicted to bind similarly. (a) Docked poses of 6 of the 15 molecules that inhibit SOD aggregation. Four of the six molecules (panels 1, 2, 4, and 5; molecules 2, 3, 5, and 7) exhibit a very similar mode of binding. These molecules represent the major class of active molecules found in our assay and bear an aromatic ring that binds in a deep pocket. Panels 3 and 6 (triamcinolone and N6-methyladenosine, respectively) are unlike the other hits and exhibit different modes of binding, although it still binds in the same cavity. (b) An overlay of the hits obtained from screening shows a conserved aromatic moiety at the hydrophobic pocket. The remaining portion of the molecules is more variable.
Even if these animal studies produce promising results, it is very unlikely that any academic laboratory will be able to marshal the resources required to convert a compound that shows activity in animal studies into a compound that could be a candidate for human clinical trials. The issue of compound “patentability” must also be solved by medicinal chemistry (the libraries screened here contain only public domain compounds) because a commercial therapeutic is the ultimate goal. It is our hope that pharmaceutical companies will use the methodology presented here to screen their own libraries of proprietary compounds or will make their libraries available to other researchers. Hits from such screens may represent the best hope for validating this target in an animal model and, ultimately, for the development of an ALS therapy.
Supplementary Material
Acknowledgments
We thank Greg Cuny for insightful discussions on the medicinal chemistry of the SOD1 aggregation inhibitors and for providing the raw compound libraries; John Hart (University of Texas, San Antonio) and Lawrence Hayward (University of Massachusetts, Worcester) for providing us with cDNA for G85R mutant of SOD; and Dr. A. Z. Mason (California State University, Long Beach) for performing inductive coupled plasma mass spectrometry analysis of the SOD samples. The inductive coupled plasma mass spectrometry facility at Long Beach is supported by National Science Foundation Grant OCE-9977564. This work was supported by National Institutes of Health Grant 085476 (to P.T.L.), a research grant from the ALS Association, and a generous gift from the Tow family. R.H.B. thanks the National Institute of Neurological Disorders and Stroke, the National Institute on Aging, ALS Therapy Alliance, Project ALS, and the A. L. Athel ALS Research Foundation.
Author contributions: S.S.R. and P.T.L. designed research; S.S.R. and R.J.N. performed research; S.S.R. contributed new reagents/analytic tools; S.S.R., R.J.N., R.H.B., and P.T.L. analyzed data; and S.S.R., R.H.B., and P.T.L. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ALS, amyotrophic lateral sclerosis; FALS, familial ALS; SOD1, superoxide dismutase 1; TTR, transthyretin.
References
- 1.Brown, R. H., Jr. (1995) Curr. Opin. Neurobiol. 5, 841-846. [DOI] [PubMed] [Google Scholar]
- 2.Brown, R. H., Jr. (1995) Cell 80, 687-692. [DOI] [PubMed] [Google Scholar]
- 3.Bruijn, L. I. & Cleveland, D. W. (1996) Neuropathol. Appl. Neurobiol. 22, 373-387. [DOI] [PubMed] [Google Scholar]
- 4.Lindberg, M. J., Tibell, L. & Oliveberg, M. (2002) Proc. Natl. Acad. Sci. USA 99, 16607-16612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston, J. A., Dalton, M. J., Gurney, M. E. & Kopito, R. R. (2000) Proc. Natl. Acad. Sci. USA 97, 12571-12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayward, L. J., Rodriguez, J. A., Kim, J. W., Tiwari, A., Goto, J. J., Cabelli, D. E., Valentine, J. S. & Brown, R. H., Jr. (2002) J. Biol. Chem. 277, 15923-15931. [DOI] [PubMed] [Google Scholar]
- 7.Tiwari, A. & Hayward, L. J. (2003) J. Biol. Chem. 278, 5984-5992. [DOI] [PubMed] [Google Scholar]
- 8.Hough, M. A., Grossmann, J. G., Antonyuk, A., Strange, R. W., Doucette, P. A., Rodriguez, J. A., Whitson, L. J., Hart, P. J., Hayward, L. J., Valentine, J. S. & Hasnain, S. S. (2004) Proc. Natl. Acad. Sci. USA 101, 5976-5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindberg, M. J., Normark, J., Holmgren, A. & Oliveberg, M. (2004) Proc. Natl. Acad. Sci. USA 101, 15893-15898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khare, S. D., Caplow, M. & Dokholyan, N. V. (2004) Proc. Natl. Acad. Sci. USA 101, 15094-15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnesano, F., Banci, L., Bertini, I., Martinelli, M., Furukawa, Y. & O'Halloran, T. V. (2004) J. Biol. Chem. 279, 47998-48003. [DOI] [PubMed] [Google Scholar]
- 12.Furukawa, Y., Torres, A. S. & O'Halloran, T. V. (2004) EMBO J. 23, 2872-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doucette, P. A., Whitson, L. J., Cao, X., Schirf, V., Demeler, B., Valentine, J. S., Hansen, J. C. & Hart, P. J. (2004) J. Biol. Chem. 279, 54558-54566. [DOI] [PubMed] [Google Scholar]
- 14.Chung, J., Yang, H., de Beus, M. D., Ryu, C. Y., Cho, K. & Colon, W. (2003) Biochem. Biophys. Res. Commun. 312, 873-876. [DOI] [PubMed] [Google Scholar]
- 15.Ray, S. S. & Lansbury, P. T., Jr. (2004) Proc. Natl. Acad. Sci. USA 101, 5701-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ray, S. S., Nowak, R. J., Strokovich, K., Brown, R. H., Jr., Walz, T. & Lansbury, P. T., Jr. (2004) Biochemistry 43, 4899-4905. [DOI] [PubMed] [Google Scholar]
- 17.Colon, W., Lai, Z., McCutchen, S. L., Miroy, G. J., Strang, C. & Kelly, J. W. (1996) Ciba. Found. Symp. 199, 228-238, and discussion 239-242. [DOI] [PubMed] [Google Scholar]
- 18.Kelly, J. W., Colon, W., Lai, Z., Lashuel, H. A., McCulloch, J., McCutchen, S. L., Miroy, G. J. & Peterson, S. A. (1997) Adv. Protein Chem. 50, 161-181. [DOI] [PubMed] [Google Scholar]
- 19.Hammarstrom, P., Schneider, F. & Kelly, J. W. (2001) Science 293, 2459-2462. [DOI] [PubMed] [Google Scholar]
- 20.Miroy, G. J., Lai, Z., Lashuel, H. A., Peterson, S. A., Strang, C. & Kelly, J. W. (1996) Proc. Natl. Acad. Sci. USA 93, 15051-15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oza, V. B., Petrassi, H. M., Purkey, H. E. & Kelly, J. W. (1999) Bioorg. Med. Chem. Lett. 9, 1-6. [DOI] [PubMed] [Google Scholar]
- 22.Baures, P. W., Oza, V. B., Peterson, S. A. & Kelly, J. W. (1999) Bioorg. Med. Chem. 7, 1339-1347. [DOI] [PubMed] [Google Scholar]
- 23.McCammon, M. G., Scott, D. J., Keetch, C. A., Greene, L. H., Purkey, H. E., Petrassi, H. M., Kelly, J. W. & Robinson, C. V. (2002) Structure (London) 10, 851-863. [DOI] [PubMed] [Google Scholar]
- 24.Baures, P. W., Peterson, S. A. & Kelly, J. W. (1998) Bioorg. Med. Chem. 6, 1389-1401. [DOI] [PubMed] [Google Scholar]
- 25.Adamski-Werner, S. L., Palaninathan, S. K., Sacchettini, J. C. & Kelly, J. W. (2004) J. Med. Chem. 47, 355-374. [DOI] [PubMed] [Google Scholar]
- 26.Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T. & Banks, J. L. (2004) J. Med. Chem. 47, 1750-1759. [DOI] [PubMed] [Google Scholar]
- 27.McCord, J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6049-6055. [PubMed] [Google Scholar]
- 28.Conway, K. A., Harper, J. D. & Lansbury, P. T. (1998) Nat. Med. 4, 1318-1320. [DOI] [PubMed] [Google Scholar]
- 29.Kleywegt, G. J. & Jones, T. A. (1999) Acta Crystallogr. D. 55, 941-944. [DOI] [PubMed] [Google Scholar]
- 30.Wallace, L. A., Burke, J. & Dirr, H. W. (2000) Biochim. Biophys. Acta 1478, 325-332. [DOI] [PubMed] [Google Scholar]
- 31.Eilers, M., Shekar, S. C., Shieh, T., Smith, S. O. & Fleming, P. J. (2000) Proc. Natl. Acad. Sci. USA 97, 5796-5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karpusas, M., Baase, W. A., Matsumura, M. & Matthews, B. W. (1989) Proc. Natl. Acad. Sci. USA 86, 8237-8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. (2001) Adv. Drug Delivery Rev. 46, 3-26. [DOI] [PubMed] [Google Scholar]
- 34.Kontoyianni, M., McClellan, L. M. & Sokol, G. S. (2004) J. Med. Chem. 47, 558-565. [DOI] [PubMed] [Google Scholar]
- 35.Santoro, M. M. & Bolen, D. W. (1992) Biochemistry 31, 4901-4907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.