

Abstract
Peritoneal metastasis of gastric cancer (PMGC) is incurable and thus has an extremely poor prognosis. We have found, however, that locoregionally administered trastuzumab armed with astatine‐211 (211At‐trastuzumab) is effective against human epidermal growth factor receptor 2 (HER2)‐positive PMGC in a xenograft mouse model. We first observed that 211At‐trastuzumab can specifically bind and effectively kill NCI‐N87 (N87) cells, which are HER2‐positive human metastatic GC cells, both in vitro and in s.c. tumors. We established a PMGC mouse model using N87 xenografts stably expressing luciferase to test α‐particle radioimmunotherapy with 211At‐trastuzumab against PMGC. Biodistribution analysis in this PMGC mouse model revealed that the i.p. administration of 211At‐trastuzumab (1 MBq) was a more efficient means of delivery of 211At into metastatic tumors than i.v. injection; the maximum tumor uptake with i.p. administration was over 60% injected dose per gram of tissue (%ID/g) compared to approximately 18%ID/g with i.v. injection. Surprisingly, a single i.p. injection of 211At‐trastuzumab (1 MBq) was sufficient to completely eradicate intraperitoneally disseminated HER2‐positive GC xenografts in two of six treated mice by inducing DNA double‐strand breaks, and to drastically reduce the tumor burden in another three mice. No bodyweight loss, leukocytopenia, or significant biochemical changes in liver or kidney function were observed in the treatment group. Accordingly, locoregionally administered 211At‐trastuzumab significantly prolonged the survival time of HER2‐positive PMGC mice compared with control treatments. Our results provide a proof‐of‐concept demonstration that locoregional therapy with 211At‐trastuzumab may offer a new treatment option for HER2‐positive PMGC.
Keywords: gastric cancer, peritoneal metastasis, radioimmunotherapy, trastuzumab, α‐Particle
Gastric cancer is one of the leading causes of cancer‐related death worldwide.1 Metastatic dissemination of GC to the peritoneal cavity frequently occurs during the progression of this disease and is the main form of GC relapse after surgical resection. The prognosis for PMGC or its associated clinical condition (peritoneal carcinomatosis) is extremely poor and, because no effective therapeutics have been established, the mean survival time is only 4 months.2
Recent advances in the molecular dissection of GC have helped to identify reliable predictive biomarkers, allowing introduction of molecular targeted drug therapy. Approximately 20% of GCs are HER2‐positive.3, 4 Therefore, HER2 is an attractive target for GC. Trastuzumab, an anti‐HER2 mAb, has been used previously as a targeted therapy for HER2‐positive GC and yields excellent treatment outcomes.4 However, its therapeutic efficacy is still limited for patients with PMGC. Novel antitumor strategies are thus still required to improve the clinical outcomes for GC patients, particularly those with PMGC.
Radioimmunotherapy is a targeted radioisotope treatment method that uses an antibody as a carrier of therapeutic radioisotopes and has considerable advantages when used against micrometastatic or disseminated tumors.5, 6, 7, 8 The selective targeting of radioisotopes to the tumor using a radiolabeled cancer‐specific antibody enables the delivery of a high dose of radiation directly to cancer cells while minimizing the exposure of normal cells. Among the radioisotopes used for RIT, 211At is of particular interest because it emits highly cytotoxic α‐particles. α‐Particles emitted through the decay of 211At (half‐life, 7.2 h) deposit a huge amount of energy (approximately 100 keV/μm) within a few cell diameters (a 55–80‐μm path length in soft tissue) to induce irreparable DNA damage but sparing the surrounding normal tissue.9, 10 A single α‐particle atom can kill a target cell, making it one of the most potent cell‐killing agents available.11, 12 Astatine‐211 is therefore particularly suited to the targeted killing of disseminated or micrometastatic solid tumors that are usually resistant to the low energy radiation used routinely in clinical settings, such as X‐rays and β‐particles.
We hypothesized that 211At‐trastuzumab emitting highly cytotoxic α‐particles would be a potent agent for the targeted ablation of HER2‐positive PMGC. We thus investigated the therapeutic efficacy of α‐RIT using 211At‐trastuzumab in preclinical mouse models of HER2‐positive PMGC.
Materials and Methods
Cells
The human GC cell line NCI‐N87 (N87) and two human breast cancer cell lines (SKBR3 and AU565) were purchased from ATCC (Manassas, VA, USA). The human GC cell line MKN45 was purchased from the Japanese Collection of Research Bioresources Cell Bank (Ibaraki, Japan). The human GC cell line MKN7 was purchased from RIKEN Cell Bank (Tsukuba, Japan). N87/Luc cells were established by the introduction of RediFect Red‐FLuc‐Puromycin lentiviral particles in accordance with the manufacturer's instructions (PerkinElmer, Waltham, MA, USA).
Antibody and reagents
Trastuzumab, an anti‐HER2 mAb, was purchased from Chugai Pharmaceutical (Tokyo, Japan). N‐Succinimidyl‐3‐(trimethylstannyl)benzoate was purchased from Santa Cruz Biotechnology (Dallas, TX,USA). N‐Chlorosuccinimide was purchased from Tokyo Chemical Industry (Tokyo, Japan).
Astatine‐211 production and radiochemistry
Astatine‐211 was produced by the 209Bi(α, 2n)211At reaction, as described previously.13 The 211At labeling of trastuzumab was carried out in accordance with a previously described direct astatination procedure.14 The activities of 211At‐trastuzumab pre/post‐isolation were measured using a dose calibrator (Capintec, Ramsey, NJ, USA) and the labeling yield was calculated by dividing the post‐isolation activity by the pre‐isolation activity.
Western blot analysis
Western blotting was carried out as described previously.8
In vitro cytotoxicity
MKN45, N87, SKBR3, MKN7, and AU565 cells (2–20 × 103/well) were seeded into 96‐well plates the day before the experiment. After the medium was removed, 100 μL culture medium containing PBS, trastuzumab, non‐carrier 211At, or 211At‐trastuzumab was applied to the cells, followed by incubation at 37°C for 24 h. The protein doses of trastuzumab and 211At‐trastuzumab were adjusted to the same amount (0.0034–0.0244 μg) by the addition of intact antibody. After the incubation, the medium was removed and the cells were washed once with PBS. Fresh medium (100 μL) was then added to each well and the cells were further incubated in a humidified atmosphere containing 5% CO2 at 37°C for 7 days.
Animal experiments
All animal experiments were approved by the Animal Care and Use Committee of the National Institute of Radiological Sciences at the National Institutes for Quantum and Radiological Science and Technology (Chiba, Japan) and were undertaken in compliance with the institutional guidelines regarding animal care and handling.
Biodistribution
The biodistribution of 211At‐trastuzumab was determined using both s.c. and PMGC xenograft mouse models. Astatine‐211‐labeled trastuzumab was injected into the tail vein (s.c. xenograft model) or the peritoneal cavity (both xenograft models). Between four and six mice were killed at 1, 3, 6, 12, and 24 h post‐injection. Tumor, whole blood, and major tissues were then sampled. All samples were weighed and the activity of 211At was measured using a γ counter (Aloka, Tokyo, Japan). The %ID/g was then calculated. The 211At activity levels were measured in both the feces and urine of the PMGC model mice up to 24 h after the i.p. injection of 211At‐trastuzumab and the %ID was thereby calculated.
Radioimmunotherapy in PMGC mice
The PMGC mouse models were established by i.p. injecting luciferase‐transfected N87/Luc cells (3 × 105) into 5‐week‐old B17/Icr‐scid/scidJcl (homo) female mice (CLEA Japan, Tokyo, Japan) 1 week before the experiment. These PMGC model mice then underwent RIT at 1 week after cell inoculation. Mice received a single i.p. injection of PBS, trastuzumab, non‐carrier 211At (1 MBq), or 211At‐trastuzumab (0.1 or 1 MBq). All protein doses were adjusted to the same amount (3.78 μg) by the addition of intact antibody except for non‐carrier 211At. Tumor growth in the PMGC mice was monitored every week using an in vivo bioluminescence imaging Fusion system (Vilber Lourmat, Marne‐la‐Valée, France). Bioluminescence from PMGC was captured for 10 s at 10 min after the injection of luciferin (10 mg/mL in PBS, 300 μL/mouse). The total bioluminescence intensity in the abdominal region was quantified using Bio‐1D software (Vilber Lourmat). The relative tumor intensity was calculated by dividing the tumor luminescence intensity on the day by the level at day 0. Bodyweights and white blood cell counts were determined at fixed intervals. Blood biochemistry on glutamic oxaloacetic transaminase, glutamic pyruvate transaminase, blood urea nitrogen, and creatinine of mice injected with 1 MBq 211At‐trastuzumab was carried out using DRI‐CHEM (Fujifilm, Tokyo, Japan) on the day before injection and at 1, 7, and 14 days post‐injection. The mice were euthanized when the tumor luciferase intensity in the abdominal region reached 1.5 × 107 photons.
Statistical analyses
Statistical analyses were carried out with JMP version 9 software (SAS Institute Japan, Tokyo, Japan) and Statcel 3 software (OMS, Tokorozawa, Japan). Tumor volume data were analyzed using two‐way repeated measures anova. The survival data were analyzed with the Kaplan–Meier method. Other data were analyzed by anova followed by the Tukey–Kramer post hoc test. A P‐value of <0.05 was considered significant.
Other materials and methods are described in detail in Doc. S1.
Results
Astatine‐211‐labeled trastuzumab specifically targets and ablates HER2‐positive human cancer cells
The labeling yield of 211At‐trastuzumab was 40.2 ± 6.9% and its radiochemical purity was consistently greater than 95%. More than 88% of the 211At‐trastuzumab yield was stable at 24 h after incubation with mouse serum (Fig. S1). We determined HER2 expression in three human GC cell lines (MKN45, N87, and MKN7), in conjunction with two human HER2‐overexpressing breast cancer cell lines (SKBR3 and AU565).8, 15 The N87, SKBR3, and AU565 cells showed considerably higher HER2 expression levels relative to MKN45 cells, in which HER2 expression was very low. Expression of HER2 was found to be approximately 25‐, 27‐, and 24‐fold higher in N87, SKBR3, and AU565 cells, respectively, than in MKN45 cells (Fig. 1a). In addition, MKN7 cells expressed an approximately 6‐fold higher HER2 level than MKN45 cells. Accordingly, we considered N87 cells to be HER2‐positive GC cells and MKN45 cells to be HER2‐negative GC cells. We next determined the cell binding of 211At‐trastuzumab to N87 and MKN45 cells. The cell binding ratios of 211At‐trastuzumab to N87 cells were much higher than those to MKN45 cells (Fig. 1b). Moreover, the addition of a 100‐fold excess of unlabeled trastuzumab markedly decreased the cell binding ratio of 211At‐trastuzumab to N87 cells, suggesting that this binding was specific (Fig. 1b).
Figure 1.
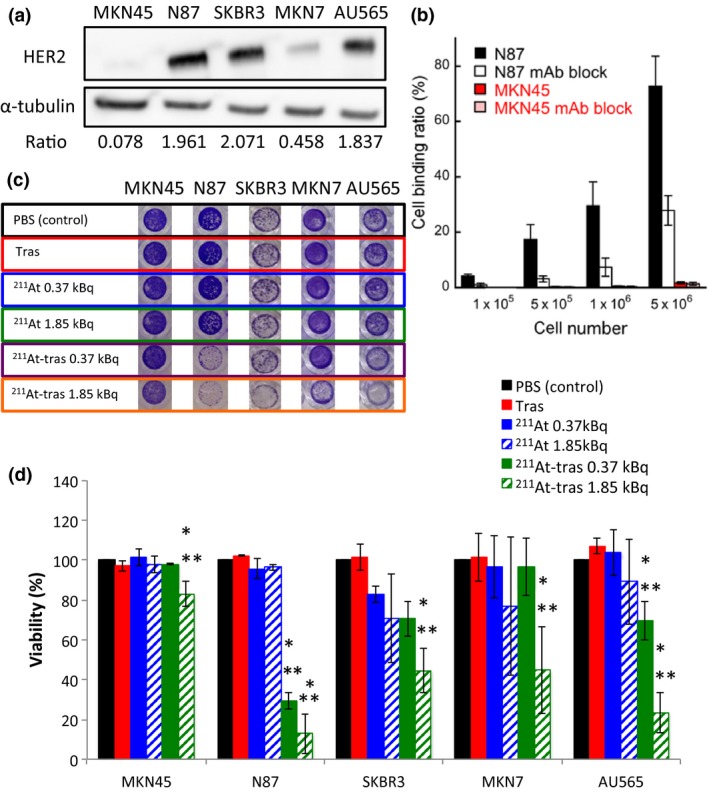
In vitro analysis of cell binding by astatine‐211‐labeled trastuzumab (211At‐tras) and subsequent cell death. (a) Human epidermal growth factor receptor 2 (HER2) expression in MKN45, N87, SKBR3, MKN7, and AU565 cells. α‐Tubulin was used as a loading control. Ratios of the band intensity of HER2 relative to α‐tubulin are indicated below the images. (b) Cell binding ratio of 211At‐trastuzumab to N87 and MKN45 cells with/without trastuzumab (mAb) block. Bars are labeled in the graph. Three independent experiments were carried out, each in triplicate (n = 3). (c) Representative images of stained surviving cells at 7 days after 24 h of treatment with PBS control, unlabeled trastuzumab (Tras), 211At (0.37 or 1.85 kBq), or 211At‐tras (0.37 or 1.85 kBq). (d) Quantification of the viability of the cells in panel (c) treated with 211At‐tras for 7 days. Bars are labeled in the graph. Three independent experiments were carried out in triplicate (n = 3). All data represent mean ± SD. *P < 0.05 versus control; **P < 0.05 versus Tras.
We next evaluated the in vitro cytotoxicity of 211At‐trastuzumab in MKN45, N87, SKBR3, MKN7, and AU565 cells by measuring cell viability after 24 h of exposure (Fig. 1c). Survival was unaffected in all cases by treatment with PBS, trastuzumab, and free 211At. In contrast, a 0.37‐ or 1.85‐kBq dose of 211At‐trastuzumab effectively killed the N87, SKBR3, and AU565 cell populations and reduced the viability of MKN7 cells (Fig. 1c). MKN45 cells were unaffected by 0.37 kBq 211At‐trastuzumab and only marginally affected by 1.85 kBq 211At‐trastuzumab (Fig. 1c). Quantitative cell viability data revealed that 211At‐trastuzumab significantly reduced the N87, SKBR3, AU565, and MKN7 cell numbers compared with the PBS and unlabeled trastuzumab controls (P < 0.05; Fig. 1d).
Astatine‐211‐labeled trastuzumab shows antitumor activity against s.c. xenografts of HER2‐positive GC cells
To investigate the potential antitumor activity of 211At‐trastuzumab against human GC cells in vivo, we examined the antitumor effects of 211At‐trastuzumab in nude mice bearing s.c. N87 tumors (Fig. S2). Prior to treatment, the tissue uptake of 211At was measured to analyze the biodistribution of 211At‐trastuzumab (0.5 MBq) in s.c. xenografts. This biodistribution analysis revealed that the uptake of i.v. injected 211At‐trastuzumab increased in the s.c. tumor over time, peaking at approximately 12.5 ± 4.0%ID/g at 24 h after the injection (Fig. S2a). The spleen showed the highest uptake (23.7 ± 8.6%ID/g) in all tissues at 1 h post‐injection, which decreased over time to 7.0 ± 1.9%ID/g after 24 h. The uptake in the whole blood peaked at 3 h after injection (27.1 ± 4.7%ID/g) and decreased thereafter. Uptake of 211At‐trastuzumab in the thyroid and stomach remained high, even at 24 h post‐injection (14.3 ± 8.4%ID/g and 18.0 ± 8.1%ID/g, respectively).
To evaluate its therapeutic efficacy in s.c. HER2‐positive GC xenografts, a single dose of 211At‐trastuzumab (0.1 or 0.5 MBq) was i.v. injected into mice bearing an s.c. N87 xenograft. An equivalent protein amount of unlabeled trastuzumab and PBS were injected as controls. There was no significant difference in the relative tumor volume between the control and trastuzumab‐treated mice (Fig. S2b). Both groups treated with 0.1 or 0.5 MBq 211At‐trastuzumab showed a significant suppression of tumor growth compared with the PBS control group (P < 0.05) and the 0.5 MBq 211At‐trastuzumab dose significantly suppressed tumor growth compared with unlabeled trastuzumab (P < 0.05) (Fig. S2b). Mice treated with 0.1 or 0.5 MBq 211At‐trastuzumab had a significantly prolonged median survival (52 and >84 days, respectively) compared with the PBS and unlabeled trastuzumab treated controls (38 and 31 days, respectively; Fig. S2c). No significant differences were found in the bodyweights among any of the treatment groups (Fig. S2d). The white blood cell counts sharply decreased only in the mice injected with 0.5 MBq 211At‐trastuzumab and recovered after 3 weeks (Fig. S2e).
Establishment of a xenograft mouse model of PMGC
We established a xenograft mouse model of PMGC that allowed tumor growth to be monitored by in vivo bioluminescence imaging (Figs. S3,S4). N87/Luc cells, which are N87 cells that stably express luciferase, were generated by lentiviral infection (Fig. S3). The bioluminescence activity of the N87/Luc cells correlated well with the cell number (Fig. S3a,b). The level of HER2 expression in the N87/Luc cells was nearly equivalent to that in N87 cells (Fig. S3c). A mouse model of PMGC was generated by i.p. injection of N87/Luc into SCID mice (Fig. S4). Peritoneal metastasis of GC could be seen around the liver, pancreas, and peritoneal region by in vivo bioluminescence imaging (Fig. S4a–c). Human epidermal growth factor receptor 2 was found to be strongly expressed in xenograft tumors in PMGC mice (Fig. S4d).
More efficient delivery of 211At‐trastuzumab to PMGC with i.p. rather than i.v. injection
To optimize the system of delivery of 211At‐trastuzumab to PMGC in our mouse model, we measured the tumor uptake of 211At in HER2‐positive PMGC mice in order to compare the biodistribution of 211At‐trastuzumab between i.p. and i.v. injected animals (Fig. 2). Astatine‐211‐labeled trastuzumab (1 MBq) was injected into the mice either by i.p. or i.v. routes, and the major tissues, including the PMGCs, were sampled at different time points. The maximum tumor uptake of 211At in the i.p. group was 63.9 ± 52.6%ID/g at 3 h, whereas it was 18.1 ± 8.6%ID/g at 12 h in the i.v. group. The overall uptakes in the whole blood and spleen were also lower in the i.p. group than in the i.v. group. These data suggest that i.p. injection more efficiently delivers 211At‐trastuzumab to HER2‐positive PMGCs without an undesirable distribution to other tissues.
Figure 2.
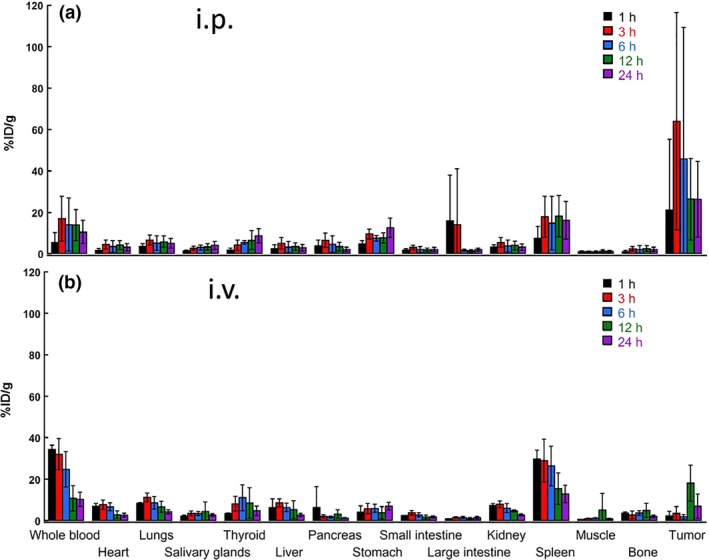
Biodistribution of astatine‐211‐labeled trastuzumab (211At‐trastuzumab) in a mouse model of peritoneal metastasis of gastric cancer. Uptakes (percent injected dose per gram of tissue [%ID/g]) of 211At in the tumor and other organs at 1, 3, 6, 12, and 24 h after i.p. (a) or i.v. (b) injection of 211At‐trastuzumab (1 MBq). Five (for i.p. injection) or four (for i.v. injection) mice were used at each time point. All data represent mean ± SD.
Locoregionally administered 211At‐trastuzumab efficiently treats HER2‐positive PMGC in a mouse model
We evaluated the therapeutic efficacy of 211At‐trastuzumab against HER2‐positive PMGC in our mouse model (Figs. 3, 4; Fig. S5; Table 1). The PMGC mice received a single i.p. injection of PBS, unlabeled trastuzumab, free 211At (1 MBq), or 211At‐trastuzumab (0.1 or 1 MBq) 8 days after N87/Luc implantation. The tumor growth was monitored longitudinally in each group by in vivo bioluminescence imaging (Fig. 3a). Representative bioluminescence images of the mice from each group are shown in Figure 3(b). Tumor growth was evident in the PMGC mice injected with PBS, unlabeled trastuzumab, free 211At, and 0.1 MBq 211At‐trastuzumab. In contrast, a single injection of 1 MBq 211At‐trastuzumab reduced the tumor burden at 3 weeks after injection, with the tumors becoming undetectable at 5–7 weeks after treatment. The relative bioluminescence signal changes in the tumors in all of the treated PMGC mice are shown in Figure 4(a). Trastuzumab and PBS injections had little to no effect on tumor growth in the PMGC model. Tumor growth appeared to be stable until 20 days after treatment with free 211At and resumed thereafter. In PMGC mice treated with 0.1 MBq 211At‐trastuzumab, some mice appeared to have stable disease but ultimately showed disease recurrence. However, in five of the six mice treated with 1 MBq 211At‐trastuzumab, the tumors shrank or stopped growing from 4 days after treatment and had disappeared or were still growth arrested at 60 days after treatment. Only a single mouse that received a 1‐MBq dose of 211At‐trastuzumab showed tumor growth, which was possibly due to misinjection.
Figure 3.
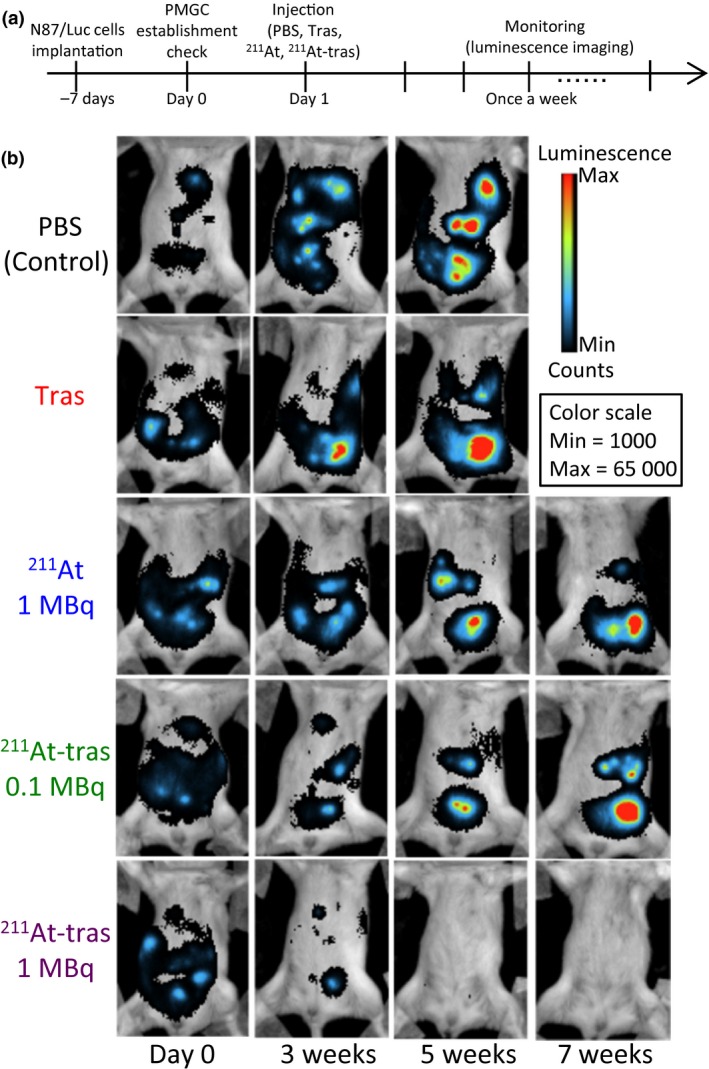
Locoregional therapy with astatine‐211‐labeled trastuzumab (211At‐tras) in a mouse model of peritoneal metastasis of gastric cancer (PMGC), and tumor monitoring. (a) Treatment schedule. (b) Representative bioluminescence images of tumor growth in the mouse model treated with PBS (control), unlabeled trastuzumab (Tras), 211At (1 MBq), or 211At‐tras (0.1 or 1 MBq). Images were captured on the day before treatment (Day 0) or at 3, 5, or 7 weeks (w) after treatment. The color scale indicates the luminescence intensity per pixel.
Figure 4.
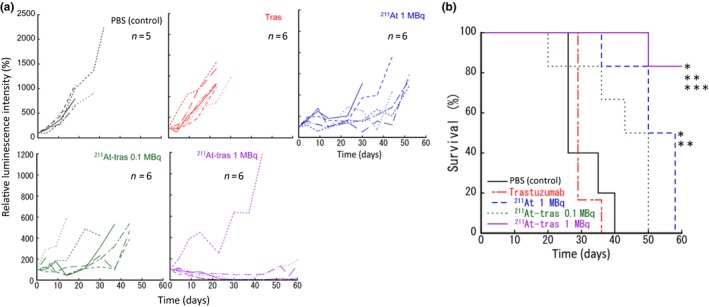
Therapeutic results of locoregional therapy with astatine‐211‐labeled trastuzumab (211At‐tras) in the xenograft mouse model of peritoneal metastasis of gastric cancer. (a) Quantified relative tumor luminescence intensities in each mouse enrolled in this study. The luminescence intensity before the treatment was considered to be 100%. Six mice were enrolled in each of the treatment groups, except the control (five mice). (b) Kaplan–Meier survival curves of mice. *P < 0.05 versus control; **P < 0.05 versus Tras; ***P < 0.05 versus 211At.
Table 1.
Tumor responses and survival in xenograft mouse models of peritoneal metastasis of gastric cancer (PMGC) up to 60 days after treatment with a single injection of PBS (control), trastuzumab (Tras), astatine‐211 (211At; 1 MBq), or 211At‐labeled trastuzumab (211At‐tras; 0.1 or 1 MBq)
| Treatment groups | Undetectable tumor† | 50% reduction‡ | 90% reduction§ | Progressing tumors | No. of survivors¶ |
|---|---|---|---|---|---|
| Control (n = 5) | 0/5 | 0/5 | 0/5 | 5/5 | 0/5 |
| Tras (n = 6) | 0/6 | 0/3 | 0/6 | 6/6 | 0/6 |
| 211At, 1 MBq (n = 6) | 0/6 | 0/6 | 0/6 | 6/6 | 0/6 |
| 211At‐tras, 0.1 MBq (n = 6) | 0/6 | 2/6 | 0/6 | 4/6 | 0/6 |
| 211At‐tras, 1 MBq (n = 6) | 2/6 | 2/6 | 1/6 | 1/6 | 5/6 |
†Numbers of mice in which the PMGC reached an undetectable level during the observation period. ‡Numbers of mice in which the total luminescence intensity of the PMGC was reduced to 50%. §Numbers of mice in which the total luminescence intensity of the PMGC was reduced to 90%, respectively. ¶Numbers of mice in which the total luminescence intensity of the PMGC did not reach the endpoint (1.5 × 107 photons).
The therapeutic results of 211At‐trastuzumab in the PMGC model are presented in Table 1. All mice treated with PBS control, unlabeled trastuzumab, and 211At (1 MBq) showed tumor progression and reached the endpoint (a tumor luciferase intensity of 1.5 × 107) within 60 days of treatment. Two of the six mice that received 0.1 MBq 211At‐trastuzumab showed a 50% reduction in the tumor but eventually also reached the endpoint. Two of the six mice that received a 1‐MBq dose of 211At‐trastuzumab had undetectable tumors; a further three of these mice showed a 50% or 90% tumor reduction, and one mouse still showed tumor growth. The five mice showing tumor regression had not reached the endpoint by 60 days after treatment. Not surprisingly, survival was significantly prolonged in the 1 MBq 211At‐trastuzumab group compared with all of the other groups (P < 0.05). The median survival outcomes were 26, 29, 50, and 43 days in the PBS control, unlabeled trastuzumab, 1 MBq 211At, and 0.1 MBq 211At‐trastuzumab groups, respectively (Fig. 4b).
Excretion and adverse effects of i.p. injected 211At‐trastuzumab in PMGC mouse model
To evaluate the possible adverse effects of 211At‐trastuzumab in our PMGC model, we monitored whether any changes occurred in the bodyweight or number of leukocytes in the treated mice. No apparent bodyweight loss was evident in any of the treated groups during the observation period (Fig. 5a). In addition, no leukocytopenia was found in any of the groups, including the 1 MBq 211At‐trastuzumab mice (Fig. 5b). To then assess the excretion of 211At from the body, we measured its levels in feces and urine. The total 211At activity in the urine (7.3%ID) of the PMGC mice up to 24 h post i.p. injection of 211At‐trastuzumab was much higher than that in the feces (1.3%ID) (Fig. 5c). Biochemical examination of glutamic oxaloacetic transaminase, glutamic pyruvate transaminase, and blood urea nitrogen at 1, 7, and 14 days post‐injection revealed no significant change compared to the pre‐treatment level (Fig. 5d). Creatinine was not detected in any of the mice pre‐injection or at 1, 7, or 14 days post‐injection.
Figure 5.
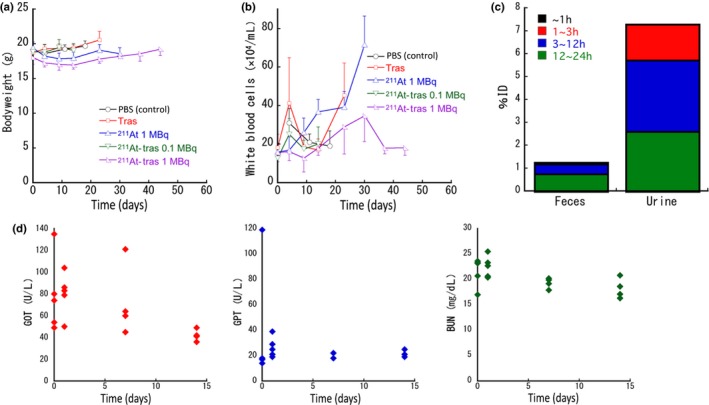
Side‐effects and excretion of astatine‐211 (211At) in a mouse model of peritoneal metastasis of gastric cancer treated with 211At‐labeled trastuzumab (211At‐tras). (a) Bodyweight of the mice after treatment. Plots were interrupted if the animal reached the defined endpoint. Data represent mean ± SD. (b) White blood cell counts in the mice after treatment. Plots were interrupted if the animal reached the defined endpoint. Data represent mean ± SD . (c) 211At levels in feces and urine of the mice up to 24 h after i.p. injection of 211At‐tras (1 MBq). Six mice were used in the experiment. The bars represent the cumulative mean values obtained over a 24‐h observation period. Bars are labeled in the graph. (d) Glutamic oxaloacetic transaminase (GOT; red), glutamic pyruvate transaminase (GPT; blue), and blood urea nitrogen (BUN; green) values up to 14 days after i.p. injection of 211At‐tras (1 MBq). Five mice were used for 0 and 1 days, and four mice were used for 7 and 14 days. Data for each mouse are shown in the graph.
DNA damage is induced in tumor cells by 211At‐trastuzumab
To better understand how 211At‐trastuzumab kills tumor cells, we examined the DNA damage in these cells both in vitro and in vivo by detecting γH2AX foci using immunostaining and Western blot analysis. Clusters of γH2AX foci were clearly observed in N87 cells treated with 211At‐trastuzumab for 24 h, but barely detectable in N87 cells treated with PBS or unlabeled trastuzumab (Fig. 6a). In addition, MKN45 cells treated with even the maximum dose of 211At‐trastuzumab (37 kBq) showed barely any γH2AX foci (Fig. 6a). The level of γH2AX, evaluated by Western blotting, also showed a clear dose‐dependency and increased in N87 cells treated with 211At‐trastuzumab compared to PBS and unlabeled trastuzumab (Fig. 6b). A slightly increased level of γH2AX was observed in MKN45 cells with a high‐dose treatment of 211At‐trastuzumab (18.5 and 37 kBq), but was still much lower than that in N87 cells (Fig. 6b). Dense clusters of γH2AX foci could clearly be observed in N87 cells treated with 18.5 kBq 211At‐trastuzumab after 24 h in an immunofluorescence image taken at a higher magnification (Fig. 6c).
Figure 6.
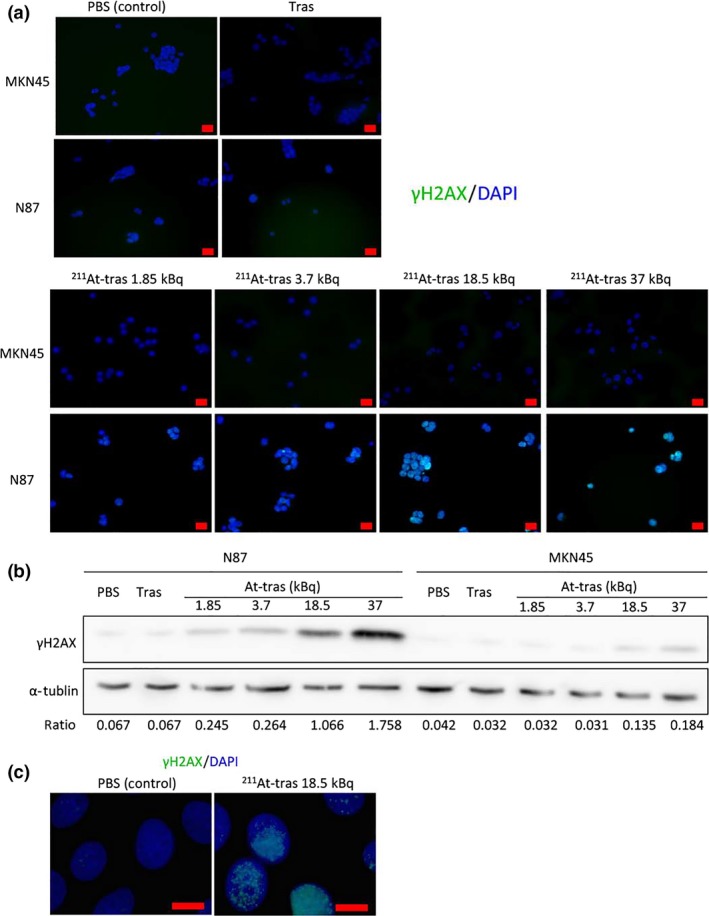
Images and quantitative analyses of γH2AX foci in cells after treatment with astatine‐211‐labeled trastuzumab (211At‐tras). (a) Immunofluorescence images of γH2AX foci in MKN45 and N87 cells treated with PBS, unlabeled trastuzumab (Tras) or 211At‐tras (1.85, 3.7, 18.5, or 37 kBq). Scale bar = 20 μm. (b) Western blot analysis of γH2AX expression in N87 and MKN45 cells treated with PBS, Tras, or 211At‐tras (1.85, 3.7, 18.5, or 37 kBq). α‐Tubulin was used as a loading control. Ratios of the band intensity of γH2AX relative to that of α‐tubulin are indicated below the images. (c) γH2AX foci in N87 cells treated with PBS or 18.5 kBq 211At‐tras. Scale bar = 10 μm.
Clusters of γH2AX foci were clearly evident in tumors dissected from the PMGC model mice at 6 h after the i.p. injection of 1 MBq 211At‐trastuzumab, compared to the control and unlabeled trastuzumab‐treated animals (Fig. 7a). As shown in Figure 7(b), clusters of γH2AX foci were located in the same area showing HER2 expression (Fig. 7b). These observations indicate that 211At‐trastuzumab specifically induces DNA DSBs in HER2‐positive PMGC cells.
Figure 7.
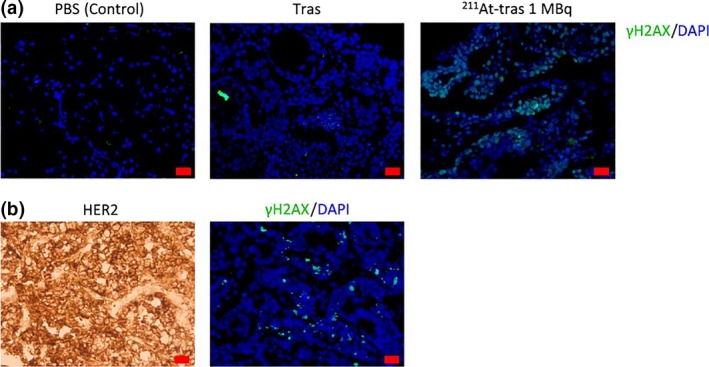
γH2AX foci in a mouse model of peritoneal metastasis of gastric cancer (PMGC). (a) γH2AX foci in PMGC cells at 6 h post‐injection with PBS, unlabeled trastuzumab (Tras), or 1 MBq of astatine‐211‐labeled trastuzumab (211At‐tras). Scale bar = 20 μm. (b) Human epidermal growth factor receptor 2 (HER2) and γH2AX foci in PMGC cells at 3 h post‐injection with 1 MBq 211At‐tras. Scale bar = 20 μm.
Discussion
Despite recent advances in chemotherapy and molecular targeted therapies directed against advanced GC, the prognosis of patients with PMGC is still miserably poor. In our current study, we show that 211At‐trastuzumab specifically targets HER2‐positive GC cells, both in vivo and in vitro, and successfully inhibits tumor growth and improves survival in PMGC model mice.
Our in vitro cytotoxicity analyses showed that 211At‐trastuzumab specifically kills HER2‐positive cells, but not MKN45 cells that have little expression of HER2. Free 211At had little effect in either cell type in vitro. Given that radiolabeled trastuzumab is internalized into the cells it binds,8 our present data suggest that targeted cell binding and internalization of 211At‐trastuzumab is required for 211At to exert its cytotoxic effects. A specific targeting strategy is critically important to maximize the cell‐killing effects of α‐particle emitters in α‐RIT.
We clearly found from our current experiments that i.p. administration is superior to i.v. injection in delivering 211At‐trastuzumab to tumors disseminated in the peritoneal cavity. Considering that 211At decays with a half‐life of 7.2 h, a faster accumulation of high‐dose 211At to the target lesion is required to maximize its antitumor effects. Intraperitoneal delivery also resulted in a lower distribution of 211At to other normal tissues, including the whole blood and spleen, compared with i.v. injection. These data strongly suggest that locoregional therapy helps to avoid adverse effects. Moreover, in this regard, we found that an i.v. injection of 211At‐trastuzumab (0.5 MBq) resulted in acute but recoverable leukocytopenia in s.c. xenografts. In contrast, no leukocytopenia or bodyweight loss was evident in i.p. injected mice, even those injected with a 1‐MBq dose of 211At‐trastuzumab. In addition, no significant biochemical changes in either the liver or kidney were observed in mice treated with 1 MBq 211At‐trastuzumab. We conclude, therefore, that i.p. delivery of 211At‐trastuzumab is far more suitable in terms of undesirable adverse effects. Recent studies have explored the utility of i.p. α‐RIT in various preclinical cancer models.16, 17, 18 Furthermore, a previous clinical trial reported that the therapeutic absorbed dose of i.p. α‐RIT in ovarian cancer at the peritoneal cavity was >100 times greater than that in the red bone marrow without significant toxicity.19 Thus, i.p. α‐RIT appears to be a clinically feasible approach.
We further showed from our current analysis that α‐RIT with 211At‐trastuzumab suppressed tumor growth in both s.c. GC xenografts and PMGC xenografts. In our PMGC mice, five of the six that received only a single locoregional treatment with 211At‐trastuzumab showed improved survival and a drastic reduction in tumor burden. We also found that 211At‐trastuzumab induced massive DNA DSBs in HER2‐positive GC cells, but not in HER2‐negative cells. Unlabeled trastuzumab of an equal protein level to that of the labeled antibody could not induce DSB in N87 cells. We thus speculate that α‐particles emitted from 211At‐trastuzumab efficiently induced irreparable DSBs in N87 cells and thereby caused tumor shrinkage.
A previous study showed that 211At‐trastuzumab is effective against radioresistant ovarian i.p. tumors in mice.20 In that report, a single i.p. injection of 0.8 MBq 211At‐trastuzumab did not achieve a tumor‐free outcome in the mice. However, 0.4 MBq radiolabeled antibody combined with unlabeled trastuzumab resulted in a complete eradication of the tumors. Additionally, 500 μg unlabeled trastuzumab, which is a more than 100‐fold higher amount of protein than that used in our present experiments, had a better therapeutic result compared to 0.8 MBq 211At‐trastuzumab monotherapy. In the present study, we did not fully investigate the effects of unlabeled trastuzumab on human GC N87 cells. Further studies are therefore needed to clarify the effects of unlabeled antibody on α‐RIT.
The success of trastuzumab has led us to develop a novel type of HER2‐targeted drug. Previously, trastuzumab emtansine, an antibody–drug conjugate carrying the cytotoxic agent emtansine bound to trastuzumab, has shown antitumor activity against HER2‐positive tumors and emerged as a powerful therapeutic option for advanced or metastatic breast cancer that is resistant to trastuzumab.21, 22 We speculate, however, that because a single α‐particle atom can kill a target cell,11, 12 the cytotoxic effects of 211At‐trastuzumab could even be superior to those of trastuzumab‐based antibody–drug conjugates.
In principle, locoregional α‐RIT reduces the risk of the hematological toxicities that are the most worrying adverse effects of this treatment approach. We propose that locoregional therapy with 211At‐trastuzumab is a safe and highly effective therapy and is a very promising future therapeutic option for PMGC.
Disclosure Statement
S. Hasegawa has received a research grant from the Astellas Foundation for research on metabolic disorders. The other authors have no conflict of interest.
Abbreviations
- %ID/g
percent injected dose per gram of tissue
- 211At
astatine‐211
- 211At‐trastuzumab
astatine‐211‐labeled trastuzumab
- DSB
double‐strand break
- GC
gastric cancer
- HER2
human epidermal growth factor receptor 2
- PMGC
peritoneal metastasis of gastric cancer
- RIT
radioimmunotherapy
- α‐RIT
α‐particle radioimmunotherapy
Supporting information
Fig. S1 Serum stability of astatine‐211‐labeled trastuzumab for up to 24 h after incubation in murine serum.
Fig. S2 Biodistribution and therapeutic results of α‐particle radioimmunotherapy with astatine‐211‐labeled trastuzumab (211At‐tras) in s.c. xenograft mouse models.
Fig. S3 Establishment of N87/Luc cells.
Fig. S4 Establishment of the xenograft mouse model of peritoneal metastasis of gastric cancer.
Fig. S5 Bioluminescence images of tumor growth in the mice.
Doc. S1 Supplementary materials and methods.
Acknowledgments
We thank Drs. Tsuneo Saga, Takako Furukawa, Atsushi Tsuji, and Masumi Abe for helpful discussions and support, and Katsuyuki Minegishi and Hisashi Suzuki for radioisotope production. This work was partly supported by the Japan Society for the Promotion of Science (KAKENHI Grant Nos. 24390296 to S.H., 16H07460 to Y.M., and 17J02307 to H.K.L.), a research grant from the Astellas Foundation for research on metabolic disorders (to S.H.), and a President's grant from the National Institute of Radiological Sciences (to S.H.).
Cancer Sci 108 (2017) 1648–1656
Funding Information
Japan Society for the Promotion of Science; Astellas Foundation for research on metabolic disorders; National Institute of Radiological Sciences
References
- 1. Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev 2014; 23: 700–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomassen I, van Gestel YR, van Ramshorst B et al Peritoneal carcinomatosis of gastric origin: a population‐based study on incidence, survival and risk factors. Int J Cancer 2014; 134: 622–8. [DOI] [PubMed] [Google Scholar]
- 3. Park DI, Yun JW, Park JH et al HER‐2/neu amplification is an independent prognostic factor in gastric cancer. Dig Dis Sci 2006; 51: 1371–9. [DOI] [PubMed] [Google Scholar]
- 4. Bang YJ, Van Cutsem E, Feyereislova A et al Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010; 376: 687–97. [DOI] [PubMed] [Google Scholar]
- 5. Pouget JP, Navarro‐Teulon I, Bardies M et al Clinical radioimmunotherapy–the role of radiobiology. Nat Rev Clin Oncol 2011; 8: 720–34. [DOI] [PubMed] [Google Scholar]
- 6. Larson SM, Carrasquillo JA, Cheung NK, Press OW. Radioimmunotherapy of human tumours. Nat Rev Cancer 2015; 15: 347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janik JE, Morris JC, O'Mahony D et al 90Y‐daclizumab, an anti‐CD25 monoclonal antibody, provided responses in 50% of patients with relapsed Hodgkin's lymphoma. Proc Natl Acad Sci USA 2015; 112: 13045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li HK, Morokoshi Y, Daino K et al Transcriptomic signatures of auger electron radioimmunotherapy using nuclear targeting (111) In‐trastuzumab for potential combination therapies. Cancer Biother Radiopharm 2015; 30: 349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Couturier O, Supiot S, Degraef‐Mougin M et al Cancer radioimmunotherapy with alpha‐emitting nuclides. Eur J Nucl Med Mol Imaging 2005; 32: 601–14. [DOI] [PubMed] [Google Scholar]
- 10. Zalutsky MR, Pruszynski M. Astatine‐211: production and availability. Curr Radiopharm 2011; 4: 177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nikula TK, McDevitt MR, Finn RD et al Alpha‐emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti‐CD33 antibodies: pharmacokinetics, bioactivity, toxicity and chemistry. J Nucl Med 1999; 40: 166–76. [PubMed] [Google Scholar]
- 12. McDevitt MR, Ma D, Lai LT et al Tumor therapy with targeted atomic nanogenerators. Science 2001; 294: 1537–40. [DOI] [PubMed] [Google Scholar]
- 13. Nagatsu K, Minegishi K, Fukada M, Suzuki H, Hasegawa S, Zhang MR. Production of (211)At by a vertical beam irradiation method. Appl Radiat Isot 2014; 94: 363–71. [DOI] [PubMed] [Google Scholar]
- 14. Lindegren S, Frost S, Back T, Haglund E, Elgqvist J, Jensen H. Direct procedure for the production of 211At‐labeled antibodies with an epsilon‐lysyl‐3‐(trimethylstannyl)benzamide immunoconjugate. J Nucl Med 2008; 49: 1537–45. [DOI] [PubMed] [Google Scholar]
- 15. Chrestensen CA, Shuman JK, Eschenroeder A, Worthington M, Gram H, Sturgill TW. MNK1 and MNK2 regulation in HER2‐overexpressing breast cancer lines. J Biol Chem 2007; 282: 4243–52. [DOI] [PubMed] [Google Scholar]
- 16. Cederkrantz E, Angenete E, Back T et al Evaluation of effects on the peritoneum after intraperitoneal alpha‐radioimmunotherapy with (211)At. Cancer Biother Radiopharm 2012; 27: 353–64. [DOI] [PubMed] [Google Scholar]
- 17. Frost SH, Back T, Chouin N et al Comparison of 211At‐PRIT and 211At‐RIT of ovarian microtumors in a nude mouse model. Cancer Biother Radiopharm 2013; 28: 108–14. [DOI] [PubMed] [Google Scholar]
- 18. Derrien A, Gouard S, Maurel C et al Therapeutic efficacy of alpha‐rit using a (213)Bi‐Anti‐hCD138 antibody in a mouse model of ovarian peritoneal carcinomatosis. Front Med (Lausanne) 2015; 2: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andersson H, Cederkrantz E, Back T et al Intraperitoneal alpha‐particle radioimmunotherapy of ovarian cancer patients: pharmacokinetics and dosimetry of (211)At‐MX35 F(ab’)2–a phase I study. J Nucl Med 2009; 50: 1153–60. [DOI] [PubMed] [Google Scholar]
- 20. Palm S, Back T, Claesson I et al Therapeutic efficacy of astatine‐211‐labeled trastuzumab on radioresistant SKOV‐3 tumors in nude mice. Int J Radiat Oncol Biol Phys 2007; 69: 572–9. [DOI] [PubMed] [Google Scholar]
- 21. Lewis Phillips GD, Li G, Dugger DL et al Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Cancer Res 2008; 68: 9280–90. [DOI] [PubMed] [Google Scholar]
- 22. Verma S, Miles D, Gianni L et al Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med 2012; 367: 1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Serum stability of astatine‐211‐labeled trastuzumab for up to 24 h after incubation in murine serum.
Fig. S2 Biodistribution and therapeutic results of α‐particle radioimmunotherapy with astatine‐211‐labeled trastuzumab (211At‐tras) in s.c. xenograft mouse models.
Fig. S3 Establishment of N87/Luc cells.
Fig. S4 Establishment of the xenograft mouse model of peritoneal metastasis of gastric cancer.
Fig. S5 Bioluminescence images of tumor growth in the mice.
Doc. S1 Supplementary materials and methods.