

Abstract
Mass spectrometry (MS) is a powerful tool to analyze complex mixtures of proteins in a high-throughput fashion. Proteome analysis has already become a routine task in biomedical research with the emergence of proteomics core facilities in most research institutions. Pst-translational modifications (PTMs) represent a mechanism by which complex biological processes are orchestrated dynamically at the systems level. MS is rapidly becoming popular to discover new modifications and novel sites of known PTMs, revolutionizing the current understanding of diverse signaling pathways and biological processes. However, MS-based analysis of PTMs has its own caveats and pitfalls that can lead to erroneous conclusions. Here, we review the most common errors in MS-based PTM analyses with the goal of adopting strategies that maximize correct interpretation in the context of biological questions that are being addressed. Finally, we provide suggestions that should help mass spectrometrists, bioinformaticians and biologists to perform and interpret MS-based PTM analyses more accurately.
Keywords: Immonium, Localization, PTM, Signature, SUMO, Technology, Ubiquitin
MS has rapidly become a method of choice for analysis of complex mixtures of proteins in a high-throughput fashion [1, 2]. PTM of proteins is a common mechanism by which the function of proteins can be precisely and dynamically regulated. There are more than 200 types of PTMs and 300 types biological and chemical modifications, as documented in the PSI-MOD [3], RESID [4], UniProt [5] and UniMod [6] databases (for representative reviews, see [7–9] for phosphorylation and [10] for O-GlcNAcylation). MS-based PTM analysis is of great interest to most biologists owing to the unique opportunity to obtain qualitative and quantitative information on a global scale [11,12]. However, PTM analysis using MS has many caveats and pitfalls that can lead to incorrect data interpretation and erroneous conclusions [13,14]. Here, we summarize a variety of common errors that can occur during MS-based analysis of PTMs.
1 Wrong PTMs assigned to a peptide
Although isobaric PTMs are defined as those PTMs that have identical molecular weights (i.e. indistinguishable by MS alone), there are a number of situations where particular PTMs are not strictly isobaric, but can nevertheless be misidentified because of mass measurement errors of the particular types of mass spectrometers. Figure 1 shows how the mass accuracy of mass spectrometers can affect the ability to distinguish PTMs with masses that are contained within specified tolerances. The impact of mass accuracy on discernibility of PTMs is dramatic when low resolution mass spectrometers (e.g. ion trap) are employed rather than high resolution mass spectrometers (e.g. Q-TOF and Orbitrap). For instance, two modifications that occur on lysine residues—tri-methylation (C3H6, 42.04695 Da) and acetylation (C2H2O, 42.01057 Da)—are very close in their mass. Further, both types of modifications frequently co-occur in vivo on the same class of molecules (e.g. histones) [15–17]. The mass difference between these two PTMs is 0.03638 Da (i.e. ~36 ppm difference for a peptide with 1000 Da). Certainly, low-resolution mass spectrometers such as ion-traps did not have capability to differentiate these. Although this type of error might not be an issue with the latest generation high-resolution mass spectrometers such as Orbitraps [18] and high-definition TOFs [15] for bottom-up proteomics, this situation indeed persists in top-down proteomics because 0.03638 Da mass difference is still ~ 1.8 ppm for a 20 000 Da intact protein (which is about the size of histones). Another example of this type is distinguishing between phosphorylation (HPO3, 79.96633 Da) and O-sulfonation (SO3, 79.95682 Da). These two PTMs also share the exact same sites of modifications (i.e. serine, threonine and tyrosine) [19] and it has been reported that sulfation could arise artifactually from the silver-staining process [20]. The mass difference between these two PTMs (sulfation and phosphorylation) is 0.00951 Da which corresponds to ~9.5 ppm for a peptide with 1000 Da. Sometimes, chemical modifications that are considered artifacts can also be observed. For example, glycinylation (C2H3NO, 57.021464 Da) can be easily confused with carbamidomethylation (C2H3NO, 57.02146 Da) and similarly the ubiquitylation remnant, di-Gly (C4H6N2O2, 114.04292 Da) modification is isobaric with di-carbamidomethylation (C4H6N2O2,114.04292 Da) [21].
Figure 1.
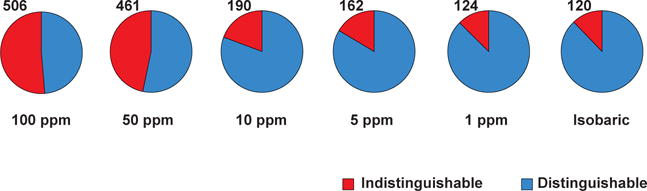
Ability of mass spectrometers to distinguish PTMs. At different levels of mass accuracy of the mass spectrometer used, the number of modifications that are uniquely identifiable varies. For example, an ion-trap mass spectrometer which has ~100 ppm of mass accuracy would not be able to uniquely distinguish almost half the modifications that are annotated in the UniMod database while a mass spectrometer with a 1 ppm mass accuracy can distinguish most of them.
Further, amino acid substitutions can also mimic PTMs if they are isobaric with the mass of other modified amino acids. For example, methylation can occur on aspartic acid and glutamic acid residues [22] and these amino acids with the added mass of methyl groups (i.e. 14.01565 Da) are isobaric with amino acid variants (e.g. V to I/L or D to E). For example, a fully tryptic peptide (LVNELTEFAK) of bovine serum albumin, which is used as a standard protein in many proteomics laboratories could be misidentified as a human peptide (LVNEVTEFAK) with methylation at E4 although y6 ion does exist and distinguish them. As another example, conversion from aspartic acid to isoaspartic acid has been described, which is an isobaric change so that it is almost impossible to differentiate them by mass unless electron-based methods such as ECD and ETD are used for fragmentation, which generates unique diagnostic ions (i.e. c + 57 or z*−57) [23,24].
In nature, there are many more examples of such isobaric PTMs in the world of glycoproteins since isomeric monosaccharides can be linked like scrambled peptide sequences. There are more than 250 glycosyltransferases in the human genome, which can potentially lead to addition of glycans to more than 50% of the human proteome [25]. Molecularly, eight different amino acids are known to be involved in glycoprotein linkages with more than 13 different monosaccharides using more than 41 different chemical bonds [26]. Isobaric glycan structures can be presented by diverse structural isomers through a combination of multiple linkage positions, anomeric centers (i.e. α or β) and ring structures (i.e. furanose or pyranose). It is not surprising that the number of glycan substructures have been estimated to be more than 10 000 [27]. Thus, newer strategies need to be developed in the future to characterize the precise nature of carbohydrate modifications. We expect that the protein lipid modifications might be as diverse as glycosylation. For example, protein lipid modifications such as N-acylation and S-acylation, often allow the proteins to anchor to cell membranes. This plays a key role in diverse signaling pathways that involve, for example, N-terminal myristoylation of glycine residue of tyrosine kinases Yes, Fyn and Lck, palmitoylation of N-terminal cysteine of the hedgehog protein family or lysine acylation by the sirtuin family [28, 29].
2 PTMs assigned to a wrong protein/gene
Many tryptic peptides are shared across distinct genes or between isoforms of the same gene because of sequence identity. A major limitation of MS is that it cannot reliably help trace back the origin of these tryptic peptides to determine which gene(s) code for the proteins that are detected in the sample. In addition, the isobaric nature of two amino acids (i.e. leucine and isoleucine) makes protein inference more complicated [30]. Although there have been efforts [31, 32] to computationally solve the problems of protein inference, this issue is inherent to MS-based proteomics and there will always be a certain level of concern regarding ‘which protein products from which genes were truly identified.’ Thus, there is still an urgent need for newer/orthogonal methods to complement the long-pursued protein inference issue that is most acute in bottom-up proteomics experiments. In the same fashion, PTM site(s) identified on peptides cannot be localized onto a protein when the peptide sequence itself is not unique to one protein. As an illustration, Fyn, Hck, Lck, Src and Yes1 all belong to the Src nonreceptor tyrosine kinase family whose phosphorylation plays an important role in various signaling pathways. However, because they have a similar domain structure and highly related amino acid sequences, it may be difficult to decide which protein is phosphorylated as illustrated in Fig. 2. Tyrosine kinase domains of the Src nonreceptor tyrosine kinases are highly homologous, thereby making some tryptic peptides identical/isobaric between proteins (Fig. 2A). For example, two isobaric phosphopeptides— IIEDNEpYTAR and LIEDNEpYTAR—identified from a phosphoproteomic study, where IIEDNEpYTAR is observed in Hck kinase while LIEDNEpYTAR is shared by Fyn, Lck, Src and Yes. Thus, identifying this phosphopeptide cannot resolve the puzzle of the corresponding phosphorylated kinase(s) even with good spectral assignment (Fig. 2B). In the studies presented by Zhong et al. [33], the authors showed an increased abundance of this peptide in TSLP-stimulated cells, from which one could conclude that one or more of these nonreceptor tyrosine kinases might be hyperphosphorylated. Thus, the exact involvement of these kinases in TSLP signaling still remains undecided based on these peptide identifications alone. In reality, this issue is intrinsic to the convention of using trypsin for bottom-up proteomics since the fully tryptic peptide sequence (I/LIEDNEYTAR) is shared by proteins encoded by five genes (Fyn, Src, Yes, Hck and Lck). To overcome this issue, one can use another enzyme such as Lys-C to generate a longer peptide sequence from the highly homologous tyrosine kinase region that may help pinpoint a single gene product or to narrow down to fewer alternatives. In the scenario presented, Lys-C would produce four different peptide sequences from the proteins as compared to one shared isobaric peptide by trypsin: one peptide (VADFGLARLIEDNEYTARQGAK) unique to Src, another peptide (IADFGLARLIEDNEYTARQGAK) shared between Fyn and Yes and two unique but isobaric peptide sequences (IADFGLARI/LIEDNEYTAREGAK) shared between Hck and Lck. Of course, the use of additional proteolytic enzymes, can resolve many more ambiguities pertaining to shared peptides.
Figure 2.
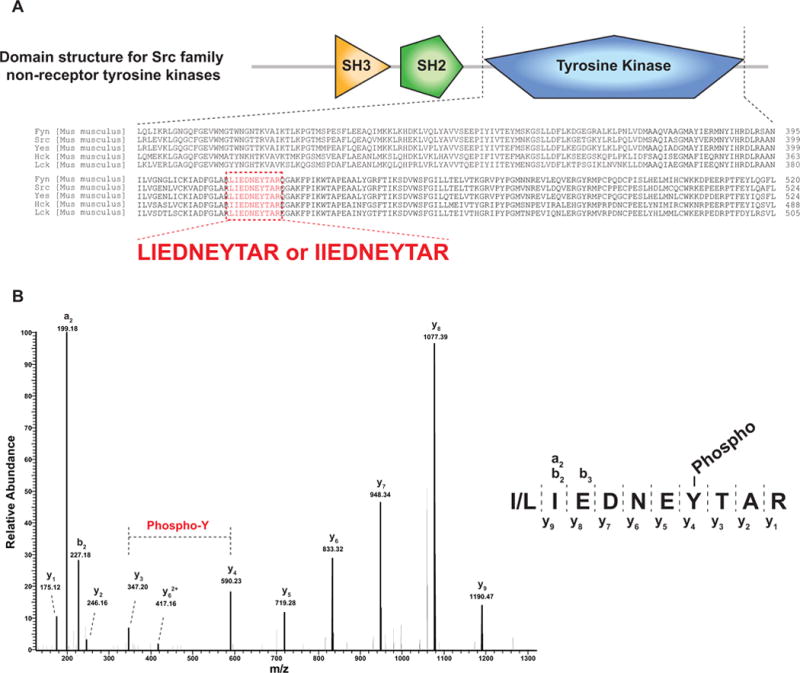
Homologous domain derived peptides are shared across many proteins. (A) Domain structure for Src family nonreceptor tyrosine kinases is shown at the top. The bottom part shows an alignment of tyrosine kinase domains of five murine members of the Src family of tyrosine kinases. The peptide sequences highlighted in red (LIEDNEYTAR or IIEDNEYTAR) are isobaric and shared by the indicated kinases. (B) A peptide sequence (I/LIEDNEYTAR) was observed to be highly phosphorylated at its tyrosine residue. However, this sequence can be derived from any of the five tyrosine kinases shown in Panel A, making it impossible to deduce the identity of the particular tyrosine kinase that is phosphorylated in the sample analyzed. Two fragment ions (y3 and y4) clearly indicate tyrosine phosphorylation.
3 PTM assigned to a wrong residue on a correctly identified peptide
In most proteomics analyses, a bottom-up approach is generally employed using trypsin. PTMs can be identified along with the corresponding peptide sequences when using database search algorithms. However, knowing that any PTMs exist on a peptide is only the first step and, an equally important second step, is to localize the modification site to one or more of often several plausible amino acids. The total number of residues in the human proteome annotated in the RefSeq protein database [34], for example, is ~20 million with the approximate size of an average tryptic peptide being ~10 amino acids long. The number of serine, threonine and tyrosine residues—the major sites of phosphorylation in eukaryotes—is ~1.5 million, 1 million and 0.5 million, respectively, resulting in an estimated 1.5 potential sites of phosphorylation per peptide that is ten amino acids long. Thus, nearly half of peptides would have an issue with site localization, implying the need for computational and statistical assessment of PTM site localization. A number of algorithms such as MD-Score [35], PhosphoRS [36], PTM score [37] and A-Score [38] have been developed to statistically assess the site of localization of phosphorylation. Although these algorithms are complementary to each other for localizing sites of phosphorylation [39], they might have been optimized for one of most abundant modifications such as phosphorylation. To improve performance of these algorithms to accurately localize sites of modifications other than phosphorylation, there is an urgent need for synthetic peptide-based standard MS/MS datasets of diverse post-translationally modified peptides (not just a few types of modifications such as phosphorylation). Here, we will discuss most frequent causes of ambiguity of PTM site assignment:
3.1 A poor fragmentation pattern observed in the MS/MS spectrum
Although many tandem mass spectra contain informative fragment ions, a number of them exhibit poor fragmentation of peptide ions leading to ambiguity in assigning the site of PTMs. In order to overcome this issue, one often employs a statistical test to see which site of all possible sites in a given peptide is modified by the PTM. Because the most heavily investigated PTM by MS is phosphorylation, many of the localization algorithms have been optimized on the basis of nature of phosphorylation. One should be aware that a PTM can be wrongly assigned to a site on a peptide sequence even when such programs are employed. When the MS/MS spectrum is too poor to localize the modification site, algorithms usually provides an even probability among all plausible sites. Thus, one could employ a probability cutoff of 75% to generally minimize false positive sites of modification. However, a simple choice of 75% as a cutoff is still arbitrary and a method should be systematically evaluated since there may be other scenarios that make the probability of a true modification site being scored at <75% (see examples below).
3.2 Failure of localizing the modification site by algorithms
Figure 3 shows an example of an MS/MS spectrum with rich fragment ions of a peptide (NIPIALCTSSNKTK) containing a di-Gly remnant on one of two lysine residues (the experimental protocol enriched for di-Gly motif containing peptides using antibody-based immunoaffinity purification). The PTM score algorithm built into the MaxQuant platform provides a higher probability score for di-Gly modification site to the C-terminal lysine by estimating probability score for the internal lysine as 0.226 and the terminal lysine as 0.774 (i.e. NIPIALCTSSNK(0.226)TK(0.774)). Currently, many groups use a threshold of 0.75 to confidently report site localizations of PTMs. However, careful manual assessment indicated the internal lysine as a modification site owing to the existence of y1-ion and y2-ion with no di-Gly and the absence of y1-ion and y2-ion containing di-Gly. Thus, in this case, it appears that the PTM score algorithm did not weigh these low abundance ions adequately thereby resulting in a slightly higher score for the terminal amino acid.
Figure 3.
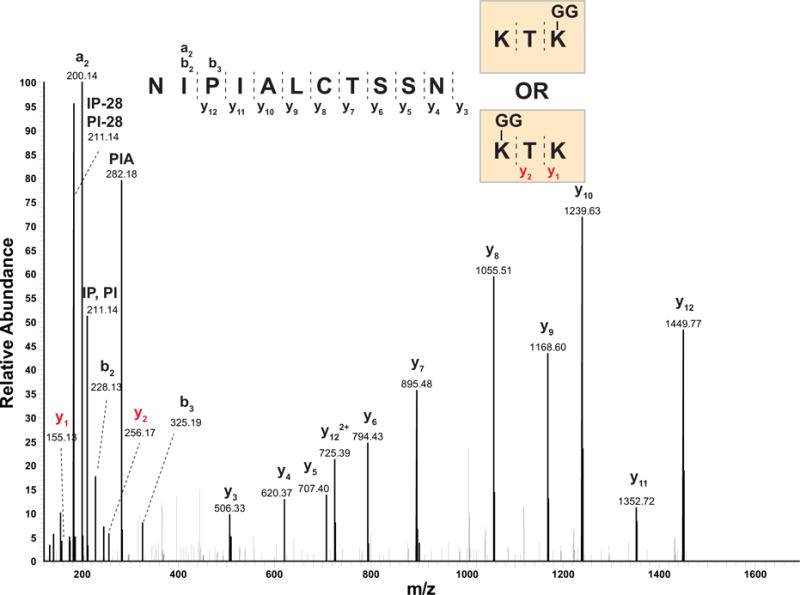
Incorrect assignment of a di-Glycine modification. A peptide sequence (NIPIALCTSSNKTK) from a hypothetical protein (YKL033W-A) of Saccharomyces cerevisiae was identified with the di-Glycine modification which serves as a signature for ubiquitylation. In the sequence, there are two lysine residues: one terminal and one internal. MaxQuant indicated a higher probability of terminal lysine being the site of ubiquitylation, but a careful manual validation revealed two signature fragment ions (y1 and y2, marked in red) indicating that the actual ubiquitylation site is likely to be the internal lysine residue instead. Note that lysine was labeled with heavy lysine (13C615N2)for a quantitative proteomic analysis.
3.3 Co-elution of peptides containing the same PTM on different residues
Another problem is co-elution of two (or more) peptides with the same PTM on different sites (since the peptide sequence is the same, the modified peptides are obviously isobaric). Here, we show one example where a mixture of two tyrosine-phosphorylated peptides that eluted closely during chromatography (Fig. 4). This peptide sequence (YYEGYYAAGPGYGGR) with one phosphorylation contains five tyrosine residues that could possibly be modified by one phosphate moiety. The site localization software (PhosphoRS) assigned a probability of 50% to Y5, 50% to Y6 and 0% to the other three sites. Since there are no sites with a higher probability (e.g. when using an arbitrary threshold of ≥75%), one would remove both of these sites (Y5 and Y6) from the final list of confidently identified sites. However, in reality, there is no doubt that two different phosphopeptides with the identical peptide sequence exist, co-eluted and were co-fragmented as evidenced by two different y10 ions–one with and the other without a phosphate (Fig. 4). In this regard, Thibault and colleagues have recently shown that ~3–6% of all identified phosphorylated peptides are found to be in this category of ‘peptides with the same PTM on different sites’ and that about half of them elute closely from the column (i.e. within 2 min) [40]. However, it is still difficult to determine all of the phosphopeptides that exist in a sample due to potential of systematic biases such as digestion efficiency of differential structural hindrance as well as because of stoichiometry of those isobaric phosphopeptides.
Figure 4.
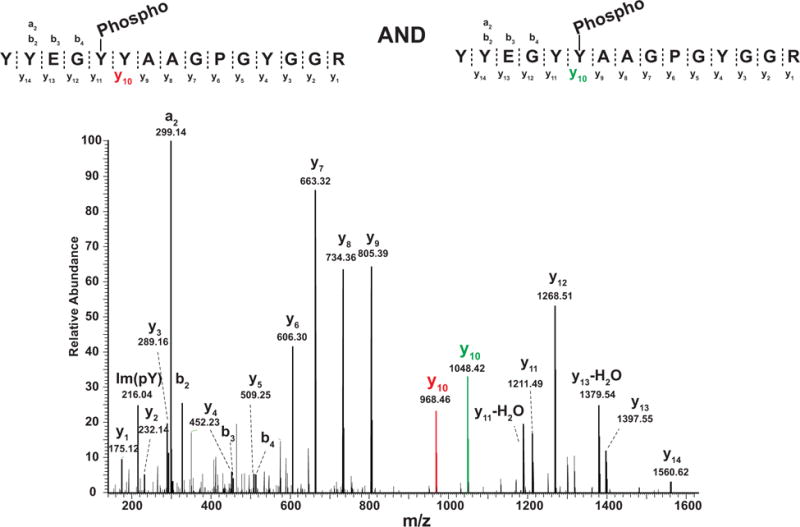
Co-fragmentation of peptides bearing a single phosphate but on two different tyrosine residues. Two different tyrosine phosphorylated peptides (YYEGYYAAGPGYGGR and YYEGYYAAGPGYGGR, the site of phosphorylation is underlined) were sampled in one MS/MS scan due to co-fragmentation. In this case, the probability of phosphorylation localization on either site is less than 0.75 which is used as a general a cutoff. However, in this case, both of these modified sites will be missed.
3.4 Multiple distinct ‘isobaric’ PTMs on a peptide
As discussed above, acetylation and tri-methylation often occur on lysine residues of histone tails leading to potential errors in identifying the exact type of PTM. Recently, it has been reported that combinatorial PTMs on histones can be observed and that a considerable fraction of peptides were found to have both modifications on a given peptide (i.e. acetylation and tri-methylation) [41]. To correctly localize the two modifications on different lysine residues on a peptide, one should acquire fragmentation data on a high resolution MS so that fragment ion masses with ~36 ppm difference can be confidently differentiated regardless of the choice of fragmentation method (e.g. CID, HCD or ETD).
4 Post-translationally modified peptides missed because of database search strategy
The majority of PTM sites are identified from bottom-up proteomic approaches by searching tandem mass spectra against theoretical protein databases using database search algorithms. The components of such computational pipelines dramatically affect the identification of PTM sites in the analysis. Currently, the identification of post-translationally modified peptides by MS mostly relies on algorithm-based analysis of real mass spectra against theoretical mass spectra generated from selected protein databases. Two computational components play critical roles in such PTM identifications–the algorithm itself and the database being searched. Here, we discuss scenarios in which peptides with PTMs were incorrectly missed from identification: (i) Database did not have the peptide sequence: i.e. protein of interest was missing, or, the protein was present but corresponded to a different variant (e.g. amino acid change owing to a mutation or SNP), (ii) Search algorithm missed the identification; and, (iii) User did not specify the appropriate modification while searching.
4.1 Database
Protein databases can be incomplete and/or inaccurate since none of the more complex genome sequences, including the human genome, is truly complete [42]. Some of the proteins are not annotated, some of annotated protein sequences are incorrect and even those that are correct reflect sequences from a given individual without accounting for SNPs. This situation will continue to improve until genome sequences and their protein coding regions are completely annotated. It has been shown that peptides containing altered amino acids by SNPs or mutations can be mined from large MS datasets [43]. If the protein sequences are not available, an option is to use publicly available transcript sequence information from related species from which a protein database can be generated [44, 45]. One could also use a personalized transcriptome data to create a personalized protein database against which PTMs may be searched–with the availability of next-generation sequencing data, such use will continue to become even more popular.
4.2 Algorithms and search parameters
There are many search algorithms available for peptide identification. Here, we do not review all the available database search algorithms but provide examples regarding some of their limitations that investigators should be aware of. Some algorithms such as MASCOT [46], X!Tandem [47] and Sequest [48] currently only allow ~10 modifications to be searched which might lead one to miss identifying certain PTMs unless an iterative search is carried out. It should be pointed out though that an increase in the number of search parameters also affects scores of peptide-spectrum matches, which can itself complicate analysis. In general, a small set of modifications are chosen in a routine proteomics analysis such as oxidation at methionine and/or protein N-termini acetylation. However, even these routine (and common) modifications (e.g. protein N-termini acetylation) cannot always be specified during a search with all search algorithms. One limitation for some algorithms such as Sequest is that they have no capability to directly search protein N-terminal acetylation although peptide N-terminal acetylation can be used at the expense of an increase in false positives. Thus, N-terminal acetylation of proteins which is quite common can easily be missed from the identification. To circumvent such limitations that are inherent in the search algorithms, one can create custom databases such that all theoretical N-terminal tryptic peptides with the desired missed cleavages are included in the search space.
4.3 PTMs not specified during the search
Most often, many of the PTMs are not specified at all during the search. For example, lipid modification and glycosylation are usually not included during most routine data analysis. Unfortunately, this means that current analysis pipelines routinely miss many interesting PTMs hidden in the data. For example, a number of novel modifications on lysine residues such as propionylation, butyrylation, succinylation, malonylation and glutarylation have recently been reported [49–52] while other modifications such as hydroxylation and methylation can also be observed frequently in samples [53]. Search for all PTMs is still an area of research and the most appropriate strategies that balance sensitivity and false discovery rates have not yet been fully explored although they are being tried.
4.4 Confidence in identifying PTMs
One of the difficult aspects of data analysis for PTM identification and localization pertains to estimating the confidence in the identified PTMs. One can use all possible PTMs as variable modifications in the same search or employ iterative searches using multiple sets of PTMs [54,55]. It has also been reported that ‘open search’ might help identify many more modified peptides [56,57]. Although there is no systematic evaluation of which strategy may work best for which PTMs, we believe that such systematic studies should be carried out in the near future using appropriate standard datasets.
5 Site of a PTM incorrectly labeled as novel
Bioinformatics analysis is often carried out to gain novel biological insights through proteomic analysis of PTMs. The first of such analyses is often to find novel PTM sites identified through proteomic analysis. Mass spectrometric analysis of PTMs has already led to accumulation of data on hundreds of thousands of PTM sites. There are many databases and repositories as resources for the scientific community that provide information on genes, proteins, peptides, PTMs and raw MS data [34,58–63]. However, none of these resources can be easily synchronized or assumed to be fully up to date thereby making the simple question of ‘whether identification of site of modifications is novel,’ an almost impossible one to answer. Thus, reporting PTMs annotated as ‘novel’ can often turn out to be erroneous. For example, Kim et al. showed that the phosphorylation level at C-terminal Y987 of receptor tyrosine kinase EphB4 was found to be heterogeneous in multiple metastases of a single patient with pancreatic cancer [64]. This phosphorylation site can be reported as ‘novel’ based on HPRD [58], but has already been annotated in PhopshoSitePlus, which contains both published datasets as well as data generated internally by Cell Signaling Technology [61]. An easier solution is that any claims of novelty of PTM sites identified could be avoided in publications as is already the policy of the Proceedings of the National Academy of Sciences, USA regarding any statements of novelty and priority.
6 Incorrect conclusions drawn because of lack of awareness of biased nature of experiments
Motif analysis is a routine bioinformatics exercise when a large number of PTM sites has been identified from a proteomics study. The analysis often requires two datasets–the foreground dataset and the background dataset. The foreground dataset often comes from PTM sites identified in the experiment and the background dataset is composed of theoretical PTM sites. However, a number of factors affect the identification of PTM sites, thus skewing the motif analysis. These factors include, but not limited to, enzyme, biased enrichment method, selective desalting on C18 tip/cartridge, uneven gaseous ion transfer efficiency, multiple and/or large PTMs, data-dependent analysis for higher abundance, fragmentation method, unequal dissociation preference among peptide bonds, incorrect database and dependence on search algorithms. Here, we will describe how some of these factors can lead to erroneous conclusions.
6.1 Sample processing and protein extraction
Cytoplasmic proteins are extracted more easily than membrane proteins in spite of methods developed to minimize this issue [65, 66]. This is because stretches of hydrophobic amino acids of membrane proteins are not optimal for current LC-MS/MS methods in that those highly hydrophobic peptides tend to strongly bind to C18 materials throughout the sample processing from basic RPLC fractionation column, stage tip, trap column and analytical column. Some of PTMs are known to be altered during sample processing itself. For example, several studies have reported that some phosphoproteins are altered by ischemia prior to protein extraction [67–69]. We anticipate that many other PTMs may be altered during the protein extraction step although they have not yet been systematically studied. Additionally, one might have to systematically assess the effect of variables such as lysis buffers on any new PTM analysis.
6.2 Enzyme
Trypsin is the first choice as an enzyme for digesting proteins into peptides in MS -based proteomics studies. Limitations of mass spectrometers often make it difficult to identify extremely short or long peptides. To use the human proteome as an example, only 60% of the protein sequence in the whole proteome is amenable to the identification, based on the assumption that peptides with length 6–25 amino acids are identified easily. Thus, it is expected that ~60% of phosphopeptides in the human proteome might be examined by current mass spectrometric analysis. There are efforts to obtain higher coverage using multiple protease for proteomic analysis as well as PTM analysis [70–74] although they are not attempted routinely.
6.3 Affinity capture
Peptides with PTMs are generally of low abundance whose detection is adversely affected by the vast excess of nonmodified peptides. Thus, current PTM analyses often employ affinity capture by enriching PTM-containing peptides, by depleting nonmodified peptides or by separating modified peptides by chromatography. In the case of antibody-based methods, most, if not all, antibodies raised against PTMs have some preference to certain sequences. This vaguely known selectivity leads to identification of capture reagent-preferred peptides, which is associated with the abundance level of the modified peptide sequences. Thus, repeated affinity-based proteomics experiments by the same laboratory or other laboratories would preferentially identify the known PTM-modified sites. Thus, it is good to continue to develop newer methods which complement current methods to minimize this type of bias.
6.4 C18-based clean-up
Mass spectrometric analysis in most proteomic setups employs desalting steps to deplete salts which interfere with stable ESI. The desalting methods used 18 carbon chains (i.e. C18) bound material such as STAGE-tip and Zip-tip. This basic principle of this method is based on hydrophobicity of peptides generated by trypsin protease such that most of peptides bind to the tips while hydrophilic molecules including small metabolites, small size peptides (i.e. monomer, dimer, etc.) and fragmented DNA/RNA molecules flow through the tip. However, highly hydrophobic or hydrophilic peptides would not be available for MS analysis after this step. In other words, only preferred sized peptides with preferred hydrophobicity profiles are routinely injected into the mass spectrometer, which is again connected online with a trap and/or an analytical column packed with C18 material. This creates a systematic bias and creates false negatives in the acquired datasets even when they are very large.
6.5 Choice of sequencing methods
Most investigators use data-dependent acquisition methods for profiling peptide mixtures and PTMs. This method is designed to perform a cycle of MS along with several MS/MS scans of the most abundant ions. Thus, low abundance ions are missed from sequencing for identification. Thus, the speed of acquisition of MS/MS with no loss of signal intensity is very important for successful proteomics analysis [75]. Alternatively, data-independent analysis can be carried out in a scheduled manner to acquire all possible fragment ions including those from low abundance ions [76–79]. This method should conceptually have the advantage to comprehensively recover most of peptide ions for sequencing.
6.6 Fragmentation method
MS-based proteomics analysis employs collision-based fragmentation methods such as CID and HCD. These two methods are also complementary to electron-based fragmentation methods (i.e. ETD and ECD). Those portions of peptides that contain higher charge states (z ≥ 3) will be better sequenced by ETD while other portions of peptides with z = 2 have a higher chance to be identified with CID and HCD. It seems that peptides with PTMs also follow this tendency, at least for phosphoproteomics analysis [80–83]. In the future, hybrid strategies of fragmentation might become more popular when a more detailed characterization of PTMs is being pursued.
6.7 Database search engine
Use of multiple database search algorithms can help increase peptide identifications as well as PTM analysis as no single search engine is perfect.
7 Incorrect PTM quantitation
A number of labeling strategies have been developed to quantify the change of PTM status on proteins including 18O, ICAT, SILAC, TMT, iTRAQ and stable isotope dimethyl labeling [84–89]. However, co-elution of peptides significantly affects the quantitation of PTM regardless of the quantitation methods employed. Figure 4 depicts an example of co-elution of isobaric phosphopeptides where one cannot quantify each phosphopeptide. Under the LC conditions of the experiment shown in Fig. 5, two isobaric peptides eluted very closely from the analytical column affecting the quantitation values. In the case of isobaric tag-based quantitative methods such as iTRAQ and TMT, this type of issue is more problematic since many interfering ions within an m/z window (e.g. 1.9 Da) covering the target ion m/z value are all selected for isolation and fragmentation. Thus, other peptide ions co-fragmented along with the target peptide ion, although non isobaric, can produce unwanted reporter ions leading to inaccurate quantitative information.
Figure 5.
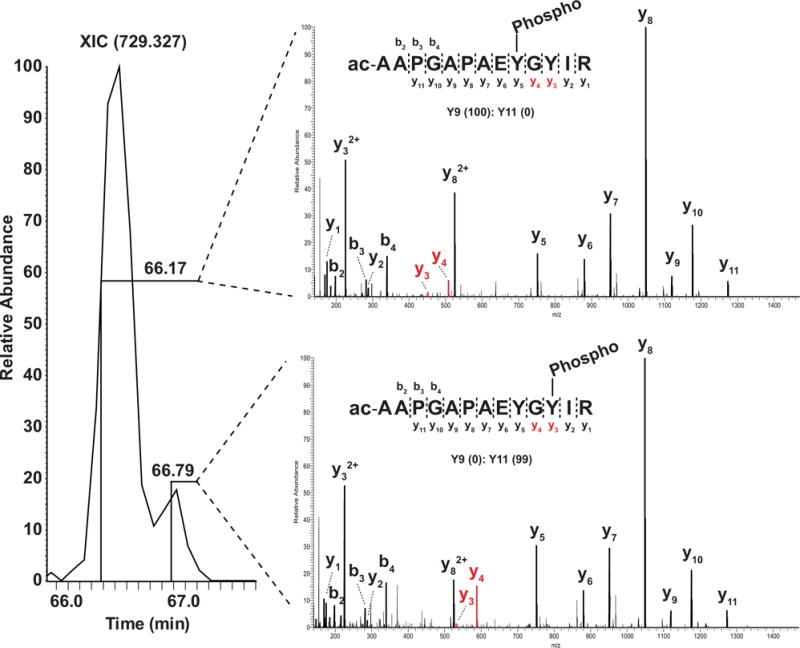
Inaccurate quantitation of co-eluting isobaric peptides. Two signature fragment ions (y3 and y4) clearly distinguish the two isobaric phosphopeptides bearing the phosphate on two different tyrosine residues. However, the isobaric phosphopeptides elute together on liquid chromatography, likely leading to inaccurate quantitation of both phosphopeptides.
8 Wrong interpretation of an increased PTM
Current measurements of PTM status in any proteome are realized through quantitation of PTM-containing peptides. However, a chance in abundance of the modified peptide does not necessarily imply a change in the extent of the PTM site. For instance, changes in abundance of protein phosphorylation could result either from changes in abundance of phosphorylation at the site or from changes in the abundance of the protein itself. Thus, it is important that protein abundance is assessed for better interpretation of phosphorylation [90]. Other potential issues to be considered for phosphoproteomics analysis on data interpretation have been reviewed elsewhere [91,92]. As shown in Fig. 6, a SILAC ratio indicating a decrease in the relative abundance of phosphopeptide can result from four different scenarios at the protein level. Although SILAC was used here to discuss the specific problems regarding interpretation, other types of quantitative methods including iTRAQ and TMT have similar issues.
Figure 6.
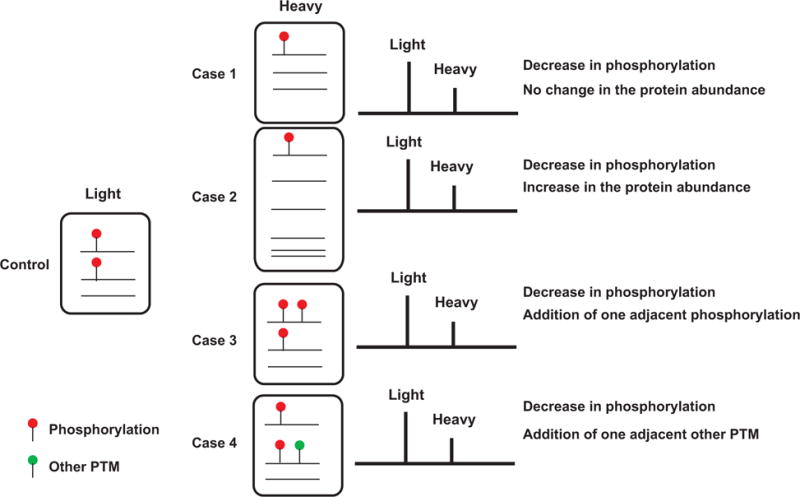
Multiple explanations of a quantitative value measured by MS. Shown in Control are two copies of modified peptide sequence and one copy of unmodified counter peptide sequence in control. After a perturbation, modified peptide was reduced to a half of Control (i.e. only one copy in Experiment). There are four possible scenarios that could explain the observation by MS: number of modified peptides was reduced by a half while total number of peptides remains unchanged (case 1) or altered (case 2). A modified peptide in control has obtained an additional modification(s) of the same (case 3) or a different kind (case 4). All four cases resulted in the identical result in MS.
9 Discussion
Here, we have discussed many aspects of current proteomics pipeline for PTM analysis in biology where we should be cautious. Before we rush to biological conclusions from mass spectrometric data, we should keep the following parameters in mind: (i) the resolution of mass spectrometer being employed; (ii) isobaric PTMs, chemical modifications and amino acids; (iii) enzymes used; (iv) search algorithm; (v) protein databases; (vi) search parameters for PTMs; (vii) site localization algorithm; (viii) co-elution of modified peptides; and (ix) interpretation of quantitation data. Some of the problems that we have listed can be solved, for example, by using different proteases, multiple algorithms, more curated protein databases or optimized searching parameters. Other problems can be solved through improvements in current bioinformatics pipelines such as integration of different PTM databases for better and more complete annotation. Some problems such as isobaric PTMs may not be solved simply by proteomic methods at all. These problems may be solved by using biochemical methods other than MS or through a combination of methods. The following list of suggestions can be used for guidance while performing MS-based PTM analyses. Suggestion #1: Know the mass errors of masses measured by the MS employed. Suggestion #2: Know alternative explanations of delta masses of PTMs of interest. Table 1 shows a list of isobaric masses. Suggestion #3: Use the presence of signature ions in MS/MS spectra to increase the confidence level of the PTMs identified. Table 2 shows a list of signature ions frequently observed in MS data. Suggestion #4: Employ a statistical analysis to assess the probability of localization at a given site for identifying the amino acid modified by PTMs more confidently. Suggestion #5: Employ enzymes other than trypsin to identify the protein of interest with PTMs. Suggestion #6: Know alternative ways to interpret a quantitative result of PTMs. Suggestion #7: Deposit the acquired MS data to public repositories such as ProteomeXchange [93] and MassIVE. We believe that public dissemination of proteomics data will ultimately enable assessment, cross-validation, integration, comparative analysis and novel discoveries.
Table 1.
A list of common modifications with isobaric delta masses
| Number | Modification | Delta mass (Da) | Composition | Alternative explanation |
|---|---|---|---|---|
| 1 | Amidation | −0.984016 | HN O(−1) | D to N, E to Q |
| 2 | Deamidation | 0.984016 | H(−1) N(−1) O | N to D, Q to E |
| 3 | Methylation | 14.01565 | H(2) C | G to A, D to E, V to I/L, S to T, N to Q |
| 4 | Formylation | 27.994915 | C O | D to S, E to T |
| 5 | di-Methylation | 28.0313 | H(4) C(2) | Ethyl, A to V, C to M |
| 6 | S-nitrosylation | 28.990164 | H(−1)NO | V to Q |
| 7 | Sulfide | 31.972071 | S | V to M, A to C |
| 8 | Di-Oxidation | 31.989829 | O(2) | P to E |
| 9 | Acetylation | 42.010565 | H(2) C(2) O | S to E |
| 10 | Tri-Methylation | 42.04695 | H(6) C(3) | Propyl, A to L/I, G to V |
| 11 | Carbamylation | 43.005814 | HCNO | A to N |
| 12 | Carboxylation | 43.989829 | C O(2) | A to D |
| 13 | Propionylation | 56.026215 | H(4) C(3) O | Acrolein adduct |
| 14 | Carbamidomethylation | 57.021464 | H(3) C(2) N O | A to Q, G to N, addition of glycine |
| 15 | Carboxymethylation | 58.005479 | H(2) C(2) O(2) | A to E, G to D |
| 16 | Methylmalonylation | 100.016044 | H(4) C(4) O(3) | Succinylation |
| 17 | Ubiquitin remnant (di-Gly) | 114.042927 | H(6) C(4) N(2) O(2) | Double carbamidomethylation, addition of asparagine |
Table 2.
A list of frequently observed signature ions of modifications
| Number | Modification | Site of modification | Signature ions (m/z) | References |
|---|---|---|---|---|
| 1 | Phosphorylation | Y | 216.04257 | [96] |
| 2 | Acetylation | K | 143.11844, 126.09189 | [97] |
| 3 | Formylation | K | 112.07624 | [98] |
| 4 | Methylation | R | 143.1291, 115.0866, 112.0869, 74.0713, 70.0651 | [99] |
| 5 | Methylation | K | 115.12352, 98.09697, 84.08132 | [99] |
| 6 | Di-methylation | R | 157.1448, 115.0866, 112.0869, 88.0869, 71.0604 | [99] |
| 7 | Di-methylation | K | 129.13917,84.08132 | [99] |
| 8 | Tri-methylation | K | 143.15482, 84.08132 | [99] |
| 9 | ADP-ribosylation | E, D, K, R | 543.07677, 428.03725, 348.07091, 250.09402, 136.06232 | [100] |
| 10 | HexNAc | N, S, T | 204.08720, 186.07663, 168.06607, 144.06607, 138.05550, 126.05550 | [101] |
| 11 | Hex | S, T, K | 163.06065, 145.05009, 127.03952, 115.03952, 109.02896 | [101] |
| 12 | Glycan signature ions | N, S, T | 366.14002 (HexHexNAc), 325.11348 (HexHex), 274.09268 (Neu5Ac-H2O), 292.10324 (Neu5Ac) | [102] |
| 13 | LRGG (Ubiquitin remnant) | K | 384.23593, 270.19300 | [98] |
| 14 | QQTGG (SUMO Q>R mutant remnant) | K | 472.21559, 455.18904, 454.20502, 398.16758,380.15701, 358.17266,341.14611, 323.13555, 241.13007, 239.11442 | [98,103] |
Last, one should understand that a standard MS-based PTM analysis does not provide occupancy of the PTM site (i.e. stoichiometry). This is because modified and unmodified peptides behave unequally in the proteomic pipeline, making it difficult to compare them directly based on their relative M S responses. Another complicating factor is that some PTMs such as S-nitrosylation and phospho(ribosyl)ation indicative of mono- or poly-ADP-ribosylation are known to be less stable during preparation of samples [94, 95], which might not permit a true measurement of the modification state of the proteins in vivo. In our experience, a careful assessment of post-translationally modified peptides is often warranted and worthwhile, especially if it is going serve as the basis of additional biological experiments that can be time consuming.
Acknowledgments
This study was supported by a grant from the National Cancer Institute (CA184165 to AP) and NCI’s Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036 to AP).
Abbreviations
- ETD
electron transfer dissociation
- HCD
high energy collision dissociation
- O-GlcNAc
O-linked beta-N-acetylglucosamine
- TSLP
Thymic stromal lymphopoietin
Footnotes
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Cravatt BF, Simon GM, Yates JR., Iii The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 3.Montecchi-Palazzi L, Beavis R, Binz PA, Chalkley RJ, et al. The PSI-MOD community standard for representation of protein modification data. Nat Biotechnol. 2008;26:864–866. doi: 10.1038/nbt0808-864. [DOI] [PubMed] [Google Scholar]
- 4.Garavelli JS. The RESID Database of Protein Modifications as a resource and annotation tool. Proteomics. 2004;4:1527–1533. doi: 10.1002/pmic.200300777. [DOI] [PubMed] [Google Scholar]
- 5.UniProt Consortium, UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creasy DM, Cottrell JS. Unimod: protein modifications for mass spectrometry. PROTEOMICS. 2004;4:1534–1536. doi: 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- 7.Engholm-Keller K, Larsen MR. Technologies and challenges in large-scale phosphoproteomics. PROTEOMICS. 2013;13:910–931. doi: 10.1002/pmic.201200484. [DOI] [PubMed] [Google Scholar]
- 8.Harsha HC, Pandey A. Phosphoproteomics in cancer. Mol Oncol. 2010;4:482–495. doi: 10.1016/j.molonc.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boersema PJ, Mohammed S, Heck AJR. Phospho-peptide fragmentation and analysis by mass spectrometry. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 10.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci. 2010;35:547–555. doi: 10.1016/j.tibs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 12.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 13.Palmisano G, Melo-Braga MN, Engholm-Keller K, Parker BL, Larsen MR. Chemical deamidation: a common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J Proteome Res. 2012;11:1949–1957. doi: 10.1021/pr2011268. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen ML, Vermeulen M, Bonaldi T, Cox J, et al. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- 15.Zhang K, Yau PM, Chandrasekhar B, New R, et al. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: An application for determining lysine 9 acetylation and methylation of histone H3. PROTEOMICS. 2004;4:1–10. doi: 10.1002/pmic.200300503. [DOI] [PubMed] [Google Scholar]
- 16.Garcia BA, Shabanowitz J, Hunt DF. Characterization of histones and their post-translational modifications by mass spectrometry. Curr Opin Chem Biol. 2007;11:66–73. doi: 10.1016/j.cbpa.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 17.Karch KR, DeNizio JE, Black BE, Garcia BA. Identification and interrogation of combinatorial histone modifications. Epigenom Epigenet. 2013;4:264. doi: 10.3389/fgene.2013.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen JV, Godoy LMF, de Li G, Macek B, et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Medzihradszky KF, Darula Z, Perlson E, Fainzilber M, et al. O-sulfonation of serine and threonine mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol Cell Proteomics. 2004;3:429–440. doi: 10.1074/mcp.M300140-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Gharib M, Marcantonio M, Lehmann SG, Courcelles M, et al. Artifactual sulfation of silver-stained proteins implications for the assignment of phosphorylation and sulfation sites. Mol Cell Proteomics. 2009;8:506–518. doi: 10.1074/mcp.M800327-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boja ES, Fales HM. Overalkylation of a protein digest with iodoacetamide. Anal Chem. 2001;73:3576–3582. doi: 10.1021/ac0103423. [DOI] [PubMed] [Google Scholar]
- 22.Sprung R, Chen Y, Zhang K, Cheng D, et al. Identification and validation of eukaryotic aspartate and glutamate methylation in proteins. J Proteome Res. 2008;7:1001–1006. doi: 10.1021/pr0705338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cournoyer JJ, Lin C, O’Connor PB. Detecting deamidation products in proteins by electron capture dissociation. Anal Chem. 2006;78:1264–1271. doi: 10.1021/ac051691q. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor PB, Cournoyer JJ, Pitteri SJ, Chrisman PA, McLuckey SA. Differentiation of aspartic and isoaspartic acids using electron transfer dissociation. J Am Soc Mass Spectrom. 2006;17:15–19. doi: 10.1016/j.jasms.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 25.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 26.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 27.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E. Global profiling of protein lipidation using chemical proteomic technologies. Curr Opin Chem Biol. 2015;24:48–57. doi: 10.1016/j.cbpa.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta. 2011;1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Nesvizhskii AI, Aebersold R. Interpretation of Shotgun Proteomic Data The Protein Inference Problem. Mol Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Li YF, Radivojac P. Computational approaches to protein inference in shotgun proteomics. BMC Bioinformas. 2012;13:S4. doi: 10.1186/1471-2105-13-S16-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi J, Wu FX. Protein inference by assembling peptides identified from tandem mass spectra. Curr Bioinforma. 2009;4:226–233. [Google Scholar]
- 33.Zhong J, Kim MS, Chaerkady R, Wu X, et al. TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol Cell Proteomics. 2012;11:M112.017764. doi: 10.1074/mcp.M112.017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savitski MM, Lemeer S, Boesche M, Lang M, et al. Confident phosphorylation site localization using the MASCOT Delta Score. Mol Cell Proteomics. 2011;10:M110.003830. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taus T, Köcher T, Pichler P, Paschke C, et al. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 37.Mortensen P, Gouw JW, Olsen JV, Ong SE, et al. MSQuant, an open source platform for mass spectrometry-based quantitative proteomics. J Proteome Res. 2010;9:393–403. doi: 10.1021/pr900721e. [DOI] [PubMed] [Google Scholar]
- 38.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 39.Marx H, Lemeer S, Schliep JE, Matheron L, et al. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat Biotechnol. 2013;31:557–564. doi: 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- 40.Courcelles M, Bridon G, Lemieux S, Thibault P. Occurrence and detection of phosphopeptide isomers in large-scale phosphoproteomics experiments. J Proteome Res. 2012;11:3753–3765. doi: 10.1021/pr300229m. [DOI] [PubMed] [Google Scholar]
- 41.Jung HR, Sidoli S, Haldbo S, Sprenger RR, et al. Precision mapping of coexisting modifications in histone H3 tails from embryonic stem cells by ETD-MS/MS. Anal Chem. 2013;85:8232–8239. doi: 10.1021/ac401299w. [DOI] [PubMed] [Google Scholar]
- 42.Rappsilber J, Mann M. What does it mean to identify a protein in proteomics? Trends Biochem Sci. 2002;27:74–78. doi: 10.1016/s0968-0004(01)02021-7. [DOI] [PubMed] [Google Scholar]
- 43.Ahn JM, Kim MS, Kim YI, Jeong SK, et al. Proteogenomic analysis of human chromosome 9-encoded genes from human samples and lung cancer tissues. J Proteome Res. 2014;13:137–146. doi: 10.1021/pr400792p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pawar H, Sahasrabuddhe NA, Renuse S, Keerthikumar S, et al. A proteogenomic approach to map the proteome of an unsequenced pathogen – Leishmania donovani. Proteomics. 2012;12:832–844. doi: 10.1002/pmic.201100505. [DOI] [PubMed] [Google Scholar]
- 45.Renuse S, Harsha HC, Kumar P, Acharya PK, et al. Proteomic analysis of an unsequenced plant — Mangifera indica. J Proteomics. 2012;75:5793–5796. doi: 10.1016/j.jprot.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinforma Oxf Engl. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 48.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Sprung R, Tang Y, Ball H, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Tan M, Xie Z, Dai L, et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie Z, Dai J, Dai L, Tan M, et al. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan M, Peng C, Anderson KA, Chhoy P, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the Swiss-prot database. Sci Rep. 2011;1:90. doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol. 2005;23:1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 55.Ning K, Fermin D, Nesvizhskii AI. Computational analysis of unassigned high-quality MS/MS spectra in proteomic data sets. Proteomics. 2010;10:2712–2718. doi: 10.1002/pmic.200900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chick JM, Kolippakkam D, Nusinow DP, Zhai B, et al. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol. 2015;33:743–749. doi: 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Na S, Bandeira N, Paek E. Fast multi-blind modification search through tandem mass spectrometry. Mol Cell Proteomics MCP. 2012;11:M111.010199. doi: 10.1074/mcp.M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prasad TSK, Goel R, Kandasamy K, Keerthikumar S, et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaudet P, Argoud-Puy G, Cusin I, Duek P, et al. neXtProt: organizing protein knowledge in the context of human proteome projects. J Proteome Res. 2013;12:293–298. doi: 10.1021/pr300830v. [DOI] [PubMed] [Google Scholar]
- 60.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9:429–434. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim MS, Zhong Y, Yachida S, Rajeshkumar NV, et al. Heterogeneity of pancreatic cancer metastases in a single patient revealed by quantitative proteomics. Mol Cell Proteomics MCP. 2014;13:2803–2811. doi: 10.1074/mcp.M114.038547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 66.Masuda T, Tomita M, Ishihama Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res. 2008;7:731–740. doi: 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- 67.Zahari MS, Wu X, Pinto SM, Nirujogi RS, et al. Phosphoproteomic profiling of tumor tissues identifies HSP27 Ser82 phosphorylation as a robust marker of early ischemia. Sci Rep. 2015;5:13660. doi: 10.1038/srep13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mertins P, Yang F, Liu T, Mani DR, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics MCP. 2014;13:1690–1704. doi: 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gajadhar AS, Johnson H, Slebos RJC, Shaddox K, et al. Phosphotyrosine signaling analysis in human tumors is confounded by systemic ischemia-driven artifacts and intra-specimen heterogeneity. Cancer Res. 2015;75:1495–1503. doi: 10.1158/0008-5472.CAN-14-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choudhary G, Wu SL, Shieh P, Hancock WS. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J Proteome Res. 2003;2:59–67. doi: 10.1021/pr025557n. [DOI] [PubMed] [Google Scholar]
- 71.Schlosser A, Vanselow JT, Kramer A. Mapping of phosphorylation sites by a multi-protease approach with specific phosphopeptide enrichment and NanoLC-MS/MS analysis. Anal Chem. 2005;77:5243–5250. doi: 10.1021/ac050232m. [DOI] [PubMed] [Google Scholar]
- 72.Biringer RG, Amato H, Harrington MG, Fonteh AN, et al. Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief Funct Genomic Proteomic. 2006;5:144–153. doi: 10.1093/bfgp/ell026. [DOI] [PubMed] [Google Scholar]
- 73.Meyer B, Papasotiriou DG, Karas M. 100% protein sequence coverage: a modern form of surrealism in proteomics. Amino Acids. 2011;41:291–310. doi: 10.1007/s00726-010-0680-6. [DOI] [PubMed] [Google Scholar]
- 74.Gilmore JM, Kettenbach AN, Gerber SA. Increasing phosphoproteomic coverage through sequential digestion by complementary proteases. Anal Bioanal Chem. 2012;402:711–720. doi: 10.1007/s00216-011-5466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim MS, Kandasamy K, Chaerkady R, Pandey A. Assessment of resolution parameters for CID-based shotgun proteomic experiments on the LTQ-Orbitrap mass spectrometer. J Am Soc Mass Spectrom. 2010;21:1606–1611. doi: 10.1016/j.jasms.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gillet LC, Navarro P, Tate S, Röst H, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, et al. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10:744–746. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lambert JP, Ivosev G, Couzens AL, Larsen B, et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods. 2013;10:1239–1245. doi: 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Distler U, Kuharev J, Navarro P, Levin Y, et al. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat Methods. 2014;11:167–170. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 80.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim MS, Zhong J, Kandasamy K, Delanghe B, Pandey A. Systematic evaluation of alternating CID and ETD fragmentation for phosphorylated peptides. Proteomics. 2011;11:2568–2572. doi: 10.1002/pmic.201000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, et al. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10:2377–2388. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 83.Kim MS, Pandey A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics. 2012;12:530–542. doi: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 85.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 86.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 87.Thompson A, Schafer J, Kuhn K, Kienle S, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 88.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 89.Boersema PJ, Aye TT, van Veen TAB, Heck AJR, Mohammed S. Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics. 2008;8:4624–4632. doi: 10.1002/pmic.200800297. [DOI] [PubMed] [Google Scholar]
- 90.Wu R, Dephoure N, Haas W, Huttlin EL, et al. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol Cell Proteomics. 2011;10:M111.009654. doi: 10.1074/mcp.M111.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kanshin E, Michnick S, Thibault P. Sample preparation and analytical strategies for large-scale phosphoproteomics experiments. Semin Cell Dev Biol. 2012;23:843–853. doi: 10.1016/j.semcdb.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 92.Dephoure N, Gould KL, Gygi SP, Kellogg DR. Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol Biol Cell. 2013;24:535–542. doi: 10.1091/mbc.E12-09-0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ratnayake S, Dias IHK, Lattman E, Griffiths HR. Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J Proteomics. 2013;92:160–170. doi: 10.1016/j.jprot.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 95.Daniels CM, Ong SE, Leung AKL. Phosphoproteomic approach to characterize protein mono- and poly(ADP-ribosyl)ation sites from cells. J Proteome Res. 2014;13:3510–3522. doi: 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Steen H, Küster B, Fernandez M, Pandey A, Mann M. Detection of tyrosine phosphorylated peptides by precursor ion scanning quadrupole TOF mass spectrometry in positive ion mode. Anal Chem. 2001;73:1440–1448. doi: 10.1021/ac001318c. [DOI] [PubMed] [Google Scholar]
- 97.Trelle MB, Jensen ON. Utility of immonium ions for assignment of epsilon-N-acetyllysine-containing peptides by tandem mass spectrometry. Anal Chem. 2008;80:3422–3430. doi: 10.1021/ac800005n. [DOI] [PubMed] [Google Scholar]
- 98.Kelstrup CD, Frese C, Heck AJR, Olsen JV, Nielsen ML. Analytical utility of mass spectral binning in proteomic experiments by SPectral Immonium Ion Detection (SPIID) Mol Cell Proteomics. 2014;13:1914–1924. doi: 10.1074/mcp.O113.035915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matthiesen R, Trelle MB, Højrup P, Bunkenborg J, Jensen ON. VEMS 3.0: algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J Proteome Res. 2005;4:2338–2347. doi: 10.1021/pr050264q. [DOI] [PubMed] [Google Scholar]
- 100.Rosenthal F, Nanni P, Barkow-Oesterreicher S, Hottiger MO. Optimization of LTQ-Orbitrap mass spectrometer parameters for the identification of ADP-ribosylation sites. J Proteome Res. 2015;14:4072–4079. doi: 10.1021/acs.jproteome.5b00432. [DOI] [PubMed] [Google Scholar]
- 101.Zhao P, Viner R, Teo CF, Boons G-J, et al. Combining high-energy C-trap dissociation and electron transfer dissociation for protein O-GlcNAc modification site assignment. J Proteome Res. 2011;10:4088–4104. doi: 10.1021/pr2002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toghi Eshghi S, Shah P, Yang W, Li X, Zhang H. GPQuest: a spectral library matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal Chem. 2015;87:5181–5188. doi: 10.1021/acs.analchem.5b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Galisson F, Mahrouche L, Courcelles M, Bonneil E, et al. A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol Cell Proteomics MCP. 2011;10:M110.004796. doi: 10.1074/mcp.M110.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]