

Abstract
Transcriptional activation by, and therefore the physiologic impact of, activated tyrosine-phosphorylated STATs (signal transducers and activators of transcription) may be negatively regulated by proteins termed PIAS (protein inhibitors of activated stats), as shown by previous experiments with mammalian cells in culture. Here, by using the genetic modifications in Drosophila, we demonstrate the in vivo functional interaction of the Drosophila homologues stat92E and a Drosophila PIAS gene (dpias). To this end we use a LOF allele and conditionally overexpressed dpias in JAK-STAT pathway mutant backgrounds. We conclude that the correct dpias/stat92E ratio is crucial for blood cell and eye development.
A variety of extracellular polypeptides cause tyrosine phosphorylation and activation of the mammalian STATs (signal transducers and activators of transcription; refs. 1 and 2), which participate in regulating a wide variety of events in embryology, hematopoiesis, and growth control. Several mechanisms of negative regulation of the transiently activated STATs have been discovered. (i) Induced proteins, variously called SOCS, JABS, or SSI proteins (3–5), prevent cytoplasmic STAT activation. (ii) Cytoplasmic tyrosine phosphatases (SHP 1) deactivate the JAK receptors halting STAT activation (6). (iii) Tyrosine phosphatase-mediated inactivation of tyrosine phosphorylated STATs also occurs in the nucleus (7, 8).
Finally, Chung and coworkers (9, 10) reported another possible means of directly deactivating STAT molecules. Proteins that bind to and block the in vitro DNA binding of activated STATs were found, when overexpressed in transfected cells, to strongly inhibit STAT-driven transcription. Five such mammalian proteins have now been identified and named, PIAS (for protein inhibitor of activated STAT) 1, 3, xα, xβ, and y (11). In transfected cells, overexpression of PIAS1 inhibited STAT1-induced transcription, overexpression of PIAS3 inhibited STAT3-induced transcription, and PIAS1 and PIAS3 coimmunoprecipitate with tyrosine-phosphorylated STAT1 and STAT3, respectively (9, 10). These results strongly implied a negative regulatory role of the PIAS proteins on activated STATs but did not prove a physiological relevance in animals.
The JAK-STAT pathway operates in Drosophila as well as in mammals (12). STAT92E (13, 14) is activated by hopscotch (HOP), the single Drosophila tyrosine kinase of the JAK family (15), after cells encounter outstretched (OS), a membrane-associated ligand, whose receptor is not yet identified (16). STAT92E then activates transcription, for example of the even skipped (eve) stripe 3 + 7 enhancer (13, 14). In addition to early lethality and segmentation phenotypes, LOF alleles of os and hop have uncovered widespread involvement of the pathway later in development, most well studied in eye ontogeny (17, 18).
Moreover, two hyperactive hop alleles, hopTum-l and hopT42, cause blood cell tumor formation, which can be suppressed by a LOF allele of stat92E, statHJ (19, 20). In mammals a conserved STAT5-dependent pathway is likely to be involved in leukemias of functionally similar cell types of the myeloid lineage (21, 22).
We now report functional aspects of the single Drosophila PIAS gene first identified many years ago as a suppressor of position effect variegation, termed Su(var)2–10, and shown recently by Hari et al. (23) to be a gene required for normal chromosome function. This gene, also recently described as zimp by Mohr and Boswell (24), has strong homology to the mammalian PIAS genes. Because the PIAS proteins have been well described in mammalian cell literature as interacting with STAT proteins, we believe the name for the Drosophila gene that might best serve the wider scientific community is the simple term dpias. A lethal P element insertion in the dpias locus produces a LOF allele furnishing a genetic tool to explore the interaction of dpias with genes in the JAK-STAT pathway. For this study, we also generated transgenic fly stocks overexpressing dPIAS. Our experiments demonstrate that STAT92E and dPIAS have to be correctly balanced for normal blood cell and eye development to occur.
Materials and Methods
Plasmid Constructs and Cultured Cells.
For transgenic plasmid constructs, the coding sequences of expressed sequence tag (EST) clones AA803041 [dpias(537)] and AA390747 [dpias(522)] were sequenced and cloned into the pUAST vector (EcoRI/XbaI). For the pull-down assay, a dpias coding sequence corresponding to amino acids 270–409 was cloned into pGEX 5–1(EcoRI/Sma). The construct expressing FLAG epitope-tagged STAT92E in Schneider S2 cells under the control of the actin promoter and extract preparation for these cells was described earlier (14).
Reverse Transcription (RT)–PCR from Larval Extracts.
dpias03697/dpias03697 were distinguished from dpias03697/CyO,actin-GFP,dpias+ mutants by means of the larval marker actin-GFP. First instar larvae were chosen as the source of mRNA because the maternal contribution of dpias mRNA (determined by mRNA in situ assays) was presumed to be low at this stage, and viable homozygous dpias03697 mutants could still be found. RNA was extracted from larvae of each genotype and semiquantitative RT-PCR assays were performed as described (25). Primer sequences used for dPIAS cDNA amplification were 5′-GCCGTATACCTGGTAAAGAAGCTCACC-3′ and 5′-TGGTGTGCTCCAAGATCCATCCTG-3′. For glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA amplification 5′-ACCGTCGACGGTCCCTCT-3′ and 5′-GTGTAGCCCAGGATTCCCT-3′ were used.
Control Stocks and Methods.
Control stocks and markers for Table 1: CyO was exchanged with Adv (as shown in Table 1); Sp1 and Bl. dpias03697 were exchanged with two deficiencies covering the dpias locus Df(2R)NP3 (as shown in Table 1) and Df(2Rw45-30n) with similar results (Bloomington Stock Center, Bloomington, IN). In the cross shown in Table 2, TM2 and TM3 were used instead of TM6b with similar results. In Fig. 3, TM2 was exchanged with TM6b and vice versa, and CyO was replaced with Bl, Sco, and Adv, which had no detectable influence on the eye phenotypes.
Table 1.
Decreasing dpias gene dosage enhances the tumor frequency in hopTum-l mutants
| F1 Genotype | Total number | With tumor | Without tumor | % with tumor | Fold increase |
|---|---|---|---|---|---|
| Parental cross: hopTum-l × dpias03697/CyO | |||||
| hopTum-l/+; dpias03697/+ | 323 | 257 | 66 | 80 | 2.2 |
| hopTum-l/+; CyO/+ | 236 | 88 | 148 | 37 | |
| Parental cross: hopTum-l × dpias03697/Adv | |||||
| hopTum-l/+; dpias03697/+ | 351 | 226 | 125 | 64 | 2.0 |
| hopTum-l/+; Adv/+ | 372 | 118 | 254 | 32 | |
| Parental cross: hopTum-l × Df(2R)Np3bw/CyO | |||||
| hopTum-l/+; Df(2R)Np3bw/+ | 256 | 241 | 15 | 94 | 2.4 |
| hopTum-l/+; CyO/+ | 288 | 113 | 175 | 39 | |
The difference between the two genotypes was statistically significant with a P value of P < 0.01.
Table 2.
Overexpressing dpias suppresses the tumor frequency in hopTum-l mutants
| F1 Genotype | Total number | With tumor | Without tumor | % with tumors | Fold decrease |
|---|---|---|---|---|---|
| Parental cross: hopTum-l × UAS-dpias(537); hs-Gal4/TM6b | |||||
| hopTum-l/+; UAS-dpias(537)/+; hs-Gal4/+ | 257 | 48 | 209 | 19 | 1.9 |
| hopTum-l/+; UAS-dpias(537)/+; TM6b/+ | 385 | 141 | 244 | 37 | |
The difference between the two genotypes was statistically significant with a P value of P < 0.01.
Figure 3.

Eye phenotypes and genetic interaction demonstrated by various gene dosages of dpias, stat92E, and os. (A) ey-Gal4/TM2 flies appear WT. (B) Overexpression of dpias(537) by using one copy of UAS-dpias(537) driven by ey-Gal4 results in small and rough eyes (UAS-dpias(537)/CyO;ey-Gal4/TM2). (C) The majority of flies with two copies of UAS-dpias(537) have no eyes (UAS-dpias(537)/UAS-dpias(537);ey-Gal4/TM2). (D) Eye size and texture of eyes as in B can be rescued with simultaneous overexpression of STAT92E (UAS-dpias(537)/hs-stat92E;ey-Gal4/TM2). (E) Flies heterozygous for stat06346 have phenotypically WT eyes (stat06346/TM2). (F) When heterozygous stat06346 flies are crossed to flies (as in B), an additional antennae can develop in place of the eye. The two resulting antennae (aristae) are marked by white arrows (UAS-dpias(537)/CyO;stat06346/ey-Gal4). (G) dpias03697/+ eyes have WT appearance. (H) The eyes of os1;CyO/+;stat06346/+ flies are small but can be partially rescued in size by reducing the gene dosage of dpias as shown in a sibling (I) with the genotype os1;dpias03697/+;stat06346/+.
Two independent transgenic lines, UAS-dpias(537) and UAS-dpias(522), respectively, were used in this study with equal effects in the described experiments.
Alternative splicing at the C terminus of the dpias gene results in at least three protein isoforms that share the first 515 amino acids but differ at their C termini. dpias 522AA was derived from EST AA390747 [amino acids 515–522 = TLDPFLQ (see ref. 24)] (23). dpias (526) was derived from EST AA536416: amino acids 515–526 = AVSAMNTMRKAK. dpias(537) was derived from EST AA803041: amino acids 515–537 = EDNDENCMAKAKEDSVIDLLDSP [see also Hari et al. (23)]. These sequences are available from FlyBase (http://flybase.bio.indiana.edu/seqs/).
The frequency of the eye phenotype suppression in Fig. 3 H and I was calculated in the following way. Eye sizes of os1;dpias03697/+;stat06346/+, and os1;CyO/+;stat06346/+ progeny segregating from the same parental cross were compared randomly in pairs, and flies were sorted into two pools (larger and smaller eyes), according their relative eye size. Of the flies of the larger-eyed pool, 85% were of the os1;dpias03697/+;stat06346/+ genotype. This difference was not caused by CyO, because in crosses with other dominant makers on chromosome 2, the result was similar: os1;dpias03697/+;stat06346/+ vs. os1;Adv/+;stat06346/+ with 87%, os1;dpias03697/+;stat06346/+ vs. os1;Sco/+;stat06346/+ with 89%, and os1;dpias03697/+;stat06346/+ and os1;Bl/+;stat06346/+ with 85% (over 200 flies were tested in each cross).
Fly Stocks.
L(2)03697 (Bloomington Stock Center) is a lethal P element insertion stock with no detectable dpias expression. stat06346 and hopTum-l (gifts from C. Dearolf, Massachusetts General Hospital, Harvard Medical School, Boston) were used in genetic interaction experiments as well as os1 (Bloomington Stock Center). ey-Gal4, heat shock (hs)-Gal4, and GMR-Gal4 (Bloomington Stock Center) were used to overexpress UAS-dpias(522) and UAS-dpias(537). These transgenic strains carrying stable insertions on chromosomes 1 and 2, respectively, were generated by P element-mediated transformation. Eye clones were generated as described (26), except P{ry+7.2 = ey-FLP.D}6, ry506 (Bloomington Stock center) was used as a flipase source; y{1} w[*]; P{w+mW.hs =>whs>}G13 L ! FRT 42B (Bloomington Stock Center) was recombined with L(2) 03697 and crossed to y{1} w[*]; P{w+mW.hs = >whs>}G13 P{w[+mC]-mCD*∷GFP.L}LL5 (Bloomington Stock Center). dstat−/− eye clones were identified by the absence of w expression. [The w gene on FRT 42B is flanked by two FRT cassettes, which also causes intrachromosomal recombination removing w (27).] The w gene on P{w[+mC]-mCD*∷GFP.L}LL5 is strongly expressed. Therefore, complete removal of w by intra- and extrachromosomal recombination can be seen easily.
STAT92E-dPIAS Pull-Down Assay.
It was performed as previously described (28), except that we established an S2 cell line expressing an epitope-tagged STAT92E (FLAG). Also, a glutathione S-transferase (GST) fusion protein (GST + dPIAS residues 270–409, which includes the zinc finger domain) was expressed in Escherichia coli and purified on GST beads. Nuclear extracts from untreated S2 cells or cells treated with peroxide vanadate (2 mM H2O2/1 mM for 15 min; refs. 8 and 14) were incubated with the GST-dPIAS fusion protein or GST protein alone, and then samples were exposed to FLAG Ab-conjugated beads. The beads were washed and the eluate was subjected to SDS/PAGE and Western blotting with an anti-GST Ab. The protein concentration of the nuclear extract of each sample was equalized before the addition of Flag-conjugated beads and before vanadate/H2O2 treatment by splitting the detached S2 cells into equal volumes.
Tumor-Suppression Experiments.
Abdominal tumors were identified under a dissecting microscope (Zeiss) at ×30 magnification, and adult females were scored as positive if they had at least one abdominal tumor 0–12 h after eclosion. Flies were grown at 25°C under noncrowded conditions. Pilot experiments established that at this temperature, hopTum-l female viability is comparable to wild-type (WT) siblings, and that 30–40% of hopTum-l heterozygous females have tumors in several different genetic backgrounds.
Histological Analysis.
Adult flies were prepared for scanning electron microscopy as described by Kimmel et al. (29). Eyes were sectioned and analyzed according to Tomlinson and Struhl (30). Pictures were taken by using a Zeiss Axiophot microscope.
Results
l(2)03697 Is a LOF Allele of dpias.
By matching the available flanking sequence of the P element insertion of the stock l(2)03697 (BDGP; ref. 31) with the 5′ untranslated region (UTR) of a cDNA highly homologous to the mammalian PIAS genes, we identified a putative mutant allele of the Drosophila PIAS gene before the Drosophila genomic sequence was completed.
The P element insertion at the dpias locus (the dpias03697 allele) blocked all mRNA formation. mRNA expression was assayed by semiquantitative RT-PCR of larvae homozygous and heterozygous for the insertion. Therefore, the dpias03697 allele constitutes a strong LOF or a null allele of the dpias gene (Fig. 1; see Materials and Methods for details).
Figure 1.
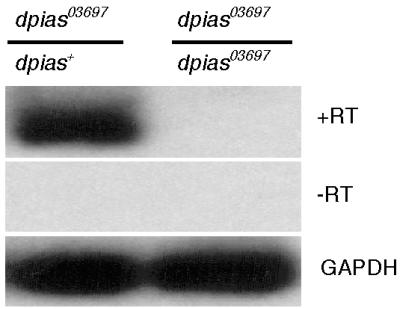
Semiquantitative RT-PCR of dpias03697/CyO;Actin-GFP,dpias+ (Left) and homozygous dpias03697 (Right) first instar larvae. +RT, reverse transcriptase added and primers amplifying dpias mRNA. −RT, no reverse transcriptase added and primers amplifying dpias mRNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), reverse transcriptase added and primers amplifying gapdh mRNA.
dPIAS Physically Interacts with STAT92E.
Coimmunoprecipitation of mammalian PIAS and tyrosine-phosphorylated STATs (9) has been established, and the interacting region of PIAS3 with STAT3 lies in the center of the molecule embracing a portion of a putative zinc finger domain (11). By using an in vitro protein association assay, we found that a similar region of a dPIAS–GST fusion molecule bound to a FLAG-tagged STAT92E protein (Fig. 2). The interaction depended on prior activation of the STAT92E protein brought about by the inhibition of tyrosine dephosphorylation with vanadate/peroxide (32), which was used because natural activation of STAT92E has not been accomplished in cell culture.
Figure 2.

The conserved central domain of dPIAS interacts directly with vanadate/H2O2-activated STAT92E (see text for details).
Suppression of Melanotic Tumor Formation by dpias.
Because the dpias03697 allele is a homozygous lethal, we designed genetic interaction crosses in which flies heterozygous for the recessive dpias03697 allele were scored for the possible enhancement or suppression of known phenotypes in JAK-STAT pathway mutants.
hopTum-l is a dominant hyperactive allele (increased HOP activity at elevated temperature; ref. 33) that causes tumor formation (19). This tumor formation, which is suppressed by stat92E LOF mutants (32), results from excessive proliferation of blood cells (plasmatocytes) that form melanotic abdominal tumors in larvae and pupae that can be scored in adults (19). At 25°C, 37% of heterozygous hopTum-l adult females had at least one abdominal tumor (Table 1 Parental Cross: hopTum-l × dpias003697/CyO, bottom row). (Hemizygous males have a much higher tumor rate and were not scored in this experiment.) Reduction of a negative activating regulator of this pathway should cause an increase in tumors. The percentage of flies with at least one tumor more than doubled in the hopTum-l/+;dpias03697/+ genotype compared with the progeny with two WT dpias alleles (hopTum-l/+;CyO/+; Table 1 Parental Cross: hopTum-l × dpias003697/CyO, top row). Control crosses in which CyO was exchanged with other marked chromosomes, such as Adv (Table 1 Parental Cross: hopTum-l × dpias003697/Adv), and a chromosomal deletion that removes the dpias gene, Df(2R)Np3, is shown in Table 1 Parental Cross: hopTum-l × Df(2R)Np3bw/CyO. Even though we found slight variations in the tumor frequency between these stocks (which could be a result of variations in population density), these controls produced a similar increase in tumor frequency compared with their siblings with two WT copies of dpias, minimizing the possibility of genetic background as a contributing factor to the elevation of tumor frequency.
If the observed increase in tumor formation was truly caused by a reduction of dpias gene dosage, then an increase of dPIAS should decrease tumor formation. The binary UAS-Gal4 expression system (34) allows for tissue-specific and conditional overexpression of transgenes in Drosophila. Transgenic animals carrying UAS-dpias constructs of two of the three identified dPIAS isoforms of 537- and 522-aa in length (see Materials and Methods for details) were prepared and used in the following genetic interaction crosses. [No significant difference in effect between transgenic lines with these two isoforms could be detected in this study and for simplicity, from now on, we refer only to the 537-aa-containing construct as UAS-dpias(537)]. A stock containing UAS-dpias(537) was crossed to a driver stock carrying a transgene expressing the transcriptional activator Gal4 under the control of a heat-shock promoter (hs-Gal4). The double transgenic progeny was crossed into the hopTum-l background and received a daily heat shock, 37°C for 30 min, from larval stages to eclosion. A significant decrease of tumor-bearing flies was observed in the hs-Gal4 genotype as compared with the control TM6b (Table 2). Other control crosses excluded TM6b as a contributing factor to this effect (see Materials and Methods). These experiments on tumor frequency support the conclusion that dPIAS interacts negatively with the JAK-STAT pathway made overactive by hopTum-l, which leads to tumor formation. We conclude that dPIAS decreases the transcriptional impact of the overactive STAT92E.
dpias and stat92E in Eye Development.
We next examined the role of dpias in eye development because hypomorphic mutants of hop and os have small eyes (17, ‖). We used two different lines, GMR-Gal4 and ey-Gal4, in which dpias overexpression depends on Gal4 activation at different times during eye development. When the GMR-Gal4 line was used to drive UAS-dpias(537), we observed no obvious effect on eye size or texture (not shown). Also, the ey-Gal4 transgene by itself had no effect on eyes (Fig. 3A). However, when we activated UAS-dpias(537) with the ey-Gal4 driver, eye size was severely reduced and the remaining small eye had a rough texture (Fig. 3B). A doubling of the transgene dosage further aggravated this phenotype and resulted in complete loss of the eyes in most of the surviving progeny (Fig. 3C). Because ey-Gal4 is active very early in eye development (before cellular differentiation) and GMR-Gal4 at later stages [during cellular differentiation (35)], we conclude that overexpression of dpias(537) has an effect primarily on cells in the early proliferating eye disk.
We further investigated whether this occurs because of a decreased activity of the JAK-STAT pathway. To this end, we crossed small-eyed UAS-dpias(537)/CyO;ey-Gal4 flies (as in Fig. 3B) to a stock carrying a heat shock-inducible stat92E gene (hs-stat92E) (32) and raised the progeny under mild heat-shock conditions (see Materials and Methods). A significant rescue of eye size and texture was observed only in progeny that carried the hs-stat92E transgene but not in genotypes without the hs-stat92E transgene segregating from the same cross (compare Fig. 3 B–D). Moreover, a similar eye-size rescue effect was achieved by crossing the hopTum-l stock with small-eyed UAS-dpias(537)/CyO;ey-Gal4 flies (not shown), further bolstering the notion that activated STAT92E is required for eye development and that dPIAS counteracts the activated STAT92E.
We went on to investigate how the stat92E LOF allele, stat06346, might affect small-eyed UAS-dpias/CyO;ey-Gal4 flies. (Heterozygous stat06346/TM2 was used because homozygotes of stat06346 die as first instar larvae). Fig. 3E depicts an eye from a heterozygote for the stat06346/TM2 mutant that is phenotypically WT. When this stock was crossed to the small-eyed UAS-dpias(537)/CyO;ey-Gal4 stock, however, the small-eyed phenotype was clearly made more severe, resulting in the loss of eyes similar to those with two copies of the UAS-dpias(537) transgene (like Fig. 3C and therefore not shown). Yet, frequently (27 of 100) the enhancement of phenotype seemed to go even further, and the progeny grew antenna in place of eyes (see Fig. 3F). Thus, stat92E has a role in the early phase of eye development and determination.
We also tried to establish whether the dPIAS–STAT92E interaction occurs naturally during eye development. [Enhancer trap lac-Z stains of dpias03697/+ mutants and detection of STAT92E by antiserum had indicated that both genes are coexpressed in developing third instar larval eye disks (data not shown and refs. 17 and 18).] To this end, we used the hypomorphic LOF mutant os1, which reduces function of the only known ligand in the JAK-STAT pathway in flies resulting in a small-eye phenotype (16, ‖). When we introduced dpias03697 into the os1 background (os1;dpias03697/+), a subtle increase in eye size in os1;dpias03697/+ progeny was observed as compared with the siblings segregating from the same cross with WT dpias (os1;CyO/+; not shown). This increase of eye size became more pronounced when JAK-STAT function of this genotype was further reduced in a background also heterozygous for the LOF stat06346 allele (os1;dpias03697/+;stat06346/+; compare Fig. 3 H–I). In contrast to the drastic eye-size differences described in Fig. 3 A–F, the increase in eye size observed in os1;dpias03697/+;stat06346/+ vs. os1;CyO/+;stat06346/+ was more modest but still significant on average (see Materials and Methods). To exclude genetic background as a contributing factor, we used several control stocks in which CyO was replaced with Adv, Sco, and Bl, but none of these substitutions had any significant influence on the difference in eye size (not shown; see Materials and Methods). Also, dpias/+ flies are phenotypically WT as shown in Fig. 3G. We conclude that by removing one copy of the WT dpias gene, suboptimal JAK-STAT activity in eyes brought about by the os1 allele can be partially compensated. Furthermore, this effect occurs without ectopic expression of transgenes and partially depends on stat92E gene dosage.
Removal of dpias Activity in LOF Eye Clones.
The effect of replacing both WT copies of the dpias gene with the mutant dpias03697 alleles in eyes was examined next. We used the yeast recombinase (flipase) system (26) to generate clonal patches of mutant homozygous dpias03697 cells (from now on called dpias−/−) within heterozygous phenotypically WT flies. Fig. 4A Left shows an example of an SEM of an eye with a dpias−/− clone extending along the dorsoventral midline. Fig. 4A Right, an enlarged view of the center of this eye, shows that the lens structure completely failed to develop and was replaced by a heterogeneous bulged-out surface lacking bristles. Partially differentiated lenses surrounded the border of the clone.
Figure 4.

(A Left) SEM of a larger equatorial dpias−/− eye clone extending along the equator (marked by an arrow), which can be seen to extend horizontally along the equator. In the blow up (A Right), normal lenses outside the clone, located dorsally and ventrally of the clone (two are marked with an asterisk), can be seen. In the center of the clone (marked by the bracket), the lens architecture is completely lost and the surface is bulged out. The Inset marks an example of a “partial lens phenotype” (37) with dotted lines, straddling the clonal border with ectopic hairs. (B Left) Diagram of a sectioned dpias−/− eye clone delineating the clonal area in gray. In the blow up (B Right), partial (arrow) or completely failed cellular differentiation (center) can be observed in the clonal area.
Sections through dpias−/− clones (Fig. 4B Right) revealed that cellular differentiation into photoreceptors and other cell types had failed, especially in the center of these clones. Along the clonal borders, partially differentiated ommatidia could be seen with incomplete sets of photoreceptors. (Fig. 4B Left indicates schematically the area of the clone in gray.) Other dpias−/− clones (not shown) had apparently undergone necrotic and/or apoptotic cell death, because scars in the eyes were found frequently. These results indicate that dpias gene function plays a critical role in proper growth, differentiation, and survival of potentially all apparent cell types in the developing eye.
Discussion
From the recent completion of the genomic sequence and annotation of the Drosophila genome, there seems to be only a single STAT gene, stat92E, and now a single PIAS homologue, dpias. We show here that the tyrosine-phosphorylated STAT92E and dPIAS can interact directly and specifically, and that a P element inserted in the dpias gene suppresses dpias mRNA formation.
Tumor Formation in the Dysregulated JAK-STAT Pathway.
The overgrowth of plasmatocytes (20) and melanotic abdominal tumor formation caused by the hopTum-l allele presumably depends on too much activated STAT92E, because stat92E LOF mutants such as statHJ suppress tumor formation (32). By the same logic, we infer that dPIAS regulates the number of active STAT92E molecules, because increased dPIAS decreases tumor formation and decreased dPIAS increases tumor formation, indicating that HOP, STAT92E, and dPIAS act together in this pathway. This type of behavior—genetic removal increasing tumorigenesis and overexpression reducing tumorigenesis—is characteristic of genes in mammals that are labeled tumor-suppressor genes. By this definition, dpias would be a tumor suppressor. Recent widespread reports of persistently active STAT3 in a variety of human tumors (22) and the demonstration of an engineered constitutively active STAT3 as an oncogene (36) coupled with the present results predict that mutations in human PIAS3 might very well allow for persistent activation of STAT3, resulting in tumor formation. This interpretation is further supported by recent findings of Hari et al. (23): Certain transheteroallelic dpias [Su(var)2–10] LOF alleles in otherwise genetically WT backgrounds caused melanotic tumors in third instar larvae.
stat92E and dpias Interact in Eye Development.
A dramatic developmental role of dpias–stat92E interaction was found in eye development. Overexpression of dPIAS early (driven by ey-Gal4) aborts eyes, but loss of stat92E function later (by overexpression of dPIAS under the control of GMR-Gal4) has no apparent detrimental effect on cell growth or survival. Similar observations were made by Papayannopoulos et al. (35), using the same Gal4 driver stocks in combination with other early and late eye genes. Therefore, factors controlling stat92E function must be normally balanced in a critical time window in early eye development. Further increased expression of dpias or coupling with heterozygosity for the stat92E LOF allele stat06346 led to transformation events with antennae frequently replacing eyes. LOF alleles of the Drosophila JAK-kinase hop of increasing severity cause the same sequence of increasing phenotypic abnormalities (17). Moreover, os1, a hypomorphic LOF allele of a JAK-STAT pathway ligand, results in small eyes (16, ‖). This phenotype could be partially suppressed and the eye size increased by reducing the dpias gene dosage, implying that with no transgenic intervention, dPIAS and STAT92E naturally interact in eye formation and eye determination.
In results to be described elsewhere, we have used a naturally occurring dominant negative stat92E to examine effects on eye development and find phenotypes similar to dpias(537) overexpression. The two sets of data substantiate a previously unrecognized role for STAT92E in growth, cell survival, and determination in early eye development.
The Consequences of Removal of dPIAS Activity on Eye Development.
Through somatic recombination, patches of eye cells were created that presumably lack dPIAS, leaving any activated STAT92E unopposed. Under these conditions, none of the apparent retinal cell types differentiated normally.
The lack of, or abnormal differentiation of, lens structure observed on the surface of dpias−/− clones, in particular in the clonal border areas (see Insert in Fig. 4A Right), appears to be phenotypically similar to Notch GOF phenotypes as reported in an earlier study (37). Overexpression of activated Notch delayed the differentiation of cone cells, the cells that secrete the lens material. Therefore, we infer that cone cell differentiation in surviving dpias−/− clones might be similarly affected.
We also found that in sections through dpias−/− clones, retinal cellular differentiation failed and was replaced by a heterogeneous cell mass. Other clones had apparently undergone either apoptotic or necrotic cell death, as indicated by frequent scars. Which of theses diverse phenotypes might be caused by unopposed overactive STAT92E remains to be seen. It will be important to learn whether members of the mammalian PIAS genes are playing related roles in STAT-dependent tumor suppression, cell death, and differentiation.
Acknowledgments
We thank all members of the M. Young Laboratory for continued technical support and discussions. We thank Cedric Wesley and Toby Lieber for assistance in in situ hybridizations. We thank Chingwen Yang for helpful discussions and Lois Cousseau for editing. We also received helpful suggestions from Ulrike Gaul. Charles Dearolf kindly provided fly stocks. We thank Ke Shuai who originally alerted us to the existence of PIAS-like sequences in the Drosophila EST database before his first PIAS publication. This work was supported by National Institutes of Health Grants AI32489 and AI32440 (to J.E.D.).
Abbreviations
- EST
expressed sequence tags
- RT
reverse transcription
- GST
glutathione S-transferase
- WT
wild type
- hs
heat shock
Footnotes
Verderosa, F. J. & Muller, H. J. (1954) Genetics 39, 999 (abstr.).
References
- 1.Darnell J E., Jr Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 2.Stark G R, Kerr I M, Williams B R, Silverman R H, Schreiber R D. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 3.Starr R, Willson T A, Viney E M, Murray L J L, Rayner J R, Jenkins B J, Gonda T J, Alexander W S, Metcalf D, Nicola N A, Hilton D J. Nature (London) 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 4.Endo T A, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, et al. Nature (London) 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 5.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, et al. Nature (London) 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 6.Klingmuller U, Lorenz U, Cantley L C, Neel B G, Lodish H. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 7.Haspel R L, Salditt-Georgieff M, Darnell J E., Jr EMBO J. 1996;15:6262–6268. [PMC free article] [PubMed] [Google Scholar]
- 8.Haspel R L, Darnell J E., Jr Proc Natl Acad Sci USA. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung C D, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 10.Liu B, Liao J, Rao X, Kushner S A, Chung C D, Chang D D, Shuai K. Proc Natl Acad Sci USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao J, Fu Y, Shuai K. Proc Natl Acad Sci USA. 2000;97:5267–5272. doi: 10.1073/pnas.97.10.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeidler M P, Bach E A, Perrimon N. Oncogene. 2000;19:2598–2606. doi: 10.1038/sj.onc.1203482. [DOI] [PubMed] [Google Scholar]
- 13.Hou X S, Melnick M B, Perrimon N. Cell. 1996;84:411–419. doi: 10.1016/s0092-8674(00)81286-6. [DOI] [PubMed] [Google Scholar]
- 14.Yan R, Small S, Desplan C, Dearolf C R, Darnell J E., Jr Cell. 1996;84:421–430. doi: 10.1016/s0092-8674(00)81287-8. [DOI] [PubMed] [Google Scholar]
- 15.Binari R, Perrimon N. Genes Dev. 1994;8:300–312. doi: 10.1101/gad.8.3.300. [DOI] [PubMed] [Google Scholar]
- 16.Harrison D A, McCoon P E, Binari R, Gilman M, Perrimon N. Genes Dev. 1998;12:3252–3263. doi: 10.1101/gad.12.20.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo H, Asha H, Kockel L, Parke T, Mlodzik M, Dearolf C R. Dev Biol. 1999;213:432–441. doi: 10.1006/dbio.1999.9390. [DOI] [PubMed] [Google Scholar]
- 18.Zeidler M P, Perrimon N, Strutt D I. Genes Dev. 1999;13:1342–1353. doi: 10.1101/gad.13.10.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dearolf C R. Acta Biochim Biophys. 1998;1377:M13–M23. doi: 10.1016/s0304-419x(97)00031-0. [DOI] [PubMed] [Google Scholar]
- 20.Luo H, Hanratty W P, Dearolf C R. EMBO J. 1995;14:1412–1420. doi: 10.1002/j.1460-2075.1995.tb07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia R, Jove R. J Biomed Sci. 1998;5:79–85. doi: 10.1007/BF02258360. [DOI] [PubMed] [Google Scholar]
- 22.Jove R. Oncogene. 2000;19:2466–2467. doi: 10.1038/sj.onc.1203549. [DOI] [PubMed] [Google Scholar]
- 23.Hari K L, Cook K R, Harpen G H. Genes Dev. 2001;15:1334–1348. doi: 10.1101/gad.877901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohr S E, Boswell R E. Gene. 1999;229:109–118. doi: 10.1016/s0378-1119(99)00033-5. [DOI] [PubMed] [Google Scholar]
- 25.Yang E, Wen Z, Haspel R L, Zhang J J, Darnell J E., Jr Mol Cell Biol. 1999;19:5106–5112. doi: 10.1128/mcb.19.7.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu T, Rubin G M. Development (Cambridge, UK) 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 27.Golic K G, Lindquist S. Cell. 1989;59:499–509. doi: 10.1016/0092-8674(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J J, Vinkemeier U, Gu W, Chakravarti D, Horvath C M, Darnell J E., Jr Proc Natl Acad Sci USA. 1996;93:15092–15096. doi: 10.1073/pnas.93.26.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimmel B E, Heberlein U, Rubin G M. Genes Dev. 1990;4:712–727. doi: 10.1101/gad.4.5.712. [DOI] [PubMed] [Google Scholar]
- 30.Tomlinson A, Struhl G. Development (Cambridge, UK) 1999;126:5725–5738. doi: 10.1242/dev.126.24.5725. [DOI] [PubMed] [Google Scholar]
- 31.Spradling A C, Stern D, Beaton A, Rehm E J, Laverty T, Mozden N, Misra S, Rubin G M. Genetics. 1999;153:135–177. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan R, Lou H, Darnell J E, Jr, Dearolf C R. Proc Natl Acad Sci USA. 1996;93:5842–5847. doi: 10.1073/pnas.93.12.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison D A, Binari R, Nahreni T S, Gilman M, Perrimon N. EMBO J. 1995;14:2857–2865. doi: 10.1002/j.1460-2075.1995.tb07285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand A H, Manoukian A S, Perrimon N. Methods Cell Biol. 1994;44:635–654. doi: 10.1016/s0091-679x(08)60936-x. [DOI] [PubMed] [Google Scholar]
- 35.Papayannopoulos V, Tomlinson A, Panin V M, Rauskolb C, Irvine K D. Science. 1998;281:2031–2034. doi: 10.1126/science.281.5385.2031. [DOI] [PubMed] [Google Scholar]
- 36.Bromberg J F, Wrzeszczynska M H, Devgan G, Zhao Y, Pestell R G, Albanese C, Darnell J E., Jr Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 37.Fortini M E, Rebay I, Caron L A, Artavanis-Tsakonas S. Nature (London) 1993;365:555–557. doi: 10.1038/365555a0. [DOI] [PubMed] [Google Scholar]