

Abstract
Cardiac arrhythmias can follow disruption of the normal cellular electrophysiological processes underlying excitable activity and their tissue propagation as coherent wavefronts from the primary sinoatrial node pacemaker, through the atria, conducting structures and ventricular myocardium. These physiological events are driven by interacting, voltage-dependent, processes of activation, inactivation, and recovery in the ion channels present in cardiomyocyte membranes. Generation and conduction of these events are further modulated by intracellular Ca2+ homeostasis, and metabolic and structural change. This review describes experimental studies on murine models for known clinical arrhythmic conditions in which these mechanisms were modified by genetic, physiological, or pharmacological manipulation. These exemplars yielded molecular, physiological, and structural phenotypes often directly translatable to their corresponding clinical conditions, which could be investigated at the molecular, cellular, tissue, organ, and whole animal levels. Arrhythmogenesis could be explored during normal pacing activity, regular stimulation, following imposed extra-stimuli, or during progressively incremented steady pacing frequencies. Arrhythmic substrate was identified with temporal and spatial functional heterogeneities predisposing to reentrant excitation phenomena. These could arise from abnormalities in cardiac pacing function, tissue electrical connectivity, and cellular excitation and recovery. Triggering events during or following recovery from action potential excitation could thereby lead to sustained arrhythmia. These surface membrane processes were modified by alterations in cellular Ca2+ homeostasis and energetics, as well as cellular and tissue structural change. Study of murine systems thus offers major insights into both our understanding of normal cardiac activity and its propagation, and their relationship to mechanisms generating clinical arrhythmias.
I. INTRODUCTION
A. Scope of Review
Cardiac arrhythmias result from disruption of the orderly physiological sequence of electrical excitation processes that initiates coordinated and effective cardiac contraction. Of the wide clinical variety of arrhythmic variants, ventricular arrhythmias, particularly ventricular fibrillation (VF), often preceded by ventricular tachycardia (VT), potentially lead to sudden cardiac death (SCD), defined as unexpected death from cardiac causes occurring <1 h after onset of symptoms (971, 1152). This major worldwide source of morbidity and mortality causes >300,000 and ∼70,000 deaths/year in the United States (USA) (535) and United Kingdom (UK) (215), respectively. Cardiac causes likely account for 56.4% of nontraumatic, sudden death in autopsies of patients aged 5–35 years. Among these, arrhythmic causes likely account for ∼30% of cases. Although most of the latter cases result from ischemic heart disease (87), autopsy fails to reveal a cause in ∼4% of SCD cases and 14% of all resuscitation attempts performed on patients aged <40 years (206, 738–740, 1149). Furthermore, such deaths often occur in the absence of structural abnormalities. This suggests the possibility of underlying channelopathy (116, 1122). Of deaths in infants <1 yr, 60–80% are autopsy-negative. They are accordingly defined as sudden infant death syndrome (SIDS) (47), and 10–20% of these cases may result from channelopathy (575). Finally, arrhythmogenesis as a possible adverse effect of pharmacotherapeutic agents constitutes an important clinical problem with significant implications for pharmaceutical drug discovery (564).
Atrial arrhythmias are similarly becoming increasingly clinically and demographically important. Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, with an overall prevalence of ∼1–2% of the general population (30, 683, 1093). AF is often associated with advancing age. It affects 4.7 and 9% of individuals of age >65 years and between 80 and 89 years, respectively (285, 1212). It predisposes to further, major, cardiac and cerebrovascular morbidity and mortality (1093). Thus it increases risks of stroke fivefold (1268).
Sinus node disorder (SND) causes sinus bradycardia, sinus pause/arrest, chronotropic incompetence, and sinoatrial node (SAN) exit block (271). Its incidence increases exponentially with age to 1 in ∼600 cardiac patients aged >65 years. It is responsible for ∼50% of the million permanent pacemaker implants per year worldwide often in otherwise healthy individuals (272, 731).
Cardiac arrhythmogenesis poses significant clinical challenges in terms of both risk stratification and management (986). The latter are limited by our current inadequate understanding of the physiological mechanisms underlying initiation, maintenance, or propagation of cardiac arrhythmias, whether in the atria, ventricles, or the conducting tissues within or between them. Much valuable data have derived from human studies. However, much of this is inevitably observational. Physiological animal models of arrhythmic disease, whether involving pharmacological interventions or targeted genetic changes, permit more analytical studies of mechanisms and their consequences. Of these systems, transgenic mouse models are a relatively recent addition to other animal, rabbit, and canine systems. They offer a means of genetic and physiological manipulation that can be effectively directed at particular molecular targets strategic to cardiac electrophysiological function.
This review describes studies using some of these approaches, relating arrhythmic phenomena to cardiac electrophysiological properties. The latter in turn bear upon the generation of atrial or ventricular action potentials (APs), any abnormal, triggered activity accompanying such events, and associated metabolic and structural changes. The analysis is made in the light of the features of different exemplars modifying the activity of specific ion channels, cellular properties, or tissue or chamber structure. This involves first summarizing the roles of the major ion channels that underlie cardiac electrophysiological excitation, and the consequent excitation-contraction coupling processes involving the release and reuptake of sarcoplasmic reticular (SR) store Ca2+. Alterations in these processes are next related to the properties of genetic murine exemplars of ion channel dysfunction. These include models for altered gap junction function compromising the spread of excitation, losses, and gains of function in the Na+, K+, and Ca2+ channels, and their subunits and associated proteins, affecting cell excitability, and ryanodine receptor (RyR2) modifications affecting cellular Ca2+ homeostasis. Finally, further upstream, metabolic, energetic, and structural changes are considered in relation to human arrhythmic conditions.
B. Normal Physiology of Cardiac Excitation
1. Ion current contributions to cardiac action potentials
Effective cardiac function depends on repetitive cycles of AP excitation followed by recovery and the propagation of these events through the myocardial or conducting tissue as coherent electrical waves. This conduction takes place successively through the SAN, atria, atrioventricular bundles, Purkinje conducting tissue, and ventricular endocardial and epicardial myocardium. Repetitive atrial and ventricular excitation cycles normally depend on SAN automaticity driven by pacemaker cells (see sect. IIIA). The typical human ventricular AP waveform begins with a rapid (∼400 V/s) initial, phase 0, depolarization. This is driven by a rapidly increasing voltage-gated Na+ current (INa) (∼400 μA/μF). This drives the myocardial membrane potential from its normal negative (approximately −90 mV) resting potential to a positive (+40 to +60 mV) voltage close to the Na+ Nernst potential (Figure 1A and Table 1). This is followed by a, phase 1, initial rapid repolarization from this positive value that results from both inactivation of INa and the activation of fast and slow transient outward (Ito), K+, Ito,f, and Ito,s currents (Figure 1, B and C) (reviews in Refs. 71, 827, 863, 1020, 1255).
Figure 1.

Basic features of cardiac electrophysiological excitation. Inward (A) and outward (B) ionic current contributions attributable to surface membrane ion channels to human (C) and mouse (D) ventricular action potential (AP) waveforms.
Table 1.
Human and murine ventricular and atrial expression of cardiac ionic currents mediating excitable activity
| Human |
Mouse |
||||||
|---|---|---|---|---|---|---|---|
| Current/Symbol | Protein | Gene | Ventricle | Atrium | Ventricle | Atrium | Action Potential Contribution |
| Voltage-gated inward currents | |||||||
| Fast Na+ current, INa | Nav1.5 | SCN5A | +++ | +++ | +++ | +++ | [0] |
| L-type Ca2+ current, ICaL (dihydropyridine receptor: DHPR) | Cav1.2 | CACNA1C | +++ | ++ | ++ | ++ | [2] |
| Voltage-gated outward currents | |||||||
| Fast transient outward K+ current, Ito,f | Kv4.2 | KCND2 | ++ | +++ | ++ | +++ | [1] |
| Kv4.3 | KCND3 | ||||||
| Slow transient outward K+ current, Ito,s, | Kv1.4 | KCNA4 | ++ | +++ | ++ | +++ | [1] |
| Delayed rectifier K+ current, IKr | Kv11.1 | KCNH2 (HERG) | +++ | + | + | + | [3] |
| Delayed rectifier K+ current, IKs | Kv7.1 | KCNQ1 | +++ | + | + | + | [3] |
| 4-Aminopyridine-sensitive, rapidly activating, slowly inactivating K+ current, IKslow1 | Kv1.5 | KCNA5 | − | − | ++ | + | |
| 4-Aminopyridine-insensitive, rapidly activating, slowly inactivating K+ current, IKslow2 | Kv2.1 | KCNB1 | − | − | ++ | + | |
| Sustained 4-aminopyridine-sensitive delayed rectifier K+ current, Iss | Kv1.5 | KCNA5 | − | − | ++ | + | |
| Atrial-specific 4-aminopyridine-sensitive ultrarapid delayed rectifier K+ current, IKur | Kv1.5 | KCNA5 | − | ++ | − | ++ | |
| Inward rectifiers | |||||||
| Inwardly rectifying current, IK1 | Kir2.1 | KCNJ2 | +++ | ++ | +++ | ++ | [3], [4] |
| Kir2.2 | KCNJ12 | ||||||
| Kir2.3 | KCNJ4 | ||||||
| Acetylcholine-activated, K+ current, IKACh | Kir3.1 | KCNJ3 | − | +++ | − | +++ | |
| Kir3.4, | KCNJ5 | ||||||
| ATP-sensitive potassium channel, IKATP | Kir6.2 | KCNJ11 | ++ | ++ | ++ | ++ | |
| Leak currents | |||||||
| Two-pore domain K+ leak current, IK2p | K2p3.1 | KCNK3 | +++ | ++ | ++ | +++ | |
| Ca2+-activated K+ currents, IKCa | KCa2.x- | KCNNx | − | +++ | − | +++ | |
| Exchange currents | |||||||
| Transient, inward, Na+-Ca2+ exchange current, Iti | NCX | SLC8A1 | ++ | ++ | ++ | ++ | |
Action potential contributions (human ventricle): [0], phase 0 rapid depolarization; [1], phase 1 initial rapid repolarization; [2], phase 2 plateau; [3], phase 3 repolarization; [4], phase 4 electrical diastole.
This partial recovery is then followed by a phase 2, plateau, phase brought about by the activation of Ca2+ current (ICaL) through voltage-gated L-type Ca2+ channels often termed dihydropyridine receptors (DHPRs), reflecting their pharmacological sensitivities (107, 131, 556, 1319).
The action potential is then terminated by a phase 3 repolarization. This returns the membrane to its resting potential. This is driven by outward currents mediated by a species-dependent variety of K+ channels. In human ventricles, these include the delayed rectifier IKr (1169) and IKs (1097), inwardly rectifying IK1 (693), and two-pore domain K+ leak currents (IK2p) (1020).
This culminates in full repolarization to electrical diastole (phase 4), in which the inward rectifying current IK1 plays a major part in maintaining a fully polarized (approximately −90 mV) resting potential. With repolarization, Nav1.5 channels recover their capacity for reexcitation. This recovery takes place over both absolute and relative relative (effective) refractory periods (ERPs). Following this, Nav1.5 channels become capable of beginning the next excitation cycle.
Atrial APs begin from more depolarized resting potentials. The latter mainly reflects their smaller IK1. They show triangular waveforms with a more prominent phase 1 recovery reflecting a larger Ito. Atrial myocytes also specifically express ultrarapid delayed rectifier K+ (IKur) (1256), acetylcholine-activated K+ currents (IKACh), and Ca2+-activated K+ currents (IKCa) (1020, 1257). They show a less prominent phase 2 plateau phases than observed in ventricular APs. This is the consequence of smaller IKr, IKs, and IK1 currents but a more prolonged phase 3 repolarization (822).
Finally, ATP-sensitive K+ current (IKATP) occurs throughout the heart but generally accounts for relatively little current due to its inhibition by intracellular ATP (320, 324). However, IKATP may be activated under conditions of energetic stress (324, 419, 486, 1024).
2. Excitation-contraction coupling
Excitation-contraction coupling in ventricular myocytes is initiated by the surface membrane depolarization described above and the transmission of this electrical change into the transverse tubules. This results in an opening of voltage-gated L-type Ca2+ channels within the membranes of the transverse tubules, which then mediate the inward Ca2+ currents (ICaL) responsible for the phase 2 plateau phase of the AP. The accompanying influx of extracellular Ca2+ produces a local elevation of cytosolic Ca2+ concentration ([Ca2+]i) in regions of the sarcolemmal-SR junctions (107). This triggers an opening of cardiac SR RyR2 Ca2+ channels by a process of Ca2+-induced Ca2+ release (CICR) (298). The resulting release of intracellularly stored SR Ca2+ produces the elevation of [Ca2+]i that drives the Ca2+-troponin binding which triggers mechanical activation. The cytosolic Ca2+-mediated regulation of cardiac, RyR2-Ca2+ release channel activity is facilitated by SR luminal Ca2+ (395, 622, 1069, 1285) and cytosolic ATP. It is inhibited by cytosolic Mg2+ (see sect. VIIA) (1313). RyR2 is also sensitive to thiol-oxidation and reactive oxygen species (ROS) which may disrupt its interdomain stability (see sect. VIIIA) (785). The CICR process in cardiac muscle contrasts with the control of RyR1 opening by the more rapid direct allosteric control by voltage-dependent configurational changes in the DHPR in skeletal muscle. It explains the contrasting dependence and relative independence of these respective excitation-contraction coupling processes upon extracellular [Ca2+] (458, 462). It also contrasts with RyR2 function in inexcitable osteoclasts in which the RyR2 assumes a surface rather than an endoplasmic membrane site (460, 1314–1316).
The smaller atrial myocytes possess less prominent transverse tubular networks, particularly in hearts of small mammals such as the mouse. Their SR membranes are differentiated into 1) corbular regions that form junctional elements close to the cell periphery. These regions are flanked by clusters of surface membrane L-type Ca2+ channels and SR membrane RyR2-Ca2+ release channels. 2) Noncorbular SR in the cell interior also contains membrane regions that express RyR2 but do not show this proximity to the cell surface. Depolarization of the cell surface membrane triggers entry of extracellular Ca2+ through the activation of ICaL, resulting in a local increase in [Ca2+]i. This induces RyR2-mediated Ca2+-induced Ca2+ release at corbular SR. The resulting local elevation of [Ca2+]i then initiates Ca2+-induced Ca2+ release and its propagation through adjacent, deeper regions of noncorbular SR. This results in an inward, centripetal, propagated Ca2+-induced Ca2+ release wave by noncorbular cytoplasmic SR within the cell interior (128, 419, 521, 710, 1335, 1338).
3. Recovery from excitation
Contractile relaxation follows reduction in the elevated [Ca2+]i back to baseline levels. This permits Ca2+-troponin dissociation. Cytosolic [Ca2+] is reduced by Ca2+ transport activity by SR Ca2+-ATPase (SERCA2), sarcolemmal Na+/Ca2+ exchange through the Na+/Ca2+ exchanger (NCX), and slow systems represented by sarcolemmal Ca2+-ATPase (PMCA) and mitochondrial Ca2+ uniport. The relative contributions of these different Ca2+ transport mechanisms to the restoration of resting levels of [Ca2+]i varies with species (110). In rabbit ventricular myocytes, SERCA2a activity eliminates 70%, NCX removes 28%, and the slow systems ∼1% of released Ca2+ with similar levels for ferret, dog, cat, guinea pig, and human ventricle (450). Corresponding contributions in rat and mouse ventricle are 92, 7, and 1%, respectively (78, 140, 653).
Of these processes, the NCX exerts potentially important effects on membrane potential through its electrogenic effects. These can contribute to arrhythmic tendency. Each cycle of NCX activity translocates 1Ca2+ from the intracellular to the extracellular space in return for a transfer of 3Na+ in the opposite direction. This results in a net current (INCX) whose reversal potential depends on both the Na+ and Ca2+ Nernst potentials, ENa and ECa, giving ENCX = 3ENa − 2ECa. ENCX therefore falls within the range of voltages traversed by normal physiological activity. Consequently, whether INCX takes an inward, depolarizing or outward, hyperpolarizing direction varies with the changes in both [Ca2+]i and membrane potential that take place through the cardiac cycle. Thus INCX takes an inward direction and exerts a depolarizing effect on membrane potential under conditions when [Ca2+]i is elevated and thereby drives Ca2+ efflux. Conversely, INCX takes an outward direction exerting a hyperpolarizing effect on membrane potential when [Ca2+]i is relatively low, thereby driving Ca2+ influx. The pattern of INCX activity also varies with species-related differences in AP waveform. The long plateau phase in rabbit ventricular APs results in a sustained INCX-mediated Ca2+ influx through the AP plateau phase (165). Following repolarization, a large outward electrochemical gradient then drives Ca2+ efflux. In contrast, the short AP in rat ventricles results in a rapid initial INCX-mediated Ca2+ influx, but this is followed by a more marked Ca2+ efflux. This results in a more rapid removal of the added Ca2+ load arising from electrical excitation and a lower subsequent diastolic NCX activity (1042). Either of these effects potentially modify the action potential duration (APD) (Figure 1,A and B) (265, 1025).
Of the overall energetic cost of contractile and excitable cardiac activity, ∼60–70% of this cellular ATP is consumed by cardiac muscle contraction. The remainder maintains Ca2+ homeostasis and the transmembrane ion gradients.
C. Propagation of Excitation
1. Electrical current flow between cardiac cells
The subsequent propagation of cellular-level events through conducting atrial or ventricular tissue first involves a local spread of electrotonic excitation currents. These are driven typically in the more rapidly conducting cardiac tissues by INa (137, 496, 1043). Cable theory classically describes this current flow along a constant intracellular, axial resistance ra from one depolarized myocyte to its quiescent neighbor (492, 577). In cardiac tissue, ra reflects the electrical resistances formed, respectively, by the cytosol and intercellular gap junctions connecting successive adjacent cells. An axial current, ia, having traversed the resistance ra, then discharges the membrane capacitance, cm, of neighboring quiescent cells. Where this resulting depolarization exceeds the activation threshold of its voltage-sensitive Na+ channels, this results in a regenerative production of further transmembrane depolarizing currents in that cell. This continues the AP propagation process. Thus, in cardiac cells, resistance to conduction through intercellular gap junction channels, the membrane capacitance, and the magnitude of INa are critical to AP propagation (958).
2. Determinants of conduction velocity
The velocity θ of AP conduction along a simple one-dimensional uniform cylindrical fiber of excitable tissue can be approximated by a nonlinear cable equation (550, 902). This has been applied to biological membrane with circuit elements that each incorporate a capacitance of unit fiber length, cm (typically expressed in units of μF/cm) in parallel with a linear membrane resistance of unit fiber length rm (kΩ·cm). The membrane additionally contains further, nonlinear, time- and voltage-dependent, membrane conductances representing the properties of its contained individual ion channels. These together generate the time (t)-dependent total membrane ionic current ii (A/cm) in unit fiber length, x (cm). Successive circuit elements are connected by terms arising from cytoplasmic resistances and the gap junction resistances between cells (see sect. IV) (262, 1181). Any one of these factors could be modified by changes including tissue fibrosis or inflammatory processes (see sects. V, E and F, and IXA).
The membrane potential V at any given membrane site then depends on the charging of its unit length by currents traversing the membrane, ii, as well as the axial current flow, ia, coming from neighboring regions along the length x of the membrane area in question through the equation
| (1) |
This equation reduces at constant conduction velocity, θ = dx/dt to
| (2) |
This simplified interpretation identifies ra, cm, and ii as key determinants of θ, although interdependences between some of the terms involved preclude analytic solution (471). Explicit prediction instead requires numerical solution of a stiff equation involving iterative estimates of θ (492). This is particularly given potential further contributions from other membrane components including transverse tubular membrane resistances and capacitances, tubular luminal geometry and its resistance, and nonlinear capacitances (9, 457, 1047). A full cable analysis of AP propagation would also be required to incorporate the three-dimensional nature of cardiac geometry.
Nevertheless, a number of useful, simple relationships between θ and its determinants arise from computational studies of one-dimensional electric current flow in skeletal muscle fibers whose APs upstrokes are similarly dominated by fast INa (330, 567, 880). These confirm the importance of iNa during the AP upstroke, and that the maximum Na+ current
| (3) |
PNa(max) is the maximum permeability produced by the fast Na+ channels. It is accordingly dependent on Na+ channel density. Of the key determinants of θ, ra does not influence the AP waveform. It thus does not influence either its rate of upstroke voltage change, as given by the first derivative (dV/dt), or its second derivative (d2V/dt2), though the cable Equation 2 predicts that θ2 α 1/ra. In contrast, increased cm does alter AP waveform. It reduces both dV/dt and d2V/dt2 as well as reducing θ, giving the simple approximations
| (4) |
| (5) |
| (6) |
The subscript max designates the maximum value of the parameter in question. Finally, it is difficult to obtain analytic relationships between iNa(max) and θ. Nevertheless, it is possible to demonstrate the straightforward empirical relationship
| (7) |
and the following effects of iNa(max) upon AP waveform
| (8) |
| (9) |
This cable analysis, confined to a simple cylindrical, geometrically one-dimensional, structure, can be generalized to a continuous electrically coupled myocyte network. This provides cable equations extended from one to three dimensions analyzing the conduction velocity resulting from a matching of current and load (577, 599). Such an approach has been used to characterize the passive cable properties of cardiac muscle including its relationships between dV/dt and macroscopic (>1 mm) propagation and altered cell to cell coupling (576, 1246).
3. Repolarization gradients and action potential wavelength
Finally, normal atrial and ventricular myocardium shows a highly ordered sequence of AP repolarization and return of the membrane to resting conditions. In the ventricular myocardium, this typically proceeds transmurally from epicardium to endocardium and from apex to base. These features reflect regional differences in K+ channel density and kinetics. This results in the normal spatial repolarization gradients that may normally protect the orderly electrical and mechanical activation sequence. This thereby ensures correctly timed and coordinated mechanical activation and relaxation of the chamber concerned. In contrast to the ventricles, the thinner walled atria do not show a marked transmural differentiation into epicardial and endocardial tissues. The geometrical distribution of excitable events then occurs only within the plane of the atrial wall.
Together, the velocity θ of AP propagation and the recovery parameters of APD or the effective refractory period (ERP) define the AP wavelength (λ = θ·APD or θ·ERP). The APD reflects the period during which the membrane deviates from its normal resting value. It may closely correlate with the ERP in normally functioning canine and human hearts (243, 329, 624). The latter provides an indication of the time during which there is reduced likelihood of ectopic or reentrant action potential activation in the membrane behind the propagating excitation wavefront. However, ERPs can be selectively affected by factors that need not similarly affect APD. These include alterations in the maximum amplitude and activation or inactivation kinetics of INa and IK, the amplitudes and durations of applied stimuli and myocyte injury and ischemia (975). Subsequent sections will consider VERP-ERP differences and their significance in proarrhythmic situations including Brugada syndrome (sect. VC5) (50, 734) and hypokalemia (sect. VE)(978).
However, measurement of ERP, typically from the shortest S1S2 interval following the last (S1) pacing stimulus at which a subsequent extrasystolic S2 stimulus elicits an AP, itself poses problems under particular experimental circumstances. Thus 1) some protocols are not amenable to such direct determinations of ERPs, yet 2) ERPs themselves vary with pacing protocol. 3) The observed ERPs critically depend on the relationship between stimulus and recording sites. A detectable AP requires its successful propagation through the entire tissue pathway from stimulating to recording electrode. The resulting ERP consequently actually reflects recovery from refractoriness over the entire line of tissue between stimulus and recording sites. 4) This condition poses further problems for determinations of spatial ERP heterogeneities between recording sites. The difficulties are compounded if stimulus and recording sites have differing ERPs. 5) ERP determinations are further affected by conduction velocities in the paths intervening between stimulus and recording sites. This was demonstrated in comparisons of ERP measurements at the stimulus site itself, ERPs, and the ERPr, given by the time interval separating AP upstrokes, at the recording site. ERPr then did not equal ERPs when the conduction velocity of the AP produced by the S1 stimulus differed from the conduction velocity of the AP produced by the S2 stimulus. These discrepancies were accentuated at high pacing rates close to refractoriness, and with increasing distance between stimulation and recording sites (734). 6) Whereas absolute refractory period may be regarded as an invariant value, ERP depends on the value of the effective stimulus intensity at the site of membrane excitation. Thus an adaptive S1S2 protocol demonstrated differing ERPs corresponding to differing magnitudes of S2 extrastimulus. Representations involving ERP, rather than APD, are thus strongly dependent on applied stimulus voltage (281).
The wavelength parameter nevertheless provides a useful description of the spatial extent of excitation by the traveling wave. Larger values of λ would result in a reduction of the likelihood that areas of depolarization and repolarization meet on encountering tissue heterogeneity. The resulting safety factor would then ensure that the traveling wave completely passes over the heterogeneity without disruption (1251). Conversely, a decreased value of λ would increase the likelihood of a wave breakup into into multiple wavelets, formation of scroll-waves (244, 866, 1317), and a positive feedback formation of further wavebreaks giving wavelets along chaotic conduction pathways corresponding to VF (1084). For example, where alterations in heart rates decrease APD, ERP, or θ, they could thereby alter AP wavelength λ and thereby potentially exert arrhythmic effects (595, 753).
Figure 2 illustrates such situations for a sequence of murine AP waveforms (753). The relevant parameters can be quantified, in terms of their basic cycle lengths (BCL), APDs at 90% repolarisation (APD90), latencies, and corresponding diastolic intervals (DIs) at 90% repolarization (DI90) separating the current and preceding action potentials. Together these yield active and resting wavelengths λ' and λ0' (Figure 2A). The latter sum together to give a basic cycle distance, BCD' (Figure 2B). When an AP with long λ' passes over a heterogeneity that can potentially cause conduction block, the back of the propagating wave blocks retrograde propagation. This leaves only an orthograde excitation wave (Figure 2C). In contrast, when λ' is short, the back of the wave passes the heterogeneity before retrograde excitation has passed through the unidirectional block. This initiates a new propagating retrograde wave which potentially sets up a sustained reentrant circuit (Figure 2D).
Figure 2.
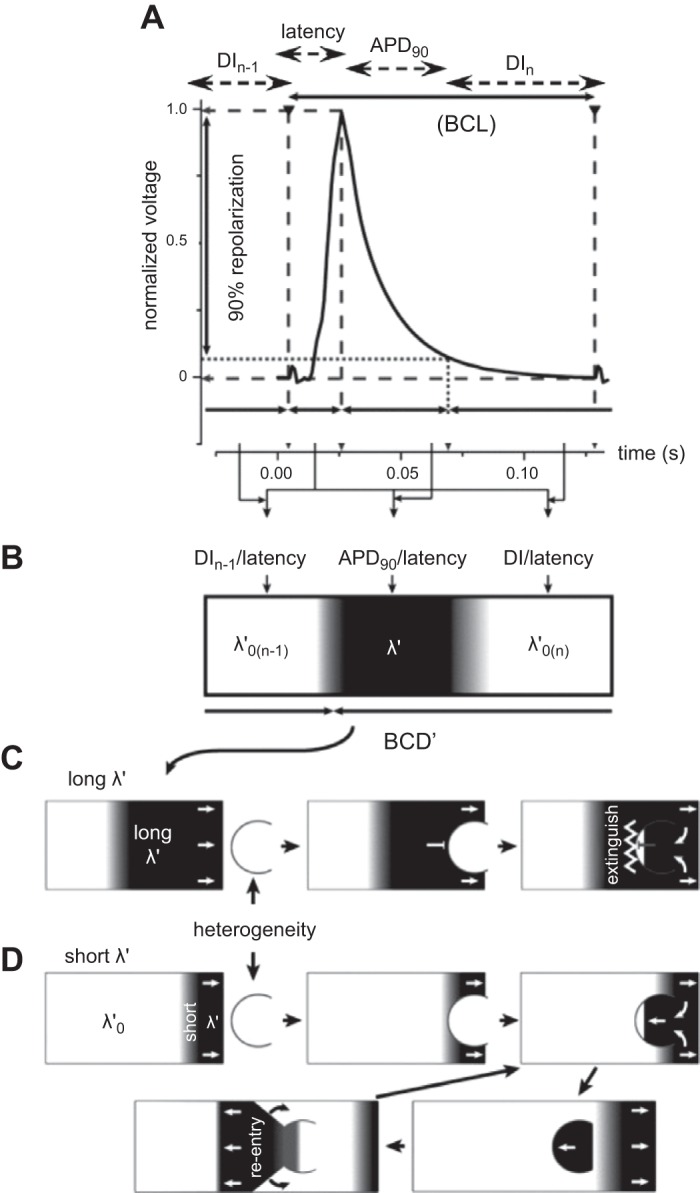
Extension of cable analysis to action potential wavelength, wave-break, and re-entry. A: typical murine monophasic right ventricular action potential (AP) waveform, indicating basic cycle length (BCL), action potential duration at 90% recovery (APD90), latency and diastolic interval (DI) of the current (nth) and preceding [(n−1)th]AP. B: these variables yield the active and resting wavelengths λ' and λ0' for which the basic cycle distance, BCD' = λ' + λ0'. C: orthograde propagation of an AP with long λ' over a heterogeneity results in the back of the propagating wave blocking retrograde propagation. D: propagation of an AP with a short λ' results in the back of the wave passing the heterogeneity before retrograde excitation has crossed the unidirectional block. This results in initiation of a new propagating retrograde wave and a re-entrant circuit. [From Matthews et al. (753).]
II. ARRHYTHMOGENESIS: DISRUPTION OF ORDERED EXCITATION AND CONDUCTION
A. Electrophysiological Conditions Initiating and Perpetuating Arrhythmia
1. Triggering events at the cellular level
Arrhythmias result from an inappropriate generation, or a breakdown in the orderly sequencing, of cardiac electrical activity. They often follow triggering events, and then take place in tissue with intrinsic instabilities resulting in arrhythmic substrate (284, 529, 931, 1111). Events reflecting such phenomena could occur at the single-cell level, during propagation of excitation at the tissue level, or at the level of entire cardiac chambers (560, 981).
At the cellular level, triggered activity results from extrasystolic membrane depolarization that could potentially generate premature APs, and therefore, triggered beats, following an otherwise normal AP, if the voltage changes that they produce are sufficiently large. Of these, early afterdepolarizations (EADs) intercept the repolarization time course of a prolonged AP. They thereby permit time for L-type Ca2+ channel recovery from inactivation in a still depolarized membrane. The reactivated inward ICaL then produces a further depolarization. This initiates a positive feedback process resulting in the afterdepolarization potentially triggering AP firing (498). In contrast, delayed afterdepolarizations (DADs) follow full AP repolarization. They can result from an enhanced SR Ca2+ release. This in turn increases activation of electrogenic NCX or Ca2+ activated Cl− transient inward (Iti) currents. DADs are associated with conditions of Ca2+ overload as occurs in digitalis toxicity and catecholaminergic polymorphic ventricular tachycardia (CPVT) (509). In the atria, triggering can also arise from the pulmonary or the superior caval veins and may thereby trigger episodes of AF (190) (see sects. VIA and VIIA).
Rarer causes of arrhythmias initiated by abnormal AP triggering at the cellular level include the enhanced automaticity resulting from accelerations in depolarization of pacemaker tissue. This might follow increased sympathetic activity, hypokalemia, or pharmacological intervention. In addition, parasystole could result from a parallel activation of two or more pacemaker regions (32).
2. Spatial electrophysiological heterogeneities at the tissue level
At the tissue level, failure of the AP wave to completely extinguish after normal activation, leading to reexcitation of regions that had hitherto recovered excitability can result in a reentrant excitation. This can occur in the presence of spatial electrophysiological heterogeneities that result in 1) an obstacle around which the AP can circulate provided 2) this occurs with slowed conduction velocities that would permit each region to recover excitability before the wave returns, in the presence of 3) a unidirectional conduction block. The latter might occur with spatial gradients in the latency separating stimulation and depolarization or the time between depolarization and the end of the ERP (see sect. VC) (740). Either would prevent the wave from self-extinguishing (778).
Figure 3 reconstructs the generation of arrhythmic substrate through such a combination of conditions. It illustrates the consequences of introduction of a slow conducting myocardial pathway passing through nonconducting myocardium (path 1; dark gray). This is bordered by a second pathway of normal myocardium (path 2; white). A normal AP (blue arrow) would propagate along path 2 following excitation (Figure 3Ai). The myocardium then becomes refractory. As indicated above (see sect. IC2), the resulting normal action potential traveling along path 2 possesses an excitation wavelength λ (yellow region). Consequently, the impulse conducting along path 1 cannot reenter the circuit as it would collide with refractory tissue in path 2 (Figure 3Aii). Similarly, when an abnormal impulse from an ectopic focus is triggered immediately following the normal AP, it cannot enter path 1 as this remains refractory (Figure 3Bi). It therefore splits at the end of path 2 to conduct retrogradely along path 1 and orthogradely along path 2 (Figure 3Bii). In contrast, a self-perpetuating reentrant excitation can result when 1) an action potential conducting retrogradely along path 1 enters the beginning of path 2 (Figure 3Ci) under conditions of 2) reduced conduction velocity (θ) and/or reduced effective refractory period (ERP). The latter result together in a reduction in excitation wavelength (λ = θ × ERP), to values smaller than the dimensions of the available circuits (Figure 3Cii). This results in persistent reentrant excitation (567).
Figure 3.

Conditions underlying generation of re-entrant arrhythmia. A: basic features of arrhythmic substrate, consisting of slow conducting myocardial pathway (path 1; dark gray), nonconducting myocardium, and second normally conducting pathway (path 2; white) (i). Normal action potential (blue arrow) propagates with velocity θ and effective refractory period (ERP) resulting in propagation wavelength (λ = θ × ERP) (yellow region) along path 2. It initiates a slow conducting impulse traveling along path 1 (i). In normal activity, the latter impulse cannot re-enter the circuit as it collides with refractory tissue in path 2 (ii). B: an abnormal triggered impulse immediately following the normal action potential cannot enter path 1 as this remains refractory. C: self-perpetuating re-entrant excitation occurs when a retrogradely conducting AP along path 1 (i) enters the beginning of path 2 with reduced conduction velocity and effective refractory period and therefore reduced excitation wavelength smaller than the dimensions of the propagation pathways. [From King et al. (567).]
Data from both canine right ventricular (RV) wedge preparations (801, 802) and the Scn5a+/ΔKPQ mouse model (see sect. VIB) further implicate reentry arising from repolarization abnormalities as exemplified by their epicardial dispersions of repolarization in triggering ventricular arrhythmia. The simplest example of this might arise from substrate for reentrant excitation arising from relative changes in two key parameters describing the recovery from excitation. Thus windows of reexcitation have been suggested in situations where critical intervals result from positive time differences between full action potential repolarization and the refractory period. These parameters are often quantified by APD90 and VERP, respectively. This could take place within, or between, adjoining areas of myocardium, and is exemplified in section VE4 (978).
At the level of individual cardiac chambers, disruption of the normal sequence of repolarization following the depolarization wave accentuates arrhythmic tendency (1012). Mammalian ventricular myocardium normally shows a consistent and regular sequence of repolarization. This proceeds from epicardium to endocardium resulting in a transmura l repolarization gradient that optimizes the normal sequence and coordination of electromechanical activation and relaxation (see sect. VIA4). These spatial differences are mainly determined by regional differences in repolarizing K+ channel densities. Disturbances in these gradients may permit regions of depolarization to reentrantly reexcite already recovered areas (see sect. VIA2).
Transmural gradients may be particularly important in producing repolarization differences across relatively short distances. They have been implicated in a number of both canine models subject to pharmacological manipulation and murine models genetically modified to reproduce Brugada syndrome (BrS) (see sect. V) and long QT syndrome (LQTS) (see sect. VI). In addition, a number of animal models show ventricular, base-to-apex, heterogeneities in AP characteristics (see sect. VI). In human clinical situations, increases in this dispersion have been associated with arrhythmogenesis in cardiomyopathies and have been related to increased incidences of T wave alternans and VT (181). Finally, left-right interventricular differences in APD have been implicated in arrhythmogenesis in BrS. BrS patients show characteristic right precordial electrocardiographic ST elevation, right bundle branch block, and changes specific to RV epicardial AP waveforms (605) (see sect. VD).
3. Temporal electrophysiological heterogeneities at the tissue level
Temporal electrophysiological heterogeneities may appear as beat-to-beat variations in AP amplitude or duration. These instabilities have been clinically associated with the appearance of alternans in electrical properties between beats. T-wave alternans reflecting alternating time courses in successive ventricular APs classically precedes breakdown of regular electrophysiological activity and an onset of major arrhythmias. Both T-wave alternans and dispersion of the QT interval thus constitute risk stratification markers in both patients susceptible to sustained ventricular arrhythmias (46, 823, 966) and experimental situations (872). At the cellular level, several hypotheses have suggested possible, potentially coexistent, mechanisms. Voltage-driven alternans can arise from instabilities in membrane voltage resulting from steep APD restitution properties (413, 838). These can reflect the properties of depolarizing, INa, or repolarizing currents including Ito (701), Kir3.x in the case of atrial alternans (126), and currents giving rise to EADs (1005). Membrane voltage is also influenced by the Ca2+-sensitive ICaL, INCX, IKs, and Ca2+-activated SK channels (1004, 1050, 1309, 1333). Nonlinearities can occur in intracellular Ca2+ cycling itself (894, 920, 1218). The latter can become unstable with SR Ca2+ overload, RyR2 sensitization, or at high pacing rates. These can result in steep SR Ca2+ release versus SR Ca load relationships giving Ca2+ restitution properties predisposing to [Ca2+]i-driven alternans (204, 1050). At the tissue level, the resulting alternans may be spatially concordant in which the alternations in adjacent regions of a tissue are in phase, or discordant, when these are out of phase.
APD alternans may be an important mechanism for the generation of arrhythmic substrate. Spatially concordant alternans is not itself arrhythmogenic. However, it may represent a stage that precedes the development of discordant alternans. Discordant alternans in adjacent tissue areas produces APD gradients across their intervening regions which are separated by a nodal line region not showing alternans (872, 1236). Discordant alternans greatly amplifies the dispersion of refractoriness, generates regions of conduction block, and predisposes to figure-of-8 reentry phenomena. Triggered activity within the area with the short APD that propagates directly to the nodal line then likely collides with the electrical activity in the area with the long APD while this is still depolarized and refractory. It will then become extinguished. However, where its propagation is less direct over a longer distance, it would reach the nodal line at a later time when the area with the long APD has recovered (Figure 3 in Ref. 1250). It would then induce reexcitation and a reentrant circuit leading to VT, wavebreak, and evolution into VF (928, 1236).
4. Heterogeneities arising from restitution phenomena
APD restitution phenomena were first reported in classic cardiac electrophysiological papers describing alternations in excitation duration, later measurable by intracellular recording as APDs, with increasing heart rate (436, 778). The subsequent classical analysis described below arose from observations that APDs recorded from canine papillary muscle decreased with increasing steady-state pacing rate. The latter findings had led to the development of a restitution theory seeking to integrate such observations (838). This theory has been recently successfully applied to murine hearts (979, 982) (see sects. VC6 and VIA5).
The restitution analysis seeks to graphically predict the occurrence and magnitude of alternans with alterations in heart rate (Figure 4A). It does so through the expected oscillatory properties of a negative feedback system. The output of such a system (O) is considered to be the result of an amplification G of an input I. The latter is itself controlled by an independent variable, X
| (10) |
Figure 4.

Temporal heterogeneity in the generation of re-entrant substrate. A: classical restitution curves in which action potential duration (APDn) of the nth AP decreases with the decreasing, preceding, (n−1)th, diastolic interval (DI) observed at successively shortened basic cycle lengths (BCL). The accompanying progressively increasing slope requires successively greater number of cycles of alternans to intervene before the system reaches a new steady-state APD (points 1 and 2). When unity slope is reached, alternans become sustained (point 3). Slopes exceeding unity result in waxing oscillations (point 4) in APD. This culminates in conduction block and/or tachyarrhythmia resulting from wave-break. B: fuller analysis of generic restitution function relating APD90 corresponding to the APD at 90% AP recovery to the corresponding DI90. In addition to conventional measures of critical diastolic interval (DIcrit) and maximum gradient (mmax), this maps the maximum APD (APDmax) at low heart rates, DI90 at the effective refractory period (DIERP), and the horizontal axis intercept of the restitution function (DIlimit), corresponding to absolute refractoriness. This permits definition of conditions for stability (unshaded), instability (gray), as well as relative (dark shading) and complete loss of capture (left shaded area). C and D: typical records reflecting arrhythmic phenotypes in monophasic action potential recordings from regularly paced (triangular markers) murine Scn5a+/− right ventricular (RV) epicardia showing nonsustained VT (BCL 134 ms) (C) and the initiation of sustained polymorphic VT (BCL 124 ms) (D). [From Martin et al. (740) and Matthews et al. (752).]
The output O in turn influences the input, to an extent which depends on a fraction of the output (F) and the independent variable (X)
| (11) |
The solution of these simultaneous Equations 10 and 11 corresponds to the points of their graphical intersection. These thereby yield the set points giving the input I and output O at any value of X. The original analysis adopted as input variable the diastolic interval (DI) over which the membrane is restored to the resting potential. Over this period, the membrane is recovering following the AP. The output variable is the APD. This is itself dependent on the preceding DI. Thus, for any given, nth, beat
| (12) |
The independent variable controlling DI is the BCL. The DI in the subsequent, (n+1)th beat, DIn+1, depends on both the BCL and the APD in the previous, nth beat, APDn
| (13) |
An A curve was obtained by plotting the output variable of APD against the input variable of DI. A family of D lines each taking the form
| (14) |
with a consistent negative unity gradient but variable ordinate intercepts set by the BCL was then plotted between the same axes. The intersection between the D line and the A curve would give the solution for the steady-state APD and DI.
A perturbation in heart rate would result in an immediate transition between two separate D-lines representing the respective BCLs. A horizontal line from the steady-state point on the A curve to the new D line would then give the magnitude of the first subsequent DI. A vertical line drawn from there to the corresponding A curve would give the corresponding subsequent APD. Graphical representations for the different cases are shown numbered 1–4 in Figure 4A. Each case would yield different predictions for the outcomes generated by continuation of this process. These would depend on the gradient of the A curve at its intersection with the D line, and its variation with DI in this region.
Where this intersection occurs in a region of the A curve where its slope is zero (Figure 4A, point 1), a final steady-state APD is immediately reached without oscillations. Where the slope at the intersection falls between zero and unity (Figure 4A, point 2), the successive projection lines converge towards and ultimately attain the set point. Each corner on the A curve then represents an individual oscillation, producing a transient alternans. With an intersection at a critical DI, DIcrit, where the the A curve assumes a unity slope, the projection lines form a square. The oscillation then does not converge (Figure 4A, point 3). This results in a sustained alternans whose magnitude is determined by the size of the square. Finally, where the intersection takes place in a region on the A curve where its slope is greater than unity, the progressive projections take a centrifugal trajectory. They thus veer away from the left-hand limit of the A curve, producing conduction block (Figure 4A, point 4). This state, particularly when heterogeneous across the myocardium, may cause reentry (754).
These different conditions can then be related to the remaining parameters describing cellular excitability in a fuller generic analysis of the restitution function. Figure 4B illustrates this development using the relationship relating APD90 to DI90 through different BCLs. In addition to the conventional measures of critical diastolic interval (DIcrit) and maximum gradient (mmax) this maps the maximum APD, APDmax, at low heart rates, DI90 at the effective refractory period (DIERP), and the horizontal axis intercept of the restitution function (DIlimit) corresponding to absolute refractoriness. These additional limits permit definition of different stability conditions within the plane of the restitution function. Thus stability would correspond to the condition when the gradient of the restitution function, m ≤ 1 as outlined above (unshaded areas). Instability would be expected under conditions when DIcrit > DI90 > DIERP (filled areas). This would manifest in occurrence of either nonsustained (Figure 4C) or sustained arrhythmia (Figure 4D). These are exemplified in the recordings from murine Scn5a+/− RV epicardia at two respective BCL values (134 and 124 ms, respectively). Relative loss of capture would be expected in the interval DIERP > DI90 > DIlim, (dotted areas) and complete loss of capture when pacing takes place at a BCL shorter than the absolute refractory period when DI90 < DIlim (hatched areas) (752).
The first restitution (A-) curves were deduced from transmembrane APs in isolated frog (Rana catesbiana) ventricles. These were paced at successively higher rates until refractoriness was reached. These gave a wide range of DIs. APD then varied minimally at low pacing rates. However, with increases in rate, the A-curve gradients became progressively but reproducibly steeper. Recordings obtained immediately following rate changes were variable. They showed a hysteresis above and below the steady-state line for accelerating and decelerating rates, respectively.
Parameters other than APD, such as voltage, may also show alternans and could be similarly graphically analyzed. However, although often occurring together, the existence or extent of voltage amplitude alternans did not parallel the magnitude of duration alternans. Furthermore, wide interspecies variations exist in the time course and the final steady states of adaptations to change in BCL. Imposed increases in heart rate most frequently result in initial reductions in APD with reduced DI, then increasing to nevertheless still reduced steady-state APD values, in human, guinea pig, mouse, and frog ventricular APs. However, APD initially lengthens then rapidly decreases to shorter values expected at shorter DI in rabbit, dog, and cat ventricles. This likely reflects incomplete Ito decay in rabbit and transient L-type Ca2+ current facilitation in dog and cat. Finally, APD actually lengthens and remains prolonged with decreased DI in rat heart. Ca2+ homeostasis has been implicated in restitution changes invoking actions of [Ca2+]i upon the activity of numerous other channels and carriers within the cell (166). Ionic mechanisms for APD accommodation to rate changes remain unclear. They almost certainly involve inactivation processes in depolarizing INa and ICa currents or enhancement of repolarizing K+ currents, with both processes accumulating with successive APs. However, there are numerous differing reports on their relative importance. This has hampered attempts at in silico modeling of APD restitution. Recent clinical reports similarly suggest complexities in the use of such plots in predicting human arrhythmia (818).
Conduction velocity restitution plots of θ against DI similarly reflect the changes in θ with DI in consecutive AP waves. Conduction velocity alternans results in a compression and rarefaction of APs as they travel through the tissue. Slowed θ also reduces the distance between nodes. This results in a higher number of nodes thus predisposing to reentry. θ restitution thus depends primarily on Na+ channel and intercellular coupling properties, whereas APD restitution is also affected by Ca2+ (212, 530), and K+ channel properties and is thus engaged at lower pacing rates (928). In silico modeling studies suggested that θ alternans initiated at higher pacing rates may be responsible for the breakdown of concordant to discordant alternans (1236). However, θ restitution, as represented by plots of θ against DI, are not amenable to the systems analysis of the kind illustrated in Figure 4 and Equations 10–14. Thus the variable θ does not directly feed into that of DI. Furthermore, θ restitution is primarily concerned with the wavefront, whereas APD restitution describes the recovery from excitation that follows (753). Nevertheless, a recent analysis has unified restitution analysis involving APD and θ, respectively, into a single λ restitution analysis. This may offer a more general approach relating restitution conditions to arrhythmic tendency (see sect. VC6) (753).
B. Experimental Models in Studies of Arrhythmic Phenomena
1. Animal models for arrhythmic disease
Genetic exemplars useful for clarification of physiological and arrhythmic effects of particular molecular modifications are available from a range of species. Rabbit ventricular cardiac APs share similar ionic currents and positive plateau phases whose durations resemble the corresponding features in human hearts. Of rabbit ion channel variants, 1) transgenic rabbits with LQTS contain pore mutations in KCNQ1 and KCNH2. They showed LQTS1 and LQTS2 phenotypes corresponding to their absence of IKs and IKr, respectively (152). LQTS2 rabbits showed high incidences of spontaneous SCD (>50% at 1 yr) due to polymorphic VT. Optical mapping studies revealed increased spatial dispersions of repolarization in LQTS2 rabbits. In both, elimination of one repolarizing current (e.g., IKr) was associated with downregulation of the other (e.g., IKs). This contrasts with the upregulation found in mouse LQTS models (383, 384). 2) Rabbits with chronic atrioventricular block showed biventricular hypertrophy, QT interval prolongation following IKs and IKr downregulation, spontaneous torsades de pointes, and shortened lifespan (1147). 3) Hypercholesterolemic rabbits showed an electrophysiological and neural cardiac remodeling including cardiac hypertrophy, QT prolongation, and a vulnerability to VF (682). Of hypertrophic variants, there are 4) hypertrophic cardiomyopathic (HCM) rabbit models affecting the β-myosin heavy chain through the β-myosin heavy chain (MyHC)-R400Q mutation (725) and a cardiac-restricted expression of the mutant β-MyHC-Q403 known to cause clinical HCM (950). These exhibited cardiac hypertrophy, myocyte disarray, interstitial fibrosis, and SCD. However, they often did not exhibit electrophysiological properties normally accompanying proarrhythmia (see sect. IXB).
The earliest described spontaneous mutation causing nonhuman HCM involved a feline G to C variant in cardiac myosin binding protein C (MYBPC3) producing a alanine to proline (A31P) substitution (771). This was followed by similar reports with a feline C820T mutation (770). Both variants resulted in a computationally predicted alteration in protein conformation.
Canine hearts show ion current and AP waveforms characteristic resembling those in human myocardium. 1) Dog hearts with chronic atrioventricular block showed complex structural hypertrophic and electrophysiological remodeling. The latter was associated with IKs and IKr downregulation (1205), NCX upregulation (1068), and predispositions to torsades de pointes and SCD (375, 858, 1068, 1206, 1207). 2) Boxer dogs provide a spontaneous model of arrhythmogenic right ventricular cardiomyopathy (ARVC) and SCD. Their clinical and pathological features closely resembled the human condition. These included VT arising from an enlarged RV showing myocyte loss with fatty or fibrofatty replacement, myocarditis, and apoptosis, in some cases accompanying altered RyR2 expression (81, 769). 3) Finally, dogs affected with X-linked Duchenne muscular dystrophy also showed calcified myocardia and surrounding dense connective tissue associated with ventricular arrhythmias (791).
2. Transgenic mouse models as disease systems
In addition to spontaneous mutations producing phenotypes recapitulating human disease, genetic modifications can be produced from insertion into the animals' genome a mutated human gene (transgene), or introduction of a mutation at a particular locus by gene targeting (160). Among mammalian models, the mouse remains the most amenable to such genetic modifications. The resulting animals often reproduced the corresponding congenital human arrhythmic disorders, particularly when they involved established monogenic mutations (200, 246, 563, 831, 995). Many of these latter genetic variants are clinically rare. Nevertheless, they may model better defined pathological mechanisms than variants representing more common and often more complex conditions. They thus potentially yield important and widely applicable pathophysiological insights. Use of genetic exemplars additionally avoids the need to replicate clinical phenotypes through the use of potentially nonspecific pharmacological manipulations (319, 594, 774, 1054, 1055).
Conventional gene targeting involves homologous recombination of an engineered exogenous DNA fragment containing a targeting vector and selection marker conferring resistance to a cytotoxic drug with the genome of an embryonic stem (ES) cell. The modified ES cell is then screened for presence of the fragment using the cytotoxic agent. It is then injected into a blastocyst that develops into a chimeric animal where the desired gene is present in the gametes. Targeted ES cells injected into wild-type (WT) mouse blastocysts can then contribute to the germline of chimeric mice. These can then be used to generate progeny containing the targeted gene. Selective breeding then creates heterozygous, then possibly homozygous, offspring. Knock-in models contain alterations in the genetic code resulting in a modified mRNA producing a protein with loss or gain of, or modified, function. In knockout models, no mRNA is produced from the altered gene (246).
Alternative methods expediting genome modification generate DNA double-strand breaks by directly injecting DNA or mRNA coding for site-specific nucleases into the one-cell embryo (660). The latter utilize zinc-finger nucleases (ZFNs) (163, 350, 1160), transcription activator-like effector nucleases (TALENs) (1101), or the RNA-guided clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) nuclease system (648, 1290). The double-stranded breaks occur at a specified genomic locus. They are then repaired by error-prone nonhomologous end joining to resulting mutant alleles (163, 350, 1101, 1216). The Cre-lox system has proven a useful technique to restricting expression of a mutated gene to the heart. It is particularly applicable where the mutation is embryonically lethal when involving the entire organism. It also makes it possible to achieve temporal control over gene expression (414, 601, 813). It ensures that recombination events only occur only in specific cell types expressing the site-specific DNA cyclization recombinase (CRE).
3. Murine hearts as models for human arrhythmic disease
Murine hearts are similar in overall anatomy to human hearts. However, there was initial uncertainty as to whether their considerably smaller tissue volumes could sustain the polymorphic arrhythmias shown by larger hearts (343, 526). In the latter event, they would not accommodate the multiple drifting rotors then thought necessary to produce the scroll waves required to generate polymorphic arrhythmia (1264). However, later reports from rabbit heart demonstrated that a single drifting rotor generated scroll waves leading to such polymorphic arrhythmia could thus exist in relatively small volumes of tissue (371). Polymorphic arrhythmia was subsequently observed in mouse ventricles (172, 301, 505, 736, 752–754, 1162). Some of these also demonstrated transitions from a monomorphic to the polymorphic character pattern (982) as observed in larger hearts (1259).
In common with findings in human hearts, murine ventricular APs show rapid depolarization phases driven by inward INa (384). Furthermore, human and murine hearts have similar transmural AP conduction velocities (428, 668). It is generally thought that the effective refractory period ends before the completion of the repolarization process (301, 582, 977, 980, 995; but see Ref. 59). These features expedited their use in studying physiological effects of SCN5a mutations that can underlie BrS and LQTS3, and of effects of class I, Na+ channel blocking drugs (see sect. V and Table 5).
Table 5.
Phenotypic similarities between arrhythmogenic properties in murine Scn5a+/− and Scn5a+/Δkpq hearts and human BrS and LQTS3
| Model | Phenotype | Mouse | Human | Reference Nos. |
|---|---|---|---|---|
| Scn5a+/− compared with BrS | Arrhythmic phenotype unmasked by flecainide | + | + | 150, 344, 914, 1270 |
| Arrhythmic phenotype relieved by quinidine | + | + | 92, 421, 792 | |
| Positive ΔAPD90 persistent with flecainide and quinidine | + | − | 743, 1095 | |
| RVOT/right ventricular initiation of arrhythmia | + | + | 734–736, 741, 754, 1336 | |
| Fibrotic change with age | + | + | 500–503 | |
| Presence of male/female phenotypic differences | + | + | 500–503 | |
| Scn5a+/Δkpq compared with LQTS3 | Arrhythmic phenotype reduced by flecainide | + | + | 94, 914, 1096, 1263 |
| Arrhythmic phenotype exacerbated by quinidine | + | + | 207 | |
| Negative ΔAPD90 rescued by flecainide | + | + | 94, 1096 | |
| Absence of male/female phenotypic differences | + | + | 684, 1096, 1321 |
However, AP waveforms in mouse ventricles show recovery phases distinct in character from those found in the ventricles of larger animals and humans. The latter show clear-cut plateau phases. These are attributable to activation, followed by inactivation, of ICaL in combination with voltage-dependent changes in the specific K+ currents. The K+ current contributions are dominated by rapid, IKr, and slow, IKs, delayed rectifier currents. Of these, IKr rapidly activates with phase 0 AP depolarization. However, its inactivation, that rapidly follows, makes the channel nonconducting during phases 0–2 of the AP. The initial phase 3 repolarization then releases the inactivation and reopens the channel. Its subsequent, slow, deactivation then permits a sustained phase 3 and early phase 4 IKr. In contrast, IKs activates slowly with depolarization to a relatively positive greater than −20 mV potential, and barely inactivates. It gradually increases over phase 2 to become a major phase 3 K+ conductance (696, 697, 1169, 1170).
In contrast, AP repolarization in mouse ventricles results in shorter, triangulated, APDs (∼30–80 ms) compared with those of humans (∼150–400 ms, respectively) (Figure 1D) (236). K+ currents are similarly central to this AP repolarization, although there are smaller ICaL contributions. However, repolarization is driven mainly by fast, Kv4.3 and Kv4.2-mediated, Ito,f, as well as the more slowly inactivating Kv1.4-mediated Ito,s, components of the rapidly activating and inactivating transient outward current Ito (Figure 1, B and D). Both Ito,f and Ito,s become activated at potentials greater than −30 mV (829). They show distinct time constants for inactivation and recovery from inactivation. For Ito,f, these fell in the range of 20–100 ms for both processes. In the case of Ito,s, these processes took hundreds of milliseconds and seconds, respectively (833). Murine hearts also showed rapidly activating but slowly inactivating 4-aminopyridine-sensitive and -insensitive, Kv1.5 (KCNA5)-mediated IK,slow1 and Kv2.1 (KCNB1)-mediated IK,slow2, as well as a steady-state Iss (829, 1284).
Recent evidence suggests murine hearts do express both IKr and IKs, but their roles are unclear (53, 64, 276). IKATP and IK1 continue to be important in repolarization and electrical diastole (324, 828, 830). Mouse ventricles also show additional repolarizing rapidly activating and sustained delayed rectifier K+ current, Iss, and slow K+, IKslow, currents (669). Murine hearts additionally exhibit inward rectification properties attributed to IK1. These strongly reduce K+ conductance at voltages greater than −20 mV in phases 0–2, permit outward currents with repolarization to greater than −40 mV late in phase 3, and stabilize the phase 4 diastolic resting potential (357, 1020).
Atrial myocytes additionally show an ultra-rapid IKur in phase 1. There is also the IKACh, which is activated through its interaction with the βγ part of acetylcholine muscarinic receptor-associated Gi proteins particularly in the SAN but also in the atria and ventricles (1020). Studies exploring effects of parasympathetic challenge in Girk4−/− mice implicate IKACh in AF (589). Finally, IKATP is normally a small current. However, its activation by reduced intracellular ATP levels associated with energetic stress exerts triangulating effects on AP waveform (320, 486).
Finally, recent evidence suggests that induced pluripotent stem cells derived from carriers of clinical conditions might replicate some of the expected cellular properties. This may have potential utility in the development of novel treatment strategies in the LQTS2 (488), LQTS3 (718), CPVT (307, 525), and some overlap syndromes (248), given the appropriate correlations between basic molecular physiology and our understanding of arrhythmia in structurally intact hearts.
C. Experimental Studies on Murine Systems
1. Studies at the organism or tissue levels
Arrhythmogenesis itself ultimately takes place at the whole heart level and likely depends on electrophysiological properties through populations of coupled cells in addition to events within single cells. These may include myocardial properties, such as reentry mechanisms and gradients of excitation or recovery from excitation operating at the tissue and whole chamber level. Culture methods that might reproduce such cell populations show initial promise (158). Studies in mouse systems bred on electrophysiologically stable, typically 129/sv or C57BL/6, genetic backgrounds, permit examination of such phenomena whether related to mechanisms of SND, and both atrial and ventricular arrhythmia. These could carry well-defined genetic modifications strategically selected to reflect genotypes associated with specific disease conditions. Such modifications could also be used to expedite experimental clarification of the role of alterations in the function of particular ion channels in arrhythmic mechanisms. Use of WT hearts also permits investigations of acute, reversible, often pharmacological, manipulations, replicating clinical situations and potentially modifying arrhythmogenicity. Investigations of either case typically begin by verifying reproducible arrhythmic phenotypes. This would relate the experimental system to the corresponding clinical condition, providing a translational background for studies proceeding to clarify their underlying mechanisms. The latter investigations are directed at successive, interacting, organism, organ, tissue, in addition to the cellular and molecular levels.
Of these, in vivo electrocardiographic (ECG) studies in ambulatory or anesthetized intact animals (e.g., Refs. 184, 1040) can demonstrate spontaneous rhythm disruptions, electrocardiographic abnormalities (742, 1340), or chronic changes in electrophysiological properties. The latter have been demonstrated in particular genetic conditions with age (e.g., sect. VE) (503). They have also been instrumental in the analysis of the effects of genetic connexin modifications on ventricular conduction and excitation reflected in their PR, QRS, and QT intervals (378, 803, 1132). They could be extended to localizations of atrioventricular conduction abnormalities to supra-Hisian and particularly, infra-Hisian, conduction tissues, in common with human clinical findings in a murine myotonic dystrophic model (976). These studies additionally established strategic conditions of temperature, anesthesia, and selection of electrocardiographic parameters as important in such determinations (111). Recent studies have successfully correlated QT intervals with simultaneously measured APDs in Langendorff-perfused murine hearts. This further makes ECG recording a useful quantitative tool (236, 782, 1219, 1341).
Ex vivo isolated Langendorff-perfused preparations permit closer investigations for triggering arrhythmic events occurring spontaneously during intrinsic activity. In addition, arrhythmic substrate may be detected through an appearance of spontaneous arrhythmia during spontaneous activity, regular epicardial pacing, or septal pacing. Arrhythmia could also be provoked by programmed electrical stimulation (PES). This typically involves imposition of extrasystolic (S2) beats following successively decrementing S1S2 time intervals following trains of regular S1 pacing beats (240, 392, 561, 1095). Alternatively, dynamic pacing protocols could apply sequences of successively incremented steady-state pacing rates (e.g., Refs. 735, 752–754, 979, 982). Some atrial studies also include burst pacing protocols involving delivery of successive high-frequency stimulus trains (245, 402, 659).
Different recording techniques can then explore for underlying pathophysiological and pharmacological processes through closer studies of AP initiation, propagation, repolarization, and refractoriness (392, 978). Of these, intracellular recordings using sharp glass microelectrodes provide definitive indications of absolute resting potentials and AP waveforms including their durations and their maximum rates of rise (dV/dt)max (e.g., sect. VIID) (569, 1340).
Extracellular recording methods offer stable and prolonged in situ estimates of such parameters and can be applied to specific cardiac regions in intact preparations. Unipolar recordings made against remotely positioned reference electrodes represent tissue electrical activity in the form of single large negative intrinsic deflections reflecting arrival of the excitation process immediately beneath the electrode contact with tissue. These are superimposed upon the extrinsic deflections produced by far-field effects. The latter cause positive deflections as the excitation wave travels from remote regions of tissue towards, and negative deflections as the wave subsequently moves away, from the recording electrode. The result is a biphasic waveform (646, 1271). The steep negative intrinsic deflection in the unipolar electrogram coincides with the transmembrane AP upstroke. This makes it possible to determine wave velocity from the time at which it assumes maximum negative slope (275).
Bipolar extracellular recordings comparing voltages at two recording electrodes positioned close to the conducting path for tissue excitation similarly yield biphasic waveforms. The latter reflect a summation of two unipolar recordings. The two terminals are affected to almost the same degree by the extrinsic potentials and far field effects. The latter are therefore minimized by taking the difference between recordings from the positive and negative poles of the amplifier (493). Comparison between biphasic waves obtained under conditions of bipolar recording with biphasic waves obtained under conditions of unipolar recording correlate the fast part of the intrinsic deflection in the unipolar recording with the top of the differential spike obtained from the bipolar recording. Thus, in bipolar, in contrast to unipolar, electrograms, the intrinsic deflection coincides with the top of the spike.
Bipolar electrogram (BEG) recordings thus yield electrogram latencies and durations (EGDs). They have been used in conjunction with PES procedures. The resulting extracellular waveforms have been analyzed by programmed electrogram fractionation analysis (PEFA). This involved plotting conduction latencies in the recorded BEG deflection components at progressively shortened S1S2 intervals in clinical studies. This analysis had first successfully assessed clinical arrhythmogenic risk in HCM and cardiac channelopathy (461, 1009, 1011, 1012). It thus associated arrhythmogenic reentrant substrates with increased EGD. The latter reflects the distribution or spread (“fractionation”) of ventricular myocardial conduction velocities at reduced S1S2 intervals. This approach proved applicable to murine systems (see sects. V, D2 and G4, and VIB3) (64, 416, 1095, 1096).
Monophasic AP (MAP) recordings additionally reproduce many features of AP waveforms without requiring cell impalement, particularly the AP repolarization phases, and their quantification as APDs and ERPs (559, 560, 562, 563, 1129). They also are potentially translatable from studies in mouse to human hearts (796). These in turn yield APD90/ERP ratios. Increases in such ratios are associated with increased arrhythmogenicity (978).
MAPs were initially recorded by suction electrodes as injury potentials (443, 512). Subsequent contact MAP recordings measured the voltage drop between a positive contact electrode firmly pressed against, and an adjacent, closely placed (∼5 mm), negative electrode in light contact with, the myocardial surface. The volume conductor hypothesis suggested that the pressure exerted by the positive electrode depolarizes the underlying cells to approximately −20 to −30 mV by opening membrane stretch-sensitive ion channels. This locally inactivates their contained voltage-gated Na+ channels. This makes the local membrane unexcitable and assuming a fixed reference potential, but leaves the adjacent cells capable of AP generation. During electrical diastole, the inactivated region acts as a current source as it is relatively depolarized compared with the surrounding myocardium. The resulting current flow between source and sink regions originates from a large number of cells, given the ∼1–2 mm tip diameter of the positive electrode. It is determined by the potential gradient and the number of cells contributing to the source-sink interface, and reverses during electrical systole. Alterations in the number of cells generating the MAP voltage by altering the contact pressure, studying the atria rather than the ventricles, or differently sized hearts, alter the amplitude of the MAP recording.
Comparisons with intracellular recordings confirm that MAP recordings faithfully represent the AP time course, particularly its repolarization phase, which largely comprises low-frequency electrical signals, where they show smooth upstrokes over <5 ms rise times to overall amplitudes >10 mV uncontaminated by intrinsic or QRS deflections and fall to smooth stable diastolic baselines (217, 328, 582). The latter condition can be affected by motion artifacts resulting from cardiac contraction, but such artifacts may be minimized by spring mounting of the electrode. MAP recordings reproduce alterations in, but not absolute values of, resting and plateau voltage. They yield significantly smaller (∼7 V/s) values of (dV/dt)max than intracellular recording (200–300 V/s). This likely reflects the lower seal resistances made between tissue and MAP compared with those made by intracellular electrodes. In addition, MAP recordings reflect the activity of large numbers of potentially sequentially activated, rather than single cells.
Electrical contact or optical mapping methods provide patterns and timings of wavefront propagation of either electrical excitation or spectrofluometrically measured Ca2+ release (292, 296, 427, 632, 736, 803, 1109). Unipolar, multi-electrode array (MEA) recording from the left or right atrial or ventricular epicardial surfaces can follow AP propagation from either stimulated or spontaneously beating hearts. They thereby yield isochronal maps displaying activation times (AT) to the point of maximal negative slope (dV/dt)max of each recorded electrogram. Recovery times (RT) can be measured as the (dV/dt)max for a negative T wave or the (dV/dt)min of a positive T wave. They thereby provide activation recovery intervals (ARI) separating their respective AT and RT. ARI proved comparable to APD values obtained from MAP electrode measurements. It is then also possible to determine activation and repolarization time differences (ATD and RTD) between the first and last ATs and RTs and the ARI differences (ARID) between the shortest and longest ARI (see sect. VC7) (736).
MEA recording also permits determination of conduction velocity parameters. It can provide both effective conduction velocities representing AP conduction through recording points in the array as a whole, or from local vector analyses giving both the magnitude and direction of AP conduction (see sects. VB2 and VIIC6) (996, 1336). Fluorescence imaging permits a spatiotemporal characterization not only of voltage, but also changes in calcium cycling in tissue labeled with voltage-sensitive (e.g., di-4-ANEPPS, RH237) or Ca2+ indicators (such as rhod-2-AM). This involves appropriate excitation of the signals, and separating and collecting emissions of the appropriate wavelengths. These give results that clarify the pattern of the activation and calcium release, and presence of diastolic calcium leak (59, 1218).
2. Studies in single cells and at the molecular level
Single-cell studies in intact or perforated whole cell patch-clamped myocardial cells enzymically isolated from Langendorff-perfused hearts have studied Ca2+ (1128) and K+ current activation (561) and activation, inactivation and recovery from inactivation of Na+ channels under both acute, including hypokalemic, conditions and in genetically modified, including Scn5a+/− and Scn5a+/ΔKPQ, myocytes (see sect. VC2) (416, 741, 867). In addition, a recent, loose patch-clamp, technique (22, 23, 1304) has been introduced to examine differences in current-voltage relationships of peak INa in intact Scn5a+/−, RyR2-P2328S, and WT atria. These have included studies of the effects of high extracellular Ca2+, caffeine, or cyclopiazonic acid (CPA). All these agents are expected to acutely increase [Ca2+]i (see sect. VIIC6) (568). This technique shows significant future promise for studies of biophysical events in intact hearts (935). Transfection methods whether involving α- or β-subunits in expression systems such as those offered by Chinese hamster ovary (CHO) or human embryonic kidney (HEK) cells have been used to study and both activation and recovery properties for subsequent modelling (696, 697, 887, 1170) as well as reconstruct channel behavior in response to simulated APs (273, 406, 1060).
Isolated atrial and ventricular fluophore-loaded myocardial cells were also used to examine Ca2+ signaling properties by themselves or following acute pharmacological interventions. These have studied measures of SR Ca2+ release or reuptake, or cellular Ca2+ entry or expulsion, and their effects upon arrhythmic properties. These were first explored in WT murine hearts (see sect. VIIB) (62, 63, 352, 446, 1335, 1338) prior to their introduction to genetically modified systems (see sect. VIIC) (e.g., Refs. 364, 387, 1334).
At the molecular level, assessments of longer term factors contributing to arrhythmia have involved examination of gene expression characteristics, by quantitative reverse transcriptase polymerase chain reaction (qPCR) and western blotting, of a wide range of ion channels and regulatory genes. These have included genes for Na+ channel subunits, Ca2+ channel subunits and Ca2+-handling proteins, K+ channel subunits, hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, gap junction proteins, transcription factors, and other selected genes, including transforming growth factor-β1 (TGF-β1) and vimentin gene expression (see sects. VIF and IXA) (245, 392, 408).
3. Studies of contributions from morphological change
Finally, explorations can be made of morphological changes extending from gross organ and tissue architecture, through hypertrophic and fibrotic tissue changes, to electronmicroscopic analysis, associated with particular arrhythmic conditions. These studies have assessed contributions of age and/or genotype to the observed arrhythmic changes. This has included studies of gross tissue architecture, as well as quantitative digital microscopy studies of the results of picrosirius red staining and vimentin fibroblast immunostaining, in different cardiac chambers of the right and left sides of the heart (see sects. V, E and F, and IX) (408, 501, 502, 968, 1180).
4. Computational modeling studies
Quantitative computational reconstructions have explored the consequences of the individual changes in properties obtained by the studies above for properties at the systems level. These could optimally link findings in murine hearts (133) to properties of human hearts under circumstances of both normal (154, 369, 489, 1153) and abnormal function (551, 552, 1154, 1155). Recent modeling approaches now potentially permit further incorporation of electroneutral solute and osmotic fluxes, and interacting alterations in the resulting ion compositions and cell volumes (331, 332). At higher levels of organization, they extend to whole chamber reconstructions of the spread and recovery of the electrophysiological change (1140), with significant promise of clinical translatability (936).
Studies analyzing experimental findings in murine hearts have initially examined situations involving relatively simple geometry as exemplified by the spread of excitation from the SAN into and through the atria. These provided models that implicated INa in AP propagation through the SAN and from SAN to atria, its effects upon heart rate, and their reconstruction of features of clinical SND. This clarified possible roles for Na+ channels in both normal SAN function and SND following targeted disruption of the murine cardiac sodium channel gene (see sect. VF3) (408, 456, 632, 634, 1276). They extended to investigations of the effects of fibrotic, resulting in morphological change (see sect. IXA3) (245). Computational reconstructions have also been used to guide interpretation of ion channel changes (e.g., Ref. 1276). They have also provided theoretical predictions of the effects of particular ion channel variants for which experimental murine models are not available as in some short QT syndrome (SQTS) variants (see sect. VIH, e.g., 6–8). Computational studies thus represent an approach that will become increasingly valuable as physiological, anatomical, and molecular data become progressively available.
5. Murine exemplars for the study of cardiac arrhythmias
The remainder of this article reviews experimental and theoretical studies of electrophysiological mechanisms underlying cardiac arrhythm ias. They make systematic examinations of specific murine exemplars variously representing ventricular, and sinoatrial arrhythmic clinical disease. In each case, these could be used to assess the relevance of the biophysical and physiological principles underlying stable and unstable atrial or ventricular excitation as outlined above. Such instabilities could disrupt the normally orderly sequence of cardiac electrophysiological activation and recovery and its propagation.
Of these, primary electrophysiological disorders could arise from reversible pharmacological manipulations or loss- or gain-of-function genetic modifications that involve proteins directly concerned with cardiac excitable activity or its modulation. They thereby influence electrophysiological stability even in the presence of normal cardiac anatomy. Such changes can occur against a background of changes in heart rate. Section III discusses exemplars bearing on sinoatrial pacing disorders. The resulting propagated electrophysiological activity can then be characterized in terms of a wavelength (λ) of excitation as well as events falling within the resting wavelength following recovery from activity (λ0) that follows (see sects. IC and IIA). By defining the spatial extent of excitation terms, λ would clarify the tendency for wave breakdown leading to arrhythmic substrate. A first term determining λ is the conduction velocity θ. This in turn is first dependent (see sect. IC, 1 and 2) on the intracellular resistance ra, which determines passive spread of local circuit currents through Cx molecules between successive myocytes. Murine hearts with expected alterations in these are explored in section IV. Local circuit current activation resulting in AP propagation relevant to cellular excitation in turn depends on INa (see sect. IB1). The consequences of compromised INa are accordingly examined using examples discussed in section V. Section VI then discusses the consequences of abnormalities in the remaining, recovery, term determining λ, viz. the APD or ERP. Sections VII and VIII then explore inputs to excitable properties from intracellular events. These often bear upon Ca2+ homeostasis and cardiac energetics. These may alter both excitation as well as triggering events following AP repolarization. Finally, development of anatomical abnormalities could potentially result in arrhythmic substrate through alterations in the relationship between the spatial indicators of electrophysiological activity, particularly λ, and the effective length of the conducting excitation paths. These are considered in section IX.
III. MODELS FOR SINOATRIAL PACING DISORDER
A. Murine Models for Sinoatrial Pacing Function
Successive cycles of cardiac activity are normally driven by SAN automaticity, effectively constituting an oscillator generated by a combination of time-dependent outward, and constant or voltage-dependent inward currents activated during AP repolarization (723). The high background SAN pacing rate in murine compared with human hearts, and the wide variation of heart rates through different species may suggest similarly varying roles of the different ion mechanisms that have been implicated in this function. Similarly, isolation of murine SAN cells is challenging owing to the small size of murine pacemaker regions (136, 137, 197, 210, 440, 632, 637, 722). Nevertheless, mouse strains carrying specific genetic modifications in particular ion channels have offered useful models for pacemaker dysfunction (Table 2) (700).
Table 2.
Murine models involving sinoatrial pacemaker function
| Gene | Genotype | Phenotype | Reference Nos. |
|---|---|---|---|
| Rapid K+ current IKr | |||
| Erg1b | Erg1b−/− | Loss of IKr; bradycardic episodes | 210, 626 |
| Hyperpolarization-activated cyclic nucleotide-gated (HCN) current If | |||
| Hcn1 | Hcn1−/− (global) | Bradycardic, reduced If | 310, 837 |
| Hcn2 | Hcn2−/− (global) | Sinus dysrhythmia and reduced If | 699 |
| Hcn2−/− (cardiac-specific) | Sinus dysrhythmia | 311 | |
| Hcn3 | Hcn3−/− (global) | Sinus rhythm, normal If | 311 |
| Hcn4 | Hcn4−/− (global) | Embryologically lethal | 1094 |
| Hcn4−/− (cardiac-specific) | Embryologically lethal; bradycardia; reduced If | 1094 | |
| Hcn4-R669Q/R669Q (global) | Embryologically lethal; bradycardia; slowed If | 410 | |
| Hcn4−/− (global; inducible) | Sinus pauses; reduced If | 426 | |
| KiT-Hcn4−/− (inducible) | Sinus pauses; reduced If | 435 | |
| Ci-Hcn4−/− (inducible, cardiac-specific) | Bradycardia; reduced If | 74 | |
| Hcn4–573X KI (inducible, cardiac-specific) | Bradycardia; normal If | 20, 425 | |
| Voltage-gated Ca2+ current ICa | |||
| Cacna1d | Cacna1d−/− | Bradycardia; 70% reduced ICa,L; residual ICa,L due to Cav1.2 | 720, 751, 1344 |
| Cacna1c | Cacna1c-DHP−/− | Dihydropyridine-insensitive Cav1.2, continued DHP effects implicating Cav1.3 | 1066 |
| Cacna1g | Cacna1g−/− | ICaT due to Cav3.1 entirely lacking; slowed atrioventricular conduction; moderate bradycardia | 724 |
| Cacna1h | Cacna1h−/− | Normal ECG characteristics | 186 |
Note: mutations involving Scn5a+/− (Scn5a) are covered in Table 4.
B. Role of the Rapid K+ Current, IKr
In rabbit SAN, IKr and IKs are activated during the AP upstroke and then deactivate relatively slowly during the end of the repolarization and diastolic depolarization phases. IKr likely controls AP repolarization and sets the maximum diastolic potential. Nanomolar E-4031 concentrations partially blocking IKr thus positively shift the maximum diastolic potential, decrease the (dV/dt)max and amplitude, and prolong the repolarization duration of the AP (855, 1189). The resulting reduction in recruitment of other depolarizing, diastolic currents (210) then slows pacing rate. Complete IKr block by micromolar E-4031 terminates automaticity and leaves a depolarized (−30 to −40 mV) resting potential (855). Mouse pacemaker cells showed similar effects of E-4031-mediated IKr block (210). Erg1b−/− hearts lack fast IKr and demonstrate bradycardic episodes (see sect. VIF2; Ref. 626).
C. Role of the Hyperpolarization-Activated Cyclic Nucleotide-Gated Current, If
The initial phase of the diastolic depolarization that follows likely includes “membrane clock” contributions from inward, depolarizing, cAMP-sensitive If carried by hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (122). If is activated within diastolic depolarization voltage ranges (−40 to −60 mV) (72, 73, 153). Diastolic depolarization may include additional contributions from time-independent background current including ICl (114) or TRPM4-mediated Ib (252) and decay of outward IKr (400). Nevertheless, studies of the If blockers Cs+ (256), UL-FS 49 (1127), ZD-7228 (733), and ivabradine on pacemaker activity demonstrated a definite role for If in controlling diastolic depolarization rates not only in rabbits (1126) but also mice (280, 643).
D. Murine Models with Altered HCN4 Channels
Genetic alterations involving If, mediated by HCN channels, are associated with both mild and severe clinical arrhythmic conditions (72). Of the four HCN isoforms, HCN4 is likely to be the major contributor to HCN-mediated pacemaker currents in adult SAN of most species, including mouse and human. Hearts with homozygous Hcn4 deletions showed marked bradycardia and decreases (75–90%) in If, and a residual current, possibly carried by Hcn1 and Hcn2, before lethality at embryonic day (ED) 9.5–11.5 (1094). This is consistent with HCN4 function becoming important only after this embryonic stage. In common with both global and cardiac-specific constitutive Hcn4−/− mice, Hcn4-R669Q/R669Q mice with altered cyclic nucleotide binding that would result in an inability to respond to adrenergic stimulation (410) were similarly bradycardic before dying at ED 11–12 (179, 906). Similarly, constitutive deletion of the transcription factor implicated in SAN, Shox2, permitted normal development until an onset of severe bradycardia and decreased Hcn4 expression at approximately ED 10.5 and lethality between ED 11.5 and 12.5. Similar findings occurred with Hcn4-KO mice (297).
However, hearts from adult mice with inducible Hcn4 knockouts, whether globally with a tamoxifen-inducible Cre construct (426) or specific to Hcn4-expressing cells (435), showed limited changes in cardiac pacemaker generation and modulation. Hearts showed normal mean basal heart rates, albeit interrupted by ∼8–16 sinus pauses/min with duration ∼320 ms. Yet patch-clamped SAN cells showed reduced (60–80%) If that nevertheless retained normal activation-voltage curves. AP activity was absent in a significant proportion of cells. However, where such activity was present, spontaneous firing rates were normal. Sympathetic challenge by isoproterenol or treadmill activity in ambulant mice produced alterations in heart rates similar to those found in control mice albeit with increased frequencies of post-exercise sinus pauses (426). However, muscarinic stimulation produced more marked bradycardic effects in Hcn4-KO than WT (426, 435).
Tamoxiphen-inducible models have been produced by crossing floxed Hcn4 mice (74) with mice carrying the Cre-recombinase controlled by the cardiac-specific α-myosin heavy chain (αMHC) promoter (1075), sparing neuronal Hcn4 channel expression. These showed a similar ∼70% reduction in If. But this now accompanied progressive, up to 50%, reductions in heart rates without sinus pauses, with reduced rate responses to isoproterenol challenge during telemetric recording. There was also an unexpected prolonged PQ interval and atrioventricular block, eventually proving lethal. Mouse models in which it was possible to induce an elimination of Hcn4-expressing cardiomyocytes and their substitution with collagen fibers were similarly bradycardic with a similarly reduced tachycardic response to isoproterenol adminstration on ambulant ECG recording (424). However, the elimination of HCN4-expressing cells additionally resulted in sinoatrial pauses, and atrioventricular node conduction changes increasing PR interval and producing complete heart block and supraventricular tachycardia or VT.
The hHCN4-573X mutation has also been correlated with clinical SAN dysfunction. It produces a dominant negative abolition of cAMP-mediated HCN4 modulation (20, 1028). Ambulant transgenic mice with an α-MHC promoter and a Tet-Off system-controlled cardiac-specific overexpression of Hcn4-573X (20) showed ∼20% reductions in resting heart rates but no changes in PQ and QTc intervals. They retained albeit reduced tachycardic effects of exercise. Their If showed slower activation kinetics and a negative, −20 mV, shift in voltage dependence. Their isolated SAN cells were either quiescent or, in a small fraction of cells, showed subthreshold membrane potential oscillations followed by regular firing at reduced rates. Isoproterenol challenge restored regular pacemaker activity but to lower rates than when applied to WT (425).
E. Murine Models With Modifications in the Remaining HCN1-3 Channels
The occurrences of HCN1 and HCN2 are species dependent, and their levels of expression are lower than that shown by HCN4. There are nevertheless reports of Hcn1 signal in the SAN (425). Recent immunohistochemical studies demonstrated Hcn1 in WT mouse SAN (310). However, a generalized knockout Hcn1−/− mouse developed normally (837). Nevertheless, Hcn1−/− mice were bradycardic with low cardiac output, sinus dysrhythmia, and recurrent sinus pauses on telemetric in vivo ECG and echocardiographic study. Single Hcn1−/− spindle and elongated SAN cells showed significantly reduced If amplitudes. If activation kinetics were slowed, consequently resembling that of cloned HCN4 channels. Isolated Hcn1−/− SAN cells accordingly showed reduced pacing frequencies (310). These findings suggest a role for Hcn1 in stabilizing SAN pacemaker function. Hcn1 was also expressed in mouse cardiac conducting tissue (425, 726). Hcn1 has also been demonstrated in rabbit SAN (804). Together with Hcn4 it could there contribute to If (25). Finally, Hcn1 has been implicated in rhythmic and resting neuronal activity in mouse brain (122).
Hcn2 occurs mainly in adult mouse ventricles. It is hardly detectable in atria and SAN. Nevertheless, global and cardiac-specific constitutive Hcn2-deficient mouse models showed sinus dysrhythmic features. Their resting heart rates were normal, but resting ambulatory ECGs showed increased RR variability (699). Isoproterenol and exercise challenge accomplished similar maximum rates as those in control mice and reduced the sinus dysrhythmia. Their isolated SAN cells showed slowed activation kinetics and ∼30% reductions in If and a −5 mV hyperpolarization in maximum diastolic potential.
Hcn3 subunits have long been regarded as little relevant to cardiac activity. Hcn3 was not observed in SAN (145, 178, 311, 425). There have been no reports of Hcn3 mRNA or protein expression in rodent SAN cells (311, 425). There are varying in situ hybridization, real-time PCR or protein expression reports in mouse and rat hearts (425). Global and constitutive Hcn3−/− knockout mice were born with normal Mendelian ratios. Ambulant ECG recording showed normal sinus rhythm. However, under bradycardic conditions, T-wave amplitudes and duration as well as QT intervals all increased. Epicardial cells showed shortened APDs and ∼30% reduced If. These findings suggest a role as a ventricular background current that opposes ventricular AP repolarization (311).
F. Role of Ca2+ Currents, ICa
Late diastolic depolarization may further involve contributions from depolarizing, ICaL, and T-type Ca2+ channel current, ICaT (998). It is compromised in mice homozygously lacking either L-type, Cav1.3 (Cacna1d), or T-type, Cav3.1 (Cacna1g), channels (768). Both genetic and pharmacological data implicated ICa,L, with its more negative threshold in SA than ventricular myocytes, in both diastolic depolarization and the AP upstroke through the actions of Cav1.3 and Cav1.2 (Cacna1c), respectively (720, 721). In vivo ECG studies in anesthetized mice demonstrated that dihydropyridine Ca2+ channel blockers induced bradycardia (620). Studies in genetically modified murine platforms suggested a role for Cav1.3 in automaticity and Cav1.2 in excitation contraction coupling.
Cav1.3 channels activated at the more negative potentials (approximately −50 mV) within the range corresponding to diastolic depolarization (720). Both intact Cav1.3−/− mice and Cav1.3−/− atria were bradycardic, the former even following autonomic block (901). This altered function was reflected in corresponding properties of isolated SAN (1344) and pacemaker cells, the latter showing erratic pacemaker function (720) The Cav1.3 inactivation produced a 70% reduction in ICa,L in pacemaker cells with the persisting ICa,L then likely arising from Cav1.2. Thus ICa in Cav1.3−/− pacemaker cells showed a greater dihydropyridine sensitivity (720) and faster inactivation kinetics (1344) than in WT. Maximal pacing rates in Cav1.3−/− hearts following isoproterenol challenge were slightly lower than those shown by WT hearts (751). This could reflect the effect of β-adrenergic activation negatively shifting thresholds for Cav1.3-mediated ICa,L, activation to approximately −55 mV (720). In contrast, in Cav1.2-DHP−/− mice with dihydropyridine-insensitive Cav1.2, dihydropyridines continued to exert in vivo bradycardic effects implicating Cav1.3 in this effect (1066).
The ICa,T inhibitors Ni2+ and tetrametrine slowed SAN cell pacemaker activity (399, 1008). Yet there is relatively little ICa,T activity through diastolic membrane voltages. Mouse SAN expresses both Cav3.1 and Cav3.2 mRNA. However, although their SAN ionic currents and automaticity have not been studied, Cav3.2−/− mice had normal ECG characteristics (186). In contrast, Cav3.1−/− SAN and AVN cells entirely lacked ICa,T with no evidence for a residual ICa,T that would be expected from the remaining Cav3.2. In contrast, only Cav3.1- and not Cav3.2-mediated ICa,T occurred in postnatal rat atria (315, 834). These findings suggest that Cav3.2 is expressed in developing heart, whereas Cav3.1 expression dominates the phenotype in adult heart. Spontaneous activity in isolated SAN pacemaker cells was slowed by ∼30%. Cav3.1−/− mice showed slowed atrioventricular conduction and a moderate bradycardia persistent even following pharmacological autonomic block reflecting the slowing of intrinsic SAN automaticity (724).
Finally, murine models for N-type (Cav2.2: Cacna1b) and R-type (Cav2.3: Cacna1e) channels have been investigated for changes in autonomic rather than intrinsic pacing mechanisms (479, 698, 1247).
G. Role of Na+ Currents, INa
The upstroke components of pacemaker cell APs are primarily driven by Ca2+. Nevertheless, the SAN expresses both TTX-sensitive neuronal Nav1.1, likely important in pacemaking (75, 77, 635, 714), and TTX-resistant Nav1.5 that may mediate intranodal conduction (632, 635). In neonatal rabbit SAN, TTX (3 μM) slowed pacemaker activity by 63% through slowing late phase diastolic depolarization, decreasing AP thresholds and reducing overshoot. INa contributed a window current that declined with age (75). INa was activated by ramp depolarizations to extents that varied with ramp slopes. This suggested an inward current contribution resulting from incomplete inactivation of the INa that was initiated during the AP upstroke during diastolic depolarization (76). Nav1.1 becomes downregulated in the adult consistent with roles in increasing basal heart rate primarily in neonates (75, 76). However, adult mouse pacemaker cells expressed both TTX-sensitive and -resistant INa. Nav1.1 may assume a pacemaker role in adult mice (197, 635, 722). INa inhibition by lidocaine reduced heart rate (620), and low, 50–100 nM, TTX concentrations reduced pacing rates in Langendorff-perfused mouse hearts (714). The roles for the effects of Nav1.5 haploinsufficiency in intranodal and sinoatrial conduction is discussed in further detail in section VF2.
H. Contributions of Ca2+ Homeostatic Processes
Further contributions to SAN pacing may arise from RyR2-mediated Ca2+ release in conjunction with the electrogenic effects of a resulting increase in INCX and the decay of delayed rectifier currents. Whereas ryanodine-mediated block of Ca2+ release reduced the rate of late diastolic depolarization, Cs+-mediated block of If reduced initial diastolic depolarization (652, 972). Distinct cytosolic, early, delayed, and late Ca2+ transients have been observed in SAN cells. These have been attributed to events reflecting action potential activation, nuclear Ca2+ change, and subsarcolemmal local Ca2+-induced Ca2+ release, respectively (523). Of these, the last has been implicated in a distinct intracellular “Ca2+ clock” driven by spontaneous SR Ca2+ release that could contribute to the pacemaker process (161, 459, 616, 719, 722, 946, 1121). Increases in RyR2-mediated Ca2+ spark activity that would drive this pacing mechanism might occur when a critical SR Ca2+ level is regained during diastole (193), or arise from particular kinetic properties of SR Ca2+ uptake or release proteins (132, 615, 1121, 1199).
The properties of SAN cells are consistent with such a mechanism. SAN cells both contain the requisite SR Ca2+ stores and express RyR2 and RyR3 (748). They also have high basal cAMP levels. This could facilitate a protein kinase A (PKA)-dependent phosphorylation of RyR2 that would enhance RyR2-mediated SR Ca2+ release. The consequent, appropriately timed elevations of local [Ca2+]i would then enhance INCX whose electrogenic effects would then contribute to the observed depolarizing potentials (1198). Increases in [Ca2+]i could also activate physiologically important enzymes such as calcium/calmodulin-dependent protein kinase II (CaMKII) (132, 616). This scheme would be consistent with observations in which the rapid Ca2+ chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) slowed and ultimately abolished AP firing in isolated guinea pig SAN myocytes (161). Such suggestions are potentially testable through the use of genetic paradigms for RyR2 or SERCA outlined in section VII, D and E.
I. Roles of Other Charge Carriers
Of the remaining charge carriers, both murine genetic exemplars and pharmacological probes are lacking for the sustained current Ist (136, 783). Its function in SAN automaticity has been studied by modeling studies (783, 1327). Further contributions to pacing may arise from Na+-K+-ATPase (INaK), INCX, and its modification by intracellular Ca2+ handling (132, 136). Preliminary reports additionally implicated transient receptor potential cation channel, subfamily C, member 3 (TrpC3), the endoplasmic reticular Ca2+ sensor stromal interacting molecule (STIM), and the surface membrane channel Ca2+ release-activated Ca2+ channel protein 1 (Orai1), involved in store-operated Ca2+ entry (SOCE) in inexcitable cells in this complex regulatory mechanism (524, 670). Finally, studies in Girk4−/− mice deficient in Kir3.4 implicate IKACh in regulation of pacemaker activity and heart rate following sympathetic stimulation (767).
IV. TISSUE CONNECTIVITY AND ARRHYTHMIC DISORDER
A. Gap Junction Proteins and Tissue Electrical Connectivity
Following initiation at the SAN, a wave of regenerative electrophysiological activity is conducted through successive cardiac structures initiating their activation (see sect. IC3). A first determinant of the conduction (θ) term in the expression for the wavelength λ of this conducted AP arises from the longitudinal resistance, ra. The latter depends on gap junction-mediated passage of the local circuit current between cells which spreads the excitation. Each gap junction comprises two apposed and connected connexon hemichannels. The latter are protein hexamers of one of a variety of genetically distinct connexon protein isoforms. The six connexin (Cx) subunits surround an aqueous, conducting, pore electrically coupling successive cells (283, 495, 578, 606, 1192).
Of the major murine cardiac Cx isoforms (Table 3), Cx40 occurs at intercalated disks in atrial myocytes, atrioventricular node, and ventricular conduction system (247). Cx43 occurs in both atrial and ventricular myocytes and distal conduction system (495, 942, 1179). Cx45 mainly occurs in SAN, atrioventricular node, and conducting bundles (1179). Altered gap junction expression slows conduction enhancing arrhythmogenecity. It can occur by itself or can accompany alterations in other AP conduction determinants such as fibrotic change or other remodeling accompanied by excitability change (496, 803, 948, 1038, 1091). Thus Cx43 distribution and expression both alter with most human ventricular remodeling following cardiac overload. This was first demonstrated with the fiber disarray in infarct border zones (888). Early compensated hypertrophic remodeling following aortic stenosis was accompanied by increased Cx43 expression (587). HCM is accompanied by either Cx43 downregulation (888) or lateralization (1036). Dilated cardiomyopathies (DCMs) also correlate with reduced and lateralized Cx43 expression.
Table 3.
Selected loss-of-function murine cardiac connexin models
| Gene | Genotype | Phenotypes | Reference Nos. |
|---|---|---|---|
| Cx30.2 | Cx30.2−/− | Reduced PQ interval | 590–592 |
| Cx40 | Cx40−/− | Prolonged ECG P-wave; prolonged PQ intervals; supraventricular arrhythmias; normal ventricular conduction | 57, 176, 396, 573, 1026, 1065, 1172, 1191 |
| Cx43 | Cx43+/− | Normal P wave durations; reduced ventricular conduction velocities; homozygote lethal | 296, 937, 1132 |
| Cx43- (inducible) | 286 | ||
| Cx43- (cardiac specific) | 389 | ||
| Cx45 | Cx45−/− | Normal AV conduction, i.e., normal PQ and QRS duration | 596, 602 |
| Cx45−/− (cardiac specific) | 326 |
B. Connexin-Deficient Murine Hearts
1. Sinus node function
Mice deficient in Cx43 (286, 389, 803), Cx40 (57, 1109, 1172, 1191), Cx30.2 (1026), combined Cx40 (596) and Cx45, or Cx30.2 (1026) showed normal sinus node function. However, Cx40−/− hearts demonstrated shifts in leading pacemaker activity from SAN to secondary sites including the sulcus terminalis, RA free wall, and right superior vena cava. Cx40−/− mice also showed prolonged ECG P-wave durations (57), PQ intervals, and QRS durations (1191). This could possibly reflect local conduction blocks prolonging intra-atrial activation path length and therefore conduction time. However, their resting membrane potentials, (dV/dt)max, AP amplitude, or APD were normal compared with WT Cx40+/+ (57).
2. Atrial arrhythmia
Both animal and human AF occur with altered atrial Cx40 expression and distribution (820). Cx40−/− atria showed reduced RA epicardial conduction velocities (57), with prolonged ECG P-wave durations in some (57, 396, 1191) but not all reports (1109, 1172). They also showed increased incidences of supraventricular arrhythmias (176, 396, 573, 1191). In contrast, Cx43-haploinsufficient or conditional Cx43 knockouts showed normal P wave durations (286, 1132) and conduction velocities (1132). Increased proportions of Cx40 decreased, while increased proportions of Cx43 increased conduction velocity in cultured atrial myocyte strands from Cx40- or Cx43-deficient mice (85).
3. Atrioventricular conduction
Mouse atrioventricular conduction systems contain Cx40, Cx30.2, and Cx45 (496, 592, 948, 1026), as well as the murine-specific Cx30.2 (590). Cx40 knockout prolonged PQ intervals (57, 1026, 1065, 1172, 1191). This was attributable to prolonged His-ventricle interval (1026) with or without increased atrial-His intervals (1172). They also showed QRS widening and fractionation (57, 596, 1026) during anterograde but not apical ventricular pacing (1191) despite normal ventricular conduction velocities. These findings are consistent with delayed bundle branch conduction (1109, 1191). This delayed conduction was directly observable in both right (947, 1109) and left bundle branches (947).
In contrast, Cx45 haploinsufficiency did not affect ECG measures of atrioventricular conduction. PQ and QRS duration then were similar to control. However, it accentuated the effects of Cx40 insufficiency on PQ and QRS duration (596). Finally, Cx30.2 knockout surprisingly was associated with reduced PQ durations. This suggested increased atrioventricular conduction velocities attributable to decreased atrial-His as opposed to His-ventricle intervals (590, 592). In contrast, combined Cx40 and Cx30.2 deficiency resulted in normal atrial-His and His-ventricular intervals consistent with opposing actions in, respectively, increasing or decreasing conduction velocity (1026). Loss or redistribution of Cx40 accompanies other murine models of AF including overexpression of Ras homolog gene family member A of RHOA, angiotensin converting enzyme, tumor necrosis factor (TNF), and cAMP response element modulator (CREM) (539, 572, 990, 1013).
4. Ventricular arrhythmia
Complete knockout of the main ventricular connexin Cx43−/− causes perinatally lethal pulmonary outflow tract malformation (937). Cx43+/− mice, 50% Cx43-haploinsufficient, were viable but showed increased ventricular activation delays reflecting reduced conduction velocities in some (296, 1132) but not all studies (803, 948). Cultured strands of perinatally obtained Cx43+/− and Cx43−/− myocytes accordingly showed 0 and 96% reductions in conduction velocities, respectively (84). Several conditional Cre/LoxP Cx43 knockout mouse models provided expression systems with >50% Cx43 reduction (237, 389). Even very low Cx43 levels then permitted conduction velocities that were ∼50% of normal. Nevertheless, a >80% reduction in Cx43 did slow ventricular conduction, particularly in the transverse as opposed to longitudinal myocardial fiber direction. These effects were greater in the RV than the left ventricle (LV) (389, 948). They accompanied arrhythmias initiating in the RV (389, 948) and lethal ventricular tachyarrhythmias during telemetric recording (286, 389).
Computational predictions correspondingly demonstrated nonlinear dependences of conduction velocity upon junctional conductance. Gap junction uncoupling enhanced both safety factor (957) and (dV/dt)max (1083). Consequently, large gap junction conductance reductions were required to slow conduction (520, 1043).
V. ELECTROPHYSIOLOGICAL EXCITATION AND ARRHYTHMIC DISORDER
The Na+ Channel, Nav1.5 (Scn5a), α-Subunit
Cardiac Na+ channel function is essential for myocardial excitation. In addition to voltage-dependent activation properties, it demonstrates transitions into and from fast and slow inactivated states, resulting in entry into and recovery from refractoriness to reexcitation (171, 970, 1184, 1200). These properties in turn are altered by Nav1.5 modification through a wide range of agents.
The main, Nav1.5, Na+ channel, α-subunit 260-kDa protein, encoded by SCN5A, comprises four homologous domains (I–IV). These are assembled around a central ion-selective pore. Each domain contains six transmembrane segments (S1–S6). Of these, S5 and S6 and their connecting loop regions form the pore module. The charged amphipathic S4 segments move towards the membrane extracellular face on depolarization. This gating transition initiates conformational changes opening the pore (878). Conformational changes in domains I–III show relatively rapid kinetics and result in ion channel opening (177). In contrast, the slower changes in domain IV permit an inactivation component connecting S6 of domain III to S1 of domain IV. This occludes the pore to cause channel inactivation (171). The α, Nav1.5, subunit thus provides the primary and often sufficient requirement for functional, voltage-gated, channel opening, ion selectivity, and channel activation and inactivation (11).
SCN5A itself shows evidence of alternative promoter usage and splicing resulting in multiple transcripts of Nav1.5 protein resulting in differing functional properties. Nav1.5 may additionally undergo posttranslational glycosylation and phosphorylation by protein kinases A and C, and modification by tyrosine kinases and phosphatases. Nav1.5 may also be a regulatory target for intracellular Ca2+, calmodulin (CaM), and CaM-dependent protein kinase II (CaMKII) (963), as well as ROS and the cell NAD+/NADH ratio (see sect. VIII, A and C). INa is also modifiable by either indirect, PKA-dependent, and direct, PKA-independent mechanisms. The PKA-independent response may involve stimulation through G protein subunit-α (Gsα)-caveolin-3 binding resulting in access to caveolar-associated Na+ channels (963).
In ventricular myocytes, Na+ channels may exist in three subpopulations. These have distinct properties, localizations, and associated molecules (889). Nav1.5 channels at intercalated disks were associated with β2 and β4-subunits, ankyrin-G, synapse-associated protein 97 (SAP97), and junctional proteins (889, 1006). Genetic variants reducing ankyrin-G/Nav1.5 affected intercalated disk but not sarcolemmal Nav1.5 channels. They reduced INa and produced human or mouse BrS (716, 787). Nav1.5 demonstrated by recordings at the lateral and tubular membranes were associated with the α1-syntrophin PDZ (postsynaptic density protein/Drosophila disc large tumor suppressor/zonula occludens-1 protein) domain of syntrophin/dystrophin through the COOH-terminal, S-I-V motif of Nav1.5 β1 and β3-subunits (1063). Mutations deleting the COOH-terminal motif of α1-syntrophin affected lateral membrane but not intercalated disk Nav1.5. However, they also reduced INa (1063). There have been recent suggestions of differing kinetic properties of INa arising from Nav1.5 at the two latter sites. There is also a tetrodotoxin (TTX)-sensitive, Na+ channel subpopulation unlikely to reflect Nav1.5. However, this likely makes only minimal INa contributions (662).
Genetic alterations involving Nav1.5 and its associated molecules are correspondingly associated with a wide variety of cardiac arrhythmic disorders. These include BrS (see sect. VB; Refs. 32, 95, 189), progressive cardiac conduction disease (see sect. VB3; PCCD; Lev-Lenegre syndrome) (737, 1021), congenital LQTS3 (see sect. VIB; Ref. 1224), SND (see sect. VF; Ref. 100), and SIDS (575, 862; editorials in Refs, 456, 633, 985). The comparative studies of these using murine models implicated abnormalities in some or all the fundamental electrophysiological processes of conduction, depolarization, and repolarization in arrhythmic substrate. Single SCN5A mutations can also accompany combinations of as opposed to single clinical symptoms. These could include BrS with cardiac conduction disorder (609, 1027, 1073) SND and/or atrial standstill (1073) and LQTS3 (see sect. VIB7; Refs. 115, 412).
B. The Brugada Syndrome
1. Occurrence and inheritance
BrS is characterized by increased risks of arrhythmogenic SCD occurring particularly in middle aged (∼40–45 yr) males (147, 289, 912). Although relatively recently identified, BrS is implicated in 4–12% of unexpected sudden deaths. It accounts for up to 20% of deaths in patients with structurally normal hearts worldwide. This suggests a population incidence of such SCD of ∼0.05% (31). There is a greater male prevalence. There are also significant worldwide variations in incidence. Thus BrS may be the most common cause of sudden death in anatomically normal hearts below age 50 yr in South Asia (749).
BrS is inherited as an autosomal dominant trait with incomplete penetrance, so genetic testing may aid diagnosis. However, the known causative mutations are demonstrable only in ∼30% of cases. The most common mutation concerns SCN5A encoding Nav1.5, in 15–30% of BrS cases. There are links to almost 300 different mutations in the gene (537). Mutations in other genes are also associated with BrS. In addition to Na+ channel α-subunits (SCN5A and SCN10A; Ref. 453) and β-subunits (SCN1B, SCN2B, and SCN3B) (see sect. VG, 2–4), these include glycerol-3-phosphate dehydrogenase 1-like (GPD1-L) (see sect. VIIIC). Other genes involved encode proteins associated with the Na+ channel. These include GTP-binding nuclear protein guanine nucleotide release factor (RANGRF), sarcolemma associated protein (SLMAP), and plakophilin 2 (PKP2). There are also K+ (ABCC9, KCNE3, KCNJ8, HCN4, KCND3, and KCNE5) and Ca2+ channel associations (CACNA1C, CACNB2B, CACNA2D1, and TRPM4) (1003). Recently, murine knockouts of the naturally occurring protein inhibitor of Kv4.3 which underlies Ito, SEMA3A, have been identified with an arrhythmic BrS-like phenotype (130). Table 4 summarizes murine models used in studies of BrS phenotypes.
Table 4.
Murine variants recapitulating conduction abnormality
| Gene/Protein | Genotype | Phenotype | Reference Nos. |
|---|---|---|---|
| Na+ channel Nav1.5 (α-subunits) | |||
| Scn5a/Nav1.5; Na+ channel α-subunit | Scn5a+/− | BrS including fibrotic conduction changes | 734, 867, 1095 |
| Scn5a-1798insD | Overlap BrS/LQT3 syndrome | 208, 939, 1184 | |
| Serine of Nav1.5 SIV motif replaced by premature stop codon causing truncated ΔSIV | Loss of binding to PDZ domain of lateral membrane α1-syntrophin; reduced lateral membrane NaV1.5 and INa | 1063 | |
| Na+ channel Navβ (β-subunits) | |||
| Scn2b/Navβ2; Na+ channel, intercalated disk, β2-subunit | Scnb2−/− | Seizures; reduced neuronal Na+ channel density; cardiac phenotypes not yet examined. | 185 |
| Scn3b/Navβ3; Na+ channel, transverse tubular, β2-subunit | Scn3b−/− | Reduced Na+ channel function; monomorphic or polymorphic VT on programmed stimulation | 304, 402, 403, 452, 713 |
| Na+ channel-associated proteins | |||
| Ankyrin-G (affecting intercalated disk Nav1.5 and and spectrin) | Cardiac-selective ankyrin-G−/− | Reduced INa; altered AP waveform; bradycardia; reduced ventricular (QRS) conduction; ventricular arrhythmias following Na+ channel antagonism | 716, 787, 790, 889, 1006 |
| Synapse-associated protein-97 (SAP97) (affecting intercalated disk Nav1.5) | αMHC-Cre/Sap97-fl/fl: cardiomyocyte-specific SAP97 deletion | Reduced IK1, Ito, IKur; intact INa; slightly increased Nav1.5 expression | 355 |
| Desmosomal proteins | |||
| Plakophilin-2 | Pkp2+/− | Desmosomal abnormalities; reduced and modified INa; ventricular arrhythmias | 173, 174 |
| Desmoglein-2 | Dsg2-N271S | Reduced INa and conduction velocities | 952. |
| Kv4.3 channel mediating Ito | |||
| Semaphorin 3a (Sema3a). | Sema3a−/− (KO of naturally occurring protein inhibitor of Kv4.3 (underlies Ito) | Arrhythmic BrS-like phenotype | 130, 476 |
Note: For desmosomal proteins, see also Table 8.
2. The clinical BrS electrophysiological phenotype
BrS is characterized electrocardiographically by right precordial ST elevation, negative T waves, RV delay, and one of three types of repolarization pattern. The diagnostic type 1 pattern shows a coved ST segment elevation >2 mm followed by a negative T wave. Type 2 refers to a saddleback appearance producing a >2 mm ST segment elevation followed by a biphasic or positive T wave. Type 3 is characterized by a saddleback or coved ST elevation of <1 mm. There is also variability in clinical severity. In some cases the classical electrocardiographic type I characteristics can be elicited by administration of the INa blockers flecainide, ajmaline, or procainamide or appear under particular clinical or pharmacological circumstances including pyrexia (554).
BrS symptoms mainly appear during adulthood. The mean age for sudden death is around 40 yr, but there is a wide range of ∼2 days to 84 yr (33). Both the syncopal episodes and SCD are often preceded by fast polymorphic VT or VF. These often originate from the RV outflow tract (RVOT) (800). They are more common in males than females. This has been suggested to partly reflect sex-related differences in IK and ICaL (987, 1057). Males consequently show a more marked epicardial Ito-mediated notch often related to testosterone levels (318). In contrast, females have longer QT intervals and therefore APDs that may protect against BrS-associated arrhythmia (847) (see sect. VE).
Detailed mechanisms by which BrS leads to ventricular arrhythmia remain under discussion. Experimental studies in canine pharmacological ventricular preparations (1288) and some small clinical studies implicated a primary repolarization disorder (1051). The deep phase 1 notch, particularly in the RV epicardial AP, makes it susceptible to effects of reduced INa. This produces a steep RV transmural voltage gradient. This potentially causes epicardial reactivation by neighboring regions of myocardium with longer APs. The outcome would be a functional, phase 2, reentry. Alternatively, reduced INa might slow AP conduction and appears to do so particularly in the RVOT, in a depolarization disorder hypothesis (619, 811). This could implicate the RVOT in both the electrocardiographic abnormalities and the delayed epicardial conduction associated with potentially fatal ventricular arrhythmia in BrS (442, 766, 907). The RVOT might then act as a site for initiation of arrhythmias. The resulting regional differences in epicardial conduction velocity between the RVOT and RV might then trigger epicardial re-entrant excitation waves (766). This is discussed in detail in section VD1.
3. Anatomical abnormalities associated with BrS
Cardiac structure in BrS patients was initially thought to be normal. Several studies then reported progressive myocardial structural abnormalities (117, 940). Some patients showed a fatty replacement and fibrosis in the RVOT, despite normal LV anatomy (220). Nav1.5 changes, exemplified by SCN5A-D1275N, were also associated with ventricular dysfunction and structural changes including DCM (346, 758). The latter is consistent with known Nav1.5 interactions with cytoskeletal components (see sect. VA).
Nav1.5 abnormalities have also been associated with a heterogeneous group of inherited progressive conduction diseases (PCCD) often of unknown cause (367). The Lev-Lenegre variant manifests as progressive His-Purkinje conduction slowing with left (LBBB) or right bundle branch block (RBBB) and QRS complex widening. This can advance to complete atrioventricular block. PCCD is sometimes associated with syncope or SCD. Postmortem studies revealed diffuse fibrotic degeneration through the fibrous skeleton of the heart (644) or localized conduction tissue fibrosis (640). The earliest association of PCCD with Nav1.5 was observed in a family whose members showed progressive RBBB, LBBB, left anterior or posterior hemiblock, and long PR intervals (1021). Subsequent studies revealed a wide range of associated SCN5A mutations, as well as mutations in a range of other genes. These were implicated in loss of channel function that could result from abnormalities in channel trafficking into the membrane or gating, activation, an d/or inactivation behavior once the protein is inserted into the membrane, some of whose effects additionally were modified by Cx gene polymorphisms (737).
C. Experimental Models for Scn5a Insufficiency
1. Genetic models for Scn5a haploinsufficiency
BrS has been modeled in canine RV wedge preparations. However, these experiments entailed use of potentially nonspecific pharmacological manipulations to replicate the phenotypes under examination. Furthermore, they did not permit localization of arrhythmias between intact cardiac chambers or regions, including in particular the RVOT. Experimental systems directly replicating genetic modifications in BrS include the murine heterozygotic Nav1.5 haplo-insufficient Scn5a+/− mouse. The Scn5a+/− heterozygotes were generated by replacing exon 2 of the Scn5a gene with a splice acceptor (SA)-Gfp-PGK-neomycin cassette (867). Homozygous Scn5a−/− embryos died at midgestation. Scn5a+/− heterozygotes survived but demonstrated compromised atrial and atrioventricular conduction even at age 8–10 wk. Myocytes from 8–10 wk mice showed a ∼50% INa reduction compared with WT. Both the Scn5a+/− and the Scn5a-1798insD variants (see sect. VIB7) (1091) also proved useful as physiological models for both atrial and ventricular arrhythmic properties in human BrS (408, 632, 1095).
Loss of Nav1.5 function following Scn5a mutations resulted in a range of murine phenotypes that directly recapitulated clinical findings. These included SND, atrial arrhythmia, and progressive conduction disorders in addition to the ventricular phenotypes. Thus Scn5a+/− hearts replicated the ventricular, atrial, and sinoatrial arrhythmogenicity, and age-dependent fibrotic, properties observed in clinical BrS (408, 503, 632, 867, 968, 1095). Programmed electrical stimulation techniques demonstrated that Scn5a+/− hearts showed increased susceptibility to ventricular arrhythmias (867). These were accompanied by accentuations of the increases in EGD normally observed with progressively shortening S1S2 intervals (1095). The latter feature had been previously clinically associated with reentrant substrates (461, 1011, 1012). These investigations also recapitulated clinical observations of increased and decreased arrhythmic incidences following challenge by the normally antiarrhythmic agents flecainide and quinidine, respectively. These were correspondingly accompanied by accentuations or reductions in the EGD alterations (743, 1095). Studies on atrial arrhythmic susceptibility then suggested that 50% of Scn5a+/− but 0% of WT hearts had spontaneous atrial arrhythmia. This was eliminated by both flecainide and quinidine (240) (see sect. VF1). Their accompanying alterations in electrophysiological (740) and pharmacological (742) properties thus throw light on mechanisms for arrhythmia particularly in relationship to altered AP conduction.
2. Biophysical features of the murine Scn5a+/− system
Combined biophysical and molecular biological analyses separately examining the LV and RV of Scn5a+/− ventricles confirmed their selective loss of Nav1.5 function. They established a basis for clarifying the arrhythmic mechanisms that follow (Figure 5) (741). Thus Scn5a+/− ventricles showed reduced Nav1.5 mRNA and protein expression. These changes were more marked in the RV than the LV. These findings concurred with results from patch-clamp analyses of INa. These revealed reduced maximum INa (Figure 5, A and B). Scn5a+/− myocytes also showed correspondingly decreased AP upstroke velocities [(dV/dt)max] and maximum INa densities than WT, changes again more noticeable in the RV compared with the LV (Figure 5C). There were concordant alterations in the density of the late Na+ currents (INaL). Despite normal steady-state dependences of activation upon test voltage (Figure 5D), INa showed a negative shift in the voltage dependence of its inactivation function (Figure 5E). These findings are consistent with depolarization abnormalities that would be expected to slow action potential conduction.
Figure 5.

Sodium (Na+) currents in Scn5a+/− hearts. A: Na+ currents (INa) normalized to cell capacitance from myocytes from left (LV) and right ventricles (RV) of wild-type and Scn5a+/− hearts. Corresponding current-voltage relationships (B), maximum INa (C), and activation (D) and inactivation (E) curves with Boltzmann fits are shown. *Effect of genotype. #Effect of cardiac ventricle. [From Martin et al. (741).]
In contrast, both Scn5a+/− and WT RVs showed similarly higher Kv4.2, Kv4.3, and KChIP2 expression levels than the LV. Both Scn5a+/− and WT ventricles accordingly showed greater RV than LV transient outward current (Ito) densities (741).
These findings correlate with studies at the organ level in Scn5a+/− hearts as described below. These further clarified the clinical evidence for BrS being primarily a RV condition. They additionally distinguished contributions to this property from alterations in ion channel expression and consequent electrophysiological abnormalities. These could result in abnormal depolarization (1091) or repolarization (39), fibrotic change, or reduced Cx expression (see sect. VE; Refs. 501–503, 1180).
3. Electrocardiographic features of Scn5a+/− hearts
Intact anesthetized Scn5a+/− mice showed ECG features also compatible with conduction abnormalities that could be directly attributed to reduced Na+ channel function. The initial studies observed a second-degree atrioventricular block and increased PR intervals (742). The changes were directly translatable to clinical observations in BrS patients with identifiable SCN5A mutations (717, 1052, 1148). Flecainide increased these PR intervals and the QRS durations particularly in the Scn5a+/− mice. This also paralleled the clinical effects of flecainide in normal and BrS patients (899, 1052). Scn5a+/− mice also showed accentuated QT dispersions, suggesting an increased repolarization heterogeneity. These were further increased by flecainide but not quinidine. This recapitulated their respective clinical proarrhythmic and antiarrhythmic effects, and their accompanying temporal QT dispersion, in clinical BrS (477, 899). The prolongations in PR and QRS intervals increased with age (968), particularly in male in contrast to female Scn5a+/− mice (501–503, 1180).
4. Action potential waveforms in Scn5a+/− ventricles
Endocardial and epicardial MAP recordings in isolated Langendorff-perfused Scn5a+/− hearts demonstrated similar RV and LV endocardial APDs that nevertheless exceeded their corresponding epicardial APDs. APDs were shorter in the Scn5a+/− compared with WT particularly in the RV. This resulted in greater RV than LV transmural repolarization gradients as quantified through ΔAPD90 values, differences particularly marked in Scn5a+/− hearts. Flecainide and quinidine exerted similar patterns of pharmacological effects in Scn5a+/− and WT hearts. In both cases, flecainide shortened whereas quinidine lengthened the APDs. It did so for all four, endocardial and epicardial, and right and left, ventricular regions but particularly so for the RV epicardium. These changes accentuated the reduced RV transmural gradients that occurred even before pharmacological challenge. They thereby resulted in strongly positive RV but not LV transmural gradients in the flecainide-treated Scn5a+/− hearts which has been suggested as being potentially arrhythmogenic (see sect. VIA2). In contrast, quinidine lengthened the APDs in all ventricular regions in the Scn5a+/− hearts, particularly in RV epicardium. This decreased the RV transmural gradients which were now similar to the gradients observed in untreated WT ventricles (743).
5. Conduction and refractoriness in Scn5a+/− ventricles
Increased RV transmural repolarization gradients in Nav1.5-deficient ventricles combined with localized AP shortening could potentially reduce the AP dome. This would potentially increase the likelihood of the early epicardial phase II reexcitation previously implicated in arrhythmia in BrS (1288). However, such early reentry mechanisms would require corresponding reductions in epicardial refractory periods, if the reexcitation were to take place. Normal canine (243, 329) and human (624) hearts indeed showed a concordance between changes in APDs and changes in ventricular effective refractory periods (VERPs). However, VERPs can be selectively affected by factors that spare APD. Thus contrasting changes in VERPs in BrS were reported in clinical (50) and cell expression studies (118). Contrasting APD and VERP changes occur under other arrhythmogenic circumstances including hypokalemia (977, 978, 980).
In murine Scn5a+/− ventricles, increased arrhythmogenicity accompanied shortened APDs and increased RV repolarization gradients. The corresponding VERPs were increased rather than decreased, particularly in the presence of arrhythmogenic Scn5a+/− phenotypes (734). Both WT and Scn5a+/− hearts also showed regional VERP heterogeneities, consistent with both clinical reports (703) and known Ito differences (1124). Computational analysis confirmed that these findings were consistent with the reduced numbers of Na+ channels available for reexcitation in Scn5a+/− hearts (930, 1090). Refractory period would depend on recovery of a critical number of then reactivatable Na+ channels. This level would be achieved at a greater interval following repolarization when total channel numbers were reduced. Furthermore, notwithstanding their contrasting proarrhythmic and antiarrhythmic effects, flecainide and quinidine both increased VERP. This is in agreement with their known effects in canine, rabbit, and human hearts (148). Quinidine exerted the greater effects, likely reflecting its K+ channel blocking actions in contrast to the predominant Na+ channel blocking effect of flecainide (1231). However, increased VERPs along the patterns found in Scn5a+/− ventricles are inconsistent with an arrhythmia initiated by early local phase 2 reexcitation (1288).
Nevertheless, the findings remained compatible with roles for VERPs, particularly their spatial variations, and the consequences of these for conduction of excitation, in reentrant substrate. The Scn5a+/− hearts, particularly their RVs, in addition to the greater VERPs, showed a greater conduction slowing following S2 stimuli than did WT, as expected from their reduced INa. This effect was accentuated by flecainide, and, although less so, by quinidine challenge, concordant with clinical findings (907). Thus, whereas flecainide produced a more marked conduction slowing, quinidine exerted greater effects in increasing VERP. Either could potentially produce conditions of a slowed conduction or even conduction block through such partially refractory tissue with heterogeneously increased VERPs sufficient to result in arrhythmic substrate. These results paralleled other findings in LQTS (59), SQTS (775) and ischemic experimental models (487), and modeling studies (1141). LQTS (1175) and arrhythmic postinfarct patients (781) similarly show increased refractory period dispersions and greater heterogeneities in tissue excitability and refractoriness (1231).
6. Steady-state analyses of action potential stability in Scn5a+/− ventricles
These differences in AP properties between Scn5a+/− and WT translated into differing ventricular electrical instabilities during protocols applying progressively incremented steady-state pacing rates. This could be observed with decreasing BCLs ranging between 130 and 30 ms (see sect. IIA4) (754). Such protocols ended with either an onset of refractoriness and a 2:1 block, or VT or VF. Scn5a+/− ventricles showed more frequent arrhythmic end points leading to VT, particularly in the RV (752, 754). Flecainide increased the frequency of such arrhythmic events. These events then took place earlier in the pacing protocol and therefore at longer BCLs. In contrast, quinidine diminished the frequencies of such events.
Scn5a+/− and WT ventricles also demonstrated differing onsets of alternans in MAP magnitude and duration at RV and LV, epicardial and endocardial sites before and after flecainide and quinidine challenge in the course of the pacing procedures. Scn5a+/− ventricles accordingly demonstrated longer BCLs and DIs corresponding to the onset of either transient (tr) or sustained (ss) APD (DI*) and AP amplitude (DI') alternans. Following flecainide challenge, the RV epicardium showed the greatest evidence for potential instability. It then showed an onset of alternans at longer BCLs and larger amplitudes than the corresponding endocardium. This would generate transmural heterogeneities and potential discordant alternans. These findings thus provided an empirical basis for implicating the RV in the observed clinical arrhythmogenesis (752, 754).
Quantitative analysis of the corresponding restitution properties (see sect. IIA4) similarly selectively implicated the RV epicardium in the Scn5a+/− proarrhythmic phenotype. Refractory properties, reflected in the DIERP associated with onset of either 2:1 block or arrhythmia, were indistinguishable between corresponding recording sites in WT and Scn5a+/− before pharmacological manipulations. However, DIERP was increased in both RV epicardium and RV endocardium in flecainide or quinidine-treated Scn5a+/− hearts. Onsets of instability, reflected in DI values corresponding to unity slope, DIcrit, in APD versus DI plots, were similar in WT and Scn5a+/−, at each recording site. However, Scn5a+/− RV ventricles showed steeper restitution functions than the corresponding WT. Flecainide then specifically destabilized Scn5a+/− RV, selectively increasing DIcrit (752, 754). This corroborated the alternans findings implicating RV epicardium in Scn5a+/− arrhythmia. This is consistent with suggestions that APD restitution is a predictor for arrhythmic tendency (274, 817).
In the above analyses, the relationship between alternans magnitude and restitution curve slope was a continuous second order one. This would contrast with the direct linear function containing an abrupt instability threshold predicted by voltage feedback mechanisms produced by AP repolarization and refractoriness properties (838). This prompted more complete analyses of electrophysiological instabilities which additionally explored for contributions from abnormalities in conduction velocities θ. Previous studies had implicated changes in the broadness of restitution functions representing effects on θ of similarly decreasing BCLs and consequently of DIs, as indicators of observed arrhythmia and alternans (929). These studies did not yield such findings. Nevertheless, they demonstrated that maximum achievable magnitudes of θ and DI corresponding to conduction failure correlated well with arrhythmic tendency, particularly in the Scn5a+/− RV epicardium. The latter was the case both before or following flecainide challenge. They also demonstrated θ alternans. Although less frequent than APD alternans, it abruptly appeared at pacing rates close to producing refractoriness. Finally, flecainide increased θ alternans specifically in Scn5a+/− RV epicardium (752).
7. Wavelength restitution analysis in Scn5a+/− hearts
The restitution analysis could then be further extended to consider indexes of excitation that reflected both θ, and recovery as represented by APD. This utilized the excitation wavelength, λ = APD × θ (see sect. IC3). Such wavelengths, λ, then fell nonlinearly with falling DI in agreement with previous reports (695). However, the variables in the plots of λ against DI were differently dimensioned in distance and time, respectively. They were consequently not amenable to feedback instability analysis. Nevertheless, a systems restitution analysis was made possible by expressing DI in terms of a resting, as opposed to excitation wavelength, λ0 = DI × θ. Comparisons of λ against λ0 gave rise to identically dimensioned plots of active and resting wavelengths. In these, the BCL variable was similarly transformed into a basic cycle distance, BCD = BCL × θ. It was then possible to analyze electrophysiological stability in terms of the wavelength parameters λ and λ0, thus incorporating the effects of BCL upon APD, latency, and DI. The resulting, more general, instability analysis then demonstrated differing, epicardial and endocardial, maximum λ in untreated hearts at low heart rates. Quinidine decreased these in WT endocardium and abolished endocardial-epicardial differences in Scn5a+/− hearts. These findings were consistent with quinidine exerting proarrhythmic effects in WT but antiarrhythmic effects in Scn5a+/− hearts (753).
Increased heart rates then resulted in an onset of λ-alternans, failed wave propagation, and re-entrant foci corresponding to a positive feedback condition at unity gradients in the λ-λ0 plots. This critical condition further corresponded to similar critical values of resting (λ0crit) and total, basic cycle distance (BCD) wavelengths through both genotypes and all recording sites and all conditions of flecainide or quinidine challenge. This analysis thus yielded a common critical instability point which would thereby correspond to achievement of both necessary and sufficient conditions for initiating arrhythmia. Common instability points also implicate particular intrinsic cellular mechanisms underlying excitation and resting wavelengths. These could arise from interactions between channel opening and voltage change (1236), or other signaling that may modulate channel properties potentially through altered Ca2+ (204, 1137) and ATP homeostasis.
Pacing rates at which instability occurred were indistinguishable between untreated Scn5a+/− and WT hearts. However, they were decreased by both flecainide and quinidine specifically in Scn5a+/− RV epicardium, and by quinidine challenge in WT RV endocardium. Finally, over the studied pacing rates, alternans magnitude agreed more closely with the gradient of the λ than the APD restitution function. Thus analysis of λ rather than its component variables of an isolated θ or APD provides an analysis closer to mechanisms underlying alternans, thereby better predicting arrhythmia (753).
These findings are consistent with simple schemes directly relating changes in wavelength parameters to the occurrence or otherwise of wavebreak reentry leading to arrhythmia when the propagating AP encounters heterogeneities. With a long wavelength, as the AP passes over the heterogeneity, the back of the propagating wave blocks and extinguishes any retrograde excitation while orthograde excitation continues. With a short wavelength, the back of the AP wave may traverse the heterogeneity before retrograde excitation passes through the unidirectional block. This generates a new propagating retrograde, potentially reentrant excitation wave (see sect. IC3).
8. Direct visualization of reentrant circuit formation in Scn5a+/− ventricles
The two main BrS arrhythmogenic mechanisms involving slowed conduction and repolarization heterogeneity proposed in the experiments above could be both directly visualized and integrated by contact multielectrode (0.5 mm) array (MEA) mapping techniques applied to the RV and LV epicardial surfaces (736). Use of spontaneously beating hearts before and following flecainide or quinidine challenge provided more realistic indications of in vivo epicardial electrophysiological patterns following physiological activation than possible with imposed pacing. Each electrode array site recorded biphasic electrograms comprising negative followed by positive deflections. Spontaneously beating WT hearts showed neither ventricular ectopic (VE) nor VT phenomena before, and only low frequencies of these following, pharmacological challenge. The low incidences of ventricular ectopic events and VT in Scn5a+/− markedly increased with flecainide but not quinidine challenge.
Electrophysiological features derived from MEA recording could be integrated with those obtained from MAP studies. Activation (ATs) and recovery times (RTs) corresponded to points of maximal negative slope (dV/dt)min of the initial and second negative deflections, respectively. Activation recovery intervals (ARIs), obtained from the AT-RT interval, correlated linearly with MAP measures of APD. Their dispersions were expressed as the AT, RT, and ARI differences (ATD, RTD, and ARID, respectively) between values of the first and last values in each set of electrograms of the arrays. Isochronal maps of these measures were then used to clarify the interactions between depolarization and repolarization abnormalities and the appearance and evolution of arrhythmic substrate through reentrant circuits.
The mapping studies directly demonstrated slowed conduction with dispersion of activation and lines of block in Scn5a+/− ventricles. LV and RV MEA recordings of excitation in both WT and Scn5a+/− hearts usually showed single planar wavefronts arriving at the epicardium with the intraepicardial vector directed from apex to base. However, WT ventricles showed similar LV and RV ATDs. Both increased in Scn5a+/− ventricles which additionally showed longer RV than LV ATDs. Flecainide, but not quinidine, increased the ATDs in the RVs of WT and LVs and particularly RVs of Scn5a+/− ventricles.
The mapping studies also directly demonstrated evidence for shorter, and increased dispersions of recovery in Scn5a+/− ventricles. Scn5a+/− ventricles thus showed shorter but more heterogeneous ARIs. ARIs and ARIDs indicating recovery time courses were similar in LVs and RVs of WT. However, ARIs were shorter and ARIDs increased in RVs of Scn5a+/− hearts. Flecainide decreased the ARIs whilst increasing the ARIDs. In contrast, quinidine increased ARIs and spared ARIDs in both RVs and LVs of both WT and Scn5a+/− hearts. Flecainide thus resulted in greater RV ARIDs in Scn5a+/− than WT hearts.
WT ventricles showed ARI maps closely similar in pattern to the corresponding activation maps. The longest and shortest ARIs, respectively, occurred in regions of earliest and latest regions of activation. In contrast, Scn5a+/− ventricles showed disorganized ARI maps without clear gradients. There was also greater RT heterogeneity with RTs spread over wider time courses in Scn5a+/− ventricles. Finally, WT hearts showed similar RTDs in the LV and RV that were spared by flecainide. In contrast, Scn5a+/− hearts showed increased RTDs in the LV and RV, with the RTDs additionally greater in the RV than LV. Flecainide increased these RTDs in the Scn5a+/− but not WT. Quinidine decreased them in both Scn5a+/− and WT.
These features promoted an initiation of VT, originating in the RV and arising from lines of conduction block leading to induction of reentrant circuits. Figure 6 illustrates the generation of a premature ventricular beat following arrival of activation at the RV epicardium. Results are shown for a flecainide-treated, spontaneously beating, Scn5a+/− heart leading to initiation of polymorphic VT. Such ventricular ectopics often occurred with increased RTDs in their preceding sinus beats. Figure 6, A–F, summarizes the sequence of events, mapped onto the ECG (Figure 6G, letters A–F). The last sinus beat shows close isochronal contours reflecting delayed arrival of epicardial activation (Figure 6A). Its corresponding repolarization map shows increased repolarization heterogeneities in all cases where premature beats led to the reentrant VT circuits that followed (Figure 6A″). A superimposed, premature ventricular activation produces a line of block. Impulse propagation flows around this (Figure 6B). A subsequent ventricular ectopic event initiates a circuit running anticlockwise (Figure 6C). This persists into the following beat, initiating VT (Figure 6D). VTs most frequently followed premature ventricular beats that were specifically synchronized with the T wave (Figure 6J). The continually changing line of block produces a nonstationary vortex accounting for the polymorphic nature of the VT (Figure 6, E and F). The VT then propagates as a wavefront across the LV from its initiation site in the RV (Figure 6I) (736).
Figure 6.

Re-entrant circuit initiation of ventricular arrhythmia in Scn5a+/− ventricle. A–F: right ventricular (RV) isochronal propagation maps in flecainide-treated Scn5a+/− heart illustrating initiation of ventricular tachycardia (VT). Thick black lines denote propagation block. Thin arrows denote lines of propagation. G: ECG trace with ventricular ectopic initiating polymorphic VT. H: part of the same ECG trace, with 8 electrogram traces, at the point of VT initiation. Electrogram numbers correspond to the channel numbers of the array marked in maps A–F. A: crowded isochronal lines in the last sinus beat and area of conduction slowing. A”: repolarization map of the last sinus beat with increased repolarization heterogeneity in the same area. B: premature ventricular beat superimposed on this leads to line of block with impulse propagation flowing around it. C: a second ventricular ectopic (VE) resulting in a reentrant circuit. D: the circuit continuing into the next beat to initiate VT. E and F: changes in the line of block that create a nonstationary vortex, causing polymorphic arrhythmia. I: propagation map, ECG, and electrogram traces of the VT propagating as a wave front across the LV from the RV. J: ECG trace of a VE occurring after the T wave. [From Martin et al. (736).]
D. Initiation Sites for Ventricular Arrhythmia in Scn5a+/− Hearts
1. The right ventricular outflow tract as a site for arrhythmia initiation in clinical BrS
In addition to outlining the basis for formation of arrhythmic substrate in BrS models, the previous section included evidence for preferential initiation of arrhythmia in the RV as opposed to the LV. Further evidence more specifically implicated the right ventricular outflow tract (RVOT) in this initiation of clinical arrhythmia in BrS. It also indicated that the murine Scn5a+/− system models both the human arrhythmia and its possible underlying mechanisms. The localization of the background depolarization and repolarization changes in RVOT as opposed to the remaining RV, and the LV, in BrS may reflect their differing embryological origins (1312). The RVOT originates from cells that also contribute to the slow conducting atrioventricular region (797). This may relate to suggestions for an existence of slow conducting tissue in the RVOT dependent on L-type Ca2+ channel rather than Nav1.5 activation (766), or of a further reduced Nav1.5 expression in the RVOT particularly with flecainide challenge (1183).
The RVOT has been implicated in both the clinical ECG abnormalities (see sect. VB) and a delayed epicardial conduction potentially associated with initiation of ventricular arrhythmia in clinical BrS (102, 218, 538, 766, 801, 907, 1148). These in turn were attributed to RVOT structural abnormalities such as fibrotic or fatty endocardial infiltration in BrS. Noninvasive clinical studies such as echocardiographic investigations for wall motion abnormalities (1148), body surface mapping (287, 491, 1148), signal-averaged ECGs (433), or tissue Doppler echocardiography indeed posed evidence for RVOT conduction delays (1148).
Some invasive RVOT endocardial studies demonstrated increased activation delays (531), electrogram prolongations, narrower ARI, and steeper restitution curves (619). A more restricted number of epicardial investigations demonstrated activation delays (811) and shortened RVOT ARIs on catheter recording in the great cardiac vein during ST elevation, as well as epicardial, but not endocardial spike-and-dome AP waveforms in BrS hearts during open chest surgery (605). Finally, explanted human BrS hearts showed VF associated with RVOT subendocardial rather than subepicardial reentry and RV activation slowing. In contrast, there was an absence of transmural repolarization gradients (218, 442). They also showed a reduced basal RV subepicardial local activation associated with the ST segment elevation (441). Both these studies also reported fibrosis and fatty infiltration in the RV. This could accentuate the current-to-load mismatch between the abnormal RVOT and more normal myocardial tissue already established by altered INa. This could explain the reduced conduction velocities and ECG patterns (441).
2. Murine Scn5a+/− hearts model the RVOT as an initiation site for arrhythmia in BrS
Comparisons of isolated Langendorff-perfused murine Scn5a+/− and WT ventricles provided a physiological basis for a possible RVOT involvement in the VT associated with BrS. These comparisons systematically compared arrhythmogenic tendency and the related electrophysiological parameters of VERPs, response latencies, electrogram durations (EGDs), APDs, the presence or absence of concordant or discordant alternans, and restitution curves. This involved making bipolar electrogram and MAP recordings at multiple RV and LV epicardial locations before and following flecainide and quinidine challenge (735). The bipolar electrogram measurements implicated the RVOT as an arrhythmogenic focus in Scn5a+/− hearts. Thus imposition of extrasystolic stimuli at progressively shorter intervals following regular pacing spikes elicited earlier occurrences of VT in the RVOT than the base of the LV in the Scn5a+/− but not the WT heart, which further showed only one VT episode. The same protocol demonstrated a greater degree of electrogram fractionation, previously used to assess the existence of arrhythmogenic reentrant pathways, in Scn5a+/− than WT ventricles. This was particularly so at the RVOT compared with the remaining recording sites.
The basis of these findings emerged from the MAP assessments of VERPs and conduction velocities. Both Scn5a+/− and WT showed marked apical-basal VERP gradients. However, these gradients were greater in Scn5a+/− than WT ventricles, specifically in the RV. Scn5a+/− ventricles also showed greater response latencies reflecting slowed conduction velocities than in WT. Both these differences were accentuated by flecainide challenge. Furthermore, the RVOTs showed shorter APDs and VERPs and steeper gradients of these values than elsewhere in both WT and Scn5a+/− hearts.
RVOTs of WT and Scn5a+/− hearts showed similar onsets of concordant alternans in incremental pacing protocols whether before or following pharmacological intervention. However, RVOTs of Scn5a+/− hearts showed more frequent transitions from concordant to discordant alternans. This in turn led to arrhythmogenesis, during pacing protocols involving progressively increased frequencies of stimulation. This tendency was exacerbated by flecainide and relieved by quinidine. In parallel with this, RVOTs of Scn5a+/− hearts showed the greatest heterogeneities in response latencies, and the largest APD and VERP gradients. The discordant alternans were always observed prior to any observed transitions into VT. Scn5a+/− RVOTs also showed steeper restitution curves. In these curves, the diastolic interval at which the gradient reached unity strongly correlated with the diastolic interval corresponding to the onset of discordant alternans (735).
3. Arrhythmic tendency, fibrotic change, and Nav1.5 expression in the RVOT of murine Scn5a+/− hearts
Further experiments correlated these arrhythmic features of the RVOT with key determinants for both arrhythmic tendency and conduction velocity. They demonstrated reduced Nav1.5 protein expression in both the RV and RVOT of Scn5a+/− compared with WT hearts. They also observed increased levels of fibrosis in the RVOT than the RV in both WT and Scn5a+/− (1336). Corresponding electrophysiological studies then first confirmed that stimulation at the RVOT as opposed to the remaining RV elicited increased arrhythmic incidences in Scn5a+/− but not WT hearts. Yet, Scn5a+/− showed greater VERPs than WT in both the RVOT and remaining RV. Furthermore, VERPs in the RVOT were greater in Scn5a+/− than WT, but were similar in the remaining RV in both Scn5a+/− and WT. These findings were subsequently paralleled in clinical reports (810). Both are at variance with a phase II reentry-induced arrhythmia, particularly if this were to take place in the RVOT of Scn5a+/− hearts (702, 1182) (see also sect. VC5).
In contrast, positive findings emerged from correlations of the arrhythmic properties with MEA assessments of AP conduction. First, overall AP conduction velocities were determined from simple regressions to planar fits to the local activation times of each MEA recording site. This correlated a reduced velocity with both the Scn5a+/− as opposed to the WT genotype, and in recordings from the RVOT as opposed to recordings from the RV. Second, the magnitudes of conduction velocities were averaged over the MEA recording sites by a local vector analysis. These magnitudes were reduced in the Scn5a+/− compared with WT, without variation between RV or RVOT recording sites. Third, dispersions in the conduction direction were assessed from the same vector analysis. The resulting dispersions were greater in the RVOT than in the RV, but there was then no influence of or interaction with, Scn5a+/− versus WT, genotype. Finally, these results were correlated with biochemical and histological results. Nav1.5 expression was correspondingly reduced in both RVOT and the remaining RV of Scn5a+/− as opposed to WT hearts. But interstitial fibrosis was greater in the RVOT than the remaining RV in both Scn5a+/− and WT hearts (1336).
E. The Scn5a+/− Genotype, Age-Dependent Fibrotic Change and Arrhythmogenesis
1. Age dependence of the BrS arrhythmic phenotype
BrS thus includes additional phenotypic features not directly explicable solely in terms of Nav1.5-related biophysical changes. As indicated above (sect. VB2) its clinical manisfestations are often postponed until adulthood giving an average age of SCD of 40 years (33). There is also a greater male prevalence (749). Nav1.5 haploinsufficiency is also associated with the fibrotic changes in the related condition of PCCD (Lev-Lenegre disease), which can also exist in an overlap syndrome with BrS (919) (see sect. VB3). Experimental studies in Scn5a+/− murine hearts recapitulated coexistent Nav1.5 deficiencies with fibrotic changes dependent on age and sex that would accentuate conduction and therefore arrhythmic disorders (500).
2. Sex- and age-dependent fibrotic phenotypes in Scn5a+/− hearts
Aged murine Scn5a+/− hearts demonstrated prolonged electrocardiographic P wave, PR and QRS durations, and evidence of spontaneous arrhythmic tendency (Figure 7A) (392). This accompanied a significant degree of ventricular fibrosis, decreased Cx43 expression, increases in the hypertrophic cardiac markers β-MHC and skeletal α-actin, and reductions in atrial levels of the atrial-specific gap junction protein Cx40 (642, 968). They showed elevated microarray and real-time PCR assays of Atf3 and Egr1 transcription factor (1180).
Figure 7.

Conduction and arrhythmic properties in ageing male Scn5a+/− hearts. A: lead II electrocardiographic traces obtained from anesthetized aged Scn5a+/− mouse showing spontaneous nonsustained ventricular tachycardia (VTs indicated by arrow). B: chest lead ECG complexes from young (a) and old (b) intact anesthetized male Scn5a+/− mice. The latter shows patterns of fragmented QRS complexes, indicating bundle branch block most frequently observed with Scn5a+/−. C and D: activation maps from five successive cardiac cycles in young male WT (C) and old male Scn5a+/− hearts (D). E: picrosirius red staining demonstrating ventricular fibrosis in 85-wk-old WT (a) and Scn5a+/− with mild (b) and severe fibrosis which appears red (c). F: corresponding INa records in ventricular myocytes from 12-wk-old WT (a) and mildly (b) and severely affected Scn5a+/− mice (c). G: frequency distributions of activation times in young male WT (a) and old male Scn5a+/− mice (b). [From Jeevaratnam and co-workers (501–503) and Leoni et al. (642).]
With epicardial MEA mapping (Figure 7, C and D), even young (3–4 mo) Scn5a+/− showed slowed RV conduction compared with the corresponding WT hearts, in an absence of demonstrable fibrosis or altered Cx43 expression. These findings were therefore primarily attributable to the direct biophysical consequences of a Nav1.5 haploinsufficiency (736). However, old WT mice showed slowed RV conduction associated with interstitial fibrosis. Old Scn5a+/− ventricles showed slowed conduction in both the RV and LV, severe reactive fibrosis, and downregulated Cx43 expression (1180). The Scn5a+/− mice then could additionally be stratified into subgroups with severe (QRS >18 ms) and mild (QRS ≤ 18 ms) ventricular conduction defects. This stratification correlated with the extent of fibrotic change and arrhythmic severity. These findings paralleled heterogeneities also observed in human SCN5A-mutated patients (Figure 7, E and F). Ageing conserved and accentuated this stratification, together with its related reductions in Scn5a mRNA expression and INa. QRS durations with even the mild Scn5a+/− phenotype then exceeded that of WT (642).
Systematic, quantitative studies then analyzed the effects of, and interactions between, genotype, age (3 vs. >12 mo) and sex upon both conduction and fibrotic change in murine Scn5a+/− hearts. Electrocardiographic PR intervals were longer in young female compared with young male WT and Scn5a+/− anaesthetized mice. However, these intervals increased with age with a development of evidence for RV conduction block on chest lead recording in male but not female Scn5a+/− hearts (Figure 7B) (502). In contrast, old female Scn5a+/− and WT hearts showed similar PR intervals (503).
In isolated Langendorff-perfused WT hearts, MEA mapping of activation times demonstrated sequential propagations of RV epicardial activation from a localized region. This suggested orderly propagation of such excitation either through the plane of the epicardium or from endocardium to epicardium. In contrast, Scn5a+/− hearts showed a more fragmented activation pattern (Figure 7, C and D; Ref. 501). This often included multiple random firing points consistent with greater likelihoods of reentrant proarrhythmic events between neighboring epicardial sites where these showed strongly different activation times (501).
Quantification of these activation times in terms of average and maximum activation delays indicated interacting effects on these of all three factors of genotype, age, and sex, with the greatest effects consequently observed in old male Scn5a+/− hearts. It was possible first to quantify temporal variations in activation time at any given recording site. These depended on genotype and not on age and sex. They specifically gave larger variations in activation times in old male Scn5a+/− than the corresponding old WT hearts. Second, it was possible to assess spatial dispersions of activation times among recording sites within any given cardiac cycle. These were influenced by interacting effects of all the three variates of genotype, age, and sex. Old male Scn5a+/− hearts then gave greater dispersions than either old male WT or the remaining Scn5a+/− groups (501).
Analysis of frequency distributions in these activation times attributed these differences to alterations in an initially bimodal activation time distribution reflecting distinct early and late activation phases (Figure 7G). Old male Scn5a+/− showed a reduced early and a correspondingly increased late component relative to distributions shown by young male Scn5a+/−, old female Scn5a+/−, and old male WT. The corresponding morphometrically determined patterns of fibrosis then closely matched these electrophysiological findings among the experimental groups when these were sorted by genotype, age, and sex (502).
3. Regulatory consequences of Nav1.5 deficiency
Studies in murine Scn5a+/− hearts also suggested that the BrS condition itself accentuates anatomical, fibrotic, change. The latter could potentially contribute to its typically adult (∼40 yr) rather than juvenile presentation and primarily male prevalence (749). Na+ channels might thus exert regulatory functions distinct from their mediation of electrophysiological excitability (3, 408). As indicated previously, Na+ channels occur clustered with other proteins. Alterations in their properties or expression could therefore impact on expression or properties of other molecules. This could potentially include agents affecting fibroblast development. Nav1.5 haploinsufficiency correlated with upregulation of the stress inducible gene, Atf3, and the early growth response gene Egr1 (642, 968, 1180). In atrial preparations, Nav1.5 deficiency was associated with increased TGF-β1 and vimentin transcript expression. These could potentially increase collagen and fibroblast abundance resulting in interstitial fibrosis (408). Such effects could accentuate the biophysical consequences of Nav1.5 downregulation particularly in Scn5a+/− RV (741).
4. Possible combined effects of Nav1.5 haploinsufficiency and fibrotic change in ventricular arrhythmogenesis
Nav1.5 haploinsufficiency in the murine Scn5a+/− system thus appears to result in both biophysical and structural change, particularly in the RV. This is consistent with its overlapping proarrhythmic clinical manifestations in the form of BrS and PCCD. This would account for the clinical evidence. Table 5 summarizes the resemblances between human BrS and the murine Scn5a+/− model. The experimental findings suggest that it is the overlap of these changes that together resulted in accentuated arrhythmic substrate sufficient to produce spontaneous arrhythmic events particularly in aged male Scn5a+/−hearts.
In such a scheme, the Nav1.5 deficiency would produce a background electrophysiological ion channel abnormality compromising conduction of excitation. This itself produces arrhythmic substrate if unmasked by flecainide or ajmaline challenge. This underlying phenotype could then interact with a coexisting tissue disruption arising from fibrotic change with age. This would be the effect of Na+ channels exerting regulatory or metabolic functions in addition to those of electrophysiological excitability particularly in male Scn5a+/− resulting in preferential expression of slowly at the expense of rapidly conducting pathways. Fibrotic change would compromise ephaptic, gap junction-mediated, myocyte-myocyte coupling. It could thereby compromise AP conduction between cells particularly with the accompanying Nav1.5 downregulation (741). It would also distort tissue geometry dispersing conduction through alternative conducting branches prolonging conduction pathlengths (see also sect. IXA3).
Arrhythmic tendency would then be the consequence of a combination of Nav1.5 haploinsufficiency and an evolving structural change in Nav1.5-associated arrhythmic disease including both progressive cardiac conduction defect and BrS. The background Nav1.5 haploinsufficiency would produce arrhythmic substrate typically unmasked by flecainide or ajmaline challenge that accentuates an underlying conduction defect (500). Cardiac fibrotic changes with age, accentuated in males, could then accentuate the arrhythmic substrate to an extent sufficient to lead to arrhythmic events (Figure 8). Such suggestions based on studies in murine hearts have proven translatable into clinical findings (810).
Figure 8.

Development of arrhythmia resulting from a combination of Nav1.5 haploinsufficiency and structural change in progressive cardiac conduction defect (PCCD) and Brugada syndrome (BrS). Nav1.5 haploinsufficiency produces a background electrophysiological defect in conduction. This results in arrhythmic substrate typically unmasked by flecainide or ajmaline challenge. Cardiac fibrotic changes occur with age, particularly in males. This further compromises action potential propagation. Superimposition of the two factors sufficiently compromises conduction, thereby accentuating arrhythmic substrate to lead to arrhythmic events. There is thus a combination of biophysical and structural change with age, particularly in males, that results in arrhythmia. [From Jeevaratnam et al. (500).]
A recently developed SCN5A-E558X porcine model provided a large animal system with features that were comparable with the arrhythmic phenomena in murine hearts (868). The porcine model showed heart rates, cardiac size, AP shape, and autonomic innervations as well as cardiac depolarization and repolarization kinetics similar to that of humans. Langendorff-perfused porcine SCN5A-E558X heart reproduced many clinical loss-of-Nav1.5 function phenotypes. They thus confirmed many features modeled by murine Scn5a+/− hearts. They shared increased incidences of pacing-induced or spontaneous VF as well as diminished Nav1.5 protein expression and INa. However, the porcine hearts did not show the macroscopic and microscopic structural changes shown by murine Scn5a+/− hearts (see sect. VB3). Such a system could nevertheless complement the mouse models. Nevertheless, their ease in genome manipulation, and shorter reproductive cycles (∼20 vs. 111 days gestation), will favor murine models particularly in disease occurring in later adulthood given longer porcine (between 15–17 yr) compared with murine (∼48 wk) life expectancies.
F. Nav1.5 Haploinsufficiency and Abnormal Sinoatrial and Atrial Electrophysiology
1. Physiological changes accompanying atrial and sinus node disorder
Loss of Na+ channel function is also associated with SND and atrial tachyarrhythmia. SAN pacemaker function in both humans and other mammals normally declines with age, decreasing intrinsic heart rates and increasing SAN conduction times (272, 731). Sinus node dysfunction (SND) causes sinus bradycardia, pause or arrest, atrial chronotropic incompetence, and SAN exit block. Its incidence increases exponentially with age, accounting for ∼50% of the million permanent pacemaker implants per year worldwide (272, 731). It may involve a range of ion channel changes (see sect. IIIA). Ageing guinea pig SANs showed reduced Cav1.2 expression reflected in a greater sensitivity of their electrical activity to L-type Ca2+ channel block. They also showed decreased Cx43 expression in the vicinity of the SAN (517). This could increase SAN conduction time and incidences of SAN exit block (518). Ageing rat SANs showed decreased Kv1.5 expression that might explain increased SAN APDs (1115). They also showed altered RyR2 expression. This could also compromise SAN pacemaking through actions on its Ca2+ clock mechanism of pacemaking (1115, 1298)
2. Sinus node disorder in murine Scn5a+/− hearts
Familial SND is also associated with ∼13 human Nav1.5 mutants (634, 637). These were variously associated with apparently normal (e.g., SCN5A-L212P, P1298L, DelF1617, and R1632H), reduced (e.g., E161K, T220I, and D1275N), or undetectable (e.g., T187I, R878C, G1408R, and the truncated variants W1421X, K1578fs/52, and R1623X; Refs. 379, 1339) peak INa in expressed heterologous systems.
Physiological evidence implicated both neuronal Nav1.1 and cardiac Nav1.5 channels in normal SAN function, suggesting that Nav1.1 was involved in pacing activity, and Nav1.5 was primarily involved in AP conduction through the SAN and from the SAN to surrounding atrial myocytes. It also suggested that these separate but related functions predominated in the smaller central pacing cells and larger conducting peripheral SAN cells, respectively. Nevertheless, Nav1.5-mediated INa can modify heart rate by modifying impulse propagation within the SAN and from the SAN to the atrium (632).
Voltage-clamp studies of INa, and SAN tissue staining with anti-Nav antibodies distinguished TTX-sensitive INa associated with Nav1.1 and TTX-resistant INa associated with Nav1.5 expression (632, 635). In intact WT mouse hearts, selective Nav1.1 block by low (50 nM) TTX concentrations reduced pacemaker rates in isolated SAN and isolated SAN pacemaker cells (635, 714). The TTX-sensitive INa could be demonstrated by action potential clamp experiments to normally occur late in diastolic depolarization and during the AP upstroke (635). Block of both Nav1.1 and the relatively TTX-resistant Nav1.5 by mM TTX concentrations additionally slowed or even blocked AP conduction through the SAN periphery from its leading pacemaker site, and to surrounding atrium (635). Additionally, although Scn5a+/− mice retained normal circadian variations in telemetrically determined heart rates, they replicated the reduced overall mean rates and SA block observed in clinical SND (52).
Langendorff-perfused Scn5a+/− hearts showed sinus bradycardia, slowed SA conduction, and sinoatrial exit block. Isolated Scn5a+/− SAN and atrial preparations showed slowed SA conduction and frequent SA conduction block in mapping experiments (Figure 9A) (632). These findings correlated with results from patch-clamped Scn5a+/− SAN cells which showed normal steady-state Nav1.5 activation and inactivation characteristics but reduced maximum INa compared with WT. These whole animal, tissue, and cellular findings could then be unified by modeling of the resulting roles for Nav1.5 in AP propagation through the SAN and from SAN to atria. These replicated the changes in pacing rate and demonstrated that these changes were brought about by effects of loss of Nav1.5 function upon the electrical coupling between peripheral SAN and atrial cells (632).
Figure 9.

Variations in conduction properties through the isolated sinoatrial node of Scn5a+/− hearts with age. A: electrograms and activation mapping in young WT (a) and young Scn5a+/− (b) as well as old WT (c) and old Scn5a+/− atria (d). B and C: sinoatrial node (SAN) cycle lengths (B) and sinoatrial conduction times in the four experimental groups (C). D–F: fibrosis-regulating, TGF-β1, vimentin, and Nav1.5 gene expression and interacting effects of ageing and genotype. D: mRNA abundance assessed by real-time polymerase chain reaction. E: correlations between gene expression of TGF-β1 and Nav1.5 (a), vimentin and Nav1.5 (b), and vimentin and TGF-β1 (c). F: TGF-β1 gene expression initiated by Nav1.5 inhibition with Nav1.5 antibody. TGF-β1 gene expression was increased by Nav1.5 antibody treatment at the transcriptional (a) and protein levels (b) in both human neonatal myocytes and fibroblasts. [From Hao et al. (408).]
Ageing and associated fibrotic processes interacted with Nav1.5 haploinsufficiency in modifying SAN electrophysiological properties. This finding paralleled the associations between Scn5a+/− and ventricular fibrotic changes outlined above (see sect. VE4). Nav1.5 disruption increased P-wave duration, RR, PR, and QRS intervals, and SAN recovery times. It interacted with ageing in decreasing heart rate variability with the greatest effects in old Scn5a+/− mice. Aging and Nav1.5 insufficiency exerted both individual and interacting effects in increasing cycle lengths and SA conduction times in ex vivo, isolated, SAN preparations (Figure 9, B and C) (408), as well as in Langendorff-perfused hearts (391).
Both the Scn5a+/− phenotype and ageing correspondingly resulted in downregulated Nav1.5 expression to the following sequence of extents: old Scn5a+/− > old WT > young Scn5a+/− > young WT. They interacted in increasing collagen and fibroblast levels and the corresponding expression of the fibrotic modulator transforming growth factor-β1 (TGF-β1) and the fibroblast marker vimentin. The greatest effects were observed in aged Scn5a+/− (Figure 9D, a and b). Nav1.5 expression levels consequently negatively correlated with TGF-β1 and vimentin levels (Figure 9E). Increased TGF-β production by both cardiac myocytes and cardiac fibroblasts could be replicated by acute application of either Nav1.5-E3 antibody or Na+ channel blocker (Figure 9F). This was consistent with previous reports that pharmacological block of electrical activity in Na+ channels upregulated TGF-β activity in rat primary myotubes (1159). Such activity could form a cell signaling pathway leading to fibrotic change and atrial electrophysiological pathology in other clinical circumstances (454). Nav1.5 haploinsufficiency in combination with aging was also accompanied by altered expression in a wide range of ion channels and regulatory genes in the SAN associated with ageing. Ageing itself accentuated the reduction in Nav1.5 expression found in Scn5a+/−. There were accompanying reductions in the tbox transcription factor Tbx3. This is known to regulate the SAN pacemaker gene expression program and decrease expression of a wide range of SAN ion channels.
3. Atrial arrhythmia in murine Scn5a+/− hearts
Finally, BrS (10–30%) is associated with increased tendency to AF. This often clinically precedes its ventricular manifestations in patients with a severe type I phenotype (26, 607). Langendorff-perfused Scn5a+/− hearts similarly showed an increased atrial arrhythmogenicity (240). This was greatest in young Scn5a+/− atria. Scn5a+/− atria particularly from aged hearts showed slower AP conduction whether measured by MEA mapping or PR intervals (391, 503). Young Scn5a+/− hearts showed the most prolonged monophasic APDs (240). However, the aged Scn5a+/− atria showed increased AERPs and therefore showed the smallest APD90/AERP. Atria from young Scn5a+/− hearts also showed the greatest prolongations in electrogram duration following extrasystolic stimulation. Slowed conduction and increased APD90/AERP ratio thus correlate with the increased AF tendency particularly shown by young Scn5a+/− hearts (390).
G. Arrhythmic Changes With Abnormalities in Na+ Channel β-Subunits
1. Features of Navβ subunits
SCN5A forms macromolecular or signaling complexes with a number of further proteins (3, 852). These could directly or indirectly modify INa, Nav1.5 localization, or both (759). Of these, Navβ subunits are prominent integral, single-spanning, membrane glycoprotein components of Nav signaling complexes (844). Their extracellular NH2-terminal region contains a V-type immunoglobulin domain with a short ”neck“ connected to a single α-helical transmembrane domain and intracellular COOH-terminal region (170, 484, 759, 1111). Navβ1 and Navβ3 have the more closely related primary sequences. They noncovalently bind to the α-subunit possibly at similar COOH-terminal sites close to domain IV. This contrasts with the disulfide bonding suggested for Navβ2 and Navβ4 (239). Atomic resolution X-ray crystallographic studies of human Navβ3 and Navβ4 suggest that Navβ3 and therefore possibly Navβ1 could trimerize. If so, Nav α-subunits could correspondingly coassemble into oligomeric complexes stabilised by Navβ3 (or Navβ1) subunits. This assembly could then potentially have effects upon electrophysiological properties through possible interactions between component Nav1.5s (765, 814).
Navβ subunits do not themselves mediate ion permeation. They rather perform multiple functions in cell adhesion, recruitment of scaffolding or anchoring proteins including ankyrins (485, 759). They may also be involved in trafficking Na+ channels to the cell membrane, thereby increasing peak INa, and negatively shifting steady-state activation and inactivation consistent with biophysical effects of providing screening charge (138, 231, 403). Navβ subunit mutations are associated with inherited conditions including epilepsy, neuropathies, cardiac conduction diseases, and some cancers (815).
Navβ1 and Navβ3 subunits occur in transverse tubular and surface membranes, whereas Navβ2 and Navβ4 subunits occur at cell membranes in the regions of the intercalated disks (713). Navβ2 and Navβ3 are discussed here in terms of the arrhythmic effects of Na+ channel loss of function. Loss of Navβ1 function has been associated with experimental LQTS, despite clinical BrS-like phenotypes, and is discussed in section VIB7. Navβ4 subunits produce negative shifts in activation (353, 1310). Their loss of function is associated with arrhythmogenesis accompanied by increased late INa (10). They are discussed in section VIB8 also in connection with LQTS.
2. Scn2b-encoded Navβ2 subunit variants
Scn2b-encoded Navβ2 subunits occur in intercalated disks particularly in the epicardium of human atria and ventricles (336, 713) in association with SCN5A (261). SCN2B-R28Q and SCN2B-R28W patients often show lone AF (1234). SCN2B-R28W patients additionally show prolonged PR intervals and right precordial ST elevation. Oocyte Navβ2 coexpression with neuronal Nav1.1 α-subunits increased INa, but left INa kinetics unchanged (484). It exerted negative shifts in voltage dependences of both activation and inactivation. These alterations were additive to the Navβ1 effects, to extents dependent on glycosylation (514). Navβ2 reduction or absence decreased INa. In CHO expression systems, SCN2B-R28G reduced INa density and shifted activation; SCN2B-R28W positively shifted inactivation. In both cases, INaL was not affected (1234). Scnb2−/− mice showed seizures accompanied by reduced neuronal Na+ channel density (185). However, the possible cardiac phenotypes have not been examined.
3. Scn3b-encoded Navβ3 subunit variants
SCN3B-encoded Navβ3 subunits occur in both ventricles (452) and atria of human hearts (852). Navβ3 subunits may function in both trafficking of SCN5A to the surface membrane and modifying its electrophysiological properties (482). Their presence increases, and absence reduces, INa. There are variable effects on INa voltage dependence and kinetics. Heterologous oocyte Navβ3 coexpression with SCN5A increased INa threefold, accelerated recovery from refractoriness, and positively shifted steady state voltage-dependent inactivation whilst sparing activation properties (304). Human SCN3B-L10P is implicated in the BrS7 variant (452), SCN3B-R6K, L10P and M161T in familial AF (852), and SCN3B-V54G in idiopathic VF (1165). Studies in TSA201, CHO-5, or HEK-293 expression systems related these conditions to reduced INa and reduced cell surface SCN5A expression. In the last case, these features were partly rescued by coexpression with Navβ1 (452, 1165). SCN3B-V36M and SCN3B-A130V are associated with SIDS and AF, respectively. Studies using HEK-293 expression systems related these to decreased INa with either increased late relative to peak INa (SCN3B-V36M) or without decreased SCN5A surface expression (SCN3B-A130V) (1110, 1221).
Scn3b-encoded Navβ3 subunits also normally occur in mouse ventricles (403, 452) and atria (402, 852) and sheep ventricular and Purkinje but not atrial tissue, where they are similarly associated with SCN5A (304). Homozygous Scn3b−/− hearts showed the expected absence of Scn3b mRNA but showed elevated Scn1b mRNA, and increased RV Scn5a mRNA. Isolated, Langendorff-perfused Scn3b−/− hearts showed increased incidences of monomorphic or polymorphic VT on programmed electrical stimulation. Their accompanying electrophysiological abnormalities showed both parallels with and contrasts from those of Scn5a+/− hearts (403). They showed a consistently reduced Na+ channel function compatible with BrS-like phenotypes (Figure 10). Whole cell patch-clamped Scn3b−/− myocytes showed reduced maximum peak INa (approximately −60 vs. −90 pA/pF in WT) (Figure 10A). This suggested reduced membrane expression or proportions of functional Na+ channels. Inactivation curves were shifted in the negative direction relative to those in WT (Figure 10B). However, the time courses of recovery from inactivation were not changed (Figure 10C).
Figure 10.

The Scn3b−/− exemplar for cardiac arrhythmogenesis. A: INa traces from WT (a) and Scn3b−/− myocytes (b) showing peak INa that is significantly smaller in Scn3b−/− myocytes. B: Boltzmann fits to steady-state voltage dependence of activation (squares) and inactivation (circles) in WT (filled symbols) and Scn3b−/− (open symbols). Differing voltage dependence of inactivation in Scn3b−/− compared with WT particularly between holding voltages of −70 and −40 mV. C: recovery from inactivation in WT (filled symbols) and Scn3b−/− (open symbols). D: bipolar electrogram waveforms from programmed electrical stimulation at the longest (a, c) and shortest (b, d) S1–S2 intervals in WT (a, b) and Scn3b−/− hearts (c, d). The latter hearts showed consistently longer waveforms. E: representative monophasic action potential (MAP) recordings from the WT (a) and their shortening in Scn3b−/− hearts (b). F: ECG recordings obtained from WT (a) and Scn3b−/− mice (b). Scn3b−/− mice showed slower heart rates and prolonged PR intervals. c: Some ECGs from Scn3b−/− mice showed ventricular QT complexes occurring independently of regularly occurring atrial P waves, i.e., third degree heart block. G: atrial tachycardia (AT) resulting in regular deflections at a higher frequency (a) and atrial fibrillation (AF) resulting in irregular deflections at a higher frequency following atrial burst pacing (ABP) in Langendorff-perfused Scn3b−/− hearts (b). The ventricular spikes result in the larger, and the atrial spikes in the smaller deflections. c: PES-induced VT beginning as a monomorphic then deteriorating into polymorphic VT in a Scn3b−/− heart preparation. [From Hakim and co-workers (402–404).]
Bipolar electrogram recordings obtained during programmed electrical stimulation yielded deflections whose latencies mapped onto demonstrated conduction curves, suggesting lengthened electrogram durations compared with WT, particularly at the shortest S1S2 intervals. This suggested reduced AP conduction velocities (Figure 10D). Scn3b−/− ventricles also showed shorter VERPs than WT. MAP recordings demonstrated reduced endocardial and epicardial APDs. However, the resulting endocardial-epicardial differences, ΔAPD90s, were indistinguishable from those of WT (Figure 10E). Finally, both flecainide and quinidine selectively reduced arrhythmic incidence in Scn3b−/− relative to WT hearts without affecting ΔAPD90, likely through increasing VERP (404).
Sinoatrial and conduction properties in Scn3b−/− complemented these findings. They also suggested a compromised Na+ channel-mediated excitability and conduction. Scn3b mRNA and protein were expressed in the atria of WT but not Scn3b−/− hearts. ECGs from Scn3b−/− mice showed slower heart rates, and prolonged P waves and PR intervals (Figure 10F). Spontaneously active Langendorff-perfused Scn3b−/− hearts showed abnormal atrial electrophysiological properties and partial or complete atrioventricular block. Scn3b−/− hearts also showed atrial tachycardia and fibrillation on atrial burst pacing and longer sinus node recovery times than WT hearts (Figure 10G, a–c) (402).
H. Nav1.5 and Scaffolding Proteins
Nav1.5 is also associated with scaffolding proteins. In addition, two PDZ domain-scaffolding proteins, SAP97 and α1-syntrophin, mediate reciprocal interactions upon cardiomyocyte membrane expression levels of Nav1.5 and Kir2.1; the latter underlies the inward rectifying IK1 (1262). Recent evidence from effects of inheritable mutations suggest similar additional interactions involving plakophilin-2, ankyrin-G, dystrophin, syntrophin, and caveolin-3 (1262).
Of these, 1) caveolin-3 (Cav-3) promotes formation of cavelike membrane structures (65) and organization and concentration of particular molecules interacting with caveolin. Its precise association with Nav1.5 is uncertain (861). It may produce an adrenergic INa upregulation mediated by G proteins (1301). Cav-3 mutations are associated with HCM (415) (see also sect. VIC3). 2) Mutations in the cytosolic scaffolding protein α1-syntrophin (SNTA1) (12) increase INaL. They are associated with LQTS12 (1158) and SIDS (194). These are considered in detail in section VIC1.
3) The scaffolding protein ankyrin-G may connect ion channels, including Nav1.5, to the spectrin-actin cytoskeleton (99). Ankyrin polypeptides are known to be critical to ion channel and transporter targeting in both excitable and nonexcitable cells. Ankyrin-G interaction with Nav1.5 is disrupted by the SCN5A-E1053K mutation resulting in BrS (787). Studies on cardiac-selective ankyrin-G−/− mice suggested that ankyrin-G is a functionally important intercalated disc receptor for both Nav1.5 and βIV spectrin. The COOH-terminal domain of βIV spectrin in turn associates with the Nav1.5 regulator CaMKIIδ. Ankyrin-G-cKO mice were bradycardic and showed cardiac conduction abnormalities, QRS prolongation, and ventricular arrhythmias following challenge by Na+ channel antagonists. Their myocytes showed reduced INa and decreased AP amplitudes and (dV/dt)max. Nav1.5 expression was decreased particularly at intercalated discs. This accompanied a reduced βIV spectrin recruitment to intercalated disc membranes. In common with mouse qv4J hearts carrying βIV spectrin lacking its COOH-terminal domain, they showed defective CaMKIIδ targeting and defective CaMKIIδ regulation of INaL. There was additionally a reorganization of the resident desmosome protein plakophilin-2. The latter is critical to intercalated disc integration with the intermediate filament-based cytoskeleton and lethal arrhythmias in response to β-adrenergic stimulation (716). However, such reduced ankyrin-G expression did not affect peripheral sarcolemmal Nav1.5.
4) The desmosomal proteins plakophilin 2 (PKP2) and desmoglein-2 (DSG2) are localized to intercalated discs associated with Nav1.5 (1007). Mutations in both are associated with human ARVC (see sect. IXD, 2 and 5) (251). However, VF and SCD often occur prior to the onset of morphological change. Experimental studies attributed this to cross-talk between desmosomes and the Na+ channel complex. Murine Pkp2+/− hearts showed ultrastructural but not histological or anatomical abnormalities, with sporadic or absent desmosomes, and unevennesses in intercellular spaces. INa was reduced in maximum amplitude in both Pkp2+/− and with a number of heterozygous missense mutations (173). Voltage dependences of activation were unchanged, but there were negative shifts in steady-state inactivation and slowed recoveries from inactivation. Flecainide affected electrocardiographic parameters in anesthetized animals and ventricular conduction in Langendorff-perfused hearts. It reduced myocyte INa to greater extents in Pkp+/− than WT. It triggered ventricular arrhythmias and death in anesthetized Pkp2+/− but not WT animals (174). Hearts from Dsg2-N271S mice similarly showed reduced INa and conduction velocities (952).
5) In common with α1-syntrophin, the membrane-associated guanylate kinase (MAGUK) protein synapse associated protein-97 (SAP97) includes PDZ domains. SAP97-M861T has recently been associated with BrS (1262). However, myocytes from a murine model with a cardiomyocyte-specific Sap97 deletion showed reduced IK1, Ito, and IKur but unaltered INa yet slightly increased NaV1.5 protein expression, indicating a requirement for further experiment (355).
I. Idiopathic Ventricular Fibrillation
A small proportion (∼5%) of sudden adult death cases had classically occurred in an absence of identifiable hereditary causes. However, recent reports indicate that a significant proportion of these share the J-point or ST-segment elevations with BrS (784). Some of these cases may be associated with abnormal Ca2+ (CACNA1C, CACNB2, and CACNA2D1) or KATP channel (KCNJ8) genes (156). Some of the remaining cases of idiopathic VF may involve the DPP6 gene which regulates Ito (932). No murine genetic models yet exist for these conditions.
VI. RECOVERY FROM EXCITATION AND ARRHYTHMIC DISORDER
A. Action Potential Repolarization and QT Prolongation
1. Arrhythmogenic clinical long QT syndromes (LQTS)
In addition to altered AP activation and conduction (see sects. IV and V), arrhythmic substrate can also arise from alterations in AP repolarization and refractoriness. The latter can arise from alterations in Na+ channel recovery from fast or slow inactivation and development of late Na+ currents, INaL. It could also result from changes in the magnitude or time course of Ca2+ and K+ currents that normally are activated following the initial Na+ channel activation that triggers the AP. AP repolarization and recovery from refractoriness can thus be prolonged by persistent or increased inward depolarizing, plateau INa or ICaL, or compromised repolarizing, IK, currents. The resulting delayed myocardial repolarization clinically results in LQTS (563, 692). The characteristic prolonged ECG QT intervals accompany an increased predisposition to atypical ventricular, torsades de pointes, arrhythmias, and SCD (549). The incidences of these phenomena appear to correlate with the extent of this QT prolongation (918). Some LQTS variants are also associated with early onset AF (515).
Acquired forms of LQTS can follow a number of clinical conditions as well as a wide range of medications (37, 313, 334). The latter often produce LQTS through reducing IKr (953). Hereditary LQTS were first described as the autosomal recessive Jervell and Lange Nielsen (JLN) syndrome accompanied by congenital sensorineural deafness (506) and the more common autosomal dominant Romano-Ward (RW) syndrome (549, 961, 1232). However, only ∼50% of LQTS patients have known mutations (1123a, 1197). The remainder may be associated with genetic conditions causing abnormal ion channel function often with incomplete penetrance and phenotypic overlaps (144, 224, 358, 1031, 1253). LQTS has been associated with genetic modifications in Na+ channel α-subunits (Nav1.5 encoded by SCN5A, giving LQTS3), their associated Navβ4 subunits (Navβ4 encoded by SCN4B: LQTS10), various K+ channel α-subunits mediating IKs (Kv7.1 encoded by KCNQ1: LQTS1), IKr (Kv11.1, encoded by KCNH2: LQTS2), IK1 (among others, Kir2.1, encoded by KCNJ2: LQTS7 or Anderson-Tawil syndrome) and IKAch (Kir3.4, encoded by KCNJ5: LQTS13), the K+ channel β-subunits MinK and MiRP (encoded by KCNE1 and KCNE2: LQTS5 and LQTS6), and Ca2+ channel α-subunits [Cav1.2 encoded by CACNA1C: LQTS8 or the Timothy Syndrome (TS)]. LQTS is also associated with abnormalities in the cyoskeletal proteins. The latter include caveolin-3 (encoded by CAV3: LQTS9), A-kinase anchor protein 9 (encoded by AKAP9: LQTS11), and α1-syntrophin (encoded by STNA1: LQTS12). All these variants can present as a Romano-Ward syndrome. Some LQTS1 and LQTS5 can also present with Jervell and Lange-Nielsen syndrome. The clinical prevalence of inherited LQTS is ∼0.01% of the population. The most common, LQTS1, LQTS2, and LQTS3, account for 45, 45, and 5%, respectively, of inherited and genotype-confirmed LQTS cases (1030).
2. Electrophysiological phenomena associated with LQTS arrhythmia
Clinical and experimental evidence associates LQTS-mediated arrhythmogenesis, exemplified in murine models of the kind listed in Table 6, with two potentially proarrhythmic electrophysiological abnormalities (827). First, the normally smooth time course of AP repolarization can be interrupted. Where this follows phase 2 plateaus, EADs result. EADs were experimentally observed following exposure to drugs implicated in acquired LQTS, or catecholaminergic or hypoxic challenge, and in genetically modified murine hearts modeling human LQTS, particularly under bradycardic conditions (1128). EADs have been clinically observed in both bradycardic hypokalemic conditions (1058, 1059) and in congenital LQTS patients (1058). EADs have been attributed to a prior conditioning by plateau currents under conditions of AP prolongation combined with L-type Ca2+ channel recovery from inactivation within the voltage-window range of such recovery. The resulting L-type Ca2+ channel reactivation then produces depolarizing current. If it involves sufficient numbers of cardiomyocytes, this produces ectopic beats. The latter can trigger polymorphic VT when they occur within a proarrhythmic myocardial substrate (498, 499). Other suggestions for mechanisms producing EADs invoked increased INaL (416) or INCX (1204).
Table 6.
Transgenic murine models associated with long QT syndromes
| Protein/Gene | Genotype | Ion Current | Clinical Condition/Phenotype | Reference Nos. |
|---|---|---|---|---|
| Channel α-subunits: Na+ channel variants | ||||
| Nav1.5/Scn5a | Scn5a-ΔKPQ/+ | INaL | LQTS3; prolonged APD | 416, 843, 1095, 1128, 1129 |
| Scn5a-F1759A | INaL | Atrial and ventricular arrhythmia | 1214 | |
| Scn5a−1798insD/+ | INaL | Overlap syndrome: SND, conduction disease, BrS and LQTS3 | 115 | |
| Channel α-subunits: K+ channel variants | ||||
| Kv4.2/Kcnd2 | Kcnd2-DN | Ito,f | Prolonged APD | 558, 687 |
| Kv1.4/Kcna4 | Kcna4−/− | Ito,s | Normal APD | 558, 687 |
| Kv4.2/Kcnd2 and Kv1.4/Kcna4 | Kv4.2-DN × Kv1.4−/− | Ito,f and Ito,s | Prolonged APD | 558, 687 |
| Kv11.1/Kcnh2 | Merg1−/− | Embryologically lethal | 53, 688 | |
| Merg1+/− | ?IKr | LQTS2; prolonged apical and basal APDs and VERPs | 994 | |
| Merg1b−/− | ?IKr | Normal APD; episodic sinus bradycardia | 627, 1307 | |
| hERG-G628S (dominant negative) expression | ?IKr | Normal QT durations | 53 | |
| Kv7.1/Kcnq1 | Kcnq1−/− | IKs | LQTS1; Jervell and Lange-Nielsen syndrome; prolonged QT intervals in intact animal | 167, 1138, 1222 |
| Human KCNQ1 isoform 2 overexpression | IKs | Prolonged QT interval (dominant negative effects on murine Kcnq1 isoform 1) | 254 | |
| Kv1.5/Kcna5 (ventricular) | Cardiac Kv1.1 NH2-terminal fragment overexpression | Islow | Prolonged QT intervals; spontaneous nonsustained VT | 321, 688 |
| Kv1.5/Kcna5 (atrial) | Kcna5-E375X/+ | IKur | Atrial AP prolongation, EADs, and AF | 853 |
| Kir2.1/Kcnj2 | Kcjn2−/− | IK1, | Anderson-Tawil syndrome (LQTS7); broader APs; spontaneous APs | 1144, 1323 |
| Channel α-subunits: Ca2+ channel variants | ||||
| Cav1.2/Cacna1c. | Cacna1c-G406R/+ | ICaL | LQTS8;Timothy Syndrome (TS) | 56, 192, 277, 1086 |
| Cacna1c-G406R/Akap−/− | Normal phenotype | 192, 1150 | ||
| Channel β-subunits | ||||
| Navβ1/Scn1b | Scn1b−/− | INa and INaL | Human: BrS; mouse: prolonged QT and RR intervals (increased Nav1.5, INa and INaL; altered Ca2+ homeostasis) | 694 |
| Navβ4/Scn4b | Scn4b−/− | INa or INaL | LQTS10; resurgent INa/increased INaL | 373, 713 |
| MinK/Kcne1 | Kcne1−/− | IKs | LQTS5; Jervell and Lange-Nielsen syndrome | 64, 276, 1130, 1195 |
| MirP/Kcne2 | Kcne2−/− | IKslow1, Ito,f | LQT6: prolonged APD | 956 |
| Cytoskeletal proteins | ||||
| Ankyrin-B | Ankyrin-B+/−; ankyrin-B +/E1426G | LQTS4; abnormal Ca2+ homeostasis; triggered events, prolonged QT intervals; VT; atrial arrhythmias | 225, 361, 788, 790 | |
| Ankyrin B−/− | Deficient Kir6.2 membrane expression and decreased IKATP | 182, 649 | ||
| Caveolin-3/Cav3 | Cav3−/− | LQTS9 (transverse tubular abnormalities) | 337, 401, 434, 779 | |
In contrast, where the membrane potential is unstable following full AP repolarization, DADs result. DADs accompany conditions of disrupted Ca2+ homeostasis. The latter in turn can follow challenge with pharmacological agents including cardiac glycosides or catecholamines, or genetic conditions such as CPVT (see sect. VIIC). DADs have been attributed to transient inward currents (Iti) resulting from increased inward INCX or Ca2+-activated Cl− current [I(Ca)Cl] following excessive SR Ca2+ release. In common with EADs, DADs potentially result in triggered beats initiating arrhythmia where the displacements in membrane potential they produce reach the threshold for reexcitation (498, 499).
Second, enhanced dispersion of repolarization can potentially generate arrhythmogenic reentrant circuits (19, 38). Normal cardiac repolarization takes spatial directions running from epi- to endocardium and apex to base. These reflect longer endo- than epicardial and basal than apical APDs (687, 1228, 1287). The latter are the consequence of differing myocardial repolarization rates reflecting varying local densities of ion channels mediating repolarizing K+ currents. The epi-/endocardial transmural gradients themselves contribute to a coordinated process of initiation of, followed by recovery from, electrophysiological excitation (558, 563, 981).
As indicated in section IIB3, mouse ventricles show shorter, triangulated, APs (∼30–80 ms), as opposed to the prolonged waveforms AP that contain distinct plateau phases in larger mammals including humans (∼150–400 ms, respectively) (Figure 1D) (236). AP repolarization is thus dominated by the fast, Kv4.3- and Kv4.2-mediated Ito,f and the more slowly inactivating Kv1.4-mediated Ito,s, components of the transient outward current Ito and includes smaller ICaL contributions (Figure 1, B and D).
The dispersions of repolarization in murine hearts correspondingly arise from spatial variations in Ito giving gradients of excitation and recovery that similarly enhance the overall cohesiveness of cardiac activation (826, 989, 1228). Of Ito components, Ito(fast), mediated by the pore-forming Kv4.3/Kv4.2 (267) and its regulatory β-subunit KChIP2 (604, 876), are expressed along the apical-basal, epicardial-endocardial, and interventricular axes of adult murine hearts (1108). In contrast, Ito(slow), mediated by Kv1.4, appears confined to the interventricular septum (151, 1284). Region-specific myocardial Kv4.2 and KChiP2 distributions were assessed using both standard manual and laser capture microdissection giving 80- to 100-μm width myocardial slices particularly useful in analyses of small murine hearts. Quantitative RT-PCR measurements were assessed against control, uniform, transmural distributions of Kir2.1. There were no apex-base expression gradients but greater RV than LV, and greater epicardial than endocardial Kv4.2 expression levels in both LV and RV (1124). The laser capture microdissection went on to demonstrate similar, though less marked, LV and RV, KChIP2 transmural gradients. These observations contrasted with an absence of spatial gradients in the other selected K+ channel genes. These Kv4.2 and KChIP2 expression differences could thus underly Ito gradients between RV and LV, and epicardium and endocardium, but not apex and base in adult mouse ventricles (151). These findings parallel the overall transmural current density gradients found in ventricles of larger mammals (875), although human and canine Ito differences may primarily reflect KChIP2 expression gradients (964, 965). In both events, alterations in such gradients have been associated with myocardial disease states (863).
Nevertheless, manipulations involving Ito accomplished alterations in membrane potential recovery, refractory periods, and their spatial variations as well as the occurrence of triggered activity that paralleled those shown by the more prolonged APs shown in larger animals. Murine hearts thus remain valuable models for understanding mechanisms of arrhythmogenesis more broadly, including arrhythmic phenomena associated with AP repolarization.
Perfused, canine ventricular wedge preparations of larger mammalian hearts (36) have been reported to contain a further, midmyocardial cell (or M-cell) layer. This is located at varying locations within the thickness of the ventricular wall (35). Such M-cells showed the greatest repolarization times. This was attributed to a decreased IKs but increased INaL and the greatest APD sensitivity to bradycardia and IKr antagonists. This property results in an overall increase in transmural dispersion of repolarization (TDR). Thus the difference between longest and shortest APD would then be the difference between M cell and epicardial APDs (1056). This property had been implicated in arrhythmogenecity in BrS and both congenital (1053) and acquired LQTS (37, 772). However, the contribution of M cells to myocardial repolarization and arrhythmogenesis in intact hearts remains under discussion (40, 216, 859). Furthermore, although observed in isolated cells and ventricular tissue preparations, the existence of M cells in intact porcine (955), canine (859), or human hearts is uncertain (1106).
Langendorff-perfused mouse hearts did show regional APD heterogeneities, as reflected in differences between epi- and endocardial APD90s in common with larger mammalian hearts. The availability of murine models with specifiable genetic modifications made it possible to directly relate mutations seen in LQTS patients to physiological mechanisms of cardiac arrhythmias (560, 563). These issues are explored in specific examples in section VI, B4, E4, F2, and F3.
B. Arrhythmic Properties and Na+ Channel Recovery From Excitation
1. The late Na+ current
Arrhythmogenic gain of Na+ channel function can result from SCN5A mutations that result in a slowing of INa inactivation and rates of its recovery from inactivation. This is thought to be the basis of clinical LQTS3 (98, 416, 843). It could also be acquired in association with epileptic conditions (125). In either case, the resulting increased APDs and QT intervals predispose the myocardium to EADs and arrhythmogenesis (235, 1223). Normally, a gradually increasing late Na+ current, INaL, accompanies the AP potential plateau. LQTS3 is associated with increases in this normally small INaL (806) whether in acute or chronic, or inherited or acquired, pathological settings. This type of INaL could result from alterations in the gating, trafficking, or cytoskeletal anchoring of the Na+ channel. It results in both triggering events and arrhythmic substrate (88).
Increased INaL thus can prolong the APD and increases its variability. This enhances the genesis of, and arrhythmic triggering by, EADs (444, 499). In addition, it produces increases in temporal and the spatial dispersions of repolarization. These in turn increase APD restitution curve gradients and the development of APD alternans, produce local conduction block, to result in reentrant arrhythmic activity (381, 891). The increased inward INaL also decreases repolarization reserve or causes diastolic depolarization. Both enhance automaticity in partially depolarized cardiomyocytes. Finally, increased INaL causes increases in [Na+]i. This potentially induces reverse-mode NCX (1062). This could elevate diastolic [Ca2+]i, and thereby promote spontaneous SR Ca2+ release, Ca2+ waves and DADs at more negative voltages, and EADs at more positive voltages (162, 444, 988). The consequent elevations in [Ca2+]i potentially activate CaMKII and its protein phosphorylation effects. These could enhance spontaneous RyR2-mediated SR Ca2+ release (1203) and INaL (51, 467).
INaL itself can be activated by CaMKII. This action is localized to a Ser-571 phosphorylation site located in the Nav1.5 DI-DII linker. Studies in Scn5a knock-in murine models whose Nav1.5 carries a phosphomimetic Ser571 (S571E) mutation, or in which the phosphorylation site is ablated (S571A), suggested that Ser-571 specifically regulates INaL but not other channel properties linked to CaMKII. The resulting increased INaL altered repolarization and intracellular Ca2+ homeostasis. These had proarrhythmic consequences (363). INaL is also modified by more direct interactions with CaMKII through Nav1.5-associated proteins. CaMKII associates with a COOH-terminal motif in actin-associated βIV-spectrin protein involved in CaMKII-mediated phosphorylation of the subpopulation of voltage-gated Na+ channels residing in the cardiomyocyte intercalated discs (467, 716). This may modulate not only steady-state Na+ channel inactivation and recovery from inactivation, but also the magnitudes of INaL through separate Nav1.5 modulation pathways (467, 1209). Modeling studies suggested positive-feedback interactions in which CaMKII causes increases in often arrhythmogenic INaL in turn increasing [Na+]i and [Ca2+]i and resulting in further activation of CaMKII (105). CaMKII may also complex with SAP97. This may define its interaction with Kv4.3 α mediating Ito (295).
2. Long QT3 syndrome, LQTS3
The arrhythmic features of LQTS3 differ from those of BrS. This is exemplified in their responses to antiarrhythmic agents (914). The class IA cardiotropic agent quinidine is proarrhythmic and the class IC agent flecainide is antiarrhythmic in LQTS3 (805, 954). This directly contrasts with the respective antiarrhythmic effects of quinidine and proarrhythmic effects of flecainide in BrS (150). LQTS3 also differs from other LQTS variants in that 39% of fatal arrhythmias occur during sleep or rest (954). In contrast, arrhythmia in LQTS1 and LQTS2 is associated with stress or arousal (1029). Furthermore, in LQTS3, β-adrenoceptor agonists paradoxically exert antiarrhythmic effects. LQTS3 patients derive less benefit from β-adrenoceptor antagonist therapy than do LQTS1 and LQTS2 patients (915).
3. Murine LQTS3 exemplars: Scn5a+/ΔKPQ hearts
In WT murine hearts, the sea anemone, Anemonia sulcata, toxin and INaL agonist ATX-II produced effects resembling an acquired LQTS3. It slowed intrinsic heart rate, prolonged PR and QT intervals, increased MAP durations, and increased sinus node recovery times. Its application resulted in an appearance of EADs, DADs, and rapid, repetitive ventricular tachyarrhythmias and sinoatrial bradyarrhythmias. ATX-II slowed sinus node pacemaking. This resulted in in bradycardic arrhythmias in isolated sinoatrial preparations. The INaL antagonist ranolazine (10 μM) inhibited such effects (1275). This finding suggests potential translational, antiarrhythmic, therapeutic application (385).
Two murine genetic, LQTS3, models were independently produced by targeted deletion of ΔKPQ1505-1507 residues in the Nav1.5 inactivation domain (416, 843). Scn5a+/ΔKPQ ventricular myocytes showed prolonged AP waveforms (Figure 11A), increased peak INa despite normal Nav1.5 expression, increased INaL (Figure 11, B and C), and EAD events (Figure 11, D and E) (416). These features were replicated in patient-specific induced pluripotent stem cells harboring a SCN5A mutation for the condition (707). They were alleviated by the INaL antagonist ranolazine (333).
Figure 11.

Separation of contributions to arrhythmogenesis from EADs and transmural APD gradients in Scn5a+/ΔKPQ hearts. A: microelectrode recordings showing action potential prolongation in Scn5a+/ΔKPQ myocytes. B and C: prolonged recovery tail currents after 100 ms test pulses to +40 mV from a −120 mV holding potential in WT (B) and Scn5a+/ΔKPQ myocytes (C). D: action potential (AP) waveforms with early afterdepolarizations (EAD) generating inward late tetrodotoxin (TTX)-sensitive INaL in myocytes in an action potential clamp (E). F and G: epicardial monophasic AP recordings from spontaneously active Scn5a+/ΔKPQ hearts showing multiple EADs and nonsustained VT (F), abolished by 1 μM nifedipine (G). H and I: patch-clamp studies in isolated LV ventricular myocytes from Scn5a+/ΔKPQ hearts demonstrating that nifedipine (300 nM) completely suppressed the inward Ca2+ current following depolarizing steps from a holding voltage of −40 mV to 10 mV (H). In contrast, nifedipine had no effect on inward Na+ currents in response to depolarizing steps from −100 mV to −40 mV (I). J–L: number of hearts showing VT arrhythmia (J), percentage of monophasic action potentials showing EADs in Scn5a+/ΔKPQ hearts (K), and mean ± SE endocardial and epicardial APD90 values and ΔAPD90 in Scn5a+/ΔKPQ (L) at different nifedipine concentrations (0 nM, 1 nM, 10 nM, 100 nM, 300 nM, and 1 μM). M: effects of nifedipine (1 μM) on VERPs of Scn5a+/ΔKPQ and WT hearts. [From Head et al. (416) and Thomas et al. (1128).]
Isolated, Langendorff-perfused, Scn5a+/ΔKPQ hearts showed increased arrhythmogenecity on programmed electrical stimulation imposing extrasystolic S2 stimuli at progressively decreased S1S2 intervals following trains of pacing S1 stimuli (Figure 11, F and G). These studies yielded conduction curves for which paced electrogram fractionation analysis demonstrated increased EGDs compared with WT (416, 1095, 1096). Similar findings were previously associated with clinical reentrant substrate and arrhythmogenesis in both HCM (1009) and LQTS (1011) (Table 5). Abrupt rate accelerations transiently increased ventricular APDs with potentially arrhythmic effects (843). Pharmacological challenge altered arrhythmic tendencies in parallel with its clinical effects. Thus, in the murine Scn5a+/ΔKPQ hearts, flecainide exerted antiarrhythmic and quinidine exerted proarrhythmic effects. This directly contrasted with the effects of these agents in Scn5a+/− hearts (1095, 1096). Arrhythmogenesis persisted with propranolol perfusion which further increased EGD, reflecting accentuated reentrant substrate (416).
4. Spatial repolarization heterogeneities and triggering events in Scn5a+/ΔKPQ hearts
Monophasic AP waveform recordings in Langendorff-perfused Scn5a+/ΔKPQ hearts related the arrhythmias at the whole-heart level to abnormal ventricular myocardial EADs that could potentially act as arrhythmic triggers. Thus Scn5a+/ΔKPQ but not WT hearts showed EADs that were often followed by VT. They also showed spatial differences in electrophysiological recovery properties that could additionally potentially result in arrhythmic substrate. The latter took the form of abnormal transmural repolarization gradients.
Thus Scn5a+/ΔKPQ hearts showed increased epicardial APD90 (∼60 vs. ∼47 ms in WT) despite normal endocardial APD90 (∼52–54 ms in both Scn5a+/ΔKPQ and WT hearts). The increased epicardial APD possibly reflected regional differences in INaL in Scn5a+/ΔKPQ hearts. This c ould provide the necessary critical voltage ”window“ that has been described for L-type Ca2+ channel reactivation (1128). In addition, the preferential prolongation of epicardial APD90 in Scn5a+/ΔKPQ resulted in abnormal transmural repolarization gradients. This was quantified by the difference, ΔAPD90. Such ΔAPD90 changes showed a pattern that was the inverse of that in WT. Epicardial APD now exceeded endocardial APD. Endocardial repolarization consequently preceded epicardial repolarization, the reverse of the transmural epicardial to endocardial repolarization sequence normally found in WT (1128). Hearts from larger mammalian models made to pharmacologically replicate LQTS3 similarly showed altered EADs and transmural repolarization gradients (1056).
It was possible separately to implicate EAD phenomena in the triggering of arrhythmia, and alterations in ΔAPD90 in the generation of reentrant substrate by examining their respective sensitivities to pharmacological agents. First, nifedipine was useful as a specific dihydropyridine L-type Ca2+ channel antagonist (1190). It exerted antiarrhythmic effects in several experimental animal models (28, 821). This contrasts with the actions of phenylalkylamine Ca2+ channel blocker verapamil used in previous pharmacological investigations (16, 773) which also affects INa and IKr (1330). Nifedipine at progressively increasing concentrations (10 nM to 1 μM) correspondingly decreased the incidences of both EADs and arrhythmias evoked by programmed electrical stimulation in Scn5a+/ΔKPQ hearts in parallel (Figure 11, J and K). However, it did so without altering either epicardial or endocardial APD90, the resulting ΔAPD90 (Figure 11L), or VERP (Figure 11M) in either Scn5a+/ΔKPQ or WT. Thus suppression of EADs alone sufficed to suppress arrhythmogenesis without alterations of underlying re-entrant substrate. Patch-clamp studies in isolated ventricular myocytes confirmed that these effects of nifedipine were the result of alterations in ICaL but not INa (Figure 11, H and I) (1128). This makes it likely that alterations in SR-Ca2+ loading might also contribute to the arrhythmic phenotype (664).
Second, treatment with varying concentrations of the β-adrenoceptor antagonist propranolol made it possible empirically to separate distinct contributions of EADs, and arrhythmic substrate arising from repolarization gradients, to arrhythmogenesis in Scn5a+/ΔKPQ hearts (1129). Thus low propranolol concentrations (100 nM) suppressed both spontaneous and provoked arrhythmias. It also both suppressed EADs and reduced epicardial APD. The latter change accordingly corrected the repolarization gradient abnormalities in Scn5a+/ΔKPQ hearts. In contrast, high (1 mM) concentrations eliminated both EADs and spontaneous arrhythmias. However, it increased susceptibility to provoked arrhythmogenesis, while also prolonging epicardial yet reducing endocardial APDs (1129). This would exacerbate the abnormalities in repolarization gradient shown by Scn5a+/ΔKPQ hearts. The latter findings concur with similar differential actions of propranolol upon ventricular epicardial and endocardial APs previously reported in canine ventricular wedge preparations (593). Together these findings provide a physiological basis for clinical reports that β-adrenoceptor block is relatively ineffective in suppressing arrhythmia in LQTS3. This is in contrast to their use to suppress arrhythmia in LQTS1 and LQTS2 (915).
Third, agents specific for K+ channel as opposed to Ca2+ homeostatic function furnished a final separation of the contributions of EADs acting to trigger, and repolarization gradients providing substrate for, persistent cardiac arrhthymia (445). Thus the KATP channel opener nicorandil exerts antiarrhythmic effects in clinical LQTS. It reduces QT intervals and spatial repolarization gradients (17, 324, 1056). It also suppreses APD alternans in idiopathic LQTS (335). Nicorandil at concentrations of ∼20 mM has been reported to increase time-independent outward currents while sparing L-type Ca2+, delayed rectifier, or transient outward currents (430). Such concentrations reduced the incidence of arrhythmia provoked by programmed electrical stimulation in Langendorff-perfused Scn5a+/ΔKPQ hearts during MAP recording. This was accompanied by a reduction of LV epicardial but not LV endocardial APD90s in both Scn5a+/ΔKPQ and WT. This resulted in a restoration of normal transmural repolarization gradients in Scn5a+/ΔKPQ ventricles. Scn5a+/ΔKPQ hearts also showed greater epicardial critical intervals for reexcitation, reflected in APD90-VERP differences, than WT hearts, but these were reduced by nicorandil (445).
Finally, alterations in electrophysiological properties of the Purkinje cell conducting system (1163) in the Scn5a+/ΔKPQ variant were consistent with increased incidences of triggering effects. Isolated ventricular and Purkinje cell myocytes could be distinguished using offspring from crosses of Scn5a+/ΔKPQ or WT, with contactin2-Egfp BAC. The latter murine variants express a fluorescent reporter gene within the Purkinje fiber network. Whole cell patch-clamp studies have proven possible in murine cardiac Purkinje cells (780). These demonstrated increased INaL in both Scn5a+/ΔKPQ (Egfp+) Purkinje cells and Scn5a+/ΔKPQ (Egfp−) ventricular myocytes. Scn5a+/ΔKPQ Purkinje cells showed the greater increases in INaL and APD, as well as frequent EADs not observed in the ventricular myocytes, features reversible by mexiletine. The latter events correlated with repetitive oscillations of microfluometrically measurable [Ca2+]i (490).
5. Temporal heterogeneities and arrhythmic phenomena in Scn5a+/ΔKPQ hearts
Electrophysiological activity in Scn5a+/ΔKPQ hearts also showed greater temporal heterogeneities in the form of epicardial and endocardial APD90 alternans, particularly at increased pacing rates, than WT. The derivation of epicardial and endocardial restitution curves from such experiments made it possible to derive the values of DIcrit at which the slopes of such curves exceeded unity. The latter point is known to correlate with an onset of arrhythmic phenomena (see sect. IIA4). In control WT hearts, both quinidine and flecainide increased DIcrit. This would agree with the observed correspondingly increased incidences of arrhythmic activity. Both Scn5a+/ΔKPQ and Scn5a+/− hearts similarly showed greater DIcrit in parallel with their increased arrhythmic activity. Furthermore, quinidine (1 μM) and flecainide (1 μM) challenge then exerted contrasting effects on DIcrit. Thus, in Scn5a+/ΔKPQ hearts, quinidine increased and flecainide decreased the DIcrit values in parallel with their observed proarrhythymic and antiarrhythmic effects. In contrast, in Scn5a+/− hearts, quinidine correspondingly decreased and flecainide increased DIcrit in parallel with decreased and increased arrhythmic activity respectively (983). Nicorandil reduced the epicardial and endocardial DIcrit values to levels indistinguishable from untreated WTs through an overall flattening of their restitution curves, it correspondingly reduced arrhythmogenicity in Scn5a+/ΔKPQ hearts (445).
6. Atrial phenotypes in Scn5a+/ΔKPQ hearts
Murine Scn5a+/ΔKPQ hearts also showed age-dependent atrial arrhythmic phenotypes. Both young and aged Scn5a+/ΔKPQ hearts showed slowed sinus rates compared with corresponding WT. They thus showed an overlap in phenotype with Scn5a+/− hearts. Atrial arrhythmic tendencies in young Scn5a+/ΔKPQ were indistinguishable from or even reduced relative to young WT. However, they were increased in aged Scn5a+/ΔKPQ compared with WT (241, 392). These findings were explicable in terms of alterations in the properties of atrial AP initiation, propagation, and recovery. MEA recordings thus demonstrated slowed intra-atrial conduction in Scn5a+/ΔKPQ (1276). Furthermore, atrial Scn5a+/ΔKPQ cardiomyocytes showed prolonged APD90 values and frequent EADs particularly with slow pacing (127, 241). Both these effects were rescued by ranolazine (<2 Hz) (639, 812). Atrial APDs and P wave durations were similarly prolonged in regularly paced Scn5a+/ΔKPQ relative to WT particularly with age (241, 392). AERPs were similar in young WT and Scn5a+/ΔKPQ but increased with ageing in WT but not Scn5a+/ΔKPQ. This left aged Scn5a+/ΔKPQ with the greatest APD90/AERP ratios and consequent arrhythmic substrate. These findings correlated with observations of a greater Nav1.5 expression in young Scn5a+/ΔKPQ relative to young WT, but increases in Nav1.5 expression with age in WT but not Scn5a+/ΔKPQ (241, 392).
Murine Scn5a-F1759A atria also showed incomplete Nav1.5 inactivation, resulting in increased INaL, prolonged APD, and spontaneous and prolonged AF episodes. It was possible to demonstrate atrial rotors and wave and wavelets in parallel with human AF. There was an accompanying fibrosis, atrial and ventricular enlargement, myofibril disarray, and mitochondrial injury. Both the atrial and ventricular arrhythmias were inhibited by acute inhibition of the NCX, likely through its effects on the increased Na+ entry (1214).
7. Multiple phenotypes associated with mutations involving Nav1.5: overlap syndromes
A final and important group of Nav1.5 variants appear to be associated with overlapping combinations of phenotypes. These were then associated with both loss of SCN5A function producing BrS and conduction disease (1111), and gain of function mutations producing increased INaL resulting in LQTS3 (115, 941, 1215). First, a number of albeit uncommon monogenic SCN5A mutations (SCN5A-D1114N and SCN5A-delF1617) are clinically associated with a overlapping combination of LQTS3 and BrS (191, 914, 1085) or impaired cardiac conduction (1322). A SCN5A-delK1500 in the intracellular DIII-DIV linker close to the LQTS3-associated mutation SCN5A-delKPQ1505-1507 similarly results in a heterogeneous clinical LQTS, BrS, and conduction disease (370). Finally, members of a family carrying SCN5A-1795insD variously showed ECG evidence for isolated or combined features of SND, conduction disease, BrS and LQTS3 (115). Expression studies demonstrated reduced INa, and negatively shifted inactivation-voltage curves in an absence of INaL, in Xenopus oocytes (115). Alternatively, there was a contrasting, increased INaL resulting from disrupted fast inactivation, and increased slow inactivation that reduced maximum INa at high pacing frequencies in HEK-293 cells (1184). However, computational analysis suggested that both sets of characteristics were required to reproduce a combined LQTS and BrS, depending on heart rate (208).
Nevertheless, heterozygous Scn5a-1798insD/+ mice recapitulated the corresponding human SCN5A-1795insD/+ phenotype (939). They showed both signs of SND, of prolonged PQ, QRS, and QTc intervals on ECG recording, and of slowed conduction, especially in the RV, on epicardial mapping in isolated hearts. Patch electrode recordings demonstrated prolonged AP repolarization phases particularly at low, and reduced INa particularly at high pacing frequencies. Accompanying marked reductions in peak INa there was a prolonged time course of fast inactivation, and an increased INaL (208, 1184).
Second, other expression system studies similarly correlated multiple biophysical defects in Nav1.5 with overlapping clinical consequences. Scn5a+/delK1500 enhanced INa inactivation but induced an increased INaL (370). Scn5a+/delF1617 was associated with reduced peak INa, slowed recovery from inactivation. It showed an increased INaL only at positive membrane potentials, and reduced open times at negative potentials. Both variants were associated with an overlapping LQTS3/BrS phenotype with conduction disease.
A third group of SCN5A-V1777M and SCN5A-V1763M mutations are associated with LQTS3 in the presence of conduction disease. These displayed an increased INaL, yet showed no decrease in peak INa or other properties suggesting loss of Nav1.5 function (180). This may reflect further contributions besides biophysical INa properties, possibly involving structural changes (see sect. VE) in producing the overlap syndromes. Nav1.5 changes, exemplified by SCN5A-D1275N, are also associated with SND and atrial arrhythmias (238, 614, 941).
C. Arrhythmic Consequences of Genetic Modifications in Na+ Channel-Associated Proteins
1. Mutations in Scn1b subunits of the Na+ channel
The general features of Navβ subunits as well as the Navβ2 and Navβ3 variants associated with BrS-like phenotypes have been discussed above (see sect. VG). Of the remaining variants, consequences of Navβ1 changes differed when studied in coexpression in vitro systems, murine hearts, and clinical studies.
Thus 1) Navβ1 coexpression with Nav1.5 in HEK293 cells and Xenopus oocytes resulted in overlapping increases in INa and decreases in INaL (842, 1166). It increased rates with which INa recovered from inactivation (304). Conversely, in CHO cells, coexpression with SCN1B-D153N and SCN1B-R85H decreased INa relative to findings with WT Navβ1 (1234).
2) Scn1b−/− mice showed prolonged QT and RR intervals expected for LQTS as opposed to BrS phenotypes. Their acutely dissociated ventricular myocytes showed increases in both peak INa and INaL (∼1.6-fold) and in Nav1.5 expression (∼1.3-fold). Gating and kinetic properties were unchanged, and AP repolarization phases were prolonged (694). Nav1.5, Navβ, ankyrin B, ankyrin G, N-cadherin, and Cx43 showed normal membrane localization on immunostaining. In juvenile mice cardiac specific for Scn1b−/−, macropatch and scanning ion conductance microscopy methods demonstrated increases in TTX-sensitive INa specific to the midsection of isolated ventricular myocytes that could even precede full transverse tubule formation. Finally, ventricular myocytes from adult Scn1b−/− mice showed indications of increased transverse tubular expression of TTX-sensitive INa as well as the increased Scn5a mRNA.
3) Clinical loss of Navβ1 function was associated with BrS rather than LQTS phenotypes, despite increased INaL observed in the experimental systems. Thus a truncated SCN1B-p.Trp179X and a SCN1B-1E87Q mutation was associated with clinical BrS and cardiac conduction disease (1235). Furthermore, SCN1B-R85H and D153N was associated with lone AF (1234) and SCN1B-R85H with the right precordial electrocardiographic ST elevation diagnostic of BrS.
The different, experimental and clinical, situations thus yielded important differences in results. Levels of INa expression were decreased in 1 yet increased in 2, while both 1 and 2 reported increased INaL. Such findings would be predictive of clinical overlap and LQTS phenotypes, respectively. The available clinical evidence 3 agreed with predictions from 1 but differed from those arising from 2 in reporting a BrS rather than a LQTS phenotype. Findings in 3 also differed from those in both 1 and 2 whose observed increases in INaL would have predicted a LQTS clinical phenotype. Further investigations into these differences might then yield fundamental insights clarifying the relationship between expression systems, murine models, and clinical effects. They would be of particular interest in view of a number of additional features associated with Scn1b−/−.
Thus 1) Scn1b-encoded Navβ1 subunits were associated with not only Nav1.5 α-subunits but also with ankyrin-B (787) in cardiac and neuronal tissue (926, 927, 1234). They also occurred as alternative Navβ1 or Navβ1b splice variants (926).
2) Scn1b−/− myoctes showed increased mRNA staining for neuronal Scn3a. This was consistent with increased TTX-sensitive Nav1.3 protein nevertheless excluded from expression around the intercalated disc.
3) Scn1b−/− hearts showed evidence for altered cellular Ca2+ homeostasis. They showed triggered beats, delayed Ca2+ transients, and frequent spontaneous Ca2+ release events accompanying the increased susceptibility to polymorphic ventricular arrhythmias. These abnormalities in Ca2+ homeostasis were prevented by TTX at concentrations (100 nM) expected to block the TTX-sensitive, Nav1.3-mediated INa (663).
4) Murine Scn1b−/−, but not Scn1b+/−, mice also showed neurophysiological changes. This took the form of an epileptic phenotype. They mimicked the severe myoclonic epilepsy of infancy normally associated with heterozygous SCN1A mutations in Dravet syndrome. Hippocampal slice recordings of CA3 neurons exhibited higher peak voltages in APs of correspondingly increased amplitude compared with WT Scn1b+/+ while not showing the altered INa exhibited by the Nav1.1, Scn1a+/−, variant of Dravet syndrome (877).
2. Mutations in Scn4b subunits of the Na+ channel: LQTS10
LQTS10 was first associated with a genetic, SCN4B-L179F, variant (138, 761). Subsequently a SCN4B-S206L mutation was associated with SIDS (1110). Both conditions were associated with increased INaL and prolonged APDs when overexpressed in rat ventricular myocytes (1110). SCN4B-V162G and SCN4B-I166L were then associated with AF (656). Navβ4 subunits have largely been studied experimentally in connection with neuronal function, permtting resurgent INa generation leading to high-frequency firing by nerve and muscle Nav isoforms (67, 159, 645). Navβ4 subunit coexpression with Nav1.2 and Nav1.4 in tsA201 cells negatively shifts their activation (1310). Navβ4 subunits also occur in mouse ventricle (713). The arrhythmic effects of Navβ4 mutations may thus result from resurgent INa (373) or increased INaL. It coimmunoprecipates with SCN5A in the HEK293 expression system. However, in the latter situation it did not affect INa kinetics or density, or INaL (761).
3. Caveolin-3 (Cav3) mutations: LQTS9
Caveolae are rounded, 50- to 100-nm-diameter, surface membrane invaginations abundant in ventricular, atrial, and nodal cells (65). Their structure depends on interactions between their contained cholesterol and specific scaffolding proteins, exemplified by caveolins 1–3. The latter are encoded by CAV1, CAV2, and CAV3, respectively. Caveolin-3 is also associated with the transverse tubules during skeletal muscle development (870). Human caveolin-3 mutations are classically associated with skeletal muscle phenotypes. These take the form of limb girdle muscular dystrophy, rippling muscle disease, distal myopathy, and hyperCKemia (elevated serum creatine kinase) (1273). Gene mutations involving caveolin-3 are also associated with LQTS9.
Cav-3, via its scaffolding domain, likely interacts with numerous signaling molecules. These include G protein-coupled receptors (GPCRs), ion channels, and receptor tyrosine kinases. CAV3 may be important for G protein-mediated adrenergic cardiac INa upregulation (1301). Cav-3 inhibits neuronal nitric oxide synthase (nNOS) (1186) and SCN5A nitrosylation by such nNOS increases INaL (195, 1158). Expression of WT Cav-3 with SCN5A in HEK293 cells did not affect either peak INa or INaL (223, 1177). However, the clinical CAV3 mutations CAV3-F97C and CAV3-S141R, that first demonstrated a LQTS, resulted in an increased INaL in HEK293 cells (1177). So did the CAV3-V14L, CAV3-T78M, and CAV3-L79R mutations found in a SIDS cohort (223). The CAV3-F97C mutation was also associated with increased SCN5A nitrosylation and increased INaL and APD in HEK293 cells (223). Cav3 mutations have also been linked to HCM through an as yet unclear mechanism (415, 1272).
Murine knockout or knock-down studies implicated Cav3 in cardiomyocyte caveolus formation (337). These may integrate ion channel and exchangers, including pacemaker Hcn4, Cav1.2, Kv1.5, Kir6.2/Sur2a and Nav1.5 channels, and NCX, into specific signaling complexes (65, 1082, 1303). Two independent groups reported generation of caveolin-3-deficient mice by targeted exon 2 disruption involving the Cav3 caveolin-scaffolding, transmembrane domains, and the COOH-terminal region (337, 401). The mice also showed mild myopathic changes resembling those of limb girdle muscular dystrophy-1C in patients with CAV3, exon 2 mutations (434, 779).
Cav3−/− mice showed an abnormal, diffuse, tubular dihydropyridine receptor DHPR1-α and RyR1 localization. Their transverse tubular systems were disorganized. They exhibited dilated and longitudinally oriented transverse tubules (337). The mice developed a progressive cardiomyopathic phenotype. Thus magnetic resonance imaging and transthoracic echocardiographic studies demonstrated hypertrophy, dilation, and reduced fractional shortening. Histological examination revealed cardiomyocyte hypertrophy, cellular infiltratation, and interstitial fibrosis. Such changes were evident even at age 4 mo. There was an accompanying increase in activity in the p42/44 mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK1/2) cascade. The latter is normally inhibited by Cav3 (1272). Conversely, cardiac-specific Cav-3 overexpression resulted in lower resting heart rates at rest, prolonged PR interval but shortened QTc intervals, and alterations in ion channel expression in an absence of alterations in metabolic background activity (727, 728, 1017).
4. Arrhythmic consequences of genetic abnormalities in α1-syntrophin (SNTA): LQTS12
The rod-shaped scaffolding protein α1-syntrophin (SNTA) forms part of the dystrophin glycoprotein complex that is involved in linking proteins to the cytoskeleton. It is associated with multiple further proteins including other syntrophins and dystrophin (12). It may be associated through its PDZ domain to the three specific residues, Ser-Ile-Val, at the distal end of the COOH terminus (1063) of those SCN5A channels that are located in the lateral membranes of cardiac myocytes (345, 889, 1158). SNTA mutations are associated with LQTS12 (1158) and SIDS (194). The LQTS12, SNTA-A390V, mutation selectively dissociates the plasma membrane calcium transporter and nNOS inhibitor PMCA4b from this complex (846). This increases SCN5A nitrosylation and thereby increases INaL (1158). In contrast, mice with genetically modified α1-syntrophin lacking the COOH-terminal motif (ΔSIV) showed altered targeting of Nav1.5 to the lateral membrane, normal Nav1.5 targeting to intercalated discs, and reduced INaL (1063).
D. Arrhythmic Properties and Altered ICaL: Timothy Syndrome (LQTS8)
1. Clinical features of Cav1.2 abnormality
Slowed Cav1.2 inactivation constitutes a further source of AP plateau prolongation that may promote lethal arrhythmias. Timothy Syndrome (LQTS8, TS) results from rare, autosomal dominant gain-of-function missense CACNA1C-G406R mutations involving the junction between DI/S6 and the I–II loop of the L-type Ca2+ channel (Cav1.2). It may occur with or without an accompanying CACNA1C-G402S mutation (1086). AP prolongation may be the result of abnormal stabilization of the open state of Cav1.2 involving the scaffolding protein AKAP79 (AKAP150 in rodents). The enhanced Ca2+ influx may be further accentuated by increased channel activity resulting from physical interactions between neighboring channels through their C-tails (268). The condition is often accompanied by congenital heart disease, syndactyly, immunodeficiency, cognitive abnormalities, and autism.
2. Murine models for Timothy syndrome
Two distinct TS mouse lines have been used respectively to illustrate its resulting neurological (56) and cardiac phenotypes (192). In the cardiospecific transgenic mouse LQTS8 model of TS, ∼30% of the Cav1.2 channels were Cav1.2-TS. The LQTS8 hearts were hypertrophied relative to WT. Their ventricular myocyte ICaL showed a slowed inactivation. This likely accounts for the prolonged AP waveforms and longer QT intervals. It likely also accounts for the increased incidences of exercise-induced arrythmogenic events including premature ventricular depolarizations and torsades de pointes, in intact hearts. Cell-attached patch recordings attributed this to greater opening probabilities, open time durations, and coupled gating frequencies in the Cav1.2 (192). Cav1.2-TS channel expression also appeared to increase background sarcolemmal Ca2+ leak in resting ventricular myocytes, increasing diastolic intracellular [Ca2+] relative to WT. This was attributed to an increased SR Ca2+ load and Ca2+ spark activity. The latter also resulted in larger amplitudes of evoked Ca2+ transients and Ca2+ wave frequencies (277). Crossing LQTS8 with AKAP150−/− giving LQTS8/AKAP150−/− rescued all these phenotypes (192, 1150).
E. Hypokalemic Murine Models for Acquired LQTS
1. Arrhythmic consequences of acquired LQTS
Inheritable LQTS-mediated arrhythmias account for only 1–2% of lethal clinical ventricular arrhythmias. The remaining LQTS are associated with acquired rather than congenital risk factors. These include metabolic conditions, electrolyte abnormalities, particularly hypokalemia, and bradycardic situations (37, 288, 953, 1037, 1171). A significant number of drugs not initially associated with cardiac effects also exert significant cardiotoxic profiles that include QT prolongation. In some cases these predispose to polymorphic VT and torsades de pointes (953). Even therapeutic levels of some cardiac drugs such as dofetilide and ibutilide can noticeably (∼50 ms) increase QT interval and risk of torsades de pointes (513, 953). However, even agents slightly increasing QT interval (5–10 ms) can increase risk of torsades de pointes (288, 910). These effects often have been attributed to inhibition of K+ current, particularly IKr (1171).
2. Murine hearts as models for clinical hypokalemia
Experimental murine cardiac models permitted a modeling of acquired LQTS following reversible imposition of their causative conditions in physiological studies of often common, clinically important, situations (562). For example, both increased and decreased [K+]o clinically predispose to major arrhythmias (230). Decreased [K+]o increases risks of torsades de pointes (37). This may involve a delayed repolarization prolonging the QT interval that predisposes to EADs in turn triggering premature beats (953). These appeared within a substrate resulting from heterogeneous transmural ion channel distributions altering transmural dispersions of repolarization (TDR) and therefore refractoriness, particularly when accompanied by increased APD (494). Reduced [K+]o has been used to assess arrhythmic effects of pharmacological agents implicated in acquired LQTS or generation of arrhythmias (288, 772, 774).
Mouse hearts replicate the [K+]o sensitivity shown by the major voltage-dependent repolarizing K+ currents shown in hearts of larger mammals (561, 829, 1294). In particular, IK1 similarly contributes membrane current through the voltage sensitivity arising from its inward rectification properties. These reduce K+ conductance at membrane potentials positive to −20 mV during phases 0–2 of the action potential, but permit outward currents following repolarization to negative to −40 mV late in phase 3, and stabilize the diastolic, phase 4, resting potential (357, 1020). In ventricles, these effects are augmented by currents arising from its tubular localization (211, 322). Ventricular transverse tubules form a restricted extracellular space permitting K+ accumulation following depolarization where this results in outward K+ current activation. The tubular K+ accumulation shifts the local tubular K+ Nernst potential, activating IK1, thereby modulating AP recovery. This activation was demonstrated in single adult mouse ventricular myocytes in the form of transient inward tail currents observed following termination of depolarizing voltage steps sufficient to elicit outward current by repolarizations to holding potentials close to the normal cell resting potential (211, 317). The latter effect was modified by both increased and decreased [K+]o (see sect. VIE). Murine hearts thus therefore can usefully physiologically model the effects of clinical hypokalemia (301, 559, 563, 979, 982, 983).
3. Triggering and arrhythmic substrate associated with hypokalemia
Studies that examined the electrophysiological effects of decreasing [K+]o in murine cardiomyocytes (559–561, 978) confirmed previous reports in other systems of decreased Ito, IKr, and IK1 despite increased driving forces on outward K+ currents (164, 317, 1001). Under normokalemic (5.4 mM [K+]o) conditions, patch-clamped epicardial myocytes showed greater Ito than endocardial myocytes (561) (Figure 12A). This was in keeping with the higher epicardial than endocardial levels of their corresponding proteins (151, 967). The resulting transmural Ito differences were in turn predictive of the normally observed transmural APD differences. Hypokalemic conditions decreased epicardial (Figure 12A, a and b) but not endocardial Ito (Figure 12A, c and d). This would predict preferential epicardial over endocardial increases in APD, and diminished repolarization gradients. In contrast, epicardial and endocardial myocytes under normokalemic conditions showed similar IK1 obtained in response to hyperpolarizing steps. These were reduced to similar extents by hypokalemia. This would be consistent with effects in increasing APD but not to the resulting transmural repolarization gradients.
Figure 12.

Electrophysiological features contributing to arrhythmogenesis in hypokalemic murine ventricles. A: outward and inward K+ currents (a, c) and representations of their respective maximum currents (b, d) from whole cell patch-clamped epicardial (a, b) and endocardial myocytes (c, d) under normokalemic (dark lines) and hypokalemic (3 mM) conditions (pale lines). Under normokalemic conditions, epicardial myocytes (a) showed greater early outward IK than endocardial myocytes (c). However, whereas hypokalemia reduced early outward IK in epicardial but not endocardial cells (b), it reduced inward IK1 in epi- and endocardial cells by similar extents (b, d). B: monophasic AP recordings from LV endocardial and epicardial Langendorff-perfused WT murine hearts paced at 125 ms BCL, under control (5.2 mM [K+]; a) and hypokalemic conditions (3 mM [K+]; b). C: steady-state epicardial (white columns) and endocardial APD90s (gray columns) and the resulting ΔAPD90 (black columns) at [K]o = 5.2 mM (a), 4 mM (b), and 3 mM (c), respectively. D: programmed electrical stimulation (PES) of isolated, WT Langendorff-perfused mouse hearts under normokalemic (a) and hypokalemic 4 mM (b) and 3 mM [K+]o (c) conditions did not induce VT in (a) but induced VT in 2 of 7 hearts in 4 mM [K+]o (b) and 9 of 11 hearts in 3 mM [K+]o (c). E: LV epicardial monophasic action potential (MAP) recordings during intrinsic pacing (a), and programmed electrical stimulation (PES) (b) in the presence of 3 mm [K+]o and 2 μm KN-93. KN-93 reduced the occurrence of early afterdepolarizations (EADs), triggered beats and ventricular tachycardia (VT) in spontaneously beating hearts (a). It failed to protect against arrhythmia provoked by PES in 6 of 6 hearts (b). [From Killeen and co-workers (559, 561).]
AP recordings in intact, Langendorff-perfused mouse hearts fulfilled these predictions from this cellular analysis of changes in Ito and IK1 (Figure 12, B and C) (561). Under normokalemic conditions, they thus showed longer ventricular endocardial than epicardial MAPs. This led to transmural APD gradients of ∼14 ms. This difference closely correlates with previous studies of in vivo murine MAP recordings (Figure 12Ba). Reduced [K+]o prolonged both the epicardial and the endocardial MAPs. However, it did so to differing extents. This led to a reduction in the transmural APD gradients as quantified by ΔAPD90 values (Figure 12, Bb and C, a–c). In addition, the more bradycardic of intrinsically active hearts showed EADs even with reductions in [K+]o, from 5 to 4 mM. They also showed a 29% incidence of VT (Figure 12D). These findings directly parallel clinical findings in male hypertensive patients on diuretic therapy in which each 1 mM reduction in [K+]o increases ventricular arrhythmic risk by 28% (214). Further reduced, 3 mM, [K+]o additionally resulted in triggered beats followed by episodes of nonsustained VT (Figure 12Dc). This would implicate L-type Ca2+ channels as a possible depolarizing charge carrier at low slow stimulation rates (1325).
Pharmacological maneuvers using the L-type Ca2+ channel-blocker nifedipine and CaMKII inhibitor KN-93 made it possible to separate the contributions made by EADs and transmural repolarization gradients to the arrhythmic process. They suggested that the mechanisms responsible for triggering and those producing reentrant substrate leading to persistent arrhythmia were separate (Figure 12E) (559). Thus both nifedipine (100 nM) and KN-93 reduced EADs and spontaneous arrhythmias (Figure 12Ea). However, neither prevented arrhythmias provoked by programmed electrical stimulation (Figure 12Eb). Nor did they alter either epicardial or endocardial APD. They thus preserved the abnormal repolarization gradients associated with the hypokalemic conditions. In contrast, higher (1 μM) nifedipine concentrations abolished all arrhythmic phenomena, whether spontaneous or provoked. It now correspondingly shortened the epicardial APDs and restored the transmural repolarization gradients ΔAPD90 to control values. This implicated the alterations in ΔAPD90 in arrhythmic substrate (559). Together these findings were also compatible with a prolongation of the AP repolarization thereby inducing EADs through Ca2+ channel reactivation. This in turn would trigger premature APs and triggered beats (Figure 13, A and B) (301, 497). These findings concurred with findings in murine (KChIP2−/−) cardiomyocytes lacking KChIP2, which encodes three auxiliary subunits of Kv4.2 and Kv4.3. These cells similarly lacked Ito, showed increased APD, and were susceptible to induced, but not spontaneous ventricular arrhythmias (604).
Figure 13.

Development of arrhythmia resulting from a combination of triggering activity and recovery abnormality exemplified by the gain of Nav1.5 function Scn5a+/ΔKPQ, or loss of K+ channel function through genetic modification or hypokalemic challenge in murine exemplars. Emerging from studies of murine exemplars (A), triggered activity results from the prolonged APD predisposing to L-type Ca2+ channel reexcitation, producing triggered action potentials (B). Recovery abnormality, particularly in the epicardium, produces a background electrophysiological defect measurable as a LQTS, resulting in arrhythmic substrate involving alterations in recovery gradients arising from APD and VERP changes in epicardial and endocardial myocardium (C). Together, B and C predispose to arrhythmic events and SCD (D).
Other pharmacological manipulations similarly culminating in altered APD and the appearance of EAD phenomena yielded concordant results. For example, NS1643 (30 μM) is known to reduce HERG inactivation (169). It reduces APD in isolated guinea pig ventricular myocytes (407). The KATP channel activator nicorandil (20 μM) was reported as antiarrhythmic in LQTS (1056). Both of these agents reduced the occurrences of both spontaneous, unprovoked VT, and VT following premature stimulation in murine hearts studied under hypokalemic conditions. This was accompanied by reduced incidences of EADs and reductions in epicardial APD that thereby restored the normal transmural repolarization gradients (562).
Evidence from murine systems thus attributed arrhythmic tendencies in both Scn5a+/ΔKPQ (Section VIB) and hypokalemic hearts to alterations in AP repolarization and refractoriness. This would be in direct contrast to the altered conduction velocity implicated in Scn5a+/− hearts (see sect. V, B and C). Such a distinction was confirmed in investigations of arrhythmic or refractory end points in adaptive programmed electrical stimulation protocols that consisted of regular pacing (S1) stimulus trains that were followed by S2 extrastimuli. S1S2 intervals were progressively decreased, but S2 amplitudes were progressively increased to maintain stimulus capture. Control normokalemic WT, test hypokalemic WT, and Scn5a+/ΔKPQ hearts all showed similar relationships between conduction velocity and S1S2 coupling interval and between reexcitation thresholds and S1S2 coupling interval. Yet the onset of arrhythmia did not correspond to a given change in conduction velocity. When arrhythmias were induced by extrasystolic APs in the LQTS, hypokalemic WT, and Scn5a+/ΔKPQ groups, these took place at higher AP conduction velocities and lower S2 stimulus amplitudes than in the normokalemic WT controls. Furthermore, where there were arrhythmic as opposed to refractory outcomes, the APs initiating such arrhythmias showed lower conduction velocities than when there were refractory outcomes. These conduction velocities were higher and occurred at longer S1S2 coupling intervals and smaller stimulus amplitudes in the LQTS groups compared with controls (281).
4. Transmural heterogeneities and arrhythmic substrate in hypokalemic hearts
Section IIA2 discussed situations in which substrate for reentrant excitation arose from relative changes in parameters describing AP repolarization and refractoriness. This suggested that windows of reexcitation could result from an appearance of critical intervals produced by positive time differences between measures of action potential repolarization such as APD90, and recovery from refractoriness as exemplified by VERP. Such differences could either take place within a given cardiac region or involve adjoining, electrotonically coupled regions of myocardium (978). These predictions were demonstrated in isolated hypokalemic (∼3 mM [K+]o) Langendorff-perfused hearts (Figure 13, C and D). These made it possible to test the effects of conditions under which alterations in transmural repolarization gradients would give rise to reentrant excitation in LQTS models (Figure 12C).
In this analysis, hypokalemia increased the epicardial but not the endocardial APD90. It also decreased both the epicardial and endocardial VERPs. However, it left differences in endocardial and epicardial AP latencies unchanged. Lignocaine abolished the arrhythmogenecity associated with the hypokalemia. It also abolished these alterations in recovery parameters. The differences in the endocardial and the epicardial intervals between stimulation and 90% repolarization remained unchanged. These findings gave rise to quantititave predictors of arrhythmic behavior involving AP latency, APD90 and VERP. Risks of local reexcitation in either the epicardium or endocardium could thus be obtained from critical intervals that were based on their respective (APD90-VERP) differences. Risks of transmural reentrant epicardial excitation by endocardium, or endocardial excitation by the epicardium could be predicted by critical intervals given by (endocardial APD90 + Δlatency − epicardial VERP) and (epicardial APD90 + Δlatency − endocardial VERP), respectively. The Δlatency term corrected for delays between endocardial and epicardial excitation. These findings were corroborated by computer modeling of AP waveforms using established data on the effects of hypokalemia on ionic conductivities in ventricular myocytes (978).
The relevance of incorporating a VERP term reflecting recovery from refractoriness was further justified by extending the analysis of arrhythmic tendency in hypokalemic hearts through a range (80–180 ms) of, as opposed to single, BCLs. These extended to bradycardic conditions. This gave a graphical analysis of local and transmural relationships plotting epicardial and endocardial APD90, appropriately corrected for conduction times, against VERP. Hearts studied under normokalemic (5.2 mM [K+]) conditions then yielded straight line loci close to lines of equality. These lines thus subtended an angle ∼45° with the abscissa. Hypokalemia shifted the APD90 values above these reference lines. These differences increased with BCL. The latter was correctly predictive of correspondingly increasing arrhythmic tendency. It also direct paralleled the corresponding increases in critical (APD90-VERP) and in the subtended angle. The antiarrhythmic effects of lignocaine directly correlated with its effect in modifying this relationship. It decreased the subtended angle and (APD90-VERP) in hearts studied under both normokalemic and hypokalemic conditions (977).
5. Temporal heterogeneities and arrhythmic substrate in hypokalemic hearts
Hypokalemia also contributed a dynamic component to the arrhythmic substrate. This was apparent first in the features of extrasysolic APs that were obtained in response to premature (S2) stimuli. The APs that followed subsequent (S3) stimuli then showed alterations in their transmural repolarization gradients ΔAPD90 under hypokalemic conditions. This effect was not observed under normokalemic conditions (980). Furthermore, in parallel with its antiarrhythmic effects both in murine preparations and in clinical situations (938), lignocaine abolished these ΔAPD90 changes. Second, hypokalemia also contributed changes to alterations in restitution properties with progressively decreasing BCL. Isolated, Langendorff-perfused hearts studied under hypokalemic (3 mM) but not normokalemic (5.2 mM [K+]) conditions showed APD alternans and arrhythmia with progressive increases in heart rates. This was associated with increased restitution curve slopes. Both such effects were prevented by lidocaine (10 μM). Hypokalemia increased the values of both epicardial and endocardial maximum gradients and DIcrit corresponding to unity gradient identified with the onset of arrhythmia. In contrast, lidocaine decreased such gradients and DIcrit under hypokalemic but not normokalemic conditions (979, 982).
F. Genetic Modifications in Voltage-Dependent K+ Channels and Their Associated Subunits
1. Loss of function in transient outward currents, Ito
The pore-forming Kv4.3 α-subunit coassembles with a range of modulatory β-subunits, particularly KChIP2, but also KCNE2 (MiRP1) and DPP6 (2, 294). Coassembly of Kv4.3/KChIP2 channels with MiRP1 slowed activation and inactivation in whole cell patch-clamped CHO cells (933). Murine Kcne2−/− ventricles showed prolonged APDs, accompanied by 25% reductions in Ito,f and 50% reductions in IK,slow1, implicating associations between Kcne2 and Kv1.5 for the first time. There was an accompanying coimmunoprecipitation of ventricular MiRP1 protein with native Kv1.5 and Kv4.2 but not Kv1.4 or Kv4.3 (956). Kcne2 knockdown by RNA interference similarly prolonged APD and decreased Ito in both neonatal and adult mouse myocytes. In contrast, Kcne2 overexpression produced by adenoviral gene delivery shortened APD and increased Ito in neonatal but not adult myocytes (679).
The magnitude of Ito normally falls with the LV epicardial-endocardial gradient. This transmural gradient is likely important in normal ventricular repolarization (151, 383, 604). Studies in Nfat−/− mice, deficient in nuclear factor of activated T cell, suggest that this may reflect control of Kv4 expression by calcineurin and NFATc3 (967). APD is consequently shorter in their LV epicardial than their LV endocardial myocytes (209, 604). Transgenic mice with altered Ito components can show APD and QT prolongation. This may correlate with arrhythmic tendency where these involve regional Ito,f and Ito,s variations. The latter in turn may alter regional apex-base, endocardium-epicardium, or septum-ventricular free wall patterns of ventricular repolarization (70, 151, 383, 691, 1284).
The consequences of reduced Ito for APD, apical-basal APD gradients, and arrhythmogenecity varied with the extent to which these reductions affected Ito,f, Ito,s, or both. Thus Kv4.2 (KCND2) dominant negative transgenic (Kv4.2-DN) mice lacking Ito,f, but not Kv1.4 (KCNA4)−/− mice lacking Ito,s, showed increased APD and prolonged QT. Neither showed spontaneous arrhythmias. However, double transgenic (Kv4.2-DN × Kv1.4−/−) mice lacking both Ito,f and Ito,s showed both AP and QT prolongation, and spontaneous ventricular tachyarrhythmias. With optical mapping using di-4-ANEPPS, Langendorff-perfused Kv4.2-DN, Kv1.4−/− and Kv4.2-DN × Kv1.4−/− hearts showed similar activation patterns and conduction velocities, but longer APD75 (∼28, ∼28, and ∼34.3 ms) than WT (∼20.3 ms). However, Kv4.2-DN hearts showed reduced apicobasal dispersions of refractoriness compared with WT. They did not show VT on premature stimulation. Voltage-clamped Kv4.2-DN myocytes showed 30% greater apical than base Ito,f. In contrast, both Kv1.4−/− and Kv4.2-DN × Kv1.4−/− hearts showed increased dispersions, and VT, on premature stimulation (558, 687).
2. Loss of function in the rapid delayed rectifier K+ current, IKr: LQTS2 and LQTS6
The Kv11.1 (KCNH2)-mediated IKr is important both for completing phase 3 repolarization and preventing arrhythmias induced by EADs in hearts of larger mammals (1070). Phase 0 depolarization rapidly activates initially resting IKr channels into their open state. But this is rapidly succeeded by channel inactivation. However, repolarization produces a rapid recovery from inactivation involving transitions through the open state preceding return to the resting state. The latter produces a tail current (696, 697, 1000, 1070, 1170). Loss-of-function KCNH2 (formerly termed HERG, the human form of ERG) mutations affect IKr (229), particularly in the S5/pore region (472, 1342). The consequent clinical LQTS2 results in low-amplitude bifid T waves in the ECG (806), reflecting increased transmural dispersions of repolarization, cardiac arrhythmic events following sudden arousal, and episodic sinus bradycardia (1002, 1307).
IKr is also the dominant repolarizing current in fetal (∼day 18) mouse hearts. IKr block by dofetilide then caused EADs and failure of repolarization. IKr block in fetal mice produced bradycardia and arrhythmia resulting in lethal circulatory failure (1219). Simlarly, complete knockout of all the ERG1 isoforms is intrauterine lethal.
However, in adult mice, IKr may only play minor roles in ventricular repolarization. Nevertheless, it likely retains a role in SAN pacemaker function (see sect. IIIA). Thus results in mice in which the B transcript was eliminated while preserving the A transcript suggested that in adult myocytes, Erg1B is necessary for IKr surface membrane expression and that its knockout predisposes to episodic sinus bradycardia (627). However, QT intervals were normal notwithstanding loss of the rapidly decaying component of IKr. Murine LQTS2 models carrying the G628S dominant negative mutation showed normal QT durations but minor abnormalities in QRS and T waveforms. There was a loss of IKr and increased APDs in isolated myocytes, but normal AP waveforms in ventricular strips (53). Merg1b−/− mice showed episodic abrupt sinus bradycardia. This finding parallels clinical observations in some LQTS2 families where this can trigger arrhythmias (1307). However, they also showed normal QT intervals despite an abolished IKr in isolated myocytes (626), while not excluding a persistence of repolarizing K+ currents produced by other mERG isoforms (690). Nevertheless, epicardial optical mapping studies related an increased VT propensity in a Merg+/− mouse to prolonged apical and basal APDs and VERPs, despite conduction velocities similar to those found in WT (994).
Studies in murine models have also yielded important results bearing on roles of alternatively spliced isoforms. Human and mouse hearts express two, A and B, isoforms in the underlying ERG1. These have differing NH2 termini and show slow and rapid deactivation properties, respectively (627). The A transcript is relatively abundant in both human and mouse hearts. The B transcript is abundant in mouse but not human hearts (627). Most reports bearing on LQTS2 appear to reflect properties of the A isoform. Studies in Xenopus oocyte expression systems suggest that functional IKr in mouse hearts requires heteromeric assembly of different, mERG1a, mERG1a', and mERG1b isoforms. Currents from expressed mERG1a alone resembled those obtained following HERG expression. Currents from mERG1a', lacking the first 59 NH2-terminal amino acids, and mERG1b currents showed a 10-fold more rapid deactivation. Coexpression of mERG1a and mERG1b gave currents closely resembling murine IKr (690). hERG1b is also likely to be critical to normal repolarization with its deficiency resulting in proarrhythmic effects in human cardiomyocytes. sh-RNA-induced knockdown of the 1b subunit halved its corresponding mRNA and protein levels as well as IKr, increasing AP duration and variability, with an appearance of EAD phenomena in cardiomyocytes from human induced pluripotent stem cells. These features were replicated by converting hERG heteromers to hERG1a homomers by expressing a fragment representing the HERG1a-specific NH2-terminal Per-Arnt-Sim domain absent in hERG1b (516).
IKr channels may require further coassembly of auxiliary modulatory subunits with different channel isoforms to reproduce their in vivo function. Thus native IKr differs from currents recorded from hERG channels in heterologous expression systems in their gating, external K+ regulation, and drug sensitivity properties (207). Normal in vivo IKr function thus also depends on association of KCNH2 with the accessory KCNE2 (minK-related, MiRP1) protein (1, 2, 294). This results in a current with +5 to +10 mV shifts in steady-state activation, enhanced rates of deactivation, and reduced unit channel conductance from 13 to 8 pS compared with electrophysiological findings associated with expression of KCNH2 alone (696, 697). Loss-of-function KCNE2 mutations are associated with LQTS6 (1085). This has phenotypic consequences similar to those of LQTS2. Conversely, the gain-of-function KCNE2-R27C mutation has been associated with familial AF (1295).
3. Loss of function in the slowly activating delayed rectifier K+ current, IKs: LQTS1 and LQTS5
The slow, IKs, current observed in hearts of larger mammals is mediated by the K+ channel protein KCNQ1. This protein is modulated by its association with the single transmembrane domain β-subunit KCNE1 (Mink) (69, 999). Adult mouse hearts do express Kcnq1 (276), but their Kcne1 expression declines with age (276, 308) from age ∼1 wk (253). Adult mouse myocytes consequently lack typical IKs (53, 253). In addition, alternative splicing of KCNQ1 results in an isoform 1 with a long NH2-terminal end (69) and an isoform 2 with a short NH2-terminal end of only two amino acids (253). Isoform 2 does not form functional channels but exerts strong dominant-negative effects on isoform 1.
KCNQ1 mutations are associated with clinical LQTS1. Cardiac incidents typically occur during exercise (1222). Kcnq1−/− mice recapitulated the morphological inner ear abnormalities, auditory defects, and the shaker/waltzer and cardiac phenotypes of Jervell and Lange-Nielsen syndrome, with which clinical LQTS1 often presents. Intact Kcnq1−/− mice showed abnormal in vivo electrocardiographic P and T waves, prolonged QT intervals, and episodic nonsustained arrhythmias. In contrast, isolated perfused murine Kcnq1−/− hearts showed normal QT intervals and APDs. In Kcnq1−/− but not WT hearts, these were nevertheless prolonged by nicotinic or sympathomimetic stimulation. This implicates extrinsic cardiac innervation in reproducing the clinical phenotype (1138). Nevertheless, mice overexpressing human KCNQ1 isoform 2, which exerts dominant negative effects on murine Kcnq1 isoform 1, showed marked QT prolongation. They also showed SND and atrioventricular block. The latter was associated with prolonged atrial-His but normal His-ventricular intervals in His bundle recordings. Their patch-clamped mutant cardiomyocytes showed increased APD. However, there was also evidence for additional electrophysiological remodeling. This took the form of reductions in Ito and Iss through downregulation of Kv4.2 and Kv1.5 and upregulation of Kv4.3 (254). Thus complex physiological changes add to those directly arising from the genetic changes in vivo.
Loss-of-function mutations involving the β-subunit KCNE1 are associated with LQTS5 (1085, 1087). Its phenotype resembles that of LQTS1, with prolonged APs and increased arrhythmic risk (121, 999). Homozygous Kcne1−/− mice with a deletion in the entire KCNE1 coding sequence recapitulated both the sensorineural and the arrhythmic effects clinically associated with LQTS5 (64, 1195). They showed circular movements, repetitive falling, head nodding, and bilateral deafness attributed to deficient transepithelial endolymphatic K+ transport (1195).
Kcne1−/− mice showed increased QT intervals at slow heart rates that paradoxically shortened at increased heart rates (276). Spontaneously beating Langendorff-perfused Kcne1−/− hearts showed frequent EADs, closely coupled triggered beats and spontaneous VT. Regular pacing demonstrated increased bipolar electrogram durations. Premature stimuli during programmed electrical stimulation frequently provoked monomorphic VT episodes (64, 447). Prolonged epicardial and endocardial APDs resulted in reduced critical intervals as given by APD90-VERP differences. They also resulted in reduced transmural repolarization gradients, consistent with spatial reentrant substrate (1130). These changes took place in an absence of epicardial apical-basal APD90s measured by optical mapping (994). Temporal re-entrant substrate appeared as increased APD90 alternans and steeper epicardial and endocardial APD90 restitution curves during dynamic pacing. Nifedipine suppressed the EADs, triggered beats, and repolarization alternans. It selectively shortened epicardial APD, restoring the transmural repolarization gradient (1130).
Pharmacological manipulations directed at K+ channel rather than Ca2+ homeostatic function using nicorandil yielded concordant results (562). Nicorandil increases K+ conductance (478, 528), enhancing repolarization reserve (478), and shortening APD90 in atrial (1289) and Purkinje fibers (478), and isolated guinea pig and rabbit ventricular myocytes (528). These effects were independent of external [Na+] (478). They occurred despite normal ICa (528), (dV/dt)max (478, 528), or maximum diastolic membrane potentials. Nicorandil (20 μM) rescued both the spontaneous and provoked arrhythmogenic phenomena. It also restored the AP parameters reflecting reentrant substrate towards the normal values in untreated WT, further shifting them in WT hearts in similar directions (447).
4. Loss of function in Islow
In addition to rapidly decaying outward Ito, mouse ventricular myocytes show gradually decaying contributions from a rapidly activating but slowly inactivating, 4-aminopyridine-sensitive Islow. This is likely at least partially carried by Kv1.5. There is also a constant current component (1348). Murine studies demonstrated potentially arrhythmic contributions arising from abolition of Islow. These employed a dominant negative transgenic mouse overexpressing a cardiac Kv1.1 NH2-terminal fragment controlled by the α-myosin heavy chain promoter (688). The truncated channel fragment coassembles with WT Kv1.x. This traps heteromeric channels in the endoplasmic reticulum. The latter accounts for the dominant negative reduction in Kv1.x currents in heterologous expression systems (321).
The transgenic mice showed prolonged QT intervals and spontaneous nonsustained VT on ambulatory telemetry. Programmed stimulation applied to the RV induced polymorphic VT in anesthetized, open-chest preparations. The patch-clamped ventricular myocytes showed prolonged APDs attributable to reduced Islow. Studies using voltage-sensitive dyes and optical mapping techniques demonstrated the normal apical-basal activation in apically paced hearts. Mean AP conduction velocities (∼0.5 m/s) were also similar to those showed by WT. Transgenic hearts showed increased APD75 and APD90 (59). They showed altered restitution properties. Thus APD did not show the normal decrease with decreased BCL. There was instead a flattened dependence on decreased BCL. These findings implicate Islow in the adaptation of APD to altered heart rate, particularly at short BCLs. These properties together culminated in marked reentrant substrate. Premature apical stimuli triggered prolonged (≥30 min) reentrant VT episodes in transgenic but not WT hearts.
5. Other K+ channel variants
Deficiencies in inward rectifying K+ current, IK1, resulting from loss-of-function KCNJ2 mutations encoding Kir2.1 are associated with Anderson-Tawil syndrome (LQTS7) (1143, 1144). In common with Jervell and Lange Nielsen syndrome, there are accompanying extracardiac phenotypes. In LQTS7, these include periodic paralysis, dysmorphic facial features, and syndactyly (1144). Kcjn2−/− mouse neonates lacking Kir2.1 did not demonstrate ectopic beats or reentry arrhythmias but were bradycardic on ECG recording. Their ventricular myocytes lacked detectable IK1 while showing persistent sustained outward K+ currents and L- and T-type channel-mediated Ba2+ currents. They showed broader APs and more frequent spontaneous APs. Kcjn12−/− mice lacking Kir2.2 showed 50% reductions in IK1 (1323). This contrasts with the association between the gain of IK1 function KCNJ2-V93I mutation and familial AF (1278).
Murine models with decreased ultrarapid delayed rectifier potassium current (IKur), mediated by Kv1.5, resulting from loss of Kcna5 function showed atrial AP prolongation, increased incidences of EADs, and AF (853).
G. K+ Channels and Scaffolding Proteins
1. Genetic abnormalities in ankyrin-B (Ank2): LQTS4
In common with Na+ channels, K+ channels are associated with scaffolding proteins (1071). Of these, ankyrin-B, ANK2, is critical in regulating cardiac membrane protein expression. ANK2 dysfunction can follow myocardial infarction (377, 469). Genetic ANK2 variants are clinically associated with LQTS4 but may correlate with arrhythmic susceptibility in general (1034). ANK2 loss-of-function results in complex phenotypic abnormalities including sinus node and conduction disorder, ventricular arrhythmia, and SCD (788–790, 1033). LQTS4 patients showed increased QT intervals and arrhythmias with additional features including sinus bradycardia and paroxysmal AF (1023).
Ankyrin-B+/− mice recapitulated the latter findings. Mice with heterozygotic, loss-of-function, ankyrin-B +/E1426G mutations showed reduced transverse tubular targeting and protein levels in Na+-K+-ATPase, NCX, and inositol 1,4,5-trisphosphate receptors. All the latter proteins are known to bind ankyrin-B (789). Abnormalities in Na+-K+-ATPase and NCX expression would potentially increase intracellular Na+ overload and reduce Ca2+ extrusion. The consequent SR Ca2+ overloading might then account for the abnormal SR Ca2+ release events following adrenergic stimulation. The adult cardiomyocytes correspondingly showed potentially arrhythmic abnormal intracellular SR Ca2+ transients and EADs and DADs following adrenergic challenge (788). In vivo ECGs showed prolonged QT intervals and polymorphic VT episodes.
Subsequent studies of further mutational variants reported a wide range of clinical phenotypes. These included bradycardia, and AF, although not always QT interval prolongation (790). Ankyrin-B also associates with Kir6.2, in the complex that includes Na+-K+-ATPase (579). This is consistent with its possible regulation of IKATP gating which in turn may cardioprotect from ischemia. Ankyrin B−/− hearts and cardiomyocytes showed deficient Kir6.2 membrane expression and decreased IKATP. (649). Ankyrin-B in turn is recruited to the cardiac dyad by the actin associated βII spectrin. The ankyrin-B-p.R990Q mutation disrupts this interaction causing a severe human arrhythmia phenotype. Correspondingly, βII spectrin−/− mice showed SND and ventricular arrhythmia associated with afterdepolarization phenomena and abnormal Ca2+ waves (1072).
Disruption of ankyrin B may also clinically predispose to atrial arrhythmias. Ambulant ankyrin-B+/− mice developed spontaneous atrial arrhythmias. This was attributed to altered interactions with Cav1.2 reducing ICaL and APD (225). They also show evidence of altered pacemaker function (361).
2. Genetic abnormalities in A-kinase anchoring protein-9 (AKAP-9): LQTS11
Scaffolding A-kinase anchoring protein subtypes (AKAPs) structurally and functionally link particular enzyme molecules and their end targets. The latter include ion channels, thereby affecting cardiac myocyte excitation and contraction (755). Genetic abnormalities in AKAP9, also known as Yotiao, are associated with LQTS11. AKAP9 complexes with KCNQ1, the type II regulatory subunit (RII) of PKA, and protein phosphatase 1 (PP1). It thereby conveys PKA to the vicinity of the channel where PKA phosphorylates serine residue S27 on the KCNQ1 NH2 terminus. AKAP9 possess two KCNQ1 binding sites located respectively close to its NH2 and COOH termini. Thus the AKAP9-G589D mutation near its LZ motif disrupted both AKAP9-KCNQ1 interaction and functional regulation of IKs by PKA-mediated phosphorylation. A clinically observed AKAP9-S1570L mutation in the latter region similarly reduced KCNQ1 binding and phosphorylation. It also diminished cAMP-mediated enhancement of IKs channel activity and prolonged APD on computational modeling particularly following isoproterenol stimulation (188). AKAP9 thus mediates sympathetic regulation of IKs channel regulation. Disruption of this produces pathological effects that may include LQTS (187, 746).
H. Short QT Syndromes
Short QT syndromes (SQTSs) (388) are characterized by often familial occurrences of shortened electrocardiographic QT intervals (less than ∼320 ms) and peaked T-waves despite normal cardiac anatomy (874). They are clinically associated with syncope, shortened AERPs and VERPs, and increased atrial and ventricular arrhythmogenicity potentially causing SCD (359). The known genetic SQTS variants complement some of the genetic changes in LQTS. SQTS1-3 result from gain-of-function K+ channel mutations in KCNH2, KCNQ1, and KCNJ2 encoding α-subunits mediating IKr (149), IKs (93), and IK1, respectively (257). SQTS4-SQTS6 result from loss-of-function CACNA1C, CACNB2b (34), and CACNA2D1 mutations (1116). These involve L-type Ca2+ channel α1C, β2b, and α2δ-1-subunits, respectively. Future studies on these will likely yield important insights on arrhythmogenic mechanisms complementing those derived from LQTS models. Available physiological analysis applying K+ channel openers in LV wedge preparations implicated increased transmural repolarization dispersions and shortened VERP as arrhythmogenic substrate (873).
A murine SQTS model with increased Kir2.1 expression and therefore increased IK1 showed shortened QT intervals and APDs (841). This was accompanied by bradycardia, extrasystoles, atrioventricular block, and atrial flutter (651) as well as ventricular arrhythmias (893). These included prolonged episodes of high-frequency VT (841). Recent computational analysis replicated QT interval shortening in hERG-N588K SQTS1 and Kir2.1-D172N SQTS3 mutations, modeling experimental data from recombinant WT and hERG-N588K channels (7, 8, 257, 464, 911, 1326, 1328). These predicted increased tissue vulnerability to premature stimuli and an increased tendency to formation of reentrant excitation waves (7).
VII. ARRHYTHMIC CONSEQUENCES OF ALTERED Ca2+ HOMEOSTASIS
A. Effects of Cellular Ca2+ Homeostasis on Myocyte Electrophysiology
1. Modulation of the excitation-contraction coupling process
The process of contractile activation following membrane excitation brought about by the ion channel function discussed in previous sections begins with a voltage-triggered, L-type Ca2+ channel-mediated, influx of extracellular Ca2+ (Figure 1, A and C). This locally increases [Ca2+]i in the vicinity of each individual L-type Ca2+ channel and its nearby RyR2s. A Ca2+-induced Ca2+ release then initiates Ca2+ release sparks. The amplitudes of these are graded with that local Ca2+ current flow (193, 1092). A nonregenerative, spatial and temporal, graded, summation of such unit events makes up the resulting [Ca2+]i transient (480, 1226). The consequent increase in [Ca2+]i turn drives the Ca2+ binding to troponin that initiates myofilament mechanical activity (see sect. IB2).
The released Ca2+ is subsequently returned from the cytosol to the SR for resequestration by calsequestrin. This transport process is mediated by a phospholamban (PLN)-regulated SERCA2a. Ca2+ is also returned to the extracellular space by two major routes. The electrogenic NCX provides a low-affinity and a high-capacity transporter (129, 265). In contrast, the PMCA, for which translocation of one Ca2+ is coupled to hydrolysis of one ATP, acts as a high-affinity (Km 100–200 nM) low-capacity transporter effective even at the normally very low [Ca2+]i (143) (see sect. IB3).
The molecules involved in the above sequence can undergo modifications in response to extrinsic physiological demands. Kinase-mediated protein phosphorylation exerts strategic regulatory effects on myocyte excitation-contraction coupling, metabolism, intracellular Ca2+ homeostasis, mitochondrial function, and protein transcription. This can be driven by upstream β-adrenergic signaling. This stimulates G proteins (Gs) that activate adenylate cyclase. The latter in turn increases cellular cAMP levels. cAMP dissociates the inhibitory regulatory (R) subunit (R subunit) from PKA. The resulting PKA activation phosphorylates multiple proteins involved in cardiac excitation, contraction, and relaxation dynamics (see sect. VIIB1) (544). This may account for the multiple, inotropic, chronotropic, and lusitropic effects of cAMP signaling (103) (Figure 14A).
Figure 14.

Signalling pathways underlying excitation-contraction coupling. β-Adrenergic receptor (βAR) activation through stimulatory guanine nucleotide-binding (Gs) proteins increases cellular cAMP levels (A). This in turn drives a phosphokinase-A (PKA)-mediated phosphorylation and activation of L-type Ca2+ channels and cardiac ryanodine receptor (RyR2) SR-Ca2+ release channels (A) and the exchange protein directly activated by cAMP (Epac) pathway producing a calmodulin kinase II (CaMKII)-mediated RyR2 activation (B). Either action on the Ca2+-induced Ca2+ mechanism impinges on the level of SR Ca2+ release (C), and consequent alterations in cytosolic Ca2+ (D). Experimentally used agonists (+) and antagonist (−) agents used on these pathways include isoproterenol, H-89, 8-pCPT-2-O-Me-cAMP (8-CPT), and KN-93.
Thus PKA-mediated phosphorylation of the COOH-terminal tail region of the Cav1.2, L-type Ca2+ channel modifies both the ventricular AP plateau phase as well as SAN pacemaker potentials. That of RyR2 reduces the binding of a highly expressed regulatory cis-trans peptidyl prolyl isomerase, FK506 binding protein type 12.6 (FKBP12.6), whose binding to RyR2 normally stabilizes its closed state. This FKBP12.6-RyR2 dissociation increases the Ca2+ sensitivity of RyR2-mediated release of SR Ca2+. Hyperphosphorylation inducing leaky channels may occur in pathological situations of sympathetic overactivity, as may occur in cardiac failure (see sect. VIIC7) (729, 730, 747, 1239, 1241). Phosphorylation of PLN removes its inhibition of SERCA2 in its reuptake of previously released cytosolic Ca2+ (544).
The protein phosphatases PP1 and PP2A conversely mediate protein dephosphorylation. This often takes place at the same protein substrates and serine/threonine sites as catalyzed by PKA (704). PP2A acts preferentially on L-type Ca2+ channels reversing the β-adrenergic effects of PKA (405). Its actions are upregulated with stimulation of inhibitory G proteins, Gi, through other signaling processes (547). In saponin-permeabilized cardiomyocytes, both PP1 and PP2A increased frequencies of spontaneous Ca2+ sparks. This was followed by a disappearance of such events, reflecting the consequent SR Ca2+ store depletion. These effects were inhibited by the PP1 and PP2A phosphatase inhibitors okadaic acid and calyculin A (89, 1119). PP1 caused dephosphorylation of the SERCA2a inhibitor PLN with a consequent reduction in SERCA2a activity. PP2A also reduced Cx43 conductivity (see sect. VIIG) (14, 636).
An alternative or coexistent, PKA-independent, mechanism of cAMP-dependent catecholaminergic signaling may offer a further level of adrenergic control. This occurs downstream of β-adrenergic receptor-dependent cAMP generation but upstream of Ca2+-induced Ca2+ release (see sect. VIIB2) (Figure 14B). It involves signaling by cAMP-dependent, exchange proteins directly activated by cAMP (Epac) (541, 962). Of Epac isoforms, Epac1 contains a single cAMP-binding domain and appears to be ubiquitously expressed. Epac2 contains two cAMP-binding sites and occurs preferentially in brain, pituitary, and adrenal gland (134, 962). Cardiac Epac1 forms part of the macromolecular regulatory complex for RyR2 possibly through RyR2-Epac functional coupling along with the muscle-specific A-kinase anchoring protein (mAKAP) and PKA (437). Use of a novel fluorescent Epac analog has differentially localized Epac1 and Epac2 to the nucleus and the Z lines, respectively, in cardiomyocytes, consistent with their having separate intranuclear and calcium homeostatic roles (885).
2. Direct and indirect physiological effects of modified Ca2+ homeostasis
Modifications in [Ca2+]i and Ca2+ homeostasis in turn affect strategic molecules that may further modify the Ca2+ homeostatic processes themselves, alter potentially electrogenic transmembrane ion fluxes, or modify particular enzymic signaling sequences. These in turn potentially directly or indirectly modify membrane excitation and stability with potential arrhythmogenic consequences (556, 1210). These modifications can result from direct actions of Ca2+ itself. Alternatively, they could involve mechanisms directly or indirectly involving a wide possible range of intracellular targets. Besides PKA and protein kinase C (PKC), the latter include calmodulin (CaM) and calcium/calmodulin kinase II (CaMKII) (166). Both CaM and CaMKII may sense local [Ca2+] through Ca2+ binding to EF-hand motifs at the NH2 and COOH terminals of CaM. The binding results in formation of a Ca2+/CaM complex. This then binds to the CaMKII regulatory domain. The binding relieves the baseline action of the CaMKII autoinhibitory domain. Thr-287 autophosphorylation then preserves the resulting activated state even following return of [Ca2+] to its resting level, until a phosphatase-mediated dephosphorylation (29).
CaMKII is a multifunctional serine/threonine kinase that exists in cardiomyocytes as various splice variants of its δ isoform, CaMKIIδ (29). These variants have a wide range of downstream targets. Cytoplasmic CaMKIIδ2 (CaMKIIδC) is localized close to, and associates with, L-type Ca2+ channels and RyR2. Further CaMKII subpopulations occur in the intercalated disc and mitochondria (1103). These act upon PLN, Nav1.5, and multiple voltage-gated and ATP-sensitive K+ and Cl− channels (468, 898). Kinase-mediated protein phosphorylation thus exerts strategic regulatory effects in myocyte excitation-contraction coupling, metabolism, intracellular Ca2+ homeostasis, mitochondrial function, and protein transcription (see sect. VII, B5 and F). As detailed below, these actions perturb the balance between inward ICaL and outward IK currents determining cardiac AP plateau durations (24) and modify intracellular Ca2+ in turn modifying CaM and CaMKII activity (898).
3. Effects on Ca2+ release and its triggering
These direct and indirect regulatory mechanisms potentially alter both the triggering and the Ca2+ release process itself through effects on ICa (see sect. VIIB1) and on the RyR2-Ca2+ release channel (Figure 14C) (see sect. VIIB3). Increased subsarcolemmal [Ca2+] exerts rapid negative feedback inhibitory effects on ICaL. These take place in addition to, and over much more rapid time courses compared with, the slow voltage-mediated inactivation of ICaL following its initial activation by the AP upstroke (1067). The feedback effects likely entail Ca2+ binding to CaM itself prebound to the L-type Ca2+ channel COOH terminal (641). In contrast, Ca2+-free CaM reduces L-type Ca2+ channel inactivation (897). The resulting Ca2+/CaM then interacts with a closely located IQ domain. This in turn blocks the inner pore of the L-type Ca2+ channel (712). This mechanism potentially provides a mechanism by which Ca2+ either entering from the extracellular fluid or released from the SR exerts a normal negative feedback inactivating L-type Ca2+ channel activity. Such an action would normally prevent Ca2+ overload.
Increased [Ca2+]i may also potentiate ICaL. This may also involve a Ca2+ binding to CaM. This in turn activates CaMKII which then promotes phosphorylation of the COOH terminal of the L-type Ca2+ channel α1-subunit (27, 1311). Moderate increases in [Ca2+]i following repetitive stimulation do produce such a calmodulin-mediated CaMKII activation (1311). Recovery from Ca2+-mediated ICaL inactivation then requires a fall in subsarcolemmal [Ca2+] typically brought about by the NCX, whose activity is also voltage-dependent. These mechanisms together result in complex interactions between L-type Ca2+ channel and NCX activity, membrane potential, and intracellular Ca2+ (341).
RyR2s also show Ca2+-dependent adaptation and inactivation properties dependent on separate mechanisms sensing both local cytosolic and SR intraluminal [Ca2+]. These may involve luminal (L-) (KD = ∼60 μM), cytoplasmic (A-) (KD = ∼0.9 μM) activation sites, and a cytoplasmic Ca2+ inactivation (I2-) site (KD = ∼1.2 μM). Regulation by luminal and cytoplasmic [Ca2+] are closely related. L-site activation produces brief (1 ms) <10/s openings. These nevertheless give luminal Ca2+ access to the A-site whose occupancy by Ca2+ produces considerable longer openings. They also give access to the I2 site, which inactivates the RyR2 at high Ca2+ turnovers. In addition, luminal Mg2+ (at ∼1 mM) is essential for the control of SR excitability in inhibiting cardiac muscle RyR2 but not skeletal muscle RyR1. It competes with Ca2+ for the L-site (review in Ref. 622). In consequence, alterations in cytosolic Ca2+ influence the triggering of RyR2-mediated Ca2+ release. SR luminal Ca2+ levels also influence the Ca2+ available for, and the extent of SR Ca2+ release, and the likelihood of spontaneous and propagated release phenomena. RyR2-Ca2+ release channels also show inactivation/adaptation phenomena dependent on the level and the rate of Ca2+ increase (394). Finally, Ca2+-free CaM decreases RyR2 open channel probabilities by ∼60%, effects reversible by Ca2+ itself (898), and CaMKII action results in RyR2 phosphorylation promoting release of SR-Ca2+ (1243).
Finally, significant increases in [Ca2+]i (to >320–560 nM) could close gap junctions mediating intercellular coupling (389, 756, 839, 1074).
4. Effects on cellular Ca2+ efflux processes
As indicated above, NCX extrudes much of the L-type Ca2+ channel-mediated Ca2+ entry. A smaller part is transported by plasma membrane Ca2+-ATPase. NCX activity is electrogenic: it transports three Na+ in exchange for one Ca2+ (129, 265). The direction of its current flow depends on the Na+ and Ca2+ gradients and the membrane potential Em. Thus INCX is inwards and depolarizing when Ca2+ is extruded, and outwards and hyperpolarizing with Ca2+ influx. Consequently, increased diastolic [Ca2+]i potentially activates transient inward currents (Iti) mediated by a forward-mode NCX-mediated Na+ influx (101, 354). This could cause premature sarcolemmal depolarization and a consequent triggered activity which causes systolic dysfunction and increases arrhythmic tendency (see sect. VII, B4 and C4) (903). It is also possible that increased SR Ca2+ leak into the restricted space of the dyadic cleft could increase local NCX activity removing the resulting increased local Ca2+. This in turn would correspondingly increase local [Na+]i potentially reducing the driving force for Na+ entry through nearby Na+ channels, and the resulting INa (124, 666).
Both heart failure and hypertrophic cardiomyopathies are associated with increased spontaneous SR Ca2+ leak (13, 226, 864). An increased SR Ca2+ would then activate inward depolarizing NCX-mediated current. If sufficiently large, such transient inward currents (Iti) could initiate proarrhythmic spontaneous DAD phenomena (106). Human cardiac heart failure is also associated with a decreased INa (1164). The consequent reductions in CV could similarly increase susceptibility to AF (362).
Finally, PMCA activity is enhanced by Ca2+/calmodulin and calmodulin binding (143).
5. Effects on Nav1.5 function and expression
Several studies suggest that altered cytosolic Ca2+ also more directly modifies either Nav1.5 function (15, 51, 1112) or expression (196, 282, 1114) (see sect. VIIC6). The functional effects may involve direct and/or indirect Ca2+ binding to Nav1.5 itself. Direct Ca2+ binding can take place at an EF hand motif close to the Nav1.5 COOH terminal. This shifted the voltage dependence of Na+ channel inactivation in a positive direction, potentially increasing Na+ channel activity (1265). Indirect Ca2+ binding involves an additional binding site, the ”IQ“ domain, for Ca2+/CaM in the Nav1.5 COOH-terminal region, or multiple phosphorylatable sites, including serines 516 and 571, and threonine 594, in the DI-II linker region which is targeted by CaMKII (368, 799, 1211).
These two mechanisms both require prior Ca2+ binding to the EF hand motifs of Ca2+/CaM or CaMKII. They shift INa activation in a positive direction along the voltage axis (51). They also enhance slow INa inactivation (1112). Thus increased pipette [Ca2+] reduced INa density and (dV/dt)max in patch-clamped myocytes (168). Increased and decreased [Ca2+]i following increasing extracellular [Ca2+] and the acetomethoxy ester, BAPTA-AM, respectively increased and decreased INa densities in cultured neonatal rat myocytes while sparing unit Na+ channel conductance or gating (196). The Ca2+ channel antagonist verapamil and the Ca2+ ionophore calcimycin respectively increased and decreased Nav1.5 mRNA and Nav1.5 protein expression in rat cardiomyocytes (282, 850, 1114). Such properties appeared specific to cardiac as opposed to skeletal muscle Na+ channels (260, 1308). However, recent rapid Ca2+ photorelease methods did not induce Ca2+ modulation in Nav1.5, but did so in Nav1.4 (96).
Furthermore, more indirect interactions, which may in particular influence INaL (see sect. VIB1), may involve protein subunits associated with the Na+ channel. CaMKII associates with a COOH-terminal motif in actin-associated βIV-spectrin protein. The latter is strongly homologous to a binding domain within the L-type Ca2+ channel β subunit that has been invoked in the increased open probability and mean open time following channel phosphorylation (105, 1103). The CaMKII/βIV-spectrin interaction also operated in CaMKII-mediated phosphorylation of those voltage-gated Na+ channels residing at the cardiomyocyte intercalated disc (467, 716). This may mediate the modulation of steady-state Na+ channel inactivation, recovery from inactivation, and the magnitudes of INaL components observed in heterologous cells and primary myocytes. These actions likely involve separate Nav1.5 modulation pathways (467, 1209).
6. Effects on repolarization processes
Alterations in both [Ca2+]i and its consequent phosphorylation levels thus modulate both ICaL and INa and therefore action potential propagation and plateau duration, respectively. These alterations also affect membrane processes related to recovery from excitation. These may similarly have potential arrhythmic effects. Guinea pig ventricular APs do not involve K+-mediated Ito: its function may be replaced by a Ca2+-activated Cl− current in turn activated by SR Ca2+ release (1067). In addition, IKs is enhanced by increased [Ca2+]i (1136), α- or β-adrenergic stimulation, or PKC activation (1213). IKr is increased by PKA likely through increased [Ca2+]i and PKC activation, primarily through altering its inward rectification properties (418). Modeling studies suggested that changes in IKs modify APD restitution to a greater extent than changes in IKr, although the ratio between the two currents is likely important. Spatial variations in the magnitude of these currents across the myocardium may also influence their restitution properties. This would be particularly the case in areas with steeper AP repolarization slopes including subendocardial M-cell layer, with possible implications for altered Ca2+ homeostasis upon restitution properties (752, 753, 1201).
Of membrane transporters, the Na+-K+-ATPase is maximally activated at physiologically occurring K+ concentrations. Instead, it is intracellular Na+ concentration, in turn dependent on INa and NCX activity, that may limit its activation. However, Na+-K+-ATPase activity is also increased by its phosphorylation by PKC following adrenergic stimulation or increased Ca2+ (340). Na+-K+-ATPase activity is thus stimulated by β- and α-adrenergic receptor activation and increased [Ca2+]i (340). NCX in turn is energetically dependent on transmembrane Na+ gradients and therefore Na+-K+-ATPase activity.
B. Acquired Arrhythmic Disorders Associated With Altered Cellular Ca2+ Homeostasis
1. Ventricular arrhythmic effects of β-adrenergic activation
Abnormal cellular Ca2+ homeostasis is accordingly implicated in a wide range of arrhythmic pathologies. Cardiac failure and hypertrophy exemplify acquired conditions associated with arrhythmic phenomena (1244, 1299). Of these, cardiac failure is associated with chronically elevated adrenergic receptor-mediated cellular signaling (1238, 1242). In common with the inherited condition of CPVT, it has been analyzed in terms of schemes involving a PKA-mediated phosphorylation of RyR2 serine 2809 (or serine 2808, depending on species). This increases its open probability (Po) by dissociating FKBP12.6 from RyR2 (1238). However, there remains uncertainty in the exact phosphorylatable RyR2 sites and the individual protein kinases involved, whether involving CaMKII or protein kinase G (PKG), and the functional roles of such phosphorylation (109, 1279).
At all events, murine systems provide useful models for the acute arrhythmic effects of altered Ca2+ homeostasis. This extends their utility in studies of the arrhythmic effects of altered ion channel expression (see sects. V and VI). For example, isolated murine hearts recapitulated the effects of β-adrenergic stimulation and L-type Ca2+ channel blockade both on ventricular arrhythmogenesis in intact Langendorff-perfused hearts and Ca2+ homeostasis in isolated myocytes. Programmed stimulation thus demonstrated arrhythmogenic tendencies predisposing to VT following both isoproterenol challenge and elevated extracellular [Ca2+]. Programmed electrophysiological fractionation analysis suggested that this took place with normal AP conduction, in contrast to the increased EGDs at shortened S1–S2 intervals in arrhythmic Kcne1−/− and Scn5a+/− hearts. The latter had then suggested reentrant as opposed to triggered arrhythmia (64, 416, 867, 1095).
These comparisons were consistent with the existence of an arrhythmogenic tendency that could result from triggered activity. Accordingly, in isolated ventricular myocytes, both isoproterenol and elevated extracellular [Ca2+] increased the amplitudes of electrically evoked Ca2+ transients as reported in other cardiac systems (104). Pretreatment with the dihydropyridine or benzothiazepine L-type Ca2+ channel blockers nifedipine or diltiazem both suppressed the VT and decreased, and prevented the isoproterenol-induced increases in Ca2+ transient amplitudes (63). Murine hearts thus recapitulate the increased inward ICaL and consequently SR Ca2+ uptake, in turn producing spontaneous SR Ca2+ release and increased Iti with β-adrenergic stimulation (905).
2. Ventricular arrhythmic effects of Epac activation
Murine systems also permitted an examination of the effects of Epac pathway activation on cardiac Ca2+ homeostasis, electrophysiological properties, and arrhythmic tendency (see sect. VIIA1). Of Epac isoforms, Epac1 is localized and functionally involved in nuclear signaling. Epac2 is located at the transverse tubules and may regulate arrhythmogenic SR Ca2+ leak (885). Baseline basal cAMP concentration and Epac activation levels appeared low in adult ventricular myocytes in the absence of β-adrenergic activation. Murine Epac1−/−, Epac2−/−, and CaMKIIδ−/− hearts showed normal in vivo baseline cardiac structure, ratios of heart to body weight, or early pressure overload-induced hypertrophy. They also showed normal physiological indexes of ventricular ejection, heart rate, and LV pressure indexes. All these indexes furthermore showed normal responses to dobutamine challenge. Western blot studies revealed normal SERCA and NCX expression levels. Isolated Epac1−/− and Epac2-/1 myocytes showed normal Ca2+ transient amplitudes and decline time courses, SR Ca2+ content, NCX function, and diastolic Ca2+ spark frequency when normalized to SR Ca2+ load (883).
RyR2 activation produced by Epac activation that is independent of PKA action was attempted using 8-(4-chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate (8-CPT). Low (1 μM) as opposed to high (100 μM to 1 mM) 8-CPT concentrations provide a specific >300-fold efficacy in activating Epac rather than PKA (437). In Langendorff-perfused mouse WT hearts, this produced arrhythmic effects often resulting in triggered activity and VT (446). 8-CPT also increased the frequencies of spontaneous cytosolic Ca2+ transients in neonatal rat cardiac myocytes (798), amplitudes of electrically evoked Ca2+ transients in mouse ventricular myocytes (849), and frequencies of Ca2+ sparks in isolated rat ventricular myocytes (884). There were also associated Ca2+ homeostatic abnormalities observable in isolated cardiomyocytes (446). There was thus a propensity to generate the spontaneous Ca2+ waves previously associated with cardiac arrhythmogenesis triggered by Iti resulting in DADs (Figure 15, A and B) (101, 903). There were also increased incidences of ectopic Ca2+ release in both paced and resting cells (Figure 15C, a and b). These were abolished by treatment with the CaMKII inhibitor KN-93 (Figure 15Cc).
Figure 15.

Epac-induced RyR2 activation as an exemplar for Ca2+-mediated arrhythmia. A: propagation of a Ca2+ wave from one end of the cell to the other following 8-CPT challenge shown in successive confocal microscope frame scan images. B: Ca2+ fluorescence signal from six successive 2 μm × 2 μm regions of interest (a–f), placed along the long axis of a myocyte demonstrating progressively increasing delays in onset of the Ca2+ transient. C: Ca2+ signals in regularly stimulated ventricular myocytes (triangles mark timing of pacing stimuli) showing irregularly occurring ectopic Ca2+ transients during 8-CPT treatment (a, b) abolished by KN-93 (c). D: persistent ventricular tachycardia (VT) following programmed electrogram stimulation observed during perfusion with 8-CPT prevented by CaMKII inhibition with KN-93. E: triggered activity (*) during intrinsic activity observed during perfusion with 8-CPT, prevented by KN-93 pretreatment. F: epicardial (a) and endocardial APD90 (b), ΔAPD90 (c), and VERP (d) under control conditions (clear bars), during 1 μM 8-CPT treatment in the absence (black bars) and following 1 μM KN-93 pretreatment (striped bars). [From Hothi et al. (446).]
At the level of intact Langendorff-perfused murine hearts, challenge by either 8-CPT or a combination of isoproterenol and the PKA inhibitor H-89 (808) increased the incidences of VT provoked by programmed electrical stimulation (Figure 15D). It also increased incidences of triggering events following AP activation (Figure 15E). In contrast, spatial electrophysiological ventricular heterogeneities whether in the form of epicardial or endocardial APD90, transmural or apico-basal gradients of repolarization, or of VERP, remained unchanged (Figure 15F). Temporal AP properties in the form of maximum APD90 restitution gradients also remained normal (446). These findings provide clearcut contrasts with the altered, negative, ΔAPD90s, and altered restitution properties thought to mediate arrhythmogenesis through the resulting reentrant substrate in the murine congenital LQTS and hypokalemic models described above (see sect. VI, B–E) (561, 1128, 1130).
Both the cellular effects of Epac on Ca2+ homeostasis and its arrhythmic effects in intact hearts were reduced by CaMKII inhibition using 1 μM KN-93. This was consistent with a dependence of Epac action on CaMKII activity (446). Finally, genetic ablation of Epac2, β1-AR, CaMKIIδ, and RyR2-S2814 phosphorylation all abolished Epac-dependent arrhythmogenic effects. These findings implicate Epac2 in β1-adrenergic arrhythmias through mechanisms involving CaMKIIδ and RyR2-S2814 phosphorylation (883).
3. Ventricular arrhythmic effects of direct RyR2-SR Ca2+ release channel activation
Interventions directly opening RyR2-Ca2+ release channels (1139) employed either caffeine or the immunosuppressant FK506 known to alter RyR2-FKBP12.6 binding (142, 527, 618, 1282, 1300). These also altered myocyte Ca2+ homeostasis and exerted arrhythmogenic effects in murine hearts (62). This agrees with previous reports in canine and rabbit hearts in vivo (481, 762). Programmed electrical stimulation confirmed that these proarrhythmic effects took place under conditions of normal conduction (62).
Caffeine either conserved or reduced the amplitudes of regularly evoked cytosolic [Ca2+] transients in isolated mouse myocytes consistent with a partial SR Ca2+ depletion (62). The latter would parallel the reduced myocyte SR Ca2+ in human heart failure (665). Both caffeine and FK506 increased the frequencies of spontaneous periodic peaks resulting from propagating Ca2+ waves (62) associated with SR Ca2+ loading by increased extracellular [Ca2+] (263, 1139, 1258). The latter effect is thought to result from increased Ca2+ spark frequency and to trigger arrhythmia (325) through activated NCX (760). These effects were abolished by the RyR inhibitor tetracaine.
However, diltiazem but not nifedipine prevented VT following programmed electrical stimulation in caffeine-treated hearts (1300). It also abolished the effects of both caffeine and FK506 upon the frequency of spontaneous Ca2+ release events. These findings correlate well with findings that diltiazem but not nifedipine inhibited RyR2-mediated SR Ca2+ leak in canine SR vesicles after addition of FK506 (1300) in addition to their L-type Ca2+ channel block (64). Such Ca2+ wave phenomena have also been observed in skeletal muscle under conditions of dihydropyridine receptor-RyR1 decoupling. These showed similar pharmacological sensitivities to caffeine, L-type Ca2+ channel block, and tetracaine challenge (183).
Cyclopiazonic acid (CPA) inhibits the activity of SR Ca2+-ATPase (SERCA) (1035) by blocking its Ca2+ channel (794). It inhibits SERCA activity in myocardial vesicle preparations (1032) while sparing Ca2+ sensitivity in contractile myofilaments, Ca2+ currents, and NCX activity (1107, 1302). It would therefore be expected to produce delayed reductions in SR Ca2+. CPA (100 and 150 nM) accordingly produced delayed rather than immediate reductions in Ca2+ transients in regularly stimulated fluo 3-loaded isolated myocytes. It correspondingly antagonized arrhythmic effects of isoproterenol, elevated extracellular [Ca2+], or caffeine. However, it did so only when the latter agents were added following rather than simultaneously with introduction of CPA. Both this introduction of arrhythmia and its rescue occurred without appearance of reentrant substrate, MAP waveforms, or VERPs (351).
4. Ventricular arrhythmic effects of purinergic receptor activation
Murine models also modeled the arrhythmic effects of extracellular fluid alterations resulting from metabolic change in surrounding tissue. A number of these may produce arrhythmia through Ca2+-dependent mechanisms. For example, both in normal and ischemic cardiac cells, sympathetic nerve terminals release ATP (934). Conversely, ventricular myocytes express both ionotropic P2X receptors and G protein-coupled P2Y receptors (1176). Cardiac ischemia is often accompanied by elevated interstitial [ATP] (165) and is often also associated with ectopic beats and VT. Extracellular ATP induced oscillatory contractions and ectopic APs (203) in isolated ventricular myocytes which additionally then showed afterdepolarizations with isoprenaline challenge (1079). Similarly, Langendorff-perfused murine hearts showed ectopic beats and VT (386) with challenge by extracellular ATP levels (∼100 μM) close to those previously reported under pathological conditions. Such findings did not occur with adenosine and UTP (213).
These findings implicated P2 purinergic rather than adenosine-activated P1 or UTP-activated P2 receptors. Thus ATP correspondingly induced sustained increases in diastolic Ca2+ and triggered Ca2+ waves that possessed relatively early (10–20 s) rather than prolonged (minutes) onsets. Single-cell voltage-clamp experiments demonstrated ATP-activated multiple cationic inward currents. These showed positive reversal potentials as expected of the Ca2+ (1 mM) rather the monovalent (Na+ and Cs+) ion concentrations in which the isolated ventricular myocytes were studied. All these arrhythmic, Ca2+ homeostatic, and electrophysiological effects were antagonized by the P2 receptor antagonists suramin and pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS). These observations are consistent with a Ca2+ influx following P2X receptor opening. These currents could induce arrhythmogenic changes in resting membrane potentials. APs induced by repetitive (2 Hz) current-clamp steps in the presence of ATP (100 μM) were followed by oscillatory resting membrane potential changes, DADs, and triggered ectopic APs and EADs, and themselves were frequently prolonged (386).
In contrast, metabotropic P2Y receptors are mainly coupled to Gq proteins. They then activate PKC through phospholipase C-dependent production of diacylglycerol (DAG) (157). PKC in turn can target L-type Ca2+ channels, RyR2s, and voltage-gated Na+ and K+ channels. ADP (100 μM), associated with P2Y receptor activation, did not produce even delayed (>3 min) diastolic Ca2+ elevations. However, its application was followed by gradual declines in diastolic Ca2+ and [Ca2+]i transients. Its sustained application (>3–5 min) resulted in prolonged waveforms of Ca2+ transients induced by field stimulation. There was then an ultimate failure of subsequent stimulation to induce [Ca2+]i transients. These changes persisted even following ADP washout (386).
5. Ventricular arrhythmic effects of acidotic stress
Murine hearts also recapitulated the acute arrhythmic effects that followed the imposition and withdrawal of acidotic challenge. These effects showed features consistent with altered Ca2+ homeostasis involving CaMKII action (860). This may be important in ischemia/reperfusion injury. Intracellular acidosis is then a component of the ischemia. The reperfusion that follows produces rapid return of pH to its background value. Cell acidification is also an early accompaniment to acute myocardial ischemia (1168). Such acidication produced both arrhythmogenic effects (860) and increased CaMKII activity (466, 586).
MAPs recorded from Langendorff-perfused rat and mouse hearts subject to imposition followed by withdrawal of respiratory acidosis showed ectopic beats. Their frequency was reduced by the CaMKII kinase inhibitor KN-93, the SR uptake inhibitor thapsigargin, and the RyR2 SR Ca2+ release blockers ryanodine and dantrolene (992). They were not observed in transgenic mice expressing a CaMKII inhibitory peptide targetted to cardiac longitudinal SR (507). The acidosis increased phosphorylation of the Thr17 site of phospholamban (PT-PLN) increasing SR Ca2+ load. KN-93 inhibited the latter effect. The ectopic activity was triggered by DADs, particularly in epicardial myocytes. The latter was also prevented by KN-93.
Acidification also produced spontaneous SR Ca2+ release, spontaneous AP firing, and triggered arrhythmias (264). It increased the amplitudes of caffeine-induced Ca2+ transients that were abolished by KN-93 (586). These were attributed to increased NCX activity in response to Ca2+ leak from an overloaded SR likely due to CaMKII-dependent increase in SR Ca2+ uptake (860). Thus arrhythmias following acidosis result from a CaMKII activation that increases SR Ca2+ load occurring mainly through from increased PT-PLN activity.
MAP recordings from Langendorff-perfused whole murine heart preparations could be performed in parallel with single whole-ventricular myocyte AP and Ca2+ fluorescence measurements under acidotic stress conditions. These made it possible to separate CAMKII-dependent and CAMKII-independent contributions to the arrhythmic changes following metabolic acidification (879). Lactate challenge induced spontaneous arrhythmogenesis in both intrinsically active and regularly paced Langendorff-perfused murine hearts. It also induced spontaneous Ca2+ waves in isolated intact fluo 4-loaded myocytes. Both these effects were reversibly abolished by KN-93. Acidification also produced spontaneous AP firing and membrane potential oscillations in isolated ventricular myocytes whose patch pipette solutions permitted [Ca2+]i to increase. Both KN-93 and use of EGTA-buffered pipette solutions holding cytosolic [Ca2+]i constant during the acidosis abolished the latter effects. Together these findings implicate CaMKII-dependent waves of SR Ca2+ release in spontaneous arrhythmic events during metabolic acidification.
However, programmed electrical stimulation similarly induced VT during metabolic acidification. This correlated with appearance of an arrhythmic substrate resulting from reduced LV transmural repolarization gradients that resulted from prolongations of epicardial but not endocardial APD90. However, these latter parameters were KN-93-resistant both before and after acidification. Thus agents inhibiting CaMKII action reduced AP triggering while sparing the associated arrhythmic substrate produced by cell acidification (879).
6. Roles of Ca2+ in acute atrial arrhythmogenesis
Mechanisms initiating AF are incompletely understood. Nevertheless, significant contributions to these may arise from alterations in cellular Ca2+, particularly diastolic, signaling. These could involve release of Ca2+ from nevertheless finite SR Ca2+ stores, a process induced by an initial extracellular Ca2+ entry (1202). The resulting increase in NCX activity might then result in EAD or DAD phenomena initiating focal firing. Acute episodes of acquired atrial arrhythmias can follow cardiothoracic, particularly coronary artery bypass grafting, surgery, typically at ∼2 but rarely beyond 7–15 postoperative days. They are reduced by either β-adrenoceptor or Ca2+ channel antagonism (60). Sustained arrhythmia may require reentrant substrate that might arise from altered balances between conduction velocity (θ) and refractory period (269, 819, 822). Such substrate may develop following physiological and anatomical remodeling (184, 269, 270, 451, 658, 923, 1194).
Murine WT Langendorff-perfused hearts recapitulated these roles of altered Ca2+ homeostasis in producing acute acquired atrial arrhythmias. They related these arrhythmias to an occurrence of diastolic Ca2+ events in isolated atrial myocytes. It was then possible to pharmacologically separate the contributions from altered SR Ca2+ storage or release, and extracellular Ca2+ entry, in this initiation (1338). These studies thus implicated an increased Ca2+-induced Ca2+ release from a nevertheless finite SR Ca2+ store ultimately dependent upon entry of extracellular Ca2+ in such arrhythmogenesis.
Thus, in isolated Langendorff-perfused hearts, caffeine exerted atrial proarrhythmic effects immediately following but not >5 min after application. Regularly stimulated atrial myocytes then correspondingly showed diastolic Ca2+ waves and progressive reductions in their evoked Ca2+ transients and these diastolic events with prolonged exposure (1338). Both findings are directly explicable in terms of increased Ca2+ release through sensitized SR RyR2-Ca2+ release channels from a finite and therefore depletable intracellular Ca2+ store (1187). The arrhythmic phenomena associated with caffeine challenge could thus be abolished by pretreatment with either the SERCA inhibitor CPA or the ICaL blocker nifedipine (1338). At the cardiomyocyte level, CPA applied by itself produced successive reductions in the amplitudes of evoked Ca2+ transients. Use of nifedipine as a ICaL blocker implicated a dependence of extracellular Ca2+ entry on these effects (1048, 1142). Thus nifedipine by itself promptly reduced the evoked Ca2+ transients. These again increased then declined in amplitude with caffeine challenge but in an absence of diastolic Ca2+ events (1338).
The dependence of acute atrial arrhythmogenesis upon extracellular Ca2+ entry was contrastingly accentuated by the Ca2+ channel opening agent FPL-64176. This is known to prolong depolarization-induced L-type Ca2+ channel opening and slow repolarization-induced L-type Ca2+ channel closure (306, 1347). FPL-64176 challenge elicited diastolic Ca2+ events in isolated myocytes and atrial arrhythmogenesis intact perfused hearts. It did so without changing AERP. Both effects were inhibited by pretreatment with nifedipine, caffeine, or CPA. Enhanced extracellular Ca2+ entry thus exerts acute atrial arrhythmogenic effects nevertheless dependent on diastolic Ca2+ release (1335).
C. Genetic Abnormalities in the RyR2-SR Ca2+-Release Channel: CPVT
1. Clinical features of CPVT
Genetic disorders have also provided useful systems to study how abnormal Ca2+ homeostasis may cause cardiac arrhythmogenesis (676, 917). One such exemplar of these is offered by the CPVT typically associated with RyR2 or calsequestrin (CASQ) mutations (786, 1241). An autosomal dominant CPVT is associated with cardiac RyR2 (916). A recessive variant is associated with homozygous cardiac calsequestrin (CASQ2) gene mutations (610). CPVT-like conditions are also associated with ankyrin-B mutations (790). RyR2 mutations are also implicated in ARVC type 2 (82, 234, 621, 1135).
CPVT presents as an occurrence of acute VT on adrenergic stimulation that may degenerate into potentially fatal VF (175, 913). It typically presents earlier, at age ∼7–9 yr, than BrS (625). CPVT patients show relatively normal resting ECGs. However, exercise or catecholaminergic challenge elicits arrhythmic features that include bidirectional VT with 180 degree beat-to-beat alternations in the QRS axis. Clinical management involves β-blocker therapy but ∼30% of patients require implantable cardioverter defibrillators (ICDs) (913). Left cardiac sympathetic denervation is considered for intractable arrhythmias (1261). CPVT has a poor prognosis, particularly in males with a RyR2 mutation. It likely contributes ∼15% of SCD in structurally normal hearts (678, 1123). A third of CPVT patients have family histories of premature sudden death or stress-related syncope (625). Of these, 50–70% show a genetic abnormality (58, 913).
2. Human and murine RyR2 mutations in CPVT
Two groups of investigators made the initial identifications of RyR2 mutations in CPVT (613, 916). Five clinically affected carriers proved to have RyR2-S2246L, RyR2-R2427S, RyR2-N4104K, and RyR2-R4497C. Three unrelated Finnish families carried RyR2-P2328S, RyR2-Q4201R, and RyR2-V4653F. The latter abnormalities were accompanied by a 30–33% mortality by age 35 yr. RyR2-P2328S was associated with normal resting ECGs including normal QTc intervals. Ten of 12 such patients showed exercise-induced VT. Two were asymptomatic but exhibited abnormalities on clinical testing.
A large proportion of the >70 RyR2 mutations reported subsequently in CPVT, a number of which have been replicated in murine models (Table 7), have similarly been base-pair substitutions involving highly conserved residues. The mutations mainly cluster in three, NH2-terminal (176–433), central (2246–2504), and COOH-terminal regions (4104–4653) of the RyR2 gene (1240). Approximately 20 have been studied in vitro (676). All three regions are represented in murine studies of ventricular properties associated with the RyR2-R420W (851), RyR2-R176Q/+ (534), RyR2-R4496C/+ (172), RyR2-R2474S/+ (1157), and heterozygotic and homozygotic RyR2-P2328S exemplars, respectively (364, 1340). All the murine variants showed normal structure and histology, despite known associations of RyR2-R176Q with human ARVC2. Finally, studies in HEK293 expression systems indicate that large deletions encompassing exon 3 in the NH2-terminal RyR2 region remain compatible with continued RyR2 stability and function through insertion of a flexible loop into the β-trefoil domain (119). Ca2+ release then terminates at a lower luminal Ca2+ (1113).
Table 7.
Transgenic murine models for catecholaminergic polymorphic ventricular tachycardia (CPVT)
| Gene | Genetic Modification | Phenotype | Reference Nos. |
|---|---|---|---|
| RyR2 (NH2-terminal region) | RyR2-R176Q/+ | CPVT (ARVC2) | 534 |
| RyR2-R420W | CPVT | 851 | |
| RyR2 (central region) | RyR2-P2328S/+ and RyR2-P2328S/P2328S | CPVT | 364, 1340 |
| RyR2-P2328S/P2328S | AF | 569, 832, 996, 1334, 1337 | |
| RyR2-R2474S/+ | CPVT | 585, 629, 1156 | |
| RyR2 (COOH-terminal region) | RyR2-R4496C/+ | CPVT | 172, 673 |
| Casq2 | Casq2-D307H overexpression | CPVT | 266 |
| Casq2−/− | CPVT and cardiac hypertrophy | 581 | |
| Casq2+/− | No phenotype | 199 | |
| Casq2-D307H/D307H | Spontaneous arrhythmias | 1078 | |
| Casq2-ΔE9/ΔE9 | Spontaneous arrhythmias; cardiac hypertrophy with age | 1077 | |
| Casq2-R33Q/R33Q | Spontaneous arrhythmias | 951 |
3. Electrocardiographic features of murine models with genetically modified RyR2
Genetically modified RyR2 mouse models recapitulated electrophysiological phenomena associated with initiation of arrhythmogenesis in CPVT. In vitro studies associated gain-of-function RyR2, and loss-of-function Casq2, mutations with increased SR Ca2+ leakage thereby increasing [Ca2+]i (509, 631). This would give a situation resembling clinical digitalis toxicity (625). In either case there would be an expected increase in NCX activity. This would increase Iti that in turn drives DADs. The latter have been suggested to potentially cause triggered activity where they produce membrane depolarizations attaining the INa reactivation threshold (581, 673). This would particularly affect Purkinje fibers owing to their greater susceptibility than ventricular myocytes to Ca2+ overload due to their greater Na+ loads and longer APDs (1174). Clinical arrhythmic activity in both CPVT and digitalis toxicity is thus accompanied by bidirectional VT reflected in alternating 180° rotations of the electrocardiographic QRS axis between beats (625, 674). This can then be followed by degeneration into rapid polymorphic VT and VF.
Electrocardiographic properties of a significant number of these mice expressing variant RyR2 recapitulated these clinical arrhythmic and ECG features. Polymorphic VT and/or bidirectional VT was observed in ambulant RyR2-R4996C/+ (172, 673) and RyR2-P176Q/+ mice (534) upon combined or individual epinephrine and caffeine administration, and in RyR2-R2474S/+ mice during treadmill testing (585, 629, 1157). Endocardial optical potentiometric mapping localized the arrhythmic foci in the RyR2-R4496C/+ to triggered firing alternating between the left and left Purkinje bundles. Thus chemical ablation of the RV Purkinje network converted the bidirectional VT to a monomorphic, wide-QRS, VT (175).
Episodes of bigeminy and BVT also occurred in anesthetized heterozygotic RyR2-P2328S/+ (RyR2S/+) hearts following isoproterenol challenge. They further occurred in anesthetized homozygotic, RyR2-P2328S/P2328S (RyR2S/S) hearts in association with multiple ventricular ectopic beats, both before and following isoproterenol challenge (1334). These findings potentially implicated an involvement of Purkinje system conduction in CPVT-associated arrhythmia (1016). Thus, before pharmacological challenge, there was only a single BVT episode among 12 RyR2S/S mice. Pharmacological, epinephrine and caffeine, challenge selectively increased the number of RyR2S/S, but not RyR2S/+ or WT mice showing arrhythmias in the form of bigeminy, bidirectional, monomorphic, or polymorphic VT. In common with clinical findings, the bidirectional VT appeared as salvos of four or more beat-to-beat 180° QRS axis alternations and bigeminy. They formed repeated occurrences of a ventricular premature complex succeeded by a normal beat. Polymorphic VT or VF appeared as series of three or more ventricular ectopics with irregular QRS axes. Monomorphic VT was identified in runs of abnormally shaped ECG complexes nevertheless occurring with regular rates and rhythms (1340).
4. Altered Ca2+ homeostasis in murine models of altered RyR2
The murine models for altered RyR2 consistently showed evidence of abnormal Ca2+ homeostasis. Murine RyR2-R176Q/+ cardiomyocytes showed higher incidences of spontaneous, nontriggered Ca2+ oscillations compared with WT both before and following isoproterenol stimulation. Isoproterenol (100 nM) induced a range of Ca2+ transients. These included slowed decays, spontaneous diastolic transients, and prolonged evoked transients (534). A comparison of Ca2+ transients from regularly stimulated WT, heterozygotic (RyR2S/+), and homozygotic (RyR2S/S), RyR2-P2328S myocytes similarly suggested altered Ca2+ release patterns and a resulting SR Ca2+ depletion (364). Thus WT, RyR2S/+, and RyR2S/S myocytes showed indistinguishable peak amplitudes in their evoked Ca2+ transients. RyR2S/S myocytes showed subsidiary events, as well as irregular Ca2+ signaling patterns. Isoproterenol (100 nM) elicited ectopic peaks and subsidiary events in WT and RyR2S/S but not RyR2S/+. However, it reduced the amplitudes of evoked peaks in the RyR2S/+ myocytes. Isoproterenol-treated RyR2S/S myocytes additionally showed spontaneous Ca2+ propagating waves (Figure 16A). Such waves have been previously associated with arrhythmogenic properties under conditions where they were produced by caffeine (62). RyR2-R2474S/+ myocytes showed markedly greater Ca2+ spark frequencies with isoproterenol addition (584). They also showed SR Ca2+ thresholds for spontaneous Ca2+ transients, cAMP-induced Ca2+ sparks and waves following PKA-mediated RyR2 phosphorylation that were greatly reduced compared with WT (1157).
Figure 16.

Proarrhythmic features in RyR2-P2328S hearts. A: sequential gallery of confocal microscope images showing Ca2+ waves in a single fluo3-loaded ventricular homozygotic RyR2-P2328S (RyR2S/S) myocyte following isoproterenol (100 nM) challenge. Arrowed: path taken by typical Ca2+ wave. B–D: epicardial monophasic action potentials in intrinsically active RyR2s/s hearts. B: spontaneous early afterdepolarizations (EADs) (*) followed by episodes of sustained monomorphic VT (sVT). C: coupled beats. D: persistent ventricular fibrillation (VF) following the cessation of regular S1 pacing after two intrinsic MAPs. E: S2 extra-stimuli during PES typically producing limited episodes of nonsustained VT in the absence (left trace) but sustained (>30 s) VT in the presence of 100 nm isoproterenol (right trace). F–I: electrophysiological assessment of Na+ channel function in RyR2S/S, Scn5a+/−, and WT atria. F: left atrial intracellular APs showing conduction latencies from WT, Scn5a+/−, and RyR2S/S myocytes and their corresponding (dV/dt)max (G). H and I: loose patch-clamp recordings of currents during a 100 mV 50-ms activation step following 50-ms prepulses between 20 and 100 mV for WT (H) and RyR2S/S atria (I). [From Goddard et al. (364) and King et al. (568).]
5. Spontaneous and provoked arrhythmia related to electrophysiological properties
Electrophysiological studies in Langendorff-perfused RyR2S/+ and RyR2S/S hearts went on to distinguish between phenotypes reflecting tendencies to provoked, and both provoked and spontaneous arrhythmia. These employed conditions of either regular or programmed electrical stimulation. They were performed both before and following isoproterenol challenge (364) (Figure 16, B–E). Unchallenged RyR2S/S though not RyR2S/+ hearts showed extrasystolic events. These were often followed by spontaneous sVT during intrinsic activity (Figure 16, B and C). RyR2S/+ hearts correspondingly showed nonsustained VT with programmed electrical stimulation but not regular pacing. However, following isoproterenol treatment, RyR2S/+ hearts showed both nonsustained and sustained VT with both regular pacing and programmed electrical stimulation. RyR2S/S hearts then showed higher incidences of nonsustained VT before and mainly sustained VT during both regular pacing and programmed electrical stimulation, particularly at higher pacing frequencies (Figure 16, D and E). Correspondingly, isolated RyR2-R4496C/+ but not WT myocytes showed DAD and triggered activity. These increased in frequency with isoproterenol administration and were abolished by ryanodine (673). This increased Ca2+ release could extend to compromising SR Ca2+ stores sufficiently to compromise both atrial and ventricular contractility (314).
6. Evidence for reentrant substrate in RyR2-P2328S hearts
In addition to demonstrating such potential triggering events, Langendorff-perfused RyR2S/S hearts additionally demonstrated evidence for arrhythmic substrate (816, 1340). Regularly paced RyR2S/+ and RyR2S/S hearts showed ventricular AP recovery characteristics of ventricular MAP duration, refractory period (364), and AP restitution properties indistinguishable from WT. This was the case whether before or following catecholaminergic challenge (984). Ventricular AP conduction velocities, assessed by vectorial analysis of local epicardial activation times using multiarray electrode array recordings, were indistinguishable in untreated WT and RyR2S/S. However, isoproterenol and caffeine challenge that would elicit arrhythmia specifically in RyR2S/S ventricles correspondingly slowed these conduction velocities. Intracellular microelectrode recordings demonstrated correspondingly reduced values of (dV/dt)max. The latter is known directly to correlate with conduction velocity and peak INa (567, 1047). Such findings directly parallelled alterations in membrane excitability following manipulations of Ca2+ homeostasis in skeletal muscle (1161).
Further studies corroborated this existence of potential arrhythmic substrate. They revealed evidence for reduced membrane Nav1.5 expression and a correspondingly reduced INa. These could account for the slowed ventricular conduction velocities in RyR2S/S ventricles (832). Thus Western blot analyses demonstrated that both whole tissue and membrane fractions of RyR2S/S ventricles showed similar Cx43, but reduced Nav1.5 protein expression compared with WT. Similarly, loose patch-clamp studies demonstrated a reduced INa. Extrasystolic S2 stimulation following 8-Hz S1 pacing at progressively decremented S1S2 intervals correspondingly demonstrated increases in ventricular arrhythmic tendency despite unchanged VERPs in Langendorff-perfused RyR2S/S hearts. Similarly, dynamic pacing employing progressively reduced BCLs produced greater reductions in conduction velocity at equivalent BCLs and diastolic intervals in RyR2S/S compared with WT ventricles. Yet both showed comparable changes in APD90 and its alternans.
These findings were compatible with both short- and long-term consequences of abnormal cytosolic Ca2+ homeostasis for Nav1.5 function. Thus ectopic activity and possible effects on Nav1.5 activation may follow transient [Ca2+]i elevations consequent upon catecholaminergic stimulation. Chronic elevations in [Ca2+]i might downregulate either Nav1.5 expression or activity. Either effect would reduce AP conduction velocity potentially generating arrhythmic substrate. Furthermore, the short-term events could potentially initiate triggering arrhythmic events then perpetuated by a longer term preexisting arrhythmic substrate reflected in decreased conduction. Such hypotheses merit further exploration. They have broader implications for potential therapeutic interventions involving Ca2+ homeostasis in management of potentially Nav1.5-dependent arrhythmic conditions (832).
Other situations related to disturbed Ca2+ homeostasis also caused conduction disturbance and major cardiac arrhythmia related to compromised INa. FKBP12 shares an 85% homology with FKBP12.6. However, it has mainly been associated with a modulation of skeletal rather than cardiac muscle RyR1-mediated excitation-contraction coupling (1133). Nevertheless, FKBP12 has reported effects on RyR2. Its haploinsufficiency is lethal in utero, and this is accompanied by ventricular hypertrabeculation, noncompaction, and septal defect (1061). Transgenic αMyHC-FKBP12 mice overexpressing FKBP12.6 showed high incidences (38%) of SCD. This accompanied ECG, electrophysiological, and optical mapping evidence of slowed conduction, slowed AP upstrokes, and prolonged APDs. Whole cell patch-clamp analyses attributed these findings to an 80% reduction in peak INa, its slowed recovery from inactivation, positive shifts of its steady-state activation and inactivation curves, and enhanced INaL. Conversely, cardiomyocyte-restricted conditional FKBP12 knockout (FKBP12-f/f/αMyHC-Cre) ventricular cardiomyocytes showed faster AP upstrokes and greater than twofold increased peak INa. These effects were reversed by dialysis of exogenous recombinant FKBP12 protein. This resulted in INa changes seen in the overexpression αMyHC-FKBP12 myocytes (744).
7. RyR2 phosphorylation and arrhythmic tendency
Murine hearts have also been used to model the mechanisms relating RyR2 protein abnormalities to alterations in RyR2-Ca2+ release function that would potentially trigger arrhythmia, although these remain controversial. These include the arrhythmic changes in acquired conditions including cardiac failure, which have been attributed to increased RyR2 open probabilities. However, the RyR2 is a large and complex molecule. Its monomer is associated with numerous potentially regulatory proteins including CaM, FKBP12.6, PKA, PP1 and PP2A, and CaMKII (107). Hence, altered regulation of RyR2 gating by processes such as phosphorylation is likely critical to altered diastolic SR Ca2+ release. One initial line of inquiry explored suggestions that PKA could alter RyR2-Ca2+ release function through a RyR2 hyperphosphorylation that dissociates a normally bound FKBP12.6 (745, 747, 856). This specifically implicated RyR2-S2808, as opposed to other possible PKA and CaMKII targets in cardiac failure. Mice with the nonphosphorylatable substitution RyR2-S2808A ablating the RyR2-S2808 phosphorylation site were correspondingly not susceptible to effects of adrenergic agonists or other pathological stressors known to induce congestive cardiac failure (747, 1242). Furthermore, selectively preventing PKA-mediated RyR2-S2808 hyperphosphorylation improved postinfarction or exercise cardiac performance in the genetically modified mouse (630, 1039, 1241, 1242).
These findings led to suggestions for a physiological role for PKA-mediated RyR2-S2809 phosphorylation in the increased cardiac contractility following sympathetic stimulation during exercise. Persistent pathological increases in such adrenergic activity would cause RyR2-S2809 hyperphosphorylation. This would dissociate FKBP12.6 normally bound to and stabilizing the RyR2 and thereby cause an arrhythmogenic diastolic leakage of SR Ca2+ (747). However, subsequent studies along similar or independent lines did not confirm critical predictions of this RyR-S2809 hyperphosphorylation hypothesis (449).
A first group of experiments concerned the physiological consequences of RyR2-S2809 phoshorylation. Channel function in recombinant phosphorylated and dephosphorylated RyR1 and RyR2 (RyR1-S2843D and RyR2-S2809D, and RyR1-S2843A and RyR2-S2809A, respectively) expressed in HEK293 cells did not show alterations in their responses to applied Ca2+, Mg2+, and ATP in single-channel and [3H]ryanodine binding measurements, or to caffeine in Ca2+ imaging measurements, that differed from findings in WT. Coimmunoprecipitation and Western blot analysis demonstrated similar binding of FK506-binding proteins. Metabolic labeling generated evidence for RyR2 phosphorylation sites other than S2809 (1089). Excitation-contraction coupling in whole hearts and isolated cardiomyocytes from a RyR2-S2808A mouse model showed normal β-adrenergic responses with a persistent maladaptive cardiac remodeling following chronic stress. Single-channel RyR2-S2808A activity showed only minor alterations and then only in their responses to applied Ca2+ (97, 356, 708).
Second, FKPB12.6 binding or dissociation did not modify RyR2 sensitivities to applied Ca2+ or caffeine, their tendencies to spontaneous or store overload-induced Ca2+ release, or the effect upon these of FK506, in HEK293 cells either coexpressing RyR2 and FKBP12.6 or expressing RyR2 alone. In these particular studies, FKBP12.6-null mice did not show altered tendencies for stress-induced ventricular arrhythmias. The properties of their RyR2 channels were indistinguishable from WT (1281).
Third, a series of experiments in cardiomyocytes indicated that PKA phosphorylation did not dissociate FKBP12.6 from RyR2. FKBP12.6 occurred in lower abundance than RyR2 therefore binding only to ∼15% of available RyR2s, in contrast to the more abundant FKBP12. However, PKA-mediated RyR-S2809 phosphorylation dissociated neither FKBP12.6 nor FKBP12 from RyR2 (382).
Fourth, Ser-2808 phosphorylation was often relatively insensitive to either PKA activation or inhibition (13, 109, 657). PKA-dependent increases in Ca2+ spark frequency in ventricular myocytes were instead attributable to enhanced SR Ca2+ load following phospholamban B phosphorylation rather than altered resting RyR2 function even under conditions of controlled [Ca2+]i (657). CaMKII- rather than PKA-dependent phosphorylation of RyR2 was implicated in the enhanced SR diastolic Ca2+ leak and reduced SR Ca2+ load consequent upon adrenergic stimulation or cardiac failure (13, 227). Finally, where CaMKII, or S-nitrosylation, mediated phosphorylation did enhance RyR2-mediated Ca2+ sparks and SR Ca2+ leak (232, 382, 884), it did so possibly at an alternative, S2814, site (943).
Nevertheless, phosphorylation events in general could still be expected to increase RyR2 Ca2+ sensitivity (747, 1241). This would then increase SR Ca2+ leak, with potential arrhythmogenic triggering effects (325). This would also ultimately deplete SR Ca2+ and thereby compromise Ca2+ transients and the resulting contraction in ventricular myocytes (411, 665, 892, 895). Thus both RyR2-P2328S and RyR2-Q4201R mutations were associated with decreased RyR2-FKBP12.6 binding, increased Ca2+ opening and positive shifts in half-maximal inhibitory Mg2+ concentrations after PKA phosphorylation. In contrast, the benzothiazepine derivative JTV519 (K201) appeared to both enhance RyR2-FKBP12.6 binding and normalize channel function (631). The RyR2-G230C mutant showed similar biophysical defects and demonstrated decreased FKBP12.6 binding and in common with RyR2-P2328S, normal luminal Ca2+ activation, but a negative shift in the cytosolic Ca2+ dependence of activation (763). Similar hyperphosphorylation hypotheses have been postulated for the triggering of arrhythmia in other systems, including a rabbit LQTS2 model (1118).
Furthermore, murine hearts with a heterozygous FKBP12.6 knockout correspondingly showed polymorphic VT with adrenergic challenge. This was rescued by JTV518 (631, 1241). RyR2-R2474S hearts also demonstrated reduced FKBP12.6-RyR2 binding on adrenergic stimulation. Rycal (S107) stabilized the binding, and therefore also stabilized the RyR2 closed state. It thus rescued the VT (629). The RyR2-S2246L, RyR2-R2474S, and RyR2-R4497C mutations similarly impaired RyR2-FKBP12.6 binding in parallel with their CPVT phenotype (1241). Ca2+ release by RyR2-P2328S, RyR2-Q4201R, and RyR2-V4653F expressed in HEK293 cells similarly showed rightward shifts in half-maximal inhibitory [Mg2+] and decreased FKBP12.6 binding (631, 1239).
8. Store overload-induced Ca2+ release
A store overload-induced Ca2+ release mechanism has been suggested as an alternative or complementary mechanism to altered FKBP12.6 binding in producing CPVT phenotypes. Murine RyR2-R4496C, equivalent to human RyR2-R4497C (511), showed enhanced Ca2+ and caffeine sensitivitivity when expressed in HEK293 cells. Lipid bilayer and HL1-cardiomyocyte studies similarly suggested increased diastolic SR Ca2+ release with sympathetic activation (348, 631, 1241). Despite this, interaction between RyR2-R4496C and FKBP12.6 was normal both before and following such adrenergic challenge (348, 509). Furthermore, JTV519 did not inhibit either in vitro DADs or in vivo ventricular arrhythmias induced by adrenergic challenge (673). In lipid bilayers, JTV519 did suppress both spontaneous Ca2+ release and [3H]ryanodine-RyR2 binding. However, it did so independently of FKBP12.6 association (470), likely through a JTV519 binding site in the RyR2 2114–2149 domain (1286). The latter may further bind the central RyR2 protein domain (2234–2750) and thereby stabilize the RyR2.
These findings suggested that the increased SR-Ca2+ release was alternatively attributable to an increased RyR2 channel sensitivity specifically to SR luminal Ca2+ that would thereby decrease cytosolic Ca2+ release thresholds (509, 510). Such a store overload-induced Ca2+ release mechanism would be consistent with observed diastolic Ca2+ leaks associated with the RyR2-R176Q/T2504M and RyR2-L433P mutations that involve the channel domains of RyR2 (1131). It would also be compatible with the loss of such luminal activation with the RyR2-A4860G mutation (508).
It is nevertheless possible that these detailed differences in arrhythmogenic mechanisms could reflect genuine differences between phenotypic consequences of the various genetic RyR2 variants. Thus in vitro assays associated mutations in the RyR2 central region not only with decreased (RyR2-R2427S, RyR2-S2246L, and RyR2-P2328S) but also with increased (RyR2-N2386I and RyR2-Y2392C) FKBP12.6 binding affinities (1135, 1241). COOH-terminal RyR2 mutations (RyR2-Q4201R, RyR2-R4496C, and RyR2-V4653F) were associated with decreased FKBP12.6 affinities (1241).
D. Genetic Abnormalities in the RyR2-SR Ca2+-Release Channel: Atrial and SAN Arrhythmic Phenotypes
1. Genetic RyR2 abnormalities associated with acute AF phenotypes
A number of RyR2 mutations are associated with SND and atrial arrhythmic in addition to CPVT phenotypes. Atrial arrhythmia can occur as a first clinical manifestation of CPVT (623). Both murine and clinical reports suggest that this could increase the risk of VT in this condition (302). The RyR2 exon 3 deletion is associated with a broad range of atrial, ventricular, and SND phenotypes. Some single RyR2 mutations associated with CPVT are also associated with exercise-associated atrial arrhythmias. These include RyR2-P2328S (613, 1099, 1104), RyR2-G3946A (900), RyR2-S4153R (542), and RyR2-A7420G. Rapid AF culminating in CPVT has been associated with RyR2-M4109R and RyR2-I406T (835). VT and AT following isoproterenol infusion has been associated with RyR2-W4645R (86).
Murine hearts provided realistic models for the acute initiation of atrial arrhythmias in the presence of genetic RyR2 abnormalities classically associated with CPVT. They similarly related these to specific SR Ca2+ release abnormalities (1337). Studies in a selected group of mouse models with modified RyR2 genotypes demonstrated that, in common with the ventricular arrhythmia, intracellular diastolic SR Ca2+ leak via RyR2 can trigger, and inhibiting this leak can prevent, AF (184, 824, 1040, 1098, 1334).
Increased RyR2-dependent Ca2+ leakage due to enhanced CaMKII activity may be an important downstream effect of CaMKII that generates susceptibility to AF. Thus rapid atrial pacing in RyR2-R176Q/+ mice increased atrial arrhythmic tendency compared with WT mice (184). This atrial arrhythmia in RyR2-R176Q/+ hearts accompanied increased CaMKII-mediated RyR2 phosphorylation. Both the arrhythmia and increased SR leak of Ca2+ were prevented by pharmacological inhibition of CaMKII. Similarly, preventing CaMKII-mediated RyR2 phosphorylation by a RyR2-S2814A mutation prevented AF in a vagotonic experimental model (534). Atrial biopsies from mice with atrial enlargement and spontaneous AF showed increased CaMKII-mediated RyR2 phosphorylation (184).
AF can also follow alterations in RyR2-associated proteins. These include FKBP12.6 deletion (655, 1081), overexpression of the Ca2+ binding junctate 1 (438, 439), or the sarcoplasmic transmembrane protein junctin (438), as well as deletion of the Ca2+- and GTP-dependent membrane-associated annexin Aa7 (Anxa7) (423). Intraesophageal burst pacing induced AF and diastolic SR Ca2+ leaks at the myocyte level in RyR2-R2474S+/−, RyR2-N2386I+/−, and RyR2-L433P+/− mice (1040). The RyR2-R2474S+/− RyR2s showed an increased oxidation and calstabin2 depletion compared with WT. S107 is known to stabilize RyR2-calstabin2 interactions by inhibiting oxidation/phosphorylation-induced calstabin2 dissociation from RyR2. It rescued both the increased SR-Ca2+ release and AF phenotype. It contrastingly failed to do so in calstabin2-deficient mice (1040).
The RyR2-P2328S mutation is clinically associated with atrial in addition to ventricular arrhythmia (613, 1104). RyR2S/+ and RyR2S/S mice showed normal ECG parameters whether before or following isoproterenol challenge apart from decreased cycle lengths. However, both regular and programmed stimulation resulted in a greater atrial arrhythmogenic incidence in isolated perfused RyR2S/S, though not RyR2S/+, compared with WT. Isoproterenol challenge increased arrhythmic incidences in all three groups. This was despite unchanged APD90 and AERPs on MAP recording. Regularly stimulated, fluo 3-loaded RyR2S/S, but not RyR2S/+ or WT atrial myocytes, showed diastolic Ca2+ release phenomena. These were made more frequent by isoproterenol (1334). Floating microelectrode recordings in isolated perfused RyR2S/S but not WT atria correspondingly demonstrated DADs and ectopic APs potentially providing arrhythmic trigger (569).
2. Altered atrial AP conduction in RyR2-P2328S hearts
In parallel with findings reporting on their ventricular arrhythmic properties (1340), murine RyR2S/S atria also demonstrated potential arrhythmic substrate potentially important for perpetuation of atrial arrhythmias. MEA recordings showed reduced epicardial AP conduction velocities despite normal MAP recovery measurements of both APD90 and AERP. Floating intracellular electrode recordings demonstrated increased interatrial conduction delays and correspondingly reduced (dV/dt)max (Figure 16, F and G). In contrast, AP recovery parameters of atrial effective refractory periods (AERPs), APDs, and AP amplitudes were indistinguishable from WT. Furthermore, despite persistent CV differences with extrasystolic S2 stimulation at progressively decreasing S1S2 intervals, the onset of arrhythmia coincided with similar CVs in WT and RyR2S/S (569).
These findings accompanied loose-patch clamp findings of compromised Nav1.5 activity in both RyR2S/S atria and positive Scn5a+/− controls. These took the form of reduced peak INa despite similar normalised current-voltage relationships (Figure 16, H and I) and reduced Nav1.5 but normal Cx40 or Cx43 protein expression. WT hearts showed similar reductions in INa under conditions of acutely increased [Ca2+]i resulting from increased extracellular [Ca2+], caffeine, or CPA. These findings together suggest a slowed conduction resulting from downregulated Nav1.5 expression that could be further exacerbated by the effects of elevated [Ca2+]i upon Na+ channel function in RyR2-P2328S atria (568).
The RyR2-P2328S system thus provided an experimental platform for an examination of recent clinical reports suggesting that flecainide may provide an approach to antiarrhythmic therapies through actions mediated through the RyR2 (1233). These reported that flecainide exerted marked antiarrhythmic effects in CPVT. This was attributed to tetracaine-like actions resulting in its acting as a potent RyR2 inhibitor (429, 455, 458, 473, 1233, 1252). As indicated above, flecainide acts both as a class 1C INa (66, 675, 1064) and RyR2 inhibitor (IC50 values of respectively 5–11 and 2–7 μM) (338, 417).
MEA recording confirmed decreased CVs in isolated intact RyR2S/S atria. This directly paralleled findings of a decreased INa relative to WT and the higher incidences of atrial arrhythmia observed in these mice. Low flecainide (1 μM) concentrations both paradoxically restored CV and reduced the frequency of arrhythmia. This sharply contrasted with the effect of flecainide in decreasing CV and promoting arrhythmia in WT. Thus low flecainide concentrations correspondingly restored INa to normal levels in intact RyR2S/S atria from a likely inhibited baseline level. These effects were replicated by the RyR2 blocker dantrolene (228, 996). These findings suggested that by inhibiting the increased SR Ca2+ leak, flecainide reduces Ca2+-dependent inhibition of INa. This could restore conduction velocity in RyR2S/S hearts. Such findings target diastolic SR Ca2+ leak in potential therapeutic antiarrhythmic strategies for heart failure (1318).
3. Ca2+ signaling and remodeling associated with chronic AF
Established as opposed to acute episodic clinical AF is associated with additional, anatomical or functional, atrial remodeling and congestive cardiac failure. There is often an accompanying intracellular Ca2+ overload and increased spontaneous diastolic SR Ca2+ release (309, 451, 1088, 1269). However, precise cause-and-effect relationships between these changes remain uncertain. It is also difficult to isolate contributions of disease entities associated with AF to the changes in Ca2+ signaling in AF. Thus short-term, sustained high atrial activation rates in a rabbit model appeared to produce an antiarrhythmic “Ca2+ signaling silencing.” This was accompanied by normal CaMKII expression levels but reduced CaMKII-mediated RyR2 phosphorylation and [Na+]i (372).
Atrial myocytes from chronic AF patients show potentially proarrhythmic, increased frequencies in spontaneous Ca2+ release events (61, 451, 1194). These took place despite reduced ICaL (202, 1274). These features might reflect increased single RyR2-Ca2+ channel open probabilities as observed in canine hearts with persistent AF. There was then an increased protein kinase A-dependent RyR2-S2808 phosphorylation and decreased FKBP12.6-RyR2 binding. Such changes would be expected to compromise RyR2 channel closure (1194, 1238, 1244). However, the direct relevance of these mechanisms to AF remains uncertain (290).
Murine hearts provide experimental paradigms appropriate to the analysis of remodeling changes accompanying and potentially exacerbating atrial arrhythmia. The transgenic mouse model overexpressing the cardiac transcriptional repressor cAMP-response element M, CREM-IbΔC-X (CREM), recapitulates this clinical progression (572). Telemetric ECG recordings first showed spontaneous atrial ectopy at ∼3 mo. This then progressed to paroxysmal and chronic AF. These changes followed CaMKII-dependent increases in diastolic Ca2+ release (184). The latter could be attributed to increased RyR2 open probabilities and Ca2+ spark frequencies. There were accompanying atrial dilatation and atrial conduction abnormalities (654). The latter conduction changes were related to Cx40 downregulation (572). RyR2-S2814 phosphorylation accompanied, and the RyR2-S2814A mutation prevented, this remodeling. There was no accompanying fibrotic change. Increased RyR2-S2814 phosphorylation correspondingly increased AF inducility by programmed electrical stimulation in mutant mouse models (655). These findings together paralleled clinical observations of increased CaMKII-activity and RyR2-S2814 phosphorylation in long-term AF patients.
Thus abnormal RyR2-mediated SR Ca2+ release, related to RyR2-S2814 phosphorylation in CREM-mice, contributes to remodeling in AF. It results in arrhythmic substrate, in addition to triggering events. These changes directly correlated with hyperactive RyR2 channels directly stimulating the Ca2+-dependent hypertrophic nuclear factor of activated T cell (NFAT)/Rcan1-4 pathway, detected through Rcan1-4 mRNA expression, implicating it in developing substrate for longlasting AF (654).
Chronic AF is also clinically associated with reduced sarcolipin (SLN) mRNA and protein expression (1041). The transmembrane protein SLN regulates SERCA2a activity in common with PLN (54, 777). It likely does so through CaMKII rather than PKA-mediated mechanisms (120). SLN inhibits SERCA at low [Ca2+] (1151). Aged homozygous Sln−/− mice (54) developed atrial fibrosis and spontaneous AF under anesthesia (1283), paradoxically suggesting that increased SERCA function promotes AF.
4. RyR2 function and SAN pacemaker activity
Continuing discussions bear on possible important roles for RyR2-mediated Ca2+ release in SAN pacemaker activity (see sect. IIIA) (615, 723, 1121). Enhanced SR Ca2+ release should activate inward INCX (522, 616). This would potentially facilitate AP firing in a “Ca2+ clock” mechanism (132). Ryanodine-mediated RyR2 block thus reduced SAN activation frequencies (132, 522, 972). Roles of SR-Ca2+ release in regulating normal SAN pacemaker function thus merit closer study (615, 723).
However, human CPVT is associated with reduced SAN automaticity resulting in basal bradycardia, sinus pauses, and impaired chronotropic β-adrenergic responses (625, 909). In vivo telemetry revealed that RyR2-R4496C mice did not show basal bradycardia. Nevertheless, they similarly showed sinus pauses followed by atrial and junctional escapes and impaired SAN automaticity with isoproterenol challenge. This did reproduce findings in CPVT patients following exercise (824). RyR2-R4496C SAN cells showed markedly reduced ICaL on whole cell patch-clamping. Spontaneous [Ca2+]i transients studied by confocal microscopy in isolated SAN cells correspondingly showed slowed pacemaker activity, impaired chronotropic β-adrenergic responses, and sinus pauses. Isoproterenol produced 5- to 10-fold increases in the frequencies of Ca2+ sparks and Ca2+ waves (824).
E. Genetic Abnormalities in Cardiac Calsequestrin 2: CPVT
1. Action of CASQ2 on Ca2+ homeostasis
The calsequestrin 2 (CASQ2) protein buffers SR Ca2+. This action would result in a reduction of free SR Ca2+ concentrations potentially below those expected to inhibit SERCA2a. This would then enhance SR Ca2+ reuptake. CASQ2 thus acts both as the major SR Ca2+ reservoir and a local intra-SR Ca2+ buffer. CASQ2 also functions as a Ca2+-dependent RyR2 channel regulator in cardiac myocytes (108). Thus CASQ2 is anchored to RyR2 via junctin and triadin. It may modulate RyR2 sensitivity to luminal Ca2+ through functional interactions with these molecules. It has been suggested that CASQ2 binding to triadin and junctin inhibits RyR2 channel activity at low luminal [Ca2+]. Increasing luminal [Ca2+] would then relieve this inhibition. This binding would be accompanied by a CASQ2 transition from a monomeric to a polymeric form with the greater Ca2+ binding capacity. The resulting occupation of CASQ2 Ca2+ binding sites would then weaken the interactions between CASQ2 and triadin and/or junctin. This would increase RyR2 sensitivity to luminal Ca2+. The result would be an increased open probability in the RyR2-Ca2+ channel (581).
2. CASQ2 mutations and the CPVT phenotype
Around 3% of CPVT patients show one of seven homozygous CASQ2 mutations (836). Four such mutations constitute premature stop codons. This generates truncated forms of the protein, lowering CASQ2 levels. This would be expected to shorten the time required for functional SR Ca2+ store recharging thereby increasing the likelihood of diastolic RyR2-mediated Ca2+ leaks and premature RyR2 activation. This would produce phenotypic similarities to CPVT (1120).
The remaining three mutations are single nucleotide replacements involving single amino acid substitutions. CASQ2-D307H was first described in seven consanguinous Bedouin families (611). This results in a substitution of the negatively charged aspartic acid for histidine in a highly conserved putative Ca2+ binding region between the second and third thioredoxin-like domains. This likely affects the CASQ Ca2+ affinity in its monomeric form and impairs its Ca2+ binding and its interactions with junctin and triadin (448, 565). The CASQ2-R33Q mutation was first demonstrated in a CPVT patient with history of syncope and death of a male sibling at a young age (908). When overexpressed in rat cardiac myocytes it resulted in abnormal spontaneous diastolic Ca2+ release. However, it did not appear to alter SR Ca2+ store capacity. CASQ2-R33Q is associated with impaired CASQ2 dimerization (565) or polymerization which consequently only takes place at a higher [Ca2+] (1167). This reduces the CASQ2-mediated RyR2 inhibition at low luminal Ca2+ (924, 925). Finally, CASQ2-L167H was identified in a young patient with stress-induced ventricular arrhythmia and cardiac arrest. In rat cardiomyocyte expression systems, CASQ2-L167H appeared to behave like a functionally inert protein (565, 924, 925).
These features are consistent with similar mechanisms for CPVT arrhythmias whether these are associated with RYR2 or CASQ2 mutations. They suggest similar parallels for the malignant hyperthermia associated with RYR1 or CASQ1 mutations in skeletal as opposed to cardiac muscle. One could thus postulate that arrhythmia would be initiated when levels of free SR luminal [Ca2+] exceeded a threshold for a store-overflow-induced Ca2+ release (SOICR). In such a hypothesis, RyR2 mutations would reduce the threshold for such SOICR. SOICR would then take place with the increases in the SERCA-mediated Ca2+ uptake that results from β-adrenergic stimulation. In contrast, the reduced CASQ2 Ca2+ binding and buffering resulting from CASQ2 mutations would increase the free luminal Ca2+ levels beyond those of a normal RyR2 SOICR threshold (711). However, studies in murine systems suggest greater complexities in relating the genetic changes to their phenotypic consequences.
3. Murine hearts with modified Casq2
Features of mice with genetic Casq2 modifications, exemplified in Table 7, were consistent with reductions in Casq2 being involved in their observed catecholaminergic stress-induced polymorphic VT and bidirectional VT phenotypes. Casq2−/− mice also showed increased tendencies to AF. This was associated with increased diastolic spontaneous Ca2+ elevations and DADs. All these features were inhibited by the RyR2 inhibitor R-propafenone (303). However, whereas even heterozygous (Casq2+/−) mice showed positive phenotypes (199), heterozygous carriers of premature CASQ2 truncations were asymptomatic. Furthermore, the precise mechanisms by which the Casq2 deficiency causes the causative diastolic SR Ca2+ leak leading to premature spontaneous Ca2+ release and arrhythmogenic triggered beats remains uncertain. Casq2−/− mice appeared to show normal SR Ca2+ content (581). However, they showed compensatory increases in SR volume and posttranscriptional reductions of junctin and triadin. Casq2 thus appears not to be essential for maintaining SR Ca2+ stores (581). Yet the Casq2 deficiency appeared central to the stress-induced polymorphic and bidirectional VT phenotype. In bilayer experiments, removal of CASQ2 at fixed intraluminal [Ca2+] increased RyR2 open probabilities in bilayer experiments (393). In heterozygous Casq+/− mice, even 25% decreases in Casq2 protein increased diastolic SR Ca2+ leak despite unchanged triadin and junctin levels and SR volume (199).
Two transgenic models with positive CPVT phenotypes showed evidence for increased expression of both RyR2 and the fetal SR Ca2+ binding protein calreticulin. The latter might then produce a compensatory increase in SR Ca2+ binding capacity. In common with Casq2−/− and homozygous Casq2-D307H/D307H mice, the homozygous deletion carrier Casq2-ΔE9/ΔE9 showed a 95% reduction in Casq2 protein. However, this accompanied other changes including increased RyR2 and calreticulin though normal calstabin levels (1077). Casq2-D307H/D307H mice were used to explore verapamil as a possible therapeutic antiarrhythmic agent (540). Homozygous, Casq2-R33Q/R33Q, mice also showed stress-induced polymorphic VT and bidirectional VT. Their myocytes showed diastolic depolarization phenomena and decreased SR Ca2+ (951). However, despite normal Casq2 mRNA, they showed a 50% reduction in Casq2 protein possibly reflecting a greater degree of Casq2-R33Q protein digestion by trypsin (951). However, calreticulin and RyR2 protein levels then were normal.
F. CaMKII Dysfunction, Altered Ca2+ Homeostasis, and Cardiac Arrhythmogenesis
Murine systems have thrown light on CaMKII function and consequences of its modification under conditions of both genetic and pharmacological manipulation. In the latter connection, the CaMKII inhibitor KN-93 has been useful as a blocker of CaMKII activation. However, KN-93 may also act upon voltage-gated K+ and L-type Ca2+ channels. Inhibition through endogenous CaMKII, autocamtide-3 inhibitor (AC3-I), and autocamtide-2 inhibitor proteins (AIP) may offer greater specificity and therefore future promise (882).
Transgenic mice overexpressing either CaMKIIδ2 or CaMKIIδ3 developed DCM and cardiac failure (1331, 1332). CaMKII overexpression also altered Ca2+ homeostasis and cellular excitability. This resulted in proarrhythmic AP afterdepolarizations (291). In contrast, CaMKII deletion prevented the heart failure that follows transaortic constriction (55, 667). Similarly, overexpression of the CaMKII inhibitory peptide AC3-I enhanced postinfarction cardiac function and Ca2+ handling. It reduced LV hypertrophy and dysfunction following isoproterenol challenge. It also decreased PLN phosphorylation at Thr17 and SR Ca2+ load (1329). Murine AC3-I overexpression systems also demonstrated feedback relationships between CaMKII, PLN, and sarcolemmal K+ channels. They showed upregulated inward rectifier IK1 and transient outward currents Ito,f and consequent APD shortening. This was in contrast to the decreased Ito and IK1 and APD prolongation in cardiac failure (295). These changes accompanied normal channel RNA and protein expression consistent with either altered membrane expression or posttranslational modification. This remodeling was prevented with crossing of Ac3-I and Pln−/− (650). Finally, both KN-93- or AC3-I-mediated CaMKII suppression in a murine calcineurin overexpression cardiac failure model reduced spontaneous ventricular arrhythmias and improved LV function (557).
CaMKII dysfunction has also been associated with AF and SND (654, 1277, 1297), CPVT (677), LQTS3 (588), and the ankyrin-B related (LQTS4) (249) and Timothy (LQTS8) syndromes (1125). A recent study in transgenic mice with cardiomyocyte-specific expression of the SR-targeted CaMKII inhibitor aryl-hydrocarbon receptor-interacting protein (AIP) implicated RyR2 phosphorylation produced by ROS-activated CaMKII, in arrhythmogenic changes following impairment of glucose tolerance (857, 1076). Studies in permeabilized cardiomyocytes suggested that whereas the small proportion of CaM variants associated with a CPVT could evoke arrhythmogenic perturbations in Ca2+ homeostasis, this did not appear to be the case with variants associated with LQTS (474, 1306). Finally, mice haploinsufficient in Purkinje cell protein-4 (Pcp4) which may regulate CaM and CaMKII signaling specifically within His-Purkinje cells showed proarrhythmic ventricular phenotypes, increased ICa, positive shifts in ICa inactivation, and altered evoked Ca2+ transients accompanying CaMKII activation (566).
G. Genetic Modifications in Dephosphorylation Pathways
1. Cardiac expression of p21-activated kinase-1
Increasing evidence implicates phosphatases in dampening kinase-mediated cardiac excitation and contraction mechanisms. This potentially exerts cardioprotective effects (548). The serine/threonine protein kinase p21-activated kinase-1 (Pak1) is highly expressed in different cardiac regions. It is localized to the Z-disc, cell and nuclear membrane, and intercalated discs in rat ventricular myocytes (547). It is directly activated by the small GTPases, cell division control protein 42 homolog (Cdc42), and Ras-related C3 botulinum toxin substrate 1 (Rac1). Coimmunoprecipitation studies suggest that Pak1 is associated with the similarly localized cellular cardiac PP2A for which it might form an upstream activator in both SAN and ventricular cells (546, 1254). PP2A removes phosphate groups at PKA phosphorylation sites on the cardiac isoform of the cTnI troponin subunit that inhibits actin-myosin ATPase activity (547, 709), MYBPC3 (547), PLN (709), inwardly rectifying K+ channels (543), as well as L-type Ca2+ channels (242) and RyR2s (730), increasing Ca2+ spark generation (1119). It also regulates transcription factors including cAMP response element-binding protein (CREB) (1208) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (1291). The latter effects influence cardiac remodeling (312, 327).
Both Pak1 and its upstream activators Cdc42 and Rac1 also stimulate cytoskeletal changes. They reduce stress fibers and focal adhesion complexes, filapodia and lamelliopodia formation, and membrane ruffling (544, 545, 1102, 1229). These features are compatible with a dynamic balance between phosphatase and kinase activity in regulating aspects of Ca2+ homeostasis ion channel activity influencing pacemaker and excitable activity in sinoatrial and ventricular cells, respectively, and their modulation by autonomic stimulation.
2. Loss of Pak-1 function and atrial arrhythmic phenotype
Mice with either cardiac-specific conditional (Pak1-cko) or total (Pak1−/−) Pak1 deletions showed increased sinus heart rates compared with control homozygous flox (Pak1-f/f) littermates. This was despite similar electrocardiographic, P, PR, QRS, and QT waveforms (1230). Conversely, infection of cultured guinea pig SAN cells with recombinant adenovirus expressing constitutively active Pak1 (CA-Pak1) depressed L-type Ca2+ and delayed rectifier K+ currents. It reduced repetitive AP frequencies in response to isoproterenol-induced β-adrenergic stimulation compared with findings in SAN cells infected with Ad-LacZ controls (545). The PP2A inhibitor okadaic acid partly reversed this suppression of L-type Ca2+ channel responses to isoproterenol in Ad-Pak1-infected cells (543). These findings led to suggestions for a dynamic balance between kinase and phosphatase activities in modulating both L-type Ca2+ channel and delayed rectifier IK activity. This could furnish a potentially important mechanism for alterations in pacemaker activity following autonomic stimulation (545).
3. Loss of Pak-1 function, SERCA function, and ventricular arrhythmic phenotype
Pak1-cko and Pak1−/− hearts also showed increased incidences of polymorphic VT, despite normal refractory periods, following either acute or chronic β-adrenergic isoproterenol challenge, relative to controls (Figure 17A) (259, 1044, 1230). Pak1-cko and control, Pak1-f/f, ventricular myocytes showed comparable baseline ICaL similarly reduced by chronic β-adrenergic stress. However, Pak1-cko myocytes showed higher incidences of irregular, alternating Ca2+ transients with repetitive stimulation, and of spontaneous Ca2+ waves, both before and particularly following imposition of chronic β-adrenergic stress (Figure 17B). This appeared at least partly to reflect decreased SERCA function. The latter would reduce SR Ca2+ content and increase diastolic [Ca2+], effects induced or accentuated by either chronic or acute β-adrenergic stress (Figure 17, C and D). The latter target offers potential future therapeutic targets for novel agents directed at SERCA activity through PLN such as istaroxime and CDN1163 (463, 533). Such changes were suggested by assessments of SR Ca2+ levels through observed time constants and integrals of INCX, as well as of SERCA2 activity from decay constants of systolic Ca2+ transients, following caffeine challenge. It also emerged from comparisons of the decays of systolic Ca2+ transients, and increases in diastolic and decreases in systolic [Ca2+], and reductions in systolic contraction amplitude, despite unchanged ICaL, following regularly applied stimulation (1230).
Figure 17.

Physiological features mediating arrhythmogenesis in Pak1-deficient hearts. A: monophasic action potential (AP) recordings showing AP alternans (a), torsades de pointes (TdP) (b), and polymorphic VT (c) following burst pacing (horizontal line below trace) in ex vivo Pak1-cko hearts, features not observed in Pak1-f/f hearts. B: Ca2+ transients in field-stimulated Pak1-f/f (a) and Pak1-cko myocytes (b) at a 1-Hz stimulation frequency under baseline (left traces; i) and chronic β-adrenergic stress conditions (right traces; ii). Increased pacing frequencies increase the occurrence of Ca2+ waves to greater extents in Pak1-cko than Pak1-f/f myocytes particularly with chronic β-adrenergic stress. C and D: recovery of SR Ca2+ stores from after previous depletion by caffeine challenge, after which regular stimulation resumed. i: Ca2+ transients indicating recovery of SR Ca2+ in Pak1-f/f (C) and Pak1-cko myocytes (D) under baseline conditions. a–c: Comparison of increasing Ca2+ transients (ii), constant ICaL (iii), and increasing INCX (iii) at different stages (a–c) of SR Ca2+ recovery. [From Wang et al. (1230).]
More direct assessments of effects on SERCA2 activity first interposed a caffeine challenge to deplete SR Ca2+ in regularly stimulated ventricular myocytes. They then followed the subsequent recovery of evoked Ca2+ signals to assess recovery of SR Ca2+ stores. Pak1-cko myocytes showed more prolonged recovery time courses compared with Pak1-f/f whether before or following chronic β-adrenergic stress (Figure 17, C and D, i). Such chronic β-adrenergic stress increased both these differences and the recovery times. In contrast, ICaL and INCX magnitudes and time courses were indistinguishable through these conditions and the duration of recovery from caffeine challenge (Figure 17, C and D, ii–iv) (1230).
Pak1-cko hearts correspondingly showed reduced SERCA2a mRNA and protein expression. However, they showed normal PLN mRNA, protein and phosphorylation levels, Nav1.5 protein levels, and RyR2 and Cacna1c mRNA levels whether before or following imposition of chronic β-adrenergic stress. Pak1-cko contrasted with Pak1-f/f hearts in failing to upregulate NCX protein or NCX1 mRNA following hypertrophic stress. Control Ad-shC2 virus-infected neonatal rat cardiomyocytes correspondingly showed increased SERCA2a protein and mRNA expression with phenylephrine challenge. This was abolished by Pak1-knockdown in Ad-shPak1-infected neonatal rat cardiomyocytes (NRCMs). It was accentuated by Pak1 overexpression produced by Ad-CAPak1 (1230).
In parallel with these results, myocytes expressing constitutively active Pak1 (CA-Pak1) showed faster time to peak shortenings but slower [Ca2+]i decays and relengthening times compared with LacZ-expressing controls. CA-Pak1 expression also inhibited the effect of isoproterenol in increasing [Ca2+]i transient amplitude. It increased SR Ca2+ content, slowed [Ca2+]i decay rates, and increased cell shortening. It also reduced Ca2+ spark amplitude and slowed propagation of spontaneous Ca2+ waves. Although it did not affect PLN phosphorylation, it reduced cardiac troponin I phosphorylation (1044).
Pak1 additionally downregulates NADPH-oxidase 2 activity in electron transfer from NADPH to O2 to generate superoxide. Simulated ischemia thus increased [Ca2+]i and induced spontaneous Ca2+ release and Ca2+ waves in Pak1−/− ventricular myocytes. These effects were rescued not only by the reverse-mode NCX blocker KB-R7943 and the Ras-related C3 botulinum toxin substrate 1 (RAC1) inhibitor (NSC23766) but also the ROS scavenger TEMPOL. Furthermore, both Pak1−/− and WT myocytes treated with the Pak inhibitor IPA3 showed increased cellular ROS production not exhibited by either p47-phox−/−, NADPH oxidase 2 (NOX2) deficient, myocytes or WT myocytes exposed to the NADPH oxidase peptide inhibitor gp91ds-tat (258).
4. Loss of Pak-1 function and hypertrophic phenotype
Pak1-cko hearts showed an accentuated cardiac hypertrophy and heart failure following either chronic pressure overload or angiotensin II infusion. There was a reduced activation of c-Jun N-terminal kinase (JNK). The latter is known to phosphorylate and inactivate the transcription factor NFAT, important in activating hypertrophic genes (793). The Pak1 activator FTY720 prevented these effects in WT but not Pak1-cko hearts (681). FTY720 also exerted cardioprotective effects against ischemia/reperfusion injury in both rat and mouse models (545).
Pak1 activating peptide (PAP) is known to increase phosphorylated Pak1 levels in cultured neonatal rat ventricular myocytes. It both reduced such angiotensin II-induced hypertrophy in neonatal rat ventricular myocytes (NRVMs) and C57BL/6 ventricles and inhibited the arrhythmias that followed such ventricular hypertrophy (1225). Intriguing studies in Pak1−/− mice attributed such effects of Pak1 deficiency to decreased amplitudes and slowed kinetics in Ca2+ transients coinciding with decreased transverse tubular densities and capacitances. All these changes were rescued by overexpressing constitutively active Pak1. The resulting alterations in cytosolic Ca2+ homeostasis might then modify regulation of transcription factors involved in hypertrophic remodelling (258).
H. Causal Schemes Relating Altered Ca2+ Homeostasis to Cardiac Arrhythmia
The above exemplars can be organized into a hypothetical scheme (Figure 18) that could form a basis for further explorations of causal relationships between modifications in cellular Ca2+ homeostasis and cardiac arrhythmias. Such modifications might arise from acquired or genetic alterations that might increase RyR2-mediated release of SR Ca2+ or decrease SERCA2a-mediated Ca2+ reuptake from cytosol to SR Ca2+ store. These could take place both through direct modifications of the molecules concerned, or indirectly through other signaling mechanisms (Figure 18, A and B). This in turn could potentially alter NCX activity whose electrogenic effects might lead to diastolic triggering phenomena (Figure 18C). Results from some of the exemplars above also suggest that it could reduce synthesis or membrane trafficking and therefore membrane expression of Nav1.5 or acutely alter its biophysical properties (Figure 18D). These effects could either chronically or acutely slow AP conduction, resulting in arrhythmic substrate (Figure 18E) even under conditions of normal action potential recovery as reflected in normal APD/ERP ratios.
Figure 18.

Scheme exploring relationship between perturbations in Ca2+ homeostasis, triggering, arrhythmic substrate, and generation of arrhythmia. A: acquired or genetic perturbations resulting in increased release of sarcoplasmic reticular (SR) Ca2+ or decreased Ca2+ reuptake from cytosol to store both perturb cytosolic Ca2+ (B). This in turn alters (C) Na+-Ca2+ exchange (NCX) electrogenic activity leading to diastolic triggering phenomena. It can also (D) reduce Nav1.5 synthesis or membrane trafficking and therefore its membrane expression, or directly alter Nav1.5 biophysical properties. Both effects potentially slow conduction, resulting in arrhythmic substrate even under conditions of normal action potential recovery as reflected in action potential duration/effective refractory period (APD/ERP) ratios. Combination of C and D culminates in E, potentially fatal ventricular arrhythmia. Possible direct actions of intermediates arising from metabolic change on INa are not shown for simplicity.
VIII. CARDIAC ENERGETIC DYSFUNCTION AND ARRHYTHMIA
A. Energetic Consequences of Cardiac Disease
1. Metabolic stress and mitochondrial dysfunction
Murine exemplars containing specific membrane ion channel modifications directly bearing upon cell excitability and Ca2+ homeostasis thus offer relatively straightforward physiological insights into the relationship between biophysical channel modification and whole-heart arrhythmogenesis. In so doing, they usefully model a range of relatively rare arrhythmic conditions. However, the clinically more common causes of arrhythmia are often less clearly genetically characterized. Significant insights into their arrhythmia can nevertheless be obtained through a strategic physiological targeting of particular aspects of cell function. For example, both the oxidative stress associated with hypertrophic change, cardiac failure, ischemia-reperfusion (90, 201, 374, 1117), and metabolic conditions including obesity, insulin resistance and type 2 diabetes, and increasing age are accompanied by energetic, in particular, mitochondrial, dysfunction (600, 1005, 1196). Mitochondria constitute the main cardiac cellular source of ATP (845). Dysfunctional mitochondria have been implicated in ventricular arrhythmia with altered ion channel function, AP heterogeneity, and cell excitability (49, 146, 483). They are also associated with the oxidative stress, possibly associated with L-type Ca2+ channel activity in atrial tachyarrhythmias (155).
2. Mitochondrial dysfunction and biophysical properties
A number of mechanisms link such alterations in metabolism to altered membrane excitability. These often take place under conditions also associated with increased frequencies of Ca2+-mediated triggers (41, 42). Mitochondrial inner membranes sustain a ∼200 mV potential that drives electrons down the electron transport chain. The latter ultimately drives ATP synthesis. This process is tightly regulated to match energy supply to demand. Mitochondrial oxidation destabilizes this potential through a mitochondrial permeability transition. This is likely produced by activation of inner membrane anion channels (IMAC) among possible others (845, 881). This destabilization initiates ROS-induced ROS release producing alternans in both mitochondrial and surface electrical activity (41). Damaged or dysfunctional mitochondria can thus increase ROS production by up to 10-fold (374).
ROS exert numerous, potentially arrhythmogenic, actions. They decrease INa (671) and IK (1217), activate sarcolemmal KATP channels (1249), modify Na+ and L-type Ca2+ channel inactivation kinetics, increase INaL, and oxidize RyR2. These result in an increased leak of SR Ca2+ leak and a consequent modulation of intracellular Ca2+ cycling (135, 146, 1117). They also reduce Cx43 trafficking and function (18, 1292, 1293). These actions together also increase [K+]i, [Na+]i, and [Ca2+]i. This produces a wide range of potentially proarrhythmic physiological effects bearing upon cell-cell coupling (1074), AP conduction (671), repolarization (1217) and alternans (91), and Ca2+-mediated triggers (1117).
ATP depletion or increased ADP increases open probabilities in sarcolemmal ATP-sensitive K+ channels (sarcKATP) (18). These occur at relatively high densities in myocyte surface membranes. This opening may physiologically protect myocyte viability during ischemia (380, 532). However, even opening 1% of such channels shortens APD, and therefore ERP and AP wavelength. This can potentially produce proarrhythmic reentrant substrate (305, 323). In addition, opening of sufficient channels generates heterogeneous current sinks driving the cell membrane potential towards EK. This can compromise cell excitability and AP propagation (18).
B. The Nuclear Receptor Family of Peroxisome Proliferator-Activated Receptors
1. Role of PGC-1 species
The nuclear receptor family of peroxisome proliferator-activated receptors (PPARs) include the PPAR-α, PPAR-γ, and PPAR-δ isoforms. Modifications in these offer one possible genetic access to studying actions of mitochondrial-related factors on cardiac function (944). All are subject to transcriptional coactivation by peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) species. The latter include PGC-1α, PGC-1β, and PGC-1-related coactivator (922). All have been increasingly implicated in regulation of energy metabolism in both health and disease (316, 661).
Both PGC-1α and PGC-1β regulate expression of genes involved in oxidative phosphorylation (555, 825, 1014, 1015). High PGC-1α and PGC-1β expression levels thus occur in actively oxidative tissues often characterized by abundant mitochondria. These include brown adipose tissue and cardiac and skeletal muscle. Nevertheless, mouse models with deletions of either PGC-1α or PGC-1β showed normal baseline cardiac contractile function. However, mutations involving both are postnatally lethal through cardiac failure. This is consistent with redundancies or overlaps between the functions of PGC-1α and PGC-1β (612). Both PGC-1α and PGC-1β deletions accentuated cardiac failure following transverse aortic constriction (44).
2. Effects of genetic modification of PGC-1α
PGC-1α appears important to cardiac adjustments that require increased ATP and work output in response to physiological stimuli. Embryonic mouse hearts showed large increases in mitochondrial biogenesis and oxidative metabolism close to birth. This followed dramatically increased PGC-1α expression (628). Ectopic PGC-1α expression drives mitochondrial biogenesis in cultured cardiac myocytes. Oxidative phosphorylation in these mitochondria was tightly coupled to ATP production (628). One of the two available PGC-1α knockout models showed normal baseline cardiac contractility, and the other showed age-dependent contractile dysfunction (43, 628). Both showed normal mitochondrial volume density. However, both also showed impaired expression of genes involved in oxidative phosphorylation and fatty acid oxidation. These were associated with an impaired maximal mitochondrial oxygen consumption using palmitoyl-l-carnitine substrate and mitochondrial ATP synthesis accompanied by mitochondrial uncoupling (43). In vivo and Langendorff-perfused PGC-1α-KO hearts showed impaired inotropic and chronotropic exercise and dobutamine responses, respectively.
3. Effects of genetic modification of PGC-1β
The four independently generated PGC-1β-KO models (638, 1080, 1196) showed normal inotropic responses in contrast to, but reduced chronotropic responses in common with, PGC-1α hearts following dobutamine challenge (638). PGC-1β−/− models showed impaired expression of genes involved in oxidative phosphorylation but not fatty acid oxidation (945). PGC-1β−/− mice showed irregular heartbeats and increased incidences of polymorphic VTs following isoproterenol challenge. They showed an increased Scn5a, Kcna5, RyR2, and CaMKII RNA expression. Langendorff-perfused PGC-1β−/− hearts showed AP alternans, EADs, and VT on monophasic AP recording (Figure 19, A and B). Myocyte patch electrode studies (Figure 19, C and E–H) demonstrated unstable oscillatory resting potentials and early and delayed afterdepolarizations accompanying their APs (Figure 19C). Sustained current injection elicited burst firing (Figure 19H). These changes were accompanied by diastolic Ca2+ transients exaerbated by isoprenaline (Figure 19D), increased inwardly and outward rectifying K+ currents (Figure 19, F and G), and a negatively shifted Ca2+ current inactivation (Figure 19E) (387).
Figure 19.

Arrhythmogenic features of PGC1β−/− hearts. A: increased heart rate following isoproterenol challenge accompanied by polymorphic ventricular tachycardia (VT) in PGC1β−/− mice during ECG recording. B: monophasic action potential (AP) recordings of VT following programmed electrical stimulation in Langendorff-perfused PGC1β−/− hearts. C: APs from PGC1β−/− ventricular myocytes showing early (EADs) and delayed afterdepolarizations (DADs) and ectopic APs. Inset magnifies voltage trace 40-fold. D: abnormal Ca2+ homeostasis with intermittent elevations in diastolic Ca2+ and Ca2+ waves increased in amplitude and frequency in isoproterenol challenged PGC1β−/− ventricular myocytes. E: voltage-gated ICa and transient and sustained outward IK (F) in response to depolarizing pulses from −40 to +50 mV altered in successive 10-mV increments from a −40 mV holding potential. G: inwardly rectifying currents obtained in response to hyperpolarizing steps from −40 to −100 mV incremented in 10-mV intervals. E–G shown for PGC1β−/− (a) and WT ventricular myocytes (b), with voltage step protocols shown in c. H: step current injections produced single APs with prolonged plateaus with burst AP firing in PGC1β−/− (a) but not WT myocytes (b). [From Gurung et al. (387).]
C. Effects of Cellular Redox Potential
1. Importance of NAD+/NADH
Studies in aging skeletal muscle suggested that reduced nuclear NAD+ levels variously attributable to altered NAD+/NADH ratios (921) or altered NAD+ consumption or synthesis (205) may also compromise mitochondrial function. This may involve mechanisms independent of PGC-1α/β. Thus restoring the NAD+ levels restored the mitochondrial function (365). In addition, these pyridine nucleotides themselves have independent cellular actions. Some of these could potentially interact with those of altered mitochondrial function. Thus NADH enhanced smooth muscle superoxide production which could result in and be further reinforced by PKC activation (221). NAD+ caused PKA activation in osteoblastic cells (960). In turn, PKA upregulated (1349) and PKC downregulated Nav1.5 function (809). Pyridines also modified voltage-dependent K+ channel gating (1134), Ca2+-activated K+ channel (869), and TrypC currents (420). NAD+ enhanced and NADH inhibited RyR2-mediated Ca2+ release (1352). In addition, NAD+ is convertible, by RyR2-associated CD38 (4, 1100), to cyclic ADP-ribose, known to trigger RyR2-mediated Ca2+ release (5).
Finally, mutations in GPD1-L protein, highly expressed in cardiac tissue, are associated with reduced INa and clinically with both BrS and SIDS. GPD1-L is >80% homologous in amino acid sequence with glycerol-3-phosphate dehydrogenase. The latter enzyme family is directly involved in NAD-dependent energy metabolism potentially altering cellular redox state as reflected in NAD+/NADH levels. This would bridge alterations in cellular energetic state with modifications in INa (689). Coexpression of Scn5a with the implicated Gpd1-l-A280V, E83K, I124V, and R273C reduced INa in expression systems and neonatal myocytes (689, 840).
2. Effects of NAD+/NADH on INa
Altered NADH might modify Nav1.5 function and expression giving changes that might explain the effects of GPD1-L mutations on INa. Such studies first demonstrated that additional expression of GPD1-L-A280V but not of WT-GPD-1L markedly increased [NADH]i in HEK cell lines stably expressing human Nav1.5 alone. Second, [NADH]i produced marked dose-dependent (20–100 μM) rapid and stable, ≤50%, reductions in maximum INa in HEK cells expressing human Nav1.5. It also did so to a lesser extent in rat neonatal cardiomyoctyes. In contrast, the overall voltage dependences of both activation and inactivation of Nav1.5 and Nav1.5 mRNA expression were preserved. These effects were reversed by NAD+, the PKC antagonist chelerythrine, and superoxide dismutase. They were mimicked by the PKC activator phorbol 12-myristate 13-acetate (PMA). The rescue effects of NAD+ were antagonized by PKA inhibition by PKAI6–22 (100 nM) (360). Conversely, the PKA activator (1 μM) forskolin blocked the NADH effect on INa while not by itself affecting INa. Thus the mutually antagonistic effects of internally or externally applied NAD+ and NADH likely involved separate signaling pathways. In contrast, surface Nav1.5 expression was conserved through these manipulations (672). Finally, WT murine hearts exposed to increased [NADH]i through perfusion using external 10:1 lactate:pyruvate showed a reversible arrhythmogenesis on programmed electrical stimulation. Conversely, application of [NAD+]o perfusion reversibly abolished the arrhythmogenesis shown by Scn5a+/− hearts (672).
Figure 20 simplifies some of the factors outlined in section VIII, A–C, and their possible relationships to biophysical events leading to arrhythmogenic effects.
Figure 20.

Energetic dysfunction and arrhythmic phenotype. Possible simplified relationships between (A) energetic dysfunction associated with ischemic conditions, cardiac failure, ageing and diabetes, (B) mitochondrial dysfunction associated with ROS production, altered NAD+/NADH, and ATP/ADP and their possible consequences for (C) RyR2-mediated release of SR Ca2+ leading to increased [Ca2+]i, and NCX and DAD triggering activity, and (D) Na+ and K+ channel activity affecting AP excitation, propagation, and recovery potentially resulting in (E) substrate that can be potentially triggered to give arrhythmic phenotypes.
IX. ARRHYTHMIC SUBSTRATE RESULTING FROM STRUCTURAL CHANGE
A. Fibrotic Change and Arrhythmic Phenotype
1. Cytokine signaling and fibrotic change
In addition to ion channel and cellular homeostatic changes, structural changes, whether at the subcellular, cellular, tissue, or even organ levels, could alter the conditions of excitation, its conduction, and recovery from such excitation. They thus would also contribute to arrhythmic phenotype. Table 8 lists examples to be considered below (465). Previous sections had alluded to the association between Nav1.5 haploinsufficiency in the murine Scn5a+/− system and structural in addition to biophysical change (see sect. VE). This particularly involved a cardiac fibrotic phenotype induced by increased levels of TGF-β1 (see sect. VF). This may be a manifestation of a more general regulation of fibrotic change with possible implications for arrhythmia (432, 959).
Table 8.
Murine genetic models of structural disorders associated with ventricular or atrial arrhythmia
| Gene Product | Model | Features | Reference Nos. |
|---|---|---|---|
| Fibrotic change | |||
| TGF-β1 | Cardiac-specific TGF-β1 overexpression (MHC-TGFcys33 ser) | Atrial fibrosis and AF inducibility | 1193 |
| TNF-α | Cardiac-specific TNF-α overexpression | Atrial dilation, fibrosis, thrombosis, spontaneous AF in ambulant mice | 598, 974 |
| Angiotensin I converting enzyme (ACE) | ACE 8/8 | Atrial dilatation, fibrosis, and spontaneous AF in ambulant mice | 1280 |
| Cardiac-specific overexpression of ACE | |||
| Adenosine (A1 or A3) receptors | Cardiac-specific A3AR overexpression of at high (A3high) or low (A3low) adenosine levels | Bradycardiomyopathy | 300 |
| Ras homolog gene family member A (RHOA) | Cardiac-specific (αMhc promoter) overexpression of WT or activated form of RHOA | Atrial enlargement, cellular hypertrophy, interstitial fibrosis, AF susceptibility, atrioventricular block, premature death | 991 |
| Mitogen-activated protein kinase kinase 4 (Mkk4), | Atrial cardiomyocyte-specific conditional knock-out Mkk4-acko-Nppa-Cre4 | AF, atrial fibrosis, upregulated TGF-β1 signaling and dysregulation of matrix metalloproteinases | 245 |
| Hypertrophic cardiomyopathy (HCM) | |||
| α-Myosin heavy chain (Myh6) | Mhc-R403Q/+ | QT dispersion, conduction heterogeneity, inducible VT, sudden death, variable degree of hypertrophy | 112, 347, 1266 |
| Myosin binding protein C (Mybp-C3) | MyBP-C-t/+ and MyBP-C-t/t; (t = truncated Mybp-C3) | Hypertrophy, fibrosis, ventricular ectopic activity | 113 |
| Troponin T (Tnnt2) | Severity: Tnnt2-I79N >TnnT-F110I>TnnT-R278C | Altered Ca2+ sensitivity, spontaneous VT, reduced IK1 | 83, 580, 583 |
| Myosin light chain (coded by Mlc17) | Mlc-17-M149V | Hypertrophy | 1185 |
| Ras (HRAS) | Targeted, chronic Ras-Raf–MAPK pathway activation and αMHC-loxp-GFP-loxp-H-RasV12 | Hypertrophy, ventricular arrhythmia, atrial fibrillation, conduction block | 969, 1346 |
| Dilated cardiomyopathy (DCM): sarcomeric protein mutations | |||
| α-Myosin heavy chain (Myh6) | Mhc-F764L/+, Mhc-S532P/+, Mhc-F764L/F764L and Mhc-S532P/S532P) | Dilatation | 1018 |
| Mammalian-enabled protein and vasodilator-stimulated phosphoprotein (Vasp) | Cardiac myocyte-restricted, α-myosin heavy chain promoter-directed expression of the dominant-negative VASP-EVH1 domain | Dilatation, hypertrophy, bradycardia, sudden death | 293 |
| Neuron-restrictive silencer factor (Nsrf) | α-MHC-dnNRSF (dominant-negative knock-in) | Dilatation, heart block, sudden death | 608 |
| Dilated cardiomyopathy (DCM): cytoskeletal protein mutations | |||
| Lamin (coded by LMNA) | LMNA+/− | AV nodal disease, atrial arrhythmia, VT | 1267 |
| Lmna-N195K | Dilatation, fibrosis bradycardia, SA node exit block | 807 | |
| Lmna-H222P/H222P | Models Emery-Dreifuss syndrome; dilatation, increased PR interval, variable conduction block, QRS prolongation, ventricular extrasystoles | 45 | |
| Dilated cardiomyopathy (DCM): SR Ca2+-cycling protein mutation | |||
| Phospholamban (Pln) | Pln-R14Del | Dilation, fibrosis, VT sudden death | 397 |
| Pln-R9C/R9C | Reduced protein kinase A activity, delayed SERCA-Ca2+ reuptake, dilated cardiomyopathy, congestive heart failure | 1019 | |
| Dilated cardiomyopathy (DCM): developmental gene mutations | |||
| Cardiac homeobox gene product (Nkx-2.5) | Ventricle-restricted deficiency: Nkx2-5-f/f/cre+ | Hypertrabeculation, noncompaction, conduction disturbance | 871 |
| Vinculin (VCL) | Cardiac-restricted Vclfl/fl MLC-2vCre/+ knockout | Dilatation, VT, sudden death | 1324 |
| Troponin I (TNNI3) | cTnI-R192H | Atrial dilatation, decreased LV volume, decreased EF, delayed relaxation | 278, 279 |
| Arrhythmogenic right ventricular dysplasia (ARVD): desmosomal protein gene mutations | |||
| Plakophilin-2 (Pkp2) | Pkp2−/− | Lethal | 376 |
| Pkp2+/− | Normal cardiac phenotype | 376 | |
| Plakoglobin (Pg) | Pg−/− | Lethal | 647 |
| Cardiac specific Pgf/f-αMHCcre | RV enlargement, fibrosis, slowed RV conduction, spontaneous VT of right ventricular origin. Phenotype exacerbated by training | 571, 647, 973 | |
| Desmoplakin (Dsp) | Dsp−/− | Embryonic lethal | 339, 475 |
| Dsp+/− | Slowed conduction and inducible VT prior to adult structural changes | 366 | |
| Cardiac overexpression of flag-tagged Dsp cDNA carrying Dsp-R2834H-Tg COOH-terminal mutation | Apoptosis, fibrosis, RV and LV dilatation, reduced ventricular function, widened intercalated disks | 1296 | |
| Cardiac specific Dsp-f/f- (MLC2v-Cre) | Cardiomyopathic changes, ventricular arrhythmias accentuated by exercise and catecholaminergic challenge, slowed RV conduction | 706 | |
| Desmoglein (Dsg2) | Cardiac overexpression of flag-tagged dominant Dsg2-N271S mutant (homologous with human DSG2–N266S ARVC mutation) | Myocyte necrosis, calcification and fibrous tissue replacement, spontaneous ventricular arrhythmias, conduction slowing, ventricular dilatation and aneurysms, replacement fibrosis and SCD | 896 |
| Arrhythmogenic right ventricular dysplasia (ARVD): laminin receptor mutations | |||
| Laminin receptor 1 (Lamr1) | KK/Rvd (knock-down) | Right ventricular fibrosis, QRS prolongation | 48 |
| Left ventricular noncompaction cardiomyopathy (LVNC) | |||
| Cypher/Zasp | Mutations in Cypher/Zasp | LVNC with DCM and HCM phenotype, early postnatal death | 1345, 1350 |
TGF-β1 is a key regulator of fibrosis in adult hearts. Increased fibrosis, to which atria are particularly susceptible (949, 1305), reduces cardiomyocyte-cardiomyocyte coupling potentially resulting in conduction defects (21). Increased TGF-β1 expression is clinically associated with chronic AF (222). In murine hearts, cardiac-specific TGF-β1 overexpression was followed by atrial fibrosis, atrial conduction disturbances, and AF (1193). The latter was attributed to triggered activity following reductions in APD and an appearance of spontaneous Ca2+ release events (198). Angiotensin II and reactive oxygen species may also increase TGF-β1 promoter activity, connective tissue growth factor (Ctgf), and collagen in fibroblasts causing a profibrotic environment (408, 1146). Similarly cardiac-specific TNF-α overexpression produced at rial dilation, fibrosis, thrombus development, and spontaneous AF in ambulant mice (598, 974). In some models, it resulted in Cx40 downregulation (1013).
2. G protein signaling and fibrotic change
Cardiac G protein-coupled receptors serve various signaling pathways involving epinephrine, angiotensin, endothelin, and adenosine as messengers. Thus Gαq associates with α1-adrenergic, endothelin (ETA), and angiotensin II type I (AT1) receptors. They may thereby exert metabolic effects that impact on tissue fibrosis. Thus AF patients show increased atrial angiotensin II expression (519). Rapidly paced in vitro atrial myocytes show increased angiotensin II secretion (1145, 1280). Cardiac-specific overexpression of angiotensin I converting enzyme caused atrial dilatation, fibrosis, and spontaneous AF in ambulant mice (1280). Similarly, transgenic models overexpressing adenosine A1 or A3 receptors showed atrial bradycardia and atrioventricular block and increased susceptibility to AF dependent on the degree of bradycardia (300, 570). This could potentially lead to bradycardiomyopathy (300). Both available transgenic mouse lines overexpressing active Gαq subunit under αMhc promoter control developed cardiac hypertrophy followed by diffuse atrial and ventricular fibrosis and heart failure (233, 764) and spontaneous AF in ambulant animals (431).
The membrane-associated GTPase, Ras homolog gene family member A (RHOA), is involved in actin cytoskeleton organization. Mice overexpressing either WT RHOA or an activated form of RHOA under cardiac-specific αMhc promoter control died prematurely. They then showed cardiac, particularly atrial, enlargement, cellular hypertrophy, and interstitial fibrosis. This accompanied AF, atrioventricular block, and heart failure (991). ECG performed under anesthesia suggested a susceptibility to AF and atrioventricular block (991). A murine overexpression system for the rho protein GDP dissociation inhibitor RhoGDIα showed atrial arrhythmias and mild ventricular hypertrophy and bradycardia, atrioventricular block, and atrial arrhythmias with a downregulation of Cx40. These were followed by cardiac hypertrophy and cardiac failure (1245).
3. Mitogen-activated protein kinases, fibrotic change, and atrial arrhythmic phenotype
Mitogen-activated protein kinases (MAPKs) are serine/threonine kinases central to diverse cellular stimuli including environmental stress, proinflammatory cytokine action, and developmental cues. Of these, mitogen-activated protein kinase kinase 4 (MKK4) and MKK7 are critical components in the stress-activated MAPK signaling pathway (1227). Both MKK4 and MKK7 are kinases for c-Jun NH2-terminal kinase (JNK) (255), reported to suppress TGF-β1 gene expression (1188). MKK4 additionally activates p38 (139). Studies in genetically modified murine platforms suggested that attenuation of cardiac p38 activity resulted in a progressive growth response and myopathy particularly following aortic banding (141). MKK4 may also regulate Cx43 remodeling by either phosphorylation- or transcription-dependent mechanisms (890). MKK4 may thus inhibit maladaptive pathological hypertrophy through its activation of the JNK pathway (680).
Genetic modifications in Mkk4 expression offered an experimental platform for investigating the consequences of alterations in Cx43 expression. The kinetics of Cx43 synthesis, trafficking, and degradation results in a rapid turnover (half-life ∼1–5 h) (948). This in turn potentially influences its expression in directions that could contribute arrhythmic substrate. Both knockdown of Mkk4 expression by silencer siMkk4 and inhibition of its kinase activity by infection with a dominant negative, Ad-dnMkk4, adenovirus reduced the Cx43 expression induced by phenylephrine challenge in NRCMs. This likely involved two specific activator protein-1 binding elements in the Cx43 promoter region.
Cardiomyocyte-specific knockout mice (Mkk4-cko) showed reduced and heterogeneous Cx43 expression and zonula occludens-1 (ZO-1) protein content following transverse aortic constriction. Their ECG recordings demonstrated widened QRS durations (∼16 vs. 10 ms), and increased yet normal QT intervals. Programmed electrical stimulation induced VT in 6 of 13 Mkk4-cko but none of the studied WT mice. Finally, epicardial activation mapping indicated ventricular activation delays in Mkk4-cko hypertrophied hearts. Mathematical modeling simulated a slowed, fragmented, and potentially proarrhythmic ventricular conduction (1351).
MKK4-based systems also permitted analysis of the effects of fibrotic change on connectivity between atrial myocytes. The clinical prevalence of AF and that of atrial fibrosis increase in parallel with age. Clinical evidence implicated MKK4 in this AF pathogenesis. Thus qPCR comparisons of MAPK components and profibrotic signaling molecules in atrial tissues demonstrated MKK4, but not MKK7, P38 or JNK, downregulation, in AF relative to age-matched control patients in sinus rhythm. These findings were further confirmed as decreased MKK4 protein levels. qPCR analysis in the AF patients further suggested increased profibrotic gene expression. There were thus increased levels of Col1a1 and Col3a1 collagen, and the profibrotic connective tissue growth factor (CTGF), in contrast to downregulated matrix metalloprotein 2 (MMP2) expression. MKK4 mRNA downregulation was the more marked in older (>75 yr) compared with younger age AF groups.
These clinical observations could be correlated with findings in a Mkk4-acko conditional knockout mouse model with an atrial cardiomyocyte-selective deletion of Mkk4 using the natriuretic peptide precursor A (Nppa) promoter-driven Cre transgene. Young (3–4 mo), adult (6 mo), and old (1 yr) Mkk4-acko mice were compared with age-matched Mkk4-f/f controls. Aging Mkk4-acko mice were more susceptible to atrial tachyarrhythmias. These were associated with slowed and dispersed atrial conduction (Figure 21, Aa, Ab, and B). There were increased incidences of arrhythmia particularly in old Mkk4-acko hearts (Figure 21C). Yet, Langendorff-perfused Mkk4-acko and Mkk4-f/f hearts showed similar APD90 and ERP values, and Cx levels and distribution through all age groups. Studies of atrial proteins that might contribute to slowed conduction and atrial arrhythmogenesis demonstrated no differences in mRNA or protein expression level expression of Nav1.5, RyR2, NCX, inositol 1,4,5-trisphosphate receptor (IP3R), the transient receptor potential channel proteins, TRPC1, TRPC3, and TRPC6, and the gap junction proteins, Cx40 and Cx43 between young or old, Mkk4-f/f and Mkk4-acko mice.
Figure 21.

The arrhythmic mitogen-activated protein kinase kinase 4 knockout heart as an arrhythmic exemplar for fibrotic change. In vivo and ex vivo cardiac electrophysiological characterizations of mice carrying an atrial cardiomyocyte specific mitogen-activated protein kinase kinase 4 knockout, Mkk4-acko, compared with Mkk4-flox/flox, Mkk4-f/f, controls. A: representative in vivo ECG recordings showing (a) normal rhythm in Mkk4-f/f (a) in contrast to polymorphic atrial ectopic beats and spontaneous atrial tachycardic episodes in Mkk4-acko mice (b). B: ex vivo atrial epicardial monophasic action potential (AP) recordings in Langendorff-perfused hearts during programmed electrical stimulation interposing extrasystolic S2 stimuli following trains of pacing S1 stimuli. These show contrasting persistent sinus rhythm in Mkk4-f/f (a) with observations of frequent AF in Mkk4-acko (b). C: occurrence of atrial arrhythmic events (AT and AF) in young (3 mo) and old (12 mo), Mkk4-f/f and Mkk4-acko, hearts. D: picrosirius red-stained atrial tissue, fibrotic areas dark red, from 3- and 12-mo-old, Mkk4-f/f and Mkk4-acko, mice. E: percentage fibrotic area in 3- and 12-mo-old Mkk4-f/f (white) and Mkk4-acko atria (black bar). F: epicardial multielectrode array (MEA) activation maps resulting from differing AP conduction velocities following pacing in the center of the array in right (RA) and left atria (LA) of Mkk4-f/f and Mkk4-acko mice (a). G: computer modeling of the effects of fibroblast-cardiomyocyte coupling resulting in reentry following a premature beat. A standard S1S2 stimulation protocol is applied at the left edge of the 2D model containing randomly distributed fibroblast populations in which between one and five fibroblasts are coupled to any given cardiomyocyte. Subsequent snapshots demonstrate a breaking down of the wavefront of atrial excitation wave leading to formation of reentrant excitation waves. [From Davies et al. (245).]
The increased arrhythmogenecity thus more likely resulted from a development of an increased interstitial fibrosis (Figure 21D). This was accompanied by upregulated TGF-β1 signaling and dysregulation of matrix metalloproteinases (Figure 21E). Mkk4 inactivation also increased the sensitivity of TGF-β1 signaling to angiotensin II-induced activation in cultured cardiomyocytes. It thus increased the expression of profibrotic molecules in cultured cardiac fibroblasts. Thus Mkk4 likely downregulates the TGF-β1 signaling associated with atrial remodeling and arrhythmogenesis with age (245). Mkk4 deletion thus appeared to increase atrial arrhythmic tendency by altering conduction properties especially in old Mkk4-acko mice. MEA recordings of atrial AP arrival times, following stimulus application at a point in the center of the MEA array, demonstrated similar conduction velocities in both LAs and RAs of young (3 mo) Mkk4-f/f and Mkk4-acko hearts. However, conduction velocity was slower in old (12 mo) Mkk4-acko atria than old Mkk4-f/f hearts (Figure 21F).
Modeling studies confirmed that such fibrotic processes could potentially lead to atrial arrhythmogenic phenotypes in Mkk4-acko mice through formation of electrical fibroblast-cardiomyocyte couplings (574). This decreased AP overshoots and (dV/dt)max values, prolonged APDs, and depolarized resting potentials to extents dependent on the number of fibroblasts coupled to each myocyte. This would fulfil expectations from the coupled fibroblasts variously acting as passive electrical loads and electrical drivers through the cardiomyocyte AP time course. Two-dimensional modeling of the consequences of these loading features demonstrated that such fibroblast-cardiomyocyte couplings slowed the conduction velocities of atrial excitation waves. Furthermore, it did so to extents increasing with fibroblast density. It thereby replicated differences in conduction velocity in young and old Mkk4-acko or Mkk4-f/f atria, particularly the greater negative dependence of conduction velocity on fibroblast density in the old than the young group. It further demonstrated wavebreaks in propagated atrial excitation resulting from such coupling. Figure 21G illustrates such effects following S1S2 stimulation protocol applied at the left edge of the two-dimensional map representing the situation in which one to five fibroblasts had been coupled to any particular cardiomyocyte. Illustrations of the subsequent S2-induced excitation waves at a S1S2 coupling interval of 60 ms demonstrated breakdown of the excitation wavefront and re-entrant excitation.
B. Hypertrophic Cardiomyopathy
1. Mutations involving sarcomeric proteins
Cardiomyopathies are characterized by structural myocardial abnormalities not accountable for by acquired, ischemic, valvular, hypertensive, or congenital conditions (732). These conditions together account for most cases of SCD. Cardiomyopathies are broadly categorized into hypertrophic (HCM), dilated (DCM), arrhythmogenic RV, restrictive, and LV noncompaction cardiomyopathies (715, 732) with multiple genotypes occasionally existing through families (1220). The most common, HCM (prevalence 1:500), presents as asymmetric or symmetric myocyte hypertrophy, fibrosis, and myofiber disarray (1260). It is associated with a wide variety of often sarcomeric mutations (732). The condition is clinically heterogeneous. The different mutant alleles involved are associated with different hypertrophic severities and clinical outcomes (1237). The anatomical abnormalities provide potential reentrant excitation circuits leading to high arrhythmia risk (1010). Some sarcomeric mutations appear to result in high SCD risk even without cardiac hypertrophy (1173).
Seventy-five percent of HCM cases involve mutations in genes encoding myosin α- and β-chains, MYBPC3, and troponin T. The murine models available provide broad phenotypic parallels relating to their accompanying structural changes or alterations in Ca2+ homeostasis that, however, merit further more detailed study. Heterozygous, particularly male, Mhc-R403Q/+ mice that contain a clinically relevant, myosin α-heavy chain mutation, developed the classical histology of disarray, hypertrophic myocytes, and interstitial fibrosis between 5 and 15 wk (347). These changes accompanied prolonged ECG repolarization intervals and right axis deviation. There were also heterogeneous ventricular electrophysiological conduction properties, prolonged sinus node recovery times, and ventricular ectopic events (112). However, their arrhythmic susceptibility varied more with the extent of ventricular hypertrophy than it did with the extent or location of myocyte disarray or cardiac fibrosis (1266).
Heterozygous, MyBP-C-t/t, defects truncating myosin-binding protein C in its cardiac myosin heavy chain-binding and titin-binding domains also cause human HCM. Mice with analogous heterozygous, MyBP-C-t/+ and homozygous MyBP-C-t/t showed mild dilated and severe hypertrophic cardiomyopathic phenotypes, respectively. However, they showed normal ECG intervals, and sinus node, atrial, and ventricular, conduction, and refractoriness. MyBP-C-t/t but not MyBP-C-t/+ mice were more susceptible to induction of VT than WT and showed ventricular ectopic activity, to extents that, however, did not correlate with their degrees of interstitial fibrosis (113).
Mutations in TNNT2 which encodes cTnT involve the thin filament sarcomeric protein component troponin T. These are associated with ventricular arrhythmia and SCD. In the case of TNNT2-R92T, SCD occurs typically at ∼17 ± 9 yr. There is then minimal hypertrophy or fibrosis but significant myofilament disarray (795, 1173). Similarly, Tnnt2-I79N, clinically linked to human HCM, is associated with increased incidence of SCD despite a mild cardiac hypertrophic phenotype. However, TNNT2 mutations might cause their associated stress-induced VT through electrophysiological remodeling of Ca2+ transients and reducing IK1 rather than hypertrophy and/or fibrosis. Ambulant Tnnt2-I79N mice were more susceptible to nonsustained VT produced by behavioral stress. Intact mouse hearts and ventricular myocytes carrying Tnnt2-I79N showed Ca2+ transients with slowed decay kinetics, suggesting increased Ca2+ sensitivities in their TnT. They also showed shortened APD70, and normal ICa and Ito but decreased IK1. The APD reductions were reversed by increasing intracellular Ca2+ buffering or blocking NCX function. Higher pacing rates or isoproterenol challenge resulted in diastolic Ca2+ levels higher than those found in control myocytes and increased susceptibility to ventricular ectopic events (583).
Phenotypic comparisons of HCM-linked murine TnT mutations resulting in strong (TnT-I79N), intermediate (TnT-F110I), and an absence of Ca2+ sensitization (TnT-R278C) respectively in human cardiac fibers directly implicated myofilament Ca2+ sensitivity in this arrhythmic susceptibility which takes place in the absence of hypertrophy, fibrosis, or myofibrillar disarray (83, 422, 580). All three groups showed similar low baseline incidences of ventricular ectopic activity that were increased by isoproterenol challenge. However, fast pacing triggered sustained VT with incidences in the sequence: TnT-I79N > TnT-F110I > TnT-R278C = WT. Epifluorescence imaging demonstrated greater spatial variability in activation times in TnT-I179N than TnT-R278C hearts. Finally, the Ca2+-sensitizing agent EMD 57033 and the myofilament Ca2+ desensitizing agent blebistatin respectively reproduced and diminished these arrhythmic susceptibilities. Ca2+ sensitization altered ventricular APD, increased its beat-to beat variation, and shortened VERPs. It also increased the dispersion of ventricular conduction velocities at high heart rates. All these changes potentially create arrhythmic substrate (83).
Mutations in other sarcomeric proteins, such as the regulatory (RLC) or essential (ELC) myosin light chain, constitute rarer causes of HCM (902a). However, even genetically modified mice carrying ELC or RLC mutations known to result in human HCM with midventricular cavity obstruction frequently did not show a hypertrophic phenotype, even in the presence of myofilament and cellular level increases in Ca2+ sensitivity and relaxation rates (997).
2. Abnormalities in nonsarcomeric cellular components
HCM can also follow abnormalities in nonsarcomeric cellular components. Syrian hamsters carrying spontaneous δ-sarcoglycan mutations show HCM, DCM (993), and stress-induced SCD (750) in common with human sarcoglycan mutation-related HCM. Gain-of-function mutations involving the Ras-Raf protein kinase pathway classically associated with development disorders including the Noonan and LEOPARD syndromes are also associated with HCM. This likely takes place through an upregulation of RAS signaling that leads to a pathological cardiomyocyte hypertrophy (865). Genetically modified mice with a targeted and chronic Ras-Raf-MAPK pathway activation developed diastolic dysfunction, SR Ca2+ defects, and increased early sudden cardiac mortality (1346). Inducible gene-switch activation of the hypertrophic Ras-Raf-mitogen-activated protein kinase pathway in adult murine hearts elicited ventricular hypertrophy and ventricular arrhythmias. The isolated ventricular myocytes showed an electrophysiological remodeling that paralleled changes frequently reported in hypertrophic and failing hearts. The latter took the form of AP prolongation, increased NCX activity, reduced IK, altered SR Ca2+, and deficient PKA-dependent PLN phosphorylation. The changes accompanied induction of Gα-inhibiting subunit 1 (Giα1) expression and were partly rescued by Gi/o inhibitor pertussis toxin (969).
C. Dilated Cardiomyopathy
1. General features
DCM results in LV dilatation and consequent systolic dysfunction despite normal loading conditions and coronary function. Twenty-five to 40% of cases are familial, often as autosomal dominant characteristics with variable penetrances. DCM can also be acquired following infection, toxin exposure, or metabolic abnormalities. Its associated electrophysiological abnormalities include sinus node dysfunction, atrioventricular block, and atrial or ventricular arrhythmias often following tissue scarring (603, 904). DCM is associated with over 30 different cytoskeletal, sarcomeric protein/Z-band, nuclear membrane, and intercalated disc proteins (603). The murine systems replicating DCM may also demonstrate its accompanying alterations in contractile function and/or its arrhythmic phenotypes.
2. Ion channel genes involved in DCM
Of ion channel genes associated with DCM (504, 617), SCN5A has been implicated as a candidate gene in screening studies of DCM families with cardiac conduction disorders. These include the SCN5A-D1275N (758), and SCN5A-T220I, R814W, and D1595H missense and 2550-2551insTG truncation mutations (854). SCN5A-D1275N cosegregated with age-dependent DCM phenotypes with variable penetrances associated with AF, impaired automaticity, and conduction delay. A Chinese family carrying SCN5A-A1180V showed a mild in vitro INa phenotype accompanied by DCM preceded by atrioventricular block (346). Hypertrophic change has also been associated with Na+ channel-related, LQTS phenotypes (553, 1049). Murine Scn5a+/− hearts while similarly showing conduction defects, AF and ventricular arrhythmia, and progressive fibrotic change (see sect. VE) did not exhibit features of DCM. This finding is in direct contrast to the remaining close phenotypic similarities between murine Scn5a+/− and human BrS phenotypes. This may prompt further investigation for differences that might be related to the effects of the intraventricular systolic pressures developed within the small murine as opposed to the relatively large human ventricular chambers.
3. Sarcomeric protein mutations
A substantial proportion of clinical DCM cases are associated with sarcomeric protein mutations. However, the two murine myosin heavy chain (MYH6) models were not arrhythmic (1018). In contrast, murine mutations modulating myosin function showed arrhythmia, thus recapitulating the corresponding clinical phenotypes. Transgenic mice containing dominant-negative mutations altering cardiac-specific expression of the intercalated disk mammalian-enabled protein (Mena) and vasodilator-stimulated phosphoprotein (VASP) showed bradycardic and sudden death phenotypes. These were associated with reduced cell-to-cell interactions (293). Similarly, mice overexpressing a dominant-negative transgenic copy of the transcriptional myosin regulator, neuron-restrictive silencer factor, showed VT and heart block associated with upregulation of If and ICaT, normally specific to pacemaker and Purkinje cells (608).
4. Cytoskeletal protein mutations
Of mutations disrupting cytoskeletal function, those involving the lamin A/C (LMNA) gene cause a wide range of clinical conditions. Lamin A/C, LMNA+/−, mutations producing 50% reductions in normal cardiac lamin A/C resulted in a development of atrioventricular node myocyte apoptosis and conduction loss, atrial arrhythmias, and VT by age 4 wk, and DCM with age (1267). Mice carrying the missense Lmna-N195K murine variant known to cause human DCM, similarly showed DCM, a profound and progressive sinus bradycardia reflecting sinoatrial exit block, and a range of conduction abnormalities. These included progressive PR interval prolongations with a premature, arrhythmic, death on continuous ECG monitoring. They also showed misexpression of transcription factor Hf1b/Sp4 and of Cx40 and Cx43, and disorganized sarcomeres and intercalated disks.
The cardiomyopathy thus likely arises from this disruption of internal cardiomyocyte organization and possibly an abnormal expression of transcription factors ensuring normal cardiac development (807). The LMNA-H222P missense mutation occurred in a family with the autosomal dominant Emery-Dreifuss muscular dystrophy. Homozygotic Lmna-H222P mice showed apparently normal embryonic and sexual development. However, adult male, and at a later stage, female, mice showed abnormal voluntary locomotor activity and an echocardiographically detected DCM. They also showed ECG evidence of increased PR intervals and QRS durations, episodes of sinoatrial block and ventricular extrasystoles, and premature mortality at ∼9 mo. There was an accompanying cardiac and skeletal muscle degeneration and fibrosis and an activated Smad signaling (45).
5. SR Ca2+-cycling protein mutations
Genetic abnormalities in SR Ca2+-cycling proteins, particularly those affecting the SERCA regulator PLN, also cause familial cardiomyopathies. PLN dephosphorylation reduces apparent SERCA2a Ca2+ affinity. The resulting SERCA2a inhibition can be abolished by PKA-mediated phosphorylation. Of naturally occuring PLN mutations, one carrying a homozygotic lysine-39 stop codon (PLN-L39stop) abolished PLN expression and caused a lethal DCM and heart failure at teenage. However, this clinical phenotype was not reproduced in Pln-deficient mice (398). A R9C mutation in the Pln coding region seemed to preclude PKA-mediated phosphorylation of even WT PLN producing a chronic inhibition of SERCA2a (1019). This was similarly associated with a clinical DCM recapitulated in transgenic mice overexpressing Pln-R9C.
Mutations affecting Pln transcription levels also result in inhibited SERCA2a activity. One of 87 of a series of hypertrophic patients showed an A to G mutation in this region (776). Infections of in vitro rat myocytes suggested that such a mutation markedly increased PLN promoter activity consistent with a role in producing DCM. Finally, a heterozygotic Pln-R14Del variant involving PLN coding demonstrated by genetic screening of DCM patients was clinically associated with LV dilatation, contractile dysfunction, episodic ventricular arrhythmias, and heart failure by middle age. These features were reproduced in transgenic mice overexpressing Pln-R14Del. WT and Pln-R14Del coexpression in HEK-293 cells demonstrated a SERCA2a superinhibition that might provide a mechanism for the DCM (397).
6. Developmental gene mutations
Of developmental genes, the cardiac homeobox gene Nkx2-5 is central to early cardiogenesis (409). Mutations produce a wide range of malformations including septal and outflow tract defects, cardiomyopathy and LV hypoplasia, and associated arrhythmias (1022). Neonatal mice with ventricle-restricted Nkx2-5 deficiency appeared structurally normal. They displayed no arrhythmias on ECG recording. They subsequently developed a trabecular muscle hypertrophy and progressive, first degree (by 12 wk), and complete heart block (at 6–12 mo) recapitulating clinical findings in some Nkx2-5 patients. This accompanied a cardiomyocyte dropout and fibrosis following their sarcomeric and cellular disorganization in the central conduction system. This produced a distinct form of cardiomyopathy characterized by extensive trabeculae and myocardial noncompaction. There was a progression to dilatation of both chambers, their marked trabeculation, and heart failure. There was a dysregulation of genes associated with myocardial cell proliferation and trabeculation. This included an increased expression of bone morphogenic protein-10 (BMP-10) and a downregulated cardiac homeodomain-only protein, HOP. These findings likely represented a distinct pathway for progressive cardiomyopathy associated with conduction defects in congenital heart disease (871).
7. Cytoskeletal protein mutations
Of cytoskeletal proteins, vinculin anchors the actin cytoskeleton to the cell membrane. It is thus likely required to preserve cell-cell and cell-matrix adhesion. Murine cVcl-KO models with Cre-loxP generated cardiac-myocyte-specific vinculin (Vcl) gene inactivation showed telemetric ECG evidence of VT leading to sudden death, and optical mapping evidence of reduced myocardial conduction even at <3 mo. This was followed by development of DCM often fatal at <6 mo. This accompanied abnormal adherens junction and intercalated disc structure, reduced cadherin and beta1D integrin expression, and mislocalization of Cx43 to the lateral myocyte border (1324).
D. Arrhythmogenic Right Ventricular Cardiomyopathy
1. Clinical features
ARVC is associated with ventricular arrhythmias and SCD, particularly in young athletes (79, 219), giving a phenotype unmasked by exercise testing (886). It is characterized by a fibro-fatty infiltration leading to RV dilatation. Dilatation may extend to areas of the LV but characteristically spares the septum (732). Desmosomal protein gene mutations occur in 50% of such patients (685). Desmosomes participate in end-to-end connections transmitting contractile force between cardiac myocytes (250). Abnormal junctional organization leads to myocardial damage and replacement fibrosis culminating in focal scars isolating cardiomyocytes within nonconducting fibrous tissue (80). This slows conduction, generating reentrant substrate.
2. Plakophilin-2 (Pkp2) deletions
Murine models exist for the transmembrane desmosomal cadherins, plakoglobin (γ-catenin) and plakophilin 2, and the plakin protein desmoplakin, which links the junctional complex to the intermediate filament, desmin, thereby maintaining mechanical integrity (685, 1046). Mutation of the gene encoding plakophilin-2 may occur in up to one third of ARVC families (349). However, Pkp2+/− mice with a heterozygotic plakophilin-2 targeted deletion showed no cardiac morphological phenotype. Nevertheless, human subjects with plakophilin-2 (PKP2) mutations showed more frequent VT episodes than those without detected PKP2 mutations (68). Furthermore, the homozygous deletion was lethal at postfertilization day 11 with evidence for endothelial discontinuities in heart and vessels (376).
3. Consequences of plakoglobin (Pg) deletion and overexpression
One of the two initially available heterozygous murine plakoglobin deletion models showed RV enlargement, increased spontaneous ventricular arrhythmias, and RV conduction slowing (123, 973). This took place despite an absence of replacement fibrosis, junctional remodeling, or alterations in Cs43 localization and distribution, at 10 mo. These abnormalities were exacerbated by exercise (571) and relieved by load reduction regimes (299). In contrast, homozygous targeted plakoglobin deletion was lethal between ED 9.5 and ED 16 with thinning of the cardiac walls, reduced trabeculation, and epicardial discontinuities. Nevertheless, a cardiac-specific targeted Pg deletion controlled by αMHCcre partially recapitulated human ARVC. Pgf/f-αMHCcre mice showed ∼30% of WT protein but no Pg in cardiac sections. They showed a tendency to SCD, progressive RV and LV dilatation, and fibrosis though no cardiac fat deposition from ∼2 mo. Desmosomal ultrastructure was disrupted (647). There was evidence of myocyte apoptosis in addition to myocyte necrosis, in contrast to the necrosis observed with desmoglein (Dsg2)-N271S overexpression. The intercalated disks also lacked other desmosomal proteins, including Dsg2 (647). However, β-catenin expression appeared increased and may have partially rescued the sudden death phenotype. The latter was suggested in crosses of double-targeted mice carrying both floxed Pg and floxed β-catenin loci (Pg-f/f; β-catenin-f/f) loci with αMHC/MerCreMer mice. Tamoxifen challenge resulting in specific targeted deletion produced a strong arrhythmogenic SCD phenotype in all animals between 3 and 5 mo after tamoxifen injections (1105). Finally, mice with both WT or mutant Pg overexpression showed increased incidences of SCD (686).
Similarly, a two-base pair plakoglobin gene deletion is associated with the recessive Naxos syndrome, associated with further, keratotic, manifestations (757, 973). Yet overexpression of the resulting, truncated, Naxos-associated plakoglobin also produces an arrhythmogenic cardiomyopathic phenotype. However, it is likely that it is an insufficiency of either the normal or truncated plakoglobin that produces disease phenotypes. One of two knockin murine models expressing a Naxos-associated plakoglobin that bypassed the nonsense-mediated mRNA decay mechanism thereby producing normal levels of truncated plakoglobin showed normal cardiac function (1343).
4. Desmoplakin (Dsp) targeted deletion and overexpression
Homozygous loss-of-function desmoplakin (Dsp) mutations are clinically associated with a biventricular form of ARVC. Heterozygous Dsp+/− mice developed normally. However, adults then showed attenuated ventricular walls, increased LV diameters, reduced LV ejection fractions, spontaneous arrhythmias on ECG recordings, and extrastimulus-induced ventricular arrhythmias. The structural changes were preceded by increased activation times and arrhythmic tendency measurable in Langendorff-perfused hearts (366). Homozygous, Dsp−/−, desmoplakin targeted deletions were lethal at ED 6.5 (475). Postponing this lethality to ED 11 by tetraploid aggregation permitted demonstration of severe cardiac malformation though persistent desmosomal-like ultrastructures (339). Embryos with a cardiac-specific, αMHCcre induced, targeted Dsp−/− deletion revealed poorly formed hearts (342).
Mice with cardiac overexpression of flag-tagged Dsp cDNA carrying the Dsp-R2834H-Tg COOH-terminal mutation demonstrated increased heart-to-body weight ratios compared with mice whether with human WT (WT-Tg) Dsp cDNA or WT littermates (1296). Dsp-R2834H-Tg hearts also showed increased apoptosis and fibrosis, RV and LV dilatation, reduced ventricular function, widened intercalated disks, and disrupted plakoglobin-desmoplakin interaction. Overexpression of Dsp with NH2-terminal, Dsp-V30M, and Dsp-Q90R mutations was lethal after ED 13.5, resulting in reduced ventricular wall attenuation and dilatation.
Finally, studies with crosses of desmoplakin-floxed mice (Dsp-f/f) with cardiomyocyte-specific ventricular myosin light chain-2-Cre recombinase (MLC2v-Cre) knock-in mice yielded viable animals. But these showed early ultrastructural desmosomal defects and cardiomyopathic changes including the cell death, ventricular fibro-fatty replacement, biventricular dysfunction, and failure and premature death. Hearts showed ventricular arrhythmias accentuated by exercise and catecholaminergic challenge. They also showed slowed RV conduction. These were attributable to reduced Cx40 and Cx43 expression, suggesting roles for desmoplakin in stabilizing Cx integrity (706).
5. Desmoglein (Dsg) overexpression models
Mice with cardiac overexpression of flag-tagged Dsg2-N271S mutant, homologous with the human DSG2-N266S ARVC mutation, showed spontaneous ventricular arrhythmias, conduction slowing, ventricular dilatation and aneurysms, replacement fibrosis, and SCD at age <2 wk. This was associated with myocyte necrosis, calcification, and fibrous tissue replacement. In contrast, mice with flag-tagged Dsg2-WT showed normal phenotypes indistinguishable from WT littermates even at age 2 mo (896). Mice with a targeted deletion in the extracellular adhesion domain of Dsg2 similarly showed RV and LV dilatation, fibrosis, calcification, and SCD (597). Hearts showed intercellular widenings at the intercalated disk and loss of desmosomal ultrastructure (536).
6. Laminin receptor mutations
A line of KK/Rvd mice with right ventricular dysplasia (RVD) contain a gene laminin receptor 1 (Lamr1) mutation containing an intron-processed retroposon whose gene product binds to heterochromatin protein 1 potentially modifying transcriptional regulation. The hearts showed a marked fibrosis in the RV free wall and cardiomyocyte cell death. Electrocardiography revealed prolonged QRS durations, suggesting intraventricular conduction disturbance potentially increasing susceptibility to arrhythmia (48).
E. Left Ventricular Noncompaction Cardiomyopathy
LV noncompaction cardiomyopathy (LVNC) is a rare, frequently autosomal dominant condition characterized by a histologically distinct noncompacted myocardial layer containing deep and prominent trabeculae (1248, 1320). It is associated with cardiac failure, angina pectoris and arrhythmia in the form of AF, ventricular tachyarrhythmias, and SCD in some series (848). The condition has been related to mutations in the neurogenic locus notch homolog protein (NOTCH) pathway regulator E3 ubiquitin-protein ligase (MIB1) (705) as well as various genes overlapping those associated with other cardiomyopathies. These include the cypher/Z-band alternatively spliced PDZ-motif protein in the sarcomeric Z line (1178). This binds to α-actinin likely contributing cytoskeletal structural integrity during contraction, and may be involved in PKC signaling through its LIM domains (1045). Mice with disrupted Z-line proteins as a result of mutations in the CYPHER/ZASP gene show phenotypes in common not only with LVNC but also DCM and HCM and suffer early postnatal death (1345, 1350).
X. SUMMARY AND CONCLUSIONS
Cardiac arrhythmias result from a breakdown of the ordered successive patterns of excitation through the SAN, atria, or ventricles essential for normal contractile function. They constitute a major public health burden (see sect. IA). Their analysis requires understanding of the process of normal electrical excitation, conduction, recovery, and membrane stability. At the molecular level, this process depends on normal function in a large range of individual ion channels with specific ion permeabilities and voltage dependences in their steady state and kinetic properties. Successful function at the cellular level requires a sequential and interdependent activation and inactivation of their resulting ion channel currents Ii (see sect. IB). At the tissue level, the resulting AP propagates through electric current flow along the intracellular resistance ra to depolarize the membrane capacitance cm. It thereby initiates excitation in hitherto quiescent connected membrane effectively along a multidimensional cable. Regularly paced APs thus acquire their characteristic duration, APD, and refractory period, ERP, separated by diastolic interval DI, through an integration of molecular mechanisms into cellular level events. The APs then propagate through myocardial tissue with velocity θ, accordingly forming active regions with wavelength λ = θ × APD, or θ × ERP, followed by a quiescent wavelength determined by θ and the diastolic interval DI, defined by an integration of cellular events and tissue properties. These fundamental characteristics define the conditions necessary for orderly excitation patterns initiated from the pacemaker and propagating through the remaining cardiac structures (see sect. IC).
Arrhythmias following perturbations of these parameters may thus take place through their temporal heterogeneities resulting from transient membrane potential change, or alterations in steady-state pacing rates. They may also be the consequence of spatial heterogeneities potentially resulting from varying tissue properties in different organ regions, or pathological anatomical change, particularly those impairing the processes of conduction or excitation (see sect. IIA). Analysis of cardiac excitation and its alterations producing arrhythmias thus requires experimental systems amenable to study at different levels of biological organization. Studies translatable to human arrhythmic conditions would require such systems to parallel human hearts in structure and function, and share a significant number of molecular species underlying their electrophysiological activation.
Mouse hearts fulfil a significant number of such criteria. In addition, they are amenable to genetic modification modifying the expression or function of particular, physiologically important, biological molecules. Murine systems further provide a range of translationally relevant exemplars for the clarification of arrhythmic processes arising from abnormalities in the initiation, propagation, and recovery of electrical activity or upon the potential for spontaneous triggering of arrhythmia (see sect. IIB). Each examplar was amenable to experimental and theoretical analysis at varied levels of biological organization and function through application and collation of results from a wide range of techniques. These extended from electrocardiography in entire organisms, through extracellular and intracellular AP recording from isolated hearts subject to varied transient perturbations or steady-state pacing conditions, biophysical ion channel and spectrophotometric Ca2+ homeostasis studies at the cellular level, to characterizations of protein and mRNA expression and localization at the molecular level (see sect. IIC).
Arrhythmic phenomena ultimately arise from biophysical events, beginning with ion channel-mediated processes in the cell surface membrane. A first group of studies examined a series of defined monogenic surface ion channel variants (see sects. III–VI). These recapitulated the arrhythmic phenotype in their corresponding clinical genetic arrhythmic conditions. This gave credence to their use as disease exemplars in physiological studies including explorations for possible therapeutic maneuvers. They fell into canonical groups that could be classified into effects relating to pacing (sect. III), cellular connectivity (sect. IV), AP excitation (sect. V), and recovery from depolarization as well as refractoriness (sect. VI). The last two recapitulated features of the BrS and the LQTS, respectively. The analysis accordingly began with the effects of compromised or prolonged Na+ channel function whether through alterations in the conducting Nav1.5 subunit itself, or its associated β-subunits or intracellular regulatory or structural regulatory proteins (sects. V and VI). These were followed by considering the physiological consequences of alterations in other ion channels carrying depolarizing Ca2+ or repolarizing K+ currents, and their different variants occurring variously in ventricular or atrial myocytes. The analysis of the murine systems recapitulating BrS further demonstrated longer term structural alterations recapitulating those found in human BrS and reproduced the time development of conduction change and arrhythmia in BrS and PCCD.
In each group, a systematic survey identified the observed arrhythmic properties with alterations in pacing, initiation, and conduction of, as well as, recovery from excitation. They quantified critical conditions in heart rate (HR), conduction velocity θ, APDs, and refractory periods ERPs. Detailed alterations in APD waveforms or recovery could modify the likelihood of reexcitation phenomena, potentially through effects upon Ca2+ current mediating EADs, and through effects on refractoriness. Arrhythmic substrate could arise from both temporal variations of these physiological parameters with extrasystolic provocation or with varying steady-state pacing rates, and spatial variations over the epicardial surface, across myocardial walls, and different cardiac regions and chambers. Genetic modifications of any particular molecular species impacted upon more than one of these physiological parameters. Conversely, any one physiological parameter was affected by alterations in more than one species of channel. These complex physiological dependences have translational implications for the application of therapeutic agents directed at specific molecular species in the management of arrhythmia.
A second group of canonical variants concerned intracellular, as opposed to surface ion channel, changes. Intracellular Ca2+ is the predominant ion species involved in excitation-contraction coupling. The consequent alterations in Ca2+ homeostasis in turn impact upon surface membrane channel action (sect. VII). Such alterations could take place through abnormal L-type Ca2+ channel-mediated Ca2+ entry, RyR2-mediated SR Ca2+ release, SERCA-mediated SR Ca2+ reuptake, and SR store levels. A range of acquired acute or chronic conditions could potentially alter the Ca2+ release process in both atria and ventricles. Similarly, murine systems with modified RyR2 or calsequestrin 2 provided genetic exemplars for an abnormal release of SR Ca2+. The resulting variations in [Ca2+]i could in turn influence the stability of surface membrane potentials through actions on electrogenic NCX mediating DAD phenomena potentially triggering extrasystolic activity. It could also potentially modify Na+ channel activity either acutely or through downregulation of its membrane expression reducing AP conduction velocity and thereby causing arrhythmic substrate. Modified intracellular Ca2+ could in turn drive metabolic alterations through its action on intracellular Ca2+ activated enzymes.
A third group of variations concerned alterations in cell metabolic state, with alterations in levels of particular intermediates affecting surface KATP and Na+ channels as well as RyR2-Ca2+ release channels (sect. VIII). The latter intermediates include cellular levels of ATP, redox [NAD+]/[NADH] state, and altered mitochondrial function generating reactive oxygen species. These could be exemplified in genetic models modifying the mitochondrial upregulator PGC1, as well as models manipulating [NAD+]/[NADH].
The final group of examplars involve pathological anatomical, cardiomyopathic, changes that would be expected to modify passive membrane capacitative, cm, or intracellular resisistive, ra, cable properties determining electrophysiological conduction (sect. IX). In addition to the fibrotic changes in the Na+ channels outlined previously, these could be exemplified by hearts containing variant signaling mechanisms.
Study of murine systems themselves thus provide insights concerning the function of particular ion channels at the tissue and organ levels, and the effects of their deficiencies or overexpression. They thereby contribute substantially to our understanding of the physiological basis of normal electrical activity and its propagation. This leads to developments in our fundamental understanding of the genesis and perpetuation of arrhythmia in terms of perturbations of these processes. A substantial number of exemplars additionally show phenotypic features that parallel the corresponding human clinical conditions. In such situations, they may then be useful in fundamental explorations of both diagnostic and risk assessment criteria and possible therapeutic interventions. However, as with all physiological models, species-related differences would be expected between mouse and humans. This concerns not only normal physiology, but also the pathological changes either leading to or resulting from phenotypic changes at the whole animal level. This is reflected in the small but important group of murine exemplars whose phenotypes showed marked contrasts from their corresponding human conditions. These continue to offer opportunities for specific investigations of such differences, and assessing the applicability or otherwise of specific biomarkers useful in risk stratification in mice to clinical management in humans. Nevertheless, particular readouts from gene targeted mouse models provide insights into broad physiological mechanisms underlying electrophysiological stability or arrhythmogenesis. It is the latter that may be the more useful for the identification of approaches to direct translation.
GRANTS
I thank the Medical Research Council, Wellcome Trust, McVeigh Benefaction, the British Heart Foundation, Helen Kirkland Trust, and Sudden Arrhythmic Death Syndrome-SADS UK for research support.
DISCLOSURES
No conflict of interest, financial or otherwise are declared by the author.
ACKNOWLEDGMENTS
I gratefully acknowledge the mentorship in my early physiological studies from Prof. Richard Adrian, to whose memory this review honors. The review benefited from discussions with Drs. Andrew Grace (Cambridge), Ming Lei (Oxford), Yanmin Zhang (Xi'an), Antonio Vidal-Puig (Cambridge), and Antony Jackson (Cambridge). Particular topics were discussed with M. Killeen, G. Thomas, I. N. Sabir, S. S. Hothi, L. Guzadhur, C. A. Martin, K. Jeevaratnam, G. D. K. Matthews, S. C. Salvage, S. Ahmad, H. Valli, H. R. Matthews, and J. A. Fraser.
Adddress for reprint requests and other correspondence: C. L.-H. Huang, Physiological Laboratory, Univ. of Cambridge, Downing Street, Cambridge CB2 3EG, UK (e-mail: clh11@cam.ac.uk).
REFERENCES
- 1.Abbott G. The KCNE2 K+ channel regulatory subunit: ubiquitous influence, complex pathobiology. Gene 569: 162–172, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97: 175–187, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Abriel H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol 48: 2–11, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Adebanjo OA, Anandatheerthavarada HK, Koval AP, Moonga BS, Biswas G, Sun L, Sodam BR, Bevis PJ, Huang CLH, Epstein S, Lai FA, Avadhani NG, Zaidi M. A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol 1: 409–414, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Adebanjo OA, Shankar VS, Pazianas M, Simon BJ, Lai FA, Huang CLH, Zaidi M. Extracellularly applied ruthenium red and cADP ribose elevate cytosolic Ca2+ in isolated rat osteoclasts. Am J Physiol Renal Fluid Electrolyte Physiol 270: F469–F475, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Adeniran I, Hancox J, Zhang H. In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Card Electrophysiol 4: 166, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adeniran I, El Harchi A, Hancox JC, Zhang H. Proarrhythmia in KCNJ2-linked short QT syndrome: insights from modelling. Cardiovasc Res 94: 66–76, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Adeniran I, McPate MJ, Witchel HJ, Hancox JC, Zhang H. Increased vulnerability of human ventricle to re-entrant excitation in hERG-linked variant 1 short QT syndrome. PLoS Comput Biol 7: e1002313, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adrian RH, Peachey LD. Reconstruction of the action potential of frog sartorius muscle. J Physiol 235: 103–131, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adsit GS, Vaidyanathan R, Galler CM, Kyle JW, Makielski JC. Channelopathies from mutations in the cardiac sodium channel protein complex. J Mol Cell Cardiol 61: 34–43, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahern CA, Payandeh J, Bosmans F, Chanda B. The hitchhiker's guide to the voltage-gated sodium channel galaxy. J Gen Physiol 147: 1–24, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn A, Freener C, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem 271: 2724–2730, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97: 1314–1322, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Ai X, Jiang A, Ke Y, Solaro RJ, Pogwizd SM. Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43. Cardiovasc Res 92: 106–114, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O'Rourke B, Akar FG, Tomaselli GF. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res 85: 454–463, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding WG, Zankov DP, Toyoda F, Matsuura H, Horie M, Sunagawa K. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J Am Coll Cardiol 45: 300–307, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Aizawa Y, Uchiyama H, Yamaura M, Nakayama T, Arita M. Effects of the ATP-sensitive K channel opener nicorandil on the QT interval and the effective refractory period in patients with congenital long QT syndrome. J Electrocardiol 31: 117–123, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Akar FG, O'Rourke B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol Ther 131: 287–294, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation 105: 1247–1253, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME, Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci USA 106: 12189–12194, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allessie M, Schotten U, Verheule S, Harks E. Gene therapy for repair of cardiac fibrosis: a long way to Tipperary. Circulation 111: 391–393, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Almers W, Stanfield PR, Stühmer W. Lateral distribution of sodium and potassium channels in frog skeletal muscle: measurements with a patch-clamp technique. J Physiol 336: 261–284, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almers W, Stanfield PR, Stühmer W. Slow changes in currents through sodium channels in frog muscle membrane. J Physiol 339: 253–271, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci USA 99: 17185–17190, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altomare C, Terragni B, Brioschi C, Milanesi R, Pagliuca C, Viscomi C, Moroni A, Baruscotti M, DiFrancesco D. Heteromeric HCN1-HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. J Physiol 549: 347–359, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflügers Arch 460: 223–237, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res 75: 854–861, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther 287: 996–1006, 1998. [PubMed] [Google Scholar]
- 29.Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther 106: 39–55, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ Res 114: 1453–1468, 2014. [DOI] [PubMed] [Google Scholar]
- 31.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference. Heart Rhythm 2: 429–440, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Antzelevitch C, Brugada P, Brugada J, Brugada R, Towbin JA, Nademanee K. Brugada syndrome: 1992–2002: a historical perspective. J Am Coll Cardiol 41: 1665–1671, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Brugada P, Brugada J, Brugada R. Brugada syndrome: from cell to bedside. Curr Probl Cardiol 30: 9–54, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115: 442–449, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov a Di Diego J, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 10: 1124–1152, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 69: 1427–1449, 1991. [DOI] [PubMed] [Google Scholar]
- 37.Antzelevitch C, Sun Z. Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J Am Coll Cardiol 28: 1836–1848, 1996. [DOI] [PubMed] [Google Scholar]
- 38.Antzelevitch C, Yan GX, Shimizu W. Transmural dispersion of repolarization and arrhythmogenicity: the Brugada syndrome versus the long QT syndrome. J Electrocardiol 32 Suppl: 158–165, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Antzelevitch C. Brugada syndrome: clinical, genetic, molecular, cellular and ionic aspects. Expert Rev Cardiovasc Ther 1: 177–185, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Anyukhovsky EP, Sosunov EA, Gainullin RZ, Rosen MR. The controversial M cell. J Cardiovasc Electrophysiol 10: 244–260, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Aon MA, Cortassa S, Akar FG, O'Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta 1762: 232–240, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aon MA, Cortassa S, Marbán E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278: 44735–44744, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab 1: 259–271, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci USA 103: 10086–10091, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacène E, Fromes Y, Toussaint M, Mura AM, Kelle DI, Amthor H, Isnard R, Malissen M, Schwartz K, Bonne G. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 14: 155–169, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Armoundas A, Osaka M, Mela T, Rosenbaum D, Ruskin J, Garan H, Cohen R. T-wave alternans and dispersion of the QT interval as risk stratification markers in patients susceptible to sustained ventricular arrhythmias. Am J Cardiol 82: 1127–1129, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Arnestad M, Vege A, Rognum TO. Evaluation of diagnostic tools applied in the examination of sudden unexpected deaths in infancy and early childhood. Forensic Sci Int 125: 262–268, 2002. [DOI] [PubMed] [Google Scholar]
- 48.Asano Y, Takashima S, Asakura M, Shintani Y, Liao Y, Minamino T, Asanuma H, Sanada S, Kim J, Ogai A, Fukushima T, Oikawa Y, Okazaki Y, Kaneda Y, Sato M, Miyazaki J, Kitamura S, Tomoike H, Kitakaze M, Hori M. Lamr1 functional retroposon causes right ventricular dysplasia in mice. Nat Genet 36: 123–130, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Asghar O, Alam U, Hayat SA, Aghamohammadzadeh R, Heagerty AM, Malik RA. Diabetes, obesity and atrial fibrillation: epidemiology, mechanisms and interventions. Curr Cardiol Rev 8: 253–264, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashino S, Watanabe I, Kofune M, Nagashima K, Ohkubo K, Okumura Y, Nakai T, Kasamaki Y, Hirayama A. Abnormal action potential duration restitution property in the right ventricular outflow tract in Brugada syndrome. Circ J 74: 664–670, 2010. [DOI] [PubMed] [Google Scholar]
- 51.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem 287: 19856–19869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asseman P, Berzin B, Desry D, Vilarem D, Durand P, Delmotte C, Sarkis EH, Lekieffre J, Thery C. Persistent sinus nodal electrograms during abnormally prolonged postpacing atrial pauses in sick sinus syndrome in humans: sinoatrial block vs overdrive suppression. Circulation 68: 33–41, 1983. [DOI] [PubMed] [Google Scholar]
- 53.Babij P, Askew G, Nieuwenhuijsen B, Su C. Inhibition of cardiac delayed rectifier K+ current by overexpression of the long-QT. Circ Res 83: 668–678, 1998. [DOI] [PubMed] [Google Scholar]
- 54.Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, Periasamy M. Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc Natl Acad Sci USA 104: 17867–17872, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson AJ, Hill AJ, Katus AH, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA 106: 2342–2347, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Bett GCL, Tsien RW, Rasmusson RL, Shamloo M. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci 108: 15432–15437, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bagwe S, Berenfeld O, Vaidya D, Morley GE, Jalife J. Altered right atrial excitation and propagation in connexin40 knockout mice. Circulation 112: 2245–2253, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies how to prioritize access to genetic testing. Circ Arrhythmia Electrophysiol 2: 6–15, 2009. [DOI] [PubMed] [Google Scholar]
- 59.Baker LC, London B, Choi BR, Koren G, Salama G. Enhanced dispersion of repolarization and refractoriness in transgenic mouse hearts promotes reentrant ventricular tachycardia. Circ Res 86: 396–407, 2000. [DOI] [PubMed] [Google Scholar]
- 60.Baker WL, White CM. Post-cardiothoracic surgery atrial fibrillation: A review of preventive strategies. Ann Pharmacother 41: 587–598, 2007. [DOI] [PubMed] [Google Scholar]
- 61.de Bakker JMT, Ho SY, Hocini M. Basic and clinical electrophysiology of pulmonary vein ectopy. Cardiovasc Res 54: 287–294, 2002. [DOI] [PubMed] [Google Scholar]
- 62.Balasubramaniam R, Chawla S, Grace AA, Huang CLH. Caffeine-induced arrhythmias in murine hearts parallel changes in cellular Ca2+ homeostasis. Am J Physiol Heart Circ Physiol 289: H1584–H1593, 2005. [DOI] [PubMed] [Google Scholar]
- 63.Balasubramaniam R, Chawla S, Mackenzie L, Schwiening CJ, Grace AA, Huang CLH. Nifedipine and diltiazem suppress ventricular arrhythmogenesis and calcium release in mouse hearts. Pflügers Arch 449: 150–158, 2004. [DOI] [PubMed] [Google Scholar]
- 64.Balasubramaniam R, Grace A, Saumarez R, Vandenberg J, Huang CLH. Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552: 535–546, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol 98: 149–160, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bannister ML, Thomas NL, Sikkel MB, Mukherjee S, Maxwell C, MacLeod KT, George CH, Williams AJ. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ Res 116: 1324–1335, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci USA 107: 12357–12362, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bao J, Wang J, Yao Y, Wang Y, Fan X, Sun K, He DS, Marcus FI, Zhang S, Hui R, Song L. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet 6: 552–556, 2013. [DOI] [PubMed] [Google Scholar]
- 69.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384: 78–80, 1996. [DOI] [PubMed] [Google Scholar]
- 70.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 subunit. Circ Res 83: 560–567, 1998. [DOI] [PubMed] [Google Scholar]
- 71.Bartos DC, Grandi E, Ripplinger CM. Ion channels in the heart. Compr Physiol 5: 1423–1464, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baruscotti M, Bottelli G, Milanesi R, DiFrancesco JC, DiFrancesco D. HCN-related channelopathies. Pflügers Arch 460: 405–415, 2010. [DOI] [PubMed] [Google Scholar]
- 73.Baruscotti M, Bucchi A, DiFrancesco D. Physiology and pharmacology of the cardiac pacemaker (”funny“) current. Pharmacol Ther 107: 59–79, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A, DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci USA 108: 1705–1710, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baruscotti M, DiFrancesco D, Robinson RB. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. J Physiol 492: 21–30, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baruscotti M, DiFrancesco D, Robinson RB. Na+ current contribution to the diastolic depolarization in newborn rabbit SA node cells. Am J Physiol Heart Circ Physiol 279: H2303–H2309, 2000. [DOI] [PubMed] [Google Scholar]
- 77.Baruscotti M, Westenbroek R, Catterall WA, DiFrancesco D, Robinson RB. The newborn rabbit sino-atrial node expresses a neuronal type I-like Na+ channel. J Physiol 498: 641–648, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol 476: 279–293, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basso C, Corrado D, Bauce B, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythmia Electrophysiol 5: 1233–1246, 2012. [DOI] [PubMed] [Google Scholar]
- 80.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 373: 1289–1300, 2009. [DOI] [PubMed] [Google Scholar]
- 81.Basso C, Fox PR, Meurs KM, Towbin JA, Spier AW, Calabrese F, Maron BJ, Thiene G. Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: a new animal model of human disease. Circulation 109: 1180–1185, 2004. [DOI] [PubMed] [Google Scholar]
- 82.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, Turrini P, Thiene G, Danieli GA, Nava A. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: Early diagnosis of asymptomatic carriers. J Am Coll Cardiol 40: 341–349, 2002. [DOI] [PubMed] [Google Scholar]
- 83.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118: 3893–3903, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beauchamp P, Choby C, Desplantez T, de Peyer K, Green K, Yamada KA, Weingart R, Saffitz JE, Kléber AG. Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ Res 95: 170–178, 2004. [DOI] [PubMed] [Google Scholar]
- 85.Beauchamp P, Yamada KA, Baertschi AJ, Green K, Kanter EM, Saffitz JE, Kléber AG. Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ Res 99: 1216–1224, 2006. [DOI] [PubMed] [Google Scholar]
- 86.Beery TA, Shah MJ, Benson DW. Genetic characterization of familial CPVT after 30 years. Biol Res Nurs 11: 66–72, 2009. [DOI] [PubMed] [Google Scholar]
- 87.Behr E, Wood DA, Wright M, Syrris P, Sheppard MN, Casey A, Davies MJ, Mckenna W. Cardiological assessent of first-degree relatives in sudden arrhythmic death syndrome. Lancet 362: 1457–1459, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC. Cardiac late Na+ current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 12: 440–448, 2015. [DOI] [PubMed] [Google Scholar]
- 89.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One 6: e28324, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes a C., Billman GE, Györke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res 110: 569–577, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 84: 387–395, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 110: 1731–1737, 2004. [DOI] [PubMed] [Google Scholar]
- 93.Bellocq C, Van Ginneken ACG, Bezzina CR, Alders M, Escande D, Mannens MMAM, Baró I, Wilde AAM. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109: 2394–2397, 2004. [DOI] [PubMed] [Google Scholar]
- 94.Benhorin J, Taub R, Goldmit M, Kerem B, Kass RS, Windman I, Medina A. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation 101: 1698–1706, 2000. [DOI] [PubMed] [Google Scholar]
- 95.Benito B, Brugada R, Brugada J, Brugada P. Brugada Syndrome. Prog Cardiovasc Dis 51: 1–22, 2008. [DOI] [PubMed] [Google Scholar]
- 96.Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R, Yue DT. Conservation of Ca2+/calmodulin regulation across Na+ and Ca2+ channels. Cell 157: 1657–1670, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact β-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res 101: 819–829, 2007. [DOI] [PubMed] [Google Scholar]
- 98.Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature 376: 683–685, 1995. [DOI] [PubMed] [Google Scholar]
- 99.Bennett V, Baines A. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81: 1353–1392, 2001. [DOI] [PubMed] [Google Scholar]
- 100.Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 112: 1019–1028, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Berlin JR, Cannell MB, Lederer WJ. Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ Res 65: 115–126, 1989. [DOI] [PubMed] [Google Scholar]
- 102.Berruezo A, Mont L, Nava S, Chueca E, Bartholomay E, Brugada J. Electrocardiographic recognition of the epicardial origin of ventricular tachycardias. Circulation 109: 1842–1847, 2004. [DOI] [PubMed] [Google Scholar]
- 103.Bers D. Excitation-Contraction Coupling and Cardiac Contractile Force (2nd ed). Dordrecht, The Netherlands: Kluwer Academic, 2001. [Google Scholar]
- 104.Bers D. Calcium and cardiac rhythms: physiological and pathophysiological. Circ Res 90: 14–17, 2002. [PubMed] [Google Scholar]
- 105.Bers DM, Morotti S. Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front Pharmacol 5: 144, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol 97 Suppl 1: I36–42, 2002. [DOI] [PubMed] [Google Scholar]
- 107.Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002. [DOI] [PubMed] [Google Scholar]
- 108.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol 37: 417–429, 2004. [DOI] [PubMed] [Google Scholar]
- 109.Bers DM. Cardiac ryanodine receptor phosphorylation: target sites and functional consequences. Biochem J 396: e1–3, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70: 23–49, 2008. [DOI] [PubMed] [Google Scholar]
- 111.Berul C. A gap in understanding the connection between connexins and cardiac conduction. J Cardiovasc Electrophysiol 10: 1376–1379, 1999. [DOI] [PubMed] [Google Scholar]
- 112.Berul CI, Christe ME, Aronovitz MJ, Seidman CE, Seidman JG, Mendelsohn ME. Electrophysiological abnormalities and arrhythmias in alpha MHC mutant familial hypertrophic cardiomyopathy mice. J Clin Invest 99: 570–576, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berul CI, McConnell BK, Wakimoto H, Moskowitz IP, Maguire CT, Semsarian C, Vargas MM, Gehrmann J, Seidman CE, Seidman JG. Ventricular arrhythmia vulnerability in cardiomyopathic mice with homozygous mutant Myosin-binding protein C gene. Circulation 104: 2734–2739, 2001. [DOI] [PubMed] [Google Scholar]
- 114.Bescond J, Bois P, Petit-Jacques J, Lenfant J. Characterization of an angiotensin-II-activated chloride current in rabbit sino-atrial cells. J Membr Biol 140: 153–161, 1994. [DOI] [PubMed] [Google Scholar]
- 115.Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van der Hout AH, Mannens MM, Wilde A. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res 85: 1206–1213, 1999. [DOI] [PubMed] [Google Scholar]
- 116.Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res 116: 1919–1936, 2015. [DOI] [PubMed] [Google Scholar]
- 117.Bezzina CR, Rook MB, Groenewegen WA, Herfst LJ, Van der Wal AC, Lam J, Jongsma HJ, Wilde AAM, Mannens MMAM. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res 92: 159–168, 2003. [DOI] [PubMed] [Google Scholar]
- 118.Bezzina CR, Rook MB, Wilde AM. Cardiac sodium channel and inherited arrhythmia syndromes. Cardiovasc Res 49: 257–271, 2001. [DOI] [PubMed] [Google Scholar]
- 119.Bhuiyan ZA, Van Den Berg MP, Van Tintelen JP, Bink-Boelkens MTE, Wiesfeld ACP, Alders M, Postma AV, Van Langen I, Mannens MMAM, Wilde AAM. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 116: 1569–1576, 2007. [DOI] [PubMed] [Google Scholar]
- 120.Bhupathy P, Babu GJ, Ito M, Periasamy M. Threonine-5 at the N-terminus can modulate sarcolipin function in cardiac myocytes. J Mol Cell Cardiol 47: 723–729, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bianchi L, Shen Z, Dennis AT, Priori SG, Napolitano C, Ronchetti E, Bryskin R, Schwartz PJ, Brown AM. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum Mol Genet 8: 1499–1507, 1999. [DOI] [PubMed] [Google Scholar]
- 122.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89: 847–885, 2009. [DOI] [PubMed] [Google Scholar]
- 123.Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol 180: 780–785, 1996. [DOI] [PubMed] [Google Scholar]
- 124.Biesmans L, MacQuaide N, Heinzel FR, Bito V, Smith GL, Sipido KR. Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS One 6: e25100, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Biet M, Morin N, Lessard-Beaudoin Graham RM, Duss S, Gagné J, Sanon N, Carmant L, Dumaine R. Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol 8: 912–920, 2015. [DOI] [PubMed] [Google Scholar]
- 126.Bingen BO, Neshati Z, Askar SFA, Kazbanov IV, Ypey DL, Panfilov AV, Schalij MJ, de Vries AAF, Pijnappels DA. Atrium-specific Kir3.x determines inducibility, dynamics and termination of fibrillation by regulating restitution-driven alternans. Circulation 128: 2732–2744, 2013. [DOI] [PubMed] [Google Scholar]
- 127.Blana A, Kaese S, Fortmller L, Laakmann S, Damke D, Van Bragt K, Eckstein J, Piccini I, Kirchhefer U, Nattel S, Breithardt G, Carmeliet P, Carmeliet E, Schotten U, Verheule S, Kirchhof P, Fabritz L. Knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm 7: 1862–1869, 2010. [DOI] [PubMed] [Google Scholar]
- 128.Blatter LA, Kockskämper J, Sheehan KA, Zima AV, Hüser J, Lipsius SL. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol 546: 19–31, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Blaustein M, Lederer W. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999. [DOI] [PubMed] [Google Scholar]
- 130.Boczek NJ, Ye D, Johnson EK, Wang W, Crotti L, Tester DJ, Dagradi F, Mizusawa Y, Torchio M, Alders M, Giudicessi JR, Wilde AAM, Schwartz PJ, Nerbonne JM, Ackerman MJ. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ Res 115: 460–469, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest 115: 3306–3317, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res 88: 1254–1258, 2001. [DOI] [PubMed] [Google Scholar]
- 133.Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 287: H1378–H1403, 2004. [DOI] [PubMed] [Google Scholar]
- 134.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev cell Biol 4: 733–738, 2003. [DOI] [PubMed] [Google Scholar]
- 135.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 590: 3291–3304, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res 47: 658–687, 2000. [DOI] [PubMed] [Google Scholar]
- 137.Boyett MR. “And the beat goes on.” The cardiac conduction system: the wiring system of the heart. Exp Physiol 94: 1035–1049, 2009. [DOI] [PubMed] [Google Scholar]
- 138.Brackenbury WJ, Isom LL. Na+ channel β subunits: overachievers of the ion channel family. Front Pharmacol September: 1–11, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev 17: 1969–1978, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brandes R, Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res 80: 82–87, 1997. [DOI] [PubMed] [Google Scholar]
- 141.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest 111: 1475–1486, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brillantes AMB, Ondriaš K, Scott A, Kobrinsky E, Ondriašová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77: 513–523, 1994. [DOI] [PubMed] [Google Scholar]
- 143.Brini M, Calì T, Ottolini D, Carafoli E. The plasma membrane calcium pump in health and disease. FEBS J 280: 5385–5397, 2013. [DOI] [PubMed] [Google Scholar]
- 144.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 112: 2602–2610, 2005. [DOI] [PubMed] [Google Scholar]
- 145.Brioschi C, Micheloni S, Tellez JO, Pisoni G, Longhi R, Moroni P, Billeter R, Barbuti A, Dobrzynski H, Boyett MR, DiFrancesco D, Baruscotti M. Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J Mol Cell Cardiol 47: 221–227, 2009. [DOI] [PubMed] [Google Scholar]
- 146.Brown DA, O'Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res 88: 241–249, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation 105: 73–78, 2002. [DOI] [PubMed] [Google Scholar]
- 148.Brugada J, Sassine A, Escande D, Masse C, Puech P. Effects of quinidine on ventricular repolarization. Eur Hear J 8: 1340–1345, 1987. [DOI] [PubMed] [Google Scholar]
- 149.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109: 30–35, 2004. [DOI] [PubMed] [Google Scholar]
- 150.Brugada R. Use of intravenous antiarrhythmics to identify concealed Brugada syndrome. Curr Control Trials Cardiovasc Med 1: 45–47, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brunet S, Aimond F, Li H, Guo W, Eldstrom J, Fedida D, Yamada a K, Nerbonne JM. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol 559: 103–120, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest 118: 2246–2259, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bucchi A, Barbuti A, DiFrancesco D, Baruscotti M. Funny current and cardiac rhythm: Insights from HCN knockout and transgenic mouse models. Front Physiol 3 July: 1–10, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bueno-Orovio A, Cherry EM, Fenton FH. Minimal model for human ventricular action potentials in tissue. J Theor Biol 253: 544–560, 2008. [DOI] [PubMed] [Google Scholar]
- 155.Bukowska A, Schild L, Keilhoff G, Hirte D, Neumann M, Gardemann A, Neumann KH, Röhl FW, Huth C, Goette A, Lendeckel U. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med 233: 558–574, 2008. [DOI] [PubMed] [Google Scholar]
- 156.Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpn E, Hu D, Desai M, Borggrefe M, Hissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laio R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C, Antzelevitch C. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7: 1872–1882, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58: 58–86, 2006. [DOI] [PubMed] [Google Scholar]
- 158.Bursac N, Parker KKK, Iravanian S, Tung L. Cardiomyocyte cultures with controlled macroscopic anisotropy: A model for functional electrophysiological studies of cardiac muscle. Circ Res 91: e45–e54, 2002. [DOI] [PubMed] [Google Scholar]
- 159.Cannon SC, Bean BP. Sodium channels gone wild: Resurgent current from neuronal and muscle channelopathies. J Clin Invest 120: 80–83, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 6: 507–512, 2005. [DOI] [PubMed] [Google Scholar]
- 161.Capel R, Terrar D. Cytosolic calcium ions exert a major influence on the firing rate and maintenance of pacemaker activity in guinea-pig sinus node. Front Physiol 6: 23, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res 61: 498–503, 1987. [DOI] [PubMed] [Google Scholar]
- 163.Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, Cui X. Targeted genome modification in mice using zinc-finger nucleases. Genetics 186: 451–459, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Carmeliet E. Induction and removal of inward-going rectification in sheep cardiac Purkinje fibres. J Physiol 327: 285–308, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 79: 917–1017, 1999. [DOI] [PubMed] [Google Scholar]
- 166.Carmeliet E. Intracellular Ca2+ concentration and rate adaptation of the cardiac action potential. Cell Calcium 35: 557–573, 2004. [DOI] [PubMed] [Google Scholar]
- 167.Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci USA 98: 2526–2531, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Casini S, Verkerk AO, van Borren MMGJ, van Ginneken ACG, Veldkamp MW, de Bakker JMT, Tan HL. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc Res 81: 72–81, 2009. [DOI] [PubMed] [Google Scholar]
- 169.Casis O, Olesen S, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol 69: 658–665, 2006. [DOI] [PubMed] [Google Scholar]
- 170.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13–25, 2000. [DOI] [PubMed] [Google Scholar]
- 171.Catterall WA. Voltage-gated sodium channels at sixty: structure, function and pathophysiology. J Physiol 590: 2577–2589, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cerrone M, Colombi B, Santoro M, di Barletta M, Scelsi M, Villani L, Napolitano C, Priori S. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res 96: e77–82, 2005. [DOI] [PubMed] [Google Scholar]
- 173.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HSV, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 129: 1092–1103, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, Van Der Nagel R, Hund T, Birchmeier W, Mohler P, Van Veen TA, Van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 95: 460–468, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 101: 1039–1048, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chaldoupi SM, Loh P, Hauer RNW, De Bakker JMT, Van Rijen HVM. The role of connexin40 in atrial fibrillation. Cardiovasc Res 84: 15–23, 2009. [DOI] [PubMed] [Google Scholar]
- 177.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 120: 629–645, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, DiFrancesco D, Baruscotti M, Longhi R, Anderson RH, Billeter R, Sharma V, Sigg DC, Boyett MR, Dobrzynski H. Molecular architecture of the human sinus node insights into the function of the cardiac pacemaker. Circulation 119: 1562–1575, 2009. [DOI] [PubMed] [Google Scholar]
- 179.Chandra R, Portbury AL, Ray A, Ream M, Groelle M, Chikaraishi DM. Beta1-adrenergic receptors maintain fetal heart rate and survival. Biol Neonate 89: 147–158, 2006. [DOI] [PubMed] [Google Scholar]
- 180.Chang CC, Acharfi S, Wu MH, Chiang FT, Wang JK, Sung TC, Chahine M. A novel SCN5A mutation manifests as a malignant form of long QT syndrome with perinatal onset of tachycardia/bradycardia. Cardiovasc Res 64: 268–278, 2004. [DOI] [PubMed] [Google Scholar]
- 181.Chauhan VS, Downar E, Nanthakumar K, Parker JD, Ross HJ, Chan W, Picton P. Increased ventricular repolarization heterogeneity in patients with ventricular arrhythmia vulnerability and cardiomyopathy: a human in vivo study. Am J Physiol Heart Circ Physiol 290: H79–H86, 2006. [DOI] [PubMed] [Google Scholar]
- 182.Chauhan VS, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res 86: 441–447, 2000. [DOI] [PubMed] [Google Scholar]
- 183.Chawla S, Skepper JN, Hockaday AR, Huang CLH. Calcium waves induced by hypertonic solutions in intact frog skeletal muscle fibres. J Physiol 536: 351–359, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XHT. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 119: 1940–1951, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 24: 4030–4042, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA, Campbell KP. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels, Supporting Material [Online]. Science 302: 1416–1418, 2003. [DOI] [PubMed] [Google Scholar]
- 187.Chen L, Kass RS. A-kinase anchoring protein 9 and IKS channel regulation. J Cardiovasc Pharmacol 58: 1–6, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci USA 104: 20990–20995, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392: 293–296, 1998. [DOI] [PubMed] [Google Scholar]
- 190.Chen SA, Hsieh MH, Tai CT, Tsai CF, Prakash VS, Yu WC, Hsu TL, Ding YA, Chang MS. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 100: 1879–1886, 1999. [DOI] [PubMed] [Google Scholar]
- 191.Chen T, Inoue M, Sheets MF. Reduced voltage dependence of inactivation in the SCN5A sodium channel mutation delF1617. Am J Physiol Heart Circ Physiol 288: H2666–H2676, 2005. [DOI] [PubMed] [Google Scholar]
- 192.Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintrón M, Scott JD, Santana LF. Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res 109: 255–261, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev 88: 1491–1545, 2008. [DOI] [PubMed] [Google Scholar]
- 194.Cheng J, Van Norstrand DW, Medeiros-Domingo A, Valdivia C, Tan B, Ye B, Kroboth S, Vatta M, Tester DJ, January CT, Makielski JC, Ackerman MJ. Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol 2: 667–676, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ, Makielski JC. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J Mol Cell Cardiol 61: 102–110, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chiamvimonvat N, Kargacin ME, Clark RB, Duff HJ. Effects of intracellular calcium on sodium current density in cultured neonatal rat cardiac myocytes. J Physiol 483: 307–318, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cho HS, Takano M, Noma A. The electrophysiological properties of spontaneously beating pacemaker cells isolated from mouse sinoatrial node. J Physiol 550: 169–180, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Choi EK, Chang PC, Lee YS, Lin SF, Zhu W, Maruyama M, Fishbein MC, Chen Z, Rubart-von der Lohe M, Field LJ, Chen P-S. Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-β1. Circ J 76: 1354–1462, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC. Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res 101: 617–626, 2007. [DOI] [PubMed] [Google Scholar]
- 200.Chopra N, Knollmann BC. Genetics of sudden cardiac death syndromes. Curr Opin Cardiol 26: 196–203, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Christ T, Boknik P, Wöhrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110: 2651–2657, 2004. [DOI] [PubMed] [Google Scholar]
- 203.Christie A, Sharma VK, Sheu SS. Mechanism of extracellular ATP-induced increase of cytosolic Ca2+ concentration in isolated rat ventricular myocytes. J Physiol 445: 369–388, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J 77: 2930–2941, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem 280: 17227–17234, 2005. [DOI] [PubMed] [Google Scholar]
- 206.Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol Rev 85: 33–47, 2005. [DOI] [PubMed] [Google Scholar]
- 207.Clancy CE, Kurokawa J, Tateyama M, Wehrens XHT, Kass RS. K(+) channel structure-activity relationships and mechanisms of drug-induced QT prolongation Annu Rev Pharmacol Toxicol 43: 441–461, 2003. [DOI] [PubMed] [Google Scholar]
- 208.Clancy CE, Rudy Y. Na(+) channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation 105: 1208–1213, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Clark RB, Bouchard RA, Salinas-Stefanon E, Sanchez-Chapula J, Giles WR. Heterogeneity of action potential waveforms and potassium currents in rat ventricle. Cardiovasc Res 27: 1795–1799, 1993. [DOI] [PubMed] [Google Scholar]
- 210.Clark RB, Mangoni ME, Lueger A, Couette B, Nargeot J, Giles WR. A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am J Physiol Heart Circ Physiol 286: H1757–H1766, 2004. [DOI] [PubMed] [Google Scholar]
- 211.Clark RB, Tremblay A, Melnyk P, Allen BG, Giles WR, Fiset C. T-tubule localization of the inward-rectifier K(+) channel in mouse ventricular myocytes: a role in K(+) accumulation. J Physiol 537: 979–992, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Clusin WT. Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am J Physiol Heart Circ Physiol 294: H1–H10, 2008. [DOI] [PubMed] [Google Scholar]
- 213.Coade SB, Pearson JD. Metabolism of adenine nucleotides in human blood. Circ Res 65: 531–537, 1989. [DOI] [PubMed] [Google Scholar]
- 214.Cohen JD, Neaton JD, Prineas RJ, Daniels KA. Diuretics, serum potassium and ventricular arrhythmias in the Multiple Risk Factor Intervention Trial. Am J Cardiol 60: 548–554, 1987. [DOI] [PubMed] [Google Scholar]
- 215.Colquitt JL, Mendes D, Clegg AJ, Harris P, Cooper K, Picot J, Bryant J. Implantable cardioverter defibrillators for the treatment of arrhythmias and cardiac resynchronisation therapy for the treatment of heart failure: systematic review and economic evaluation. Health Technol Assess 18: 1–560, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Conrath CE, Wilders R, Coronel R, De Bakker JMT, Taggart P, De Groot JR, Opthof T. Intercellular coupling through gap junctions masks M cells in the human heart. Cardiovasc Res 62: 407–414, 2004. [DOI] [PubMed] [Google Scholar]
- 217.Coronel R, de Bakker JMT, Wilms-Schopman FJG, Opthof T, Linnenbank AC, Belterman CN, Janse MJ. Monophasic action potentials and activation recovery intervals as measures of ventricular action potential duration: Experimental evidence to resolve some controversies. Heart Rhythm 3: 1043–1050, 2006. [DOI] [PubMed] [Google Scholar]
- 218.Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJG, Verkerk AO, De Groot JR, Bhuiyan Z, Bezzina CR, Veldkamp MW, Linnenbank AC, Van Der Wal AC, Tan HL, Brugada P, Wilde AAM, De Bakker JMT. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112: 2769–2777, 2005. [DOI] [PubMed] [Google Scholar]
- 219.Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 339: 364–369, 1998. [DOI] [PubMed] [Google Scholar]
- 220.Corrado D, Nava A, Buja G, Martini B, Fasoli G, Oselladore L, Turrini P, Thiene G. Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J Am Coll Cardiol 27: 443–448, 1996. [DOI] [PubMed] [Google Scholar]
- 221.Costa ATD, Pierre SV, Cohen MV, Downey JM, Garlid KD. cGMP signaling in pre- and post-conditioning: the role of mitochondria. Cardiovasc Res 77: 344–352, 2008. [DOI] [PubMed] [Google Scholar]
- 222.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res 89: 265–272, 2011. [DOI] [PubMed] [Google Scholar]
- 223.Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm 4: 161–166, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL, Schwartz PJ. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation 112: 1251–1258, 2005. [DOI] [PubMed] [Google Scholar]
- 225.Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P, Koval OM, Li J, Gudmundsson H, Gumina RJ, Karck M, Schott JJ, Probst V, Le Marec H, Anderson ME, Dobrev D, Wehrens XHT, Mohler PJ. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124: 1212–1222, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol 49: 25–32, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res 100: 391–398, 2007. [DOI] [PubMed] [Google Scholar]
- 228.Curran J, Louch W. Linking ryanodine receptor Ca2+ leak and Na(+) current in heart: a day in the life of flecainide. Acta Physiol 214: 300–302, 2015. [DOI] [PubMed] [Google Scholar]
- 229.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80: 795–803, 1995. [DOI] [PubMed] [Google Scholar]
- 230.Curtis MJ, Pugsley MK, Walker MJ. Endogenous chemical mediators of ventricular arrhythmias in ischaemic heart disease. Cardiovasc Res 27: 703–719, 1993. [DOI] [PubMed] [Google Scholar]
- 231.Cusdin FS, Nietlispach D, Maman J, Dale TJ, Powell AJ, Clare JJ, Jackson AP. The sodium channel β3-subunit induces multiphasic gating in NaV1.3 and affects fast inactivation via distinct intracellular regions. J Biol Chem 285: 33404–33412, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Cutler MJ, Plummer BN, Wan X, Sun QA, Hess D, Liu H, Deschenes I, Rosenbaum DS, Stamler JS, Laurita KR. Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc Natl Acad Sci USA 109: 18186–18191, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 94: 8121–8126, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke a D., Calkins H. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation 112: 3823–3832, 2005. [DOI] [PubMed] [Google Scholar]
- 235.Damiano BP, Rosen MR. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation 69: 1013–1025, 1984. [DOI] [PubMed] [Google Scholar]
- 236.Danik S, Cabo C, Chiello C, Kang S, Wit AL, Coromilas J. Correlation of repolarization of ventricular monophasic action potential with ECG in the murine heart. Am J Physiol Heart Circ Physiol 283: H372–H381, 2002. [DOI] [PubMed] [Google Scholar]
- 237.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res 95: 1035–1041, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117: 1927–1935, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Das S, Gilchrist J, Bosmans F, Van Petegem F. Binary architecture of the Nav1.2-β2 signaling complex. Elife 19: e10960, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Dautova Y, Zhang Y, Grace AA, Huang CLH. Atrial arrhythmogenic properties in wild-type and Scn5a+/− murine hearts. Exp Physiol 95: 994–1007, 2010. [DOI] [PubMed] [Google Scholar]
- 241.Dautova Y, Zhang Y, Sabir I, Grace AA, Huang CLH. Atrial arrhythmogenesis in wild-type and Scn5a+/ΔKPQ murine hearts modelling LQT3 syndrome. Pflügers Arch 458: 443–457, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Davare MA, Horne MC, Hell JW. Protein phosphatase PP2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem 275: 39710–39717, 2000. [DOI] [PubMed] [Google Scholar]
- 243.Davidenko JM, Antzelevitch C. Electrophysiological mechanisms underlying rate-dependent changes of refractoriness in normal and segmentally depressed canine Purkinje fibers. The characteristics of post-repolarization refractoriness. Circ Res 58: 257–268, 1986. [DOI] [PubMed] [Google Scholar]
- 244.Davidenko JM, Salomonsz R, Pertsov AM, Baxter WT, Jalife J. Effects of pacing on stationary reentrant activity. Theoretical and experimental study. Circ Res 77: 1166–1179, 1995. [DOI] [PubMed] [Google Scholar]
- 245.Davies L, Jin J, Shen W, Tsui H, Shi Y, Wang Y, Zhang Y, Hao G, Wu J, Chen S, Fraser JA, Dong N, Christoffels V, Ravens U, Huang CLH, Zhang H, Cartwright EJ, Wang X, Lei M. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc 3: 1–19, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Davis J, Maillet M, Miano JM, Molkentin JD. Lost in transgenesis: a user's guide for genetically manipulating the mouse in cardiac research. Circ Res 111: 761–777, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Davis LM, Rodefeld ME, Green K, Beyer EC, Saffitz JE. Gap junction protein phenotypes of the human heart and conduction system. J Cardiovasc Electrophysiol 6: 813–822, 1995. [DOI] [PubMed] [Google Scholar]
- 248.Davis RP, Casini S, Van Den Berg CW, Hoekstra M, Remme CA, Dambrot C, Salvatori D, Oostwaard Van DW, Wilde AAM, Bezzina CR, Verkerk AO, Freund C, Mummery CL. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125: 3079–3091, 2012. [DOI] [PubMed] [Google Scholar]
- 249.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XHT, Anderson ME, Hund TJ, Mohler PJ. CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 9: 2034–2041, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 107: 700–714, 2010. [DOI] [PubMed] [Google Scholar]
- 251.Delmar M. Desmosome-ion channel interactions and their possible role in arrhythmogenic cardiomyopathy. Pediatr Cardiol 33: 975–979, 2012. [DOI] [PubMed] [Google Scholar]
- 252.Demion M, Bois P, Launay P, RG. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res 73: 531–538, 2007. [DOI] [PubMed] [Google Scholar]
- 253.Demolombe S, Baro I, Pereon Y, Bliek J, Mohammad PR, Pollard H, Morid S, Mannens M, Wilde A, Barhanin J, Charpentier F, Escande D. A dominant negative isoform of the long QT syndrome 1 gene product. J Biol Chem 273: 6837–6843, 1998. [DOI] [PubMed] [Google Scholar]
- 254.Demolombe S, Lande G, Charpentier F, van Roon M, van den Hoff Toumaniantz G MJ, Baro I, Guihard G, Le Berre N, Corbier A, de Bakker J, Opthof T, Wilde A, Moorman A, Escande D. Transgenic mice overexpressing human KvLQT1 dominant-negative isoform. Part I: phenotypic characterisation. Cardiovasc Res 50: 314–327, 2001. [DOI] [PubMed] [Google Scholar]
- 255.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115: 61–70, 2003. [DOI] [PubMed] [Google Scholar]
- 256.Denyer JC, Brown HF. Pacemaking in rabbit isolated sino-atrial node cells during Cs+ block of the hyperpolarization-activated current if. J Physiol 429: 401–409, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori SG. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci USA 110: 4291–4296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.DeSantiago J, Bare DJ, Ke Y, Sheehan KA, Solaro RJ, Banach K. Functional integrity of the T-tubular system in cardiomyocytes depends on p21-activated kinase 1. J Mol Cell Cardiol 60: 121–128, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.DeSantiago J, Bare DJ, Xiao L, Ke Y, Solaro RJ, Banach K. p21-Activated kinase1 (Pak1) is a negative regulator of NADPH-oxidase 2 in ventricular myocytes. J Mol Cell Cardiol 67: 77–85, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Deschênes I, Neyroud N, DiSilvestre D, Marbán E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ Res 90: E49–E57, 2002. [DOI] [PubMed] [Google Scholar]
- 261.Dhar MJ, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channel alpha and beta subunits in rat and mouse cardiac myocytes. Circulation 103: 1303–1310, 2001. [DOI] [PubMed] [Google Scholar]
- 262.Dhillon PS, Gray R, Kojodjojo P, Jabr R, Chowdhury R, Fry CH, Peters NS. Relationship between gap-junctional conductance and conduction velocity in mammalian myocardium. Circ Arrhythmia Electrophysiol 6: 1208–1214, 2013. [DOI] [PubMed] [Google Scholar]
- 263.Diaz ME, Trafford AW, O'Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol 501: 3–16, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dibb KM, Eisner DA, Trafford AW. Regulation of systolic [Ca2+]i and cellular Ca2+ flux balance in rat ventricular myocytes by SR Ca2+, L-type Ca2+ current and diastolic [Ca2+]i. J Physiol 585: 579–592, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Dipolo R. Sodium/calcium exchanger: influence of metabolic regulation on ion carrier interactions. Physiol Rev 86: 155–203, 2006. [DOI] [PubMed] [Google Scholar]
- 266.Dirksen WP, Lacombe VA, Chi M, Kalyanasundaram A, Viatchenko-Karpinski S, Terentyev D, Zhou Z, Vedamoorthyrao S, Li N, Chiamvimonvat N, Carnes CA, Franzini-Armstrong C, Györke S, Periasamy M. A mutation in calsequestrin, CASQ2D307H, impairs Sarcoplasmic Reticulum Ca2+ handling and causes complex ventricular arrhythmias in mice. Cardiovasc Res 75: 69–78, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4.3 K+ channel in ventricular muscle A molecular correlate for the transient outward current. Circ Res 79: 659–668, 1996. [DOI] [PubMed] [Google Scholar]
- 268.Dixon RE, Cheng EP, Mercado JL, Santana LF. L-Type Ca2+ channel function during Timothy Syndrome. Trends Cardiovasc Med 22: 72–76, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Dobrev D, Nattel S. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J Cardiovasc Pharmacol 52: 293–299, 2008. [DOI] [PubMed] [Google Scholar]
- 270.Dobrev D, Wehrens XHT. Calmodulin kinase II, sarcoplasmic reticulum Ca2+ leak, and atrial fibrillation. Trends Cardiovasc Med 20: 30–34, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Dobrev D. Editorial: ion channel portrait of the human sinus node: useful for a better understanding of sinus node function and dysfunction in humans? Circulation 119: 1556–1558, 2009. [DOI] [PubMed] [Google Scholar]
- 272.Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115: 1921–1932, 2007. [DOI] [PubMed] [Google Scholar]
- 273.Doerr T, Denger R, Doerr A, Trautwein W. Ionic currents contributing to the action potential in single ventricular myocytes of the guinea pig studied with action potential clamp. Pflügers Arch 416: 230–237, 1990. [DOI] [PubMed] [Google Scholar]
- 274.Dorenkamp M, Morguet AJ, Sticherling C, Behrens S, Zabel M. Long-term prognostic value of restitution slope in patients with ischemic and dilated cardiomyopathies. PLoS One 8: e54768, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Dower G. In defence of the intrinsic deflection. Br Heart J 24: 55–60, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Drici MD, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G, Barhanin J. Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ Res 83: 95–102, 1998. [DOI] [PubMed] [Google Scholar]
- 277.Drum BML, Dixon RE, Yuan C, Cheng EP, Santana LF. Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J Mol Cell Cardiol 66: 63–71, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Du J, Liu J, Feng HZ, Hossain MM, Gobara N, Zhang C, Li Y, Jean-Charles PY, Jin JP, Huang XP. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol 294: H2604–H2613, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Du J, Zhang C, Liu J, Sidky C, Huang XP. A point mutation (R192H) in the C-terminus of human cardiac troponin I causes diastolic dysfunction in transgenic mice. Arch Biochem Biophys 456: 143–150, 2006. [DOI] [PubMed] [Google Scholar]
- 280.Du XJ, Feng X, Gao XM, Tan TP, Kiriazis H, Dart AM. I(f) channel inhibitor ivabradine lowers heart rate in mice with enhanced sympathoadrenergic activities. Br J Pharmacol 142: 107–112, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Duehmke RM, Pearcey S, Stefaniak JD, Guzadhur L, Jeevaratnam K, Costopoulos C, Pedersen TH, Grace AA, Huang CLH. Altered re-excitation thresholds and conduction of extrasystolic action potentials contribute to arrhythmogenicity in murine models of long QT syndrome. Acta Physiol 206: 164–177, 2012. [DOI] [PubMed] [Google Scholar]
- 282.Duff HJ, Offord J, West J, Catterall WA. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol Pharmacol 42: 570–574, 1992. [PubMed] [Google Scholar]
- 283.Duffy HS, Fort AG, Spray DC. Cardiac connexins: genes to nexus. Adv Cardiol 42: 1–17, 2006. [DOI] [PubMed] [Google Scholar]
- 284.Dumotier BM. A straightforward guide to the basic science behind arrhythmogenesis. Heart 100: 1907–1915, 2014. [DOI] [PubMed] [Google Scholar]
- 285.Dun W, Boyden PA. Aged atria: electrical remodeling conducive to atrial fibrillation. J Interv Card Electrophysiol 25: 9–18, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Eckardt D, Theis M, Degen J, Ott T, Van Rijen HVM, Kirchhoff S, Kim JS, De Bakker JMT, Willecke K. Functional role of connexin43 gap junction channels in adult mouse heart assessed by inducible gene deletion. J Mol Cell Cardiol 36: 101–110, 2004. [DOI] [PubMed] [Google Scholar]
- 287.Eckardt L, Bruns HJ, Paul M, Kirchhof P, Schulze-Bahr E, Wichter T, Breithardt G, Borggrefe M, Haverkamp W. Body surface area of ST elevation and the presence of late potentials correlate to the inducibility of ventricular tachyarrhythmias in Brugada syndrome. J Cardiovasc Electrophysiol 13: 742–749, 2002. [DOI] [PubMed] [Google Scholar]
- 288.Eckardt L, Haverkamp W, Borggrefe M, Breithardt G. Experimental models of torsade de pointes. Cardiovasc Res 39: 178–193, 1998. [DOI] [PubMed] [Google Scholar]
- 289.Eckardt L. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada Syndrome. Circulation 111: 257–263, 2005. [DOI] [PubMed] [Google Scholar]
- 290.Eckstein J, Verheule S, de Groot N, Allessie M, Schotten U. Mechanisms of perpetuation of atrial fibrillation in chronically dilated atria. Prog Biophys Mol Biol 97: 435–451, 2008. [DOI] [PubMed] [Google Scholar]
- 291.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD. Non-Equilibrium Reactivation of Na(+) Current Drives Early Afterdepolarizations in Mouse Ventricle. Circ. Arrhythm Electrophysiol 7: 1205–1213, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Efimov IR, Nikolski VP, Salama G. Optical imaging of the heart. Circ Res 95: 21–33, 2004. [DOI] [PubMed] [Google Scholar]
- 293.Eigenthaler M, Engelhardt S, Schinke B, Kobsar A, Schmitteckert E, Gambaryan S, Engelhardt CM, Krenn V, Eliava M, Jarchau T, Lohse MJ, Walter U, Hein L. Disruption of cardiac Ena-VASP protein localization in intercalated disks causes dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 285: H2471–H2481, 2003. [DOI] [PubMed] [Google Scholar]
- 294.Eldstrom J, Fedida D. The voltage-gated channel accessory protein KCNE2: multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev Mol Med 13: e38, 2011. [DOI] [PubMed] [Google Scholar]
- 295.El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A, Hatem SN. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res 104: 758–769, 2009. [DOI] [PubMed] [Google Scholar]
- 296.Eloff BC, Lerner DL, Yamada a K, Schuessler RB, Saffitz JE, Rosenbaum DS. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc Res 51: 681–690, 2001. [DOI] [PubMed] [Google Scholar]
- 297.Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev Biol 327: 376–385, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Cell Physiol 245: C1–C14, 1983. [DOI] [PubMed] [Google Scholar]
- 299.Fabritz L, Hoogendijk MG, Scicluna BP, Van Amersfoorth SCM, Fortmueller L, Wolf S, Laakmann S, Kreienkamp N, Piccini I, Breithardt G, Ruiz Noppinger P, Witt H, Ebnet K, Wichter T, Levkau B, Franke WW, Pieperhoff S, De Bakker JMT, Coronel R, Kirchhof P. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J Am Coll Cardiol 57: 740–750, 2011. [DOI] [PubMed] [Google Scholar]
- 300.Fabritz L, Kirchhof P, Fortmüller L, Auchampach JA, Baba HA, Breithardt G, Neumann J, Boknik P, Schmitz W. Gene dose-dependent atrial arrhythmias, heart block, and brady-cardiomyopathy in mice overexpressing A3 adenosine receptors. Cardiovasc Res 62: 500–508, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Fabritz L, Kirchhof P, Franz MR, Eckardt L, Mönnig G, Milberg P, Breithardt G, Haverkamp W. Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol 98: 25–32, 2003. [DOI] [PubMed] [Google Scholar]
- 302.Faggioni M, Hwang HS, van der Werf C, Nederend I, Kannankeril PJ, Wilde AAM, Knollmann BC. Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res 112: 689–697, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D, Knollmann BC. Suppression of Spontaneous Ca Elevations Prevents Atrial Fibrillation in Calsequestrin 2-Null Hearts. Circ Arrhythm Electrophysiol 7: 313–320, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Fahmi AI, Patel M, Stevens EB, Fowden AL, John JE, Lee K, Pinnock R, Morgan K, Jackson AP, Vandenberg JI. The sodium channel beta-subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J Physiol 537: 693–700, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Faivre JF, Findlay I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim Biophys Acta 1029: 167–172, 1990. [DOI] [PubMed] [Google Scholar]
- 306.Fan JS, Yuan Y, Palade P. Kinetic effects of FPL 64176 on L-type Ca2+ channels in cardiac myocytes [Online]. Naunyn-Schmiedebergs Arch Pharmacol 361: 465–476, 2000. [DOI] [PubMed] [Google Scholar]
- 307.Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnáiz-Cot JJ, Rosa AO, Matzkies M, Dittmann S, Stone SL, Linke M, Zechner U, Beyer V, Christian H, Rosenkranz S, Klauke B, Abdul S, Haverkamp W, Pfitzer G, Farr M, Morad M, Milting H, Hescheler J, Šaric T, Nguemo F, Matzkies M, Dittmann S, Stone SL, Linke M, Zechner U, Beyer V, Hennies HC, Rosenkranz S, Klauke B, Parwani AS, Haverkamp W, Pfitzer G, Farr M, Cleemann L, Morad M, Milting H, Hescheler J, Saric T, Christian H, Rosenkranz S, Klauke B, Abdul S, Haverkamp W, Pfitzer G, Farr M, Morad M, Milting H, Hescheler J, Šaric T. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem 28: 579–592, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Felipe A, Knittle TJ, Doyle KL, Snyders DJ, Tamkun MM. Differential expression of Isk mRNAs in mouse tissue during development and pregnancy. Am J Physiol Cell Physiol 267: C700–C705, 1994. [DOI] [PubMed] [Google Scholar]
- 309.Fenelon G, Shepard R, Stambler B. Focal origin of atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol 14: 1093–1102, 2003. [DOI] [PubMed] [Google Scholar]
- 310.Fenske S, Krause SC, Hassan SIH, Becirovic E, Auer F, Bernard R, Kupatt C, Lange P, Ziegler T, Wotjak CT, Zhang H, Hammelmann V, Paparizos C, Biel M, Wahl-Schott CA. Sick sinus syndrome in HCN1-deficient mice. Circulation 128: 2585–2594, 2013. [DOI] [PubMed] [Google Scholar]
- 311.Fenske S, Mader R, Scharr A, Paparizos C, Cao-Ehlker X, Michalakis S, Shaltiel L, Weidinger M, Stieber J, Feil S, Feil R, Hofmann F, Wahl-Schott C, Biel M. HCN3 contributes to the ventricular action potential waveform in the murine heart. Circ Res 109: 1015–1023, 2011. [DOI] [PubMed] [Google Scholar]
- 312.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest 101: 2415–2426, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov 2: 439–447, 2003. [DOI] [PubMed] [Google Scholar]
- 314.Ferrantini C, Coppini R, Scellini B, Ferrara C, Pioner JM, Mazzoni L, Priori S, Cerbai E, Tesi C, Poggesi C. R4496C RyR2 mutation impairs atrial and ventricular contractility. J Gen Physiol 147: 39–52, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Ferron L, Capuano V, Deroubaix E, Coulombe A, Renaud JF. Functional and molecular characterization of a T-type Ca2+ channel during fetal and postnatal rat heart development. J Mol Cell Cardiol 34: 533–546, 2002. [DOI] [PubMed] [Google Scholar]
- 316.Finck BN, Kelly DP. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Fiset C, Clark RB, Larsen TS, Giles WR. A rapidly activating sustained K(+) current modulates repolarization and excitation-contraction coupling in adult mouse ventricle. J Physiol 504: 557–563, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Fish J, Antzelevitch C. Cellular and ionic basis for the sex-related difference in the manifestation of the Brugada syndrome and progressive conduction disease phenotypes. J Electrocardiol 36 Suppl: 173–179, 2003. [DOI] [PubMed] [Google Scholar]
- 319.Fish JM, Welchons DR, Kim YS, Lee SH, Ho WK, Antzelevitch C. Dimethyl lithospermate B, an extract of Danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation 113: 1393–1400, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Flagg TP, Kurata HT, Masia R, Caputa G, Magnuson MA, Lefer DJ, Coetzee WA, Nichols CG. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ Res 103: 1458–1465, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Folco E, Mathur R, Mori Y, Buckett P, Koren G. A cellular model for long QT syndrome. Trapping of heteromultimeric complexes consisting of truncated Kv11 potassium channel polypeptides and native Kv14 and Kv15 channels in the endoplasmic reticulum. J Biol Chem 272: 26505–26510, 1997. [DOI] [PubMed] [Google Scholar]
- 322.Forbes M, Hawkey L, Sperelakis N. The transverse-axial tubular system (TATS) of mouse myocardium: its morphology in the developing and adult animal. Am J Anat 170: 143–162, 1984. [DOI] [PubMed] [Google Scholar]
- 323.Fosset M, De Weille JR, Green RD, Schmid-Antomarchi H, Lazdunski M. Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. J Biol Chem 263: 7933–7936, 1988. [PubMed] [Google Scholar]
- 324.Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev 96: 177–252, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Fozzard HA. Afterdepolarizations and triggered activity. Basic Res Cardiol 87 Suppl 2: 105–113, 1992. [DOI] [PubMed] [Google Scholar]
- 326.Frank M, Eiberger B, Janssen-Bienhold U, de Sevilla Muller LP, Tjarks A, Kim JS, Maschke S, Dobrowolski R, Sasse P, Weiler R, Fleischmann BK, Willecke K. Neuronal connexin-36 can functionally replace connexin-45 in mouse retina but not in the developing heart. J Cell Sci 123: 3605–3615, 2010. [DOI] [PubMed] [Google Scholar]
- 327.Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, Bauersachs J. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res 57: 749–756, 2003. [DOI] [PubMed] [Google Scholar]
- 328.Franz M. Current status of monophasic action potential recording: theories, measurements and interpretations. Cardiovasc Res 41: 25–40, 1999. [DOI] [PubMed] [Google Scholar]
- 329.Franz MR, Chin MC, Sharkey HR, Griffin JC, Scheinman MM. A new single catheter technique for simultaneous measurement of action potential duration and refractory period in vivo. J Am Coll Cardiol 16: 878–886, 1990. [DOI] [PubMed] [Google Scholar]
- 330.Fraser JA, Huang CLH, Pedersen TH. Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J Gen Physiol 138: 95–116, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Fraser JA, Huang CLH. A quantitative analysis of cell volume and resting potential determination and regulation in excitable cells. J Physiol 559: 459–478, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Fraser JA, Huang CLH. Quantitative techniques for steady-state calculation and dynamic integrated modelling of membrane potential and intracellular ion concentrations. Prog Biophys Mol Biol 94: 336–372, 2007. [DOI] [PubMed] [Google Scholar]
- 333.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol 148: 16–24, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Frommeyer G, Eckardt L. Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol 13: 36–47, 2016. [DOI] [PubMed] [Google Scholar]
- 335.Fujimoto Y, Morita H, Fukushima KK, Ohe T. Nicorandil abolished repolarisation alternans in a patient with idiopathic long QT syndrome. Heart 82: e8, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, Demolombe S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 582: 675–693, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem 276: 21425–21433, 2001. [DOI] [PubMed] [Google Scholar]
- 338.Galimberti ES, Knollmann BC. Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin. J Mol Cell Cardiol 51: 760–768, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Gallicano GI, Bauer C, Fuchs E. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development 128: 929–941, 2001. [DOI] [PubMed] [Google Scholar]
- 340.Gao J, Mathias R, Cohen I, Baldo G. Isoprenaline, Ca2+ and the Na+-K+ pump in guinea-pig ventricular myocytes. J Physiol 449: 689–704, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Gao J, Mathias RT, Cohen IS, Shi J, Baldo GJ. The effects of beta-stimulation on the Na+-K+ pump current-voltage relationship in guinea-pig ventricular myocytes. J Physiol 494: 697–708, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 116: 2012–2021, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Garrey WE. The nature of fibrillary contraction of the heart. Its relation to tissue mass and form. Am J Physiol 33: 397–414, 1914. [Google Scholar]
- 344.Gasparini M, Priori SG, Mantica M, Napolitano C, Galimberti P, Ceriotti C, Simonini S. Flecainide test in Brugada syndrome: a reproducible but risky tool. Pacing Clin Electrophysiol 26: 338–341, 2003. [DOI] [PubMed] [Google Scholar]
- 345.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 99: 407–414, 2006. [DOI] [PubMed] [Google Scholar]
- 346.Ge J, Sun A, Paajanen V, Wang SS, Su C, Yang Z, Li Y, Jia J, Wang KK, Zou Y, Gao L, Fan Z. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol 1: 83–92, 2008. [DOI] [PubMed] [Google Scholar]
- 347.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science 272: 731–734, 1996. [DOI] [PubMed] [Google Scholar]
- 348.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res 93: 531–540, 2003. [DOI] [PubMed] [Google Scholar]
- 349.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott a D, Lerman BB, Markowitz SM, Ellinor PT, MacRae a C, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36: 1162–1164, 2004. [DOI] [PubMed] [Google Scholar]
- 350.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ghais N, Zhang Y, Mistry B, Grace A, Huang CH. Anti-arrhythmic effects of cyclopiazonic acid in Langendorff-perfused murine hearts. Prog Biophys Mol Biol 98: 281–288, 2008. [DOI] [PubMed] [Google Scholar]
- 352.Ghais NS, Zhang Y, Grace AA, Huang CLH. Arrhythmogenic actions of the Ca2+ channel agonist FPL-64176 in Langendorff-perfused murine hearts. Exp Physiol 94: 240–254, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Gilchrist J, Das S, Van Petegem F, Bosmans F. Crystallographic insights into sodium-channel modulation by the β4 subunit. Proc Natl Acad Sci USA 110: E5016–5024, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Giles W, Shimoni Y. Comparison of sodium-calcium exchanger and transient inward currents in single cells from rabbit ventricle. J Physiol 417: 465–481, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Gillet L, Rougier JS, Shy D, Sonntag S, Mougenot N, Essers M, Shmerling D, Balse E, Hatem SN, Abriel H. Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart Rhythm 1: 1–12, 2014. [DOI] [PubMed] [Google Scholar]
- 356.Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol 556: 463–480, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Giudicessi JR, Ackerman MJ. Potassium-channel mutations and cardiac arrhythmias-diagnosis and therapy. Nat Rev Cardiol 9: 319–332, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res 161: 1–14, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Giustetto C, Di Monte F, Wolpert C, Borggrefe M, Schimpf R, Sbragia P, Leone G, Maury P, Anttonen O, Haissaguerre M, Gaita F. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 27: 2440–2447, 2006. [DOI] [PubMed] [Google Scholar]
- 360.Glass DB, Lundquist LJ, Katz BM, Walsh DA. Protein kinase inhibitor-(6–22)-amide peptide analogs with standard and nonstandard amino acid substitutions for phenylalanine 10. Inhibition of cAMP-dependent protein kinase. J Biol Chem 264: 14579–14584, 1989. [PubMed] [Google Scholar]
- 361.Glukhov AV, Fedorov VV, Anderson ME, Mohler PJ, Efimov IR. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am J Physiol Heart Circ Physiol 299: H482–H491, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Györke S, Fedorov VV. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur Heart J 36: 686–697, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132: 567–577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Goddard CA, Ghais NS, Zhang Y, Williams AJ, Colledge WH, Grace AA, Huang CLH. Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol 194: 123–140, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, De Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155: 1624–1638, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J 33: 1942–1953, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Gourraud JB, Kyndt F, Fouchard S, Rendu E, Jaafar P, Gully C, Gacem K, Dupuis JM, Longueville A, Baron E, Karakachoff M, Cebron JP, Chatel S, Schott JJ, Le Marec H, Probst V. Identification of a strong genetic background for progressive cardiac conduction defect by epidemiological approach. Heart 98: 1305–1310, 2012. [DOI] [PubMed] [Google Scholar]
- 368.Grandi E, Herren AW. CaMKII-dependent regulation of cardiac Na+ homeostasis. Front Pharmacol 5: 1–10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol 48: 112–121, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Grant AO, Carboni MP, Neplioueva V, Frank Starmer C, Memmi M, Napolitano C, Priori S. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest 110: 1201–1209, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Gray RA, Jalife J, Panfilov A, Baxter WT, Cabo C, Davidenko JM, Pertsov AM. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation 91: 2454–2469, 1995. [DOI] [PubMed] [Google Scholar]
- 372.Greiser M, Kerfant B, Williams GSB, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, Velden Van Der J, Lederer WJ, Dobrev D, Schotten U. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest 124: 4759–4772, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45: 233–244, 2005. [DOI] [PubMed] [Google Scholar]
- 374.Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria? Biochim Biophys Acta 1797: 939–944, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.de Groot SH, Schoenmakers M, Molenschot MM, Leunissen JD, Wellens HJ, Vos MA. Contractile adaptations preserving cardiac output predispose the hypertrophied canine heart to delayed afterdepolarization-dependent ventricular arrhythmias. Circulation 102: 2145–2151, 2000. [DOI] [PubMed] [Google Scholar]
- 376.Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 167: 149–160, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ. EH domain proteins regulate cardiac membrane protein targeting. Circ Res 107: 84–95, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest 99: 1991–1998, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Gui J, Wang T, Jones RPO, Trump D, Zimmer T, Lei M. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5: 16–20, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Gumina RJ, O'Cochlain DF, Kurtz CE, Bast P, Pucar D, Mishra P, Miki T, Seino S, Macura S, Terzic A. KATP channel knockout worsens myocardial calcium stress load in vivo and impairs recovery in stunned heart. Am J Physiol Heart Circ Physiol 292: H1706–H1713, 2007. [DOI] [PubMed] [Google Scholar]
- 381.Guo D, Lian J, Liu T, Cox R, Margulies K, Kowey P, Yan G. Contribution of late sodium current (I(Na-L)) to rate adaptation of ventricular repolarization and reverse use-dependence of QT-prolonging agents. Heart Rhythm 8: 762–769, 2011. [DOI] [PubMed] [Google Scholar]
- 382.Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res 106: 1743–1752, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Guo W, Li H, London B, Nerbonne J. Functional consequences of elimination of i(to,f) and i(to,s): early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ Res 87: 73–79, 2000. [DOI] [PubMed] [Google Scholar]
- 384.Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol 521: 587–599, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Gupta T, Khera S, Kolte D, Aronow WS, Iwai S. Antiarrhythmic properties of ranolazine: a review of the current evidence. Int J Cardiol 187: 66–74, 2015. [DOI] [PubMed] [Google Scholar]
- 386.Gurung IS, Kalin A, Grace AA, Huang CLH. Activation of purinergic receptors by ATP induces ventricular tachycardia by membrane depolarization and modifications of Ca2+ homeostasis. J Mol Cell Cardiol 47: 622–633, 2009. [DOI] [PubMed] [Google Scholar]
- 387.Gurung IS, Medina-Gomez G, Kis A, Baker M, Velagapudi V, Neogi SG, Campbell M, Rodriguez-Cuenca S, Lelliott C, McFarlane I, Oresic M, Grace AA, Vidal-Puig A, Huang CLH. Deletion of the metabolic transcriptional coactivator PGC1beta induces cardiac arrhythmia. Cardiovasc Res 92: 29–38, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P. Idiopathic short QT interval: a new clinical syndrome? Cardiology 94: 99–102, 2000. [DOI] [PubMed] [Google Scholar]
- 389.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res 88: 333–339, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Guzadhur L, Jeevaratnam K, Matthews G, Grace A, Huang CLH. Electrophysiological mechanisms underlying the initiation of atrial arrhythmia in genetically modified murine hearts. Trends Comp Biochem Physiol 17: 59–80, 2013. [Google Scholar]
- 391.Guzadhur L, Jiang W, Pearcey SM, Jeevaratnam K, Duehmke RM, Grace AA, Lei M, Huang CLH. The age-dependence of atrial arrhythmogenicity in Scn5a+/− murine hearts reflects alterations in action potential propagation and recovery. Clin Exp Pharmacol Physiol 39: 518–527, 2012. [DOI] [PubMed] [Google Scholar]
- 392.Guzadhur L, Pearcey SM, Duehmke RM, Jeevaratnam K, Hohmann AF, Zhang Y, Grace AA, Lei M, Huang CLH. Atrial arrhythmogenicity in aged Scn5a+/DeltaKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflüügers Arch 460: 593–601, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Györke I, Hester N, Jones LR, Györke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 86: 2121–2128, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Györke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca2+-induced Ca2+ release in heart. Science 260: 807–809, 1993. [DOI] [PubMed] [Google Scholar]
- 395.Györke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res 77: 245–255, 2008. [DOI] [PubMed] [Google Scholar]
- 396.Hagendorff A, Schumacher B, Kirchhoff S, Lüderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 99: 1508–1515, 1999. [DOI] [PubMed] [Google Scholar]
- 397.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 103: 1388–1393, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111: 869–876, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol 395: 233–253, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Hagiwara N, Irisawa H, Kasanuki H. Background current in sino-atrial node cells of the rabbit heart. J Physiol 448: 53–72, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet 9: 3047–3054, 2000. [DOI] [PubMed] [Google Scholar]
- 402.Hakim P, Brice N, Thresher R, Lawrence J, Zhang Y, Jackson AP, Grace AA, Huang CLH. Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol 198: 47–59, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Hakim P, Gurung IS, Pedersen TH, Thresher R, Brice N, Lawrence J, Grace AA, Huang CLH. Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog Biophys Mol Biol 98: 251–266, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Hakim P, Thresher R, Grace AA, Huang CLH. Effects of flecainide and quinidine on action potential and ventricular arrhythmogenic properties in Scn3b knockout mice. Clin Exp Pharmacol Physiol 37: 782–789, 2010. [DOI] [PubMed] [Google Scholar]
- 405.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45: 3448–3459, 2006. [DOI] [PubMed] [Google Scholar]
- 406.Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native “rapid” delayed rectifier K current during the cardiac ventricular action potential. Pflügers Arch 436: 843–853, 1998. [DOI] [PubMed] [Google Scholar]
- 407.Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen S, Grunnet M. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643). Mol Pharmacol 69: 266–277, 2006. [DOI] [PubMed] [Google Scholar]
- 408.Hao X, Zhang Y, Zhang X, Nirmalan M, Davies L, Konstantinou D, Yin F, Dobrzynski H, Wang X, Grace A, Zhang H, Boyett M, Huang CH, Lei M. TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ Arrhythm Electrophysiol 4: 397–406, 2011. [DOI] [PubMed] [Google Scholar]
- 409.Harvey R, Lai D, Elliott D, Biben C, Solloway M, Prall O, Stennard F, Schindeler A, Groves N, Lavulo L, Hyun C, Yeoh T, Costa M, Furtado MKE. Homeodomain factor Nkx2-5 in heart development and disease. Cold Spring Harb Symp Quant Biol 67: 107–114, 2002. [DOI] [PubMed] [Google Scholar]
- 410.Harzheim D, Pfeiffer KH, Fabritz L, Kremmer E, Buch T, Waisman A, Kirchhof P, Kaupp UB, Seifert R. Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J 27: 692–703, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34: 951–969, 2002. [DOI] [PubMed] [Google Scholar]
- 412.Havakuk O, Viskin S. A tale of 2 diseases. The history of long-QT syndrome and Brugada Syndrome. J Am Coll Cardiol 67: 100–108, 2016. [DOI] [PubMed] [Google Scholar]
- 413.Hayashi H, Shiferaw Y, Sato D, Nihei M, Lin SF, Chen PS, Garfinkel A, Weiss JN, Qu Z. Dynamic origin of spatially discordant alternans in cardiac tissue. Biophys J 92: 448–460, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244: 305–318, 2002. [DOI] [PubMed] [Google Scholar]
- 415.Hayashi T, Arimura T, Ueda K, Shibata H, Hohda S, Takahashi M, Hori H, Koga Y, Oka N, Imaizumi T, Yasunami M, Kimura A. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun 313: 178–184, 2004. [DOI] [PubMed] [Google Scholar]
- 416.Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CLH. Paced electrogram fractionation analysis of arrhythmogenic tendency in deltaKPQ Scn5a mice. J Cardiovasc Electrophysiol 16: 1329–1340, 2005. [DOI] [PubMed] [Google Scholar]
- 417.Heath BM, Cui Y, Worton S, Lawton B, Ward G, Ballini E, Doe CP, Ellis C, Patel BA, McMahon NC. Translation of flecainide- and mexiletine-induced cardiac sodium channel inhibition and ventricular conduction slowing from nonclinical models to clinical. J Pharmacol Toxicol Methods 63: 258–268, 2011. [DOI] [PubMed] [Google Scholar]
- 418.Heath BM, Terrar DA. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J Physiol 522: 391–402, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 114: 1483–1499, 2014. [DOI] [PubMed] [Google Scholar]
- 420.Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jüngling E, Zitt C, Lückhoff A. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J 371: 1045–1053, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, Guicheney P, Di Fusco S, Rey JL, Cauchemez B, Leenhardt A. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol 43: 1853–1860, 2004. [DOI] [PubMed] [Google Scholar]
- 422.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao J, Sirenko SG, Diaz Z, Guzman G, Xu Y, Wang Y, Kerrick WGL, Potter JD. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J Biol Chem 280: 37183–37194, 2005. [DOI] [PubMed] [Google Scholar]
- 423.Herr C, Smyth N, Ullrich S, Yun F, Sasse P, Hescheler J, Fleischmann B, Lasek K, Brixius K, Schwinger RH, Fässler R, Schröder R, Noegel AA. Loss of annexin A7 leads to alterations in frequency-induced shortening of isolated murine cardiomyocytes. Mol Cell Biol 21: 4119–4128, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A. Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc Res 90: 38–48, 2011. [DOI] [PubMed] [Google Scholar]
- 425.Herrmann S, Layh B, Ludwig A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol 51: 997–1006, 2011. [DOI] [PubMed] [Google Scholar]
- 426.Herrmann S, Stieber J, Stöckl G, Hofmann F, Ludwig A. HCN4 provides a “depolarization reserve” and is not required for heart rate acceleration in mice. EMBO J 26: 4423–4432, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Herron TJ, Lee P, Jalife J. Optical imaging of voltage and calcium in cardiac cells & tissues. Circ Res 110: 609–623, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Higuchi T, Nakaya Y. T wave polarity related to the repolarization process of epicardial and endocardial ventricular surfaces. Am Heart J 108: 290–295, 1984. [DOI] [PubMed] [Google Scholar]
- 429.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol 48: 293–301, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Hiraoka M, Fan Z. Activation of ATP-sensitive outward K(+) current by nicorandil (2-nicotinamidoethyl nitrate) in isolated ventricular myocytes. J Pharmacol Exp Ther 250: 278–285, 1989. [PubMed] [Google Scholar]
- 431.Hirose M, Takeishi Y, Niizeki T, Shimojo H, Nakada T, Kubota I, Nakayama J, Mende U, Yamada M. Diacylglycerol kinase zeta inhibits G(alpha)q-induced atrial remodeling in transgenic mice. Heart Rhythm 6: 78–84, 2009. [DOI] [PubMed] [Google Scholar]
- 432.Hirsh BJ, Copeland-Halperin RS, Halperin JL. Fibrotic atrial cardiomyopathy, atrial fibrillation, and thromboembolism: mechanistic links and clinical inferences. J Am Coll Cardiol 65: 2239–2251, 2015. [DOI] [PubMed] [Google Scholar]
- 433.Hisamatsu K, Kusano KF, Morita H, Takenaka S, Nagase S, Nakamura K, Emori T, Matsubara H, Mikouchi H, Nishizaki Y, Ohe T. Relationships between depolarization abnormality and repolarization abnormality in patients with Brugada syndrome: using body surface signal-averaged electrocardiography and body surface maps. J Cardiovasc Electrophysiol 15: 870–876, 2004. [DOI] [PubMed] [Google Scholar]
- 434.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv 3: 445–464, 2003. [DOI] [PubMed] [Google Scholar]
- 435.Hoesl E, Stieber J, Herrmann S, Feil S, Tybl E, Hofmann F, Feil R, Ludwig A. Tamoxifen-inducible gene deletion in the cardiac conduction system. J Mol Cell Cardiol 45: 62–69, 2008. [DOI] [PubMed] [Google Scholar]
- 436.Hoffman B, Suckling E. Effect of heart rate on cardiac membrane potentials and the unipolar electrogram. Am J Physiol 179: 123–130, 1954. [DOI] [PubMed] [Google Scholar]
- 437.Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol 577: 5–15, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS, Kwon YK, Chae SW, Kim do H. Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J 16: 1310–1312, 2002. [DOI] [PubMed] [Google Scholar]
- 439.Hong CS, Kwon SJ, Cho MC, Kwak YG, Ha KC, Hong B, Li H, Chae SW, Chai OH, Song CH, Li Y, Kim JC, Woo SH, Lee SY, Lee CO, Kim DH. Overexpression of junctate induces cardiac hypertrophy and arrhythmia via altered calcium handling. J Mol Cell Cardiol 44: 672–682, 2008. [DOI] [PubMed] [Google Scholar]
- 440.Honjo H, Boyett MR, Kodama I, Toyama J. Correlation between electrical activity and the size of rabbit sino-atrial node cells. J Physiol 496: 795–808, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Hoogendijk MG, Potse M, Linnenbank AC, Verkerk AO, den Ruijter HM, van Amersfoorth SCM, Klaver EC, Beekman L, Bezzina CR, Postema PG, Tan HL, Reimer AG, van der Wal AC, ten Harkel ADJ, Dalinghaus M, Vinet A, Wilde AAM, de Bakker JMT, Coronel R. Mechanism of right precordial ST-segment elevation in structural heart disease: Excitation failure by current-to-load mismatch. Heart Rhythm 7: 238–248, 2010. [DOI] [PubMed] [Google Scholar]
- 442.Hoogendijk MG, Potse M, Vinet A, de Bakker JMT, Coronel R. ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart Rhythm 8: 111–118, 2011. [DOI] [PubMed] [Google Scholar]
- 443.Horner SM, Vespalcova Z, Lab MJ. Electrode for recording direction of activation, conduction velocity, and monophasic action potential of myocardium. Am J Physiol Heart Circ Physiol 272: H1917–H1927, 1997. [DOI] [PubMed] [Google Scholar]
- 444.Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, Izu LT, Chen-izu Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J Mol Cell Cardiol 64: 59–68, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Hothi SS, Booth SW, Sabir IN, Killeen MJ, Simpson F, Zhang Y, Grace AA, Huang CLH. Arrhythmogenic substrate and its modification by nicorandil in a murine model of long QT type 3 syndrome. Prog Biophys Mol Biol 98: 267–280, 2008. [DOI] [PubMed] [Google Scholar]
- 446.Hothi SS, Gurung IS, Heathcote JC, Zhang Y, Booth SW, Skepper JN, Grace AA, Huang CLH. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflügers Arch 457: 253–270, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Hothi SS, Thomas G, Killeen MJ, Grace AA, Huang CLH. Empirical correlation of triggered activity and spatial and temporal re-entrant substrates with arrhythmogenicity in a murine model for Jervell and Lange-Nielsen syndrome. Pflügers Arch 458: 819–835, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Houle TD, Ram ML, Cala SE. Calsequestrin mutant D307H exhibits depressed binding to its protein targets and a depressed response to calcium. Cardiovasc Res 64: 227–233, 2004. [DOI] [PubMed] [Google Scholar]
- 449.Houser S. Does protein kinase A-mediated phosphorylation of the cardiac ryanodine receptor play any role in adrenergic regulation of calcium handling in health and disease. Circ Res 106: 1672–1674, 2010. [DOI] [PubMed] [Google Scholar]
- 450.Hove-Madsen L, Bers DM. Sarcoplasmic reticulum Ca2+ uptake and thapsigargin sensitivity in permeabilized rabbit and rat ventricular myocytes. Circ Res 73: 820–828, 1993. [DOI] [PubMed] [Google Scholar]
- 451.Hove-Madsen L, Llach A, Bayes-Genís A, Roura S, Font ER, Arís A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 110: 1358–1363, 2004. [DOI] [PubMed] [Google Scholar]
- 452.Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2: 270–278, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM, Rajamani S, DeAntonio HJ, Myerburg RJ, Ito H, Deshmukh P, Marieb M, Nam GBB, Bhatia A, Hasdemir C, Haïssaguerre M, Veltmann C, Schimpf R, Borggrefe M, Viskin S, Antzelevitch C. Mutations in SCN10A are responsible for a large fraction of cases of Brugada Syndrome. J Am Coll Cardiol 64: 66–79, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol 12: 230–243, 2015. [DOI] [PubMed] [Google Scholar]
- 455.Huang CLH. Dual actions of tetracaine on intramembrane charge in amphibian striated muscle. J Physiol 501: 589–606, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Huang CLH, Lei L, Matthews GDK, Zhang Y, Lei M. Pathophysiological mechanisms of sino-atrial dysfunction and ventricular conduction disease associated with SCN5A deficiency: insights from mouse models. Front Physiol 3: 234, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Huang CLH, Peachey LD. A reconstruction of charge movement during the action potential in frog skeletal muscle. Biophys J 61: 1133–1146, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Huang CLH, Pedersen TH, Fraser JA. Reciprocal dihydropyridine and ryanodine receptor interactions in skeletal muscle activation. J Muscle Res Cell Motil 32: 171–202, 2011. [DOI] [PubMed] [Google Scholar]
- 459.Huang CLH, Solaro R, Ke Y, Lei M. Ca2+ signalling and cardiac rhythm. Front Physiol 6: 423, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Huang CLH, Sun L, Fraser JA, Grace AA, Zaidi M. Similarities and contrasts in ryanodine receptor localization and function in osteoclasts and striated muscle cells. Ann NY Acad Sci 1116: 255–270, 2007. [DOI] [PubMed] [Google Scholar]
- 461.Huang CLH, Turner I, Saumarez RC. Numerical simulation of paced electrogram fractionation: relating clinical observations to changes in fibrosis and action potential duration. J Cardiovasc Electrophysiol 16: 151–161, 2005. [DOI] [PubMed] [Google Scholar]
- 462.Huang CLH. Intramembrane Charge Movements in Striated Muscle. Monographs of the Physiological Society, No. 44. Oxford, uk: Clarendon, 1993. [Google Scholar]
- 463.Huang CLH. SERCA2a stimulation by istaroxime: a novel mechanism of action with translational implications. Br J Pharmacol 170: 486–488, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Huang CLH. Computational analysis of the electromechanical consequences of short QT syndrome. Front Physiol 6: 44, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Hubbard ML, Henriquez CS. Microscopic variations in interstitial and intracellular structure modulate the distribution of conduction delays and block in cardiac tissue with source-load mismatch. Europace 14 Suppl 5: v3–v9, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Hulme JT, Colyer J, Orchard CH. Acidosis alters the phosphorylation of Ser16 and Thr17 of phospholamban in rat cardiac muscle. Pflügers Arch 434: 475–483, 1997. [DOI] [PubMed] [Google Scholar]
- 467.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 120: 3508–3519, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Hund TJ, Mohler PJ. Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc Med 25: 392–397, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Hund TJ, Wright PJ, Dun W, Snyder JS, Boyden PA, Mohler PJ. Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc Res 81: 742–749, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, Shimoni Y, Chen SR. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J 404: 431–438, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Hunter P, McNaughton P, Noble D. Analytical models of propagation in excitable cells. Prog Biophys Mol Biol 30: 99–144, 1975. [DOI] [PubMed] [Google Scholar]
- 472.Huo J, Zhang Y, Huang N, Liu P, Huang C, Guo X, Jiang W, Zhou N, Grace AA, Huang CLH, Ma A. The G604S-hERG mutation alters the biophysical properties and exerts a dominant-negative effect on expression of hERG channels in HEK293 cells. Pflügers Arch 456: 917–928, 2008. [DOI] [PubMed] [Google Scholar]
- 473.Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H, Knollmann BC. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 4: 128–135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL, Cornea RL, Bers DM, Knollmann BC. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res 114: 1114–1124, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Ian Gallicano G, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 143: 2009–2022, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Taniguchi M, Lee JK, Matsumura K, Tomita Y, Miyoshi S, Shimoda K, Makino S, Sano M, Kodama I, Ogawa S, Fukuda K. Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nat Med 13: 604–612, 2007. [DOI] [PubMed] [Google Scholar]
- 477.Ikeda T, Minai K, Matsumoto T, Horie H, Ohira N, Takashima H, Yokohama H, Kinoshita M. Assessment of noninvasive markers in identifying patients at risk in the Brugada syndrome: insight into risk stratification. J Am Coll Cardiol 37: 1628–1634, 2001. [DOI] [PubMed] [Google Scholar]
- 478.Imanishi S, Arita M, Kiyosue T, Aomine M. Effects of SG-75 (nicorandil) on electrical activity of canine cardiac Purkinje fibers: possible increase in potassium conductance. J Pharmacol Exp Ther 225: 198–205, 1983. [PubMed] [Google Scholar]
- 479.Ino M, Yoshinaga T, Wakamori M, Miyamoto N, Takahashi E, Sonoda J, Kagaya T, Oki T, Nagasu T, Nishizawa Y, Tanaka I, Imoto K, Aizawa S, Koch S, Schwartz A, Niidome T, Sawada K, Mori Y. Functional disorders of the sympathetic nervous system in mice lacking the alpha 1B subunit (Cav 2.2) of N-type calcium channels. Proc Natl Acad Sci USA 98: 5323–5328, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Isenberg G, Han S. Gradation of Ca2+-induced Ca2+ release by voltage-clamp pulse duration in potentiated guinea-pig ventricular myocytes. J Physiol 480: 423–438, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Ishida S, Ito M, Takahashi N, Fujino T, Akimitsu T, Saikawa T. Caffeine induces ventricular tachyarrhythmias possibly due to triggered activity in rabbits in vivo. Jpn Circ J 60: 157–165, 1996. [DOI] [PubMed] [Google Scholar]
- 482.Ishikawa T, Takahashi N. Novel SCN3B mutation associated With Brugada Syndrome affects intracellular trafficking and function of Nav1.5. Circ J 77: 959–967, 2012. [DOI] [PubMed] [Google Scholar]
- 483.Isik T, Tanboga IH, Kurt M, Kaya A, Ekinci M, Ayhan E, Uluganyan M, Ergelen M, Guvenc TS, Altay S, Uyarel H. Relation of the metabolic syndrome with proarrhythmogenic electrocardiographic parameters in patients without overt diabetes. Acta Cardiol 67: 195–201, 2012. [DOI] [PubMed] [Google Scholar]
- 484.Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83: 433–442, 1995. [DOI] [PubMed] [Google Scholar]
- 485.Isom LL. Sodium channel subunits: anything but auxiliary. Neuroscience 7: 42–54, 2001. [DOI] [PubMed] [Google Scholar]
- 486.Isomoto S, Kurachi Y. Function, regulation, pharmacology, and molecular structure of ATP-sensitive K+ channels in the cardiovascular system. J Cardiovasc Electrophysiol 8: 1431–1446, 1997. [DOI] [PubMed] [Google Scholar]
- 487.Isomura S, Toyama J, Kodama I, Yamada K. Epicardial activation patterns and dispersion of refractoriness initiating ventricular tachycardia in the canine left ventricle during acute ischemia. Jpn Circ J 47: 342–350, 1983. [DOI] [PubMed] [Google Scholar]
- 488.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471: 225–229, 2011. [DOI] [PubMed] [Google Scholar]
- 489.Iyer V, Mazhari R, Winslow RL. A computational model of the human left-ventricular epicardial myocyte. Biophys J 87: 1507–1525, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Iyer V, Roman-Campos D, Sampson KJ, Kang G, Fishman GI, Kass RS. Purkinje cells as sources of arrhythmias in Long QT Syndrome type 3. Sci Rep 5: 13287, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Izumida N, Asano Y, Doi S, Wakimoto H, Fukamizu S, Kimura T, Ueyama T, Sakurada H, Kawano S, Sawanobori T, Hiraoka M. Changes in body surface potential distributions induced by isoproterenol and Na channel blockers in patients with the Brugada syndrome. Int J Cardiol 95: 261–268, 2004. [DOI] [PubMed] [Google Scholar]
- 492.Jack J, Noble D, Tsien R. Electric Current Flow in Excitable Cells. New York: Oxford Univ. Press, 1983. [Google Scholar]
- 493.Janse M, Rosen M. History of arrhythmias. Handb Exp Pharmacol 171: 1–39, 2006. [DOI] [PubMed] [Google Scholar]
- 494.Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev 69: 1049–1169, 1989. [DOI] [PubMed] [Google Scholar]
- 495.Jansen JA, Noorman M, Musa H, Stein M, De Jong S, Van Der Nagel R, Hund TJ, Mohler PJ, Vos MA, Van Veen TA, De Bakker JM, Delmar M, Van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9: 600–607, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Jansen JA, van Veen TAB, de Bakker JMT, van Rijen HVM. Cardiac connexins and impulse propagation. J Mol Cell Cardiol 48: 76–82, 2010. [DOI] [PubMed] [Google Scholar]
- 497.January CT, Gong Q, Zhou Z. Long QT syndrome: cellular basis and arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol 11: 1413–1418, 2000. [DOI] [PubMed] [Google Scholar]
- 498.January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ Res 62: 563–571, 1988. [DOI] [PubMed] [Google Scholar]
- 499.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res 64: 977–990, 1989. [DOI] [PubMed] [Google Scholar]
- 500.Jeevaratnam K, Guzadhur L, Goh Y, Grace A, Huang CLH. Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol 216: 186–202, 2016. [DOI] [PubMed] [Google Scholar]
- 501.Jeevaratnam K, Poh Tee S, Zhang Y, Rewbury R, Guzadhur L, Duehmke R, Grace AA, Lei M, Huang CLH. Delayed conduction and its implications in murine Scn5a+/− hearts: independent and interacting effects of genotype, age, and sex. Pflügers Arch 461: 29–44, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Jeevaratnam K, Rewbury R, Zhang Y, Guzadhur L, Grace AA, Lei M, Huang CLH. Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/− hearts: the effects of ageing and sex. Mech Ageing Dev 133: 591–599, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Jeevaratnam K, Zhang Y, Guzadhur L, Duehmke RM, Lei M, Grace AA, Huang CLH. Differences in sino-atrial and atrio-ventricular function with age and sex attributable to the Scn5a+/− mutation in a murine cardiac model. Acta Physiol 200: 23–33, 2010. [DOI] [PubMed] [Google Scholar]
- 504.Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet 375: 752–762, 2010. [DOI] [PubMed] [Google Scholar]
- 505.Jeron A, Mitchell GF, Zhou J, Murata M, London B, Buckett P, Wiviott SD, Koren G. Inducible polymorphic ventricular tachyarrhythmias in a transgenic mouse model with a long Q-T phenotype. Am J Physiol Heart Circ Physiol 278: H1891–H1898, 2000. [DOI] [PubMed] [Google Scholar]
- 506.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 54: 59–68, 1957. [DOI] [PubMed] [Google Scholar]
- 507.Ji Y, Li B, Reed TD, Lorenz JN, Kaetzel MA, Dedman JR. Targeted inhibition of Ca2+/calmodulin-dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. J Biol Chem 278: 25063–25071, 2003. [DOI] [PubMed] [Google Scholar]
- 508.Jiang D, Chen W, Wang R, Zhang L, Chen SRW. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci USA 104: 18309–18314, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SRW. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res 97: 1173–1181, 2005. [DOI] [PubMed] [Google Scholar]
- 510.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SRW. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci USA 101: 13062–13067, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Jiang D. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res 91: 218–225, 2002. [DOI] [PubMed] [Google Scholar]
- 512.Jochim K, Katz L, Mayne W. The monophasic electrogram obtained from the mammalian heart. Am J Physiol 111: 177–186, 1935. [Google Scholar]
- 513.Johannesen L, Vicente J, Mason JW, Sanabria C, Waite-Labott K, Hong M, Guo P, Lin J, Sørensen JS, Galeotti L, Florian J, Ugander M, Stockbridge N, Strauss DG. Differentiating drug-induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin Pharmacol Ther 96: 549–558, 2014. [DOI] [PubMed] [Google Scholar]
- 514.Johnson D, Bennett ES. Isoform-specific effects of the beta2 subunit on voltage-gated sodium channel gating. J Biol Chem 281: 25875–25881, 2006. [DOI] [PubMed] [Google Scholar]
- 515.Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 5: 704–709, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC, Robertson AG. hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci USA 111: 18073–18077, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Jones SA, Boyett MR, Lancaster MK. Declining into failure: The age-dependent loss of the L-type calcium channel within the sinoatrial node. Circulation 115: 1183–1190, 2007. [DOI] [PubMed] [Google Scholar]
- 518.Jones SA, Lancaster MK, Boyett MR. Ageing-related changes of connexins and conduction within the sinoatrial node. J Physiol 560: 429–437, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.de Jong AM, Maass AH, Oberdorf-Maass SU, Van Veldhuisen DJ, Van Gilst WH, Van Gelder IC. Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc Res 89: 754–765, 2011. [DOI] [PubMed] [Google Scholar]
- 520.Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res 86: 1193–1197, 2000. [DOI] [PubMed] [Google Scholar]
- 521.Jorgensen AO, Shen ACY, Arnold W, McPherson PS, Campbell KP. The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol 120: 969–980, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Ju YK, Allen DG. Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J Physiol 508: 153–166, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Ju YK, Allen DG. The distribution of calcium in toad cardiac pacemaker cells during spontaneous firing. Pflügers Arch 441: 219–227, 2000. [DOI] [PubMed] [Google Scholar]
- 524.Ju YK, Lee BH, Trajanovska S, Hao G, Allen DG, Lei M, Cannell MB. The involvement of TRPC3 channels in sinoatrial arrhythmias. Front Physiol 6: 86, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, Laugwitz K- L. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med 4: 180–191, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Kaese S, Verheule S. Cardiac electrophysiology in mice: a matter of size. Front Physiol 3: 345, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Kaftan E, Marks AR, Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca2+-release channels from cardiac muscle. Circ Res 78: 990–997, 1996. [DOI] [PubMed] [Google Scholar]
- 528.Kakei M, Yoshinaga M, Saito K, Tanaka H. The potassium current activated by 2-nicotinamidoethyl nitrate (nicorandil) in single ventricular cells of guinea pigs. Proc R Soc Lond B Biol Sci 229: 331–343, 1986. [DOI] [PubMed] [Google Scholar]
- 529.Kalin A, Usher-Smith J, Jones VJ, Huang CLH, Sabir IN. Cardiac arrhythmia: a simple conceptual framework. Trends Cardiovasc Med 20: 103–107, 2010. [DOI] [PubMed] [Google Scholar]
- 530.Kanaporis G, Blatter LA. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ Res 116: 846–856, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Kanda M, Shimizu W, Matsuo K, Nagaya N, Taguchi A, Suyama K, Kurita T, Aihara N, Kamakura S. Electrophysiologic characteristics and implications of induced ventricular fibrillation in symptomatic patients with Brugada syndrome. J Am Coll Cardiol 39: 1799–1805, 2002. [DOI] [PubMed] [Google Scholar]
- 532.Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu XK, Hodgson DM, Reyes S, Miki T, Seino S, Terzic A. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet 15: 2285–2297, 2006. [DOI] [PubMed] [Google Scholar]
- 533.Kang S, Dahl R, Hsieh W, Shin AC, Zsebo KM, Buettner C, Hajjar RJ, Lebeche D. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J Biol Chem 291: 5185–5198, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, de Biasi M, Wehrens XHT, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci USA 103: 12179–12184, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Kannel WB, Kannel C, Paffenbarger RS, Cupples LA. Heart rate and cardiovascular mortality: the Framingham Study. Am Heart J 113: 1489–1494, 1987. [DOI] [PubMed] [Google Scholar]
- 536.Kant S, Krull P, Eisner S, Leube RE, Krusche CA. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res 348: 249–259, 2012. [DOI] [PubMed] [Google Scholar]
- 537.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AAM, Brugada R, Schott JJ, Ackerman MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7: 33–46, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Kasanuki H, Ohnishi S, Ohtuka M, Matsuda N, Nirei T, Isogai R, Shoda M, Toyoshima Y, Hosoda S. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 95: 2277–2285, 1997. [DOI] [PubMed] [Google Scholar]
- 539.Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, Jiao Z, Gallego CJ, Bernstein KE, Dudley SC. Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol 293: H182–H192, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Katz G, Khoury A, Kurtzwald E, Hochhauser E, Porat E, Shainberg A, Seidman JG, Seidman CE, Lorber A, Eldar M, Arad M. Optimizing catecholaminergic polymorphic ventricular tachycardia therapy in calsequestrin-mutant mice. Heart Rhythm 7: 1676–1682, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Kawasaki H, Springett GM, Mochizuki N, Toki S , Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science 282: 2275–2279, 1998. [DOI] [PubMed] [Google Scholar]
- 542.Kazemian P, Gollob MH, Pantano A, Oudit GY. A novel mutation in the RYR2 gene leading to catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation: dose-dependent arrhythmia-event suppression by β-blocker therapy. Can J Cardiol 27: 870.e7–10, 2011. [DOI] [PubMed] [Google Scholar]
- 543.Ke Y, Lei M, Collins TP, Rakovic S, Mattick PAD, Yamasaki M, Brodie MS, Terrar DA, Solaro RJ. Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ Res 100: 1317–1327, 2007. [DOI] [PubMed] [Google Scholar]
- 544.Ke Y, Lei M, Solaro RJ. Regulation of cardiac excitation and contraction by p21 activated kinase-1. Prog Biophys Mol Biol 98: 238–250, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Ke Y, Lei M, Wang X, Solaro RJ. Novel roles of PAK1 in the heart. Cell Logist 2: 89–94, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Ke Y, Lum H, Solaro RJ. Inhibition of endothelial barrier dysfunction by P21-activated kinase-1. Can J Physiol Pharmacol 85: 281–288, 2007. [DOI] [PubMed] [Google Scholar]
- 547.Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ. Intracellular localization and functional effects of p21-activated kinase 1 (Pak1) in cardiac myocytes. Circ Res 94: 194–200, 2004. [DOI] [PubMed] [Google Scholar]
- 548.Ke Y, Wang X, Jin XY, Solaro RJ, Lei M. PAK1 is a novel cardiac protective signaling molecule. Front Med 8: 399–403, 2014. [DOI] [PubMed] [Google Scholar]
- 549.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104: 569–580, 2001. [DOI] [PubMed] [Google Scholar]
- 550.Keener J, Sneyd J. Mathematical Physiology. I: Cellular Physiology (Interdisciplinary Applied Mathematics) (2nd ed). New York: Springer, 2009. [Google Scholar]
- 551.Keldermann RH, ten Tusscher KHWJ, Nash MP, Bradley CP, Hren R, Taggart P, Panfilov AV. A computational study of mother rotor VF in the human ventricles. Am J Physiol Heart Circ Physiol 296: H370–H379, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Keldermann RH, ten Tusscher KHWJ, Nash MP, Hren R, Taggart P, Panfilov AV. Effect of heterogeneous APD restitution on VF organization in a model of the human ventricles. Am J Physiol Heart Circ Physiol 294: H764–H774, 2008. [DOI] [PubMed] [Google Scholar]
- 553.Keller DI, Acharfi S, Delacrétaz E, Benammar N, Rotter M, Pfammatter JP, Fressart V, Guicheney P, Chahine M. A novel mutation in SCN5A, delQKP 1507–1509, causing long QT syndrome: Role of Q1507 residue in sodium channel inactivation. J Mol Cell Cardiol 35: 1513–1521, 2003. [DOI] [PubMed] [Google Scholar]
- 554.Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, Madle A, Fromer M, Schläpfer J, Abriel H. Brugada syndrome and fever: Genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res 67: 510–519, 2005. [DOI] [PubMed] [Google Scholar]
- 555.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368, 2004. [DOI] [PubMed] [Google Scholar]
- 556.ter Keurs HEDJ, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev 87: 457–506, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Khoo MSC, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ, Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation 114: 1352–1359, 2006. [DOI] [PubMed] [Google Scholar]
- 558.Killeen M, Sabir I. Repolarization gradients and arrhythmogenicity in the murine heart. J Physiol 583: 419–420, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Killeen MJ, Gurung IS, Thomas G, Stokoe KS, Grace AA, Huang CLH. Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol 191: 43–58, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Killeen MJ, Sabir IN, Grace AA, Huang CLH. Dispersions of repolarization and ventricular arrhythmogenesis: Lessons from animal models. Prog Biophys Mol Biol 98: 219–229, 2008. [DOI] [PubMed] [Google Scholar]
- 561.Killeen MJ, Thomas G, Gurung IS, Goddard CA, Fraser JA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CLH. Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol 189: 33–46, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Killeen MJ, Thomas G, Olesen SP, Demnitz J, Stokoe KS, Grace AA, Huang CLH. Effects of potassium channel openers in the isolated perfused hypokalaemic murine heart. Acta Physiol 193: 25–36, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Killeen MJ, Thomas G, Sabir IN, Grace AA, Huang CLH. Mouse models of human arrhythmia syndromes. Acta Physiol 192: 455–469, 2008. [DOI] [PubMed] [Google Scholar]
- 564.Killeen MJ. Drug-induced arrhythmias and sudden cardiac death: implications for the pharmaceutical industry. Drug Discov Today 14: 589–597, 2009. [DOI] [PubMed] [Google Scholar]
- 565.Kim E, Youn B, Kemper L, Campbell C, Milting H, Varsanyi M, Kang C. Characterization of human cardiac calsequestrin and its deleterious mutants. J Mol Biol 373: 1047–1057, 2007. [DOI] [PubMed] [Google Scholar]
- 566.Kim EE, Shekhar A, Lu J, Lin X, Liu F, Zhang J, Delmar M, Fishman GI. PCP4 regulates Purkinje cell excitability and cardiac rhythmicity. J Clin Invest 124: 5027–5036, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.King JH, Huang CLH, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol 4: 154, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.King JH, Wickramarachchi C, Kua K, Du Y, Jeevaratnam K, Matthews HR, Grace AA, Huang CLH, Fraser JA. Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc Res 99: 751–759, 2013. [DOI] [PubMed] [Google Scholar]
- 569.King JH, Zhang Y, Lei M, Grace AA, Huang CLH, Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol 207: 308–323, 2013. [DOI] [PubMed] [Google Scholar]
- 570.Kirchhof P, Fabritz L, Fortmuller L, Matherne GP, Lankford A, Baba HA, Schmitz W, Breithardt G, Neumann J, Boknik P. Altered sinus nodal and atrioventricular nodal function in freely moving mice overexpressing the A1 adenosine receptor. Am J Physiol Heart Circ Physiol 285: H145–H153, 2003. [DOI] [PubMed] [Google Scholar]
- 571.Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, Breithardt G, Ruiz P, Wichter T, Levkau B. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 114: 1799–1806, 2006. [DOI] [PubMed] [Google Scholar]
- 572.Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T, Schulte K, Hanstein J, Schulte JS, Vogel M, Mougenot N, Laakmann S, Fortmueller L, Eckstein J, Verheule S, Kaese S, Staab A, Grote-Wessels S, Schotten U, Moubarak G, Wehrens XHT, Schmitz W, Hatem S, Müller FU. Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol 166: 366–374, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Kirchhoff S, Nelles E, Hagendorff A, Krüger O, Traub O, Willecke K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr Biol 8: 299–302, 1998. [DOI] [PubMed] [Google Scholar]
- 574.Kizana E, Ginn SL, Allen DG, Ross DL, Alexander IE. Fibroblasts can be genetically modified to produce excitable cells capable of electrical coupling. Circulation 111: 394–398, 2005. [DOI] [PubMed] [Google Scholar]
- 575.Klaver EC, Versluijs GM, Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol 152: 162–170, 2011. [DOI] [PubMed] [Google Scholar]
- 576.Kléber AG, Riegger CB. Electrical constants of arterially perfused rabbit papillary muscle. J Physiol 385: 307–324, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84: 431–488, 2004. [DOI] [PubMed] [Google Scholar]
- 578.Kleber AG, Saffitz JE, Billman GE, State TO. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front Physiol 5: 404, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Kline CF, Kurata HT, Hund TJ, Cunha SR, Koval OM, Wright PJ, Christensen M, Anderson ME, Nichols CG, Mohler PJ. Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci USA 106: 16669–16674, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Knollmann BC, Blatt SA, Horton K, De Freitas F, Miller T, Bell M, Housmans PR, Weissman NJ, Morad M, Potter JD. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J Biol Chem 276: 10039–10048, 2001. [DOI] [PubMed] [Google Scholar]
- 581.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116: 2510–2520, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Knollmann BC, Katchman AN, Franz MR. Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12: 1286–1294, 2001. [DOI] [PubMed] [Google Scholar]
- 583.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res 92: 428–436, 2003. [DOI] [PubMed] [Google Scholar]
- 584.Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, Xu X, Uchinoumi H, Okuda S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol 53: 1993–2005, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Yamamoto T, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J 74: 2579–2584, 2010. [DOI] [PubMed] [Google Scholar]
- 586.Komukai K, Pascarel C, Orchard CH. Compensatory role of CaMKII on ICa and SR function during acidosis in rat ventricular myocytes. Pflügers Arch 442: 353–361, 2001. [DOI] [PubMed] [Google Scholar]
- 587.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res 62: 426–436, 2004. [DOI] [PubMed] [Google Scholar]
- 588.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126: 2084–2094, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37: 2136–2143, 2001. [DOI] [PubMed] [Google Scholar]
- 590.Kreuzberg MM, Liebermann M, Segschneider S, Dobrowolski R, Dobrzynski H, Kaba R, Rowlinson G, Dupont E, Severs NJ, Willecke K. Human connexin31.9,unlike its orthologous protein connexin30.2 in the mouse, is not detectable in the human cardiac conduction system. J Mol Cell Cardiol 46: 553–559, 2009. [DOI] [PubMed] [Google Scholar]
- 591.Kreuzberg MM, Schrickel JW, Ghanem A, Kim JS, Degen J, Janssen-Bienhold U, Lewalter T, Tiemann K, Willecke K. Connexin30.2 containing gap junction channels decelerate impulse propagation through the atrioventricular node. Proc Natl Acad Sci USA 103: 5959–5964, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Kreuzberg MM, Söhl G, Kim JS, Verselis VK, Willecke K, Bukauskas FF. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ Res 96: 1169–1177, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Krishnan SC, Antzelevitch C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ Res 69: 277–291, 1991. [DOI] [PubMed] [Google Scholar]
- 594.Krishnan SC, Antzelevitch C. Flecainide-induced arrhythmia in canine ventricular epicardium. Phase 2 reentry? Circulation 87: 562–572, 1993. [DOI] [PubMed] [Google Scholar]
- 595.Krogh-Madsen T, Abbott GW, Christini DJ. Effects of electrical and structural remodeling on atrial fibrillation maintenance: a simulation study. PLoS Comput Biol 8: e1002390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Krüger O, Maxeiner S, Kim JS, van Rijen HVM, de Bakker JMT, Eckardt D, Tiemann K, Lewalter T, Ghanem A, Lüderitz B, Willecke K. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J Mol Cell Cardiol 41: 787–797, 2006. [DOI] [PubMed] [Google Scholar]
- 597.Krusche CA, Holthöfer B, Hofe V, Van De Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol 106: 617–633, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res 81: 627–635, 1997. [DOI] [PubMed] [Google Scholar]
- 599.Kucera JP, Kléber AG, Rohr S. Slow conduction in cardiac tissue, II: effects of branching tissue geometry. Circ Res 83: 795–805, 1998. [DOI] [PubMed] [Google Scholar]
- 600.Kucharska-Newton AM, Couper DJ, Pankow JS, Prineas RJ, Rea TD, Sotoodehnia N, Chakravarti A, Folsom AR, Siscovick DS, Rosamond WD. Diabetes and the risk of sudden cardiac death, the Atherosclerosis Risk in Communities study. Acta Diabetol 47 Suppl 1: 161–168, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science 269: 1427–1429, 1995. [DOI] [PubMed] [Google Scholar]
- 602.Kumai M, Nishii K, Nakamura K, Takeda N, Suzuki M, Shibata Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 127: 3501–3512, 2000. [DOI] [PubMed] [Google Scholar]
- 603.Kumar S, Stevenson W, John R 2nd. Arrhythmias in dilated cardiomyopathy. Card Electrophysiol Clin 7: 221–233, 2015. [DOI] [PubMed] [Google Scholar]
- 604.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyêñ-Trân VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107: 801–813, 2001. [DOI] [PubMed] [Google Scholar]
- 605.Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, Aihara N, Kamakura S, Kobayashi J, Kosakai Y. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol 40: 330–334, 2002. [DOI] [PubMed] [Google Scholar]
- 606.Kurtenbach S, Kurtenbach S, Zoidl G. Gap junction modulation and its implications for heart function. Front Physiol 5 February: 1–10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Kusano KF, Taniyama M, Nakamura K, Miura D, Banba K, Nagase S, Morita H, Nishii N, Watanabe A, Tada T, Murakami M, Miyaji K, Hiramatsu S, Nakagawa K, Tanaka M, Miura A, Kimura H, Fuke S, Sumita W, Sakuragi S, Urakawa S, Iwasaki J, Ohe T. Atrial fibrillation in patients with Brugada syndrome relationships of gene mutation, electrophysiology, and clinical backgrounds. J Am Coll Cardiol 51: 1169–1175, 2008. [DOI] [PubMed] [Google Scholar]
- 608.Kuwahara K, Saito Y, Takano M, Arai Y, Yasuno S, Nakagawa Y, Takahashi N, Adachi Y, Takemura G, Horie M, Miyamoto Y, Morisaki T, Kuratomi S, Noma A, Fujiwara H, Yoshimasa Y, Kinoshita H, Kawakami R, Kishimoto I, Nakanishi M, Usami S, Saito Y, Harada M, Nakao K. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J 22: 6310–6321, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 104: 3081–3086, 2001. [DOI] [PubMed] [Google Scholar]
- 610.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Lorber A, Kastner DL, Goldman B, Pras E. Autosomal recessive catecholamine- or exercise-induced clinical features and assignment of the disease gene. Circulation 103: 2822–2827, 2001. [DOI] [PubMed] [Google Scholar]
- 611.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 69: 1378–1384, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev 22: 1948–1961, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan a D, Kontula K. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103: 485–490, 2001. [DOI] [PubMed] [Google Scholar]
- 614.Laitinen-Forsblom PJ, Makynen P, Makynen H, Yli-Mayry S, Virtanen V, Kontula K, Aalto-Setala K. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J Cardiovasc Electrophysiol 17: 480–485, 2006. [DOI] [PubMed] [Google Scholar]
- 615.Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol 47: 157–170, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res 106: 659–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Lakdawala N, Winterfield J, Funke B. Dilated cardiomyopathy. Circ Arrhythm Electrophysiol 6: 228–237, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Lam E, Martin MM, Timerman AP, Sabers C, Fleischer S, Lukas T, Abraham RT, O'Keefe SJ, O'Neill EA, Wiederrecht GJ. A novel FK506 binding protein can mediate the immunosuppressive effects of FK506 and is associated with the cardiac ryanodine receptor. J Biol Chem 270: 26511–26522, 1995. [DOI] [PubMed] [Google Scholar]
- 619.Lambiase PD, Ahmed AK, Ciaccio EJ, Brugada R, Lizotte E, Chaubey S, Ben-Simon R, Chow AW, Lowe MD, McKenna WJ. High-density substrate mapping in Brugada Syndrome: combined role of conduction and repolarization heterogeneities in arrhythmogenesis. Circulation 120: 106–117, 2009. [DOI] [PubMed] [Google Scholar]
- 620.Lande G, Demolombe S, Bammert A, Moorman A, Charpentier F, Escande D. Transgenic mice overexpressing human KvLQT1 dominant-negative isoform. Part II: Pharmacological profile. Cardiovasc Res 50: 328–334, 2001. [DOI] [PubMed] [Google Scholar]
- 621.Lanner JT. Ryanodine receptor physiology and its role in disease. Adv Exp Med Biol 740: 217–234, 2012. [DOI] [PubMed] [Google Scholar]
- 622.Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Clin Exp Pharmacol Physiol 34: 889–896, 2007. [DOI] [PubMed] [Google Scholar]
- 623.Lawrenz W, Krogmann ON, Wieczorek M. Complex atrial arrhythmias as first manifestation of catecholaminergic polymorphic ventricular tachycardia: an unusual course in a patient with a new mutation in ryanodine receptor type 2 gene. Cardiol Young 24: 741–744, 2014. [DOI] [PubMed] [Google Scholar]
- 624.Lee RJ, Liem LB, Cohen TJ, Franz MR. Relation between repolarization and refractoriness in the human ventricle: cycle length dependence and effect of procainamide. J Am Coll Cardiol 19: 614–618, 1992. [DOI] [PubMed] [Google Scholar]
- 625.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91: 1512–1519, 1995. [DOI] [PubMed] [Google Scholar]
- 626.Lees-Miller JP, Guo J, Somers JR, Roach DE, Sheldon RS, Rancourt DE, Duff HJ. Selective knockout of mouse ERG1 B potassium channel eliminates I(Kr) in adult ventricular myocytes and elicits episodes of abrupt sinus bradycardia. Mol Cell Biol 23: 1856–1862, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Lees-Miller JP, Kondo C, Wang L, Duff HJ. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res 81: 719–726, 1997. [DOI] [PubMed] [Google Scholar]
- 628.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED, Kelly DP. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol 295: H185–H196, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest 118: 2230–2245, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Lehnart SE, Terrenoire C, Reiken S, Wehrens XHT, Song LS, Tillman EJ, Mancarella S, Coromilas J, Lederer WJ, Kass RS, Marks AR. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci USA 103: 7906–7910, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Lehnart SE, Wehrens XHT, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 109: 3208–3214, 2004. [DOI] [PubMed] [Google Scholar]
- 632.Lei M, Goddard C, Liu J, Léoni AL, Royer A, Fung SSM, Xiao G, Ma A, Zhang H, Charpentier F, Vandenberg JI, Colledge WH, Grace AA, Huang CLH. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J Physiol 567: 387–400, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Lei M, Grace AA, Huang CLH. Translational models for cardiac arrhythmogenesis. Prog Biophys Mol Biol 98: 119, 2009. [DOI] [PubMed] [Google Scholar]
- 634.Lei M, Huang CL-H, Zhang Y. Genetic Na(+) channelopathies and sinus node dysfunction. Prog Biophys Mol Biol 98: 171–178, 2008. [DOI] [PubMed] [Google Scholar]
- 635.Lei M, Jones SA, Liu J, Lancaster MK, Fung SSM, Dobrzynski H, Camelliti P, Maier SKG, Noble D, Boyett MR. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J Physiol 559: 835–848, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Lei M, Wang X, Ke Y, Solaro R. Regulation of Ca2+ transient by PP2A in normal and failing heart. Front Physiol 6: 13, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Lei M, Zhang H, Grace AA, Huang CLH. SCN5A and sinoatrial node pacemaker function. Cardiovasc Res 74: 356–365, 2007. [DOI] [PubMed] [Google Scholar]
- 638.Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, Zhang D, Litwin SE, Zaha VG, Fountain KT, Boudina S, Jimenez-Linan M, Blount M, Lopez M, Meirhaeghe A, Bohlooly -YM, Storlien L, Strömstedt M, Snaith M, Orešič M, Abel ED, Cannon B, Vidal-Puig A. Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol 4: 2042–2056, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Lemoine MD, Duverger JE, Naud P, Chartier D, Qi XY, Comtois P, Fabritz L, Kirchhof P, Nattel S. Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc Res 92: 67–74, 2011. [DOI] [PubMed] [Google Scholar]
- 640.Lenegre J. Etiology and pathology of bilateral bundle branch block in relation to complete heart block. Prog Cardiovasc Dis 6: 409, 1964. [DOI] [PubMed] [Google Scholar]
- 641.de Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science 270: 1502-6, 1995. [DOI] [PubMed] [Google Scholar]
- 642.Leoni AL, Gavillet B, Rougier JS, Marionneau C, Probst V, Le Scouarnec S, Schott JJ, Demolombe S, Bruneval P, Huang CLH, Colledge WH, Grace AA, Le Marec H, Wilde AA, Mohler PJ, Escande D, Abriel H, Charpentier F. Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/−) mouse model. PLoS One 5: e9298, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Leoni AL, Marionneau C, Demolombe S, Le Bouter S, Mangoni ME, Escande D, Charpentier F. Chronic heart rate reduction remodels ion channel transcripts in the mouse sinoatrial node but not in the ventricle. Physiol Genomics 24: 4–12, 2005. [DOI] [PubMed] [Google Scholar]
- 644.Lev M. The pathology of complete atrioventricular block. Prog Cardiovasc Dis 6: 317, 1964. [DOI] [PubMed] [Google Scholar]
- 645.Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. J Physiol 592: 4825–4838, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Lewis T. Lectures on the Heart. New York: Hoeber, 1915. [Google Scholar]
- 647.Li D, Liu Y, Maruyama M, Zhu W, Chen H, Zhang W, Reuter S, Lin SF, Haneline LS, Field LJ, Chen PS, Shou W. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum Mol Genet 20: 4582–4596, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, Liu M. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31: 681–683, 2013. [DOI] [PubMed] [Google Scholar]
- 649.Li J, Kline CF, Hund TJ, Anderson ME, Mohler PJ. Ankyrin-B regulates Kir6.2 membrane expression and function in heart. J Biol Chem 285: 28723–28730, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Li J, Marionneau C, Zhang R, Shah V, Hell JW, Nerbonne JM, Anderson ME. Calmodulin kinase II inhibition shortens action potential duration by upregulation of K(+) currents. Circ Res 99: 1092–1099, 2006. [DOI] [PubMed] [Google Scholar]
- 651.Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol 287: H2790–H2802, 2004. [DOI] [PubMed] [Google Scholar]
- 652.Li J, Qu J, Nathan RD. Ionic basis of ryanodine's negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. [Online] Am J Physiol Heart Circ Physiol 273: H2481–H2489, 1997. [DOI] [PubMed] [Google Scholar]
- 653.Li L, Chu G, Kranias EG, Bers DM. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am J Physiol Heart Circ Physiol 274: H1335–H1347, 1998. [DOI] [PubMed] [Google Scholar]
- 654.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Müller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XHT. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129: 1276–1285, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XHT. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res 110: 465–470, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, Fang WY, Yang YQ. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med 32: 144–150, 2013. [DOI] [PubMed] [Google Scholar]
- 657.Li Y, Kranias E, Mignery G, Bers D. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res 90: 309–316, 2002. [DOI] [PubMed] [Google Scholar]
- 658.Liang X, Xie H, Zhu PH, Hu J, Zhao Q, Wang CS, Yang C. Ryanodine receptor-mediated Ca2+ events in atrial myocytes of patients with atrial fibrillation. Cardiology 111: 102–110, 2008. [DOI] [PubMed] [Google Scholar]
- 659.Liao R, Podesser BK, Lim CC. The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol 303: H156–H167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Liberali P, Snijder B, Pelkmans L. Single-cell and multivariate approaches in genetic perturbation screens. Nat Rev Genet 16: 18–32, 2014. [DOI] [PubMed] [Google Scholar]
- 661.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1: 361–370, 2005. [DOI] [PubMed] [Google Scholar]
- 662.Lin X, Liu N, Lu J, Zhang J, Anumonwo JMB, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 8: 1923–1930, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Lin X, O'Malley H, Chen C, Auerbach D, Foster M, Shekhar A, Zhang M, Coetzee W, Jalife J, Fishman GI, Isom L, Delmar M. Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J Physiol 593: 1389–1407, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Lindegger N, Hagen BM, Marks AR, Lederer WJ, Kass RS. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol 47: 326–334, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol 30: 743–749, 1998. [DOI] [PubMed] [Google Scholar]
- 666.Lines GT, Sande JB, Louch WE, Mørk HK, Grøttum P, Sejersted OM. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys J 91: 779–792, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH, Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest 119: 1230–1240, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Liu G, Iden JB, Kovithavongs K, Gulamhusein R, Duff HJ, Kavanagh KM. In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J Physiol 555: 267–279, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Liu J, Kim KH, London B, Morales MJ, Backx PH. Dissection of the voltage-activated potassium outward currents in adult mouse ventricular myocytes: I(to,f), I(to,s), I(K,slow1), I(K,slow2), and I(ss). Basic Res Cardiol 106: 189–204, 2011. [DOI] [PubMed] [Google Scholar]
- 670.Liu J, Xin L, Benson V, Allen D, Ju Y. Store-operated calcium entry and the localization of STIM1 and Orai1 proteins in isolated mouse sinoatrial node cells. Front Physiol 6: 69, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Liu M, Liu H, Dudley SC. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res 107: 967–974, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672. Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CL-H, Grace AA, London B, Dudley SC. Cardiac Na(+) current regulation by pyridine nucleotides. Circ Res 105: 737–745, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 99: 292–298, 2006. [DOI] [PubMed] [Google Scholar]
- 674.Liu N, Colombi B, Raytcheva-Buono EV, Bloise R, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Herz 32: 212–217, 2007. [DOI] [PubMed] [Google Scholar]
- 675.Liu N, Denegri M, Ruan Y, Avelino-Cruz JE, Perissi A, Negri S, Napolitano C, Coetzee a W, Boyden PA, Priori SG. Short communication: flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ Res 109: 291–295, 2011. [DOI] [PubMed] [Google Scholar]
- 676.Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J Mol Cell Cardiol 46: 149–159, 2009. [DOI] [PubMed] [Google Scholar]
- 677.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG. Calmodulin kinase II inhibition prevents arrhythmias in RyR2R4496C+/− mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol 50: 214–222, 2011. [DOI] [PubMed] [Google Scholar]
- 678.Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis 51: 23–30, 2008. [DOI] [PubMed] [Google Scholar]
- 679.Liu W, Deng J, Wang G, Gao K, Lin Z, Liu S, Wang Y, Liu J. Manipulation of KCNE2 expression modulates action potential duration, and Ito and IK in rat and mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 309: H1288–H1302, 2015. [DOI] [PubMed] [Google Scholar]
- 680.Liu W, Zi M, Jin J, Prehar S, Oceandy D, Kimura TE, Lei M, Neyses L, Weston AH, Cartwright EJ, Wang X. Cardiac-specific deletion of Mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ Res 104: 905–914, 2009. [DOI] [PubMed] [Google Scholar]
- 681.Liu W, Zi M, Naumann R, Ulm S, Jin J, Taglieri DM, Prehar S, Gui J, Tsui H, Xiao RP, Neyses L, Solaro RJ, Ke Y, Cartwright EJ, Lei M, Wang X. Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart/clinical perspective. Circulation 124: 2702–2715, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Liu YB, Wu CC, Lu LS, Su MJ, Lin CW, Lin SF, Chen LS, Fishbein MC, Chen PS, Lee YT. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res 92: 1145–1152, 2003. [DOI] [PubMed] [Google Scholar]
- 683.Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D'Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime risk for development of atrial fibrillation: The Framingham heart study. Circulation 110: 1042–1046, 2004. [DOI] [PubMed] [Google Scholar]
- 684.Locati E, Zareba W, Moss A, Schwartz P, Vincent G, Lehmann M, Towbin J, Priori S, Napolitano C, Robinson J, Andrews M, Timothy K, Hall W. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation 97: 2237–2244, 1998. [DOI] [PubMed] [Google Scholar]
- 685.Lodder EM, Rizzo S. Mouse models in arrhythmogenic right ventricular cardiomyopathy. Front Physiol 3 June: 1–5, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686.Lombardi R, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res 109: 1342-53, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.London B, Baker LC, Petkova-Kirova P, Nerbonne JM, Choi BR, Salama G. Dispersion of repolarization and refractoriness are determinants of arrhythmia phenotype in transgenic mice with long QT. J Physiol 578: 115–129, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.London B, Jeron A, Zhou J, Buckett P, Han X, Mitchell GF, Koren G. Long QT and ventricular arrhythmias in transgenic mice expressing the N terminus and first transmembrane segment of a voltage-gated potassium channel. Proc Natl Acad Sci USA 95: 2926–2931, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116: 2260–2268, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K(+) current. Circ Res 81: 870–878, 1997. [DOI] [PubMed] [Google Scholar]
- 691.London B, Wang DW, Hill JA, Bennett PB. The transient outward current in mice lacking the potassium channel gene Kv1.4. J Physiol 509: 171–182, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.London B. Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 12: 1089–1091, 2001. [DOI] [PubMed] [Google Scholar]
- 693.Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1). J Mol Cell Cardiol 33: 625–638, 2001. [DOI] [PubMed] [Google Scholar]
- 694.Lopez-Santiago LF, Meadows LS, Ernst SJ, Chen C, Malhotra JD, McEwen DP, Speelman A, Noebels JL, Maier SKG, Lopatin AN, Isom LL. Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J Mol Cell Cardiol 43: 636–647, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Lou Q, Li W, Efimov IR. The role of dynamic instability and wavelength in arrhythmia maintenance as revealed by panoramic imaging with blebbistatin vs. 2,3-butanedione monoxime. Am J Physiol Heart Circ Physiol 302: H262–H269, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Lu Y, Mahaut-Smith MP, Huang CLH, Vandenberg JI. Mutant MiRP1 subunits modulate HERG K+ channel gating: a mechanism for pro-arrhythmia in long QT syndrome type 6. J Physiol 551: 253–262, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Lu Y, Mahaut-Smith MP, Varghese A, Huang CLH, Kemp PR, Vandenberg JI. Effects of premature stimulation on HERG K(+) channels. J Physiol 537: 843–851, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Lu ZJ, Pereverzev A, Liu H, Weiergräber M, Henry M, Krieger A, Smyth N, Hescheler J, Schneider T. Arrhythmia in isolated prenatal hearts after ablation of the Cav2.3 (α1E) subunit of voltage-gated Ca2+ channels. Cell Physiol Biochem 14: 11–22, 2004. [DOI] [PubMed] [Google Scholar]
- 699.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216–224, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Ludwig A, Herrmann S, Hoesl E, Stieber J. Mouse models for studying pacemaker channel function and sinus node arrhythmia. Prog Biophys Mol Biol 98: 179–185, 2008. [DOI] [PubMed] [Google Scholar]
- 701.Lukas A, Antzelevitch C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current. Circulation 88: 2903–2915, 1993. [DOI] [PubMed] [Google Scholar]
- 702.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res 32: 593–603, 1996. [PubMed] [Google Scholar]
- 703.Luo J, Pripp CM, Hertervig E, Kongstad O, Ljungström E, Olsson SB, Yuan S. Non-invasive evaluation of ventricular refractoriness and its dispersion during ventricular fibrillation in patients with implantable cardioverter defibrillator. BMC Cardiovasc Disord 4: 8, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Luss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, Muller FU, Scheld HH, Schmid C, Schmitz W, Neumann J. Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J Mol Cell Cardiol 32: 2349–2359, 2000. [DOI] [PubMed] [Google Scholar]
- 705.Luxán G, Casanova JC, Martínez-Poveda B, Prados B, D'Amato G, MacGrogan D, Gonzalez-Rajal A, Dobarro D, Torroja C, Martinez F, Izquierdo-García JL, Fernández-Friera L, Sabater-Molina M, Kong YY, Pizarro G, Ibañez B, Medrano C, García-Pavía P, Gimeno JR, Monserrat L, Jiménez-Borreguero LJ, de la Pompa JL. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med 19: 193–201, 2013. [DOI] [PubMed] [Google Scholar]
- 706.Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, Gu Y, Evans SM, Omens JH, Peterson KL, McCulloch AD, Sheikh F. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet 23: 1134–1150, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Ma D, Wei H, Zhao Y, Lu J, Li G, Sahib NBE, Tan TH, Wong KY, Shim W, Wong P, Cook SA, Liew R. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol 168: 5277–5286, 2013. [DOI] [PubMed] [Google Scholar]
- 708.MacDonnell SM, Garcia-Rivas G, Scherman JA, Kubo H, Chen X, Valdivia H, Houser SR. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res 102: e65–e72, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.MacDougall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 196: 725–734, 1991. [DOI] [PubMed] [Google Scholar]
- 710.Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol 530: 417–429, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.MacLennan D, Chen S. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J Physiol 587: 3113–3115, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol 34: 919–939, 2002. [DOI] [PubMed] [Google Scholar]
- 713.Maier S, Westenbroek R, McCormick K, Curtis R, Scheuer T, Catterall W. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109: 1421–1427, 2004. [DOI] [PubMed] [Google Scholar]
- 714.Maier SKG, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, Catterall WA, Scheuer T. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA 100: 3507–3512, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Maisch B, Noutsias M, Ruppert V, Richter A, Pankuweit S. Cardiomyopathies: classification, diagnosis, treatment. Heart Fail Clin 8: 53–78, 2012. [DOI] [PubMed] [Google Scholar]
- 716.Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA, Wright PJ, Unudurthi SD, Snyder J, Bennett V, Hund TJ, Mohler PJ. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res 115: 929–938, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Makiyama T, Akao M, Tsuji K, Doi T, Ohno S, Takenaka K, Kobori A, Ninomiya T, Yoshida H, Takano M, Makita N, Yanagisawa F, Higashi Y, Takeyama Y, Kita T, Horie M. High risk for bradyarrhythmic complications in patients with Brugada Syndrome caused by SCN5A gene mutations. J Am Coll Cardiol 46: 2100–2106, 2005. [DOI] [PubMed] [Google Scholar]
- 718.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with Long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res 109: 841–847, 2011. [DOI] [PubMed] [Google Scholar]
- 719.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ 16: 335–348, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100: 5543–5548, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Mangoni ME, Couette B, Marger L, Bourinet E, Striessnig J, Nargeot J. Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol 90: 38–63, 2006. [DOI] [PubMed] [Google Scholar]
- 722.Mangoni ME, Nargeot J. Properties of the hyperpolarization-activated current (If) in isolated mouse sino-atrial cells. Cardiovasc Res 52: 51–64, 2001. [DOI] [PubMed] [Google Scholar]
- 723.Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev 88: 919–982, 2008. [DOI] [PubMed] [Google Scholar]
- 724.Mangoni ME, Traboulsie A, Leoni A, Couette B, Marger L, Le Quang K, Kupfer E, Cohen-Solal A, Vilar J, Shin H, Escande D, Charpentier F, Nargeot J, Lory P. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ Res 98: 1422–1430, 2006. [DOI] [PubMed] [Google Scholar]
- 725.Marian AJ, Wu Y, Lim DS, McCluggage M, Youker K, Yu QT, Brugada R, DeMayo F, Quinones M, Roberts R. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest 104: 1683–1692, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol 562: 223–234, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Markandeya YS, Fahey JM, Pluteanu F, Cribbs LL, Balijepalli RC. Caveolin-3 regulates protein kinase A modulation of the Ca(V)3.2 (alpha1H) T-type Ca2+ channels. J Biol Chem 286: 2433–2444, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Markandeya YS, Phelan LJ, Woon MT, Keefe AM, Reynolds CR, August BK, Hacker TA, Roth DM, Patel HH, Balijepalli RC. Caveolin-3 overexpression attenuates cardiac hypertrophy via inhibition of T-type Ca2+ current modulated by protein kinase C in cardiomyocytes. J Biol Chem 290: 22085–22100, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Marks A, Reiken S, Marx S. Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation 105: 272–275, 2002. [PubMed] [Google Scholar]
- 730.Marks AR. Ryanodine receptors/calcium release channels in heart failure and sudden cardiac death. J Mol Cell Cardiol 33: 615–624, 2001. [DOI] [PubMed] [Google Scholar]
- 731.de Marneffe M, Gregoire JM, Waterschoot P, Kestemont MP. The sinus node function: normal and pathological. Eur Heart J 14: 649–654, 1993. [DOI] [PubMed] [Google Scholar]
- 732.Maron F, Towbin J, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss A, Seidman C, Young J. Contemporary definitions and classification of the cardiomyopathies. Circulation 113: 1807–1816, 2006. [DOI] [PubMed] [Google Scholar]
- 733.Marshall P, Rouse W, Briggs I, Hargreaves R, Mills S, McLoughlin B. ICI D7288, a novel sinoatrial node modulator. J Cardiovasc Pharmacol 21: 902–906, 1993. [DOI] [PubMed] [Google Scholar]
- 734.Martin CA, Grace AA, Huang CLH. Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a (+/−) mouse model. Pflügers Arch 462: 495–504, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Martin CA, Grace AA, Huang CLH. Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am J Physiol Heart Circ Physiol 300: H605–H616, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Martin CA, Guzadhur L, Grace AA, Lei M, Huang CLH. Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/− mouse model. Am J Physiol Heart Circ Physiol 300: H1853–H1862, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Martin CA, Huang CLH, Grace AA. Progressive Conduction Diseases. Card Electrophysiol Clin North Am Saunders 2: 509–519, 2010. [DOI] [PubMed] [Google Scholar]
- 738.Martin CA, Huang CLH, Matthews GD. Recent developments in the management of patients at risk for sudden cardiac death. Postgrad Med 123: 84–94, 2011. [DOI] [PubMed] [Google Scholar]
- 739.Martin CA, Huang CLH, Matthews GDK. The role of ion channelopathies in sudden cardiac death: implications for clinical practice. Ann Med 45: 364–374, 2013. [DOI] [PubMed] [Google Scholar]
- 740.Martin CA, Matthews GDK, Huang CLH. Sudden cardiac death and inherited channelopathy: the basic electrophysiology of the myocyte and myocardium in ion channel disease. Heart 98: 536–543, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Martin CA, Siedlecka U, Kemmerich K, Lawrence J, Cartledge J, Guzadhur L, Brice N, Grace AA, Schwiening C, Terracciano CM, Huang CL-H. Reduced Na(+) and higher K(+) channel expression and function contribute to right ventricular origin of arrhythmias in Scn5a+/− mice. Open Biol 2: 120072, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Martin CA, Zhang Y, Grace AA, Huang CLH. In vivo studies of Scn5a+/− mice modeling Brugada syndrome demonstrate both conduction and repolarization abnormalities. J Electrocardiol 43: 433–439, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Martin CA, Zhang Y, Grace AA, Huang CLH. Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada Syndrome. J Cardiovasc Electrophysiol 21: 1153–1159, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Maruyama M, Li BYBY, Chen H, Xu X, Song LSLS, Guatimosim S, Zhu W, Yong W, Zhang W, Bu G, Lin SFSF, Fishbein MC, Lederer WJ, Schild JH, Field LJ, Rubart M, Chen PSPS, Shou W. FKBP12 is a critical regulator of the heart rhythm and the cardiac voltage-gated sodium current in mice. Circ Res 108: 1042–1052, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88: 1151–1158, 2001. [DOI] [PubMed] [Google Scholar]
- 746.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295: 496–499, 2002. [DOI] [PubMed] [Google Scholar]
- 747.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks a R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365–376, 2000. [DOI] [PubMed] [Google Scholar]
- 748.Masumiya H, Yamamoto H, Hemberger M, Tanaka H, Shigenobu K, Chen SR, Furukawa T. The mouse sino-atrial node expresses both the type 2 and type 3 Ca2+ release channels/ryanodine receptors. FEBS Lett 553: 141–144, 2003. [DOI] [PubMed] [Google Scholar]
- 749.Matsuo K, Akahoshi M, Nakashima E, Suyama A, Seto S, Hayano M, Yano K. The prevalence, incidence and prognostic value of the Brugada-type electrocardiogram: a population-based study of four decades. JACC 38: 765–770, 2001. [DOI] [PubMed] [Google Scholar]
- 750.Matsuoka N, Arakawa H, Kodama H, Yamaguchi I. Characterization of stress-induced sudden death in cardiomyopathic hamsters. J Pharmacol Exp Ther 284: 125–135, 1998. [PubMed] [Google Scholar]
- 751.Matthes J, Yildirim L, Wietzorrek G, Reimer D, Striessnig J, Herzig S. Disturbed atrio-ventricular conduction and normal contractile function in isolated hearts from Cav1.3-knockout mice. Naunyn-Schmiedebergs Arch Pharmacol 369: 554–562, 2004. [DOI] [PubMed] [Google Scholar]
- 752.Matthews GDK, Guzadhur L, Grace AA, Huang CLH. Nonlinearity between action potential alternans and restitution, which both predict ventricular arrhythmic properties in Scn5a+/− and wild-type murine hearts. J Appl Physiol 112: 1847–1863, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Matthews GDK, Guzadhur L, Sabir IN, Grace AA, Huang CLH. Action potential wavelength restitution predicts alternans and arrhythmia in murine Scn5a+/− hearts. J Physiol 591: 4167–4188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754.Matthews GDK, Martin CA, Grace AA, Zhang Y, Huang CLH. Regional variations in action potential alternans in isolated murine Scn5a (+/−) hearts during dynamic pacing. Acta Physiol 200: 129–146, 2010. [DOI] [PubMed] [Google Scholar]
- 755.Mauban JRH, O'Donnell M, Warrier S, Manni S, Bond M. AKAP-scaffolding proteins and regulation of cardiac physiology. Physiology 24: 78–87, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Maurer P, Weingart R. Cell pairs isolated from adult guinea pig and rat hearts: effects of [Ca2+]i on nexal membrane resistance. Pflügers Arch 409: 394–402, 1987. [DOI] [PubMed] [Google Scholar]
- 757.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355: 2119–2124, 2000. [DOI] [PubMed] [Google Scholar]
- 758.McNair WP, Ku L, Taylor MRG, Fain PR, Dao D, Wolfel E, Mestroni L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110: 2163–2167, 2004. [DOI] [PubMed] [Google Scholar]
- 759.Meadows LS, Isom LL. Sodium channels as macromolecular complexes: Implications for inherited arrhythmia syndromes. Cardiovasc Res 67: 448–458, 2005. [DOI] [PubMed] [Google Scholar]
- 760.Mechmann S, Pott L. Identification of Na-Ca exchange current in single cardiac myocytes. Nature 319: 597–599, 1986. [DOI] [PubMed] [Google Scholar]
- 761.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusié-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation 116: 134–142, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Mehta A, Jain AC, Mehta MC, Billie M. Caffeine and cardiac arrhythmias. An experimental study in dogs with review of literature. Acta Cardiol 52: 273–283, 1997. [PubMed] [Google Scholar]
- 763.Meli AC, Refaat MM, Dura M, Reiken S, Wronska A, Wojciak J, Carroll J, Scheinman MM, Marks AR. A novel ryanodine receptor mutation linked to sudden death increases sensitivity to cytosolic calcium. Circ Res 109: 281–290, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc Natl Acad Sci USA 95: 13893–13898, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Mercier A, Clément R, Harnois T, Bourmeyster N, Faivre JF, Findlay I, Chahine M, Bois P, Chatelier A. The β1-subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through α-α interaction. PLoS One 7: e48690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Meregalli PG, Wilde AAM, Tan HL. Pathophysiological mechanisms of Brugada syndrome: Depolarization disorder, repolarization disorder, or more? Cardiovasc Res 67: 367–378, 2005. [DOI] [PubMed] [Google Scholar]
- 767.Mesirca P, Marger L, Toyoda F, Rizzetto R, Audoubert M, Dubel S, Torrente AG, Difrancesco ML, Muller JC, Leoni AL, Couette B, Nargeot J, Clapham DE, Wickman K, Mangoni ME. The G-protein-gated K(+) channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J Gen Physiol 142: 113–126, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768.Mesirca P, Torrente AG, Mangoni ME. Functional role of voltage gated Ca2+ channels in heart automaticity. Front Physiol 6: 19, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769.Meurs KM, Lacombe VA, Dryburgh K, Fox PR, Reiser PR, Kittleson MD. Differential expression of the cardiac ryanodine receptor in normal and arrhythmogenic right ventricular cardiomyopathy canine hearts. Hum Genet 120: 111–118, 2006. [DOI] [PubMed] [Google Scholar]
- 770.Meurs KM, Norgard MM, Ederer MM, Hendrix KP, Kittleson MD. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 90: 261–264, 2007. [DOI] [PubMed] [Google Scholar]
- 771.Meurs KM, Sanchez X, David RM, Bowles NE, Towbin JA, Reiser PJ, Kittleson a J., Munro MJ, Dryburgh K, MacDonald AK, Kittleson MD. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet 14: 3587–3593, 2005. [DOI] [PubMed] [Google Scholar]
- 772.Milberg P, Eckardt L, Urgen H, Biertz J, Ramtin S, Reinsch N, Fleischer D, Kirchhof P, Fabritz L, Unter G, Haverkamp W. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther 303: 218–225, 2002. [DOI] [PubMed] [Google Scholar]
- 773.Milberg P, Reinsch N, Osada N, Wasmer K, Mönnig G, Stypmann J, Breithardt G, Haverkamp W, Eckardt L. Verapamil prevents torsade de pointes by reduction of transmural dispersion of repolarization and suppression of early afterdepolarizations in an intact heart model of LQT3. Basic Res Cardiol 100: 365–371, 2005. [DOI] [PubMed] [Google Scholar]
- 774.Milberg P, Reinsch N, Wasmer K, Mönnig G, Stypmann J, Osada N, Breithardt G, Haverkamp W, Eckardt L. Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res 65: 397–404, 2005. [DOI] [PubMed] [Google Scholar]
- 775.Milberg P, Tegelkamp R, Osada N, Schimpf R, Wolpert C, Breithardt G, Borggrefe M, Eckardt L. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol 18: 658–664, 2007. [DOI] [PubMed] [Google Scholar]
- 776.Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun 304: 1–4, 2003. [DOI] [PubMed] [Google Scholar]
- 777.Minamisawa S, Wang Y, Chen J, Ishikawa Y, Chien KR, Matsuoka R. Atrial chamber-specific expression of sarcolipin is regulated during development and hypertrophic remodeling. J Biol Chem 278: 9570–9575, 2003. [DOI] [PubMed] [Google Scholar]
- 778.Mines G. On dynamic equilibrium in the heart. J Physiol 46: 349–383, 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 779.Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati MA, Volonte D, Galbiati F, Cordone G, Bricarelli FD, Lisanti MP, Zara F. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 18: 365–368, 1998. [DOI] [PubMed] [Google Scholar]
- 780.Miquerol L, Meysen S, Mangoni M, Bois P, Van Rijen HVM, Abran P, Jongsma H, Nargeot J, Gros D. Architectural and functional asymmetry of the His-Purkinje system of the murine heart. Cardiovasc Res 63: 77–86, 2004. [DOI] [PubMed] [Google Scholar]
- 781.Misier AR, Opthof T, van Hemel NM, Vermeulen JT, de Bakker JM, Defauw JJ, van Capelle FJ, Janse MJ. Dispersion of ”refractoriness“ in noninfarcted myocardium of patients with ventricular tachycardia or ventricular fibrillation after myocardial infarction. Circulation 91: 2566–2572, 1995. [DOI] [PubMed] [Google Scholar]
- 782.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol Heart Circ Physiol 274: H747–H751, 1998. [DOI] [PubMed] [Google Scholar]
- 783.Mitsuiye T, Shinagawa Y, Noma A. Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Circ Res 87: 88–91, 2000. [DOI] [PubMed] [Google Scholar]
- 784.Miyazaki S, Shah AJ, Haïssaguerre M. Early repolarization syndrome: a new electrical disorder associated with sudden cardiac death. Circ J 74: 2039–2044, 2010. [DOI] [PubMed] [Google Scholar]
- 785.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol 49: 1722–1732, 2007. [DOI] [PubMed] [Google Scholar]
- 786.Mohamed U, Napolitano C, Priori SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol 18: 791–797, 2007. [DOI] [PubMed] [Google Scholar]
- 787.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci USA 101: 17533–17538, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogné K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421: 634–639, 2003. [DOI] [PubMed] [Google Scholar]
- 789.Mohler PJ, Le Scouarnec S, Denjoy I, Lowe JS, Guicheney P, Caron L, Driskell IM, Schott JJ, Norris K, Leenhardt A, Kim RB, Escande D, Roden DM. Defining the cellular phenotype of ”ankyrin-B syndrome“ variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation 115: 432–441, 2007. [DOI] [PubMed] [Google Scholar]
- 790.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci USA 101: 9137–9142, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Moise NS, Valentine BA, Brown CA, Erb HN, Beck KA, Cooper BJ, Gilmour RF. Duchenne's cardiomyopathy in a canine model: electrocardiographic and echocardiographic studies. J Am Coll Cardiol 17: 812–820, 1991. [DOI] [PubMed] [Google Scholar]
- 792.Mok NS, Chan NY, Chiu ACS. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol 27: 821–823, 2004. [DOI] [PubMed] [Google Scholar]
- 793.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res 63: 467–475, 2004. [DOI] [PubMed] [Google Scholar]
- 794.Moncoq K, Trieber CA, Young HS. The molecular basis for cyclopiazonic acid inhibition of the sarcoplasmic reticulum calcium pump. J Biol Chem 282: 9748–9757, 2007. [DOI] [PubMed] [Google Scholar]
- 795.Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J Am Coll Cardiol 29: 549–555, 1997. [DOI] [PubMed] [Google Scholar]
- 796.Moore HJ, Franz MR. Monophasic action potential recordings in humans. J Cardiovasc Electrophysiol 18: 787–790, 2007. [DOI] [PubMed] [Google Scholar]
- 797.Moorman AF, Schumacher CA, de Boer PA, Hagoort J, Bezstarosti K, van den Hoff MJ, Wagenaar GT, Lamers JM, Wuytack F, Christoffels VM, Fiolet JW. Presence of functional sarcoplasmic reticulum in the developing heart and its confinement to chamber myocardium. Dev Biol 223: 279–290, 2000. [DOI] [PubMed] [Google Scholar]
- 798.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompré AM, Vandecasteele G, Lezoualc'h F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res 97: 1296–1304, 2005. [DOI] [PubMed] [Google Scholar]
- 799.Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: Does VDSC acquire calmodulin-mediated Ca2+-sensitivity? Biochemistry 39: 1316–1323, 2000. [DOI] [PubMed] [Google Scholar]
- 800.Morita H, Morita ST, Nagase S, Banba K, Nishii N, Tani Y, Watanabe A, Nakamura K, Kusano KF, Emori T, Matsubara H, Hina K, Kita T, Ohe T. Ventricular arrhythmia induced by sodium channel blocker in patients with Brugada syndrome. J Am Coll Cardiol 42: 1624–1631, 2003. [DOI] [PubMed] [Google Scholar]
- 801.Morita H, Zipes DP, Fukushima-Kusano K, Nagase S, Nakamura K, Morita ST, Ohe T, Wu J. Repolarization heterogeneity in the right ventricular outflow tract: Correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm 5: 725–733, 2008. [DOI] [PubMed] [Google Scholar]
- 802.Morita H, Zipes DP, Morita ST, Wu J. Differences in arrhythmogenicity between the canine right ventricular outflow tract and anteroinferior right ventricle in a model of Brugada syndrome. Heart Rhythm 4: 66–74, 2007. [DOI] [PubMed] [Google Scholar]
- 803.Morley G, Vaidya D, Samie F, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol 10: 1361–1375, 1999. [DOI] [PubMed] [Google Scholar]
- 804.Moroni A, Gorza L, Beltrame M, Gravante B, Vaccari T, Bianchi ME, Altomare C, Longhi R, Heurteaux C, Vitadello M, Malgaroli A, DiFrancesco D. Hyperpolarization-activated cyclic nucleotide-gated channel 1 is a molecular determinant of the cardiac pacemaker current If. J Biol Chem 276: 29233–29241, 2001. [DOI] [PubMed] [Google Scholar]
- 805.Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS. Safety and efficacy of flecainide in subjects with long QT-3 syndrome (ΔKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol 10: 59–66, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann MH. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 92: 2929–2934, 1995. [DOI] [PubMed] [Google Scholar]
- 807.Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet 14: 2167–2180, 2005. [DOI] [PubMed] [Google Scholar]
- 808.Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal 1: re4, 2008. [DOI] [PubMed] [Google Scholar]
- 809.Murray KT, Hu NN, Daw JR, Shin HG, Watson MT, Mashburn AB, George AL Jr. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res 80: 370–376, 1997. [DOI] [PubMed] [Google Scholar]
- 810.Nademanee K, Raju H, de Noronha S, Papadakis M, Robinson L, Rothery S, Makita N, Kowase S, Boonmee N, Vitayakritsirikul V, Ratanarapee S, Sharma S, van der W AC, Christiansen M, Tan HL, Wilde AA, Nogami A, Sheppard MN, Veerakul GBE. Fibrosis, connexin-43, and conduction abnormalities in the Brugada Syndrome. J Am Coll Cardiol 66: 1976–1986, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Nagase S, Kusano K, Morita H, Fujimoto Y, Kakishita M, Nakamura K, Emori T, Matsubara HOT. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol 39: 1992–1995, 2002. [DOI] [PubMed] [Google Scholar]
- 812.Nagatomo T, January CT, Ye B, Abe H, Nakashima Y, Makielski JC. Rate-dependent QT shortening mechanism for the LQT3 ΔKPQ mutant. Cardiovasc Res 54: 624–629, 2002. [DOI] [PubMed] [Google Scholar]
- 813.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis 26: 99–109, 2000. [PubMed] [Google Scholar]
- 814.Namadurai S, Balasuriya D, Rajappa R, Wiemhöfer M, Stott K, Klingauf J, Edwardson JM, Chirgadze DY, Jackson AP. Crystal structure and molecular imaging of the Nav channel β3 subunit indicates a trimeric assembly. J Biol Chem 289: 10797–10811, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Namadurai S, Yereddi NR, Cusdin FS, Huang CLH, Chirgadze DY, Jackson AP. A new look at sodium channel β subunits. Open Biol 5: 140192, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 816.Napolitano C. Bridging the dimensions of research on cardiac ryanodine receptor mutations. J Cardiovasc Electrophysiol 24: 219–220, 2013. [DOI] [PubMed] [Google Scholar]
- 817.Narayan S, Kim J, Tate C, Berman B. Steep restitution of ventricular action potential duration and conduction slowing in human Brugada syndrome. Heart Rhythm 4: 1087–1089, 2007. [DOI] [PubMed] [Google Scholar]
- 818.Narayan SM, Franz MR, Lalani G, Kim J, Sastry A. T-wave alternans, restitution of human action potential duration, and outcome. J Am Coll Cardiol 50: 2385–2392, 2007. [DOI] [PubMed] [Google Scholar]
- 819.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythmia Electrophysiol 1: 62–73, 2008. [DOI] [PubMed] [Google Scholar]
- 820.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87: 425–456, 2007. [DOI] [PubMed] [Google Scholar]
- 821.Nattel S, Quantz MA. Pharmacological response of quinidine induced early afterdepolarisations in canine cardiac Purkinje fibres: insights into underlying ionic mechanisms. Cardiovasc Res 22: 808–817, 1988. [DOI] [PubMed] [Google Scholar]
- 822.Nattel S. Atrial electrophysiology and mechanisms of atrial fibrillation. J Cardiovasc Pharmacol Ther 8: S5–S11, 2003. [DOI] [PubMed] [Google Scholar]
- 823.Nearing BD, Huang AH, Verrier RL. Dynamic tracking of cardiac vulnerability by complex demodulation of the T wave. Science 252: 437–440, 1991. [DOI] [PubMed] [Google Scholar]
- 824.Neco P, Torrente AG, Mesirca P, Zorio E, Liu N, Priori SG, Napolitano C, Richard S, Benitah JP, Mangoni ME, Gómez AM. Paradoxical effect of increased diastolic Ca2+ release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126: 392–401, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Neels JG, Grimaldi PA. Physiological functions of peroxisome proliferator-activated receptor β. Physiol Rev 94: 795–858, 2014. [DOI] [PubMed] [Google Scholar]
- 826.Nerbonne JM, Guo W. Heterogeneous expression of voltage-gated potassium channels in the heart: roles in normal excitation and arrhythmias. J Cardiovasc Electrophysiol 13: 406–409, 2002. [DOI] [PubMed] [Google Scholar]
- 827.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253, 2005. [DOI] [PubMed] [Google Scholar]
- 828.Nerbonne JM, Nichols CG, Schwarz TL, Escande D. Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ Res 89: 944–956, 2001. [DOI] [PubMed] [Google Scholar]
- 829.Nerbonne JM. Molecular basis of functional voltage-gated K(+) channel diversity in the mammalian myocardium. J Physiol 525: 285–298, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Nerbonne JM. Studying cardiac arrhythmias in the mouse: a reasonable model for probing mechanisms? Trends Cardiovasc Med 14: 83–93, 2004. [DOI] [PubMed] [Google Scholar]
- 831.Nilles KM, London B. Knockin animal models of inherited arrhythmogenic diseases: what have we learned from them? J Cardiovasc Electrophysiol 18: 1117–1125, 2007. [DOI] [PubMed] [Google Scholar]
- 832.Ning F, Luo L, Ahmad S, Valli H, Jeevaratnam K, Wang T, Guzadhur L, Yang D, Fraser J, Huang CL, Ma A, Salvage S. The RyR2-P2328S mutation downregulates Na(v)1.5 producing arrhythmic substrate in murine ventricles. Pflügers Arch 468: 655–665, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 833.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol 48: 12–25, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 834.Niwa N, Yasui K, Opthof T, Takemura H, Shimizu A, Horiba M, Lee JK, Honjo H, Kamiya K, Kodama I. Cav3.2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am J Physiol Heart Circ Physiol 286: H2257–H2263, 2004. [DOI] [PubMed] [Google Scholar]
- 835.Nof E, Belhassen B, Arad M, Bhuiyan ZA, Antzelevitch C, Rosso R, Fogelman R, Luria D, El-Ani D, Mannens MMAM, Viskin S, Eldar M, Wilde AAM, Glikson M. Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm 8: 1546–1552, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Nof E, Lahat H, Constantini N, Luria D, Rosenfeld G, Eldar M, Pras E, Glikson M. A novel form of familial bidirectional ventricular tachycardia. Am J Cardiol 93: 231–234, 2004. [DOI] [PubMed] [Google Scholar]
- 837.Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115: 551–564, 2003. [DOI] [PubMed] [Google Scholar]
- 838.Nolasco JB, Dahlen RW. A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25: 191–196, 1968. [DOI] [PubMed] [Google Scholar]
- 839.Noma A, Tsuboi N. Dependence of junctional conductance on proton, calcium and magnesium ions in cardiac paired cells of guinea-pig. J Physiol 382: 193–211, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116: 2253–2259, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol 578: 315–326, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Nuss HB, Chiamvimonvat N, Pérez-García MT, Tomaselli GF, Marbán E. Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu 1) sodium channel alpha subunits expressed in Xenopus oocytes. J Gen Physiol 106: 1171–1191, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos a M, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med 7: 1021–1027, 2001. [DOI] [PubMed] [Google Scholar]
- 844.O'Malley HA, Isom LL. Sodium channel β subunits: emerging targets in channelopathies. Annu Rev Physiol 77: 481–504, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.O'Rourke B. Mitochondrial ion channels. Annu Rev Physiol 69: 19–49, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Oceandy D, Cartwright EJ, Emerson M, Prehar S, Baudoin FM, Zi M, Alatwi N, Venetucci L, Schuh K, Williams JC, Armesilla AL, Neyses L. Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation 115: 483–492, 2007. [DOI] [PubMed] [Google Scholar]
- 847.Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, Schofield L, Centracchio J, Zehender M, Peng X, Brunner M, Koren G. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm 9: 823–832, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann a P., Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 36: 493–500, 2000. [DOI] [PubMed] [Google Scholar]
- 849.Oestreich EA, Wang HA, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase C epsilon plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem 282: 5488–5495, 2007. [DOI] [PubMed] [Google Scholar]
- 850.Offord J, Catterall WA. Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel alpha subunits in rat muscle cells. Neuron 2: 1447–1452, 1989. [DOI] [PubMed] [Google Scholar]
- 851.Okudaira N, Kuwahara M, Hirata Y, Oku Y, Nishio H. A knock-in mouse model of N-terminal R420W mutation of cardiac ryanodine receptor exhibits arrhythmogenesis with abnormal calcium dynamics in cardiomyocytes. Biochem Biophys Res Commun 452: 665–668, 2014. [DOI] [PubMed] [Google Scholar]
- 852.Olesen MS, Jespersen T, Nielsen JB, Liang B, Møller DV, Hedley P, Christiansen M, Varró A, Olesen SP, Haunsø S, Schmitt N, Svendsen JH. Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res 89: 786–793, 2011. [DOI] [PubMed] [Google Scholar]
- 853.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet 15: 2185–2191, 2006. [DOI] [PubMed] [Google Scholar]
- 854.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293: 447–454, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855.Ono K, Ito H. Role of rapidly activating delayed rectifier K+ current in sinoatrial node pacemaker activity. Am J Physiol Heart Circ Physiol 269: H453–H462, 1995. [DOI] [PubMed] [Google Scholar]
- 856.Ono K, Yano M, Ohkusa T, Kohno M, Hisaoka T, Tanigawa T, Kobayashi S, Matsuzaki M. Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca2+ release in heart failure. Cardiovasc Res 48: 323–331, 2000. [DOI] [PubMed] [Google Scholar]
- 857.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XHT. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122: 2669–2679, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 858.van Opstal JM, Verduyn SC, Leunissen HDM, De Groot SHM, Wellens HJJ, Vos MA. Electrophysiological parameters indicative of sudden cardiac death in the dog with chronic complete AV-block. Cardiovasc Res 50: 354–361, 2001. [DOI] [PubMed] [Google Scholar]
- 859.Opthof T, Coronel R, Wilms-Schopman FJG, Plotnikov AN, Shlapakova IN, Danilo P, Rosen MR, Janse MJ. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: Tp-e interval does not reflect transmural dispersion. Heart Rhythm 4: 341–348, 2007. [DOI] [PubMed] [Google Scholar]
- 860.Orchard CH, Cingolani HE. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res 28: 1312–1319, 1994. [DOI] [PubMed] [Google Scholar]
- 861.Ostrom RS, Bundey RA, Insel PA. Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes. J Biol Chem 279: 19846–19853, 2004. [DOI] [PubMed] [Google Scholar]
- 862.Otagiri T, Kijima K, Osawa M, Ishii K, Makita N, Matoba R, Umetsu K, Hayasaka K. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr Res 64: 482–487, 2008. [DOI] [PubMed] [Google Scholar]
- 863.Oudit GY, Kassiri Z, Sah R, Ramirez RJ, Zobel C, Backx PH. The molecular physiology of the cardiac transient outward potassium current (I(to)) in normal and diseased myocardium. J Mol Cell Cardiol 33: 851–872, 2001. [DOI] [PubMed] [Google Scholar]
- 864.Oyehaug L, Loose K, Jølle GF, Røe ÅT, Sjaastad I, Christensen G, Sejersted OM, Louch WE. Synchrony of cardiomyocyte Ca2+ release is controlled by t-tubule organization, SR Ca2+ content, and ryanodine receptor Ca2+ sensitivity. Biophys J 104: 1685–1697, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 865.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, Lopez Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 39: 1007–1012, 2007. [DOI] [PubMed] [Google Scholar]
- 866.Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res 112: 849–862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 867.Papadatos GA, Wallerstein PMR, Head CEG, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AEO, Huang CLH, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 99: 6210–6215, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 868.Park D, Cerrone M, Morley GE, Vasquez C, Fowler S, Liu N, Bernstein SA, Liu FY, Zhang J, Rogers CS, Priori SG, Chinitz LA, Fishman GI. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 125: 403–412, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Park MK, Lee SH, Ho WK, Earm YE. Redox agents as a link between hypoxia and the responses of ionic channels in rabbit pulmonary vascular smooth muscle. Exp Physiol 80: 835–842, 1995. [DOI] [PubMed] [Google Scholar]
- 870.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 8: 185–194, 2007. [DOI] [PubMed] [Google Scholar]
- 871.Pashmforoush M, Lu J, Chen H, Amand T. Nkx2-5 pathways and congenital heart disease: loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart. Cell 117: 373–386, 2004. [DOI] [PubMed] [Google Scholar]
- 872.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99: 1385–1394, 1999. [DOI] [PubMed] [Google Scholar]
- 873.Patel C, Antzelevitch C. Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Heart Rhythm 5: 585–590, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 874.Patel C, Yan G, Antzelevitch C. Short QT syndrome: from bench to bedside. Circ Arrhythm Electrophysiol 3: 401–408, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 875.Patel SP, Campbell DL. Transient outward potassium current, ”Ito“, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol 569: 7–39, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Patel SP, Parai R, Parai R, Campbell DL. Regulation of Kv4.3 voltage-dependent gating kinetics by KChIP2 isoforms. J Physiol 557: 19–41, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 877.Patino GA, Claes LRF, Lopez-Santiago LF, Slat a E, Dondeti RSR, Chen C, O'Malley a H, Gray CBB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29: 10764–10778, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.Payandeh J, Scheuer T, Zheng N, Catterall AW. The crystal structure of a voltage-gated sodium channel. Nature 475: 353–358, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Pedersen TH, Gurung IS, Grace AA, Huang CLH. Calmodulin kinase II initiates arrhythmogenicity during metabolic acidification in murine hearts. Acta Physiol 197: 13–25, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 880.Pedersen TH, Huang CLH, Fraser JA. An analysis of the relationships between subthreshold electrical properties and excitability in skeletal muscle. J Gen Physiol 138: 73–93, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Peixoto PM, Ryu SY, Kinnally KW. Mitochondrial ion channels as therapeutic targets. FEBS Lett 584: 2142–2152, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol 5: 1–10, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Pereira L, Cheng H, Lao DH, Na L, Van Oort RJ, Brown JH, Wehrens XHT, Chen J, Bers DM. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127: 913–922, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Bénitah JP, Lezoualc'h F, Gómez AM. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 583: 685–694, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 885.Pereira L, Rehmann H, Lao DH, Erickson JR, Bossuyt J, Chen J, Bers DM. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc Natl Acad Sci USA 112: 3991–3996, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Perrin MJ, Angaran P, Laksman Z, Zhang H, Porepa LF, Rutberg J, James C, Krahn AD, Judge DP, Calkins H, Gollob MH. Exercise testing in asymptomatic gene carriers exposes a latent electrical substrate of arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 62: 1772–1779, 2013. [DOI] [PubMed] [Google Scholar]
- 887.Perry M, Ng C, Phan K, David E, Steer K, Hunter M, Mann S, Imtiaz M, Hill A, Ke Y, Vandenberg J. Rescue of protein expression defects may not be enough to abolish the pro-arrhythmic phenotype of long QT type 2 mutations. J Physiol 594: 4031–4049, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 888.Peters NS, Green CR, Poole-Wilson AP, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 88: 864–875, 1993. [DOI] [PubMed] [Google Scholar]
- 889.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108: 294–304, 2011. [DOI] [PubMed] [Google Scholar]
- 890.Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, Wang Y. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J Biol Chem 279: 15330–15338, 2004. [DOI] [PubMed] [Google Scholar]
- 891.Pezhouman A, Madahian S, Stepanyan H, Ghukasyan H, Qu Z, Belardinelli L, Karagueuzian HS. Selective inhibition of late sodium current suppresses ventricular tachycardia and fibrillation in intact rat hearts. Heart Rhythm 11: 492–501, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 892.Piacentino IIIV, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res 92: 651–658, 2003. [DOI] [PubMed] [Google Scholar]
- 893.Piao L, Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart is proarrhythmic. Basic Res Cardiol 102: 416–428, 2007. [DOI] [PubMed] [Google Scholar]
- 894.Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res 99: 740–748, 2006. [DOI] [PubMed] [Google Scholar]
- 895.Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation 92: 1169–1178, 1995. [DOI] [PubMed] [Google Scholar]
- 896.Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJB, de Bakker JMT, Tan HL, Valente M, Nava A, Wilde a M A, Moorman AFM, Thiene G, Bezzina CR. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med 206: 1787–1802, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 276: 30794–30802, 2001. [DOI] [PubMed] [Google Scholar]
- 898.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res 73: 641–647, 2007. [DOI] [PubMed] [Google Scholar]
- 899.Pitzalis MV, Anaclerio M, Iacoviello M, Forleo C, Guida P, Troccoli R, Massari F, Mastropasqua F, Sorrentino S, Manghisi A, Rizzon P. QT-interval prolongation in right precordial leads: an additional electrocardiographic hallmark of Brugada Syndrome. J Am Coll Cardiol 42: 1632–1637, 2003. [DOI] [PubMed] [Google Scholar]
- 900.Pizzale S, Gollob MH, Gow R, Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol 19: 1319–1321, 2008. [DOI] [PubMed] [Google Scholar]
- 901.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89–97, 2000. [DOI] [PubMed] [Google Scholar]
- 902.Plonsey R, Barr R. Bioelectricity: A Quantitative Approach (3rd ed). New York: Springer, 2007. [Google Scholar]
- 902a.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 13: 63–69, 1996. [DOI] [PubMed] [Google Scholar]
- 903.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14: 61–66, 2004. [DOI] [PubMed] [Google Scholar]
- 904.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation 98: 2404–2414, 1998. [DOI] [PubMed] [Google Scholar]
- 905.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res 88: 1159–1167, 2001. [DOI] [PubMed] [Google Scholar]
- 906.Portbury AL, Chandra R, Groelle M, McMillian MK, Elias A, Herlong JR, Rios M, Roffler-Tarlov S, Chikaraishi DM. Catecholamines act via a beta-adrenergic receptor to maintain fetal heart rate and survival. Am J Physiol Heart Circ Physiol 284: H2069–H2077, 2003. [DOI] [PubMed] [Google Scholar]
- 907.Postema PG, van Dessel PFHM, de Bakker JMT, Dekker LRC, Linnenbank AC, Hoogendijk MG, Coronel R, Tijssen JGP, Wilde AAM, Tan HL. Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ Arrhythm Electrophysiol 1: 379–386, 2008. [DOI] [PubMed] [Google Scholar]
- 908.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MMAM, Wilde AAM, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 91: e21–e26, 2002. [DOI] [PubMed] [Google Scholar]
- 909.Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, Dubosq-Bidot L, Sebillon P, Mannens MMAM, Guicheney P, Wilde AAM. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet 42: 863–870, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 910.Pratt CM, Ruberg S, Morganroth J, McNutt B, Woodward J, Harris S, Ruskin J, Moye L. Dose-response relation between terfenadine (Seldane) and the QTc interval on the scalar electrocardiogram: distinguishing a drug effect from spontaneous variability. Am Heart J 131: 472–480, 1996. [DOI] [PubMed] [Google Scholar]
- 911.Priori S, Pandit S, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, di Barletta M, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 96: 800–807, 2005. [DOI] [PubMed] [Google Scholar]
- 912.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, Bloise R, Giustetto C, De Nardis R, Grillo M, Ronchetti E, Faggiano G, Nastoli J. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105: 1342–1347, 2002. [DOI] [PubMed] [Google Scholar]
- 913.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FES, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106: 69–74, 2002. [DOI] [PubMed] [Google Scholar]
- 914.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome: the role of flecainide challenge. Circulation 102: 945–947, 2000. [DOI] [PubMed] [Google Scholar]
- 915.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, Moncalvo C, Tulipani C, Veia A, Bottelli G, Nastoli J. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 292: 1341–1344, 2004. [DOI] [PubMed] [Google Scholar]
- 916.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli AG. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103: 196–200, 2001. [DOI] [PubMed] [Google Scholar]
- 917.Priori SG, Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest 115: 2033–2038, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 918.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med 348: 1866–1874, 2003. [DOI] [PubMed] [Google Scholar]
- 919.Probst V, Kyndt F, Potet F, Trochu JN, Mialet G, Demolombe S, Schott JJ, Baró I, Escande D, Le Marec H. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary lenègre disease. J Am Coll Cardiol 41: 643–652, 2003. [DOI] [PubMed] [Google Scholar]
- 920.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res 94: 1083–1090, 2004. [DOI] [PubMed] [Google Scholar]
- 921.Pugh TD, Conklin MW, Evans TD, Polewski a M., Barbian HJ, Pass R, Anderson BD, Colman RJ, Eliceiri KW, Keely PJ, Weindruch R, Beasley TM, Anderson RM. A shift in energy metabolism anticipates the onset of sarcopenia in rhesus monkeys. Aging Cell 12: 672–681, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 922.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr Rev 24: 78–90, 2003. [DOI] [PubMed] [Google Scholar]
- 923.Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJJM, Dobrev D, Nattel S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 103: 845–854, 2008. [DOI] [PubMed] [Google Scholar]
- 924.Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P, Fill M. Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys J 97: 1961–1970, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 925.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol 131: 325–334, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 926.Qin N, D'Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem 270: 4762–4770, 2003. [DOI] [PubMed] [Google Scholar]
- 927.Qu Y, Isom LL, Westenbroek RE, Rogers JC, Tanada TN, McCormick KA, Scheuer T, Catterall WA. Modulation of cardiac Na(+) channel expression in Xenopus oocytes by beta 1 subunits. J Biol Chem 270: 25696–25701, 1995. [DOI] [PubMed] [Google Scholar]
- 928.Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation 102: 1664–1670, 2000. [DOI] [PubMed] [Google Scholar]
- 929.Qu Z, Garfinkel A, Weiss JN. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells, 2: multiple extrasystoles. Biophys J 91: 805–815, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.Qu Z, Karagueuzian HS, Garfinkel A, Weiss JN. Effects of Na+ channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol 286: H1310–H1321, 2004. [DOI] [PubMed] [Google Scholar]
- 931.Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol 77: 29–55, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932.Radicke S, Cotella D, Graf EM, Ravens U, Wettwer E. Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by Kv4.3. J Physiol 565: 751–756, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 933.Radicke S, Vaquero M, Caballero R, Gómez R, Núñez L, Tamargo J, Ravens U, Wettwer E, Delpón E. Effects of MiRP1 and DPP6 beta-subunits on the blockade induced by flecainide of Kv4.3/KChIP2 channels. Br J Pharmacol 154: 774–786, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 934.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev 50: 413–492, 1998. [PubMed] [Google Scholar]
- 935.Ramos-Franco J, Aguilar-Sanchez Y, Escobar A. Intact heart loose patch photolysis reveals ionic current kinetics during ventricular action potentials. Circ Res 118: 203–215, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Rappel W, Zaman J, Narayan S. Mechanisms for the termination of atrial fibrillation by localized ablation: computational and clinical studies. Circ Arrhythm Electrophysiol 8: 1325–1333, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 937.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science 267: 1831–1834, 1995. [DOI] [PubMed] [Google Scholar]
- 938.Rehnqvist N, Ericsson CG, Eriksson S, Olsson G, Svensson G. Comparative investigation of the antiarrhythmic effect of propafenone (Rytmonorm) and lidocaine in patients with ventricular arrhythmias during acute myocardial infarction. Acta Med Scand 216: 525–530, 1984. [DOI] [PubMed] [Google Scholar]
- 939.Remme CA, Verkerk AO, Nuyens D, van Ginneken ACG, van Brunschot S, Belterman CNW, Wilders R, van Roon MA, Tan HL, Wilde AAM, Carmeliet P, de Bakker JMT, Veldkamp MW, Bezzina CR. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114: 2584–2594, 2006. [DOI] [PubMed] [Google Scholar]
- 940.Remme CA, Wilde AAM, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med 18: 78–87, 2008. [DOI] [PubMed] [Google Scholar]
- 941.Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol 591: 4099–4116, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Remo BF, Giovannone S, Fishman GI. Connexin43 cardiac gap junction remodeling: lessons from genetically engineered murine models. J Membr Biol 245: 275–281, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 943.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skårdal K, Wisløff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XHT. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 110: 1474–1483, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Riehle C, Abel ED. PGC-1 proteins and heart failure. Trends Cardiovasc Med 22: 98–105, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 945.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina-Gomez G, Litwin SE, Lelliott CJ, Vidal-Puig A, Abel ED. PGC-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res 109: 783–793, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 946.Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp Physiol 81: 877–880, 1996. [DOI] [PubMed] [Google Scholar]
- 947.van Rijen HV, van Veen TA, van Kempen MJ, Wilms-Schopman FJ, Potse M, Krueger O, Willecke K, Opthof T, Jongsma HJ, de Bakker JM. Impaired conduction in the bundle branches of mouse hearts lacking the gap junction protein connexin40. Circulation 103: 1591–1598, 2001. [DOI] [PubMed] [Google Scholar]
- 948.van Rijen HVM, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, De Bakker JMT. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 109: 1048–1055, 2004. [DOI] [PubMed] [Google Scholar]
- 949.Riley G, Syeda F, Kirchhof P, Fabritz L. An introduction to murine models of atrial fibrillation. Front Physiol 3: 1–16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 950.Ripplinger C, Li W, Hadley J, Chen J, Rothenberg F, Lombardi R, Wickline S, Marian A, Efimov I. Enhanced transmural fiber rotation and connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res 101: 1049–1057, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res 103: 298–306, 2008. [DOI] [PubMed] [Google Scholar]
- 952.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res 95: 409–418, 2012. [DOI] [PubMed] [Google Scholar]
- 953.Roden D. Drug-induced prolongation of the QT interval. N Engl J Med 350: 1013–1022, 2004. [DOI] [PubMed] [Google Scholar]
- 954.Roden DM, Anderson ME. Proarrhythmia. Handb Exp Pharmacol 171: 73–97, 2006. [DOI] [PubMed] [Google Scholar]
- 955.Rodríguez-Sinovas A, Cinca J, Tapias A, Armadans L, Tresànchez M, Soler-Soler J. Lack of evidence of M cells in porcine left ventricular myocardium. Cardiovasc Res 33: 307–313, 1997. [DOI] [PubMed] [Google Scholar]
- 956.Roepke TK, Kontogeorgis A, Ovanez C, Xu X, Young JB, Purtell K, Goldstein PA, Christini DJ, Peters NS, Akar FG, Gutstein DE, Lerner DJ, Abbott GW. Targeted deletion of Kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f). FASEB J 22: 3648–3660, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 957.Rohr S, Kucera JP, Kléber AG. Slow conduction in cardiac tissue. I: Effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ Res 83: 781–794, 1998. [DOI] [PubMed] [Google Scholar]
- 958.Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res 62: 309–322, 2004. [DOI] [PubMed] [Google Scholar]
- 959.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm 6: 848–856, 2009. [DOI] [PubMed] [Google Scholar]
- 960.Romanello M, Padoan M, Franco L, Veronesi V, Moro L, D'Andrea P. Extracellular NAD(+) induces calcium signaling and apoptosis in human osteoblastic cells. Biochem Biophys Res Commun 285: 1226–1231, 2001. [DOI] [PubMed] [Google Scholar]
- 961.Romano C, Gemme G, Pongiglione R. Rare cardiac arrhythmias of the pediatric age. I. Repetitive paroxysmal tachycardia. Minerva Pediatr 15: 1155–1164, 1963. [PubMed] [Google Scholar]
- 962.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474–477, 1998. [DOI] [PubMed] [Google Scholar]
- 963.Rook MB, Evers MM, Vos MA, Bierhuizen MFA. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res 93: 12–23, 2012. [DOI] [PubMed] [Google Scholar]
- 964.Rosati B, Grau F, Rodriguez S, Li H, Nerbonne JM, McKinnon D. Concordant expression of KChIP2 mRNA, protein and transient outward current throughout the canine ventricle. J Physiol 548: 815–822, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, McKinnon D. Regulation of KChIP2 potassium channel β subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol 533: 119–125, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med 330: 235–241, 1994. [DOI] [PubMed] [Google Scholar]
- 967.Rossow CF, Dilly KW, Santana LF. Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circ Res 98: 1306–1313, 2006. [DOI] [PubMed] [Google Scholar]
- 968.Royer A, Van Veen TAB, Le Bouter S, Marionneau C, Griol-Charhbili V, Léoni AL, Steenman M, Van Rijen HVM, Demolombe S, Goddard CA, Richer C, Escoubet B, Jarry-Guichard T, Colledge WH, Gros D, De Bakker JMT, Grace AA, Escande D, Charpentier F. Mouse model of SCN5A-linked hereditary Lenègre's: disease age-related conduction slowing and myocardial fibrosis. Circulation 111: 1738–1746, 2005. [DOI] [PubMed] [Google Scholar]
- 969.Ruan H, Mitchell S, Vainoriene M, Lou Q, Xie LH, Ren S, Goldhaber JI, Wang Y. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation 116: 596–605, 2007. [DOI] [PubMed] [Google Scholar]
- 970.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 6: 337–348, 2009. [DOI] [PubMed] [Google Scholar]
- 971.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 115: 2305–2315, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972.Rubenstein DS, Lipsius SL. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res 64: 648–657, 1989. [DOI] [PubMed] [Google Scholar]
- 973.Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Günthert U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol 135: 215–225, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 974.Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-α. Am J Physiol Heart Circ Physiol 289: H1456–H1467, 2005. [DOI] [PubMed] [Google Scholar]
- 975.Saba S, Mehdi H, Mathier MA, Islam MZ, Salama G, London B. Effect of right ventricular versus Biventricular pacing on electrical remodeling in the normal heart. Circ Arrhythmia Electrophysiol 3: 79–87, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976.Saba S, Vanderbrink B, Luciano B, Aronovitz M, Berul C, Reddy S, Housman D, Mendelsohn M, Estes N 3rd, Wang P. Localization of the sites of conduction abnormalities in a mouse model of myotonic dystrophy. J Cardiovasc Electrophysiol 10: 1214–1220, 1999. [DOI] [PubMed] [Google Scholar]
- 977.Sabir IN, Fraser JA, Cass TR, Grace AA, Huang CLH. A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflügers Arch 454: 925–936, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 978.Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang CLH. The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflügers Arch 454: 209–222, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Sabir IN, Jones VL, Grace AA, Huang CLH. Restitution curves, alternans and ventricular arrhythmogenesis in murine hearts. Bull Br Soc Cardiovasc Res 21: 13–16, 2008. [Google Scholar]
- 980.Sabir IN, Killeen MJ, Goddard CA, Thomas G, Gray S, Grace AA, Huang CLH. Transient alterations in transmural repolarization gradients and arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. J Physiol 581: 277–289, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 981.Sabir IN, Killeen MJ, Grace AA, Huang CLH. Ventricular arrhythmogenesis: insights from murine models. Prog Biophys Mol Biol 98: 208–218, 2008. [DOI] [PubMed] [Google Scholar]
- 982.Sabir IN, Li LM, Grace AA, Huang CLH. Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflügers Arch 455: 653–666, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 983.Sabir IN, Li LM, Jones VJ, Goddard CA, Grace AA, Huang CLH. Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflügers Arch 455: 637–651, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984.Sabir IN, Ma N, Jones VJ, Goddard CA, Zhang Y, Kalin A, Grace AA, Huang CLH. Alternans in genetically modified Langendorff-perfused murine hearts modeling catecholaminergic polymorphic ventricular tachycardia. Front Physiol 1: 1–9, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Sabir IN, Matthews GDK, Huang CLH. Sudden arrhythmic death: from basic science to clinical practice. Front Physiol 4: 339, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986.Sabir IN, Usher-Smith JA, Huang CLH, Grace AA. Risk stratification for sudden cardiac death. Prog Biophys Mol Biol 98: 340–346, 2009. [DOI] [PubMed] [Google Scholar]
- 987.Sacher F, Meregalli P, Veltmann C, Field ME, Solnon A, Bru P, Abbey S, Jaïs P, Tan HL, Wolpert C, Lande G, Bertault V, Derval N, Babuty D, Lacroix D, Boveda S, Maury P, Hocini M, Clémenty J, Mabo P, Lemarec H, Mansourati J, Borggrefe M, Wilde A, Haïssaguerre M, Probst V. Are women with severely symptomatic brugada syndrome different from men? J Cardiovasc Electrophysiol 19: 1181–1185, 2008. [DOI] [PubMed] [Google Scholar]
- 988.Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, Ungeheuer N, Herting J, Shah AM, Maier LS, Sossalla S, Unsöld B. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J Mol Cell Cardiol 76: 94–105, 2014. [DOI] [PubMed] [Google Scholar]
- 989.Sah R, Ramirez RJ, Kaprielian R, Backx PH. Alterations in action potential profile enhance excitation-contraction coupling in rat cardiac myocytes. J Physiol 533: 201–214, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 990.Sah VP, Hoshijima M, Chien KR, Brown JH. Rho is required for Galpha(q) and alpha-1-adrenergic receptor signaling in cardiomyocytes. Dissociation of Ras and Rho pathways. J Biol Chem 271: 31185–31190, 1996. [DOI] [PubMed] [Google Scholar]
- 991.Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, Ross J, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 103: 1627–1634, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Said M, Becerra R, Palomeque J, Rinaldi G, Kaetzel MA, Diaz-Sylvester PL, Copello JA, Dedman JR, Mundiña-Weilenmann C, Vittone L, Mattiazzi A. Increased intracellular Ca2+ and SR Ca2+ load contribute to arrhythmias after acidosis in rat heart. Role of Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Heart Circ Physiol 295: H1669–H1683, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 993.Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-oka T, Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci USA 94: 13873–13878, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 994.Salama G, Baker L, Wolk R, Barhanin J, London B. Arrhythmia phenotype in mouse models of human long QT. J Interv Card Electrophysiol 24: 77–87, 2009. [DOI] [PubMed] [Google Scholar]
- 995.Salama G, London B. Mouse models of long QT syndrome. J Physiol 578: 43–53, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 996.Salvage SC, King JH, Chandrasekharan KH, Jafferji DIG, Guzadhur L, Matthews HR, Huang CLH, Fraser JA. Flecainide exerts paradoxical effects on sodium currents and atrial arrhythmia in murine RyR2-P2328S hearts. Acta Physiol 214: 361–375, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 997.Sanbe A, Nelson D, Gulick J, Setser E, Osinska H, Wang X, Hewett TE, Klevitsky R, Hayes E, Warshaw DM, Robbins J. In vivo analysis of an essential myosin light chain mutation linked to familial hypertrophic cardiomyopathy. Circ Res 87: 296–302, 2000. [DOI] [PubMed] [Google Scholar]
- 998.Sanders L, Rakovic S, Lowe M, Mattick PAD, Terrar DA. Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J Physiol 571: 639–649, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 999.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384: 80–83, 1996. [DOI] [PubMed] [Google Scholar]
- 1000.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81: 299–307, 1995. [DOI] [PubMed] [Google Scholar]
- 1001.Sanguinetti MC, Jurkiewicz NK. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflügers Arch 420: 180–186, 1992. [DOI] [PubMed] [Google Scholar]
- 1002.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 440: 463–469, 2006. [DOI] [PubMed] [Google Scholar]
- 1003.Sarquella-Brugada G, Campuzano O, Arbelo E, Brugada J, Brugada R. Brugada syndrome: clinical and genetic findings. Genet Med 18: 3–12, 2016. [DOI] [PubMed] [Google Scholar]
- 1004.Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A. Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ Res 99: 520–527, 2006. [DOI] [PubMed] [Google Scholar]
- 1005.Sato D, Xie LH, Sovari a A, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN, Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci USA 106: 2983–2988, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1006.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res 109: 193–201, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1007.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res 105: 523–526, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1008.Satoh H. Role of T-type Ca2+ channel inhibitors in the pacemaker depolarization in rabbit sino-atrial nodal cells. Gen Pharmacol 26: 581–587, 1995. [DOI] [PubMed] [Google Scholar]
- 1009.Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res 47: 11–22, 2000. [DOI] [PubMed] [Google Scholar]
- 1010.Saumarez RC, Pytkowski M, Sterlinski M, Bourke JP, Clague JR, Cobbe SM, Connelly DT, Griffith MJ, McKeown PP, McLeod K, Morgan JM, Sadoul N, Chojnowska L, Huang CLH, Grace AA. Paced ventricular electrogram fractionation predicts sudden cardiac death in hypertrophic cardiomyopathy. Eur Heart J 29: 1653–1661, 2008. [DOI] [PubMed] [Google Scholar]
- 1011.Saumarez RC, Pytkowski M, Sterlinski M, Hauer RNW, Derksen R, Lowe MD, Szwed H, Huang CLH, Ward DE, Camm AJ, Grace AA. Delayed paced ventricular activation in the long QT syndrome is associated with ventricular fibrillation. Heart Rhythm 3: 771–778, 2006. [DOI] [PubMed] [Google Scholar]
- 1012.Saumarez RC. Sudden death in noncoronary heart disease is associated with delayed paced ventricular activation. Circulation 107: 2595–2600, 2003. [DOI] [PubMed] [Google Scholar]
- 1013.Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292: H1561–H1567, 2007. [DOI] [PubMed] [Google Scholar]
- 1014.Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23: 459–466, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1015.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638, 2008. [DOI] [PubMed] [Google Scholar]
- 1016.Scheinman M. Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm 6: 1050–1058, 2009. [DOI] [PubMed] [Google Scholar]
- 1017.Schilling JM, Horikawa YT, Zemljic-Harpf AE, Vincent KP, Tyan L, Yu JK, McCulloch AD, Balijepalli RC, Patel HH, Roth DM. Electrophysiology and metabolism of caveolin-3-overexpressing mice. Basic Res Cardiol 111: 28, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1018.Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, Mende U, Lohse MJ, Warshaw D, Seidman CE, Seidman JG. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci USA 103: 14525–14530, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1019.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299: 1410–1413, 2003. [DOI] [PubMed] [Google Scholar]
- 1020.Schmitt N, Grunnet M, Olesen SP. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev 94: 609–653, 2014. [DOI] [PubMed] [Google Scholar]
- 1021.Schott J, Alshinawi C, Kyndt F, Probst V, Hoorntje T, Hulsbeek M, Wilde A, Escande D, Mannens M, Le Marec H. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23: 20–21, 1999. [DOI] [PubMed] [Google Scholar]
- 1022.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281: 108–111, 1998. [DOI] [PubMed] [Google Scholar]
- 1023.Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, Donnelly P, Vergnaud G, Bachner L, Moisan JP, Le Marec H, Pascal O. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am J Hum Genet 57: 1114–1122, 1995. [PMC free article] [PubMed] [Google Scholar]
- 1024.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev 91: 265–325, 2011. [DOI] [PubMed] [Google Scholar]
- 1025.Schouten VJ, ter Keurs HEDJ. The slow repolarization phase of the action potential in rat heart. J Physiol 360: 13–25, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1026.Schrickel JW, Kreuzberg MM, Ghanem A, Kim JS, Linhart M, Andrié R, Tiemann K, Nickenig G, Lewalter T, Willecke K. Normal impulse propagation in the atrioventricular conduction system of Cx30.2/Cx40 double deficient mice. J Mol Cell Cardiol 46: 644–652, 2009. [DOI] [PubMed] [Google Scholar]
- 1027.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat 21: 651–652, 2003. [DOI] [PubMed] [Google Scholar]
- 1028.Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest 111: 1537–1545, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1029.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Towbin JA, Denjoy I, Wilde A, Guicheney P, Zareba W, Robinson JL, Breithardt G, Keating MT, Schulze-Bahr E, Bloise R, Beggs AH, Brink P, Toivonen L, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Lehmann MH, Schwartz K, Coumel P. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103: 89–95, 2001. [DOI] [PubMed] [Google Scholar]
- 1030.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C. Prevalence of the congenital long-QT syndrome. Circulation 120: 1761–1767, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1031.Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med 259: 39–47, 2006. [DOI] [PubMed] [Google Scholar]
- 1032.Schwinger RH, Brixius K, Bavendiek U, Hoischen S, Müller-Ehmsen J, Bölck B, Erdmann E. Effect of cyclopiazonic acid on the force-frequency relationship in human nonfailing myocardium. J Pharmacol Exp Ther 283: 286–292, 1997. [PubMed] [Google Scholar]
- 1033.Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, Song LS, Le Marec H, Probst V, Schott JJ, Anderson ME, Mohler PJ. Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci USA 105: 15617–15622, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Sedlacek K, Stark K, Cunha SR, Pfeufer A, Weber S, Berger I, Perz S, Kaab S, Wichmann HE, Mohler PJ, Hengstenberg C, Jeron A. Common genetic variants in ANK2 modulate QT interval. Circ Cardiovasc Genet 1: 93–99, 2008. [DOI] [PubMed] [Google Scholar]
- 1035.Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem 264: 17816–17823, 1989. [PubMed] [Google Scholar]
- 1036.Sepp R, Severs NJ, Gourdie RG. Altered patterns of cardiac intercellular junction distribution in hypertrophic cardiomyopathy. Heart 76: 412–417, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1037.Sesti F, Abbott GW. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci USA 97: 10607, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1038.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 80: 9–19, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1039.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest 120: 4388–4398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1040.Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res 111: 708–717, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1041.Shanmugam M, Molina CE, Gao S, Severac-Bastide R, Fischmeister R, Babu GJ. Decreased sarcolipin protein expression and enhanced sarco(endo)plasmic reticulum Ca2+ uptake in human atrial fibrillation. Biochem Biophys Res Commun 410: 97–101, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1042.Shattock MJ, Bers DM. Rat vs. rabbit ventricle: Ca flux and intracellular Na assessed by ion-selective microelectrodes. Am J Physiol Cell Physiol 256: C813–C822, 1989. [DOI] [PubMed] [Google Scholar]
- 1043.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res 81: 727–741, 1997. [DOI] [PubMed] [Google Scholar]
- 1044.Sheehan KA, Ke Y, Wolska BM, Solaro RJ. Expression of active p21-activated kinase-1 induces Ca2+ flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am J Physiol Cell Physiol 296: C47–C58, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1045.Sheikh F, Bang ML, Lange S, Chen J. ”Z“eroing in on the role of Cypher in striated muscle function, signaling, and human disease. Trends Cardiovasc Med 17: 258–262, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1046.Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med 19: 182–190, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1047.Sheikh SM, Skepper JN, Chawla S, Vandenberg JI, Elneil S, Huang CLH. Normal conduction of surface action potentials in detubulated amphibian skeletal muscle fibres. J Physiol 535: 579–590, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1048.Shen JB, Jiang B, Pappano AJ. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J Pharmacol Exp Ther 294: 562–570, 2000. [PubMed] [Google Scholar]
- 1049.Shi R, Zhang Y, Yang C, Huang C, Zhou X, Qiang H, Grace AA, Huang CLH, Ma A. The cardiac sodium channel mutation delQKP 1507–1509 is associated with the expanding phenotypic spectrum of LQT3, conduction disorder, dilated cardiomyopathy, and high incidence of youth sudden death. Europace 10: 1329–1335, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1050.Shiferaw Y, Sato D, Karma A. Coupled dynamics of voltage and calcium in paced cardiac cells. Phys Rev E Stat Nonlin Soft Matter Phys 71: 021903, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1051.Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol 12: 1418–1421, 2001. [DOI] [PubMed] [Google Scholar]
- 1052.Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi a Aihara N, Takaki H, Sunagawa K, Kamakura S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol 11: 1320–1329, 2000. [DOI] [PubMed] [Google Scholar]
- 1053.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation 96: 2038–2047, 1997. [DOI] [PubMed] [Google Scholar]
- 1054.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99: 1499–1507, 1999. [DOI] [PubMed] [Google Scholar]
- 1055.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol 35: 778–786, 2000. [DOI] [PubMed] [Google Scholar]
- 1056.Shimizu W, Antzelevitch C. Effects of a K+ channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation 102: 706–712, 2000. [DOI] [PubMed] [Google Scholar]
- 1057.Shimizu W, Matsuo K, Kokubo Y, Satomi K, Kurita T, Noda T, Nagaya N, Suyama K, Aihara N, Kamakura S, Inamoto N, Akahoshi M, Tomoike H. Sex hormone and gender difference–Role of testosterone on male predominance in Brugada syndrome. J Cardiovasc Electrophysiol 18: 415–421, 2007. [DOI] [PubMed] [Google Scholar]
- 1058.Shimizu W, Ohe T, Kurita T, Shimomura K. Differential response of QTU interval to exercise, isoproterenol, and atrial pacing in patients with congenital long QT syndrome. Pacing Clin Electrophysiol 14: 1966–1970, 1991. [DOI] [PubMed] [Google Scholar]
- 1059.Shimizu W, Ohe T, Kurita T, Takaki H, Aihara N, Kamakura S, Matsuhisa M, Shimomura K. Early afterdepolarizations induced by isoproterenol in patients with congenital long QT syndrome. Circulation 84: 1915–1923, 1991. [DOI] [PubMed] [Google Scholar]
- 1060.Shimoni Y, Clark RB, Giles WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol 448: 709–727, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1061.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature 391: 489–492, 1998. [DOI] [PubMed] [Google Scholar]
- 1062.Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res 99: 600–611, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063.Shy D, Gillet L, Ogrodnik J, Albesa M, Verkerk AO, Wolswinkel R, Rougier JS, Barc J, Essers MC, Syam N, Marsman RF, van Mil AM, Rotman S, Redon R, Bezzina CR, Remme CA, Abriel H. PDZ domain-binding motif regulates cardiomyocyte compartment-specific NaV1.5 channel expression and function. Circulation 130: 147–160, 2014. [DOI] [PubMed] [Google Scholar]
- 1064.Sikkel MB, Collins TP, Rowlands C, Shah M, O'Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 98: 286–296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1065.Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr Biol 8: 295–298, 1998. [DOI] [PubMed] [Google Scholar]
- 1066.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renström E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschütz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig J. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430–1439, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1067.Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res 76: 102–109, 1995. [DOI] [PubMed] [Google Scholar]
- 1068.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, Vos MA. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 102: 2137–2144, 2000. [DOI] [PubMed] [Google Scholar]
- 1069.Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by luminal Ca2+. J Membr Biol 137: 215–226, 1994. [DOI] [PubMed] [Google Scholar]
- 1070.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379: 833–836, 1996. [DOI] [PubMed] [Google Scholar]
- 1071.Smith S, Curran J, Hund TJ, Mohler PJ. Defects in cytoskeletal signaling pathways, arrhythmia, and sudden cardiac death. Front Physiol 3: 1–6, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1072.Smith SA, Sturm AC, Curran J, Kline CF, Little SC, Bonilla IM, Long VP, Makara M, Polina I, Hughes LD, Webb TR, Wei Z, Wright P, Voigt N, Bhakta D, Spoonamore KG, Zhang C, Weiss R, Binkley PF, Janssen PM, Kilic A, Higgins RS, Sun M, Ma J, Dobrev D, Zhang M, Carnes CA, Vatta M, Rasband MN, Hund TJ, Mohler PJ. Dysfunction in the βII spectrin-dependent cytoskeleton underlies human arrhythmia. Circulation 131: 695–708, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1073.Smits JPP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan a Z, Mannens MMAM, Balser JR, Tan HL, Bezzina CR, Wilde AAM. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol 38: 969–981, 2005. [DOI] [PubMed] [Google Scholar]
- 1074.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DYR, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest 120: 266–279, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1075.Sohal DS, Nghiem M, Crackower a M, Witt a S, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25, 2001. [DOI] [PubMed] [Google Scholar]
- 1076.Sommese L, Valverde C, Blanco P, Castro M, Rueda O, Kaetzel M, Dedman J, Anderson ME, Mattiazzi APJ. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca2+ release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int J Cardiol 202: 394–406, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1077.Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner AD, Berul CI, Eldar M, Seidman CE, Seidman JG. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 117: 1814–1823, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner AD, Berul CI, Eldar M, Seidman CE, Seidman JG. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 117: 1814–1823, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1079.Song Y, Belardinelli L. ATP promotes development of afterdepolarizations and triggered activity in cardiac myocytes. Am J Physiol Heart Circ Physiol 267: H2005–H2011, 1994. [DOI] [PubMed] [Google Scholar]
- 1080.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci USA 104: 5223–5228, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1081.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XHT. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 5: 1047–1054, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1082.Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioce P, Sargiacomo M, Bricarelli FD, Minetti C, Sudol M, Lisantia MP. Caveolin-3 directly interacts with the C-terminal tail of β-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem 275: 38048–38058, 2000. [DOI] [PubMed] [Google Scholar]
- 1083.Spach MS, Kootsey JM. The nature of electrical propagation in cardiac muscle. Am J Physiol Heart Circ Physiol 244: H3–H22, 1983. [DOI] [PubMed] [Google Scholar]
- 1084.Spector P. Principles of cardiac electric propagation and their implications for re-entrant arrhythmias. Circ Arrhythm Electrophysiol 6: 655–661, 2013. [DOI] [PubMed] [Google Scholar]
- 1085.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss a J, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102: 1178–1185, 2000. [DOI] [PubMed] [Google Scholar]
- 1086.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19–31, 2004. [DOI] [PubMed] [Google Scholar]
- 1087.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress lKs function. Nat Genet 17: 338–340, 1997. [DOI] [PubMed] [Google Scholar]
- 1088.Stambler BS, Fenelon G, Shepard RK, Clemo HF, Guiraudon CM. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol 14: 499–507, 2003. [DOI] [PubMed] [Google Scholar]
- 1089.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem 278: 51693–51702, 2003. [DOI] [PubMed] [Google Scholar]
- 1090.Starmer CF, Colatsky TJ, Grant AO. What happens when cardiac Na channels lose their function? 1. Numerical studies of the vulnerable period in tissue expressing mutant channels. Cardiovasc Res 57: 82–91, 2003. [DOI] [PubMed] [Google Scholar]
- 1091.Stein M, van Veen TAB, Remme CA, Boulaksil M, Noorman M, van Stuijvenberg L, van der Nagel R, Bezzina CR, Hauer RNW, de Bakker JMT, van Rijen HVM. Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovasc Res 83: 52–60, 2009. [DOI] [PubMed] [Google Scholar]
- 1092.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J 63: 497–517, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1093.Stewart S, Hart CL, Hole DJ, McMurray JJV. A population-based study of the long-term risks associated with atrial fibrillation: 20-Year follow-up of the Renfrew/Paisley study. Am J Med 113: 359–364, 2002. [DOI] [PubMed] [Google Scholar]
- 1094.Stieber J, Herrmann S, Feil S, Löster J, Feil R, Biel M, Hofmann F, Ludwig A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci USA 100: 15235–15240, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1095.Stokoe KS, Balasubramaniam R, Goddard CA, Colledge WH, Grace AA, Huang CLH. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/− murine hearts modelling the Brugada syndrome. J Physiol 581: 255–275, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CLH. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Delta murine hearts modelling long QT syndrome 3. J Physiol 578: 69–84, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1097.Strutz-Seebohm N, Pusch M, Wolf S, Stoll R, Tapken D, Gerwert K, Attali B, Seebohm G. Structural basis of slow activation gating in the cardiac IKs channel complex. Cell Physiol Biochem 27: 443–452, 2011. [DOI] [PubMed] [Google Scholar]
- 1098.Suetomi T, Yano M, Uchinoumi H, Fukuda M, Hino A, Ono M, Xu X, Tateishi H, Okuda S, Doi M, Kobayashi S, Ikeda Y, Yamamoto T, Ikemoto N, Matsuzaki M. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2+ release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation 124: 682–694, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1099.Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, Saito a Kurosaki K, Jouo K, Koujiro M, Konishi S, Matsuoka S, Oono T, Hayakawa S, Miura M, Ushinohama H, Shibata T, Niimura I. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89: 66–70, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1100.Sun L, Adebanjo OA, Koval A, Anandatheerthavarada HK, Iqbal J, Wu XY, Moonga BS, Wu XB, Biswas G, Bevis PJR, Kumegawa M, Epstein S, Huang CLH, Avadhani NG, Abe E, Zaidi M. A novel mechanism for coupling cellular intermediary metabolism to cytosolic Ca2+ signaling via CD38/ADP-ribosyl cyclase, a putative intracellular NAD+ sensor. FASEB J 16: 302–314, 2002. [DOI] [PubMed] [Google Scholar]
- 1101.Sung Y, Baek I, Kim D, Jeon J, Lee J, Lee K, Jeong D, Kim J, Lee H. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31: 23–24, 2013. [DOI] [PubMed] [Google Scholar]
- 1102.Sussman MA, Welch S, Walker A, Klevitsky R, Hewett TE, Price RL, Schaefer E, Yager K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J Clin Invest 105: 875–886, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1103.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: Linking heart failure and arrhythmias. Circ Res 110: 1661–1677, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1104.Swan H, Piippo K, Viitasalo M, Heikkilä P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol 34: 2035–2042, 1999. [DOI] [PubMed] [Google Scholar]
- 1105.Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol 32: 1056–1067, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1106.Taggart P, Sutton P, Opthof T, Coronel R, Kallis P. Electrotonic cancellation of transmural electrical gradients in the left ventricle in man. Prog Biophys Mol Biol 82: 243–254, 2003. [DOI] [PubMed] [Google Scholar]
- 1107.Takahashi S, Kato Y, Adachi M, Agata N, Tanaka H, Shigenobu K. Effects of cyclopiazonic acid on rat myocardium: inhibition of calcium uptake into sarcoplasmic reticulum. J Pharmacol Exp Ther 272: 1095–1100, 1995. [PubMed] [Google Scholar]
- 1108.Takeuchi S, Takagishi Y, Yasui K, Murata Y, Toyama J, Kodama I. Voltage-gated K+ channel, Kv42, localizes predominantly to the transverse-axial tubular system of the rat myocyte J Mol Cell Cardiol 32: 1361–1369, 2000. [DOI] [PubMed] [Google Scholar]
- 1109.Tamaddon HS, Vaidya D, Simon AM, Paul DL, Jalife J, Morley GE. High-resolution optical mapping of the right bundle branch in connexin40 knockout mice reveals slow conduction in the specialized conduction system. Circ Res 87: 929–936, 2000. [DOI] [PubMed] [Google Scholar]
- 1110.Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, Makielski JC, Ackerman MJ. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7: 771–778, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1111.Tan HL, Bezzina CR, Smits JPP, Verkerk AO, Wilde AAM. Genetic control of sodium channel function. Cardiovasc Res 57: 961–973, 2003. [DOI] [PubMed] [Google Scholar]
- 1112.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AAM, Anderson ME, Balser JR. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415: 442–447, 2002. [DOI] [PubMed] [Google Scholar]
- 1113.Tang Y, Tian X, Wang R, Fill M, Chen S. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res 110: 968–977, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1114.Taouis M, Sheldon RS, Duff HJ. Upregulation of the rat cardiac sodium channel by in vivo treatment with a class I antiarrhythmic drug. J Clin Invest 88: 375–378, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1115.Tellez JO, Mczewski M, Yanni J, Sutyagin P, Mackiewicz U, Atkinson A, Inada S, Beresewicz A, Billeter R, Dobrzynski H, Boyett MR. Ageing-dependent remodelling of ion channel and Ca2+ clock genes underlying sino-atrial node pacemaking. Exp Physiol 96: 1163–1178, 2011. [DOI] [PubMed] [Google Scholar]
- 1116.Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D, Barajas-Martinez H, Antzelevitch C, Lüscher TF, Abriel H, Duru F. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J 32: 1077–1088, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1117.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103: 1466–1472, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1118.Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley JM, Bist K, Choi BR, Karma A, Koren G. Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 Syndrome. Circ Res 115: 919–928, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol 552: 109–118, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Terentyev D, Viatchenko-Karpinski S, Györke I, Volpe P, Williams SC, Györke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA 100: 11759–11764, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1121.Terrar D, Rigg L. What determines the initiation of the heartbeat? J Physiol 524: 316, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1122.Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol 49: 240–246, 2007. [DOI] [PubMed] [Google Scholar]
- 1123.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clin Proc 79: 1380–1384, 2004. [DOI] [PubMed] [Google Scholar]
- 1123a.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol 47: 764–768, 2006. [DOI] [PubMed] [Google Scholar]
- 1124.Teutsch C, Kondo RP, Dederko DA, Chrast J, Chien KR, Giles WR. Spatial distributions of Kv4 channels and KChip2 isoforms in the murine heart based on laser capture microdissection. Cardiovasc Res 73: 739–749, 2007. [DOI] [PubMed] [Google Scholar]
- 1125.Thiel WH, Chen B, Hund TJ, Koval OM, Purohit A, Song LS, Mohler PJ, Anderson ME. Proarrhythmic defects in timothy syndrome require calmodulin kinase II. Circulation 118: 2225–2234, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126.Thollon C, Bidouard JP, Cambarrat C, Lesage L, Reure H, Delescluse I, Vian J, Peglion JL, Vilaine JP. Stereospecific in vitro and in vivo effects of the new sinus node inhibitor (+)-S 16257. Eur J Pharmacol 339: 43–51, 1997. [DOI] [PubMed] [Google Scholar]
- 1127.Thollon C, Cambarrat C, Vian J, Prost JF, Peglion JL, Vilaine JP. Electrophysiological effects of S 16257, a novel sino-atrial node modulator, on rabbit and guinea-pig cardiac preparations: comparison with UL-FS 49. Br J Pharmacol 112: 37–42, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128.Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CLH. Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiol 578: 85–97, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1129.Thomas G, Killeen MJ, Grace AA, Huang CLH. Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in deltaKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol 192: 505–517, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1130.Thomas G, Killeen MJ, Gurung IS, Hakim P, Balasubramaniam R, Goddard CA, Grace AA, Huang CLH. Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J Physiol 578: 99–114, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1131.Thomas NL, George CH, Lai FA. Functional heterogeneity of ryanodine receptor mutations associated with sudden cardiac death. Cardiovasc Res 64: 52–60, 2004. [DOI] [PubMed] [Google Scholar]
- 1132.Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME, Saffitz JE. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction. Circulation 97: 686–691, 1998. [DOI] [PubMed] [Google Scholar]
- 1133.Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem 271: 20385–20391, 1996. [DOI] [PubMed] [Google Scholar]
- 1134.Tipparaju SM, Liu SQ, Barski OA, Bhatnagar A. NADPH binding to beta-subunit regulates inactivation of voltage-gated K(+) channels. Biochem Biophys Res Commun 359: 269–276, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1135.Tiso N, Salamon M, Bagattin A, Danieli GA, Argenton F, Bortolussi M. The binding of the RyR2 calcium channel to its gating protein FKBP12.6 is oppositely affected by ARVD2 and VTSIP mutations. Biochem Biophys Res Commun 299: 594–598, 2002. [DOI] [PubMed] [Google Scholar]
- 1136.Tohse N, Kameyama M, Irisawa H. Intracellular Ca2+ and protein kinase C modulate K+ current in guinea pig heart cells. Am J Physiol Heart Circ Physiol 253: H1321–H1324, 1987. [DOI] [PubMed] [Google Scholar]
- 1137.Tohse N. Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am J Physiol Heart Circ Physiol 258: H1200–H1207, 1990. [DOI] [PubMed] [Google Scholar]
- 1138.Tosaka T, Casimiro MC, Rong Q, Tella S, Oh M, Katchman AN, Pezzullo JC, Pfeifer K, Ebert SN. Nicotine induces a long QT phenotype in Kcnq1-deficient mouse hearts. J Pharmacol Exp Ther 306: 980–987, 2003. [DOI] [PubMed] [Google Scholar]
- 1139.Trafford AW, Sibbring GC, Díaz ME, Eisner DA. The effects of low concentrations of caffeine on spontaneous Ca release in isolated rat ventricular myocytes. Cell Calcium 28: 269–276, 2000. [DOI] [PubMed] [Google Scholar]
- 1140.Trayanova NA. Mathematical approaches to understanding and imaging atrial fibrillation: Significance for mechanisms and management. Circ Res 114: 1516–1531, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Trenor B, Romero L, Ferrero J, Saiz J, Molto G, Hernandez V. Dispersion of refractoriness in a simulated ischemic 2D tissue and implications in vulnerability to reentry. Comput Cardiol 34: 313–316, 2007. [Google Scholar]
- 1142.Triggle DJ. 1,4-Dihydropyridines as calcium channel ligands and privileged structures. Cell Mol Neurobiol 23: 293–303, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1143.Tristani-Firouzi M, Etheridge SP. Kir2.1 channelopathies: The Andersen-Tawil syndrome. Pflügers Arch 460: 289–294, 2010. [DOI] [PubMed] [Google Scholar]
- 1144.Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest 110: 381–388, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1145.Tsai C, Tseng C, Hwang J, Wu C, Yu C, Wang Y, Chen W, Lai L, Chiang F, Lin J. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor β1. Cardiovasc Res 89: 805–815, 2011. [DOI] [PubMed] [Google Scholar]
- 1146.Tsai CT, Lai LP, Kuo KT, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Tseng YZ, Chiang FT, Lin JL. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodeling. Circulation 117: 344–355, 2008. [DOI] [PubMed] [Google Scholar]
- 1147.Tsuji Y, Opthof T, Yasui K, Inden Y, Takemura H, Niwa N, Lu Z, Lee JK, Honjo H, Kamiya K, Kodama I. Ionic mechanisms of acquired QT prolongation and torsades de pointes in rabbits with chronic complete atrioventricular block. Circulation 106: 2012–2018, 2002. [DOI] [PubMed] [Google Scholar]
- 1148.Tukkie R, Sogaard P, Vleugels J, De Groot IKLM, Wilde AAM, Tan HL. Delay in right ventricular activation contributes to Brugada Syndrome. Circulation 109: 1272–1277, 2004. [DOI] [PubMed] [Google Scholar]
- 1149.Tung RT, Shen WK, Hammill SC, Gersh BJ. Idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors. Pacing Clin Electrophysiol 17: 1405–1412, 1994. [DOI] [PubMed] [Google Scholar]
- 1150.Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci USA 105: 12557–12562, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1151.Tupling AR, Asahi M, MacLennan DH. Sarcolipin overexpression in rat slow twitch muscle inhibits sarcoplasmic reticulum Ca2+ uptake and impairs contractile function. J Biol Chem 277: 44740–44746, 2002. [DOI] [PubMed] [Google Scholar]
- 1152.Turakhia M, Tseng ZH. Sudden cardiac death: Epidemiology, mechanisms, and therapy. Curr Probl Cardiol 32: 501–546, 2007. [DOI] [PubMed] [Google Scholar]
- 1153.ten Tusscher KH, Noble D, Noble PJ, Panfilov AV. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol 286: H1573–H1589, 2004. [DOI] [PubMed] [Google Scholar]
- 1154.ten Tusscher KHWJ, Bernus O, Hren R, Panfilov AV. Comparison of electrophysiological models for human ventricular cells and tissues. Prog Biophys Mol Biol 90: 326–345, 2006. [DOI] [PubMed] [Google Scholar]
- 1155.ten Tusscher KHWJ, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol 291: H1088–H1100, 2006. [DOI] [PubMed] [Google Scholar]
- 1156.Uchinoumi H, Yano M, Ohno M, Xu X, Tateishi H, Kobayashi S. Enhanced sensitivity of the cardiac ryanodine receptor to activation by luminal Ca2+ as a primary cause of catecholaminergic polymorphic ventricular tachycardia. Circulation 116: II-153, 2007. [Google Scholar]
- 1157.Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res 106: 1413–1424, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1158.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci USA 105: 9355–9360, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Ugarte G, Brandan E. Transforming growth factor beta (TGF-beta) signaling is regulated by electrical activity in skeletal muscle cells. TGF-beta type I receptor is transcriptionally regulated by myotube excitability. J Biol Chem 281: 18473–18481, 2006. [DOI] [PubMed] [Google Scholar]
- 1160.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646, 2010. [DOI] [PubMed] [Google Scholar]
- 1161.Usher-Smith JA, Xu W, Fraser JA, Huang CLH. Alterations in calcium homeostasis reduce membrane excitability in amphibian skeletal muscle. Pflügers Arch 453: 211–221, 2006. [DOI] [PubMed] [Google Scholar]
- 1162.Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart. A challenge to the critical mass hypothesis. Circ Res 85: 174–181, 1999. [DOI] [PubMed] [Google Scholar]
- 1163.Vaidyanathan R, O'Connell RP, Deo M, Milstein ML, Furspan P, Herron TJ, Pandit SV, Musa H, Berenfeld O, Jalife J, Anumonwo JMB. The ionic bases of the action potential in isolated mouse cardiac Purkinje cell. Heart Rhythm 10: 80–87, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Moll Cell Cardiol 38: 475–483, 2005. [DOI] [PubMed] [Google Scholar]
- 1165.Valdivia CR, Medeiros-Domingo A, Ye B, Shen WK, Algiers TJ, Ackerman MJ, Makielski JC. Loss-of-function mutation of the SCN3B-encoded sodium channel 3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res 86: 392–400, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166.Valdivia CR, Nagatomo T, Makielski JC. Late Na currents affected by alpha subunit isoform and beta1 subunit co-expression in HEK293 cells. J Mol Cell Cardiol 34: 1029–1039, 2002. [DOI] [PubMed] [Google Scholar]
- 1167.Valle G, Galla D, Nori A, Priori SG, Gyorke S, de Filippis V, Volpe P. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem J 413: 291–303, 2008. [DOI] [PubMed] [Google Scholar]
- 1168.Vandenberg JI, Metcalfe JC, Grace AA. Mechanisms of pHi recovery after global ischemia in the perfused heart. Circ Res 72: 993–1003, 1993. [DOI] [PubMed] [Google Scholar]
- 1169.Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function, and clinical significance. Physiol Rev 92: 1393–1478, 2012. [DOI] [PubMed] [Google Scholar]
- 1170.Vandenberg JI, Varghese A, Lu Y, Bursill JA, Mahaut-Smith MP, Huang CLH. Temperature dependence of human ether-a-go-go-related gene K+ currents. Am J Physiol Cell Physiol 291: C165–C175, 2006. [DOI] [PubMed] [Google Scholar]
- 1171.Vandenberg JI, Walker BD, Campbell TJ. HERG K+ channels: friend and foe. Trends Pharmacol Sci 22: 240–246, 2001. [DOI] [PubMed] [Google Scholar]
- 1172.VanderBrink BA, Sellitto C, Saba S, Link MS, Zhu W, Homoud MK, Estes NA 3rd, Paul DL, Wang PJ. Connexin40-deficient mice exhibit atrioventricular nodal and infra-Hisian conduction abnormalities. J Cardiovasc Electrophysiol 11: 1270–1276, 2000. [DOI] [PubMed] [Google Scholar]
- 1173.Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease. Circulation 104: 1380–1384, 2001. [DOI] [PubMed] [Google Scholar]
- 1174.Vassalle M, Lin CI. Calcium overload and cardiac function. J Biomed Sci 11: 542–565, 2004. [DOI] [PubMed] [Google Scholar]
- 1175.Vassallo JA, Cassidy DM, Kindwall KE, Marchlinski FE, Josephson ME. Nonuniform recovery of excitability in the left ventricle. Circulation 78: 1365–1372, 1988. [DOI] [PubMed] [Google Scholar]
- 1176.Vassort G. Adenosine 5'-triphosphate: a P2-purinergic agonist in the myocardium. Physiol Rev 81: 767–806, 2001. [DOI] [PubMed] [Google Scholar]
- 1177.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin AJ. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 114: 2104-12, 2006. [DOI] [PubMed] [Google Scholar]
- 1178.Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 42: 2014–2027, 2003. [DOI] [PubMed] [Google Scholar]
- 1179.van Veen TAB, Van Rijen HVM, Van Kempen MJA, Miquerol L, Opthof T, Gros D, Vos MA, Jongsma HJ, de Bakker JMT. Discontinuous conduction in mouse bundle branches is caused by bundle-branch architecture. Circulation 112: 2235–2244, 2005. [DOI] [PubMed] [Google Scholar]
- 1180.van Veen TAB, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, Huang CLH, Wilders R, Grace AA, Escande D, de Bakker JMT, van Rijen HVM. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112: 1927–1935, 2005. [DOI] [PubMed] [Google Scholar]
- 1181.Veeraraghavan R, Gourdie RG, Poelzing S. Mechanisms of cardiac conduction: a history of revisions. Am J Physiol Heart Circ Physiol 306: H619–H627, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1182.Veeraraghavan R, Larsen AP, Torres NS, Grunnet M, Poelzing S. Potassium channel activators differentially modulate the effect of sodium channel blockade on cardiac conduction. Acta Physiol 207: 280–289, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1183.Veeraraghavan R, Poelzing S. Mechanisms underlying increased right ventricular conduction sensitivity to flecainide challenge. Cardiovasc Res 77: 749–756, 2008. [DOI] [PubMed] [Google Scholar]
- 1184.Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA, Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ Res 86: E91–E97, 2000. [DOI] [PubMed] [Google Scholar]
- 1185.Vemuri R, Lankford EB, Poetter K, Hassanzadeh S, Takeda K, Yu ZX, Ferrans VJ, Epstein ND. The stretch-activation response may be critical to the proper functioning of the mammalian heart. Proc Natl Acad Sci USA 96: 1048–1053, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1186.Venema VJ, Ju H, Zou R, Venema RC. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle: Identification of a novel caveolin scaffolding/inhibitory domain. J Biol Chem 272: 28187–28190, 1997. [DOI] [PubMed] [Google Scholar]
- 1187.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res 100: 105–111, 2007. [DOI] [PubMed] [Google Scholar]
- 1188.Ventura JJ, Kennedy NJ, Flavell RA, Davis RJ. JNK regulates autocrine expression of TGF-beta1. Mol Cell 15: 269–278, 2004. [DOI] [PubMed] [Google Scholar]
- 1189.Verheijck EE, van Ginneken a C, Bourier J, Bouman LN. Effects of delayed rectifier current blockade by E-4031 on impulse generation in single sinoatrial nodal myocytes of the rabbit. Circ Res 76: 607–615, 1995. [DOI] [PubMed] [Google Scholar]
- 1190.Verheijck EE, van Ginneken ACG, Wilders R, Bouman LN. Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am J Physiol Heart Circ Physiol 276: H1064–H1077, 1999. [DOI] [PubMed] [Google Scholar]
- 1191.Verheule S, van Batenburg CA, Coenjaerts FE, Kirchhoff S, Willecke K, Jongsma HJ. Cardiac conduction abnormalities in mice lacking the gap junction protein connexin 40. J Cardiovasc Electrophysiol 10: 1380–1389, 1999. [DOI] [PubMed] [Google Scholar]
- 1192.Verheule S, Kaese S. Connexin diversity in the heart: insights from transgenic mouse models. Front Pharmacol 4: 81, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1193.Verheule S, Sat T, Everett IVT, Engle SK, Otten D, Rubart-Von Der Lohe M, Nakajima HHO, Nakajima HHO, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-β1. Circ Res 94: 1458–1465, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1194.Vest JA, Wehrens XHT, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111: 2025–2032, 2005. [DOI] [PubMed] [Google Scholar]
- 1195.Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron 17: 1251–1264, 1996. [DOI] [PubMed] [Google Scholar]
- 1196.Vianna CR, Huntgeburth M, Coppari R, Choi CS, Lin J, Krauss S, Barbatelli G, Tzameli I, Kim YB, Cinti S, Shulman GI, Spiegelman BM, Lowell BB. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab 4: 453–464, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1197.Vincent GM, Timothy K, Fox J, Zhang L. The inherited long QT syndrome: from ion channel to bedside. Cardiol Rev 7: 44–55, 1999. [PubMed] [Google Scholar]
- 1198.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res 98: 505–514, 2006. [DOI] [PubMed] [Google Scholar]
- 1199.Vinogradova TM, Zhou YY, Maltsev V, Lyashkov A, Stern M, Lakatta EG. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ Res 94: 802–809, 2004. [DOI] [PubMed] [Google Scholar]
- 1200.Viswanathan PC, Balser JR. Inherited sodium channelopathies: a continuum of channel dysfunction. Trends Cardiovasc Med 14: 28–35, 2004. [DOI] [PubMed] [Google Scholar]
- 1201.Viswanathan PC, Shaw RM, Rudy Y. Effects of I Kr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation 99: 2466–2674, 1999. [DOI] [PubMed] [Google Scholar]
- 1202.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129: 145–156, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1203.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XHT, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+-leak and increased Na+-Ca2+-exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125: 2059–2070, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1204.Volders P, Vos M, Szabo B, Sipido K, de Groot S, Gorgels A, Wellens H, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res 46: 376–392, 2000. [DOI] [PubMed] [Google Scholar]
- 1205.Volders PG, Sipido KR, Vos MA, Kulcsár A, Verduyn SC, Wellens HJ. Cellular basis of biventricular hypertrophy and arrhythmogenesis in dogs with chronic complete atrioventricular block and acquired torsade de pointes. Circulation 98: 1136–1147, 1998. [DOI] [PubMed] [Google Scholar]
- 1206.Vos MA, de Groot SH, Verduyn SC, van der Zande J, Leunissen HD, Cleutjens JP, van Bilsen M, Daemen MJ, Schreuder JJ, Allessie MA, Wellens HJ. Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation 98: 1125–1135, 1998. [DOI] [PubMed] [Google Scholar]
- 1207.Vos MA, Verduyn SC, Gorgels AP, Lipcsei GC, Wellens HJ. Reproducible induction of early afterdepolarizations and torsade de pointes arrhythmias by d-sotalol and pacing in dogs with chronic atrioventricular block. Circulation 91: 864–872, 1995. [DOI] [PubMed] [Google Scholar]
- 1208.Wadzinski BE, Wheat WH, Jaspers S, Peruski LF, Lickteig RL, Johnson GL, Klemm DJ. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol 13: 2822–2834, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1209.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127–3138, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1210.Wagner S, Maier LS, Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res 116: 1956–1970, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1211.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res 108: 555–565, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1212.Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 121: 2955–2968, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science 242: 67–69, 1988. [DOI] [PubMed] [Google Scholar]
- 1214.Wan E, Abrams J, Weinberg R, Katchman A, Bayne J, Zakharov S, Yang L, Morrow J, Garan H, Marx S. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest 126: 112–122, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1215.Wang DW, Yazawa K, George AL, Bennett PB. Characterization of human cardiac Na(+) channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci USA 93: 13200–13205, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1216.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 153: 910–918, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1217.Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K(+) channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem 279: 13289–13292, 2004. [DOI] [PubMed] [Google Scholar]
- 1218.Wang L, Myles RC, De Jesus NM, Ohlendorf AKP, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res 114: 1410–1421, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1219.Wang L, Swirp S, Duff H. Age-dependent response of the electrocardiogram to K+ channel blockers in mice. Am J Physiol Cell Physiol 278: C73–C80, 2000. [DOI] [PubMed] [Google Scholar]
- 1220.Wang L, Zuo L, Hu J, Shao H, Lei C, Qi W, Liu Y, Miao Y, Ma X, Huang CLH, Wang B, Zhou X, Zhang Y, Liu L. Dual LQT1 and HCM phenotypes associated with tetrad heterozygous mutations in KCNQ1, MYH7, MYLK2, and TMEM70 genes in a three-generation Chinese family. Europace 18: 602–609, 2016. [DOI] [PubMed] [Google Scholar]
- 1221.Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F, Liao Y, Fang F, Chen Q, Tu X, Wang QK. Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophys Res Commun 398: 98–104, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1222.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17–23, 1996. [DOI] [PubMed] [Google Scholar]
- 1223.Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 4: 1603–1607, 1995. [DOI] [PubMed] [Google Scholar]
- 1224.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80: 805–811, 1995. [DOI] [PubMed] [Google Scholar]
- 1225.Wang R, Wang Y, Lin WK, Zhang Y, Liu W, Huang K, Terrar DA, Solaro RJ, Wang X, Ke Y, Lei M. Inhibition of angiotensin II-induced cardiac hypertrophy and associated ventricular arrhythmias by a p21 activated kinase 1 bioactive peptide. PLoS One 9: e101974, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1226.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410: 592–596, 2001. [DOI] [PubMed] [Google Scholar]
- 1227.Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta 1773: 1349–1357, 2007. [DOI] [PubMed] [Google Scholar]
- 1228.Wang Y, Cheng J, Joyner RW, Wagner MB, Hill JA. Remodeling of early-phase repolarization: a mechanism of abnormal impulse conduction in heart failure. Circulation 113: 1849–1856, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1229.Wang Y, Tsui H, Bolton EL, Wang X, Huang CLH, Solaro RJ, Ke Y, Lei M. Novel insights into mechanisms for Pak1-mediated regulation of cardiac Ca2+ homeostasis. Front Physiol 6: 1–5, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1230.Wang Y, Tsui H, Ke Y, Shi Y, Li Y, Davies L, Cartwright EJ, Venetucci L, Zhang H, Terrar DA, Huang CLH, Solaro RJ, Wang X, Lei M. Pak1 is required to maintain ventricular Ca2+ homeostasis and electrophysiological stability through SERCA2a regulation in mice. Circ Arrhythm Electrophysiol 7: 938–948, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1231.Wang ZG, Pelletier LC, Talajic M, Nattel S. Effects of flecainide and quinidine on human atrial action potentials. Role of rate-dependence and comparison with guinea pig, rabbit, and dog tissues. Circulation 82: 274–283, 1990. [DOI] [PubMed] [Google Scholar]
- 1232.Ward O. A new familial cardiac syndrome in children. J Ir Med Assoc 54: 103–106, 1964. [PubMed] [Google Scholar]
- 1233.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AAM, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15: 380–383, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1234.Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol 2: 268–275, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1235.Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld ACP, Pfeufer A, Kääb S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AAM, Roden DM, Bezzina CR. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118: 2260–2268, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1236.Watanabe MA, Fenton FH, Evans SJ, Hastings HM, Karma A. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol 12: 196–206, 2001. [DOI] [PubMed] [Google Scholar]
- 1237.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 332: 1058–1064, 1995. [DOI] [PubMed] [Google Scholar]
- 1238.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol 67: 69–98, 2005. [DOI] [PubMed] [Google Scholar]
- 1239.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304: 292–296, 2004. [DOI] [PubMed] [Google Scholar]
- 1240.Wehrens XH, Marks AR. Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends Biochem Sci 28: 671–678, 2003. [DOI] [PubMed] [Google Scholar]
- 1241.Wehrens XHT, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113: 829–840, 2003. [DOI] [PubMed] [Google Scholar]
- 1242.Wehrens XHT, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA 103: 511–518, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1243.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 94: e61–e70, 2004. [DOI] [PubMed] [Google Scholar]
- 1244.Wehrens XHT, Marks AR. Novel therapeutic approaches for heart failure by normalizing calcium cycling. Nat Rev Dr ug Discov 3: 565–573, 2004. [DOI] [PubMed] [Google Scholar]
- 1245.Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, Delaughter MC, Klevitsky R, Hewett TE, Robbins J, Michael LH, Schneider MD, Entman ML, Schwartz RJ. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J 18: 857–859, 2004. [DOI] [PubMed] [Google Scholar]
- 1246.Weidmann S. Electrical constants of trabecular muscle from mammalian heart. J Physiol 210: 1041–1054, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1247.Weiergräber M, Henry M, Südkamp M, de Vivie ER, Hescheler J, Schneider T. Ablation of Cav2.3/E-type voltage-gated calcium channel results in cardiac arrhythmia and altered autonomic control within the murine cardiovascular system. Basic Res Cardiol 100: 1–13, 2005. [DOI] [PubMed] [Google Scholar]
- 1248.Weiford B, Subbarao V, Mulhern K. Noncompaction of the ventricular myocardium. Circulation 109: 2965–2971, 2004. [DOI] [PubMed] [Google Scholar]
- 1249.Weiss J, Lamp S, Shine K. Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. Am J Physiol Heart Circ Physiol 256: H1165–H1175, 1989. [DOI] [PubMed] [Google Scholar]
- 1250.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98: 1244–1253, 2006. [DOI] [PubMed] [Google Scholar]
- 1251.Weiss JN, Qu Z, Chen PS, Lin SF, Karagueuzian HS, Hayashi H, Garfinkel A, Karma A. The dynamics of cardiac fibrillation. Circulation 112: 1232–1240, 2005. [DOI] [PubMed] [Google Scholar]
- 1252.van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, Shimizu W, Sumitomo N, Fish FA, Bhuiyan ZA, Willems AR, Van Der Veen MJ, Watanabe H, Laborderie J, Hassaguerre M, Knollmann BC, Wilde AAM. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 57: 2244–2254, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1253.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation 109: 1834–1841, 2004. [DOI] [PubMed] [Google Scholar]
- 1254.Westphal RS, Coffee RL, Marotta A, Pelech SL, Wadzinski BE. Identification of kinase-phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J Biol Chem 274: 687–692, 1999. [DOI] [PubMed] [Google Scholar]
- 1255.Wettwer E, Amos G, Gath J, Zerkowski HR, Reidemeister JC, Ravens U. Transient outward current in human and rat ventricular myocytes. Cardiovasc Res 27: 1662–1669, 1993. [DOI] [PubMed] [Google Scholar]
- 1256.Wettwer E, Hála O, Christ T, Heubach JF, Dobrev D, Knaut M, Varró A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: Influence of chronic atrial fibrillation. Circulation 110: 2299–2306, 2004. [DOI] [PubMed] [Google Scholar]
- 1257.Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20: 103–114, 1998. [DOI] [PubMed] [Google Scholar]
- 1258.Wier WG, Kort AA, Stern MD, Lakatta EG, Marban E. Cellular calcium fluctuations in mammalian heart: direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc Natl Acad Sci USA 80: 7367–7371, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1259.Wiggers C, Bell J, Paine M. Studies of ventricular fibrillation caused by electric shock. II. Cinematographic and electrocardiographic observations of the natural process in the dog's heart. Its inhibition by potassium and the revival of coordinated beats by calcium. Ann Noninvasive Electrocardiol 8: 252–261, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1260.Wigle ED, Rakowski H, Kimball BP, Williams WG. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 92: 1680–1692, 1995. [DOI] [PubMed] [Google Scholar]
- 1261.Wilde AA, Bhuiyan ZA, Crotti L, Facchini M, De Ferrari GM, Paul T, Ferrandi C, Koolbergen DR, Odero A, Schwartz PJ. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med 358: 2024–2029, 2008. [DOI] [PubMed] [Google Scholar]
- 1262.Willis BC, Ponce-Balbuena D, Jalife JJ. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am J Physiol Heart Circ Physiol 308: H1463–H1473, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1263.Windle JR, Geletka RC, Moss a J, Zareba W, Atkins DL. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A:DeltaKPQ mutation. Ann Noninvasive Electrocardiol 6: 153–158, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1264.Winfree AT. Electrical turbulence in three-dimensional heart muscle. Science 266: 1003–1006, 1994. [DOI] [PubMed] [Google Scholar]
- 1265.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol 11: 219–225, 2004. [DOI] [PubMed] [Google Scholar]
- 1266.Wolf CM, Moskowitz IP, Arno S, Branco DM, Semsarian C, Bernstein SA, Peterson M, Maida M, Morley GE, Fishman G, Berul CI, Seidman CE, Seidman JG. Somatic events modify hypertrophic cardiomyopathy pathology and link hypertrophy to arrhythmia. Proc Natl Acad Sci USA 102: 18123–18128, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1267.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner AD, Berul CI, Seidman CE, Seidman JG. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 44: 293–303, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1268.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: The Framingham study. Stroke 22: 983–988, 1991. [DOI] [PubMed] [Google Scholar]
- 1269.Wolkowicz PE, Grenett HE, Huang J, Wu HC, Ku DD, Urthaler F. A pharmacological model for calcium overload-induced tachycardia in isolated rat left atria. Eur J Pharmacol 576: 122–131, 2007. [DOI] [PubMed] [Google Scholar]
- 1270.Wolpert C, Echternach C, Veltmann C, Antzelevitch C, Thomas GP, Spehl S, Streitner F, Kuschyk J, Schimpf R, Haase KK, Borggrefe M. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm 2: 254–260, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1271.Woodbury J. Potentials in a volume conductor. In: Physiology and Biophysics, edited by Ruch T, Patton H. New York: Saunders, 1965, p. 85–90. [Google Scholar]
- 1272.Woodman SE, Park DS, Cohen AW, Cheung MWC, Chandra M, Shirani J, Tang B, Jelicks LA, Kitsis RN, Christ GJ, Factor SM, Tanowitz HB, Lisanti MP. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem 277: 38988–38997, 2002. [DOI] [PubMed] [Google Scholar]
- 1273.Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology 62: 538–543, 2004. [DOI] [PubMed] [Google Scholar]
- 1274.Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res 52: 226–235, 2001. [DOI] [PubMed] [Google Scholar]
- 1275.Wu J, Cheng L, Lammers WJ, Wu L, Wang X, Shryock JC, Belardinelli L, Lei M. Sinus node dysfunction in ATX-II-induced in-vitro murine model of long QT3 syndrome and rescue effect of ranolazine. Prog Biophys Mol Biol 98: 198–207, 2008. [DOI] [PubMed] [Google Scholar]
- 1276.Wu J, Zhang Y, Zhang X, Cheng L, Lammers WJ, Grace AA, Fraser JA, Zhang H, Huang CLH, Lei M. Altered sinoatrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/KPQ hearts suggest an overlap syndrome. Am J Physiol Heart Circ Physiol 302: H1510–H1523, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1277.Wu Y, Anderson ME. CaMKII in sinoatrial node physiology and dysfunction. Front Pharmacol 5: 48, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1278.Xia M, Jin Q, Bendahhou S, et al. Kir2.1. A gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 332: 1012–1019, 2006. [DOI] [PubMed] [Google Scholar]
- 1279.Xiao B, Sutherland C, Walsh MP, Chen SRW. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP126). Circ Res 94: 487–495, 2004. [DOI] [PubMed] [Google Scholar]
- 1280.Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 165: 1019–1032, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1281.Xiao J, Tian X, Jones P, Bolstad J, Kong H, Wang R, Zhang L, Duff H, Gillis A, Fleischer S, Kotlikoff¶ M, Copello J, Wayne Chen S. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem 282: 34828–34838, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1282.Xiao RP, Valdivia HH, Bogdanov K, Valdivia C, Lakatta EG, Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J Physiol 500: 343–354, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1283.Xie LH, Shanmugam M, Park JY, Zhao Z, Wen H, Tian B, Periasamy M, Babu GJ. Ablation of sarcolipin results in atrial remodeling. Am J Physiol Cell Physiol 302: C1762–C1771, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1284.Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J Gen Physiol 113: 661–678, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1285.Xu L, Meissner G. Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+. Biophys J 75: 2302–2312, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1286.Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, Oda T, Kobayashi S, Ikemoto N, Matsuzaki M. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation 117: 762–772, 2008. [DOI] [PubMed] [Google Scholar]
- 1287.Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long-QT syndrome. Circulation 98: 1928–1936, 1998. [DOI] [PubMed] [Google Scholar]
- 1288.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100: 1660–1666, 1999. [DOI] [PubMed] [Google Scholar]
- 1289.Yanagisawa T, Taira N. Effect of 2-nicotinamidethyl nitrate (SG-75) on the membrane potential of left atrial muscle fibres of the dog. Naunyn-Schmiedebergs Arch Pharmacol 312: 69–76, 1980. [DOI] [PubMed] [Google Scholar]
- 1290.Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9: 1956–1968, 2014. [DOI] [PubMed] [Google Scholar]
- 1291.Yang J, Fan GH, Wadzinski BE, Sakurai H, Richmond A. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem 276: 47828–47833, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1292.Yang KC, Bonini MG, Dudley SC. Mitochondria and arrhythmias. Free Radic Biol Med 71: 351–361, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1293.Yang KC, Kyle JW, Makielski JC, Dudley SC. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res 116: 1937–1955, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1294.Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation 93: 407–411, 1996. [DOI] [PubMed] [Google Scholar]
- 1295.Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet 75: 899–905, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1296.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res 99: 646–655, 2006. [DOI] [PubMed] [Google Scholar]
- 1297.Yaniv Y, Maltsev VA. Numerical modeling calcium and CaMKII effects in the SA node. Front Pharmacol 5: 58, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1298.Yanni J, Tellez JO, Sutyagin PV, Boyett MR, Dobrzynski H. Structural remodelling of the sinoatrial node in obese old rats. J Mol Cell Cardiol 48: 653–662, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1299.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest 115: 556–564, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1300.Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Matsuzaki M. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 107: 477–484, 2003. [DOI] [PubMed] [Google Scholar]
- 1301.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res 90: 443–449, 2002. [DOI] [PubMed] [Google Scholar]
- 1302.Yard NJ, Chiesi M, Ball HA. Effect of cyclopiazonic acid, an inhibitor of sarcoplasmic reticulum Ca2+-ATPase, on the frequency-dependence of the contraction-relaxation cycle of the guinea-pig isolated atrium. Br J Pharmacol 113: 1001–1007, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1303.Ye B, Balijepalli RC, Foell JD, Kroboth S, Ye Q, Luo YH, Shi NQ. Caveolin-3 associates with and affects the function of hyperpolarization- activated cyclic nucleotide-gated channel 4. Biochemistry 47: 12312–12318, 2008. [PMC free article] [PubMed] [Google Scholar]
- 1304.Yee Chin J, Matthews HR, Fraser JA, Skepper JN, Chawla S, Huang CLH. Detubulation experiments localise delayed rectifier currents to the surface membrane of amphibian skeletal muscle fibres. J Muscle Res Cell Motil 25: 389–395, 2004. [DOI] [PubMed] [Google Scholar]
- 1305.Yeh YH, Kuo CT, Chan TH, Chang GJ, Qi XY, Tsai F, Nattel S, Chen WJ. Transforming growth factor-β and oxidative stress mediate tachycardia-induced cellular remodelling in cultured atrial-derived myocytes. Cardiovasc Res 91: 62–70, 2011. [DOI] [PubMed] [Google Scholar]
- 1306.Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, George AL, Satin J. Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc 3: e000996, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1307.Yoshida H, Horie M, Otani H, Kawashima T, Onishi Y, Sasayama S. Bradycardia-induced long QT syndrome caused by a de novo missense mutation in the S2–S3 inner loop of hERG. Am J Med Genet 98: 348–352, 2001. [DOI] [PubMed] [Google Scholar]
- 1308.Young KA, Caldwell JH. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J Physiol 565: 349–370, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1309.Yu CC, Corr C, Shen C, Shelton R, Yadava M, Rhea I, Straka S, Fishbein M, Chen Z, Lin SF, Lopshire JC, Chen PS. Small conductance calcium-activated potassium current is important in transmural repolarization of failing human ventricles. Circ Arrhythmia Electrophysiol 8: 667–676, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1310.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick a K, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci 23: 7577–7585, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1311.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol Heart Circ Physiol 267: H982–H993, 1994. [DOI] [PubMed] [Google Scholar]
- 1312.Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res 95: 261–268, 2004. [DOI] [PubMed] [Google Scholar]
- 1313.Zahradníková A, Valent I, Zahradník I. Frequency and release flux of calcium sparks in rat cardiac myocytes: a relation to RYR gating. J Gen Physiol 136: 101–116, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1314.Zaidi M, Blair HC, Moonga BS, Abe E, Huang CLH. Osteoclastogenesis, bone resorption, and osteoclast-based therapeutics. J Bone Miner Res 18: 599–609, 2003. [DOI] [PubMed] [Google Scholar]
- 1315.Zaidi M, Moonga BS, Huang CLH. Calcium sensing and cell signaling processes in the local regulation of osteoclastic bone resorption. Biol Rev Camb Philos Soc 79: 79–100, 2004. [DOI] [PubMed] [Google Scholar]
- 1316.Zaidi M, Shankar VS, Tunwell R, Adebanjo OA, Mackrill J, Pazianas M, O'Connell D, Simon BJ, Rifkin BR, Venkitaraman AR, Huang CLH, Lai FA. A ryanodine receptor-like molecule expressed in the osteoclast plasma membrane functions in extracellular Ca2+ sensing. J Clin Invest 96: 1582–1590, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1317.Zaitsev AV, Berenfeld O, Mironov SF, Jalife J, Pertsov AM. Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ Res 86: 408–417, 2000. [DOI] [PubMed] [Google Scholar]
- 1318.Zamiri N, Massé S, Ramadeen A, Kusha M, Hu X, Azam MA, Liu J, Lai PFH, Vigmond EJ, Boyle PM, Behradfar E, Al-Hesayen A, Waxman MB, Backx P, Dorian P, Nanthakumar K. Dantrolene improves survival after ventricular fibrillation by mitigating impaired calcium handling in animal models. Circulation 129: 875–885, 2014. [DOI] [PubMed] [Google Scholar]
- 1319.Zamponi G, Striessnig J, Koschak A, Dolphin A. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1320.Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic etiology. Curr Opin Pediatr 19: 619–627, 2007. [DOI] [PubMed] [Google Scholar]
- 1321.Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, Schwartz P, Vincent GM, Priori S, Benhorin J, Towbin JA, Robinson JL, Andrews ML, Napolitano C, Timothy K, Zhang L, Medina A. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 42: 103–109, 2003. [DOI] [PubMed] [Google Scholar]
- 1322.Zareba W, Sattari M, Rosero S, Couderc J, Moss A. Altered atrial,atrioventricular, and ventricular conduction in patients with the long QT syndrome caused by the DeltaKPQ SCN5A sodium channel gene mutation. Am J Cardiol 88: 1311–1314, 2001. [DOI] [PubMed] [Google Scholar]
- 1323.Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. The consequences of disrupting cardiac inwardly rectifying K(+) current (I(K1)) as revealed by the targeted deletion of the murine Kir2.1 and Kir22 genes. J Physiol 533: 697–710, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1324.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol 27: 7522–7537, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1325.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J 68: 949–964, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1326.Zhang H, Hancox JC. In silico study of action potential and QT interval shortening due to loss of inactivation of the cardiac rapid delayed rectifier potassium current. Biochem Biophys Res Commun 322: 693–699, 2004. [DOI] [PubMed] [Google Scholar]
- 1327.Zhang H, Holden AV, Boyett MR. Sustained inward current and pacemaker activity of mammalian sinoatrial node. J Cardiovasc Electrophysiol 13: 809–812, 2002. [DOI] [PubMed] [Google Scholar]
- 1328.Zhang H, Kharche S, Holden AV, Hancox JC. Repolarisation and vulnerability to re-entry in the human heart with short QT syndrome arising from KCNQ1 mutation–A simulation study. Prog Biophys Mol Biol 96: 112–131, 2008. [DOI] [PubMed] [Google Scholar]
- 1329.Zhang R, Khoo MSC, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya a T, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11: 409–417, 2005. [DOI] [PubMed] [Google Scholar]
- 1330.Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res 84: 989–998, 1999. [DOI] [PubMed] [Google Scholar]
- 1331.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Brown JH. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem 277: 1261–1267, 2002. [DOI] [PubMed] [Google Scholar]
- 1332.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross JJ, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 92: 912–919, 2003. [DOI] [PubMed] [Google Scholar]
- 1333.Zhang X, Lieu D, Chiamvimonvat N. Small-conductance Ca2+-activated K+ channels and cardiac arrhythmias. Heart Rhythm 12: 1845–1851, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1334.Zhang Y, Fraser JA, Jeevaratnam K, Hao X, Hothi SS, Grace AA, Lei M, Huang CLH. Acute atrial arrhythmogenicity and altered Ca2+ homeostasis in murine RyR2-P2328S hearts. Cardiovasc Res 89: 794–804, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1335.Zhang Y, Fraser JA, Schwiening C, Killeen MJ, Grace AA, Huang CLH. Acute atrial arrhythmogenesis in murine hearts following enhanced extracellular Ca2+ entry depends on intracellular Ca2+ stores. Acta Physiol 198: 143–158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1336.Zhang Y, Guzadhur L, Jeevaratnam K, Salvage SC, Matthews GD, Lammers WJ, Lei M, Huang CLH, Fraser JA. Arrhythmic substrate, slowed propagation and increased dispersion in conduction direction in the right ventricular outflow tract of murine Scn5a+/− hearts. Acta Physiol 211: 559–573, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1337.Zhang Y, Matthews GDK, Lei M, Huang CLH. Abnormal Ca2+ homeostasis, atrial arrhythmogenesis, and sinus node dysfunction in murine hearts modeling RyR2 modification. Front Physiol 4: 150, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1338.Zhang Y, Schwiening C, Killeen MJ, Zhang Y, Ma A, Lei M, Grace AA, Huang CLH. Pharmacological changes in cellular Ca2+ homeostasis parallel initiation of atrial arrhythmogenesis in murine Langendorff-perfused hearts. Clin Exp Pharmacol Physiol 36: 969–980, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1339.Zhang Y, Wang T, Ma A, Zhou X, Gui J, Wan H, Shi R, Huang C, Grace AA, Huang CLH, Trump D, Zhang H, Zimmer T, Lei M. Correlations between clinical and physiological consequences of the novel mutation R878C in a highly conserved pore residue in the cardiac Na(+) channel. Acta Physiol 194: 311–323, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1340.Zhang Y, Wu J, Jeevaratnam K, King JH, Guzadhur L, Ren X, Grace AA, Lei M, Huang CLH, Fraser JA. Conduction slowing contributes to spontaneous ventricular arrhythmias in intrinsically active murine RyR2-P2328S hearts. J Cardiovasc Electrophysiol 24: 210–218, 2013. [DOI] [PubMed] [Google Scholar]
- 1341.Zhang Y, Wu J, King JH, Huang CLH, Fraser JA. Measurement and interpretation of electrocardiographic QT intervals in murine hearts. Am J Physiol Heart Circ Physiol 306: H1553–H1557, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1342.Zhang Y, Zhou N, Jiang W, Peng J, Wan H, Huang C, Xie Z, Huang CLH, Grace AA, Ma A. A missense mutation (G604S) in the S5/pore region of HERG causes long QT syndrome in a Chinese family with a high incidence of sudden unexpected death. Eur J Pediatr 166: 927–933, 2007. [DOI] [PubMed] [Google Scholar]
- 1343.Zhang Z, Stroud MJ, Zhang J, Fang X, Ouyang K, Kimura K, Mu Y, Dalton ND, Gu Y, Bradford WH, Peterson KL, Cheng H, Zhou X, Chen J. Normalization of Naxos plakoglobin levels restores cardiac function in mice. J Clin Invest 125: 1708–1712, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1344.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional roles of Cav1.3 (alpha1D) calcium channel in sinoatrial nodes: Insight gained using gene-targeted null mutant mice. Circ Res 90: 981–987, 2002. [DOI] [PubMed] [Google Scholar]
- 1345.Zheng M, Cheng H, Li X, Zhang J, Cui L, Ouyang K, Han L, Zhao T, Gu Y, Dalton ND, Bang ML, Peterson KL, Chen J. Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum Mol Genet 18: 701–713, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1346.Zheng M, Dilly K, Dos Santos Cruz J, Li M, Gu Y, Ursitti JA, Chen J, Ross J, Chien KR, Lederer JW, Wang Y. Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am J Physiol Heart Circ Physiol 286: H424–H433, 2004. [DOI] [PubMed] [Google Scholar]
- 1347.Zheng W, Rampe D, Triggle DJ. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol Pharmacol 40: 734–741, 1991. [PubMed] [Google Scholar]
- 1348.Zhou J, Jeron A, London B, Han X, Koren G. Characterization of a slowly inactivating outward current in adult mouse ventricular myocytes. Circ Res 83: 806–814, 1998. [DOI] [PubMed] [Google Scholar]
- 1349.Zhou J, Yi J, Hu NN, George AL, Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res 87: 33–38, 2000. [DOI] [PubMed] [Google Scholar]
- 1350.Zhou Q, Chu PH, Huang C, Cheng CF, Martone ME, Knoll G, Shelton GD, Evans S, Chen J. Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J Cell Biol 155: 605–612, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1351.Zi M, Kimura T, Liu W, Jin J, Higham J, Kharche S, Hao G, Shi Y, Shen W, Prehar S, Mironov A, Neyses L, Bierhuizen M, Boyett M, Zhang H, Lei M, Cartwright E, Wang X. Mitogen-activated protein kinase kinase 4 deficiency in cardiomyocytes causes connexin 43 reduction and couples hypertrophic signals to ventricular arrhythmogenesis. J Biol Chem 286: 17821–17830, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1352.Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD(+) levels on sarcoplasmic reticulum Ca2+ release in permeabilized rat ventricular myocytes. J Physiol 555: 727–741, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]