

Abstract
Kinetic and structural studies of wild-type proteins such as prions and amyloidogenic proteins provide suggestive evidence that proteins may adopt multiple long-lived states in addition to the native state. All of these states differ structurally because they lie far apart in configuration space, but their stability is not necessarily caused by cooperative (nucleation) effects. In this study, a minimalist model protein is designed to exhibit multiple long-lived states to explore the dynamics of the corresponding wild-type proteins. The minimalist protein is modeled as a 27-monomer sequence confined to a cubic lattice with three different monomer types. An order parameter—the winding index—is introduced to characterize the extent of folding. The winding index has several advantages over other commonly used order parameters like the number of native contacts. It can distinguish between enantiomers, its calculation requires less computational time than the number of native contacts, and reduced-dimensional landscapes can be developed when the native state structure is not known a priori. The results for the designed model protein prove by existence that the rugged energy landscape picture of protein folding can be generalized to include protein “misfolding” into long-lived states.
Do proteins assume long-lived states that are not near the native state in configuration space? Kinetic and structural studies of some wild-type proteins provide evidence that proteins can adopt multiple long-lived conformations in addition to the native state conformation (1–3). Prion proteins exhibit two long-lived states—a native (cellular) form and a nonnative scrapie form (1). Because the secondary structures of these two prion forms differ, these forms may arise from different “molten globules.” Similar two-state behavior has been observed in bacterial luciferase (2) and amyloidogenic proteins (3). These proteins assume long-lived states that are not near the native state in configuration space, and our understanding of the protein folding landscape must be extended to include them.
Perhaps these nonnative long-lived states are only formed through an aggregative mechanism. In a templating aggregation, the nonnative states are formed in the presence of other misfolded nonnative states as seen in those prions and amyloidogenic proteins, which form aggregates or plaques that stabilize the nonnative long-lived folded state (1). In a nucleating aggregation, multiple proteins fold in tandem as seen in bacterial luciferase, which has been isolated in its monomeric form as well as in its structurally different dimer (2). Recent theoretical work has discussed (4) and studied (5) the specific case in which the nonnative long-lived folded state is stabilized in the presence of another model protein. Although the interactions between the individual proteins in these aggregates play a role in stabilizing the nonnative long-lived folded state in vivo, Jackson et al. (6) recently have shown that human prion protein can convert between its native and fibrilogenic conformations in monomeric form by altering the solvent pH in vitro. Thus, the generic behavior of a system with multiple long-lived states can be seen by the direct study of single (model) proteins without suffering the slow time scales of templated or nucleating systems.
Much of the current theoretical understanding of protein folding dynamics has arisen from the analytical studies and simulations of minimalist models (7, 8). In those proteins that fold, the rugged energy landscape has the primary structure of a funnel (9, 10). The protein is driven to lower free energy conformations by increasing its hydrophobic contacts (11, 12) while minimizing frustration (9). Nonnative, quasi-stable structures that give rise to the ruggedness are long-lived when they are kinetically trapped by high-energy barriers (10). However, fast-folding proteins necessarily have few kinetic traps along the folding funnel. This condition is satisfied if there are few to no interactions, leading to lower energies in directions opposite to folding, and is tantamount to imposing the minimal frustration principle (9). Thus, for fast-folding proteins to assume a long-lived, nonnative folded structure, the misfolded structure must not lie on the native folding funnel. This finding suggests that there may exist alternate folding funnels leading to nonnative folded structures that are somehow inaccessible under typical folding conditions.
Monte Carlo dynamics simulations of minimal protein models reveal the possibility that proteins may become trapped in long-lived states that are not the global energy minimum of the protein (13, 14). Some minimalist proteins exhibiting this behavior are classified as slow folders, although it is still an open question whether they correspond to naturally occurring proteins. The metastability hypothesis further postulates that globular proteins are found in nature in one of many possible metastable states with differing energies, but similar structural characteristics (7, 10, 15). Because the metastable states typically considered in the kinetic partitioning scheme reside on the same overall folding funnel, they are necessarily conformationally similar (in the sense of the disconnectivity graph to be discussed below). Thus when a protein gives rise to multiple long-lived states, each with differing functions and structural characteristics, it is no longer in the regime described by the metastability hypothesis. However, these could be compatible with the rugged energy landscape perspective for protein folding (10) if only the energy landscape is extended to include multiple folding funnels. For example, the two long-lives states in prions correspond to conformations that are far from each other in configuration space (due to their dramatically different secondary structures). Within the proposed generalization of the protein folding landscape, these long-lived “folded” states would arise from separate folding funnels.
This question can be recast within the language of disconnectivity graphs (16–18). In such graphs or trees, local and global minima are separated (disconnected) according to the energy at which they are no longer accessible to each other on the energy landscape. In the single-funnel picture, all of the disconnected subtrees are essentially small and thus it is easy for the protein to quickly bypass off-route basins on the way to folding. Although usually not discussed, it has been recognized that the minimalist protein models always admit a degenerate native structure (19). In most of the sequences observed to date, these native enantiomeric states are separated by low energetic barriers, and hence lie on the same final branches of the disconnectivity graph. In the present work, we construct a minimalist model sequence in which the energetic barrier is high, and hence the disconnectivity occurs early along the route to folding. It further affords us the possibility of studying the trapping dynamics or lack thereof that may be a consequence of the early disconnectivity.
The minimalist model and its design are reviewed in the next section. The long-lived structures chosen for the minimalist protein (illustrated in Fig. 1) are notable because of their high degree of winding about a central axis. The winding index introduced in this article is capable of distinguishing between the two long-lived enantiomers, whereas the number of native contacts is not. Equilibrium properties, such as the potentials of mean force (PMFs) with respect to various order parameters and the phase diagram, have been obtained. The energy landscape for the designed minimalist model sequence clearly contains multiple folding funnels as seen in the projected PMFs. This designed minimalist model, therefore, provides an explicit example of a (minimalist) protein with multiple long-lived states in a nonaggregating environment. The characteristic rates for various parts of the folding process also have been calculated directly from Monte Carlo simulations using the mean first-passage time (MFPT) approach. Thus, this study generalizes the rugged energy landscape perspective to allow for the possibility that even fast-folding proteins may accommodate multiple long-lived folded structures beyond the native state that are inaccessible under typical folding conditions.
Figure 1.

The folded enantiomeric target structures for DS1, with the left helix (Left) and the right helix (Right). (The winding index, wz, for the left and right helix are −11 and +11, respectively.) The three monomer types are distinguished by red (H), blue (P), and fuchsia-green (N) spheres.
A Minimalist Model
Protein Model.
The minimalist protein model is defined in this work as a discrete heteropolymer—i.e., all monomers (effective amino acid residues) are restricted to a three-dimensional rectilinear lattice—with a sequence consisting of 27 monomers. This model obeys a “law of corresponding states” between the space of all possible 27-mer configurations and that of a helical protein with a 60-aa residue sequence (10). The energy for a given conformation of the minimalist protein is
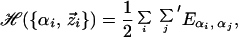 |
1 |
where {αi} is the ordered sequence of monomer types, Eα,β is the contact energy between the α-type monomer and β-type monomer, {z⃗i} is a sequence of the positions z⃗i of the monomers in a given structure, and the prime on the second sum restricts j to only those values that correspond to nonbonded (nearest neighbor) sites adjacent to zi. For simplicity, the monomer types α are restricted to a set of three values, H, P, and N, corresponding to the hydrophobic, polar, and neutral regions of the protein, respectively. All of the contact energies are favorable, i.e., lower in energy than contact to the uniform solvent. The native state is necessarily maximally compact; the native 27-mer fills a cube (13, 20). The numerical values for the contact energies are chosen so that an increase in the number of hydrophobic contacts is the dominant driving force for folding. Specifically, these energies are EH,H = −4, EH,P = −1, EH,N = EP,N = −2, and EP,P = EN,N = −3. (Note that all energies and inverse temperatures reported throughout this article are reported in dimensionless units relative to a standard temperature T0).
Protein Structure and Design.
In an attempt to construct sequences that give rise to multiple nontrivial funnels, one can imagine a native structure with a mirror symmetry or chirality where the landscape is symmetric with respect to the corresponding enantiomeric folded states. Target structures with this symmetry are presented in Fig. 1. Because both states contain the same contacts (because the structures are mirror images), a designed sequence (DS) will fold to one or the other. In most studies to date these structures have been treated as the same, and the order parameter characterizing the number of native contacts does not distinguish between them. The target structures in Fig. 1 are chosen because they are strongly separated in configuration space. Hence, the two states are separated by a large enthalpic barrier, ensuring that the target structures will correspond to distinct folding funnels.
Several algorithms exist for the design of sequences that are fast folders within a single funnel (13, 21–26). In particular, the energy landscape perspective suggests that if a sequence gives rise to a fast-folding protein, then the energy for the corresponding native structure will be a minimum (8). Given a compact target structure and sequence composition, a DS (that folds to the target structure) is obtained by minimizing the energy (as a function of the sequence identity) (13, 21). In this work, the energy is minimized by using the Metropolis Monte Carlo method with simulated annealing varying the sequence for a fixed structure. The primary sequence (DS1) in Fig. 1 is designed for a 27-monomer sequence with equal concentrations of H-, P-, and N-type monomers. The optimization yields three distinct, but similar, sequences (DS1, DS2, and DS3) that fold to the target structures with the same minimum energy, ENS = −89. The DS1 sequence,
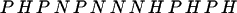 |
2 |
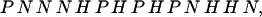 |
is used in all of the reported simulations of this work and was chosen arbitrarily. If the DS resulted in an energy minimum with optimal contacts (no unlike or unfavorable monomer contacts), then the sequence would necessarily contain the target structure as an energy minimum, although it could be kinetically inaccessible. However, DS1 gives a target structure with some energetic frustration. Simulated annealing within a Metropolis Monte Carlo chain dynamics algorithm (provided below) nonetheless confirms that the energy minimum of DS1 is the target structure. Exhaustive enumeration of all maximally compact structures further confirms that the target structures of Fig. 1 are the lowest-energy maximally compact configurations for DS1.
The Winding Index.
The analysis of the folding dynamics is greatly simplified if there exists a one-dimensional order parameter—namely, a projection of the configuration space—that can uniquely and uniformly distinguish between different stages along the folding process. For this criterion to be satisfied, the inverse images of the order parameter must be uniformly characteristic of the extent of folding. Each such inverse image is consequently likely to be a connected space that contains configurations with similar free energies. The subsequent results suggest that the winding index is a suitable order parameter for the folding of DS1. Because the winding index differentiates between the left-handed and righ-handed minimum-energy enantiomers of DS1, whereas the number of native contacts (and related indices) does not, the winding index is used as an order parameter to analyze both the thermodynamics and kinetics of the folding process for DS1 with multiple nontrivial funnels.
On a lattice, there exist three canonical (rectilinear) axes about which the winding index may be defined. For a given axis, q ∈ {x,y,z}, the winding index (in units such that an increase of wq by one corresponds to a {1/2}}π turn) is herein defined as
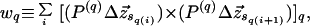 |
3 |
where Δz⃗i ≡ (z⃗i+1 − z⃗i), P(q) projects onto the plane orthogonal to the q-axis, {sq(i)} is an ordered sequence of the indices for which P(q)Δz⃗sq(i) is nonzero, the q-subscript in the bracketed summand denotes the q-component of the cross product, and i runs across the members of the {sq(i)} sequence. In a straight chain configuration, for example, the set {sq(i)} is the null set, and the winding indices, w⃗, are zero.
The construction of the winding index in Eq. 3, readily generalizes to arbitrary axes through the use of arccosines. However, the canonical (rectilinear) axes are used exclusively throughout this work because of the computational ease in calculating Eq. 3, and because the winding indices are maximal on these axes as a consequence of the lattice geometry of the minimalist model. There is a fair amount of indeterminacy in the definition of the winding index given by Eq. 3. The indeterminacy arising from the use of each end as the initial monomer leads to opposite signs in the winding indices. This is eliminated by calculating w⃗ starting from a specified end that is uniquely identifiable through its sequence. The remaining indeterminacy caused by the rotational symmetry may be eliminated by choosing wz as the one with the largest, in magnitude, component of w⃗, and wy as the second largest, in magnitude, component of w⃗. In those cases where there is a degeneracy in the magnitude but not the sign of several components of w⃗, the positively signed component is arbitrarily designated as “larger.” In what follows, these additional constraints will be implicitly included in the definition of the winding index in Eq. 3.
Although the winding index may be used as the primary order parameter only when the long-lived structures have a large degree of winding, the choice of wz as the order parameter for folding has some advantages when compared with the number of native contacts Q or related indices: (i) the native structure is not needed to calculate wz, which is of particular advantage in large proteins where the native state structure is not known a priori; (ii) wz is calculated from mostly local geometric properties and is thus significantly faster to compute than Q; and (iii) wz can distinguish between structures that differ by an inversion symmetry operation.
Equilibrium and Dynamics
Phase Transitions.
The DS1 protein exhibits both a folding and glass transition that are sufficiently separated so as to admit a nontrivial folding regime. A Metropolis Monte Carlo dynamics algorithm (see below) is used to collect the equilibrium statistics necessary to obtain this transition temperatures for the DS1 protein. The probability that DS1 is in one of the two minimum-energy structures—namely, the long-lived states—for a series of inverse temperatures, β ≡ 1/kBT, is shown in Fig. 2. The folding transition temperature is defined as in ref. 13 to be the temperature at which half of the population is in either minimum-energy configuration. According to Fig. 2, this criterion is met by DS1 at a βf approximately equal to 0.86. The glass transition temperature is estimated to be the temperature at which the dynamics of the system are slow enough that a significant portion of the ensemble will not fold to the native state in the simulation time. For DS1, this occurs at βg near 0.95. Thus the inverse temperature region, βf ≲ β ≲ βg, for DS1 is broad enough so that the sequence will fold to a long-lived structure. (Note that the emphasis here is on the long-lived structures and not on the native state. Because DS1 gives rise to two equally stable long-lived structures, it is not clear which is the unique native state. Nonetheless, as shown here, dynamic and thermodynamic processes may still be well-defined with respect to the degenerate long-lived structures. The term “native structure” is used to describe both enantiomers in this work to better conform with the usual language.)
Figure 2.

The probability of DS1 being in the native state for a Monte Carlo dynamics simulation is displayed at various inverse temperatures, β. The sigmoidal curve is drawn to guide the eye. The folding temperature is estimated as the temperature when half of the population is in the native state.
The Role of Winding Index in the Folding Path.
To visualize the many-dimensional energy landscape, reduced dimensional coordinates—either order parameters or folding-path coordinates—have been used to characterize folding on projected spaces (16, 17). The use of an order parameter is a weaker requirement for this characterization because it need only distinguish between the important separable (and identifiable) regions or phases of the folding protein such as the denatured and native structures. The number of native contacts Q has routinely been used as an order parameter because it generally satisfies this requirement, but in the case of the DS1 protein, it fails to distinguish between the structurally different enantiomers. The stronger requirement of a folding-path coordinate is that it be both an order parameter and that it be characteristic of the actual folding paths of the protein. If such a folding-path coordinate exists, then the projection of the energy landscape onto a PMF with respect to this coordinate will minimize its coupling to the projected modes (27, 28). As the determination of this coupling is nontrivial, in what follows, we use heuristic arguments to discuss the role of wz as a possible folding-path coordinate in contrast to Q.
The thermal average of a good order parameter for folding should exhibit a change from the expectation value for an unfolded state to the expectation value of the long-lived states. In particular, as described in the previous section, an increasing percentage of the protein population corresponds to the long-lived states with increasing β. In DS1, the symmetry-broken long-lived structure thus determines the expectation values at high β whereas denatured states determine them at low β. Coordinates that measure structural features characteristic of the native structure should consequently average to different expectation values as function of temperature. This is indeed seen for the thermal average of a conventional order parameter 〈Q〉 as shown in Fig. 3 for the DS1 protein as a function of effective inverse temperature, as long as the symmetry of the enantiomers is ignored. This is compared with the behavior of the thermal average of the maximal winding index 〈|wz|〉 for the DS1 protein over the same temperature range. Both coordinates seem to characterize the extent of folding in the same way as shown by a similar sigmoidal temperature dependence. As β→0, 〈Q〉, and 〈|wz|〉→0 and as β→∞, 〈Q〉→28 (the maximum possible number of native contacts) and 〈|wz|〉>→11 (the magnitude of the winding index of the native state structures). Thus, in this averaged sense, wz is as good an order parameter as Q and it offers additional information because it can distinguish between the distinct long-lived enantiomers of DS1.
Figure 3.

The ensemble-averaged number of native contacts Q (black) and the ensemble-averaged winding index wz (red)—defined in Eq. 3—of DS1 is shown as a function of inverse temperature, β ≡ (1/kBT), in arbitrary units.
Having established that wz is a useful order parameter for the folding dynamics, it remains to be shown that it is also a reasonable folding-path coordinate in the sense of a reaction path. In principle, this question should be addressed by optimizing the rate along all possible paths on the full-dimensional energy landscape (29, 30), and it is doubtful that wz would emerge as the optimum path. However, for it to be a reasonable path on the full-dimensional energy landscape, it need only give rise to a partitioning of the full-dimensional space into the correct “reactant” and “product” regions necessary for transition-state theory estimates of the rate to be accurate. Unfortunately, this softer criteria is difficult to determine for a given order parameter as it requires a full-dimensional analysis. Instead, we soften the requirement for order parameters to be a reasonable folding coordinate further by requiring that it need only give rise to a smooth partitioning of a reduced dimensional space into reactant and product regions.
The projection of the folding funnel of the right helix in Fig. 1 onto the reduced two-dimensional space of Q and wz at the inverse temperature β = 0.875 is the PMF shown in Fig. 4. All PMFs in this work have been obtained by the use of umbrella sampling combined with the weighted histogram analysis method (31). Although not obvious from the two-dimensional PMF, the surface is smooth along wz for fixed values of Q whereas the surface is rough and singular along Q for fixed values of wz. The latter failing in Q has its origin in the geometry of the lattice model. For example, there is a singularity at Q = 27 because there are no configurations with this value, although it lies on the downward path to the native structure at Q = 28. Some of the roughness visible on the PMF along Q exhibits an alternating pattern that also may be a consequence of the geometric constraints. Interestingly, wz seemingly exhibits no manifestations of the geometric constraints. The reactant and product regions are clearly visible on the PMF. Furthermore, there is a clear intermediate formed at wz = 5,6 and Q = 14. This is partially obstructed by a spike on the PMF at wz = 11 and Q = 14 in Fig. 4, but the intermediate is clear in one-dimensional PMF plots as a function of both order parameters. (The PMF as a function of wz is shown in Fig. 5.) Thus both Q and wz parametrize the important dynamic subspaces of the full-dimensional lattice space, but wz is a better choice as the reaction-path coordinate for the particular case of DS1 because it smoothes out artifacts caused by the lattice geometry and differentiates between the long-lived structures.
Figure 4.

The two-dimensional PMF U(Q,wz) obtained at β = 0.875 is displayed as a surface plot over the number of contacts Q and the maximal winding index wz. For the domain as defined in the figure, the denatured states are found toward the rear of the surface at low Q and wz values and the native state is located at the minimum toward the front of the surface. To enhance the view of the native state, the values of wz greater than 11, and all U(Q,wz) points that were not explored because of the constraints imposed by the lattice are not shown.
Figure 5.

The PMF with respect to the winding index, wz, of the minimalist protein DS1 is shown at four different inverse temperatures, β ≡ (1/kBT), in arbitrary units. Note that the inverse folding temperature, βf, is about 0.86, whereas the inverse glass transition temperature, βg, is above 0.9.
The PMF as a function of the winding index wz of DS1 is displayed in Fig. 5 for several effective inverse temperatures. The PMFs display behavior that is consistent with the various thermodynamic regimes of proteins, from denatured through folding to glass dynamics. At low β(≲0.8), the PMF has a minimum at a low value of the winding index and remains faraway from the native structure. At β∼βf (≈0.86), the PMF has minima at the value of the winding index of the native structures, with no other false minima trapping the protein. At higher values of β(≳0.875), a second minimum at a much lower value of the winding index develops. This minimum can trap the protein in a nonnative state for long times that are comparable to the inverse of the rate caused by the barrier. Thus the PMF with respect to the wz order parameter exhibits the features that would be expected from a useful order parameter characterizing the folding process.
Folding Dynamics with Multiple Deep Minima.
A Metropolis Monte Carlo algorithm is used to study the folding dynamics of the optimized heteropolymer, DS1 (13, 21). Moves along the lattice are chosen from a set of three possible types—a one-monomer corner flip, a one-monomer end pivot, and a two-monomer crankshaft move (13). This is not a true dynamics algorithm, and some caution need be applied in interpreting the time scale of these “dynamics.” However, in cases with fast folding, the dynamics are directed primarily by free energy differences driving the system toward lower and lower energy states. This is precisely the driving force in the Monte Carlo algorithm, and as such it does provide a reasonable approximation to the relative folding times of the minimalist models.
The Monte Carlo dynamics of DS1 is displayed in Fig. 6 for two representative trajectories. Both of these trajectories fold quickly to either the right or left helix as can be seen by how quickly they each achieve the energy minimum and the winding index wz of the target structure. These two trajectories suggest that DS1 is a relatively fast folder, and the folding rates calculated in the subsequent section from an ensemble of unfolded proteins confirm that DS1 is a fast folder. The trajectories are displayed for a long time beyond the first folding event to make it clear that once the protein has folded to the structure of a given winding index, it “locks” into that structure with respect to the winding index. This provides corroborative evidence that the DS1 does in fact have two long-lived states. That is, the left and right helical (long-lived) states are separated strongly in space by an enthalpic barrier as illustrated in Fig. 5, and they are separated strongly in time (dynamically) as suggested by Fig. 6.
Figure 6.

Two different (stochastic) folding trajectories for a minimalist protein with the sequence of Fig. 1, with a straight chain conformation as the initial configuration, and propagated at an inverse temperature of 0.95 (between βf and βg) are shown as a function of the Monte Carlo move number. In both panels, the black curve is the energy of the minimalist protein as a function of move number and corresponds to the energy axis on the left. The red curve is the winding index wz of the minimalist protein as a function of move number and corresponds to the winding index axis on the left. The dot-dashed lines help to guide the eye to the expected positive or negative values of the winding index for the right or left helical long-lived structures, respectively.
Transition Rates.
Several methods exist for calculating reaction rates in canonical systems (32–49). Among these, the MFPT approach has the advantage that it does not require a knowledge of velocities. As such, it is amenable to the calculation of rates for the pseudodynamics of a Monte Carlo simulation. The MFPT approach is reviewed in ref. 49, and several important references may be found therein. An illustrative case in which this procedure provides exact rates is the exponential decay of reactants from the region Ω to a product region Ω′—where Ω ∩ Ω′ = ⊘. The normalized probability distribution of the reactants is given by
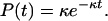 |
4 |
The rate for this process is κ as calculated from the expression, κ = −{d/dt}lnP(t), for any t. The same rate is obtained by calculating the mean survival time,
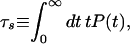 |
5 |
which turns out to be κ−1. The MFPT may be defined as in ref. 49 as
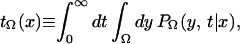 |
6 |
where PΩ(y,t|x) is the conditional probability of finding the trajectory at y at time t given that it was at x at time 0, and x ∈ Ω. For activated processes, there usually exists a subset Ω* ⊂ Ω for which tΩ(x) is invariant to x ∈ Ω* because the relaxation time in Ω* is much faster than the reaction time. Thus tΩ is the MFPT for an implicit Ω*. The inner integral in Eq. 6 is precisely equal to tP(t), and hence for the exponential case, one finds that the rate is given in terms of the MFTP as
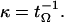 |
7 |
This rate expression is accurate for general canonical processes and is easy to calculate directly from an ensemble of Monte Carlo simulations.
To calculate MFPTs for the DS1 Monte Carlo dynamics, one must first define the regions Ω for which the rates are to be measured. According to Fig. 5, roughly three regions of interest exist. These are associated with the completely unfolded state (u), the intermediate (molten globular) state (i), and the folded (native or long-lived) state (f). Any pairing of these regions will give rise to a rate process, such as the folding rate kf←u or the isomerization rate kf'←f between the two long-lived states of DS1.
The MFPT folding rates for DS1, kf←u, are shown in Fig. 7. There is a turnover in the rates from low to high β, which can be easily understood through the use of the PMFs of Fig. 5. At low β(≤0.5), the PMF has a broad minimum faraway from the native structure and hence the protein finds the native structure slowly, and when it does, it is not trapped. In this regime, the increase in β leads to further broadening of this minimum, and hence faster rates. At larger β, stable intermediates are formed that slow down the folding rate, but once a folded structure is reached it is stable for long times. The temperature dependence on the rate is thus consistent with the analysis of the PMFs in section and the present physical understanding of protein dynamics when the possibility of multiple long-lived states is included.
Figure 7.

The logarithm of the folding rates for a Monte Carlo dynamics simulation of DS1 are displayed at various inverse temperatures, β. These are calculated as the inverse of the MFPT from 100 initial unfolded configurations u to the folded protein f. Throughout this temperature regime, the equilibration time within either the reactant or product basins is faster than the reported MFPTs, and consequently this choice of initial and final product state does not adversely affect the results. Although the error bars are about 10% of the actual rate, the error in the logarithm is significantly smaller, and hence 100 trajectories are sufficient to provide accurate results at the resolution of the figure.
The peak in the folding rates for DS1 occurs near β ≈0.6, which is somewhat lower than the observed inverse folding temperature. This finding suggests that the ground-state stabilization of the long-lived structure is more important than the folding rate in determining the folding temperature. Although the minimalist model does not include effects from solvent stabilization vis-a-vis fluctuations beyond their mean-field effects, it does include intramolecular effects and these are the likely source for the noted disparity between the folding temperature and the temperature for a maximum in the folding rate. This point should be explored in future work using stochastic models with respect to the winding index as the chosen variable, and the remaining (projected) modes leading to a dissipative environment (35, 46, 48–59). Such an approach should lead to comparisons between the MFPT rates for the Monte Carlo dynamics and various rate expressions for the corresponding real-time stochastic representation.
Concluding Remarks
The statistical mechanics of minimalist lattice models of protein folding have led to a qualitative understanding of protein folding for small, fast-folding proteins that is consistent with the bulk of experiments on protein dynamics. However, there is additional experimental evidence that some proteins adopt conformations that are long-lived, but that are not structurally similar to the native conformation. This implies that proteins can adopt conformations that are not represented within a single native folding funnel. The extension of the energy landscapes to include multiple deep funnels thus seems to be a natural development in the theory.
In this article, a specific minimalist model protein, DS1, has been constructed that provides an existence proof for such a generalization, at least to the extent that the specific behavior of such models may be isomorphic to proteins in the physical world. The thermodynamics of this model protein has been studied with the use of the winding index, a one-dimensional order parameter introduced in this work. The winding index is an ideal order parameter for the study of DS1. Because of the large degree of “winding” in the DS1 long-lived structures, the winding index distinguishes between them as well as the intermediates along the “folding reaction” path. A unique feature of this work is that the PMFs at various temperatures were obtained explicitly with respect to the winding index wz, thereby providing a foundation for a detailed analysis of the thermodynamic and kinetic behavior. In particular, it is useful in visualizing the multiple deep funnels of DS1 albeit through the projected space of wz. It is also argued that this order parameter may, in fact, be a useful folding-path coordinate for certain minimalist proteins such as that of this model study. Although the PMFs with respect to wz have been obtained, they do not lead directly to rates. The temperature-dependent folding rates obtained by calculating the MFPTs for the full dynamics exhibit a turnover behavior. This confirms that the dynamics on the projected space must contain a dissipative term and stochastic forces to account for the environment consisting of other protein modes. What exactly this friction should be is presently an open question, but the existence of detailed PMFs as well as the full-dimensional folding rates provides a starting point for addressing it.
Although the DS1 protein is equivalent to a small peptide, the dynamics that it has displayed indicate that it does lock inside a given funnel before it folds to the corresponding native structure. Such behavior suggests that the initial structure found in the denatured protein, either by happenstance or through its synthesis, may affect the folding times and the final structure it gives rise to.
In summary, this article provides suggestive evidence that the experimentally observed two-state behavior of some proteins may be observed through the universality class of a minimalist model that gives rise to multiple long-lived states on separate folding funnels.
Acknowledgments
This work has been partially supported by a National Science Foundation CAREER Award under Grant No. NSF 97-03372. R.H. is an Alfred P. Sloan Fellow, a Cottrell Scholar of the Research Corporation, and Blanchard Assistant Professor of Chemistry at Georgia Tech. The Center for Computational Molecular Science and Technology is presently funded through a Shared University Research (SUR) grant from IBM and also by Georgia Tech.
Abbreviations
- DS
designed sequence
- PMF
potential of mean force
- MFPT
mean first-passage time
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Prusiner S B. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinclair J F, Ziegler M M, Baldwin T O. Nat Struct Biol. 1994;1:320–326. doi: 10.1038/nsb0594-320. [DOI] [PubMed] [Google Scholar]
- 3.Kelly J W. Curr Opin Struc Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 4.Abkevich V, Gutin A M, Shakhnovich E I. Proteins Struct Funct Genet. 1998;31:335–344. [PubMed] [Google Scholar]
- 5.Narrison P, Chan H, Prusiner S, Cohen F. J Mol Biol. 1999;286:593–606. doi: 10.1006/jmbi.1998.2497. [DOI] [PubMed] [Google Scholar]
- 6.Jackson G S, Hosszu L L P, Power A, Hill A G, Kenney J, Saibil H, Craven C J, Waltho J P, Clarke A R, Collinge J. Science. 1999;283:1935–1937. doi: 10.1126/science.283.5409.1935. [DOI] [PubMed] [Google Scholar]
- 7.Go N. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 8.Onuchic J, Wolynes P, Luthey-Schulten Z. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 9.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 10.Onuchic J, Wolynes P, Luthey-Schulten Z, Socci N. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kauzmann W. Adv Protein Chem. 1959;16:1–64. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 12.Dill K. Biochemistry. 1990;29:7133–7151. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 13.Socci N D, Onuchic J N. J Chem Phys. 1994;101:1519–1528. [Google Scholar]
- 14.Dinner A R, Karplus M. J Phys Chem B. 1999;103:7976–7994. [Google Scholar]
- 15.Honeycutt J, Thirumalai D. Biopolymers. 1992;32:695–709. doi: 10.1002/bip.360320610. [DOI] [PubMed] [Google Scholar]
- 16.Czerminski R, Elber R. J Chem Phys. 1990;92:5580–6001. [Google Scholar]
- 17.Becker O, Karplus M. J Chem Phys. 1997;106:1495–1517. [Google Scholar]
- 18.Miller M A, Wales D J. J Chem Phys. 1999;111:6610–6616. [Google Scholar]
- 19.Wolynes P G. Proc Natl Acad Sci USA. 1996;93:14249–14255. doi: 10.1073/pnas.93.25.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 21.Shakhnovich E I, Gutin A. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramanathan S, Shakhnovich E I. Phys Rev E. 1994;50:1303–1312. doi: 10.1103/physreve.50.1303. [DOI] [PubMed] [Google Scholar]
- 23.Pande V S, Grosberg A Y, Tanaka T. Proc Natl Acad Sci USA. 1994;91:12976–12979. doi: 10.1073/pnas.91.26.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun S, Thomas P, Dill K. Protein Eng. 1995;8:769–778. doi: 10.1093/protein/8.8.769. [DOI] [PubMed] [Google Scholar]
- 25.Sun S, Brem R, Chan H, Dill K. Protein Eng. 1995;8:1205–1213. doi: 10.1093/protein/8.12.1205. [DOI] [PubMed] [Google Scholar]
- 26.Deutsch J, Kurosky T. Phys Rev Lett. 1996;76:323–326. doi: 10.1103/PhysRevLett.76.323. [DOI] [PubMed] [Google Scholar]
- 27.Grote R F, Hynes J T. J Chem Phys. 1980;73:2715–2732. [Google Scholar]
- 28.Pollak E. J Chem Phys. 1986;85:865–867. [Google Scholar]
- 29.Dellago C P, Bolhuis P, Chandler D. J Chem Phys. 1999;110:6617–6625. [Google Scholar]
- 30.Bolhuis P G, Dellago C P, Chandler D. Proc Natl Acad Sci USA. 2000;97:5877–5882. doi: 10.1073/pnas.100127697. . (First Published May 9, 2000; 10.1073/pnas.100127697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar S, Bouzida D, Swendsen R H, Kollman P A, Rosenburg J M. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 32.Eyring H. J Chem Phys. 1935;3:107–115. [Google Scholar]
- 33.Kassel L S. J Chem Phys. 1935;3:399–400. [Google Scholar]
- 34.Wigner E P. J Chem Phys. 1937;5:720–725. [Google Scholar]
- 35.Kramers H A. Physica. 1940;7:284–304. [Google Scholar]
- 36.Yamamoto T. J Chem Phys. 1960;33:281–289. [Google Scholar]
- 37.Langer J S. Ann Phys. 1967;41:108–157. [Google Scholar]
- 38.Fischer S. J Chem Phys. 1970;53:3195–3207. [Google Scholar]
- 39.Pechukas P. In: Modern Theoretical Chemistry. Miller W H, editor. Vol. 2. New York: Plenum; 1976. pp. 269–322. [Google Scholar]
- 40.Miller W H. J Chem Phys. 1974;61:1823–1834. [Google Scholar]
- 41.Chandler D. J Chem Phys. 1978;68:2959–2970. [Google Scholar]
- 42.Affleck I. Phys Rev Lett. 1981;47:388–391. [Google Scholar]
- 43.Truhlar D G, Hase W L, Hynes J T. J Phys Chem. 1983;15:2664–2682. [Google Scholar]
- 44.Truhlar D G, Garrett B C. Annu Rev Phys Chem. 1984;35:159–189. [Google Scholar]
- 45.Truhlar D G, Issacson A D, Garrett B C. In: Theory of Chemical Reaction Dynamics. Baer M, editor. Vol. 4. Boca Raton, FL: CRC; 1985. pp. 65–137. [Google Scholar]
- 46.Hynes J T. In: Theory of Chemical Reaction Dynamics. Baer M, editor. Vol. 4. Boca Raton, FL: CRC; 1985. pp. 171–234. [Google Scholar]
- 47.Pollak E, Grabert H, Hänggi P. J Chem Phys. 1989;91:4073–4087. [Google Scholar]
- 48.Pollak E. In: Dynamics of Molecules and Chemical Reactions. Wyatt R E, Zhang J, editors. New York: Dekker; 1996. pp. 617–669. [Google Scholar]
- 49.Hänggi P, Talkner P, Borkovec M. Rev Mod Phys. 1990;62:251–341. [Google Scholar]
- 50.Zwanzig R. J Chem Phys. 1960;33:1338–1341. [Google Scholar]
- 51.Zwanzig R. In: Lectures in Theoretical Physics (Boulder) Britton W E, Downs B W, Downs J, editors. Vol. 3. New York: Wiley; 1961. pp. 106–141. [Google Scholar]
- 52.Prigogine J, Resibois P. Physica. 1961;27:629–646. [Google Scholar]
- 53.Ford G W, Kac M, Mazur P. J Math Phys. 1965;6:504–515. [Google Scholar]
- 54.Mori H. Prog Theor Phys. 1965;33:423–455. [Google Scholar]
- 55.Hynes J T. Annu Rev Phys Chem. 1985;36:573–597. [Google Scholar]
- 56.Nitzan A. Adv Chem Phys. 1988;70:489–555. [Google Scholar]
- 57.Berne B J, Borkovec M, Straub J E. J Phys Chem. 1988;92:3711–3725. [Google Scholar]
- 58.Tucker S C. J Phys Chem. 1993;97:1596–1609. [Google Scholar]
- 59.Hernandez R, Somer F L. J Phys Chem B. 1999;103:1064–1069. [Google Scholar]