

Abstract
Polycystic kidney disease (PKD) involves progressive hepatorenal cyst expansion and fibrosis, frequently leading to end-stage renal disease. Increased vasopressin and cAMP signaling, dysregulated calcium homeostasis, and hypertension play major roles in PKD progression. The guanylyl cyclase A agonist, B-type natriuretic peptide (BNP), stimulates cGMP and shows anti-fibrotic, anti-hypertensive, and vasopressin-suppressive effects, potentially counteracting PKD pathogenesis. Here, we assessed the impacts of guanylyl cyclase A activation on PKD progression in a rat model of PKD. Sustained BNP production significantly reduced kidney weight, renal cystic indexes and fibrosis, in concert with suppressed hepatic cystogenesis in vivo. In vitro, BNP decreased cystic epithelial cell proliferation, suppressed fibrotic gene expression, and increased intracellular calcium. Together, our data demonstrate multifaceted effects of sustained activation of guanylyl cyclase A on polycystic kidney and liver disease. Thus, targeting the guanylyl cyclase A-cGMP axis may provide a novel therapeutic strategy for hepatorenal fibrocystic diseases.
Keywords: ARPKD, ADPKD, Congenital hepatic fibrosis, gene therapy, adeno-associated virus
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is characterized by focal development and expansion of cysts, frequently leading to end-stage renal disease (ESRD).1, 2 Autosomal recessive PKD (ARPKD) is characterized by diffuse fusiform dilatation of the collecting ducts causing massive renal enlargement at birth, followed by interstitial fibrosis, early onset ESRD,3 and congenital hepatic fibrosis. Arginine vasopressin (AVP), epithelial proliferation, and hypertension are major contributors in PKD progression. At a molecular level, causative-PKD proteins are involved in calcium (Ca2+) signaling and alterations in intracellular Ca2+ [Ca2+]i homeostasis may play a role in the upregulation of cyclic AMP (cAMP) and protein kinase A (PKA) signaling observed in cystic tissues.4, 5
B-type natriuretic peptide (BNP) binds guanylyl cyclase A (GC-A/NPRA) to stimulate the cyclic GMP (cGMP) pathway. Through activation of cGMP-dependent kinases (PKGs) and modulation of [Ca2+]i, BNP elicits vasodilating, natriuretic, and anti-hypertrophic effects in vivo.6 BNP also inhibits fibroblast proliferation and down-regulates fibrosis-associated genes such as Tgfβ, Col1a1, and Fn1, and suppresses AVP release.7 We showed BNP knockout Nppb−/− rats developed interstitial fibrosis and glomerulosclerosis prior to the development of hypertension, suggesting an intrinsic renoprotective role, separate from BNPs’ anti-hypertensive propensity.8 Further, BNP-transgenic mice are resistant to glomerular injury upon renal ablation, glomerulonephritis, and chemically induced diabetic nephropathy.9, 10
Since destruction of renal parenchyma is principally responsible for declining kidney function, early interventions aimed at preventing cyst expansion are critical for PKD therapy. BNPs’ properties, and the potential of the GC-A/cGMP pathway, promise multiple points of intervening PKD pathobiology. However, the role of cGMP in PKD is poorly understood. Increased levels of cGMP have been observed in the kidneys of PKD animals,11 however 8-Br-cGMP does not stimulate human PKD cyst-derived cell proliferation.12 In this study, we assessed BNP overexpression in vitro, and in vivo on the development of hepatorenal cysts and fibrosis in the PCK rat, an orthologous rat model of ARPKD, with human ADPKD-like hepatorenal pathology.13, 14
RESULTS
Adeno-associated viral (AAV) vector-mediated systemic BNP overexpression stimulates cGMP, suppresses circulating AVP, and improves cardiac function
Due to a short in vivo half-life, long-term BNP therapy has been challenging. To address this, we used AAV serotype 9 (AAV9) vectors, carrying CMV-driven codon-optimized proBNP cDNA.15 BNP transgene expression was determined in a rat model of PKD (PCK rats) treated with 1013 vg/kg AAV9-BNP. Three months post injection, RT-PCR detected significantly increased transgene BNP transcripts in the heart, liver, and kidney (Fig. 1a). In contrast, endogenous BNP transcripts were comparable between AAV9-BNP and control littermates (Fig. 1b). There was no significant increase in circulating BNP levels between low-dose AAV9-BNP (1013 vg/kg) and littermate controls, while High-Dose (HD) AAV9-BNP (1014 vg/kg) demonstrated significantly elevated circulating BNP (Fig. 1c). Unenhanced serum levels of BNP with low-dose AAV9-BNP may be due to rapid enzymatic degradation (e.g. by neprilysin) and/or clearance through NPR-C.16 Nevertheless, excreted cGMP was elevated (Fig. 1d), indicating in vivo cGMP stimulation by low-dose AAV9-BNP. Similarly, blood from terminal cardiac puncture demonstrated reduced CT-proAVP, an indicator of AVP levels17, with low-dose AAV9-BNP (Fig. 1e). At 3 months of age, PCK rats remained normotensive (Fig. 1f). Although effects were modest, left ventricular ejection fraction and percent fractional shortening were significantly higher with treatment (Fig. 1g), suggesting improved cardiac function upon BNP treatment.
Fig. 1. Characterization of single intraperitoneal AAV9 vector administration in neonatal female PCK rats.
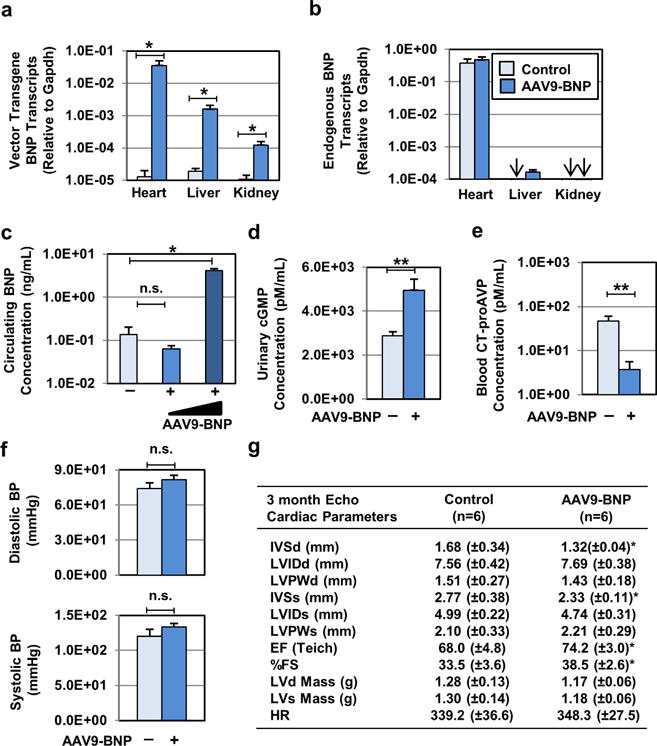
(a) Biodistribution of AAV9 vector upon neonatal AAV9 vector administration was monitored. Codon-optimized vector transgene BNP and (b) endogenous BNP transcript levels in the heart, kidney, and liver samples were determined by RT-PCR, followed by normalization to Gapdh transcript levels. Control (n=8) and AAV9-BNP vector-treated (n=6, 1.0×1013 vg/kg) PCK rats were used. Arrows indicate where transcripts assayed were below detection. (c) Circulating levels of BNP from littermate control PCK (n=6), AAV9-BNP (n=9, 1.0×1013 vg/kg) and High-Dose AAV9-BNP (n=5, 1.0×1014 vg/kg) were determined by rat BNP45 ELISA. (d) Urinary excreted cyclic GMP (cGMP) and (e) plasma levels of CT-proAVP from control (n=10) and AAV9-BNP (n=10, 1.0×1013 vg/kg) were determined. (f) Noninvasive diastolic and systolic blood pressure measurements were determined in AAV9-BNP-treated (n=15, 1.0×1013 vg/kg) and control littermate PCK rats (n=10). Both groups remained normotensive 3 months post treatment. (g) Echocardiographic assessment (Echo) of cardiac structure and function was performed in PCK (n=7) and AAV9-BNP-treated (n=10, 1.0×1013 vg/kg) groups. Improved cardiac function and reduced cardiac mass were associated with AAV9-BNP vector administration. Intraventricular septum, diastole (IVSd), Left ventricular internal diameter, diastole (LVIDd), Left ventricular posterior wall, diastole (LVPWd), Intraventricular septum, systole (IVSs), Left ventricular internal diameter, systole (LVIDs), Left ventricular posterior wall, systole (LVPWs), Ejection Fraction (EFteich), Percent fractional shortening (%FS), Left ventricular Mass, diastole (LVd Mass), Left ventricular mass, Systole (LVs Mass). *P < 0.05 and **P < 0.01, vs. PCK by t test. Data represent the mean ± SEM.
BNP renal effects in PCK rats
Three months after low-dose AAV9-BNP administration treated PCK displayed reduced 24-hour urine excretion. Although total urine metabolites per 24-hour collection were relatively uninfluenced, sodium and potassium concentrations were significantly increased with AAV9-BNP (Fig. 2a). Further, total urinary protein was reduced (Fig. 2a), and plasma albumin levels were significantly increased with treatment (Sup. Fig. S1). Creatinine clearance was significantly higher with AAV9-BNP, comparable to Sprague Dawley (SD), non-PKD controls (Fig. 2b). Together, this suggests that BNP treatment increases urine concentrating capacity and glomerular filtration rate. At sacrifice, total kidney weight per body weight (TKW/BW) was reduced with AAV9-BNP compared to PCK controls (12.6 ± 1.7 vs 14.5 ± 2.0 mg/g), without a difference in body weight between treatment (Fig. 2c). Histology revealed renal cysts in PCK controls, and a reduced (40%) cystic index with AAV9-BNP (Fig. 2d, 2e). Additionally, fewer injured glomeruli were observed by H&E staining with BNP treatment (Fig. 2f). Reduced urinary excretions of kidney injury molecule-1 (Fig. 2g) and neutrophil gelatinase-associated lipocalin (262.5±6.6 vs. 293.2±11.6 ng/ml in controls) were observed, suggesting reduced renal injury with AAV9-BNP. AAV9-BNP treated animals exhibited reduced fibroblast growth factor 2 (Fgf2), and inflammation marker, Desmin, expression as indicated by immunofluorescence (IF) staining (Fig. 2h, 2i), and trends in reduced renal fibrosis, assessed by Trichrome staining (Fig. 2j, 2k). RT-PCR of profibrotic gene expression in renal tissues were significantly reduced collagen type 1 (Col1a1), fibronectin (Fn1), and Tgfβ transcripts in treated PCK (Fig. 2l). Despite renoprotective effects with BNP treatment, total renal cAMP levels were increased in treated PCK (Fig. 2m).
Fig. 2. BNP overexpression reduces kidney size, renal cyst growth, interstitial fibrosis and glomerular injury in female PCK rats.
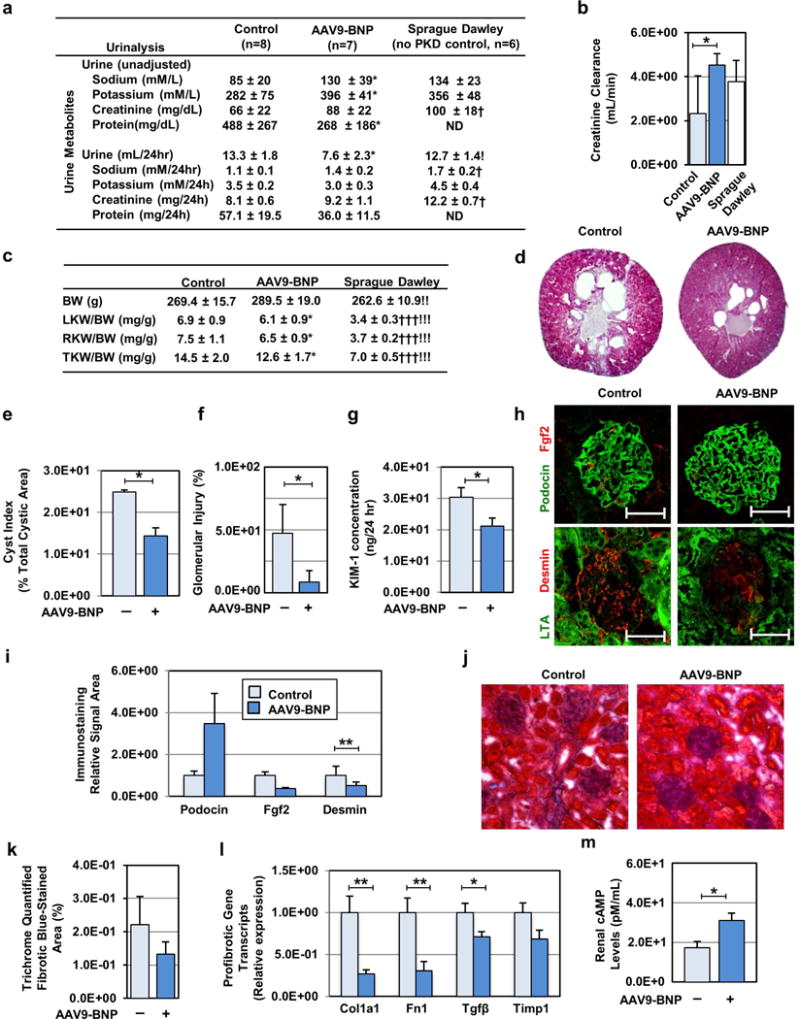
(a) Urine volume and urinary test parameters were determined in AAV9-BNP-treated, control PCK and SD rats. (b) Estimated creatinine clearance was determined by blood and urinary creatinine concentrations, and compared among the AAV9-BNP-treated, control PCK and SD rat groups. Control (n=10), AAV9-BNP (n=10, 1.0×1013 vg/kg) treated PCK and SD (n=6) rats were used (a–c). (c) Total body weight (BW) and left, right and total kidney weight to BW ratios at 3 months post injection are shown. (d) Representative Hematoxylin and Eosin (H&E)-staining images of renal sections from AAV9-BNP-treated and untreated PCK rats are shown. (e) Renal cystic index was determined using the H&E-staining images of control (n=7) and AAV9-BNP vector-treated (n=7, 1.0×1013 vg/kg) PCK rats. (f) Percent glomerular injury was quantified using H&E staining images of kidney sections of control and treated PCK rats (n=5 each, 4 images/animal). (g) Urinary excretion of tubular epithelial injury marker Kidney Injury Molecule-1 (KIM-1) was determined by ELISA. (h) Representative images of immunofluorescent microscopy of the glomeruli of control and AAV9-BNP vector-treated PCK rats are shown. Anti-basic fibroblast growth factor 2 (Fgf2, Red) and anti-podocin (Blue) antibodies (upper panels), and anti-Desmin (Red) and anti-lotus tetragonolobus (LTA, Green) antibodies were used. Nuclei were counterstained with DAPI (White). (i) Relative staining of immunofluorescent images of control and AAV9-BNP-treated PCK rats were determined (n=5 each, 4 images/animal). (j) Representative images (20×, zoomed in 25% in Adobe Photoshop CC 2015) of trichrome stained renal sections from PCK treated and control rats. (k) Interstitial fibrosis quantified from trichrome stained kidney sections (n=5 treated, n=4 PCK control). (l) Levels of Collagen type 1a1 (Col1a1), Fibronectin-1 (Fn1), Transforming growth factor-beta (Tgfβ) and Tissue inhibitor metalloprotease-1 (Timp1) transcripts were analyzed by RT-PCR. Relative transcript levels were determined using Gapdh transcript levels, and compared to littermate controls. Total kidney RNA samples from control (n=7) and AAV9-BNP vector-treated (n=7, 1.0×1013 vg/kg) PCK rats were used. (m) Renal cyclic AMP (cAMP) levels in control and AAV9-BNP vector-treated and PCK rats were determined (n=10 each). (l) *P < 0.05, **P < 0.01, and ***P < 0.001 (AAV9-BNP treated vs. PCK controls, by t test). †P<0.05, ††P<0.01, and †††P<0.001 (PCK controls vs. SD, by t test). !P<0.05, !!P<0.01, !!!P<0.001 (AAV9-BNP treated vs. SD by t test). Data represent the mean ± SEM.
To better define the pathways affected by AAV9-BNP treatment, we performed genome-wide transcriptome analysis in renal RNA of treated and control PCK rats. Transcriptome analysis revealed suppression of collagen genes, Col3a1, Col5a2, Col6a3, Col6a1 and Col8a2, with BNP treatment (Sup. Fig. S2). Intriguingly, bioinformatic analysis identified the top 10 differentially expressed signaling pathways as PTEN, aldosterone, PDGF, ErbB2-ErbB3, IL-8, PI3K/AKT, Her2/ErbB2, and NFAT (Sup. Fig. S2, S3, S4, S5 and S6), pathways routinely implicated in PKD pathogenesis.18, 19 Wnt signaling and negative regulators for calcineurin/NFAT were also identified as influenced pathways with BNP treatment, whereas no significant changes were observed in natriuretic peptide system genes (Sup. Fig. S2).
Reduced hepatic cystic lesions, fibrosis, and profibrotic gene expression in PCK rats with sustained BNP overexpression
In addition to renal cystogenesis and fibrosis, PCK rats exhibit congenital liver fibrosis, characterized by progressive fibropolycystic liver disease. We therefore characterized the influence of BNP overexpression in the liver of PCK rats. Pathological analysis of liver tissue demonstrated reduced hepatic remodeling (Fig. 3a, upper panels) and cystic indices (Fig. 3b) in AAV9-BNP treated PCK. When fibrotic regions were visualized by picrosirius red (PSR) staining, we found a trend of reduced connective tissue areas (Fig. 3a, lower panels and Fig. 3c). However, collagen content, quantified by hydroxyproline, detected no notable difference between groups (Fig. 3d). IF staining demonstrated reduced Desmin-positive hepatic stellate cells, reduced fibrosis regulator galectin-3, and CK7-positive cholangiocytes in BNP treated PCK hepatic sections (Fig. 3e, 3f). Desmin-positive cells were predominantly negative for αSMA staining in both groups (Fig. 3e). RT-PCR showed reduced transcripts of Fn1, Tgfβ, and Desmin with BNP treatment, although Col1a1 and Timp1 expression was not affected (Fig. 3g). This suggests that BNP provides liver protection through reduced hepatic cyst expansion, restrained expansion of Desmin-positive stellate cells and CK7-positive cholangiocytes, and suppression of a subset of profibrotic genes, such as Tgfβ.
Fig. 3. Sustained BNP treatment reduces cystic lesions, fibrosis and profibrotic gene expression in the liver of female PCK rats.
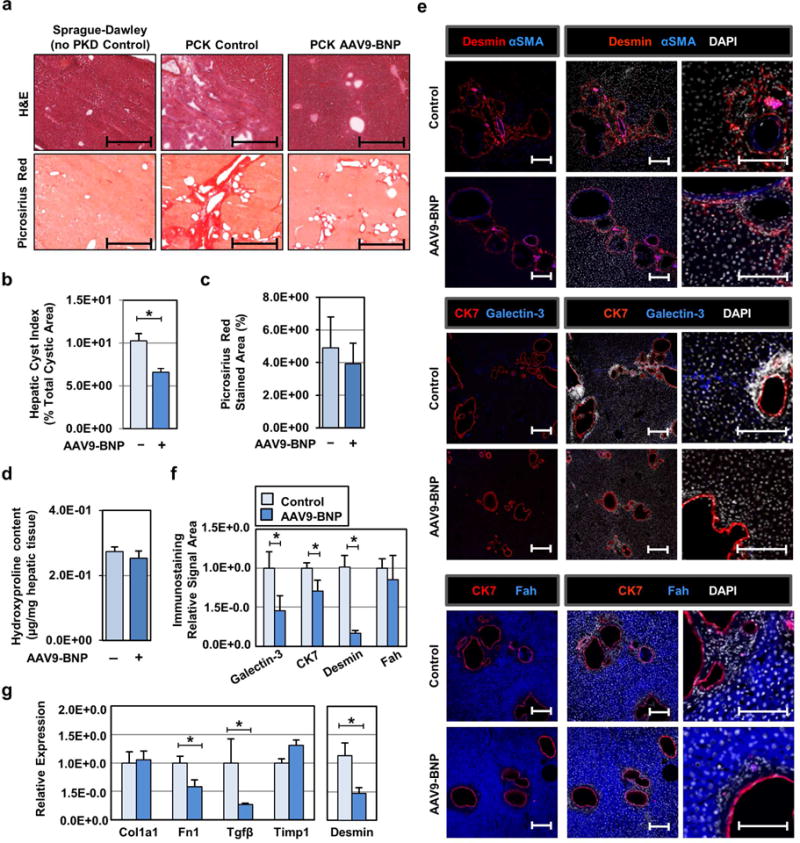
(a) Representative hematoxylin and eosin (H&E) staining images of hepatic sections 3 months’ post AAV9-BNP vector administration (top panels), and representative picosirius red staining images are shown (bottom panels). Age-matched livers from non PKD control (Sprague-Dawley rat), control PCK and AAV9-BNP vector-treated PCK rats were used. Scale bars; 1 mm. (b) Hepatic cystic index was determined using the H&E-staining images of control (n=7) and AAV9-BNP vector-treated (n=7, 1.0×1013 vg/kg) PCK rats. (c) We determined fibrotic areas as measured from picosirius red stained liver sections (n=5 treated, n=4 PCK controls). (d) Hepatic hydroxyproline levels in PCK control (n=4) and AAV9-BNP treated (n=6) PCK rats were determined to estimate collagen content. (e) Representative images of immunofluorescent staining of hepatic sections are shown. Anti-Desmin(red) and anti-alpha-Smooth Muscle Actin (blue) antibodies (top), anti-CK7 (red) and anti-Galectin-3 (green) antibodies (middle) and anti-fumarylacetoacetate hydrolase (Fah, blue) and anti-CK7 (red) antibodies (bottom) were used for immunostaining. Nuclei were counterstained by DAPI (white). Scale bars; 1.0 μm. (f) Individual immunofluorescent signal areas in (e) were quantified and shown as mean fluorescent signal areas, relative to control PCK littermates. Immunofluorescent images of control (n=4) and AAV9-BNP vector-treated (n=7, 1.0×1013 vg/kg) PCK rats were used. (g) Levels of Collagen type 1a1 (Col1a1), Fibronectin-1 (Fn1), Transforming growth factor-β (Tgfβ), Tissue inhibitor metalloprotease-1 (Timp1) and Desmin transcripts were determined by RT-PCR, corrected for Gapdh transcripts, and presented relative to those found in control littermates. Control (n=8) and AAV9-BNP vector-treated (n=6, 1.0×1013 vg/kg) PCK rats were used. *P < 0.05. (vs. PCK controls, by t test) Data represent the mean ± SEM.
Influence of a higher dose AAV9-BNP in PCK rats
To further understand tolerability of the AAV9-BNP vector, we administered a 10-fold higher dose (HD AAV9-BNP, 1014 vg/kg), resulting in 40-fold increased circulating levels of BNP (Fig. 1c). Supraphysiological BNP did not induce hypotension (Sup. Fig. S7a) or notable differences in blood chemistry parameters (Sup. Fig. S7b). Four months after AAV administration, rats were sacrificed. Compared to 3 month-old PCK controls (Fig. 2c, d, e), 4 month-old PCK rats exhibited advanced cystogenesis and increased TKW/BW (Sup. Fig. S8a, b, c). HD AAV9-BNP resulted in trends of better preserved renal architecture, reduced TKW/BW, and smaller cystic indices (Sup. Fig. S8a, b, c and d), though statistical significance was not achieved. We also found trends of reduced hepatic remodeling and cystic indices in HD AAV9-BNP rats (Sup Fig. S8e, 8f, 8g, Sup. Fig. S9). At this time point, control PCK rats showed αSMA-positive cells in the liver, a population which appeared less prominent in HD AAV9-BNP-treatment (Sup. Fig. S8h). Lastly HD AAV9-BNP treatment seemed to have increased mortality within 2 months of vector administration (Sup. Fig. S8i); however, no statistical significance was found in survival between groups.
BNP treatment reduces proliferation and increases [Ca2+]i in epithelial cells
To better characterize direct cellular effects of BNP, we treated cholangiocytes from PCK and SD with BNP. Non- and cystic cholangiocytes treated with BNP for 72 hours exhibited significantly reduced proliferation compared to vehicle (Fig. 4a, left panel). Levels of [Ca2+]i assessed with Fura-2AM were increased with BNP in cystic cholangiocytes (Fig. 4a, right panel). BNP- or GFP-lentiviral transduction of ADPKD, ARPKD, and normal human renal epithelial (HRE) cells were then assayed to determine effects on proliferation or [Ca2+]I. BNP transduced cell lines responded with significantly reduced proliferation (Fig. 4b). Reduced HRE proliferation was associated with reduced levels of phosphorylated ERK, i.e. ERK activation, as demonstrated by reduced protein levels (Fig. 4c), and PKD HRE cells exhibited increased levels of [Ca2+]i with BNP (Fig. 4d). Media supplemented with Ca2+ showed no notable influence on [Ca2+]i, suggesting that BNP-mediated increases were primarily due to a release of [Ca2+]i stores. Since the endoplasmic reticulum (ER) is the largest store of [Ca2+]i we assessed effects of two ER Ca2+ channel inhibitors on our transduced cell lines; a ryanodine receptor (RyR) inhibitor, JTV 519 fumarate, and an inhibitor of inositol triphosphate receptor (IP3R), 2-aminoethoxydiphenyl borate (2-APB). Diminished [Ca2+]i was observed in the presence of 2-APB, but not JTV 519 fumarate, suggesting that BNP increases [Ca2+]i partly through IP3R in cystic epithelial cells (Fig. 4e). Lastly, BNP overexpression in normal and cystic HRE cells significantly suppressed COL1A1 and TGFβ transcripts relative to controls (Sup. Fig. S10).
Fig. 4. BNP treatment reduces proliferation and increases intracellular calcium in primary PKD epithelial cells.
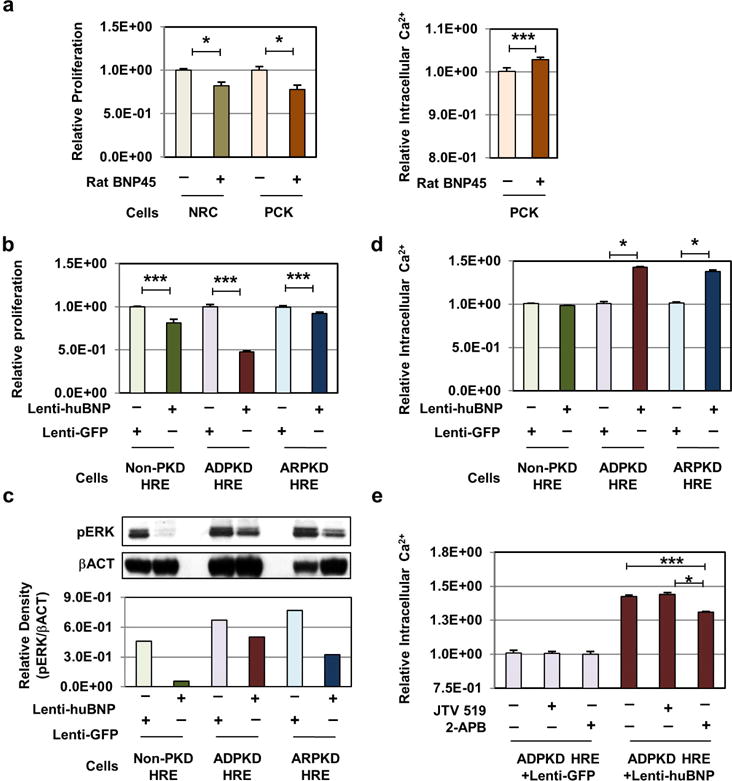
(a) In vitro proliferation studies of normal (NRC) and cystic (PCK) rat cholangiocytes derived from Sprague Dawley and PCK rats. Cells were treated with normal growth media (light grey/pink bars) or 270 nmol rat-BNP peptide-spiked media (green/orange bars), and assayed for relative proliferation 72 hours post plating (left panel). Intracellular calcium levels were determined in PCK rat-derived cholangiocytes cultured in media supplemented with (orange bar) and without (pink bar) 270nmol rat-BNP peptide, assayed by Fura-2 AM calcium quantification (right panel). Data represents a mean of 6 individual assays, each with 24 replicates ± SEM. (b) Patient-derived noncystic, normal human renal epithelial cells (Non-PKD HRE), and ADPKD and ARPKD cystic human renal epithelial cells (ADPKD HRE, ARPKD HRE) were transduced with lentiviral vectors expressing control Green Fluorescent Protein (Lenti-GFP), or codon-optimized human BNP (Lenti-huBNP). GFP- and human BNP-expressing HRE cells were assayed for relative proliferative ability, 48–72 hours post-plating. Proliferation rates are expressed as relative to Lenti-GFP vector-treated control cells. Data represents a minimum mean of 3 individual assays, with a minimum of 20 replicates ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, vs. Lenti-GFP by t test. (c) Levels of phosphorylated-extracellular-signal-regulated-kinase (PERK) protein were determined by Western blotting using non-cystic and PKD-derived cystic HRE cells transduced with Lenti-GFP, or Lenti-huBNP. (d) Intracellular calcium levels were determined by Fura-2 AM calcium quantification assay. Data represents a mean of 6 individual assays, each with 24 replicates ± SEM. (e) Intracellular calcium levels were quantified in the presence of ER-calcium channel inhibitors, 2-ABP (100 μM) or JTV 519 (1μm), or vehicle. ADPKD HRE cells transduced with Lenti-GFP or Lenti-huBNP were used for the assay. Data points represent the mean of a minimum of 24 replicates ± SEM. *P < 0.05 and ***P < 0.001, vs. Lenti-GFP by t test.
DISCUSSION
Mechanisms of BNP therapy on PKD progression
BNP-transgenic mice are resistant to various experimental renal injuries with notable suppression of renal Tgfβ and pERK.9, 10 PKD progression is known to be influenced by genetic mutations, genetic background, and hypertension.20 BNP suppressed AVP release, demonstrated by reduced serum CT-proAVP levels. In accordance with potent anti-fibrotic effects of BNP,21–24 we observed a trend of reduced fibrosis and significant suppression of profibrotic genes, including Tgfβ. Of note, Tgfβ signaling plays in cyst progression and fibrogenesis of both ADPKD murine models and ADPKD patients,25, 26 while the inhibition of the activin/Tgfβ signaling pathway retards PKD progression.27 BNP treatment also reduced glomerular injury and expression of inflammation marker Desmin in PCK rats. Gene expression profiling indicated that BNP treatment altered key PKD pathogenesis-associated pathways, such as PTEN, Aldosterone, PDGF, ErbB2/3, Her2, PI3K and calcineurin/NFAT. Additionally, in vitro BNP supplementation demonstrated a direct effect on epithelial cells, reduced proliferation, suppressed ERK activation, and increased [Ca2+]i. Thus, observed renal protective effects in BNP-treated PCK rats are likely due to multifaceted effects induced by BNP directly or indirectly.
It is known that cAMP inhibits proliferation of noncystic cells, while enhancing proliferation in cyst derived epithelial cells in a Src, Ras, B-Raf/ERK and mTOR dependent manner.12, 28 This proliferative response has been linked to reduced [Ca2+]i, as illustrated by Ca2+ channel blockers inducing a proliferative response to cAMP in normal human kidney or murine collecting duct cells, whereas Ca2+ channel activators or Ca2+ ionophores restores a normal antimitogenic response to cAMP in ADPKD or ARPKD cyst-derived cells.28, 29 Unexpectedly, BNP treatment did not reduce the total cAMP levels in kidney lysates. This may suggest that BNP does not directly suppress cAMP. Since Src inhibition has demonstrated reduced cystogenesis independent from renal cAMP levels,30 it is plausible that BNP targets effectors downstream of the cAMP pathway, such as suppression of the Ras/B-Raf/ERK pathway through PKG-II-mediated ErbB2/Her2 suppression.31, 32 Since increased intracellular Ca2+ restores inhibitory effects of cAMP against cell proliferation and ERK phosphorylation in primary cultured PKD cells,29 it is also possible that the ‘switching’ of cAMP-responsive characteristics, regulated by intracellular Ca2+ concentration, may also play an inhibitory role, as BNP treatment restores intracellular Ca2+ levels in cystic epithelia cells. Further studies will elucidate a potential anti-proliferative and ERK inhibitory roles of cAMP in BNP treatment.
Roles of the cGMP pathways in PKD
cGMP is synthesized by natriuretic peptide-activated particulate guanylyl cyclases (pGCs) or by NO-activated soluble guanylyl cyclase (sGC).33 Although recent studies have implicated crosstalk between cAMP and cGMP pathways in PKD, through cAMP- and/or cGMP-degrading phosphodiesterases (PDEs),34 the role of cGMP signaling on the development, or progression, of PKD is poorly understood. cGMP levels are increased in cystic kidneys of PKD animals.11 In vitro studies have demonstrated that cGMP supplementation modestly increases in vitro MDCK cyst expansion.11 In contrast, 8-Br-cGMP treatment does not affect the proliferation of primary human PKD cyst-derived cells.12 Our data demonstrates cGMP induction through the BNP/GC-A axis reduces cystogenesis in the PCK rat. This discrepancy may be due to compartmentalized and unique GCs, which then stimulate distinct effectors.35 For instance, GC-A-induced cGMP is degraded primarily by PDE2 while GC-B-induced cGMP is degraded by PED3.36, 37 PDE1 is largely responsible for renal cGMP PDE activity,11 suggesting that renal cGMP is produced through sGC activation and degraded by PDE1. Thus, GC-A-mediated cGMP may activate effectors, distinct from those activated by the NO/sGC axis in cystic kidneys. Another possibility is that cGMP elevation is compensatory. Multiple studies demonstrate marked renoprotective effects of sustained activation of GC-A and GC-B by BNP and CNP, respectively.,9, 10, 38 Moreover, the endothelial NO synthase Glu298Asp mutation responsible for impaired NO production39 is associated with a 5-year lower mean age at ESRD in ADPKD patients,39 supporting the renoprotective effects of sGC-induced cGMP in PKD. Thus, further studies are warranted to define specific effectors and chronic GC-A activation in PKD.
BNP antiproliferative effects and increased [Ca2+]i in renal and hepatic epithelial cells
In cardiomyocytes, cGMP activates PKG-I to reduce cytoplasmic [Ca2+]i through inhibition of Ca2+ influx by L-type Ca2+ channels, leading to relaxation and inhibition of cardiac hypertrophy.40, 41 Here, BNP increased [Ca2+]I in renal and hepatic epithelial cells, mediated partly through IP3R. This discrepancy may be due to differentially expressed PKG isoforms. PKG-II is ubiquitous throughout the nephron, with the highest levels in juxtaglomerular cells and tubules.42 Activation of PKG-II through the GC-A/cGMP axis has been shown to stimulate Ca2+ reabsorption.43 PKG-II activation can also phosphorylate IP3R, increasing cytosolic [Ca2+] in hippocampal neurons and hepatocytes.44, 45 We postulate that BNP increases [Ca2+]i levels in renal epithelium through activation of PKG-II.
Liver-protective effects
Though BNP reduced cysts with trends of reduced connective tissue, Col1a1 transcripts and hepatic collagen content were not reduced by AAV9-BNP. This suggests BNP treatment enhanced matrix degradation instead of inhibiting activation of hepatic stellate cells and/or portal fibroblasts. Conversely, we found suppression of Desmin-positive, but αSMA-negative, hepatic stellate-like cell expansion by BNP treatment. At 4 months, we saw induction of Desmin- and αSMA-double positive myofibroblasts-like cells in control rats, which appeared to be reduced by BNP treatment. Similarly, BNP-transgenic mice are resistant to hepatic damage, liver fibrosis, and suppressed stellate cell activation and Tgfβ induction.46 Further studies are warranted to define underlying collagen independent BNP anti-fibrotic effects in the liver.
pERK levels in vivo
In concert with our in vitro study, we found reduced pERK levels in the AAV-BNP-treated liver (Supplementary Fig. S11). However, we found no notable changes in pERK levels in BNP-treated kidneys. It is possible that BNP-mediated suppression of pERK in cystic cells in vivo is not strong and masked by abundant pERK in other types of renal cells.
AAV vector-mediated gene therapy for renal diseases
Previously, we demonstrated no notable toxicity 9 months post AAV9 administration in a hypertensive rat model.15 In PCK, blood chemistry revealed no detectable difference in low- or HD AAV9-BNP treatments 3 months post administration (Sup. Fig. S1 and S7B). We also found no notable toxicity, nor beneficial effects, in PCK treated with luciferase-expressing AAV9 (Sup. Fig. S12). AAV9 vectors are established for their robust transduction of variety cell types; heart, liver, kidney, and endothelial cells in adult animals, while neonatal administration of AAV9 leads to near whole-body transduction.47–50 Consistent with previous reports, AAV9 in our PCK, exhibited widespread transgene expression in the heart, liver, and kidneys. However, BNP renal transgene transcripts were still approximately 100-fold less than cardiac upon neonatal AAV9 administration. Kidney targeted optimization of AAV will further improve BNP gene therapy for PKD.
Sample numbers
We maintained multiple PCK breeding pairs, with a study design allowing for sufficient numbers for treated and control groups. However, an unexpectedly high incidence of maternal cannibalism, associated with neonatal AAV transduction, limited the study. Additionally, we observed significant gender-bias in litters, with reduced numbers of male PCK pups, possibly due to increased embryonic lethality in males. Thus, our data set focuses on females. All animal manipulations were carried out for both treatment groups with preserved time intervals. For echocardiography, blood pressure, and metabolic tests, all animals from each treatment group were assayed and included in the figure, baring animals that didn’t produce analyzable data. For assays on stored biological material, we used samples from treated and control rats from the same litters to reduce possible inter-litter variations.
Conclusions
Our study demonstrates long-term therapeutic, and multifaceted effects of BNP in a clinically relevant PKD model, the PCK rat, on hepatorenal cystogenesis.13, 14 While the role of cAMP signaling in PKD has received much attention,51 our study suggests the influence of cGMP signaling on the progression of PKD in vivo. Together with cardioprotective effects, sustained BNP and/or GC-A-stimulating strategies may present a novel avenue for preventing PKD progression and associated complications.
METHODS
Animals, Plasmids, Vectors, Data, and Statistical Analysis are detailed in the Supplemental Text.
Supplementary Material
Acknowledgments
We thank Mayo’s Translational Polycystic Kidney Disease Center, Jim Tarara, Shawna Cooper, Vlad Gainullin, Katharina Hopp, and Maria Lorenzo Pisarello for their time, reagents, and invaluable advice.
Funding source(s): National Institute of Health R01 HL098502 (to AC and YI), Mayo PKD Center Pilot Study Award (to AC and YI, parent grant NIDDK P30DK90728, PI: VET), Mayo Foundation (to Y.I.), NIDDK DK024031 (to NFL), and Mayo Graduate School (to SJH)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONTRIBUTIONS
SJH – designed research studies, conducted experiments, analyzed data, wrote and edited the manuscript
CAS – conducted experiments, analyzed data, edited the manuscript
AC, PCH, NFLR - edited the manuscript
MRA, VET - designed research studies, offered insight in the analysis of data, edited the manuscript
YI - designed research studies, offered insight in the analysis of data, wrote and edited the manuscript
DISCLOSURE
None.
References
- 1.Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015;88:17–27. doi: 10.1038/ki.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ong AC, Devuyst O, Knebelmann B, et al. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2. [DOI] [PubMed] [Google Scholar]
- 3.Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD) Medicine (Baltimore) 2006;85:1–21. doi: 10.1097/01.md.0000200165.90373.9a. [DOI] [PubMed] [Google Scholar]
- 4.Cowley BD., Jr Calcium, cyclic AMP, and MAP kinases: dysregulation in polycystic kidney disease. Kidney Int. 2008;73:251–253. doi: 10.1038/sj.ki.5002695. [DOI] [PubMed] [Google Scholar]
- 5.Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20:251–260. doi: 10.1016/j.molmed.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zellner C, Protter AA, Ko E, et al. Coronary vasodilator effects of BNP: mechanisms of action in coronary conductance and resistance arteries. Am J Physiol. 1999;276:H1049–1057. doi: 10.1152/ajpheart.1999.276.3.H1049. [DOI] [PubMed] [Google Scholar]
- 7.Fujisaki H, Ito H, Hirata Y, et al. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest. 1995;96:1059–1065. doi: 10.1172/JCI118092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holditch SJ, Schreiber CA, Nini R, et al. B-Type Natriuretic Peptide Deletion Leads to Progressive Hypertension, Associated Organ Damage, and Reduced Survival: Novel Model for Human Hypertension. Hypertension. 2015;66:199–210. doi: 10.1161/HYPERTENSIONAHA.115.05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suganami T, Mukoyama M, Sugawara A, et al. Overexpression of brain natriuretic peptide in mice ameliorates immune-mediated renal injury. J Am Soc Nephrol. 2001;12:2652–2663. doi: 10.1681/ASN.V12122652. [DOI] [PubMed] [Google Scholar]
- 10.Makino H, Mukoyama M, Mori K, et al. Transgenic overexpression of brain natriuretic peptide prevents the progression of diabetic nephropathy in mice. Diabetologia. 2006;49:2514–2524. doi: 10.1007/s00125-006-0352-y. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Ward CJ, Harris PC, et al. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2010;77:129–140. doi: 10.1038/ki.2009.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 13.Lager DJ, Qian Q, Bengal RJ, et al. The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001;59:126–136. doi: 10.1046/j.1523-1755.2001.00473.x. [DOI] [PubMed] [Google Scholar]
- 14.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 15.Cataliotti A, Tonne JM, Bellavia D, et al. Long-term cardiac pro-B-type natriuretic peptide gene delivery prevents the development of hypertensive heart disease in spontaneously hypertensive rats. Circulation. 2011;123:1297–1305. doi: 10.1161/CIRCULATIONAHA.110.981720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsukawa N, Grzesik WJ, Takahashi N, et al. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A. 1999;96:7403–7408. doi: 10.1073/pnas.96.13.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riphagen IJ, Boertien WE, Alkhalaf A, et al. Copeptin, a surrogate marker for arginine vasopressin, is associated with cardiovascular and all-cause mortality in patients with type 2 diabetes (ZODIAC-31) Diabetes Care. 2013;36:3201–3207. doi: 10.2337/dc12-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee EJ, Song SA, Mun HW, et al. Blockade of interleukin-8 receptor signalling inhibits cyst development in vitro, via suppression of cell proliferation in autosomal polycystic kidney disease. Nephrology (Carlton) 2014;19:471–478. doi: 10.1111/nep.12261. [DOI] [PubMed] [Google Scholar]
- 19.Wilson SJ, Amsler K, Hyink DP, et al. Inhibition of HER-2(neu/ErbB2) restores normal function and structure to polycystic kidney disease (PKD) epithelia. Biochim Biophys Acta. 2006;1762:647–655. doi: 10.1016/j.bbadis.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358:1439–1444. doi: 10.1016/S0140-6736(01)06531-X. [DOI] [PubMed] [Google Scholar]
- 21.Sonoyama T, Tamura N, Miyashita K, et al. Inhibition of hepatic damage and liver fibrosis by brain natriuretic peptide. FEBS Lett. 2009;583:2067–2070. doi: 10.1016/j.febslet.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 22.Huntley BK, Ichiki T, Sangaralingham SJ, et al. B-type natriuretic peptide and extracellular matrix protein interactions in human cardiac fibroblasts. J Cell Physiol. 2010;225:251–255. doi: 10.1002/jcp.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichiki T, Schirger JA, Huntley BK, et al. Cardiac fibrosis in end-stage human heart failure and the cardiac natriuretic peptide guanylyl cyclase system: regulation and therapeutic implications. J Mol Cell Cardiol. 2014;75:199–205. doi: 10.1016/j.yjmcc.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura N, Ogawa Y, Chusho H, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4239–4244. doi: 10.1073/pnas.070371497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassane S, Leonhard WN, van der Wal A, et al. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222:21–31. doi: 10.1002/path.2734. [DOI] [PubMed] [Google Scholar]
- 26.Liu D, Wang CJ, Judge DP, et al. A Pkd1-Fbn1 genetic interaction implicates TGF-beta signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2014;25:81–91. doi: 10.1681/ASN.2012050486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonhard WN, Kunnen SJ, Plugge AJ, et al. Inhibition of Activin Signaling Slows Progression of Polycystic Kidney Disease. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2015030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaguchi T, Wallace DP, Magenheimer BS, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. The Journal of biological chemistry. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi T, Hempson SJ, Reif GA, et al. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–187. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- 30.Sweeney WE, Jr, von Vigier RO, Frost P, et al. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen WS, Leung CM, Pan HW, et al. Silencing of miR-1-1 and miR-133a-2 cluster expression by DNA hypermethylation in colorectal cancer. Oncol Rep. 2012;28:1069–1076. doi: 10.3892/or.2012.1899. [DOI] [PubMed] [Google Scholar]
- 32.Cao ZH, Tao Y, Sang JR, et al. Type II, but not type I, cGMP-dependent protein kinase reverses bFGF-induced proliferation and migration of U251 human glioma cells. Mol Med Rep. 2013;7:1229–1234. doi: 10.3892/mmr.2013.1319. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Ruoho AE, Rao VD, et al. Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13414–13419. doi: 10.1073/pnas.94.25.13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto CS, Raman A, Reif GA, et al. Phosphodiesterase Isoform Regulation of Cell Proliferation and Fluid Secretion in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27:1124–1134. doi: 10.1681/ASN.2015010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moltzau LR, Aronsen JM, Meier S, et al. Different compartmentation of responses to brain natriuretic peptide and C-type natriuretic peptide in failing rat ventricle. J Pharmacol Exp Ther. 2014;350:681–690. doi: 10.1124/jpet.114.214882. [DOI] [PubMed] [Google Scholar]
- 36.Cai YL, Sun Q, Huang X, et al. cGMP-PDE3-cAMP signal pathway involved in the inhibitory effect of CNP on gastric motility in rat. Regul Pept. 2013;180:43–49. doi: 10.1016/j.regpep.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Wen JF, Quan HX, Zhou GH, et al. Altered role of C-type natriuretic peptide-activated pGC-cGMP-PDE3-cAMP signaling in hyperthyroid beating rabbit atria. Regul Pept. 2007;142:123–130. doi: 10.1016/j.regpep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Kimura T, Nojiri T, Hosoda H, et al. Protective effects of C-type natriuretic peptide on cisplatin-induced nephrotoxicity in mice. Cancer Chemother Pharmacol. 2015;75:1057–1063. doi: 10.1007/s00280-015-2734-7. [DOI] [PubMed] [Google Scholar]
- 39.Noiri E, Satoh H, Taguchi J, et al. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension. 2002;40:535–540. doi: 10.1161/01.hyp.0000033974.57407.82. [DOI] [PubMed] [Google Scholar]
- 40.Mery PF, Lohmann SM, Walter U, et al. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiedler B, Lohmann SM, Smolenski A, et al. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A. 2002;99:11363–11368. doi: 10.1073/pnas.162100799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gambaryan S, Hausler C, Markert T, et al. Expression of type II cGMP-dependent protein kinase in rat kidney is regulated by dehydration and correlated with renin gene expression. J Clin Invest. 1996;98:662–670. doi: 10.1172/JCI118837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoenderop JG, Vaandrager AB, Dijkink L, et al. Atrial natriuretic peptide-stimulated Ca2+ reabsorption in rabbit kidney requires membrane-targeted, cGMP-dependent protein kinase type II. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6084–6089. doi: 10.1073/pnas.96.11.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bredt DS, Snyder SH. Nitric oxide, a novel neuronal messenger. Neuron. 1992;8:3–11. doi: 10.1016/0896-6273(92)90104-l. [DOI] [PubMed] [Google Scholar]
- 45.Guihard G, Combettes L, Capiod T. 3′:5′-cyclic guanosine monophosphate (cGMP) potentiates the inositol 1,4,5-trisphosphate-evoked Ca2+ release in guinea-pig hepatocytes. Biochem J. 1996;318(Pt 3):849–855. doi: 10.1042/bj3180849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sonoyama T, Tamura N, Miyashita K, et al. Inhibition of hepatic damage and liver fibrosis by brain natriuretic peptide. FEBS Lett. 2009;583:2067–2070. doi: 10.1016/j.febslet.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh A, Yue Y, Long C, et al. Efficient whole-body transduction with trans-splicing adeno-associated viral vectors. Mol Ther. 2007;15:750–755. doi: 10.1038/sj.mt.6300081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yue Y, Ghosh A, Long C, et al. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16:1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saunders NR, Joakim Ek C, Dziegielewska KM. The neonatal blood-brain barrier is functionally effective, and immaturity does not explain differential targeting of AAV9. Nat Biotechnol. 2009;27:804–805. doi: 10.1038/nbt0909-804. author reply 805. [DOI] [PubMed] [Google Scholar]
- 50.Gadalla KK, Bailey ME, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21:18–30. doi: 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.