

Abstract
For more than two decades, sepsis was defined as a microbial infection that produces fever (or hypothermia), tachycardia, tachypnoea and blood leukocyte changes. Sepsis is now increasingly being considered a dysregulated systemic inflammatory and immune response to microbial invasion that produces organ injury for which mortality rates are declining to 15–25%. Septic shock remains defined as sepsis with hyperlactataemia and concurrent hypotension requiring vasopressor therapy, with in-hospital mortality rates approaching 30–50%. With earlier recognition and more compliance to best practices, sepsis has become less of an immediate life-threatening disorder and more of a long-term chronic critical illness, often associated with prolonged inflammation, immune suppression, organ injury and lean tissue wasting. Furthermore, patients who survive sepsis have continuing risk of mortality after discharge, as well as long-term cognitive and functional deficits. Earlier recognition and improved implementation of best practices have reduced in-hospital mortality, but results from the use of immunomodulatory agents to date have been disappointing. Similarly, no biomarker can definitely diagnose sepsis or predict its clinical outcome. Because of its complexity, improvements in sepsis outcomes are likely to continue to be slow and incremental.
Sepsis has been recognized in some form or another since at least 1,000 BC — when it was first described by the Islamist philosopher Ibn Sīnā (also known as Avicenna) as putrefaction of blood and tissues with fever1. Further described by Boerhaave, von Liebig, Semmelweis, Pasteur, Lister, Lennhartz and, most recently, Bone, sepsis and its treatment have confounded investigators for nearly 3,000 years. Since 1991, the consensus definition of sepsis has been the ‘systemic inflammatory response (SIRS) to a microbial infection’ (REFS 2,3) (BOX 1), with SIRS defined as at least two of the following: tachypnoea (rapid breathing), tachycardia (rapid heartbeat), pyrexia (fever) or hypothermia, and leukocytosis, leukopaenia or neutrophilia. Efforts have recently focused on eliminating the SIRS requirement entirely4 (BOX 2) because fever, tachycardia, tachypnoea and white blood cell changes reflect infection only and have proven to be too broadly applied in critically ill patients to be useful in the definition of sepsis. In its place, sepsis is now defined as an infection associated with organ injury distant from the site of infection. Septic shock remains defined as a subset of sepsis in which the risk of mortality is substantially increased, and is characterized by hypotension that persists during volume resuscitation and requires the use of vasopressors.
Box 1. 1991 criteria for sepsis, severe sepsis and septic shock.
The following definitions derive from the 1991 Consensus Conference of the American College of Chest Physicians and Society of Critical Care Medicine2,162. Infection is defined as the presence of microorganisms or tissue invasion by those microorganisms.
Sepsis
The systemic inflammatory response (SIRS) to infection, manifested by at least two of:
Temperature of >38 °C or <36 °C
Heart rate of >90 beats per minute
Respiratory rate of >20 breaths per minute or partial pressure of CO2 of <32 mmHg
White blood cell count of >12,000 per ml or <4,000 per ml, or >10% immature (band) forms
Severe sepsis
Severe sepsis is defined as sepsis associated with organ dysfunction, hypotension or hyperfusion. Hypoperfusion abnormalities of end organs may include lactataemia, oliguria or an alteration in mental status.
Septic shock
Septic shock is defined as sepsis associated with hypotension and perfusion abnormalities despite the provision of adequate fluid (volume) resuscitation. Perfusion abnormalities include lactic acidosis, oliguria or an acute alteration in mental status. Patients with septic shock who are receiving inotropic or vasopressor therapy might still exhibit perfusion abnormalities, despite the lack of hypotension.
Box 2. Proposed criteria for sepsis and septic shock.
This proposal stems from the 2015 Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3)4, which considers infection to be an interaction between a host and a pathogen that induces a local or systemic host response.
Sepsis
Life-threatening organ dysfunction owing to a dysregulated host response to infection
Onset marked by the beginning of any organ dysfunction remote from the site of infection
Septic shock
A subset of sepsis in which underlying circulatory and cellular–metabolic abnormalities are profound enough to substantially increase mortality
Operationally defined as requiring vasopressor therapy to maintain a mean arterial blood pressure of >65 mmHg and an increased plasma lactate level of >2 mmol per l
The study of sepsis treatment reflects progress in our understanding of human pathophysiology and host– microorganism interactions. Early research focused on the microorganism and its pathogenicity. In the 1980s, with the implementation of molecular cloning and the sequencing of human inflammatory genes, research in sepsis turned towards investigations that focused less on the pathogenicity of the microorganism and more on the host response to an invading pathogen5–7. The discovery of how the host distinguishes self and non-self and the introduction of the ‘danger hypothesis’ (REF.8) have dramatic ally improved our understanding of sepsis and its pathogenesis. The danger hypothesis purports that the innate immune system recognizes microbial patterns and unique host cellular products as ‘danger signals’ or ‘alarmins’ of microbial invasion or tissue injury. However, research has also revealed that the progression of sepsis is much more complex than just inflammation or microbial or host pattern recognition; sepsis also involves effects on endothelial tissues and microcirculation, primary and secondary immune tissues, coagulation, parenchymal tissues and neurological disturbances that directly affect microglial cells and neurons9–12.
Despite a dramatic increase in our understanding of sepsis, its origins, progression and resolution (recovery or death), our ability to intervene and alter the trajectory of the disease has been only partially successful. That is, our increased understanding of the pathogenesis of the disease has generally failed to substantially improve outcomes. Although in-hospital mortality from sepsis has declined over the past decade13, this improvement is more commonly attributed to earlier recognition and better compliance with best-practice supportive therapies14,15. In this Primer, we describe the contemporary definitions and the current epidemiological picture of sepsis and septic shock, as well as the best practices for the recognition and support of patients with sepsis and the use of potential biomarkers and biological response modifiers to better identify patients and treat them effectively.
Epidemiology
Incidence and prevalence
Despite its high associated mortality, comprehensive epidemiological data on the global burden of sepsis are lacking. A tentative extrapolation of data from high-income countries suggests that 31.5 million cases of sepsis and 19.4 million cases of severe sepsis occur globally each year, with potentially 5.3 million deaths annually16. These numbers are simply estimates because knowledge about the incidence and mortality of sepsis in low-income and middle-income countries remains scarce owing to scant data and the difficulty of generating population-level estimates in these regions16–18. Sepsis is also not tracked in the Global Burden of Disease report published by the WHO and World Bank, which monitor incidence, mortality and risk factors of the most important diseases in the world19. Given the high prevalence of infectious diseases associated with an increased risk of sepsis and septicaemia, such as HIV20, non-typhoid salmonella and Streptococcus pneumoniae21, a substantial burden of sepsis should be expected in regions affected by these diseases. Indeed, in 2013, lower respiratory tract infections ranked second among the leading causes of disability-adjusted life-years and accounted for >2.5 million deaths globally, of which a considerable proportion could be considered sepsis22. Similarly, malaria and viral infections such as dengue are also major sources of systemic infections in low-income and middle-income countries, with the majority of overall deaths attributable to sepsis23.
Contemporary epidemiological studies from high-income countries suggest high incidence rates of hospital-treated sepsis, ranging from 194 per 100,000 inhabitants in Australia in 2003 (REF.24) to 580 per 100,000 inhabitants in the United States in 2006 (REF.25). In Germany, the incidence of hospital-treated sepsis cases between 2007 and 2013 increased from 256 to 335 cases per 100,000 inhabitants; the proportion of patients with severe sepsis increased from 27% to 41%26.
Furthermore, for high-income countries, several prospective and retrospective epidemiological studies have presented data on the incidence, point prevalence, period prevalence and mortality of sepsis. These reports have extrapolated their results to a population level; several have suggested dramatic increases in the occurrence of sepsis27,28. However, interpretation of these findings is hampered by the fact that many of the studies use various different methods and sepsis definitions, including the 1991 consensus criteria (BOX 1) or derivative WHO International Classification of Diseases (ICD) code abstractions for register studies. Indeed, many databases included the 1991 consensus criteria such that infection (typically characterized by fever and its accompanying tachycardia and an altered white blood cell count) and sepsis were often confounded. Accordingly, depending on the codes used to identify clinical sepsis, prevalence can differ substantially29,30. For example, one study compared four different methods of assessing sepsis using the same databases and showed that the incidence of sepsis varied more than threefold between methods31. Furthermore, the increased incidence of sepsis in some health care systems might be attributable to increased clinical awareness of sepsis and/or financial incentives for enhanced reimbursement for services by coding patients with sepsis. Thus, these variable definitions could explain the dramatic increase in the number of sepsis cases associated with a reduction in mortality rates in high-income countries.
Chart-based clinical validation of cases of sepsis identified through administrative databases has often revealed several fold higher incidence rates than observed in prospective or retrospective trials27. By contrast, other studies have suggested that, in administrative data from hospitals, septicaemia, sepsis and severe sepsis might not be coded correctly or missed32,33. Accordingly, there is an ongoing controversy on the accuracy of coding itself, especially when sepsis is less severe33,34. Furthermore, only hospitalized patients are included in these observational studies, whereas a considerable number of patients experience sepsis outside the hospital setting35. As such, concerns abound that recent epidemiological data from high-income countries are unable to capture the real burden of sepsis, but there is little controversy that sepsis remains a considerable challenge in the developed world.
Mortality
Estimates of sepsis associated with in-hospital mortality are equally confounded. Between 1999 and 2009, mortality directly ascribed to sepsis seems to have declined on the basis of data obtained from death certificates or administrative databases. However, in many cases, especially in patients with chronic diseases such as cancer, congestive heart failure and chronic obstructive pulmonary disease, the official record of death often reports the underlying disease rather than the immediate cause of death (sepsis), which might contribute to the apparent underestimation in mortality from sepsis35. Data from Australia and New Zealand, in particular, have suggested that overall mortality rates attributable to sepsis are declining13. Although the percentages of patients with sepsis who are dying in the hospital are decreasing, Martin et al.28 and Gaieski et al.31 demonstrated that overall mortality rates tend to be increasing, due to the apparent increases in the number of patients with sepsis.
Whether mortality from septic shock is declining is less clear. Kaukonen et al.13 reported from their administrative databases that mortality from septic shock has declined at rates comparable to those of sepsis. However, a cursory analysis of data from randomized controlled trials has suggested that, if mortality from septic shock is declining, it is doing so at a slower rate than for sepsis. The problem, in part, is that mortality rates from septic shock vary dramatically depending on the expertise and experience of the treating centre. In some countries, mortality from septic shock still approaches 50%, whereas in others, mortality is being reported at 20–30%36.
The overall decreases in in-hospital sepsis mortality, and possibly in septic shock, are encouraging. However, given that the overall incidence of sepsis is seemingly increasing at greater rates, overall mortality is not significantly improving, demonstrating the continuing magnitude of the challenge.
Mechanisms/pathophysiology
Inflammation
Sepsis is fundamentally an inflammatory disease mediated by the activation of the innate immune system. Two key findings characterize the innate immune response in sepsis. The first finding is that sepsis is generally initiated by simultaneous recognition of multiple infection-derived microbial products and endogenous danger signals by complement and specific cell-surface receptors on cells whose primary job is surveillance37. These cells include immune, epithelial and endothelial populations that are physically located where they can continuously sample their local environment. Binding of both pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) to complement, Toll-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors, retinoic acid-inducible gene (RIG)-like receptors, mannose-binding lectin and scavenger receptors, among others, induces a complex intracellular signalling system with redundant and complementary activities38 (FIG. 1).
Figure 1. Cell-surface and intracellular receptors that are responsible for the recognition of microbial products and endogenous danger signals (alarmins).
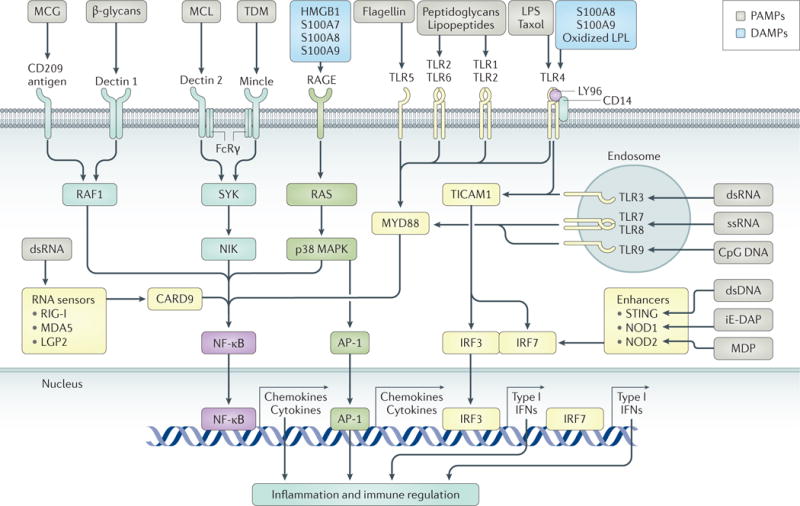
Sepsis is initiated upon host recognition of pathogen- associated molecular patterns (PAMPs) and is characterized by the activation of inflammatory signalling pathways. A large number of cell-associated and intracellular receptors are available to detect PAMPs or damage-associated molecular patterns (DAMPs), a few examples of which are illustrated here. PAMPs and DAMPs can be microbial and host glycoproteins, lipoproteins and nucleic acids. The associated pattern- recognition receptors include Toll-like receptors (TLRs), C-type lectin domain family 7 member A (dectin 1) and C-type lectin domain family 6 member A (dectin 2). At least ten different TLRs are known, and in many cases they exist as either homodimers or heterodimers. Once activated, the ensuing signalling pathways generally converge towards interferon regulatory factor (IRF) signalling and nuclear factor-κB (NF-κB). IRF is responsible for type I interferon (IFN) production. NF-κB and activator protein 1 (AP-1) signalling are predominately responsible for the early activation of inflammatory genes, such as TNF, IL1 and those encoding endothelial cell-surface molecules. CARD9, caspase recruitment domain-containing protein 9; dsDNA, double-stranded DNA; dsRNA, double-stranded RNA; FcRγ, Fcγ receptor; HMGB1, high-mobility group protein B1; iE-DAP, d-glutamyl-meso-diaminopimelic acid; LGP2, laboratory of genetics and physiology 2 (also known as DHX58); LPL, lipoprotein lipase; LPS, lipopolysaccharide; LY96, lymphocyte antigen 96; MAPK, mitogen- activated protein kinase; MCG, mannose-containing glycoprotein; MDA5, melanoma differentiation-associated protein 5 (also known as IFIH1); MDP, muramyl dipeptide; MCL, mannose-capped lipoarabinomannan; Mincle, also known as CLEC4E; MYD88, myeloid differentiation primary response protein 88; NIK, NF-κB-inducing kinase (also known as MAP3K14); NOD, nucleotide-binding oligomerization domain; RAF1, RAF proto-oncogene serine/threonine-protein kinase; RAGE, advanced glycosylation end product-specific receptor; RIG-I, retinoic acid-inducible gene 1 protein (also known as DDX58); ssRNA, single-stranded RNA; STING, stimulator of interferon genes protein; SYK, spleen tyrosine kinase; TDM, trehalose-6,6′-dimycolate; TICAM1, TIR domain-containing adaptor molecule 1.
The second key finding in sepsis is that activation of these multiple signalling pathways ultimately leads to the expression of several common gene classes that are involved in inflammation, adaptive immunity and cellular metabolism. That is, the recognition of many different components of bacteria, viruses and fungi, as well as host products of tissue injury, leads to the recruitment of pro-inflammatory intermediates that in turn result in the phosphorylation of mitogen- activated protein kinases (MAPKs), Janus kinases (JAKs) or signal transducers and activators of transcription (STATs) and nuclear translocation of nuclear factor-κΒ (NF-κΒ), to name simply a few. These intermediates initiate the expression of early activation genes. Taken together, these two characteristics of innate immunity assure a common response pattern, the intensity and direction of which can be finely regulated by the level of and variation in the repertoire of PAMPs and DAMPs and the signalling pathways activated. This complementary nature of the pathways explains the overlapping but unique early inflammatory response to common Gram-negative bacterial, Gram-positive bacterial, fungal and viral infections and tissue injury.
Early activation genes
Nuclear translocation of NF-κB and activation of its promoter in particular induce the expression of multiple early activation genes, including cytokines that are associated with inflammation (including tumour necrosis factor (TNF), IL-1, IL-12, IL-18 and type I interferons (IFNs)). These cytokines initiate a cascade of other inflammatory cytokines and chemokines (including IL-6, IL-8, IFNγ, CC-chemokine ligand 2 (CCL2), CCL3 and CXC-chemokine ligand 10 (CXCL10)), as well as the polarization and suppression of components of adaptive immunity. The activation of these inflammatory networks begins within minutes of PAMP or DAMP recognition owing to the existence of preformed inactive and active cytokine pools. Simultaneously, activation of these sentinel innate immune receptors, activation of complement and/or production of inflammatory cytokines have a profound effect on coagulation and the vascular and lymphatic endothelium, resulting in the increased expression of selectins and adhesion molecules39. The alteration in the expression of various procoagulant and anticoagulant proteins, including thrombomodulin, tissue factor, von Willebrand factor, plasminogen activator inhibitor 1 (PAI-1) and activated protein C, results in the transition of the endothelium from an anticoagulant state (in health) to a procoagulant state (in sepsis). Pro-inflammatory proteases induce the internalization of the vascular endothelial (VE)-cadherin leading to the loss of endothelial tight junctions and increased vascular permeability40.
The C5a–C5a receptor axis
Complement activation is considered to be one of the hallmarks of sepsis and is initiated immediately upon exposure to PAMPs and DAMPs. Complement activation leads to the generation of complement peptides (namely, C3a and C5a). C5a has been shown to be one of the most active inflammatory peptides produced during sepsis41 and is one of the most potent chemo attractants for neutrophils, monocytes and macro phages. In neutrophils, C5a triggers an oxidative burst leading to the generation of reactive oxygen species and the release of granular enzymes, which are thought to be crucially involved in inflammatory tissue damage. Furthermore, C5a is a stimulant for the synthesis and release of pro-inflammatory cytokines and chemokines, thereby amplifying inflammatory responses. These mechanisms are believed to contribute to vasodilation, tissue damage and multiple organ failure in settings of acute inflammation. The potential role of C5a in the development of sepsis has been linked to neutrophil dysfunction, apoptosis of lymphoid cells, exacerbation of systemic inflammation, cardiomyopathy, disseminated intra vascular coagulation (DIC) and complications associated with multiple organ failure42.
Blockade of C5a in experimental models of sepsis has been shown to be beneficial in various models from different groups. For example, inhibition of C5a by rabbit polyclonal antibodies in a primate model of sepsis induced by infusion of live Escherichia coli substantially attenuated evidence of acute sepsis-induced lung injury and failure43. Similarly, the blockade of C5a with antibodies in rats or mice with sepsis caused by caecal ligation puncture was highly effective in diminishing the severity of sepsis and improving outcome44,45. In addition, severe inflammatory responses and their associated organ damage during avian H5N1 and H1N1 viral infections have also been linked to complement activation, especially the overproduction of C5a46,47. Along these lines, a monoclonal antibody raised against human C5a greatly attenuated H7N9-induced lung damage in non-human primates, reducing the viral load and the levels of several different cytokines in this setting48. The same antibody is currently being tested in patients with early abdominal or pulmonary septic organ dysfunction49.
Immune suppression
Although the early systemic inflammatory response has been considered the hallmark of sepsis, immuno-suppression occurs both early and late in the host sepsis response. Patients who survive sepsis often have protracted clinical trajectories and exhibit both chronic immune suppression and inflammation. This finding has recently been termed the persistent inflammation/immunosuppression and catabolism syndrome (PICS)50–52 (FIG. 2). PICS-associated inflammation is characterized by markedly increased C-reactive protein concentrations (an acute phase protein), neutrophilia and the release of immature myeloid cells. Unlike the immediate inflammatory response that is presumed to be predominantly driven by PAMPs and DAMPs, the aetiology behind the persistent inflammation is unknown. PICS is probably driven by DAMPs and alarmins that are produced by injured organs and tissues, such as mitochondrial DNA and nucleosides, histones, high- mobility group protein B1 (HMGB1), protein S100A, ATP, adeno-sine and/or hyaluronan products53. Alternative explanations for how PICS progresses include opportunistic infections such as viral reactivation54, changes in the host microbiota and mechanical injury secondary to ventilation or catheter placement.
Figure 2. Current conceptual model of outcomes of sepsis.
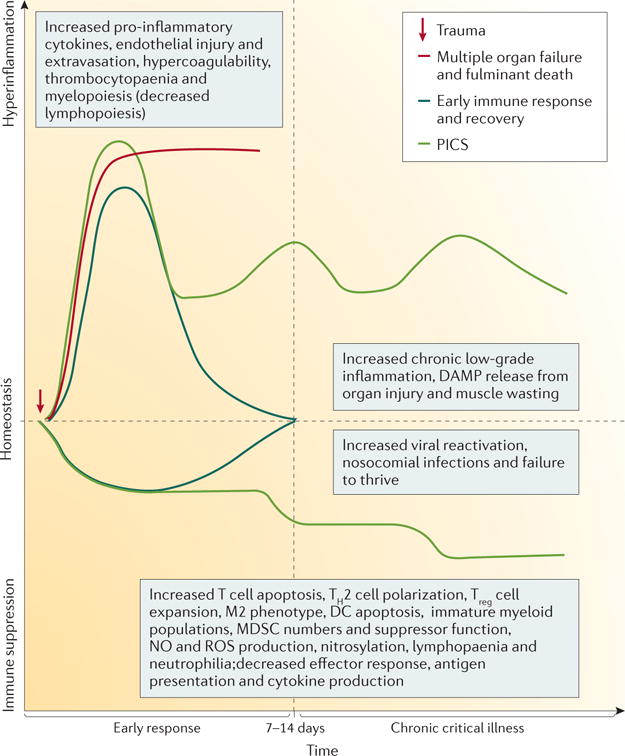
Originally conceived by Bone et al.165 in the 1990s, the current model of the clinical trajectory that patients traverse in sepsis has evolved to reflect the concurrent inflammatory and immunosuppressive responses, and the observation that fewer patients are dying in the early period owing to earlier recognition and better implementation of best clinical practices50. Successful resuscitation is occurring more frequently and the patients recover sufficiently to be discharged from the intensive care unit and hospital (blue lines). Some patients experience a pronounced early inflammatory response to the pathogen or danger signals, leading to multiple organ failure and death (red line). Other patients survive the early inflammatory response but experience chronic critical illness (green lines) that is characterized by persistent inflammation, immunosuppression and catabolism syndrome (PICS); reactivation of latent viral infections; nosocomial infections; and long-term functional and cognitive declines52. DAMP, damage-associated molecular pattern; DC, dendritic cell; MDSC, myeloid-derived suppressor cell; NO, nitric oxide; ROS, reactive oxygen species; TH2, T helper 2.
The paradoxical immunosuppression and infectious complications in patients with sepsis compound as sepsis progresses, with an increasing frequency of positive blood cultures and a shift to infection by opportunistic organisms55,56. Compared with control individuals without sepsis, patients with sepsis have increased rates of reactivation of latent viruses, with viral DNA being detected in the blood of 42% of patients with sepsis (only 5% of critically ill patients without sepsis have detectable viral DNA)54. One autopsy study confirmed the immunosuppressed state of patients with sepsis, with persistent foci of infection and microabscesses identified in 80% of cases57.
The changes in adaptive immunity in response to sepsis are profound. Lymphopaenia, an immature neutrophil (polymorphonuclear) phenotype58,59, loss of monocyte inflammatory cytokine production and antigen presentation60 and increased numbers of neutrophil- like myeloid-derived suppressor cells (MDSCs) in the circulation61 are all common consequences of sepsis. Immature myeloid cells in the circulation have characteristically defective antimicrobial activity with decreased expression of adhesion molecules and decreased formation of extracellular traps (networks of extracellular fibres composed of chromatin, DNA and granular proteins) that capture pathogens62,63. Both immature blood neutrophils and MDSCs secrete multiple anti-inflammatory cytokines, including IL-10 and transforming growth factor-β (TGFβ), which further suppress immune function. In addition, sepsis causes professional antigen-presenting cells (APCs) — including dendritic cells and macrophages — to lose expression of the activating major histocompatibility complex (MHC) class II molecule human leukocyte antigen-antigen D related (HLA-DR). In addition, loss of HLA-DR by circulating APCs has been associated with decreased responsiveness, and the failure of monocytes to recover HLA-DR levels predicts a poor outcome from sepsis64. Sepsis also causes both stromal cells and professional APCs to increase the expression of the T cell protein programmed death ligand 1 (PDL1), which binds to the inhibitory programmed death protein 1 (PD1) receptor that is expressed by T cells, further suppressing T cell function65. The combination of the increased surface expression of inhibitory T cell ligands by APCs, loss of activating MHC class II molecules and increased production of anti-inflammatory cytokines skews the T cell phenotype towards an immunosuppressive T helper 2 (TH2) phenotype, increases the suppressor activity of T regulatory cells and causes broad T cell anergy (lack of reaction) (FIG. 3). Lending further support to the notion that immune suppression occurs in sepsis, pro-inflammatory and TH1 cytokine production by lymphocytes from patients with sepsis is <10% of that of controls without sepsis65. Together, these data provide a mechanism for the well-described loss of the delayed-type hypersensitivity in patients with sepsis, a metric for the profound suppression of the adaptive immune system seen in sepsis66.
Figure 3. The late immunosuppressive effects of sepsis.
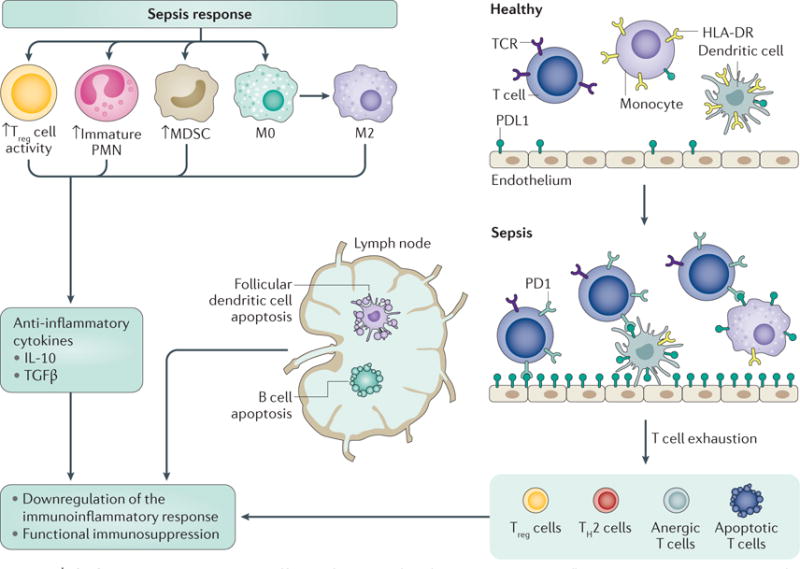
After the transitory acute inflammatory response, sepsis results in an immunocompromised state. Immunosuppressive immature polymorphonuclear leukocytes (PMNs) and myeloid-derived suppressor cells (MDSCs) mobilize from the bone marrow and monocyte differentiation skews to the production of M2 macrophages (which decrease inflammation and promote tissue repair). Although these responses can be considered normal, if the source of infection is not controlled, the continued responses rapidly become pathological and lead to chronic immune suppression. Together, immature PMNs, MDSCs and M2 macrophages produce anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β (TGFβ). Professional antigen-presenting cells, including dendritic cells and macrophages, reduce the expression of the activating major histocompatibility complex (MHC) class II molecule human leukocyte antigen-antigen D related (HLA-DR). T cells and stromal cells upregulate negative co-stimulatory molecules, including programmed death protein 1 (PD1) and programmed death ligand 1 (PDL1), respectively, to drive the expansion of regulatory T (Treg) cells and anergic (unresponsive) T cells. Follicular dendritic cells, B cells and T cells undergo apoptosis, further abrogating the immune response. TCR, T cell receptor; TH2, T helper 2.
An autopsy study of patients with sepsis identified apoptotic cell death as an underlying driver of innate and adaptive immunosuppression67. Indeed, patients with sepsis demonstrate a profound apoptotic loss of T cells, B cells and dendritic cells, an observation that is recapitulated in animal models of sepsis68. Apoptotic loss of lymphocytes is directly immunosuppressive, contributing to the lymphopaenia observed in patients with severe sepsis69. The degree of lymphocyte apoptosis correlates with the severity of sepsis and the persistent lymphopaenia predicts sepsis mortality70. Apoptotic cells also suppress immune function through interaction with other leukocytes. For instance, phagocytosis of apoptotic lymphocytes causes the release of anti-inflammatory cytokines such as IL-10 and TGFβ from macrophages and dendritic cells. This process also suppresses the production of pro-inflammatory cytokines at the level of gene transcription, thereby contributing to the paralysis of the innate inflammatory response in sepsis70. Accordingly, pharmacological or genetic manipulations that decrease sepsis-induced apoptosis improve survival in animal models of sepsis68,71–76. These data demonstrate the functional consequence of sepsis-induced apoptosis. The next generation of treatments being evaluated for sepsis includes therapies that target both lymphocyte apoptosis and sepsis-induced immunosuppression; the results of these studies are eagerly anticipated.
Endothelial barrier dysfunction
In addition to profound changes in host protective immunity, endothelial barrier function is an integral component of the sepsis response. A continuous endothelial barrier coats the vascular system and separates the fluid phase of the blood compartment from the tissues (FIG. 4). Under normal resting conditions, the endothelium serves as an anticoagulant surface that regulates the flow of gases, water, solutes, hormones, lipids, proteins and a multitude of other macro molecules within the microcirculation. Sepsis is now viewed as a dysregulation of the interacting and oscillating circuitry networks of cell–cell communication that maintain homeostasis under normal conditions9. Along these lines, endothelial barrier dysfunction is a fundamental pathophysiological event that occurs early in sepsis and septic shock in particular. The border between the blood and the interstitium is highly interactive and dynamic in both health and disease, with the endothelial cell as the principal regulatory cell type11. The endothelium functions to cover the underlying capillary basement membrane and adventitia to avoid exposing collagen fibres and tissue factor primarily to von Willebrand factor and factor VII. Collagen can immediately fix and polymerize von Willebrand factor, which activates platelets via glycoprotein 1β; at the same time, exposing tissue factor to circulating factor VII can initiate clotting via the tissue factor (formerly known as the extrinsic) pathway77.
Figure 4. Changes in the vascular endothelium in response to inflammatory stimuli during sepsis.
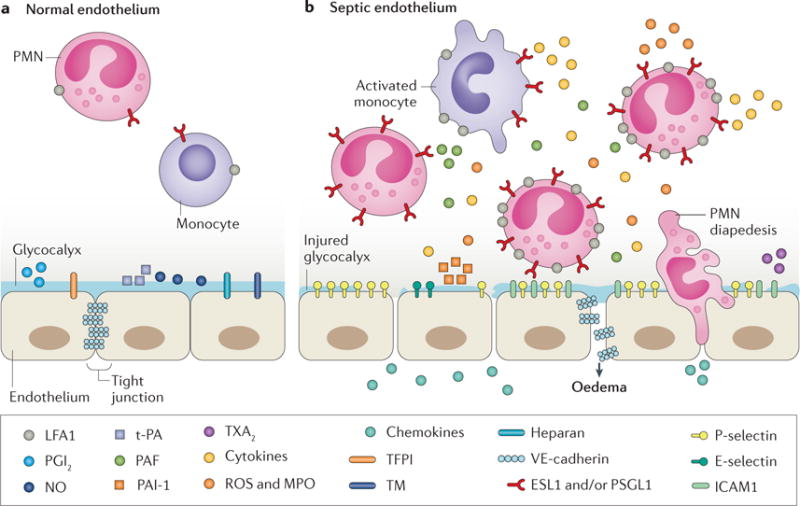
a | The resting vascular endothelium in its natural anticoagulant state. b | Sepsis produces profound changes that convert the endothelium to a procoagulant state. This disrupted endothelium expedites the loss of fluid through disengaged tight junctions and expedites the recruitment, attachment and extravasation of inflammatory cells through the endothelium. Activation of the coagulation cascade potentiates inflammation and completes a vicious cycle in which inflammation induces and exacerbates coagualopathies and endothelial injury. ESL1, E-selectin ligand 1; ICAM1, intercellular adhesion molecule 1; LFA1, lymphocyte function-associated antigen 1; MPO, myeloperoxidase; NO, nitric oxide; PAF, platelet- activating factor; PAI-1, plasminogen activator inhibitor 1; PGI2, prostaglandin I2; PMN, polymorphonuclear leukocyte; PSGL1, P-selectin ligand 1; ROS, reactive oxygen species; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; t-PA, tissue plasminogen activator; TXA2, thromboxane A2; VE, vascular endothelial.
The integrity of the endothelium is maintained by the cell cytoskeleton (actin), intercellular adhesion molecules (tight junctions) and an array of supportive proteins. In sepsis, these structures are disrupted primarily in response to platelet and neutrophil adhesion, the release of inflammatory mediators and toxic oxidative and nitrosative intermediates. Combined with the increased expression of selectins and integrins, binding of leukocytes to the endothelial surface results in the leakage of vascular fluid and migration of extravasating leukocytes across the compromised endothelial barrier. This event also provides the opportunity for collagen polymerization and tissue factor-mediated clotting to occur. Although these responses enable platelets and immune cells to reach tissue sites in response to trauma or localized infection, sepsis produces generalized, excessive and prolonged responses that can lead to considerable tissue injury.
In addition, the glycocalyx is a glycoprotein–polysaccharide layer that covers the endothelium and supports the anticoagulant state and maintains tight junctions. Sepsis alters the continuity of the glycocalyx, which also increases endothelial permeability78. In sepsis, the glycocalyx is a target for inflammatory mediators and leukocytes because it is imbedded with endothelial cell-surface receptors. The widespread presence of the glycocalyx in organ microvasculature can explain the endothelial activation and damage of tissues distant from the original site of infection via this systemic release of cytokines and other inflammatory mediators during sepsis. Inflammatory-mediated injury to the glycocalyx contributes to acute kidney injury, respiratory failure and hepatic dysfunction.
Numerous factors regulate the expression of tight junction linkers and actin polymer networks. Prominent among these regulators are the relative expression of two competing intracellular, G protein-linked GTPases known as RHOA and RAC1. RHOA generally induces actin filament breakdown and internalizes VE-cadherin, resulting in endothelial barrier breakdown. RAC1 signalling has opposing effects, stabilizing the actin cytoskeleton and preventing apoptosis. The relative concentrations of RHOA and RAC1 can be regulated, at least experimentally, by protease-activated receptors (PARs) on endothelial surfaces. Early thrombin generation in sepsis activates PAR1, which promotes RHOA GTPase signalling and induces endothelial barrier breakdown. Other proteases that activate PAR2 promote RAC1 signalling and support endothelial barrier protection79. TABLE 1 lists some of the candidate therapies that might prove to be effective in maintaining and re-acquiring endothelial barrier function in sepsis and septic shock.
Table 1.
Candidate treatment options to treat endothelial barrier injury*
| Drug | Stage of development | Proposed mechanism of action |
|---|---|---|
| ANG1–TIE2 modulators | In clinical trials for cancer166 | Reduce the loss of tight junctions and endothelial tight junction function in sepsis |
| S1P1 agonists | In clinical trials for multiple sclerosis167 and plaque psoriasis168 | Stimulate VE-cadherin and actin polymerization of endothelial junctions, preserving endothelial tight junction function in sepsis |
| Fibrinopeptide Bβ15–42 | In clinical trials for myocardial infarction169 | Fibrin cleavage product that binds to VE-cadherin and stabilizes interendothelial tight junctions to reduce endothelial permeability |
| SLIT2N agonists | Preclinical testing | Stabilize endothelial tight junctions by binding to ROBO4 to reduce p120-catenin phosphorylation and increase p120-catenin association with VE-cadherin |
| Pepducins | PAR1 pepducin in clinical trials for cardiac catheterization170 | Lipidated peptides are super agonists of PAR2, inducing RAC1-mediated endothelial barrier stabilization |
| HMGB1-specific monoclonal antibody | Preclinical testing | Blocks HMGB1-mediated loss of endothelial barrier function, upregulation of cytokine production and of adhesion molecules |
| Statins and angiotensin receptor blockers | In clinical trials for sepsis171 | Block angiotensin receptor-mediated oxidant stress on endothelial cells, regulate RAC1/RHOA ratio and prevent apoptosis |
| Selepressin | In clinical trials for sepsis172 | Vasopressin type 1a receptor antagonist121 |
ANG1, angiopoietin 1; HMGB1, high-mobility group B1; NF-κB, nuclear factor-κB; PAR, protease-activated receptor; RAC1, a subfamily of GTPases; ROBO4, roundabout homologue 4; S1P1, sphingosine 1-phosphate receptor 1; SLIT2N, slit homologue 2 protein N-product; TIE2, angiopoietin 1 receptor; VE, vascular endothelial.
See REF.11.
The leaky capillary membranes create massive loss of intravascular proteins and plasma fluids into the extravascular space. Diffuse vasodilation throughout the microcirculation alters capillary blood flow, which contributes to poor tissue perfusion and — ultimately — shock. In septic shock, events within tissue capillaries induce distributive shock in which the recovery of blood pressure is not achieved upon the administration of additional intravenous fluids, and requires a vasoconstrictive agent such as noradrenaline and/or vasopressin. The large volumes of crystalloid given to maintain central blood pressure in the presence endothelial injury frequently leads to oedema.
Coagulation
In sepsis and septic shock, the normal anticoagulative state within the vasculature is disrupted. Sepsis results in a hypercoagulable state that is characterized by microvascular thrombi, fibrin deposition, neutrophil extracellular trap (NET) formation and endothelial injury. Inflammatory cytokines as well as other mediators, such as platelet-activating factor and cathepsin G, target the endothelium and platelets. Platelet activation can itself propagate both coagulation and the inflammatory response by forming aggregates that can activate thrombin release. Thrombin is a serine protease that converts fibrinogen into insoluble strands of fibrin, as well as catalysing many other coagulation-related reactions. These strands of fibrin, along with platelets, provide the structural integrity to clot formation. In addition, inflammatory cytokines can promote coagulation by targeting the endothelium and causing endothelial injury (FIG. 5).
Figure 5. Interaction between coagulation and inflammation.
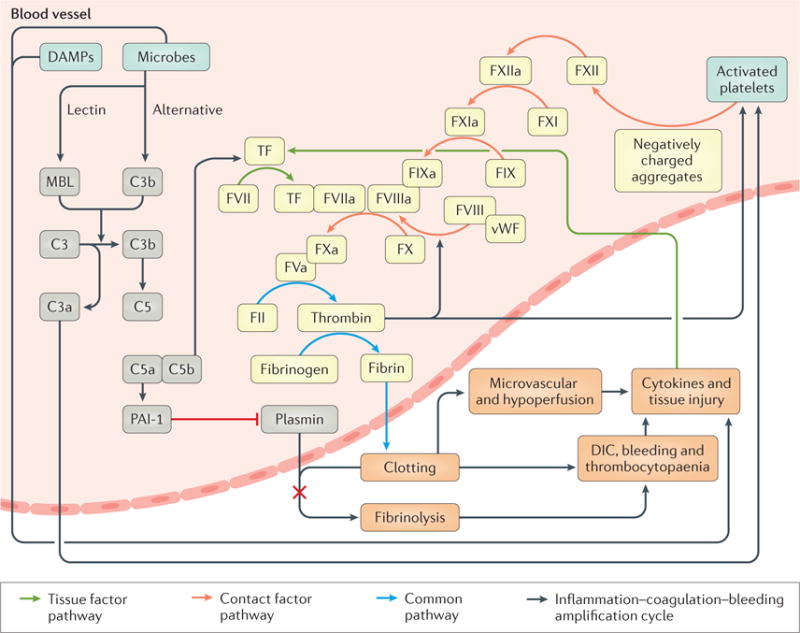
Microorganisms and damage -associated molecular patterns (DAMPs), as well as complement activation and the release of inflammatory cytokines or mediators, can initiate the coagulation cascade (involving the coagulation factors designated here as ‘F’ followed by the requisite Roman numeral). Primarily through the upregulation of procoagulant proteins such as tissue factor (TF), excessive fibrin deposition and reduced plasmin activity lead to thrombus and fibrin deposition and microcirculatory defects. The system is self-activating as complement activation and the exposure of myeloid and endothelial cells to microbial products and inflammatory cytokines increase the expression of TF. Products of complement activation, such as C3a and C5a, induce platelet-activating factor (PAF; not shown) and inflammatory cytokines. Cytokines, PAF and thrombi can also damage the endothelium, exposing collagen fibres and activating von Willebrand factor (vWF), which further increases TF expression and inflammatory cytokine production. Although not shown here, inflammatory cytokines also decrease the expression of the fibrinolytic pathway, by increasing plasminogen activator inhibitor 1 (PAI-1) activity and decreasing plasmin activity. DIC, disseminated intravascular coagulation; MBL, mannose-binding lectin.
The damaged endothelium and exposure of the underlying collagen activate von Willebrand factor, which further activates platelet aggregation and fibrin formation. Platelets might also trigger inflammation by activating dendritic cells. The activated endothelium also upregulates tissue factor, which can act directly on circulating factor VII, leading to tissue factor–factor VIIa complexes that convert factor X to factor Xa, resulting in thrombin generation, fibrin deposition, contact factor activation, clot formation, bradykinin synthesis and complement activation. Furthermore, complement activation feeds back to promote further clotting through complement-mediated shedding of cell-derived microvesicles. These microvesicles from monocytes and macrophages contain additional tissue factor, thereby exaggerating inflammation and thrombosis80.
Complement deposition on erythrocytes triggers haemolysis and the release of erythrocyte-derived microvesicles that are prothrombotic81. The resulting interaction between tissue factor and factor VIIa propagates the inflammatory process and leads to fibrin deposition on the endothelium. Microthrombi deposition, especially in the microvasculature, leads to decreased perfusion and thrombus formation. Concordantly, coagulation augments inflammation predominantly through a thrombin-induced secretion of pro- inflammatory cytokines and growth factors. Extracellular tissue factor signalling through PARs elicits cellular activation and inflammatory responses82.
Endogenous anticoagulants that inhibit different parts of the coagulation cascade (thereby inhibiting clot formation) are downregulated by the same processes that lead to the upregulation of tissue factor. For example, antithrombin and activated protein C concentrations decrease, as does endothelial glycosaminoglycans, such as heparan sulfate83. Inhibition of both thrombomodulin and endothelial cell protein C receptor contributes to the decrease in activated protein C concentrations84. Simultaneously, fibrinolysis is dramatically decreased. Increased levels of PAI-1 inhibit both tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA). This dysregulation of the PAI-1–t-PA–u-PA network results in a substantial reduction in the concentration of plasmin, which is required for dissolving intravascular fibrin clots. Thrombin generation and its binding to thrombomodulin activate thrombin-activatable fibrinolysis inhibitor — further reducing plasmin generation.
Ultimately, this exaggerated coagulopathy can lead to uncontrolled bleeding. This event might seem inconsistent with the previous statements regarding a sepsis-induced hypercoagulation and fibrin deposition, but the process is thought to occur secondary to a consumptive thrombocytopaenia and depletion of clotting factors10. The transition from a hypercoagulable state to DIC is characterized by fibrinolysis with increased circulating fibrin degradation products, thrombocytopaenia and exhaustion of liver-derived prothrombin, fibrinogen, factor X and factor V reserves.
Clot formation
Inflammation and coagulation are tightly linked defence mechanisms following injury and auto-amplify by co-stimulation85. In an anticoagulant state during health, endothelial cells generally do not express adhesion molecules that bind to leukocytes and platelets, but will do so in sepsis in response to the early inflammatory response, resulting in the activation of coagulation. The local cytokine milieu in this stage of inflammation induces cell-surface receptors for myeloid cells, lymphocytes and platelets. Platelets bind to fibrin strands and provide a ready source of P-selectin for neutrophil attachment; activated neutrophils produce NETs that provide a scaffold for more clot formation and this process self-amplifies10,86. This cooperative interaction serves to ‘wall off’ sites of injury from the rest of the host, limiting infection risk. The clot also serves to avoid blood loss and possible exsanguination by plugging the defect in the vascular system. This co-regulated clot formation and innate immune activation has an obvious survival advantage when a limited site of injury can be contained locally. However, if generalized activation of coagulation and inflammation occurs throughout the host, such as during DIC, the consequences can be devastating and lead to potentially lethal septic shock (see below).
Effect on organ systems
Sepsis is also a systemic disorder that can affect all organs of the body, probably owing to the panoply of cytokines and other mediators that are released into the general circulation during the onset of the dis order. The presenting signs and symptoms of sepsis are variable and depend on the particular organ systems that are affected. Six types of organ dysfunction predominate in sepsis: neurological (altered mental status), pulmonary (with hypoxaemia), cardiovascular (shock), renal (oliguria and/or increased creatinine concentration), haematological (decreased platelet count) and hepatic (hyperbilirubinaemia).
Neurological
Patients typically present with altered mental status manifested by lethargy, confusion or delirium. Occasionally, the mental status of the patient is so severely depressed that it is necessary to secure their airway (that is, perform endotracheal intubation). Despite this, the neurological examination at this time is typically without focal neurological findings. In the assessment, other causes of neurological disturbance (for example, hypoxaemia, hypoglycaemia, drug toxicity or central nervous system infection) should be ruled out or if present, addressed.
Pulmonary
One of the most common manifestations of sepsis is increased respiratory rate. Tachypnoea (a hallmark of sepsis-induced adult respiratory distress syndrome) can be associated with abnormal arterial blood gases, typically, a primary respiratory alkalosis. Accompanying hypoxaemia and/or hypercarbia can also occur; respiratory muscle fatigue, hypoxaemia or hypercarbia might necessitate endotracheal intubation for therapy. The aetiology of the respiratory failure in sepsis is due to inflammatory mediator- induced damage to alveolar capillary membranes. This cytokine-mediated lung injury results in noncardiogenic pulmonary oedema that can be profound and that causes decreased lung compliance and impaired oxygen uptake and carbon dioxide elimination. Decreased lung compliance and activation of juxtacapillary receptors lead to increased ventilation and are partly responsible for the tachypnoea. Chest X-ray imaging usually shows increased lung water with bilateral pulmonary infiltrates. Left ventricular heart failure must be ruled out as the cause of the pulmonary changes. Although patients with sepsis may have profound, life-threatening hypoxaemia, most patients do not die of hypoxaemia but rather of multiple organ failure.
Cardiovascular
Myocardial depression, which is characterized by hypotension or shock, is a hallmark of severe sepsis87. Several cytokines have direct cardiomyocyte toxic effects. Mild increases in circulating cardiac troponins are frequently present in sepsis and are indicative of sepsis severity. Myocardial depression affects both the right and the left ventricles and this finding distinguishes sepsis-induced myocardial depression from coronary atherosclerotic-induced myocardial ischaemic dysfunction. Sepsis-induced myocardial depression can be profound with decreases in the left and right ventricular ejection fractions, necessitating therapy with inotropic agents.
Oxidative and nitrosative stress (the build-up of reactive oxygen and nitrogen species, respectively) also contribute to cardiovascular and other organ failure, which is one of the root causes of tissue hypoxia88. Nitrosative stress is a major component of the pathophysiology of sepsis, and upregulation of inducible nitric oxide synthase (iNOS) might provide the link between inflammatory activation and cardio vascular compromise. In this context, the role of hypoxia-induced factor-α (HIFα) in sepsis also plays a major part in defining its pathophysiology89.
Renal
Renal dysfunction that progresses to frank renal failure is a major cause of sepsis-induced morbidity12. Although the exact mechanisms responsible for sepsis- induced renal failure are unknown, clinicians can reduce the incidence of severe renal failure in sepsis by aggressive and appropriate volume resuscitation in the disorder. Because of loss of intravascular volume in sepsis due to leaky capillary membranes and vasodilation, patients typically require volume resuscitation to replace these losses. Accordingly, clinicians must avoid the use of nephrotoxic agents in patients with sepsis if at all possible. For example, administration of intravenous contrast agents for radiological imaging studies can precipitate new-onset renal failure if given to a patient with sepsis who is intravascularly volume depleted. The absence of full renal recovery in sepsis is associated with poor long-term outcomes, so management of renal function during sepsis is of crucial importance. Even minor increases in the concentrations of serum creatinine are associated with increased mortality90.
Haematological
DIC is one of the most striking manifestations of severe sepsis. DIC can present in one of two contrasting clinical fashions: with overt bleeding from multiple sites or, conversely, with thrombosis of small and medium blood vessels. The reason for the striking differences in presentation of DIC is attributable to the fact that the coagulation system represents a balance between the clotting and fibrinolytic systems. In individual cases of sepsis, either system can predominate. If the fibrinolytic system is dominant, the patient will present with bleeding from multiple sites. Conversely, if the coagulation system is dominant, the patient will present with cyanotic (discoloured) fingers and toes that may progress to frank gangrene of the digits or upper and lower extremities. It is imperative to rule out heparin-induced DIC, which may masquerade as sepsis-induced DIC.
Hepatic
Liver dysfunction is common in sepsis, whereas sepsis-induced acute liver failure is rare, occurring in <2% of patients91. Sepsis-induced liver injury is indicated by increased concentrations of serum alanine transaminase and increased levels of bilirubin. The exact aetiology of liver dysfunction in sepsis is unknown. Undoubtedly, a large part of liver dysfunction in patients with septic shock is due to centrilobular necrosis of the liver secondary to poor hepatic perfusion. Autopsy studies of patients who died of sepsis have shown necrotic hepatocytes in the regions surrounding the central veins65,67. In addition to necrotic cell death in the livers of patients with sepsis, hepatocytes have also been observed to be undergo apoptotic cell death65,67. Interestingly, electron microscopy has shown that there are increased autophagic vacuoles present within hepatocytes from patients with sepsis. In rare cases, autophagic vacuoles were so extensive as to be consistent with autophagy-induced cell death92. Thus, it seems that hepatocytes undergo multiple different types of cell death in sepsis.
Diagnosis, screening and prevention
Defining sepsis
No single diagnostic test is (and will ever be) available that establishes the diagnosis of sepsis or septic shock. Sepsis and septic shock are clinical syndromes defined by a constellation of signs, symptoms, laboratory abnormalities and characteristic pathophysiological derangements. Clinicians often use these terms in an imprecise manner, which adds to the confusion when describing what is meant by the term sepsis. The 1991 SIRS criteria (BOX 1), which include parameters on temperature, heart rate and white blood cell count, have proven to be rather difficult to translate into clinical practice or even use effectively as entry criteria for clinical trials of sepsis. Using the SIRS criteria plus infection as the definition of sepsis could be applied to a large percentage of patients who are admitted with uncomplicated infections for whom the label of ‘sepsis’ seemed out of place or irrelevant. For example, most children with middle ear infections will often have two or three SIRS criteria (fever, tachycardia and leukocytosis); to consider them as ‘septic’ based on the SIRS criteria makes no clinical sense, especially when most are prescribed oral antibiotics for treatment at home. Similarly, in a large number of patients, especially those in whom antibiotics have been started empirically, the detection of bacteria in the blood or bodily fluids is often problematic. In as many as 30% of the cases of presumed sepsis, no pathogen is ever identified. In many cases, evidence of infection is inferred radiologically or from haematological measurements93.
The aforementioned proposed 2015 approach to the diagnosis of sepsis and septic shock is based on clinical realities and easily obtainable physiological and lab oratory parameters4 (BOX 2). What distinguishes sepsis from an otherwise localized microbial infection is that the host response is dysfunctional, generalized and contributes to multiple organ dysfunction and potentially septic shock94. Furthermore, sepsis is characterized by organ dysfunction in tissues that are not directly involved with the infectious process itself. A quick bedside assessment of organ injury has been proposed using readily available clinical measurements95. Indeed, early evidence of septic shock is manifested by hypoperfusion of tissues with resultant dysfunction and eventually by organ failure that occurs simultaneously or closely following the inflammatory event96.
Defining septic shock
Conceptually, septicaemia refers to sepsis with positive blood cultures, although it is an archaic term that is generally avoided. Blood cultures are not commonly positive, in part because bacteria do not need to circulate in the bloodstream to induce sepsis, and in part because some patients are being treated empirically with antibiotics at the time of testing and before the diagnosis. Thus, the term septicaemia has been abandoned. The term ‘septic shock’ remains current and is defined as a state in which sepsis is associated with cardiovascular dysfunction manifested by persistant hypotension despite an adequate fluid (volume) resuscitation to exclude the possibility of volume depletion as a cause of hypotension. Hypotension is operationally defined as the requirement for vasopressor therapy to maintain a mean arterial pressure of >65 mmHg and a plasma lactate level of >2 mmol per l. An increased level of serum lactate is a hallmark of tissue hypo perfusion and septic shock, and is helpful in early diagnosis. The usual cut-off value for an abnormally high lactate level is ≥2 mmol per l, but Casserly et al.97 have recommended the use of a lactate level of ≥4 mmol per l for inclusion in sepsis clinical trials.
Biomarkers
The ability of biomarkers to identify the presence and severity of sepsis has generally been limited. Many biomarkers based on the magnitude of the inflammatory response, such as IL-6, IL-10, CCL2, CXCL10 and HMGB1, have shown good correlation with the severity of sepsis and clinical outcome in population-based studies, but have proven less useful for individual patients — in large part because of the lack of specificity of the biomarkers and the commonality of the early inflammatory response. Our ability to distinguish sepsis from non-infectious critical illness and to prognosticate outcome is very limited.
The one exception is in the use of procalcitonin to distinguish sepsis from non-infectious critical illness and to guide the use of antibiotic therapy98. Procalcitonin is a peptide precursor of the hormone calcitonin that is produced by parafollicular cells of the thyroid and by the neuroendocrine cells of the lung and the intestine. In healthy individuals, procalcitonin levels are nearly undetectable. Initially, there was consider able enthusiasm that procalcitonin concentrations could distinguish sepsis from non-septic critical illness and to predict clinical outcomes better than inflammatory cytokines or clinical criteria. Although controversial, the general consensus to date is that procalcitonin is not an effective diagnostic measurement to rule-in or rule-out sepsis or bacterial infection, or for prognostication, in the absence of additional clinical data98,99. However, this notion has been challenged by the findings of a recent multicentre study in >1,500 critically ill patients with presumed bacterial infections and sepsis. In this study, the duration of antibiotic treatment, 28-day mortality and 1-year mortality were significantly lower in the procalcitonin-guided group than in patients who were managed without the procalcitonin measurement100. Furthermore, two recent large meta-analyses of data from patients with respiratory infections showed that procalcitonin to guide antibiotic treatment in patients with respiratory infections was not associated with higher mortality rates or treatment failure101,102. Antibiotic use was significantly reduced across different clinical settings and diagnoses.
Prevention
Prevention of sepsis and septic shock is based on good clinical practices to reduce the incidence of infections, particularly in high-risk populations. In the community setting, prevention is centred on vaccination for at-risk populations, such as for pneumococcal pneumonia in the elderly and meningococcal infections in adolescents and young adults. Other high-risk populations include those with advanced-stage cancer, type I diabetes, end-stage renal disease, congestive heart failure and chronic obstructive pulmonary disease (BOX 3). Prevention in this group of individuals entails good hygiene, maintaining mobility and reducing frailty, preserving nutritional status and adequately treating local wound infections.
Box 3. Risk factors for developing sepsis*.
Age
Very young (<2 years of age)
>55 years of age
Chronic and serious illness
Cancer
Diabetes
Chronic obstructive pulmonary disease
Cirrhosis or biliary obstruction
Cystic fibrosis
Chronic kidney disease
Congestive heart failure
Collagen vascular disease
Obesity‡
Impaired immunity
Transplantation
Chemotherapy
Radiation therapy
Drug-mediated immune suppression
Blood transfusions
Breach of natural barriers
Trauma
Surgical injury
Catheterization or intubation
Burns
Enterocolitis
Chronic infections
HIV
Urinary tract infections
Pneumonia
Decubitus or non-healing dermal wounds
Other
Protein calorie malnutrition
*See REFS 26,91,164. ‡Obesity can be associated with increased complications but improved outcomes.
Hospitalized patients pose a much greater challenge to sepsis prevention because of their concordant illness and an environment rich in pathogens. In this case, reducing primary length of stay and minimizing the frequency and duration of invasive procedures that disrupt natural barriers are often some of the most effective tools. Simple hand washing, use of devices containing antimicrobials and frequent changing of catheters can reduce incidence103. Equally important in the hospital setting is constant surveillance and immediate intervention to prevent sepsis and its progression to septic shock and multiple organ failure.
The US Agency for Healthcare Research and Quality has identified sepsis to be the most expensive condition treated in hospitals in the United States, with annual costs exceeding US$20 billion. Moreover, the US Centers for Medicare and Medicaid Services has imposed substantial financial penalties to hospitals and institutions that fail to adequately recognize and treat sepsis early. Most major academic hospitals use early warning systems to detect early infections and their systemic manifestations. These measures often include evaluation of haemodynamics, urine output, body temperature and mental function — often on an hourly basis. For suspicion of sepsis, early intervention by adequately trained health care providers using broad-spectrum antibiotics and fluid support, often necessitating a transfer to an intensive care unit (ICU), have been shown to result in significant reductions in mortality14,15.
Management
Once sepsis is identified, early and aggressive appropriate management is a priority — the timing of which is crucial. Treatment is based on three components: infection control, haemodynamic stabilization and modulation of the septic response.
Infection control
The first priority in treatment is early adequate antimicrobial administration and source control. Once a diagnosis of sepsis is suspected, a thorough search for a likely source must be conducted; clinical symptoms and signs, appropriate microbiological cultures and relevant imaging techniques must be used to try and determine the infectious source.
Adequate antimicrobial medication must be started as soon as possible and must not be delayed until culture data are obtained. The importance of initial appropriate antimicrobials has been well recognized given that it significantly reduces mortality risk104,105. Patients should be given broad-spectrum antimicrobial therapy that will cover all likely organisms, based on the likely source of infection, local microbiological flora and resistance patterns, recent antimicrobial therapy and health care facility ecology. Combination antimicrobial treatment is preferred to single-agent therapy, especially in the most severe cases106,107. Once culture results are available, the choice of antimicrobials should be re-evaluated, and de-escalation to a narrower spectrum should be performed whenever possible. This approach will optimize treatment efficacy, limit toxicity, help to prevent the development of drug resistance and reduce costs. Nevertheless, in some cases, several organisms are incriminated, and, in as many as 30% of patients, culture results will be negative108,109 such that de-escalation is not always warranted110.
The recommended doses for many of the antimicro-bials used in patients with sepsis are derived from non-critically ill patients or healthy volunteers. However, the pharmacodynamics and kinetics of many drugs could be altered in critically ill patients, especially in those with renal and hepatic dysfunction; therapies such as haemodialysis and haemofiltration can also influence drug distribution and clearance, necessitating dose adaptation. If available, daily monitoring of antimicrobial levels can help to attain therapeutic concentrations. The use of biomarkers, notably procalcitonin, to guide antimicrobial therapy has been associated with reduced antimicrobial use without major risks, but further studies are still required.
Finally, eradication of the infectious source by surgical intervention (for example, laparotomy and exploration) is sometimes necessary to remove any focus of infection, including iatrogenic causes such as drains or intravascular catheters.
Haemodynamic stabilization
The haemodynamic management of patients with sepsis and septic shock can be considered in four phases: salvage, optimization, stabilization and de-escalation111. The overall goal of these four phases is to provide immediate haemodynamic support to prevent organ injury and shock, and then to curtail therapies in a standardized manner. The amount of fluid administered will depend on the phase of shock111. In the salvage phase of treatment, fluid administration should be generous112 before monitoring is obtained. In the optimization phase, an individualized approach is needed. Signs of fluid responsiveness, including passive leg raising, can be helpful in a mechanically ventilated, deeply sedated patient. Evaluation of stroke volume changes during passive leg raising can be considered, but is not as easy. A fluid challenge technique is usually the best way to individualize fluid therapy. After the stabilization period, a de-escalation phase must be conducted, in which fluid balance should become negative.
One aspect of sepsis management that is increasingly controversial is early goal-directed therapy (EGDT). In a seminal single-centre study published in 2001, Rivers et al.113 showed that patients with septic shock in the emergency room setting benefited from EGDT to reduce mortality, as compared with standard management that did not have specific targets for determining the adequacy of response. The EGDT strategy was based on reaching a central venous oxygen saturation (ScvO2) to titrate haemodynamic resuscitation using intravenous fluids, dobutamine and packed red-cell transfusion to maintain a ScvO2 of >70%. However, these positive results could not be confirmed in more-recent larger multicentre studies, perhaps because patient management has improved in the control group114. Furthermore, the heterogeneity of patients with sepsis dictates that many different end points might be effective, making EGDT trials difficult to conduct.
When considering the immediate resuscitative phase of treatment, ‘ventilation, infusion and pump’ method proposed by Weil and Shubin115 remains a useful guide. Here, hypoxaemia should be corrected and endotracheal intubation and mechanical ventilation will be needed in severe cases; non-invasive ventilation is not recommended. On the basis of large randomized controlled trials, current guidelines recommend the use of crystalloids for fluid resuscitation and suggest the use of human albumin in cases of septic shock when patients cannot be stabilized with crystalloids alone112. The use of hydroxyethyl starch in patients with sepsis has been banned by the US FDA and the European Medicines Agency owing to increased mortality; the use of other synthetic colloids is also discouraged116. Excessive amounts of saline solutions should also be avoided, as hyperchloraemia can have adverse effects, especially on the kidneys117.
Vasoactive agents are also often required and are frequently started alongside fluid administration to avoid prolonged hypotension, which can impair tissue perfusion. Noradrenaline is recommended over dopamine owing to reduced adverse effects and mortality118,119. Dobutamine can be and is often added to noradrenaline as an inotropic agent to increase cardiac output and oxygen delivery to the tissues. Monitoring changes in blood lactate levels can help to assess the effectiveness of the resuscitation120. An inevitable consequence of large fluid administration and damaged endothelium is oedema. One of several approaches being investigated to address oedema and endothelial injury is the use of selepressin, a selective vasopressin type 1a receptor agonist that increases arterial pressure and has the potential to reduce vascular leakage and pulmonary oedema. Preclinical studies in an ovine model of septic shock showed clear survival benefits of selepressin over vasopressin and noradrenaline, especially when administered early121.
Finally, appropriate ventilator support will also be required because of the high preponderance of acute lung injury. Managing appropriate lung support should be one of the core management principles for sepsis and septic shock. The overarching goal should be achieving adequate oxygenation while minimizing the fraction of inhaled oxygen and volumes inhaled, and successfully weaning the patient from the ventilator as soon as possible.
Modulation of the septic response
This exaggerated early inflammatory response has been the predominant target of early clinical intervention using biological response modifiers in severe sepsis or septic shock. At present, there have been at least 150 clinical trials targeting either the pattern-recognition receptors, the PAMPs themselves or the early cytokines or mediators produced in response to sepsis (TABLE 2). None has proven effective to date, although several are currently still in trial. Retrospective subgroup analysis has often shown significant benefit in smaller subgroups with IL-1 receptor antagonist and TNF inhibitors, but these have not been prospectively validated. Reasons for why these clinical trials have failed have been suggested to include the timing of drug administration, failure to prospectively identify patients with sepsis who would benefit from these therapies and redundancy in patient response122. In some patient populations, treatment with these anti-inflammatory agents has actually increased mortality123–125, suggesting that, in some cases, endogenous production of these mediators might be essential for protective immunity. At present, numerous anti-inflammatory agents and immunostimulants are in clinical trials for sepsis and septic shock.
Table 2.
Selected clinical biological response modifiers used in clinical trials for severe sepsis or septic shock
| Target | Class | Study type | Drug | Finding | Refs |
|---|---|---|---|---|---|
| Endocrine abnormalities | Corticosteroids | Meta-analysis | Corticosteroid | No or modest effect | 173,174 |
| Vasopressors | Prospective RCT | Arginine vasopressin | No effect | 175 | |
|
| |||||
| Endotoxin | Monoclonal antibody | Prospective RCT | HA-1A E5 |
No effect No effect |
176,177 |
| Lipid emulsion | Prospective RCT | GR270773 | No effect | 178 | |
| Bactericidal-permeability increasing peptide | Prospective RCT | rBPI21 | Modest effect | 179 | |
|
| |||||
| TLR4 | Monoclonal antibody | Prospective RCT | TAK-242 Eritoran tetrasodium | No effect No effect |
180,181 |
|
| |||||
| CD14 | Monoclonal antibody | Prospective RCT | IC14 | Unclear | 182 |
|
| |||||
| TNF | Monoclonal antibody | Prospective RCT | BAY 1351 | No effect No effect |
183,184 |
| Immunoadhesin | Prospective RCT | Lenercept Etanercept |
No effect No effect |
124,185 | |
|
| |||||
| IL-1 | Receptor antagonist | Prospective RCT | Anakinra | No effect | 186 |
|
| |||||
| PAF | PAF antagonist | Prospective RCT | TCV-309 Lexipafant |
No effect No effect |
187,188 |
|
| |||||
| Eicosanoids | NSAIDs | Prospective RCT | Ibuprofen | No effect | 189 |
| Soluble phospholipase A2 inhibitor | Prospective RCT | Varespladib | No effect | 190 | |
|
| |||||
| C5a | Monoclonal antibody | Prospective RCT | CaCP29 | Ongoing | 49 |
|
| |||||
| Nitric oxide | l-N-methylarginine | Prospective RCT | 546C88 | Modest effect | 191 |
| Reducing agents | Prospective RCT | Methylene blue | Modest effect | 192 | |
|
| |||||
| Hypercoagulability or disseminated intravascular coagulation | Activated protein C concentrate | Prospective RCT | Drotrecogin alfa (activated) | Beneficial No effect |
141,193 |
| Tissue factor pathway inhibitor | Prospective RCT | Tifacogin | No effect | 194 | |
| Antithrombin | Prospective RCT | Antithrombin | No effect | 195 | |
| Anti-tissue factor antibody | Prospective RCT | ALT-836 | No effect | 196 | |
| Heparin | Meta-analysis | Heparin salt | Modest effect | 197 | |
| Thrombomodulin | Prospective RCT | ART-123 | Modest effect | 198 | |
|
| |||||
| Others | Intravenous immunoglobulin | Meta-analysis | Intravenous immunoglobulin | No effect | 123 |
| Selenium (antioxidant) | Prospective RCT | Selenium | No effect | 199 | |
| Bradykinin antagonists | Prospective RCT | Deltibant | No effect | 200 | |
| N-acetylcysteine | Meta-analysis | N-acetylcysteine | No effect No effect |
201,202 | |
| Statins | Meta-analysis | Atorvastatin, simvastatin and rovusastatin | No effect | 171 | |
| Extracorporeal haemoperfusion | Prospective RCT | Adsorber EN500 | No effect | 203 | |
PAF, platelet-activating factor; RCT, randomized controlled trial; TLR, Toll-like receptor; TNF, tumour necrosis factor.
The sepsis team
Despite increased awareness of the importance of early diagnosis and rapid appropriate treatment of patients with sepsis, many patients still do not receive acceptable early management. Patients with sepsis are highly complex, often with multiple comorbidities and rapidly changing haemodynamics. The management of such patients involves multiple elements, including invasive radiological procedures and setting up of haemodynamic monitoring systems, blood sampling for cultures and laboratory testing, administration of antibiotics, fluid resuscitation and administration of vasoactive agents — all of which need to be started rapidly. For initial management, the best way of being able to simultaneously perform all the necessary actions is for providers to be organized as a ‘sepsis team’, similar to the trauma teams now widely established for the management of patients with severe trauma. One member of the team would be clearly identified as the leader to direct and coordinate the overall management process. The sepsis team should be available at all times126–128.
Quality of life
Little is known about long-term mortality and quality of life following sepsis. In most cases, efficacy of intervention in sepsis has been limited to in-hospital index experiences, primarily 28-day or 30-day mortality and degree of organ injury. Only over the past decade have studies started to examine the long-term consequences of sepsis; the preliminary data are not encouraging129,130. Chronic critical illness is occurring in a large proportion of patients who survive sepsis but remain hospitalized52; >50% of patients in the ICU die within 3 months of sepsis and 60% have psychological disturbances131. Only a minority of patients return to a functional lifestyle. Few patients who survive sepsis are discharged to home; the majority of patients are discharged to long-term nursing or rehabilitative facilities. Both age and length of time in the ICU have been shown to be independent variables for long-term survival and functional recovery129,132,133. Patients >55 years of age and those who remained in the ICU for >14 days have the highest mortality rates post-discharge.
Even less is known about the long-term consequences on functional and cognitive recovery after sepsis. Intensive care specialists have recognized a syndrome in survivors of critical illness, including sepsis, termed post-ICU syndrome, which is characterized by insomnia, nightmares, fatigue, depression, loss of cognitive function and loss of self-esteem134. Almost half of the individuals who survived sepsis report at least three of these symptoms135. These individuals demonstrate cognitive deficits in verbal learning and memory up to 2 years after the hospital discharge136. Interestingly, these cognitive deficits were associated with a significant reduction of left hippocampal volume compared with healthy controls. Patients with sepsis also had more low-frequency electroencephalogram activity indicating generalized brain dysfunction and not focal damage. Sepsis has also been known to induce post-traumatic stress disorder (PTSD) in many patients137. An increased incidence of PTSD in individuals who survived sepsis is also associated with pre-sepsis depression and delirium while in intensive care137.
Several studies have used surveys to examine functionality after hospital discharge (the 36-Item Short-Form Health Survey and the EuroQual EQ-5D health questionnaire)129,138. The general consensus has been that patients who survive sepsis have a prevalence of moderate-to-severe cognitive impairment 10% higher than the general population. Equally important, patients who survived sepsis had a much higher frequency of new impairments than their age-matched counterparts.
The underlying causes of these functional and physical declines are unknown, but there are many possible reasons. Primary causes might include ICU-acquired weakness owing to both inactivity and immobilization, as well as from inflammation, corticosteroid and neuromuscular blockers commonly used in sepsis treatment. According to Poulson et al.139, in 81% of the patients who survived sepsis, loss of muscle mass at 1 year was the main cause of decreased physical function. In addition, direct neuronal damage and delirium may also contribute to lower physical functioning.
Outlook
Sepsis is not disappearing; in fact, although controversial, the number of patients with sepsis is probably increasing worldwide, as are the risks associated with increased comorbidities and an ageing population in developed countries. With that said, however, there are encouraging signs. Increased recognition of the importance of early detection and rapid intervention is most likely responsible for the declines in in-hospital mortality over the past decade. Credit for these improvements is owing in large part to the Surviving Sepsis Campaign140 for its widespread dissemination of agreed on standard of care treatment guidelines for the management of sepsis. Furthermore, the part played by the US federal government in tying reimbursement to sepsis outcomes is likely to have a major impact on sepsis recognition moving forwards. Namely, sepsis has been recognized as an unacceptable preventable patient safety index and the Centers for Medicare and Medicaid Services are penalizing providers by reducing reimbursements for care. These changes in hospital reimbursement will probably be an important driving force for the implementation of procedures for recognizing sepsis and its early intervention.
Advances in treatment
Attention is being focused not only on the in-hospital consequences of sepsis but also on the long-term outcomes, especially in terms of functional and cognitive recovery. In this regard, treatment for individuals who survive sepsis is increasingly including physical therapy, nutritional and psychological interventions.
Modulation of the septic response
To date, no single biological response modifier is available that is currently approved for use in sepsis (TABLE 3). The only immuno-modulatory agent approved for sepsis was activated protein C (drotrecogin alfa (activated))141, but this drug was withdrawn after negative post-marketing trials142. However, therapeutic approaches that target the early inflammatory response as well as the endothelial injury and coagulopathy are continuing. Two studies have indicated that the addition of steroids could be beneficial in severe community-acquired pneumonia143,144. In addition, extracorporeal techniques to remove DAMPs, PAMPs and inflammatory mediators are under investigation, and recent observations on the ‘artificial spleen’ are particularly exciting145. Using magnetic beads coupled to mannose-binding ligand, the biospleen was capable of reducing the magnitude of the inflammatory response by simultaneous removal of multiple PAMPs and DAMPs. Blockade of C5a with a monoclonal antibody is also currently being tested in patients with early septic organ dysfunction as a means to reduce an overly exuberant innate immune response49.
Table 3.
Selected potential immunomodulating agents for the treatment of severe sepsis and septic shock
| Agent | Function | Findings | Refs |
|---|---|---|---|
| GM-CSF |
|
|
147, 204–206 |
|
| |||
| G-CSF |
|
|
207, 208 |
|
| |||
| IFNγ |
|
|
205, 209, 210 |
|
| |||
| IL-7 |
|
|
146, 211, 212 |
|
| |||
| IL-15 |
|
|
146, 213–215 |
|
| |||
| Thymosin-α1 |
|
|
151, 216 |
|
| |||
| PDL1-specific antibody |
|
|
217–219 |
|
| |||
| CTLA4-specific antibody |
|
|
217, 220 |
|
| |||
| TIM3-specific antibody |
|
|
221, 222 |
|
| |||
| LAG3-specific antibody |
|
|
223, 224 |
CTLA4, cytotoxic T lymphocyte antigen 4; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; HLA-DR, human leukocyte antigen-antigen D related; ICU, intensive care unit; IFNγ, interferon-γ; LAG3, lymphocyte activating gene 3; NK cell, natural killer cell; NKT cell, natural killer T cell; PDL1, programmed death ligand 1; TH1, T helper 1; TIM3, T cell immunoglobulin and mucin-containing protein 3; Treg cell, regulatory T cell.
Acute Physiology and Chronic Health Evaluation II (APACHE II) score is a severity-of-disease classification system.
Defining the immunological state of the patient will be crucial to the success of any biological response modifier for sepsis. Application of immunotherapies will need to be targeted to the appropriate immunological state. For example, drugs that are aimed at stimulating innate and adaptive immunity might be contraindicated during the early hyperinflammatory phase of sepsis. Thus, a means of immunophenotyping patients with sepsis is needed and many approaches, such as quantification of monocyte HLA-DR expression or measuring procalcitonin concentrations, are being explored. A measured approach that is based on carefully designed clinical trials in defined populations is the crucial next step in the development of these agents.
Immunostimulatory therapies
A diverse collection of drugs are currently being evaluated to either block the exaggerated inflammatory and endothelial injury or restore an effective antimicrobial immune response146 (TABLE 3). Agents that are being evaluated as immunostimulants include leukocyte growth factors147, immunostimulatory cytokines148,149, inhibitors of negative co-stimulatory pathways (for example, PD1 and PDL1)150 and unique immunomodulators, such as the thymic peptide thymosin-α1 (REF.151). Importantly, many of these agents have been previously used as immune adjuvants in the treatment of cancer, providing preliminary data on their safety and efficacy152.
Perhaps the most promising potential immuno-therapy in sepsis is the pleiotropic cytokine IL-7 (REF.153). IL-7 acts broadly on cells of the adaptive immune system, driving proliferation and survival of naive and memory CD4+ and CD8+ T cells, which are relentlessly depleted in sepsis151,152. Studies in patients with sarcoma154 and HIV155 have shown that IL-7 is effective in patients with viral infections, which, together with ex vivo results, show that IL-7 reverses key immunological defects in patients with sepsis156, providing a compelling case for a trial of IL-7 in sepsis.
Biomarker discovery
Alongside developments in therapy, emphasis has been placed on developing biomarkers or other indices that can identify groups of patients who could benefit from individualized therapy. Indeed, given that current therapeutic approaches — targeting inflammation, immune suppression, coagulopathy, endothelial injury or organ dysfunction — must be directed at both the appropriate patient and the appropriate time, biomarkers to ‘navigate’ the temporal changes in patient responses to sepsis would be invaluable.
Continued development of biomarkers that can distinguish sepsis from inflammation alone and that can be used as prognostic indicators, also remains an area of active investigation. Although single biomarkers have achieved predictive abilities similar to anatomical and physiological scoring systems, the future may be in multiplex approaches that use biomarkers and clinical indicators. The ‘-omics revolution’ of genomics, transcriptomics, proteomics, metabolomics and inter actomics is in its infancy, but preliminary studies have identified complex signatures that might not only distinguish sepsis from non-septic critical illness but also function as prognostic indicators or indicators of response to therapy157–159. Genomic signatures and genome-wide association studies also have the potential to identify patients who might respond to specific immunomodulatory interventions through the identification of alterations of specific pathways that can be addressed by pro- inflammatory or anti-inflammatory interventions157,158,160. Although there is clear evidence that genetic polymorphisms in individual inflammatory or immunosuppressant genes are associated with a varying incidence and severity of sepsis, application of personalized medicine and individual alleles has not yet been successful161. It is still too early to tell whether these multiplex approaches will prove superior to current biomarkers and clinical indices.
Acknowledgments
This Primer does not promulgate the clinical use of a drug that is not approved by the US FDA or the off-label use of any FDA-approved drug.
Footnotes
Author contributions
Authorship is ordered alphabetically. Introduction (L.L.M. and J.-L.V.); Epidemiology (J.-L.V., K.R. and S.M.O.); Mechanisms/pathophysiology (L.L.M., R.S.H., I.R.T. and K.R.); Diagnosis, screening and prevention (L.L.M., J.-L.V. and S.M.O.); Management (J.-L.V., S.M.O., R.S.H. and I.R.T.); Quality of life (L.L.M.); Outlook (all authors); Overview of the Primer (L.L.M.).
Competing interests
R.S.H. has received no direct financial support, nor does he or his family hold patents or equity interest in any biotech or pharmaceutical company. He has received laboratory research support from Bristol-Myers Squibb, GlaxoSmithKline and Medimmune. He has served as a paid consultant to Bristol-Myers Squibb, GlaxoSmithKline, Medimmune and Merck. R.S.H. and Washington University in St Louis, Missouri, USA, have also received grant support from the US NIH, US Public Health Service for research investigations of sepsis. L.L.M. and the University of Florida College of Medicine, USA, have received financial support from the US National Institute of General Medical Sciences, US Public Health Service. No other financial support, patents or equity interest to him or his family are disclosed. S.M.O. has received no direct financial support, nor does he or his family hold patents or equity interest in any biotech or pharmaceutical company. S.M.O. and Brown University, Rhode Island, USA, have received preclinical grants in the past from the NIH, US Public Health Service, Atoxbio, GlaxoSmithKline and Arsanis, and have received financial support for assistance with clinical trial coordination from Asahi Kasei, Ferring, Cardeas and Biocartis. S.M.O. serves as a member of the Data Safety and Monitoring Board (DSMB) for Paratek (for Omadcycline), Acheogen (for Placomicin) and Bristol-Myers Squibb (anti-PDL1 monoclonal antibody). He also serves on the DSMB for two NIH-funded studies: one examining procalcitonin-guided antibiotic administration and the other investigating early intervention for community- acquired sepsis. S.M.O. serves as a paid consultant for BioAegis, Arsanis, Aridis, Batelle and Cyon on various biodefense and monoclonal antibody projects. He also receives royalty payments from Elsevier publishers for the textbook entitled, Infectious Diseases 4th edition. K.R. holds an equity interest in InflaRx and is a paid consultant for Adrenomed. J.-L.V. and I.R.T. declare no competing interests.
References
- 1.Majno G. The ancient riddle of σηψιζ (sepsis) J Infect Dis. 1991;163:937–945. doi: 10.1093/infdis/163.5.937. [DOI] [PubMed] [Google Scholar]
- 2.Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101:1481–1483. doi: 10.1378/chest.101.6.1481. This paper has laid the ground for our current understanding of sepsis by underlining the crucial role of the host response to infection for which the term SIRS was coined. Furthermore, it was pointed out that SIRS can also result from non-infectious causes. [DOI] [PubMed] [Google Scholar]
- 3.Levy MM, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 4.Singer M, et al. The third international consensus conference on sepsis and septic shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. The third consensus update of the definitions and clinical criteria for sepsis and septic shock. Although there has been an important effort to improve the understanding of sepsis, controversy remains as to whether these new criteria will be useful or practical as early warning signs, especially in low-income and middle-income countries where it is often difficult to obtain the required measures of organ injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le JM, Vilcek J. Interleukin 6: a multifunctional cytokine regulating immune reactions and the acute phase protein response. Lab Invest. 1989;61:588–602. [PubMed] [Google Scholar]
- 6.Dinarello CA. Interleukin-1. Rev Infect Dis. 1984;6:51–95. doi: 10.1093/clinids/6.1.51. [DOI] [PubMed] [Google Scholar]
- 7.Beutler B, Cerami A. The biology of cachectin/TNF — a primary mediator of the host response. Annu Rev Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 8.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 9.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–475. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Levi M, Schultz M, van der Poll T. Sepsis and thrombosis. Semin Thromb Hemost. 2013;39:559–566. doi: 10.1055/s-0033-1343894. [DOI] [PubMed] [Google Scholar]
- 11.Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Intern Med. 2015;277:277–293. doi: 10.1111/joim.12331. [DOI] [PubMed] [Google Scholar]
- 12.White LE, et al. Acute kidney injury is surprisingly common and a powerful predictor of mortality in surgical sepsis. J Trauma Acute Care Surg. 2013;75:432–438. doi: 10.1097/TA.0b013e31829de6cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA. 2014;311:1308–1316. doi: 10.1001/jama.2014.2637. This is a retrospective analysis of an administrative database from >100,000 patients with recorded sepsis or septic shock. Mortality significantly improved in patients with both severe sepsis and septic shock, but did so at rates that were comparable to other diagnoses. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer R, et al. Improvement in process of care and outcome after a multicenter severe sepsis educational program in Spain. JAMA. 2008;299:2294–2303. doi: 10.1001/jama.299.19.2294. [DOI] [PubMed] [Google Scholar]
- 15.Levy MM, et al. Surviving Sepsis Campaign: association between performance metrics and outcomes in a 7.5-year study. Crit Care Med. 2015;43:3–12. doi: 10.1097/CCM.0000000000000723. This is one of many papers to demonstrate that increasing awareness for sepsis and the initiation of quality improvement initiatives in the field of sepsis can improve patient survival. [DOI] [PubMed] [Google Scholar]
- 16.Fleischmann C, et al. Assessment of global incidence and mortality of hospital-treated sepsis — current estimates and limitations. Am J Respir Crit Care Med. 2016;193:259–272. doi: 10.1164/rccm.201504-0781OC. This population-level epidemiological data from 15 international databases over the past 36 years demonstrate a high level of sepsis incidence in developed countries. By contrast, the study emphasizes the paucity of sepsis data from the developing world. [DOI] [PubMed] [Google Scholar]
- 17.Jawad I, Luksic I, Rafnsson SB. Assessing available information on the burden of sepsis: global estimates of incidence, prevalence and mortality. J Glob Health. 2012;2:010404. doi: 10.7189/jogh.02.010404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker JU, Theodosis C, Jacob ST, Wira CR, Groce NE. Surviving sepsis in low-income and middle-income countries: new directions for care and research. Lancet Infect Dis. 2009;9:577–582. doi: 10.1016/S1473-3099(09)70135-5. [DOI] [PubMed] [Google Scholar]
- 19.Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369:448–457. doi: 10.1056/NEJMra1201534. [DOI] [PubMed] [Google Scholar]
- 20.Mayanja BN, et al. Septicaemia in a population-based HIV clinical cohort in rural Uganda, 1996–2007: incidence, aetiology, antimicrobial drug resistance and impact of antiretroviral therapy. Trop Med Int Health. 2010;15:697–705. doi: 10.1111/j.1365-3156.2010.02528.x. [DOI] [PubMed] [Google Scholar]
- 21.Gordon MA, et al. Bacteraemia and mortality among adult medical admissions in Malawi — predominance of non-typhi salmonellae and Streptococcus pneumoniae. J Infect. 2001;42:44–49. doi: 10.1053/jinf.2000.0779. [DOI] [PubMed] [Google Scholar]
- 22.Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013 a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800. doi: 10.1016/S0140-6736(15)60692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Boogaard W, Manzi M, Harries AD, Reid AJ. Causes of pediatric mortality and case-fatality rates in eight Medecins Sans Frontieres-supported hospitals in Africa. Public Health Action. 2012;2:117–121. doi: 10.5588/pha.12.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sundararajan V, Macisaac CM, Presneill JJ, Cade JF, Visvanathan K. Epidemiology of sepsis in Victoria, Australia. Crit Care Med. 2005;33:71–80. doi: 10.1097/01.ccm.0000150027.98160.80. [DOI] [PubMed] [Google Scholar]
- 25.Seymour CW, Iwashyna TJ, Cooke CR, Hough CL, Martin GS. Marital status and the epidemiology and outcomes of sepsis. Chest. 2010;137:1289–1296. doi: 10.1378/chest.09-2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleischmann C, et al. Hospital incidence and mortality rates of sepsis. Dtsch Arztebl Int. 2016;113:159–166. doi: 10.3238/arztebl.2016.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–1250. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- 28.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 29.Liu V, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA. 2014;312:90–92. doi: 10.1001/jama.2014.5804. [DOI] [PubMed] [Google Scholar]
- 30.Lagu T, et al. What is the best method for estimating the burden of severe sepsis in the United States? J Crit Care. 2012;27:414.e1–414.e9. doi: 10.1016/j.jcrc.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. The incidence and outcome of sepsis were estimated using four different published methods; depending on the methods of data abstraction, the incidence of sepsis in the United States could vary as much as 3.5-fold. [DOI] [PubMed] [Google Scholar]
- 32.Rhee C, Gohil S, Klompas M. Regulatory mandates for sepsis care — reasons for caution. N Engl J Med. 2014;370:1673–1676. doi: 10.1056/NEJMp1400276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whittaker SA, et al. Severe sepsis cohorts derived from claims-based strategies appear to be biased toward a more severely ill patient population. Crit Care Med. 2013;41:945–953. doi: 10.1097/CCM.0b013e31827466f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwashyna TJ, Angus DC. Declining case fatality rates for severe sepsis: good data bring good news with ambiguous implications. JAMA. 2014;311:1295–1297. doi: 10.1001/jama.2014.2639. [DOI] [PubMed] [Google Scholar]
- 35.McPherson D, et al. Sepsis-associated mortality in England: an analysis of multiple cause of death data from 2001 to 2010. BMJ Open. 2013;3:e002586. doi: 10.1136/bmjopen-2013-002586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vincent JL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344–353. doi: 10.1097/01.ccm.0000194725.48928.3a. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 38.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bierhaus A, Nawroth PP. Modulation of the vascular endothelium during infection — the role of NF-kappa B activation. Contrib Microbiol. 2003;10:86–105. doi: 10.1159/000068133. [DOI] [PubMed] [Google Scholar]
- 40.Parikh SM. Dysregulation of the angiopoietin–Tie-2 axis in sepsis and ARDS. Virulence. 2013;4:517–524. doi: 10.4161/viru.24906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 42.Ward PA. The harmful role of C5a on innate immunity in sepsis. J Innate Immun. 2010;2:439–445. doi: 10.1159/000317194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stevens JH, et al. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest. 1986;77:1812–1816. doi: 10.1172/JCI112506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Czermak BJ, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 45.Rittirsch D, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia CC, et al. Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS ONE. 2013;8:e64443. doi: 10.1371/journal.pone.0064443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun S, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. 2013;49:221–230. doi: 10.1165/rcmb.2012-0428OC. [DOI] [PubMed] [Google Scholar]
- 48.Sun S, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60:586–595. doi: 10.1093/cid/ciu887. This paper demonstrates the potential beneficial effects of complement inhibition in a clinically relevant monkey model of viral infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.US National Library of Medicine. Studying complement inhibition in early, newly developing septic organ dysfunction (SCIENS) ClinicalTrials.gov. 2014 http://clinicaltrials.gov/ct2/show/NCT02246595.
- 50.Gentile LF, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012;72:1491–1501. doi: 10.1097/TA.0b013e318256e000. This paper provides a description of a phenotype of individuals who survived sepsis or critical illness who exhibit PICS. The authors propose that as early treatments for sepsis and trauma improve, this phenotype will predominate in survivors, especially the elderly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu D, et al. Persistent inflammation– immunosuppression catabolism syndrome, a common manifestation of patients with enterocutaneous fistula in intensive care unit. J Trauma Acute Care Surg. 2014;76:725–729. doi: 10.1097/TA.0b013e3182aafe6b. [DOI] [PubMed] [Google Scholar]
- 52.Vanzant EL, et al. Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg. 2014;76:21–29. doi: 10.1097/TA.0b013e3182ab1ab5. discussion 29–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28:429–436. doi: 10.1016/j.it.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 54.Walton AH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE. 2014;9:e98819. doi: 10.1371/journal.pone.0098819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kollef KE, et al. Predictors of 30-day mortality and hospital costs in patients with ventilator- associated pneumonia attributed to potentially antibiotic-resistant Gram-negative bacteria. Chest. 2008;134:281–287. doi: 10.1378/chest.08-1116. [DOI] [PubMed] [Google Scholar]
- 56.Otto GP, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. 2011;15:R183. doi: 10.1186/cc10332. An important publication that documents that the majority of patients with protracted sepsis develop infections with ‘opportunistic-type pathogens’, thereby strongly supporting the concept of sepsis progressing to an immunosuppressive disorder. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Torgersen C, et al. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analg. 2009;108:1841–1847. doi: 10.1213/ane.0b013e318195e11d. [DOI] [PubMed] [Google Scholar]
- 58.Delano MJ, et al. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taneja R, Sharma AP, Hallett MB, Findlay GP, Morris MR. Immature circulating neutrophils in sepsis have impaired phagocytosis and calcium signaling. Shock. 2008;30:618–622. doi: 10.1097/SHK.0b013e318173ef9c. [DOI] [PubMed] [Google Scholar]
- 60.Munoz C, et al. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cuenca AG, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–292. doi: 10.2119/molmed.2010.00178. A study demonstrating that the expansion of MDSCs in sepsis can be associated with the preservation of innate immunity, even in the presence of adaptive immune suppression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drifte G, Dunn-Siegrist I, Tissieres P, Pugin J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med. 2013;41:820–832. doi: 10.1097/CCM.0b013e318274647d. [DOI] [PubMed] [Google Scholar]
- 63.Hashiba M, et al. Neutrophil extracellular traps in patients with sepsis. J Surg Res. 2015;194:248–254. doi: 10.1016/j.jss.2014.09.033. [DOI] [PubMed] [Google Scholar]
- 64.Hynninen M, et al. Predictive value of monocyte histocompatibility leukocyte antigen-DR expression and plasma interleukin-4 and -10 levels in critically ill patients with sepsis. Shock. 2003;20:1–4. doi: 10.1097/01.shk.0000068322.08268.b4. [DOI] [PubMed] [Google Scholar]
- 65.Boomer JS, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. This is the first study to show that immune effector cells in tissues from patients dying of sepsis have severe impairment of stimulated cytokine production. This study also demonstrated that ‘T cell exhaustion’ is a likely mechanism that contributes to immunosuppression in patients with sepsis, providing a rationale for the use of immune checkpoint inhibitors as a novel potential therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meakins JL, et al. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg. 1977;186:241–250. doi: 10.1097/00000658-197709000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hotchkiss RS, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. This is the first study to show that patients with sepsis develop profound loss of immune effector cells via apoptosis, establishing that sepsis-induced apoptosis is a major immunosuppressive mechanism in sepsis. [DOI] [PubMed] [Google Scholar]
- 68.Hotchkiss RS, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. This paper encourages more research on immune augmentatory approaches in sepsis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Drewry AM, et al. Persistent lymphopenia after diagnosis of sepsis predicts mortality. Shock. 2014;42:383–391. doi: 10.1097/SHK.0000000000000234. This is a study that demonstrated that a sustained low total lymphocyte count was associated with increased mortality. Although the mechanisms are unclear, the data reveal that a commonly obtained clinical measurement can identify the severity of sepsis and organ failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coopersmith CM, et al. Antibiotics improve survival and alter the inflammatory profile in a murine model of sepsis from Pseudomonas aeruginosa pneumonia. Shock. 2003;19:408–414. doi: 10.1097/01.shk.0000054370.24363.ee. [DOI] [PubMed] [Google Scholar]
- 71.Bommhardt U, et al. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004;172:7583–7591. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- 72.Hotchkiss RS, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- 73.Hotchkiss RS, et al. TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocyte apoptosis in vivo. J Immunol. 2006;176:5471–5477. doi: 10.4049/jimmunol.176.9.5471. [DOI] [PubMed] [Google Scholar]
- 74.Hotchkiss RS, et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. 1999;162:4148–4156. [PubMed] [Google Scholar]
- 75.Schwulst SJ, et al. Agonistic monoclonal antibody against CD40 receptor decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2006;177:557–565. doi: 10.4049/jimmunol.177.1.557. [DOI] [PubMed] [Google Scholar]
- 76.Schwulst SJ, et al. Bim siRNA decreases lymphocyte apoptosis and improves survival in sepsis. Shock. 2008;30:127–134. doi: 10.1097/shk.0b013e318162cf17. [DOI] [PubMed] [Google Scholar]
- 77.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. This review provides a concise update of what was known at the time and what has become a very prescient appraisal of the future of sepsis research. [DOI] [PubMed] [Google Scholar]
- 78.Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. 2015;19:26. doi: 10.1186/s13054-015-0741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.London NR, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karpman D, et al. Complement interactions with blood cells, endothelial cells and microvesicles in thrombotic and inflammatory conditions. Adv Exp Med Biol. 2015;865:19–42. doi: 10.1007/978-3-319-18603-0_2. [DOI] [PubMed] [Google Scholar]
- 81.Zecher D, Cumpelik A, Schifferli JA. Erythrocyte- derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc Biol. 2014;34:313–320. doi: 10.1161/ATVBAHA.113.302378. [DOI] [PubMed] [Google Scholar]
- 82.Riewald M, Ruf W. Science review: role of coagulation protease cascades in sepsis. Crit Care. 2003;7:123–129. doi: 10.1186/cc1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shorr AF, et al. Protein C concentrations in severe sepsis: an early directional change in plasma levels predicts outcome. Crit Care. 2006;10:R92. doi: 10.1186/cc4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matsumoto H, et al. Enhanced expression of cell- specific surface antigens on endothelial microparticles in sepsis-induced disseminated intravascular coagulation. Shock. 2015;43:443–449. doi: 10.1097/SHK.0000000000000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 86.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 87.Sato R, Nasu M. A review of sepsis-induced cardiomyopathy. J Intensive Care. 2015;3:48. doi: 10.1186/s40560-015-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 89.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ricci Z, Polito A, Polito A, Ronco C. The implications and management of septic acute kidney injury. Nat Rev Nephrol. 2011;7:218–225. doi: 10.1038/nrneph.2011.15. [DOI] [PubMed] [Google Scholar]
- 91.Angus DC, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 92.Watanabe E, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scerbo MH, et al. Beyond blood culture and gram stain analysis: a review of molecular techniques for the early detection of bacteremia in surgical patients. Surg Infect (Larchmt) 2016;17:294–302. doi: 10.1089/sur.2015.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet. 2013;381:774–775. doi: 10.1016/S0140-6736(12)61815-7. A plea to abandon the SIRS criteria and return to the meaning of ‘sepsis’ to common, everyday language — a ‘bad infection’ with some degree of organ dysfunction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seymour CW, et al. Assessment of clinical criteria for sepsis: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:762–774. doi: 10.1001/jama.2016.0288. This review of 1.3 million electronic health records and validation with another 700,000 records identified Sepsis-related Organ Failure Assessment (SOFA) and quick SOFA (qSOFA) scores as valuable tools to predict in-hospital mortality. The qSOFA was most predictive outside the ICU, suggesting that it might be useful as a ‘prompt’ to consider sepsis in early warning systems. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shankar-Hari M, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:775–787. doi: 10.1001/jama.2016.0289. This systematic review of 166,479 patients with defined septic shock has revealed a working consensus definition of septic shock as hypotension requiring vasopressor support to maintain a mean arterial blood pressure of >65 mmHg and a plasma lactate level of >2 mmol per l with adequate resuscitation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casserly B, et al. Lactate measurements in sepsis-induced tissue hypoperfusion: results from the Surviving Sepsis Campaign database. Crit Care Med. 2015;43:567–573. doi: 10.1097/CCM.0000000000000742. [DOI] [PubMed] [Google Scholar]
- 98.Rowland T, Hilliard H, Barlow G. Procalcitonin: potential role in diagnosis and management of sepsis. Adv Clin Chem. 2015;68:71–86. doi: 10.1016/bs.acc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 99.Bloos F, Reinhart K. Rapid diagnosis of sepsis. Virulence. 2014;5:154–160. doi: 10.4161/viru.27393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Jong E, et al. Efficacy and safety of procalcitonin guidance in reducing the duration of antibiotic treatment in critically ill patients: a randomised, controlled, open-label trial. Lancet Infect Dis. 2016 doi: 10.1016/S1473-3099(16)00053-0. http://dx.doi.org/10.1016/S1473-3099(16)00053-0. This randomized controlled trial from 15 institutions in the Netherlands demonstrated that using procalcitonin concentrations to dictate antibiotic cessation reduced antibiotic duration by 2 days and reduced in-hospital mortality. [DOI] [PubMed]
- 101.Westwood M, et al. Procalcitonin testing to guide antibiotic therapy for the treatment of sepsis in intensive care settings and for suspected bacterial infection in emergency department settings: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2015;19:1–236. doi: 10.3310/hta19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schuetz P, et al. Procalcitonin to initiate or discontinue antibiotics in acute respiratory tract infections. Cochrane Database Syst Rev. 2012;9:CD007498. doi: 10.1002/14651858.CD007498.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O’Grady NP. Guidelines for the prevention of intravascular catheter-related infections. Clin Infect Dis. 2011;52:e162–e193. doi: 10.1093/cid/cir257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kumar A, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009;136:1237–1248. doi: 10.1378/chest.09-0087. [DOI] [PubMed] [Google Scholar]
- 105.Zahar JR, et al. Outcomes in severe sepsis and patients with septic shock: pathogen species and infection sites are not associated with mortality. Crit Care Med. 2011;39:1886–1895. doi: 10.1097/CCM.0b013e31821b827c. [DOI] [PubMed] [Google Scholar]
- 106.Kumar A, et al. Early combination antibiotic therapy yields improved survival compared with monotherapy in septic shock: a propensity-matched analysis. Crit Care Med. 2010;38:1773–1785. doi: 10.1097/CCM.0b013e3181eb3ccd. [DOI] [PubMed] [Google Scholar]
- 107.Micek ST, et al. Empiric combination antibiotic therapy is associated with improved outcome against sepsis due to Gram-negative bacteria: a retrospective analysis. Antimicrob Agents Chemother. 2010;54:1742–1748. doi: 10.1128/AAC.01365-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sprung CL, et al. An evaluation of systemic inflammatory response syndrome signs in the Sepsis Occurrence In Acutely Ill Patients (SOAP) study. Intensive Care Med. 2006;32:421–427. doi: 10.1007/s00134-005-0039-8. [DOI] [PubMed] [Google Scholar]
- 109.Vincent JL, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 110.Heenen S, Jacobs F, Vincent JL. Antibiotic strategies in severe nosocomial sepsis: why do we not de-escalate more often? Crit Care Med. 2012;40:1404–1409. doi: 10.1097/CCM.0b013e3182416ecf. [DOI] [PubMed] [Google Scholar]
- 111.Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2014;370:583. doi: 10.1056/NEJMc1314999. [DOI] [PubMed] [Google Scholar]
- 112.Dellinger RP, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39:165–228. doi: 10.1007/s00134-012-2769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rivers E, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. doi: 10.1056/NEJMoa010307. This seminal clinical trial demonstrates that EGDT bundles could significantly reduce mortality in patients with severe sepsis or septic shock. This study played a major supportive part in the use of standardized treatment bundles. [DOI] [PubMed] [Google Scholar]
- 114.ProCESS Investigators et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med. 2014;370:1683–1693. doi: 10.1056/NEJMoa1401602. This is an important and somewhat controversial study showing that EGDT did not improve 60-day survival in patients with severe sepsis and septic shock. These data were inconsistent with results by Rivers et al. (reference 113) found 13 years earlier; the explanation is thought to do with the better management of the control groups receiving standard care. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Weil MH, Shubin H. The “VIP” approach to the bedside management of shock. JAMA. 1969;207:337–340. [PubMed] [Google Scholar]
- 116.Reinhart K, et al. Consensus statement of the ESICM task force on colloid volume therapy in critically ill patients. Intensive Care Med. 2012;38:368–383. doi: 10.1007/s00134-012-2472-9. [DOI] [PubMed] [Google Scholar]
- 117.Myburgh JA. Fluid resuscitation in acute medicine: what is the current situation? J Intern Med. 2015;277:58–68. doi: 10.1111/joim.12326. [DOI] [PubMed] [Google Scholar]
- 118.De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med. 2012;40:725–730. doi: 10.1097/CCM.0b013e31823778ee. [DOI] [PubMed] [Google Scholar]
- 119.De Backer D, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779–789. doi: 10.1056/NEJMoa0907118. [DOI] [PubMed] [Google Scholar]
- 120.Jones AE, et al. Lactate clearance versus central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA. 2010;303:739–746. doi: 10.1001/jama.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.He X, et al. A selective V1A receptor agonist, selepressin, is superior to arginine vasopressin and to norepinephrine in ovine septic shock. Crit Care Med. 2016;44:23–31. doi: 10.1097/CCM.0000000000001380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20:195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 123.Alejandria MM, Lansang MA, Dans LF, Mantaring JB., 3rd Intravenous immunoglobulin for treating sepsis, severe sepsis and septic shock. Cochrane Database Syst Rev. 2013;9:CD001090. doi: 10.1002/14651858.CD001090.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fisher CJ, Jr, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334:1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 125.Qiu P, et al. The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death. Expert Opin Investig Drugs. 2011;20:1555–1564. doi: 10.1517/13543784.2011.623125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cross G, et al. The epidemiology of sepsis during rapid response team reviews in a teaching hospital. Anaesth Intensive Care. 2015;43:193–198. doi: 10.1177/0310057X1504300208. [DOI] [PubMed] [Google Scholar]
- 127.Lehman KD, Thiessen K. Sepsis guidelines: clinical practice implications. Nurse Pract. 2015;40:1–6. doi: 10.1097/01.NPR.0000465120.42654.86. [DOI] [PubMed] [Google Scholar]
- 128.Zubrow MT, et al. Improving care of the sepsis patient. Jt Comm J Qual Patient Saf. 2008;34:187–191. doi: 10.1016/s1553-7250(08)34022-7. [DOI] [PubMed] [Google Scholar]
- 129.Dowdy DW, et al. Quality of life in adult survivors of critical illness: a systematic review of the literature. Intensive Care Med. 2005;31:611–620. doi: 10.1007/s00134-005-2592-6. [DOI] [PubMed] [Google Scholar]
- 130.Prescott HC, Langa KM, Liu V, Escobar GJ, Iwashyna TJ. Increased 1-year healthcare use in survivors of severe sepsis. Am J Respir Crit Care Med. 2014;190:62–69. doi: 10.1164/rccm.201403-0471OC. Using an administrative database, the authors demonstrate that individuals in hospital who survived severe sepsis spent considerable time rehospitalized and had high out-of-hospital mortality rates in the year following sepsis survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nelson JE, et al. The symptom burden of chronic critical illness. Crit Care Med. 2004;32:1527–1534. doi: 10.1097/01.ccm.0000129485.08835.5a. [DOI] [PubMed] [Google Scholar]
- 132.Baldwin MR. Measuring and predicting long-term outcomes in older survivors of critical illness. Minerva Anestesiol. 2015;81:650–661. [PMC free article] [PubMed] [Google Scholar]
- 133.Kaarlola A, Tallgren M, Pettila V. Long-term survival, quality of life, and quality-adjusted life-years among critically ill elderly patients. Crit Care Med. 2006;34:2120–2126. doi: 10.1097/01.CCM.0000227656.31911.2E. [DOI] [PubMed] [Google Scholar]
- 134.Mehlhorn J, et al. Rehabilitation interventions for postintensive care syndrome: a systematic review. Crit Care Med. 2014;42:1263–1271. doi: 10.1097/CCM.0000000000000148. [DOI] [PubMed] [Google Scholar]
- 135.Battle CE, Davies G, Evans PA. Long term health-related quality of life in survivors of sepsis in South West Wales: an epidemiological study. PLoS ONE. 2014;9:e116304. doi: 10.1371/journal.pone.0116304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Semmler A, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84:62–69. doi: 10.1136/jnnp-2012-302883. [DOI] [PubMed] [Google Scholar]
- 137.Parker AM, et al. Posttraumatic stress disorder in critical illness survivors: a metaanalysis. Crit Care Med. 2015;43:1121–1129. doi: 10.1097/CCM.0000000000000882. [DOI] [PubMed] [Google Scholar]
- 138.Hofhuis JG, et al. The impact of critical illness on perceived health-related quality of life during ICU treatment, hospital stay, and after hospital discharge: a long-term follow-up study. Chest. 2008;133:377–385. doi: 10.1378/chest.07-1217. [DOI] [PubMed] [Google Scholar]
- 139.Poulsen JB, Moller K, Kehlet H, Perner A. Long-term physical outcome in patients with septic shock. Acta Anaesthesiol Scand. 2009;53:724–730. doi: 10.1111/j.1399-6576.2009.01921.x. [DOI] [PubMed] [Google Scholar]
- 140.Dellinger RP, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med. 2004;32:858–873. doi: 10.1097/01.ccm.0000117317.18092.e4. [DOI] [PubMed] [Google Scholar]
- 141.Bernard GR, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 142.Lai PS, Thompson BT. Why activated protein C was not successful in severe sepsis and septic shock: are we still tilting at windmills? Curr Infect Dis Rep. 2013;15:407–412. doi: 10.1007/s11908-013-0358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blum CA, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo- controlled trial. Lancet. 2015;385:1511–1518. doi: 10.1016/S0140-6736(14)62447-8. [DOI] [PubMed] [Google Scholar]
- 144.Torres A, et al. Effect of corticosteroids on treatment failure among hospitalized patients with severe community-acquired pneumonia and high inflammatory response: a randomized clinical trial. JAMA. 2015;313:677–686. doi: 10.1001/jama.2015.88. [DOI] [PubMed] [Google Scholar]
- 145.Kang JH, et al. An extracorporeal blood-cleansing device for sepsis therapy. Nat Med. 2014;20:1211–1216. doi: 10.1038/nm.3640. [DOI] [PubMed] [Google Scholar]
- 146.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20:224–233. doi: 10.1016/j.molmed.2014.01.002. This paper surmises that, after many unsuccessful attempts to decrease the inflammatory response in randomized controlled trials, the possible place of immunostimulating strategies in sepsis is a reasonable option. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Meisel C, et al. Granulocyte–macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180:640–648. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 148.US National Library of Medicine. Does GM-CSF restore neutrophil phagocytosis in critical illness? (GMCSF) ClinicalTrials.gov. 2012 http://clinicaltrials.gov/ct2/show/NCT01653665.
- 149.US National Library of Medicine. The effects of interferon-gamma on sepsis-induced immunoparalysis. ClinicalTrials.gov. 2012 http://clinicaltrials.gov/ct2/show/NCT01649921.
- 150.US National Library of Medicine. Safety, pharmacokinetics and pharmacodynamics of BMS-936559 in severe sepsis. ClinicalTrials.gov. 2015 https://clinicaltrials.gov/ct2/show/NCT02576457.
- 151.Wu J, et al. The efficacy of thymosin alpha 1 for severe sepsis (ETASS): a multicenter, single-blind, randomized and controlled trial. Crit Care. 2013;17:R8. doi: 10.1186/cc11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med. 2014;371:380–383. doi: 10.1056/NEJMcibr1404664. Sepsis and cancer have many features in common, including that both are heterogeneous conditions; the host response has an essential role in the eution of the diseases. [DOI] [PubMed] [Google Scholar]
- 153.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11:330–342. doi: 10.1038/nri2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.US National Library of Medicine. CYT107 after vaccine treatment (Provenge) in patients with metastatic hormone-resistant prostate cancer. ClinicalTrials.gov. 2013 https://clinicaltrials.gov/ct2/show/NCT01881867.
- 155.Levy Y, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin Infect Dis. 2012;55:291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Venet F, et al. IL-7 restores lymphocyte functions in septic patients. J Immunol. 2012;189:5073–5081. doi: 10.4049/jimmunol.1202062. [DOI] [PubMed] [Google Scholar]
- 157.Sweeney TE, Shidham A, Wong HR, Khatri P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci Transl Med. 2015;7:287ra71. doi: 10.1126/scitranslmed.aaa5993. Investigating a large number of publically available gene expression sets, the authors identify a pattern of gene expression that can be used to diagnose sepsis in a clear demonstration that ‘-omics’ is being used to diagnose and prognose sepsis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Maslove DM, Wong HR. Gene expression profiling in sepsis: timing, tissue, and translational considerations. Trends Mol Med. 2014;20:204–213. doi: 10.1016/j.molmed.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sutherland A, et al. Development and validation of a novel molecular biomarker diagnostic test for the early detection of sepsis. Crit Care. 2011;15:R149. doi: 10.1186/cc10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Davenport EE, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4:259–271. doi: 10.1016/S2213-2600(16)00046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chung LP, Waterer GW. Genetic predisposition to respiratory infection and sepsis. Crit Rev Clin Lab Sci. 2011;48:250–268. doi: 10.3109/10408363.2011.641517. [DOI] [PubMed] [Google Scholar]
- 162.Bone RC, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 163.Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 164.Williams MD, et al. Hospitalized cancer patients with severe sepsis: analysis of incidence, mortality, and associated costs of care. Crit Care. 2004;8:R291–R298. doi: 10.1186/cc2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 166.US National Library of Medicine. Trebananib in treating patients with persistent or recurrent endometrial cancer. ClinicalTrials.gov. 2010 https://clinicaltrials.gov/ct2/show/NCT01210222.
- 167.US National Library of Medicine. ACT-128800 in relapsing-remitting multiple sclerosis. ClinicalTrials.gov. 2009 https://clinicaltrials.gov/ct2/show/NCT01006265.
- 168.US National Library of Medicine. ACT-128800 in psoriasis. ClinicalTrials.gov. 2009 https://clinicaltrials.gov/ct2/show/NCT00852670.
- 169.US National Library of Medicine. Efficacy of FX06 in the prevention of myocardial reperfusion injury. ClinicalTrials.gov. 2006 https://clinicaltrials.gov/ct2/show/NCT00326976.
- 170.US National Library of Medicine. Safety study of PZ-128 in subjects with multiple coronary artery disease risk factors. ClinicalTrials.gov. 2013 https://clinicaltrials.gov/ct2/show/NCT01806077.
- 171.Thomas G, et al. Statin therapy in critically-ill patients with severe sepsis: a review and meta-analysis of randomized clinical trials. Minerva Anestesiol. 2015;81:921–930. [PubMed] [Google Scholar]
- 172.US National Library of Medicine. Selepressin evaluation programme for sepsis-induced shock — adaptive clinical trial (SEPSIS-ACT) ClinicalTrials.gov. 2015 https://clinicaltrials.gov/ct2/show/NCT02508649.
- 173.Wang C, et al. Low-dose hydrocortisone therapy attenuates septic shock in adult patients but does not reduce 28-day mortality: a meta-analysis of randomized controlled trials. Anesth Analg. 2014;118:346–357. doi: 10.1213/ANE.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 174.Annane D, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009;301:2362–2375. doi: 10.1001/jama.2009.815. [DOI] [PubMed] [Google Scholar]
- 175.Russell JA, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358:877–887. doi: 10.1056/NEJMoa067373. [DOI] [PubMed] [Google Scholar]
- 176.Angus DC, et al. E5 murine monoclonal antiendotoxin antibody in Gram-negative sepsis: a randomized controlled trial. E5 Study Investigators. JAMA. 2000;283:1723–1730. doi: 10.1001/jama.283.13.1723. [DOI] [PubMed] [Google Scholar]
- 177.McCloskey RV, Straube RC, Sanders C, Smith SM, Smith CR. Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial CHESS Trial Study Group. Ann Intern Med. 1994;121:1–5. doi: 10.7326/0003-4819-121-1-199407010-00001. [DOI] [PubMed] [Google Scholar]
- 178.Dellinger RP, et al. Efficacy and safety of a phospholipid emulsion (GR270773) in Gram-negative severe sepsis: results of a phase II multicenter, randomized, placebo-controlled, dose-finding clinical trial. Crit Care Med. 2009;37:2929–2938. doi: 10.1097/CCM.0b013e3181b0266c. [DOI] [PubMed] [Google Scholar]
- 179.Levin M, et al. Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial. rBPI21 Meningococcal Sepsis Study Group. Lancet. 2000;356:961–967. doi: 10.1016/s0140-6736(00)02712-4. [DOI] [PubMed] [Google Scholar]
- 180.Opal SM, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 181.Rice TW, et al. A randomized, double-blind, placebo- controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 182.Axtelle T, Pribble J. An overview of clinical studies in healthy subjects and patients with severe sepsis with IC14, a CD14-specific chimeric monoclonal antibody. J Endotoxin Res. 2003;9:385–389. doi: 10.1179/096805103225003321. [DOI] [PubMed] [Google Scholar]
- 183.Abraham E, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998;351:929–933. [PubMed] [Google Scholar]
- 184.Cohen J, Carlet J. INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-α in patients with sepsis. International Sepsis Trial Study Group. Crit Care Med. 1996;24:1431–1440. doi: 10.1097/00003246-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 185.Abraham E, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med. 2001;29:503–510. doi: 10.1097/00003246-200103000-00006. [DOI] [PubMed] [Google Scholar]
- 186.Opal SM, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115–1124. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 187.Poeze M, Froon AH, Ramsay G, Buurman WA, Greve JW. Decreased organ failure in patients with severe SIRS and septic shock treated with the platelet-activating factor antagonist TCV-309: a prospective, multicenter, double-blind, randomized phase II trial. TCV-309 Septic Shock Study Group. Shock. 2000;14:421–428. doi: 10.1097/00024382-200014040-00001. [DOI] [PubMed] [Google Scholar]
- 188.Suputtamongkol Y, et al. A double-blind placebo- controlled study of an infusion of lexipafant (platelet-activating factor receptor antagonist) in patients with severe sepsis. Antimicrob Agents Chemother. 2000;44:693–696. doi: 10.1128/aac.44.3.693-696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bernard GR, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen Sepsis Study Group. N Engl J Med. 1997;336:912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 190.Zeiher BG, et al. LY315920NA/S-5920, a selective inhibitor of group IIA secretory phospholipase A2, fails to improve clinical outcome for patients with severe sepsis. Crit Care Med. 2005;33:1741–1748. doi: 10.1097/01.ccm.0000171540.54520.69. [DOI] [PubMed] [Google Scholar]
- 191.Bakker J, et al. Administration of the nitric oxide synthase inhibitor NG-methyl-L-arginine hydrochloride (546C88) by intravenous infusion for up to 72 hours can promote the resolution of shock in patients with severe sepsis: results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144–002) Crit Care Med. 2004;32:1–12. doi: 10.1097/01.CCM.0000105118.66983.19. [DOI] [PubMed] [Google Scholar]
- 192.Preiser JC, et al. Methylene blue administration in septic shock: a clinical trial. Crit Care Med. 1995;23:259–264. doi: 10.1097/00003246-199502000-00010. [DOI] [PubMed] [Google Scholar]
- 193.Annane D, et al. Recombinant human activated protein C for adults with septic shock: a randomized controlled trial. Am J Respir Crit Care Med. 2013;187:1091–1097. doi: 10.1164/rccm.201211-2020OC. [DOI] [PubMed] [Google Scholar]
- 194.Abraham E, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 195.Warren BL, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 196.Morris PE, et al. A phase I study evaluating the pharmacokinetics, safety and tolerability of an antibody-based tissue factor antagonist in subjects with acute lung injury or acute respiratory distress syndrome. BMC Pulm Med. 2012;12:5. doi: 10.1186/1471-2466-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zarychanski R, et al. The efficacy and safety of heparin in patients with sepsis: a systematic review and metaanalysis. Crit Care Med. 2015;43:511–518. doi: 10.1097/CCM.0000000000000763. [DOI] [PubMed] [Google Scholar]
- 198.Vincent JL, et al. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41:2069–2079. doi: 10.1097/CCM.0b013e31828e9b03. [DOI] [PubMed] [Google Scholar]
- 199.Kong Z, Wang F, Ji S, Deng X, Xia Z. Selenium supplementation for sepsis: a meta-analysis of randomized controlled trials. Am J Emerg Med. 2013;31:1170–1175. doi: 10.1016/j.ajem.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 200.Fein AM, et al. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo- controlled trial CP-0127 SIRS and Sepsis Study Group. JAMA. 1997;277:482–487. [PubMed] [Google Scholar]
- 201.Spapen HD, Diltoer MW, Nguyen DN, Hendrickx I, Huyghens LP. Effects of N-acetylcysteine on microalbuminuria and organ failure in acute severe sepsis: results of a pilot study. Chest. 2005;127:1413–1419. doi: 10.1378/chest.127.4.1413. [DOI] [PubMed] [Google Scholar]
- 202.Szakmany T, Hauser B, Radermacher P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst Rev. 2012;9:CD006616. doi: 10.1002/14651858.CD006616.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Reinhart K, et al. Open randomized phase II trial of an extracorporeal endotoxin adsorber in suspected Gram-negative sepsis. Crit Care Med. 2004;32:1662–1668. doi: 10.1097/01.ccm.0000132902.54925.b5. [DOI] [PubMed] [Google Scholar]
- 204.Flohe S, et al. Effect of granulocyte–macrophage colony-stimulating factor on the immune response of circulating monocytes after severe trauma. Crit Care Med. 2003;31:2462–2469. doi: 10.1097/01.CCM.0000089640.17523.57. [DOI] [PubMed] [Google Scholar]
- 205.Leentjens J, et al. Reversal of immunoparalysis in humans in vivo: a double-blind, placebo-controlled, randomized pilot study. Am J Respir Crit Care Med. 2012;186:838–845. doi: 10.1164/rccm.201204-0645OC. [DOI] [PubMed] [Google Scholar]
- 206.Woods DR, Mason DD. Six areas lead national early immunization drive. Public Health Rep. 1992;107:252–256. [PMC free article] [PubMed] [Google Scholar]
- 207.Root RK, et al. Multicenter, double-blind, placebo- controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit Care Med. 2003;31:367–373. doi: 10.1097/01.CCM.0000048629.32625.5D. [DOI] [PubMed] [Google Scholar]
- 208.Nelson S, et al. A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. CAP Study Group. J Infect Dis. 1998;178:1075–1080. doi: 10.1086/515694. [DOI] [PubMed] [Google Scholar]
- 209.Dries DJ. Interferon gamma in trauma-related infections. Intensive Care Med. 1996;22:S462–S467. doi: 10.1007/BF01743725. [DOI] [PubMed] [Google Scholar]
- 210.Dries DJ, et al. Effect of interferon gamma on infection-related death in patients with severe injuries. A randomized, double-blind, placebo-controlled trial. Arch Surg. 1994;129:1031–1041. doi: 10.1001/archsurg.1994.01420340045008. discussion 1042. [DOI] [PubMed] [Google Scholar]
- 211.Kasten KR, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through γδ T-cell IL-17 production in a murine model of sepsis. Infect Immun. 2010;78:4714–4722. doi: 10.1128/IAI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Unsinger J, et al. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis. 2012;206:606–616. doi: 10.1093/infdis/jis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Inoue S, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184:1401–1409. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pelletier M, Ratthe C, Girard D. Mechanisms involved in interleukin-15-induced suppression of human neutrophil apoptosis: role of the anti-apoptotic Mcl-1 protein and several kinases including Janus kinase-2, 38 mitogen-activated protein kinase and extracellular signal-regulated kinases-1/2. FEBS Lett. 2002;532:164–170. doi: 10.1016/s0014-5793(02)03668-2. [DOI] [PubMed] [Google Scholar]
- 215.Saikh KU, Kissner TL, Nystrom S, Ruthel G, Ulrich RG. Interleukin-15 increases vaccine efficacy through a mechanism linked to dendritic cell maturation and enhanced antibody titers. Clin Vaccine Immunol. 2008;15:131–137. doi: 10.1128/CVI.00320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chen H, He MY, Li YM. Treatment of patients with severe sepsis using ulinastatin and thymosin α1: a prospective, randomized, controlled pilot study. Chin Med J (Engl) 2009;122:883–888. [PubMed] [Google Scholar]
- 217.Chang KC, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care. 2013;17:R85. doi: 10.1186/cc12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Huang X, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA. 2009;106:6303–6308. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.West EE, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest. 2013;123:2604–2615. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Inoue S, et al. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock. 2011;36:38–44. doi: 10.1097/SHK.0b013e3182168cce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yang X, et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J Immunol. 2013;190:2068–2079. doi: 10.4049/jimmunol.1202661. [DOI] [PubMed] [Google Scholar]
- 222.Zhao Z, et al. Blockade of the T cell immunoglobulin and mucin domain protein 3 pathway exacerbates sepsis-induced immune deviation and immunosuppression. Clin Exp Immunol. 2014;178:279–291. doi: 10.1111/cei.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Workman CJ, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. 2009;182:1885–1891. doi: 10.4049/jimmunol.0800185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Durham NM, et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE. 2014;9:e109080. doi: 10.1371/journal.pone.0109080. [DOI] [PMC free article] [PubMed] [Google Scholar]