

Summary
Objective
The coincidence of autism with epilepsy is 27% in those individuals with intellectual disability1. Individuals with loss of function mutations in SHANK3 have intellectual disability, autism and variably, epilepsy2–5. The spectrum of seizure semiologies and electroencephalographic (EEG) abnormalities has never been investigated in detail. With the recent report that SHANK3 mutations are present in approximately two percent of individuals with moderate to severe intellectual disabilities and one percent of individuals with autism, determining the spectrum of seizure semiologies and electrographic abnormalities will be critical for medical practitioners to appropriately counsel the families of patients with SHANK3 mutations.
Methods
A retrospective chart review was performed of all individuals treated at the Blue Bird Circle Clinic for Child Neurology who have been identified as having either a chromosome 22q13 microdeletion encompassing SHANK3 or a loss of function mutation in SHANK3 identified through whole exome sequencing.
For each subject, the presence or absence of seizures, seizure semiology, frequency, age of onset and efficacy of therapy were determined. Electroencephalograms were reviewed by a board certified neurophysiologist. Neuroimaging was reviewed by both a board certified pediatric neuroradiologist and child neurologist.
Results
There is a wide spectrum of seizure semiologies, frequencies and severity in individuals with SHANK3 mutations. There are no specific electroencephalographic abnormalities found in our cohort, and EEG abnormalities were present in individuals diagnosed with epilepsy and those without history of a clinical seizure.
Significance
All individuals with a mutation in SHANK3 should be evaluated for epilepsy due to the high prevalence of seizures in this population. The most common semiology is atypical absence seizure which can be challenging to identify due to comorbid intellectual disability in individuals with SHANK3 mutations; however, no consistent seizure semiology, neuroimaging findings or electroencephalogram findings were present in the majority of individuals with SHANK3 mutations.
Keywords: SHANK3, Phelan-McDermid Syndrome, epilepsy, electroencephalography, autism
Introduction
Loss of function mutations in the SHANK3 gene, either through chromosomal microdeletion in Phelan-McDermid Syndrome (PMS) or through missense/nonsense mutations, are a growing area of importance for neurologists and epileptologists3. Approximately two percent of individuals with moderate to severe intellectual disability and one percent of individuals with autism are estimated to harbor such mutations6.
The protein product of the SHANK3 gene is a scaffolding protein that localizes primarily to the post-synaptic density of excitatory synapses7. At the post-synaptic density, it bridges transmembrane proteins critical for synapse formation such as Neuroligins and neurotransmitter receptors such as NMDA glutamate receptors with the underling cytoskeleton, in particular F-actin elements8; 9. Loss of SHANK3 in multiple model systems, cultured neurons to mice, has demonstrated synaptic dysfunction10–13. As such, SHANK3 is a critical protein for biogenesis and maintenance of synapses as well as synapse function.
Epilepsy has been reported in individuals found to have 22q13 deletions including SHANK3 with a prevalence ranging from 17–70% in multiple case series (Table I). Most of these studies, however, have provided limited information about the semiology, frequency or severity of seizures in this population. Nor have they provided information about the spectrum or frequency of electroencephalographic abnormalities of these patients. Here we present a detailed report of the spectrum of seizures and electroencephalographic findings in individuals with SHANK3 loss of function mutations. We find significant heterogeneity in seizure types and frequency in our cohort. Furthermore, we identify a wide spectrum of electroencephalographic abnormalities in our subjects which are present in both individuals diagnosed with epilepsy and those without.
Table I.
Seizure and EEG characterization from previous studies of Phelan-McDermid Syndrome
| Study | Prevalence of seizures |
Types of seizures described |
EEG findings | Number of patients |
|---|---|---|---|---|
| Phelan et al.21 | 27% | N/A | N/A | 37 |
| Wilson et al.22 | 70% | N/A | N/A | 51 |
| Luciani et al.23 | 24% | N/A | N/A | 33 |
| Manning et al.24 | 27% | “Petit mal and focal” | N/A | 11 |
| Lindquist et al.25 | 33% | N/A | N/A | 6 |
| Jeffries et al.26 | 17% | N/A | N/A | 30 |
| Cusmano et al.5 | 23% | N/A | N/A | 107 |
| Dhar et al.4 | 30% | N/A | N/A | 11 |
| Soorya et al.3 | 41% | “Generalized” (5/6), “partial-onset” (1/6) | 34% with abnormality | 32 |
| Sarasua et al.2 | 27% | N/A | N/A | 151 |
| Figura et al.16 | 50% | “Myoclonic, tonic, generalized tonic-clonic” | Multifocal spike and wave | 6 |
| Denayer et al.27 | 14% | N/A | N/A | 7 |
| Nesslinger et al.28 | 14% | N/A | N/A | 7 |
N/A = not available
Patients and Methods
Ethics
This study was approved by the Institutional Review Board of Baylor College of Medicine. All participants or their guardians provided informed written consent to participate in this study.
Patient Selection and Evaluation
We retrospectively collected data from a single center (Texas Children’s Hospital) through the electronic medical record. Information about the seizure characteristics of 24 individuals (10 males and 14 females) with either a chromosomal deletion encompassing chromosome 22q13 including SHANK3, point mutation, indel or microduplication of SHANK3 predicted to result in a loss of function mutation were analyzed. Data regarding the onset of first clinical seizure, seizure frequency, duration, semiology, response to pharmaceutical and non-pharmaceutical therapies and neuroimaging abnormalities were collected and analyzed.
Electroencephalogram interpretation
Electroencephalograms were obtained at a single site with review by a board certified neurophysiologist. The video-EEG recordings were obtained using the Nicolet video-EEG system (Natus Medical, Inc.) while patients were awake, drowsy and, when possible, asleep with a 21-channel EEG acquisition system using silver-chloride surface electrodes. The EEG data was reviewed in both bipolar and referential montages.
Results
Clinical findings
We retrospectively evaluated twenty-four subjects with SHANK3 mutations including twenty individuals with chromosomal deletions of 22q13.33 including SHANK3 and four individuals with point mutations identified by whole exome sequencing resulting in either frameshift or mis-splicing of the SHANK3 coding frame (Table II). We found eleven out of 24 subjects had a history of at least one lifetime seizure (46%), including two of the four individuals with point mutations. The spectrum of seizure semiologies varied significantly from atypical absence seizures (90%) being the most common seizure type to tonic (54%), atonic (18%), tonic-clonic (9%) and myoclonic (9%) in those individuals with a history of clinical seizure. Six of the eleven subjects with a history of seizure had more than one seizure type (54%), and two patients from this cohort (8%) were diagnosed with Lennox-Gastaut Syndrome based upon the combination of multiple seizure types (including tonic seizures), intractability of their seizures and characteristic EEG finding of generalized bursts of 1.5–2 Hz spike and slow wave activity. Five of our 24 subjects (20%) had a history of status epilepticus requiring emergency intravenous medication for cessation of seizure activity. The onset of first seizure ranged from 14 months to 14 years with a mean age of onset of 5.2 years ± 3.9 (SD). The frequency of seizures similarly varied from a single lifetime seizure to hundreds of seizures per day (Table II).
Table II.
Clinical Characteristics of Subjects
| Sub- ject |
Age | Genetic abnormality | Age first SZ |
Frequency of SZ prior to medication |
Frequency of SZ on medication |
SZ semi- ology |
Dura- tion of SZ |
SE | EEG findings | Neuroimaging findings |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 years | 22q deletion (44753814–51197838) hg19 | 14 month | daily | <1 per month | atypical absence, tonic | 10–15 seconds | yes | excess beta activity, foci of SW activity - left central and parietal regions, intermittent slowing (delta) - bilateral occipital regions | thinning of corpus callosum |
| 2 | 17 years | 22q13.33 (GS-99K24 X1) | 8 years | hundreds per day | daily | tonic, atypical absence, atonic, myoclonic, tonic-clonic | seconds to > 5 minute | yes | poorly organized ODR 6 Hz, frontocentral 8–10 Hz sharps evolving into 3–4 Hz spike and polyspike; bursts of generalized 1.5–2.5 Hz SW, intermittent generalized 3–5 Hz in 50% of recording | originally normal, evolved into left mesial temporal sclerosis |
| 3 | 9 years | 22q13.3 (49138472–49535360 X1) | 5 years | three times per week | 2 times per month | atypical absence, tonic | 15–20 seconds | no | ODR 7–8 Hz,, frontal 1.5–2 Hz generalized SW/polyspike | normal |
| 4 | 11 years | 22q13.33 (49177451–49525470), 2p12 (76910620–77354922 x1) hg18 | 9 years | <1X/week | <1X/week | atypical absence | 30 seconds | no | ODR 8 Hz with slower fused waveforms; multifocal SW/ polyspike most frequent in the right frontal region, rare generalized SW | increased T2 signal in centrum semiovale, small corpus callosum |
| 5 | 10 years | 22q13.33 (49484191–49525470 X1) hg18 | none | none | N/A | N/A | N/A | no | None | normal |
| 6 | 6 years | 22q13.33 (47061550–51304566 X1) hg19 | 2 years | 10–20 × per day | N/A | atypical absence | 20 sec - 1 min | no | ODR 8–8.5 Hz with some slower activity; active foci of SW in isolation and in runs in the left and right temporoparietocentral regions | brachycephalic, otherwise normal |
| 7 | 12 years | 22q13.33 (48771374–51178264 X1); 7q31.1 (111056675–111191737 X1) hg19 | none | none | N/A | N/A | N/A | no | ODR 6–7 Hz poorly developed and sustained; sharp waves and spikes over left and right temporal; anterior temporal and frontal regions independently | T2 prolongation of subcortical white matter bilateral temporal poles, slightly foreshortened corpus callosum |
| 8 | 15 years | 22q13 deletion (no data) | 5 years | hundreds per day | 1–2 per day | atonic, atypical absence | seconds to >5 minute | yes | ODR 4–5 Hz; low amplitude spike discharges - left central; polyspike at right frontoparietal | arachnoid cyst |
| 9 | 5 years | 22q13 (48533991–51178264 X1) 12p13.33 (189216–8185497 X3) hg19 | none | none | N/A | N/A | N/A | no | ODR 8–9 Hz; normal | mild increased FLAIR signal in periventricular white matter of frontal, parietal and temporal lobes, moderate reduction in cerebral white matter volume including corpus callosum thinning, mild decreased size of the left hippocampus |
| 10 | 12 years | 22q13.2 deletion by karyotype | 3 years | daily | 1/2–3 weeks | atypical absence, tonic | minute to hours | yes | no ODR, right centroparietooccipital sharps | mild thinning of the corpus callosum, generalized loss of white matter |
| 11 | 4 years | 22q13.2-3(43572964–51171678 X1) | 1 year | 2 lifetime with fever | N/A | tonic | 1 minute | no | ODR 6 Hz with slower waveforms; during sleep - bursts of generalized anterior dominant SW/polyspike | mild hypoplasia of the posterior body of the corpus callosum, large cystic cavum septum pellucidum |
| 12 | 17 years | 22q13.2-3 (43014031–51171678 X1) | none | none | N/A | N/A | N/A | no | ODR 5–6 Hz; SW in right parietal and frontal | curvilinear cortical T2 hyperintensity right extreme capsule, infratentorial/supratentorial volume loss, persistent cavum septum pallucidum et vergae, giant cisterna magna |
| 13 | 13 years | 22q13.31-3 (45680160–51178264 X1) 2q37.3 (241994956–242938241 X3) hg19 | 6 years | one | N/A | atypical absence | 30 minute | yes | ODR 8 Hz, normal | none |
| 14 | 28 years | 22q13.31q13..33 deletion by FISH | none | none | N/A | N/A | N/A | no | normal per parental report | none |
| 15 | 19 years | 22q13.33 (49451411–49525130 X1) hg18 | none | none | N/A | N/A | N/A | no | 10–11 Hz, normal | cerebellar ectopia |
| 16 | 7 years | 22q13.33 (G248p86064C8–G248p86149G7 X1) BAC array | none | none | N/A | N/A | N/A | no | ODR 10–11 Hz, excessive beta activity | normal |
| 17 | 16 years | 22q13 deletion by FISH | none | none | N/A | N/A | N/A | no | ODR 9.5–10 Hz; occasional moderate voltage sharp waves right frontocentral regions near midline and right centrotemporal regions occasionally in serial | left sylvian fissure arachnoid cyst; mild cerebral volume loss, partial empty sella |
| 18 | 12 years | 22q13.33 (49469317–49525130 X1) hg18 | none | none | N/A | N/A | N/A | no | none | normal |
| 19 | 9 years | 22q13.33 (50103257–51178264 x1) hg19 | none | none | N/A | N/A | N/A | no | none | none |
| 20 | 3 years | 22q13.33 (51137,176–51,197,838 X1) | none | none | N/A | N/A | N/A | no | poorly sustained ODR of 8 Hz | persistent cavum septum pellucidum et vergae |
| 21 | 13 years | 15q11.2 dup; SHANK3 c.2313+1G>A | none | none | N/A | N/A | N/A | no | ODR 8–9 Hz, normal | left sylvian fissure arachnoid cyst |
| 22 | 14 years | SHANK3 indel (q51160326–51160327) | 14 years | unknown | 1/week | atypical absence | 3 minute | no | no ODR | normal except mild cerebellar tonsillar ectopia |
| 23 | 14 years | SHANK3 indel (q51160326–51160327) | 7 years | multiple times per week | 2–3 times per week | atypical absence, tonic | 3–5 minutes | no | ODR 9–10 Hz, normal | normal |
| 24 | 13 years | SHANK3 c.3727dupG(p.A1212fs) | none | none | none | none | none | no | Normal | bilateral T2 hyperintensities of posterior centrum semiovale |
ODR = Occipital Dominant Rhythm; SW = spike and slow wave
Electrographic findings
Electroencephalographic abnormalities were identified in fourteen out of twenty-one subjects (67%) for which EEG data was available. The spectrum of EEG abnormalities was wide from slowing or absence of the occipital dominant rhythm (42%) to focal spike and slow wave discharges (38%) to generalized spike and slow wave discharges (19%) (Figure 1). Several patients had combinations of the above abnormalities. Of the twenty-one patients with EEG data available, five had an abnormal EEG (either slow occipital dominant rhythm or focal spike and slow wave activity or both) but no history of clinical seizure (23%).
Figure 1.
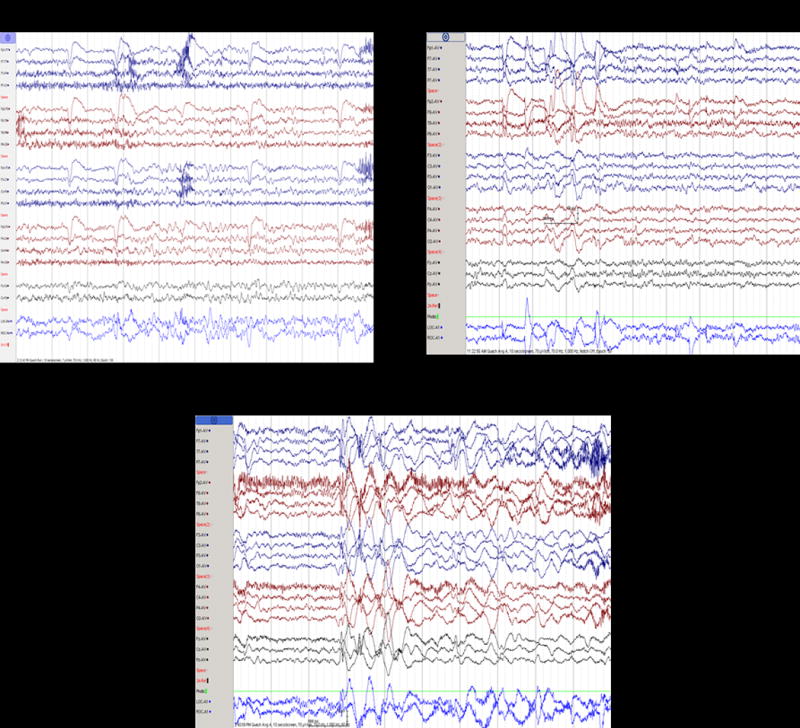
Electroencephalograms in individuals with loss of function mutations in SHANK3 (A) Subject 22 with generalized slowing of the occipital dominant rhythm (B) Subject 4 with frontal dominant generalized burst of 1.5 Hz spike and slow wave activity (C) Subject 2 with generalized burst of 1.5–2.5 Hz spike and slow wave activity.
Most individuals with multiple seizure types had an abnormally slow occipital dominant rhythm (83%) while the majority of individuals with a single seizure type were found to have an age-appropriate occipital dominant rhythm (60%). Similarly, both individuals with high seizure burden of hundreds of seizure per day prior to anti-convulsant medication were found to have a significantly slowed occipital dominant rhythm. However, it should be noted that three of the ten individuals (30%) for whom we have EEG data and without a history of seizure had abnormally slow occipital dominant rhythm.
Neuroimaging
Of our 24 subjects, 21 had an anatomic brain magnetic resonance imaging (MRI) available for review (88%). The abnormalities identified varied greatly between individuals (Figure 2). The most common abnormalities were dysmorphisms of the corpus callosum (29%) and T2 hyperintensities of the deep white matter (24%). Anatomic brain abnormalities were detected in both individuals with history of seizures (33%) and without (38%).
Figure 2.
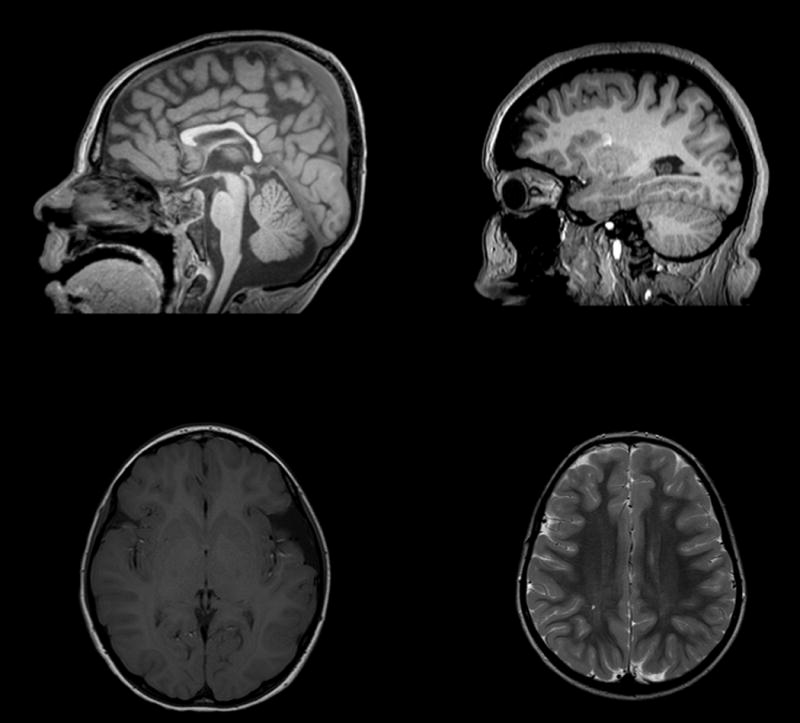
Brain Magnetic Resonance Imaging (MRI) in individuals with loss of function mutations in SHANK3 (A) Sagittal T1 weighted image of subject 9 demonstrating thinning of the corpus callosum (B) Sagittal T1 weighted image of subject 17 with mild cerebral volume loss (C) Axial T1 weighted image of subject 21 with left sylvian fissure arachnoid cyst (D) Axial T2 weighted image of subject 4 with mild T2 hyperintensity of the posterior centrum semiovale.
Treatments
The type and number of anti-convulsant medications prescribed varied greatly among subjects. The most commonly prescribed anti-convulsant was lamotrigine (17%) followed by levetiracetam and topiramate (12% each) and then several others in less than 10% of this population (rufinamide, zonisamide, perampanel, felbamate, valproate, carbamazepine, oxcarbazepine, lacosamide, phenytoin, methsuximide, chlorazepate, lorazepam, phenobarbital). While underpowered to evaluate for efficacy in this study, no medication was clearly superior for seizure prophylaxis. There was a trend for those with one seizure type to respond to fewer medications than those with multiple seizure semiologies. All individuals with only one semiology (six) were maintained on less than two anti-convulsant medications while three of five individuals with multiple seizure types required two or more maintenance anti-convulsant medications (p=0.06, Fisher’s exact test). Two individuals were found to have pharmaco-resistant epilepsy and had implantation of vagus nerve stimulators with mild improvement in seizure frequency (<50% reduction in seizures).
Longitudinal outcomes
Of the eleven patients described here with SHANK3 loss of function mutation and a history of seizure, five (subjects 1, 2, 4, 11 and 13) were evaluated at least twice in clinic.
Patient 2 developed epilepsy at eight years of age with the first seizure being generalized tonic-clonic and lasting more than five minutes thus requiring intravenous medication. Seizures evolved to include atonic and myoclonic seizures at 9 years of age and tonic seizures by 10 years of age at which time he was diagnosed with Lennox-Gastaut syndrome. Medication history included initiation of 14 different anti-convulsants in multiple combinations. Ketogenic diet was also attempted, initially with reduction of seizure burden by 90% but then return of multiple daily seizures leading to its discontinuation. A vagus nerve stimulator was also implanted at 10 years of age with initially >50% reduction in seizure burden following escalation of therapy, but subsequent increasing seizure burden. Following multiple injuries from atonic seizures despite use of a protective helmet, corpus callosotomy was performed at 15 years of age with significant reduction in frequency and severity of atonic seizures for more than two years. Initial neuroimaging was normal but developed into T2 hyperintensity of the left hippocampus suggestive of mesial temporal sclerosis.
Patient 4 developed epilepsy at two years of age. The initial seizure type was atypical absence seizure and this remains her only semiology. Initial treatment with lamotrigine was not efficacious, and she was transitioned to zonisamide therapy with less than one seizure per month.
Patient 1 had her first seizure at 14 months of age which was an episode of febrile status epilepticus. This initial event was characterized by a tonic seizure with decreased responsiveness. This was followed by multiple daily atypical absence seizures at which point she was initiated on levetiracetam therapy which reduced her seizure burden from daily to less than one per month.
Patient 11 had a history of two lifetime seizures both occurring with fever. The first was at one year of age. Both lasted less than one minute in duration. This patient was never placed on anti-convulsant medication.
Patient 13’s only seizure occurred at 6 years of age and was associated with illness. The seizure lasted for approximately 30 minutes requiring intravenous medication. She has never required daily anti-convulsant medication.
Discussion
Here we present extensive data of seizure characterization and electroencephalogram abnormalities in individuals with SHANK3 mutations. SHANK3 haploinsufficiency is believed to be the etiology of the cognitive abnormalities in individuals with Phelan-McDermid Syndrome due to a similar phenotype in an individual with a balanced translocation disrupting SHANK3 and multiple studies which have identified individuals with point mutations, indels or small duplications involving the SHANK3 gene who have similar autistic traits and intellectual disability as individuals with chromosomal deletions encompassing SHANK311; 14; 15. Epilepsy is present in a substantial number of individuals with Phelan-McDermid Syndrome with a pooled prevalence of 32% in unique studies available from the literature including this one. Whether or not mutations in SHANK3 underlie the increased propensity for epilepsy in Phelan-McDermid Syndrome is unclear. In our cohort, we identified four individuals by exome sequencing with variants in SHANK3 predicted to be deleterious. Of these, two developed epilepsy. Furthermore, the largest study to date of 22q13 deletions found no significant differences in deletion size in those with epilepsy and those without2. These data provide preliminary evidence that loss of function mutations in SHANK3 can increase the likelihood to develop epilepsy; however, confirmation will require larger cohorts of individuals with indels and nonsense mutations in SHANK3.
One previous study investigating the seizure types associated with Phelan-McDermid Syndrome suggested that epilepsy associated with this disorder is mild and easily pharmacologically controlled16. We find in our cohort some individuals with SHANK3 deletions do have infrequent and easily managed epilepsy, but we also identified individuals with intractable seizures including Lennox-Gastaut Syndrome. Lennox-Gastaut Syndrome has been previously reported in one adult with a chromosome 22q13 deletion including the SHANK3 gene17.
It is unclear exactly what distinguishes those individuals who will have a single seizure semiology and infrequent seizures from those that have multiple seizure types. There was no significant difference in deletion size, neuroimaging finding or other clinical characteristic (Table II). The only parameter that trended with multiple seizure types was slow or absent occipital dominant rhythm (five of six) in individuals with multiple seizure types versus those with a single seizure type (two of five). Together, these data demonstrate that medical practitioners must educate families caring for individuals with SHANK3 loss of function mutations that epilepsy can become pharmacoresistent and intractable.
Why haploinsufficency of SHANK3 would predispose to epilepsy is unclear. Multiple lines of mice with loss of function mutations in SHANK3 have been reported to date10; 12; 13. Only one (Shank3 alpha/beta isoform knockout) has been reported to have spontaneous seizures although these seizures were not characterized in detail10. Excitatory neurons differentiated from patient iPSCs with SHANK3 deletions have been reported18. These neurons display multiple molecular and neurophysiologic abnormalities. Importantly, they have increased input resistance compared to control neurons. This indicates a tendency to increased excitability potentially underlying the elevated prevalence of epilepsy in individuals with SHANK3 mutations compared with the general population.
Recently, a mechanism for the increased input resistance of stem cell derived neurons has been proposed19. Utilizing neurons derived from human embryonic stem cells with a targeted mutation of SHANK3, impairment of Ih currents was identified due to reduced expression of subunits of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Moreover mutations in HCN channels have been clinically associated with epilepsy20. Intriguingly while the seizures were polymorphic in individuals with HCN1 mutations, all patients reported in this study were diagnosed with atypical absence seizures. Together, these data suggest a mechanism by which hapolinsufficiency of SHANK3 might predispose to epilepsy and why there is a wide spectrum of seizure semiologies with atypical absence being the most common type of seizure in patients with SHANK3 haploinsufficiency.
There are no consistent neuroimaging abnormalities that are likely to predispose to seizures. In particular, there is no evidence in our population or from previous reports of migrational abnormalities that are likely causative of their seizures. In our cohort, we found neuroimaging abnormalities as frequently in subjects with a history of seizures as those without. What is less clear is whether individuals with SHANK3 mutations have microscopic neuroanatomic abnormalities or abnormalities in connectivity that might be responsible for both the cognitive dysfunction and propensity for epilepsy. This will require more advanced neuroimaging techniques such as diffusion tenor imaging or functional magnetic resonance imaging to determine.
Summary
Loss of function mutations in SHANK3 either through chromosomal deletion or point mutation predispose to seizures and electroencephalographic abnormalities. Atypical absence seizures are the most common seizures in individuals with SHANK3 mutations; however, there is a wide spectrum of seizure types. No specific electroencephalographic abnormality is present in our cohort, and abnormalities were seen in both individuals with a history of seizures and those without. Neuroimaging in our cohort did not identify a specific abnormality associated with seizures. No pharmacologic therapies were clearly superior for treating patients in our cohort. Individuals with loss of function mutations in SHANK3 should be monitored for seizure activity.
Key Point Box.
Seizures occur in greater than 30% of children with SHANK3 mutations
The most common seizure type is atypical absence
A subset of patients with SHANK3 mutations have medically intractable epilepsy
There are no pathognomonic abnormalities on EEG in children with SHANK3 mutations
EEG abnormalities are seen in children harboring SHANK3 mutations with and without history of seizures
Acknowledgments
We are indebted to our patients and their guardians for participating in our study. We thank Sunita Misra and James Orengo for critical review of this manuscript. Dr. Holder is supported by a grant from the Thrasher Research Fund (Early Career Award) and a K08 Clinical Scientist Career Development Award (NS091381).
Footnotes
Dr. Holder has full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
None of the authors has any conflict of interest to disclose.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Tuchman R, Hirtz D, Mamounas LA. NINDS epilepsy and autism spectrum disorders workshop report. Neurology. 2013;81:1630–1636. doi: 10.1212/WNL.0b013e3182a9f482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarasua SM, Boccuto L, Sharp JL, Dwivedi A, Chen CF, Rollins JD, Rogers RC, Phelan K, DuPont BR. Clinical and genomic evaluation of 201 patients with Phelan-McDermid syndrome. Human genetics. 2014;133:847–859. doi: 10.1007/s00439-014-1423-7. [DOI] [PubMed] [Google Scholar]
- 3.Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, Frank Y, Wang AT, Cai G, Parkhomenko E, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Molecular autism. 2014;4:18. doi: 10.1186/2040-2392-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhar SU, del Gaudio D, German JR, Peters SU, Ou Z, Bader PI, Berg JS, Blazo M, Brown CW, Graham BH, et al. 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. American journal of medical genetics Part A. 2010;152A:573–581. doi: 10.1002/ajmg.a.33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cusmano-Ozog K, Manning MA, Hoyme HE. 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. American journal of medical genetics Part C, Seminars in medical genetics. 2007;145C:393–398. doi: 10.1002/ajmg.c.30155. [DOI] [PubMed] [Google Scholar]
- 6.Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a "common" but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Molecular autism. 2013;4:17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, Gundelfinger ED. C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. Journal of neurochemistry. 2005;92:519–524. doi: 10.1111/j.1471-4159.2004.02910.x. [DOI] [PubMed] [Google Scholar]
- 8.Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, Boeckers TM, Montgomery JM, Garner CC. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, Ma K, Dietz DM, Kajiwara Y, Buxbaum JD, et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell reports. 2015;11:1400–1413. doi: 10.1016/j.celrep.2015.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cochoy DM, Kolevzon A, Kajiwara Y, Schoen M, Pascual-Lucas M, Lurie S, Buxbaum JD, Boeckers TM, Schmeisser MJ. Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Molecular autism. 2015;6:23. doi: 10.1186/s13229-015-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N, Liu S, Jaramillo TC, Bangash M, Xiao B, et al. Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Human molecular genetics. 2011;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. American journal of human genetics. 2001;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Park AR, Spiegelman D, Dobrzeniecka S, Piton A, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. American journal of human genetics. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figura MG, Coppola A, Bottitta M, Calabrese G, Grillo L, Luciano D, Del Gaudio L, Torniero C, Striano S, Elia M. Seizures and EEG pattern in the 22q13.3 deletion syndrome: clinical report of six Italian cases. Seizure. 2014;23:774–779. doi: 10.1016/j.seizure.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Lund C, Brodtkorb E, Rosby O, Rodningen OK, Selmer KK. Copy number variants in adult patients with Lennox-Gastaut syndrome features. Epilepsy research. 2013;105:110–117. doi: 10.1016/j.eplepsyres.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, Sudhof TC. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669. doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nava C, Dalle C, Rastetter A, Striano P, de Kovel CG, Nabbout R, Cances C, Ville D, Brilstra EH, Gobbi G, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nature genetics. 2014;46:640–645. doi: 10.1038/ng.2952. [DOI] [PubMed] [Google Scholar]
- 21.Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, Shaw SR, Claytor J, Willis J, Kelly DP. 22q13 deletion syndrome. American journal of medical genetics. 2001;101:91–99. doi: 10.1002/1096-8628(20010615)101:2<91::aid-ajmg1340>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 22.Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. Journal of medical genetics. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luciani JJ, de Mas P, Depetris D, Mignon-Ravix C, Bottani A, Prieur M, Jonveaux P, Philippe A, Bourrouillou G, de Martinville B, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. Journal of medical genetics. 2003;40:690–696. doi: 10.1136/jmg.40.9.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, Enns GM, Hoyme HE. Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics. 2004;114:451–457. doi: 10.1542/peds.114.2.451. [DOI] [PubMed] [Google Scholar]
- 25.Lindquist SG, Kirchhoff M, Lundsteen C, Pedersen W, Erichsen G, Kristensen K, Lillquist K, Smedegaard HH, Skov L, Tommerup N, et al. Further delineation of the 22q13 deletion syndrome. Clinical dysmorphology. 2005;14:55–60. [PubMed] [Google Scholar]
- 26.Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, Powell J. Molecular and phenotypic characterization of ring chromosome 22. American journal of medical genetics Part A. 2005;137:139–147. doi: 10.1002/ajmg.a.30780. [DOI] [PubMed] [Google Scholar]
- 27.Denayer A, Van Esch H, de Ravel T, Frijns JP, Van Buggenhout G, Vogels A, Devriendt K, Geutjens J, Thiry P, Swillen A. Neuropsychopathology in 7 Patients with the 22q13 Deletion Syndrome: Presence of Bipolar Disorder and Progressive Loss of Skills. Molecular syndromology. 2012;3:14–20. doi: 10.1159/000339119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nesslinger NJ, Gorski JL, Kurczynski TW, Shapira SK, Siegel-Bartelt J, Dumanski JP, Cullen RF, Jr, French BN, McDermid HE. Clinical, cytogenetic, and molecular characterization of seven patients with deletions of chromosome 22q13.3. American journal of human genetics. 1994;54:464–472. [PMC free article] [PubMed] [Google Scholar]