

Abstract
The development of new antimalarial therapeutics is necessary to address the increasing resistance to current drugs. Bicyclic azetidines targeting Plasmodium falciparum phenylalanyl-tRNA synthetase comprise one promising new class of antimalarials, especially due to their activities against three stages of the parasite’s life cycle, but a lengthy synthetic route to these compounds may affect the feasibility of delivering new therapeutic agents within the cost constraints of antimalarial drugs. Here, we report an efficient synthesis of antimalarial compound BRD3914 (EC50 = 15 nM) that hinges on a Pd-catalyzed, directed C(sp3)–H arylation of azetidines at the C3 position. This newly developed protocol exhibits a broad substrate scope and provides access to valuable, stereochemically defined building blocks. BRD3914 was evaluated in P. falciparum-infected mice, providing a cure after four oral doses.
Introduction
Malaria is caused by infection with Plasmodium species and transmitted by female Anopheles mosquitoes.1 While substantial progress has been made over the past two decades in reducing both mortality and burden associated with the disease,2 the emergence of parasite resistance to first-line antimalarial treatments highlights the need for safe and effective new therapies.1,3 The lifecycle of the malaria parasite includes three distinct stages—the asexual blood stage, the liver stage, and the gametocyte blood stage—and next-generation antimalarial therapies should ideally target all three.4
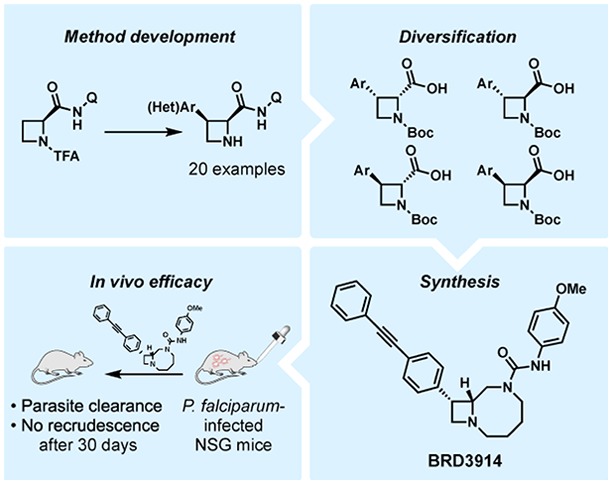
We recently identified antimalarial compounds originating from diversity-oriented synthesis (DOS), highlighted by phenylalanyl-tRNA synthetase inhibitor BRD7929 (2S,3R,4R) (Figure 1).5 BRD7929 exhibits activity in all stages of the parasite life cycle and is curative in multiple in vivo mouse models. However, a lengthy synthetic route to bicyclic azetidine BRD7929 (21 steps) hampers the ability to evaluate analogs systematically in vivo. Analysis of stereochemistry-based structure–activity relationships (SAR) revealed that two of eight possible stereoisomers of parent compound BRD3444—(2S,3R,4R) and (2R,3R,4R)—were active against a multidrug-resistant strain of malaria (P. falciparum, strain Dd2). This observation suggested that substituents at one of the stereocenters (C2) were not necessary for activity. Indeed, BRD3914, an analog lacking substitution at C2, was found to retain substantial antimalarial activity in vitro (EC50 = 13 nM, Dd2 strain).6
Figure 1.

(a) Structures and in vitro antiplasmodial activities of representative bicyclic azetidines. (b) Stereochemistry-based structure–activity relationships (SAR).
Production cost constitutes an important consideration in the development of antimalarial therapeutics.4 We recognized in BRD3914 an opportunity to achieve an efficient synthesis of this class of antimalarials that proceeds with high regio- and stereoselectivity of key transformations in fewer steps than the previous synthesis, and that minimizes the number of chromatographic purifications, which are known to drive both cost and waste output.7 To this end, we developed a stereospecific C(sp3)–H arylation of azetidines, enabling the preparation of all four stereoisomers of members of this series. The (2R,3S) stereoisomer of this building block led to an expeditious synthesis of antimalarial BRD3914. Subsequent in vivo evaluation in P. falciparum-infected humanized mice showed parasite clearance and lack of recrudescence after four oral doses, resulting in a durable cure.
A key step in our original synthesis of BRD3914 (14 steps) and other bicyclic azetidines comprises a base-mediated intramolecular cyclization from 1 that affords azetidine-2-carbonitrile 2 with little or no diastereoselectivity (ca. 1:1, Figure 2a).6a,8 To date, intramolecular cyclization from complex starting materials remains the predominant approach to access substituted azetidines.9 Following azetidine ring formation, nitrile reduction with DIBAL, protection with o-nitrobenzenesulfonyl chloride, and allylation gives the precursor for the key ring-closing metathesis to generate the eight-membered ring (Figure S1). While the previous DOS route provided a versatile starting point for an initial SAR assessment focused, for example, on the 4-methoxyurea region, it limited the ability to assess other regions of the scaffold. Notably, various aryl and heteroaryl derivatives at the C3 position of the azetidine require the execution of the entire 14-step synthesis, and the introduction of functional groups on the eight-membered ring relies on olefin chemistry and thus presents regio- and stereoselectivity challenges.
Figure 2.

(a) Comparison of ring formation and C–H arylation approaches to BRD3914. (b) Model system and optimized protocol for the C–H arylation of azetidines.
In a revised retrosynthetic analysis (Figure 2a), we reasoned that a bifunctional linker could be used in an advantageous way to form the eight-membered ring (diamine 5). This approach would rely on cis-substituted azetidine 4 as a precursor, which we envisioned arising from a directed C–H arylation using commercially available d-azetidine-2-carboxylic acid 3. Since the first examples exploiting 8-aminoquinoline as a directing group by Daugulis and co-workers,10 C(sp3)–H functionalization methods have grown11 and been applied to a number of challenging scaffolds, including cyclopropanes,12,13 cyclobutanes,13,14 cyclopentanes,10b,15 pyrrolidines,16 and piperidines.17 There are few reports of the use of azetidines as a substrate18 or product19 in C–H functionalization, and to our knowledge the C3 arylation of azetidines has not been described.
Results and Discussion
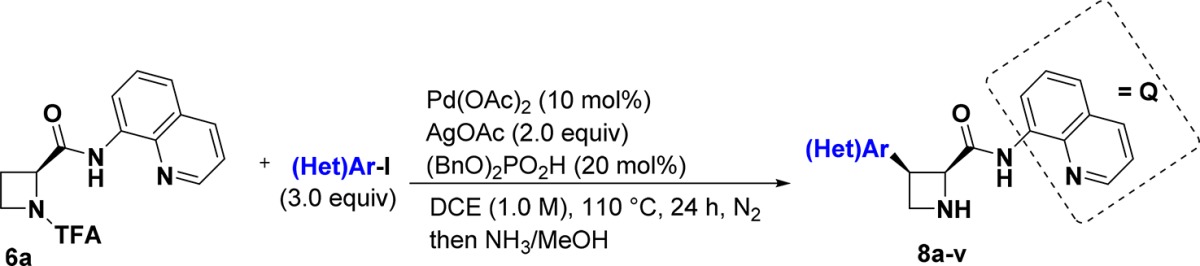
Unfortunately, initial attempts to extend C(sp3)–H arylation reactions to azetidines using known conditions exploiting 8-aminoquinoline as a directing group16a,16b resulted in low yield of desired product (Table S1). Following substantial optimization on a model system (Figure 2b, Table S2–S9), we found that an unconventional N-trifluoroacetamide (N-TFA) protecting group20a and the additive (BnO)2PO2H20b were critical to reaction efficiency. Under the optimized conditions—aryl iodide (3 equiv), Pd(OAc)2 (10 mol %), (BnO)2PO2H (20 mol %), AgOAc (2 equiv), DCE (1.0 M), 110 °C—model product 7a was formed in 72% yield as determined by 1H NMR with internal standard. The reaction exhibited optimal performance under N2 atmosphere, though it afforded product in 52–56% yield under air or O2.
N-TFA is known to undergo hydrolysis readily, releasing the corresponding amine in mild alkaline solutions,21 raising the possibility that the azetidine could be deprotected directly after the arylation. We were delighted to find that upon completion of the C–H arylation, the N-TFA group could be cleaved in the crude reaction mixture by the addition of 7.0 N NH3 in MeOH to afford the free azetidine. The one-pot C–H arylation/deprotection sequence was performed on a range of aryl iodides to demonstrate the broad applicability of this method (Table 1). The reaction tolerated a variety of 3- and 4-substituted aryl iodides (8a–m), including both electron-donating and electron-withdrawing groups. Halogenated aryl-iodide derivatives performed well and provide sites for further diversification. Exceptions include 2-fluoro-iodobenzene and 2-methyliodobenzene, which gave 32% isolated product (8n) and no product (8o), respectively, likely due to steric congestion around a palladacycle intermediate as proposed in previous studies.16a Increased yields were obtained with 4-iodobiphenyl (8p) and 2-iodonaphthalene (8q). Arylation with 2-iodothiophene (8s) proceeded in 80% yield while other heterocyclic iodides were found to react less efficiently (8t–v). Encouragingly, the homologous pyrrolidine (9) and piperidine (11) analogs yielded products in 95% and 72% isolated yields, respectively (Figure 3a), demonstrating the utility of these reaction conditions in the installation of C3 aryl groups on other cyclic amines.
Table 1. Substrate Scope of C–H Arylation Methoda.
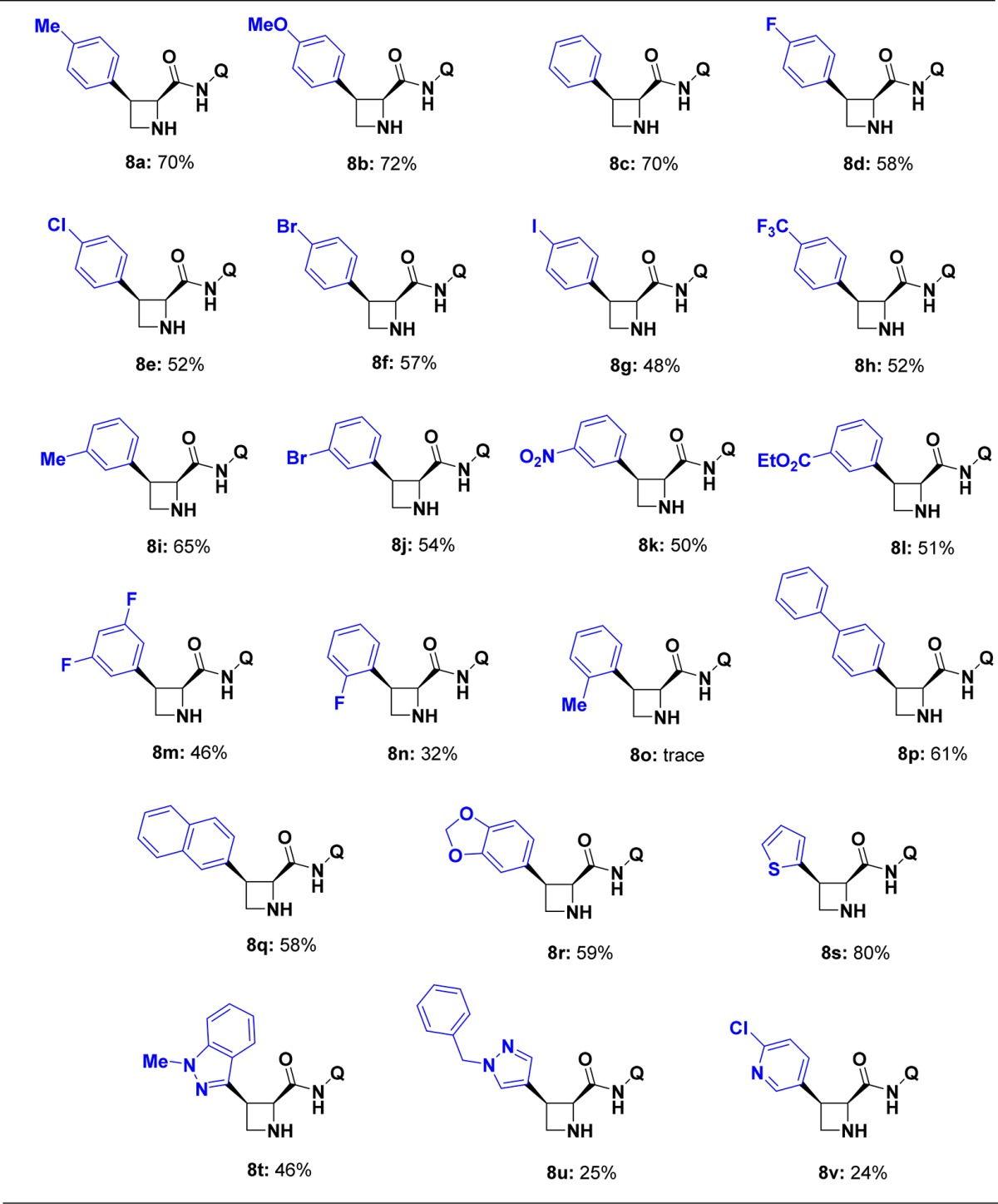
The values beneath each structure indicate the isolated yields after column chromatography.
Figure 3.

(a) C–H arylation method to access homologous series of C–H arylation products. (b) Varying conditions for directing group removal to access selectively cis and trans key building blocks; all compounds are isolated as single stereoisomers with dr > 20:1 and ee > 99%.
Next, we explored reaction conditions to remove the directing group (Figure 3b) to maximize stereochemical diversity. This would enable access to broad, well-defined chemical space in only a few steps from commercial material.22 We chose aryl bromide 8f as the substrate for these diversification studies given the possibility for further elaboration of the aromatic ring. Global Boc installation on (+)-8f (i.e., the azetidine and amide nitrogens) followed by a LiOH/H2O2-mediated cleavage14a,23 of the 8-aminoquinoline auxiliary provided cis-(+)-13 in 69% yield with retention of stereochemistry at the α-position. Alternatively, mono-Boc protection followed by treatment with NaOH/EtOH at 110 °C afforded trans-(−)-14 in 81% yield, thus achieving both removal of the auxiliary and epimerization at the α-carbon. These conditions were then applied to (−)-8f and provided (−)-13 and (+)-14 in 72% and 77% yield, respectively. All four stereoisomers can be accessed from (+)- and (−)-8f without the need for chromatographic purification. Thus, any one of the four stereoisomers could be prepared in three steps with the right choice of substrate (l- or d-azetidine-2-carboxylic acid) and auxiliary removal conditions.
Having established conditions for the C–H arylation and removal of the directing group, we turned our attention to the synthesis of BRD3914. TFA-protection and installation of 8-aminoquinoline on d-azetidine-2-carboxylic acid (+)-3 (10.0 g) afforded 26.3 g of (+)-6a in 82% yield and could be achieved in a single reaction vessel (Figure 4). The use of only 0.95 equiv of DIPEA appeared critical to avoid epimerization at the α-position during the HATU-mediated amide coupling. The key one-pot arylation and deprotection sequence was performed on a 10-g scale with 1-bromo-4-iodobenzene and furnished (−)-8f in 52% yield with both relative and absolute stereochemistry confirmed by X-ray crystallography. Of note, we found that the reaction performed equally well on 0.3, 3.0, and 30.1 mmol scale (100 mg to 10.0 g, Table S10), although incomplete TFA-deprotection under standard NH3 in MeOH conditions was observed on ≥3.0 mmol scale. In these cases, modified conditions using K2CO3 in 9:1 MeOH/H2O completely removed the protecting group. Directing group cleavage from (−)-8f with retention of cis-stereochemistry afforded gram quantities of the synthetically useful carboxylic acid building block (−)-13.
Figure 4.

Reaction conditions: a. Ethyl trifluoroacetate, DIPEA, DMF, 50 °C, 15 h; evaporate volatile materials, then 8-aminoquinoline, HATU, DIPEA, DMF, 0 °C, 2 h (82%); b. Pd(OAc)2 (10 mol %), AgOAc (2.0 equiv), (BnO)2PO2H (20 mol %), 1-bromo-4-iodobenzene (3.0 equiv), DCE, 110 °C, 24 h; evaporate volatile materials, then K2CO3 (3.0 equiv), 9:1 MeOH/H2O (52%); c. Boc2O (3.0 equiv), MeCN, 50 °C, 15 min; then DMAP (0.1 equiv), 2 h; evaporate volatile materials, then H2O2 (10.0 equiv), LiOH (6.0 equiv), THF/MeOH (2:1), 0 °C, then 50 °C, 20 h (72%); d. HCl (4.0 M in dioxane, 15.0 equiv), DCM, 2 h; evaporate volatile materials, then 15 (2.5 equiv), NaBH(OAc)3 (3.0 equiv), DCM, 12 h (71%); e. SiliaBond Piperazine (3.0 equiv), DMF, 50 °C, 2 h; then DIPEA (5.0 equiv), HATU (1.5 equiv), 20 min (89%); f. Ru3(CO)12 (10 mol %), 1,1,3,3-tetramethyldisiloxane (15.0 equiv), toluene, 90 °C, 24 h; then 4-methoxyphenylisocyanate (1.0 equiv), THF, 10 min (57%); g. XPhos Pd G3 (10 mol %), phenylacetylene (5.0 equiv), Et3N (4.0 equiv), MeCN, 70 °C, 2 h (92%).
Next, deprotection of (−)-13 using 4.0 M HCl in dioxane followed by reductive amination with bifunctional linker 15(24) proceeded smoothly in a single flask, delivering (+)-16 in 71% yield. Fmoc was removed using SiliaBond Piperazine, and sequential intramolecular amide bond formation led to lactam (−)-17 (structure confirmed by X-ray crystallography) in 89% isolated yield in a single flask. The lactam reduction proved remarkably difficult as conventional reagents such as LiAlH4 or BH3·Me2S resulted in either competing protodebromination or formation of intractable adducts. Ultimately, we found that modified conditions reported by Reeves and colleagues25 using Ru3(CO)12 and 1,1,3,3-tetramethyldisiloxane (TMDS) in toluene at 90 °C reduced the lactam; the resulting diamine being trapped in situ with 4-methoxyphenyl isocyanate to afford the corresponding urea in 57% yield. Finally, a Pd-catalyzed Heck alkynylation with phenylacetylene delivered BRD3914 in 92% isolated yield. Overall, the synthesis of BRD3914 required seven single-flask transformations (five chromatographic separations) and proceeded in 10% overall yield from commercially available (+)-3, representing a major improvement over our previous work (14 steps).6a
The short synthesis permitted further evaluation of the biological activity of BRD3914. First, the compound was confirmed to be potent in vitro (EC50 = 15 nM) against P. falciparum parasites (Dd2 strain), in line with previously measured activity.6a Pleasingly, BRD3914 exhibited low in vitro cytotoxicity to human cell lines (A549 CC50 = 38 μM, HEK293 CC50 > 50 μM, HepG2 CC50 > 50 μM). We then sought to evaluate the in vivo efficacy of this compound for the first time against blood-stage P. falciparum 3D7HLH/BRD (expressing firefly luciferase) in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) Il2rγ–/– mice engrafted with human erythrocytes (huRBC NSG). This mouse model has been shown to correlate well with human malaria challenge models.4,26 huRBC NSG mice were inoculated with parasites (−48 h) before administration with either 25 mg/kg or 50 mg/kg BRD3914 (single dose at 0 h, or multiple dosing at 0, 24, 48, and 72 h, formulated in 70% PEG300/30% solution of 5% dextrose/H2O)5 and monitored for 30 days (Figures 5, S2). Chloroquine was used as a positive control. After a single 50 mg/kg dose of BRD3914, a reduction in parasite-associated bioluminescence was observed in mice, but recrudescence occurred around day three. Gratifyingly, huRBC NSG mice treated with four q.d. (quaque die) doses27 (25 mg/kg or 50 mg/kg) of BRD3914 were parasite-free after 30 days based on bioluminescent imaging. These results are striking compared to chloroquine treatment (4 × 25 mg/kg), where recrudescence of infection was observed in one of two mice.
Figure 5.
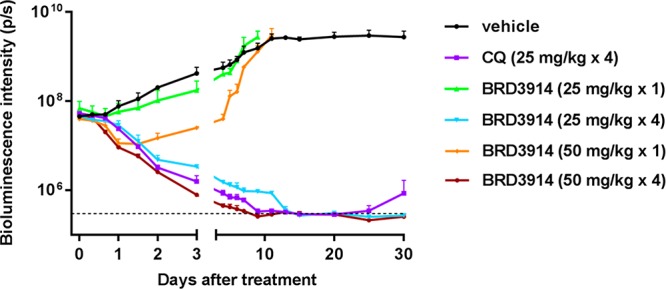
huRBC NSG mice were inoculated with P. falciparum (3D7HLH/BRD) blood-stage parasites 48 h before treatment, and BRD3914 was administered as a single dose (25 mg/kg or 50 mg/kg) at 0 h, or multiple dosing at (25 mg/kg or 50 mg/kg) at 0, 24, 48, and 72 h (n = 2 for each group, this study was conducted once). Chloroquine (CQ) was used as a positive control. Infections were monitored using the in vivo imaging system (IVIS). Bioluminescent intensity was quantified from each mouse and plotted against time. The dotted horizontal line represents the mean bioluminescence intensity level obtained from all the animals before the parasite inoculation.
While BRD3914 has a dosing shortcoming in vivo when compared to BRD7929, overall the compound succeeds in curing mice under a traditional Peters’ test,27 retains in vitro activity against P. falciparum parasites, and, most importantly, exhibits a more attractive safety profile.5 In our in vivo studies, BRD3914 outperformed the antimalarial chloroquine, one of the most successful antimalarial agents to date.28
The biological studies presented herein were facilitated by the development of a Pd-catalyzed C(sp3)–H arylation strategy, which allowed the efficient preparation of BRD3914 in seven steps with five chromatographic separations. This route represents a significant improvement over previous syntheses of this class of inhibitors with respect to step count, chromatographic separations, and waste minimization.5−7 Notable features of the synthetic approach are the stereospecificity of the arylation and the controlled cleavage of the directing group, which affords any one of four C3-arylated azetidine carboxylic acids with judicious choice of substrate and reaction conditions. The C–H arylation was performed on up to a 10-g scale without a significant decrease in yield and provides a general strategy to C3-arylated azetidines, pyrrolidines, and piperidines. Given the substrate scope and the ability to explore stereochemical space, we envision that this method can be applied to the synthesis of other promising azetidine-containing antimalarials29 and antimicrobials30 in addition to finding general use for the preparation of diverse fragments for chemical libraries22 or peptidomimetics.9
Conclusions
In summary, we developed a Pd-catalyzed, directed C(sp3)–H arylation of azetidines, which exhibited a broad substrate scope and permitted the preparation of stereochemically defined, synthetically useful carboxylic acid building blocks. We used this method in the short synthesis of antimalarial compound BRD3914, which cured P. falciparum infection in a mouse model after four oral doses. Collectively, our results arise from interplay between chemical methodology, stereochemically oriented diversification, synthesis of a structurally complex scaffold, and biological studies.
Acknowledgments
We thank the Broad Institute analytical team for high-resolution mass spectrometry data, Tom Clinckemaillie (Broad Institute) for assistance with SFC, Drs. Peter Müller and Jonathan Becker (Massachusetts Institute of Technology) for X-ray crystallographic structural analysis, and Drs. Michael F. Mesleh and Jenna B. Yehl (Broad Institute) for assistance with NMR. This work was supported by the Bill and Melinda Gates Foundation (grant OPP1032518). S.L.S. is an investigator at the Howard Hughes Medical Institute. M.M. was supported by a fellowship from the National Science Foundation (DGE1144152) and from Harvard University’s Graduate Prize Fellowship. J.Z. was supported by a postdoctoral fellowship from the German Academic Exchange Service (DAAD). B.M. was supported by a postdoctoral fellowship from Harvard University. O.V. was supported by a postdoctoral fellowship from the Wenner-Gren Foundations.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06994.
Author Contributions
∥ M.M., J.Z., B.M., and O.V. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. World Malaria Report 2016. http://www.who.int/malaria/publications/world_malaria_report_2016/en/, accessed April 1, 2017.
- Wells T. N. C.; van Huijsduijnen R. H.; Van Voorhis W. C. Nat. Rev. Drug Discovery 2015, 14, 424–442. 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Ménard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Ménard D. Nature 2014, 505, 50–55. 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; van Huijsduijnen R. H.; Möhrle J. J.; Oeuvray C.; Wells T. N. C. Malar. J. 2013, 12, 187. 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Comer E.; Sakata-Kato T.; Sharma A.; Sharma M.; Maetani M.; Bastien J.; Brancucci N. M.; Bittker J. A.; Corey J.; Clarke D.; Derbyshire E.; Dornan G.; Duffy S.; Eckley S.; Itoe M. A.; Koolen K. M. J.; Lewis T.; Lui P. S.; Lukens A. K.; Lund E.; March S.; Meibalan E.; McPhail J.; Meier B.; Mitasev B.; Moss E.; Sayes M.; VanGessel Y.; Wawer M.; Yoshinaga T.; Zeeman A.-M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K.; Duvall J. R.; Foley M. A.; Gusovsky F.; Kocken C. H.; Marti M.; Morningstar M.; Munoz B.; Neafsey D.; Sharma A.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L. Nature 2016, 538, 344–349. 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Comer E.; Kato N.; Scherer C.; Bastien J.; Duvall J.; Lewis T.; Sayes M.; Leighty M.; Pu J.; Beaudoin J.; Braibant B.; Munoz B. U.S. Patent Application NO. 15/035,044. US20160289235A1 (2014).; b Wang H. Y.; Huang K.; De Jesús M.; Espinosa S.; Piñero-Santiago L. E.; Barnes C. L.; Ortiz-Marciales M. Tetrahedron: Asymmetry 2016, 27, 91–100. 10.1016/j.tetasy.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger F.; Chase C. E.; Endo A.; Fang F. G.; Li J.; Mathieu S. R.; Wilcoxen A. Z.; Zhang H. Angew. Chem., Int. Ed. 2015, 54, 5108–5111. 10.1002/anie.201501143. [DOI] [PubMed] [Google Scholar]
- Lowe J. T.; Lee M. D. 4th; Akella L. B.; Davoine E.; Donckele E. J.; Durak L.; Duvall J. R.; Gerard B.; Holson E. B.; Joliton A.; Kesavan S.; Lemercier B. C.; Liu H.; Marié J. C.; Mulrooney C. A.; Muncipinto G.; Welzel-O’Shea M.; Panko L. M.; Rowley A.; Suh B. C.; Thomas M.; Wagner F. F.; Wei J.; Foley M. A.; Marcaurelle L. A. J. Org. Chem. 2012, 77, 7187–7211. 10.1021/jo300974j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brandi A.; Cicchi S.; Cordero F. M. Chem. Rev. 2008, 108, 3988–4035. 10.1021/cr800325e. [DOI] [PubMed] [Google Scholar]; b Couty F.; Evano G. Synlett 2009, 2009, 3053–3064. 10.1055/s-0029-1218299. [DOI] [Google Scholar]
- a Zaitsev V. G.; Shabashov D.; Daugulis O. J. Am. Chem. Soc. 2005, 127, 13154. 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; b Shabashov D.; Daugulis O. J. Am. Chem. Soc. 2010, 132, 3965–3972. 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davies H. M.; Morton D. J. Org. Chem. 2016, 81, 343–350. 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]; b Chen X.; Engle K. M.; Wang D.-H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rit R. K.; Yandav M. R.; Ghosh K.; Sahoo A. K. Tetrahedron 2015, 71, 4450–4459. 10.1016/j.tet.2015.03.085. [DOI] [Google Scholar]; d Jazzar R.; Hitce J.; Renaudat A.; Sofack-Kreutzer J.; Baudoin O. Chem. - Eur. J. 2010, 16, 2654–2679. 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]
- a Zhang S.-Y.; Li Q.; He G.; Nack W. A.; Chen G. J. Am. Chem. Soc. 2013, 135, 12135–12141. 10.1021/ja406484v. [DOI] [PubMed] [Google Scholar]; b Wasa M.; Engle K. M.; Lin D. W.; Yoo E. J.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 19598–19601. 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chan K. S.; Fu H.-Y.; Yu J.-Q. J. Am. Chem. Soc. 2015, 137, 2042–2046. 10.1021/ja512529e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wasa M.; Engle K. M.; Yu J.-Q. J. Am. Chem. Soc. 2010, 132, 3680–3681. 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen G.; Gong W.; Zhuang Z.; Andrä M. S.; Chen Y. Q.; Hong X.; Yang Y. F.; Liu T.; Houk K. N.; Yu J.-Q. Science 2016, 353, 1023–1027. 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang F.-L.; Hong K.; Li T.-J.; Park H.; Yu J.-Q. Science 2016, 351, 252–256. 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]; c He J.; Shao Q.; Wu Q.; Yu J.-Q. J. Am. Chem. Soc. 2017, 139, 3344–3347. 10.1021/jacs.6b13389. [DOI] [PubMed] [Google Scholar]
- a Gutekunst W. R.; Baran P. S. J. Am. Chem. Soc. 2011, 133, 19076–19079. 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gutekunst W. R.; Gianatassio R.; Baran P. S. Angew. Chem., Int. Ed. 2012, 51, 7507–7510. 10.1002/anie.201203897. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gutekunst W. R.; Baran P. S. J. Org. Chem. 2014, 79, 2430–2452. 10.1021/jo4027148. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chapman L. M.; Beck J. C.; Wu L.; Reisman S. E. J. Am. Chem. Soc. 2016, 138, 9803–9806. 10.1021/jacs.6b07229. [DOI] [PubMed] [Google Scholar]; e Xiao K. J.; Lin D. W.; Miura M.; Zhu R. Y.; Gong W.; Wasa M.; Yu J.-Q. J. Am. Chem. Soc. 2014, 136, 8138–8142. 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhou M.; Li X. R.; Tang J. W.; Liu Y.; Li X. N.; Wu B.; Qin H. B.; Du X.; Li L. M.; Wang W. G.; Pu J. X.; Sun H. D. Org. Lett. 2015, 17, 6062–6065. 10.1021/acs.orglett.5b03079. [DOI] [PubMed] [Google Scholar]
- a Nack W. A.; Wang B.; Wu X.; Jiao R.; He G.; Chen G. Org. Chem. Front. 2016, 3, 561–564. 10.1039/C5QO00421G. [DOI] [Google Scholar]; b Shan G.; Yang X.; Rao Y. Angew. Chem., Int. Ed. 2013, 52, 13606–13610. 10.1002/anie.201307090. [DOI] [PubMed] [Google Scholar]
- a Affron D. P.; Davis O. A.; Bull J. A. Org. Lett. 2014, 16, 4956–4959. 10.1021/ol502511g. [DOI] [PubMed] [Google Scholar]; b Reng R.; Wang B.; Liu Y.; Liu Z.; Zhang Y. Eur. J. Org. Chem. 2015, 2015, 142–151. 10.1002/ejoc.201403191. [DOI] [Google Scholar]; c Spangler J. E.; Kobayashi Y.; Verma P.; Wang D.-H.; Yu J.-Q. J. Am. Chem. Soc. 2015, 137, 11876–11879. 10.1021/jacs.5b06740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Affron D. P.; Bull J. A. Eur. J. Org. Chem. 2016, 2016, 139–149. 10.1002/ejoc.201501300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yu Q.-Y.; Zhong H.-M.; Sun W.-W.; Zhang S.-J.; Dong X.-P.; Qin H.-B.; Liu J.-K.; Wu B. Asian J. Org. Chem. 2016, 5, 608–612. 10.1002/ajoc.201600072. [DOI] [Google Scholar]; c Ye S.; Yang W.; Coon T.; Fanning D.; Neubert T.; Stamos D.; Yu J.-Q. Chem. - Eur. J. 2016, 22, 4748–4752. 10.1002/chem.201600191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P.; Verma P.; Xia G.; Yu J.-Q. Nat. Chem. 2017, 9, 140–144. 10.1038/nchem.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a He G.; Zhao Y.; Zhang S.; Lu C.; Chen G. J. Am. Chem. Soc. 2012, 134, 3–6. 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]; b He G.; Lu G.; Guo Z.; Liu P.; Chen G. Nat. Chem. 2016, 8, 1131–1136. 10.1038/nchem.2585. [DOI] [Google Scholar]
- a A similar observation was made by:Watanabe T.; Oishi S.; Fujii N.; Ohno H. Org. Lett. 2008, 10, 1759–1762. 10.1021/ol800425z. [DOI] [PubMed] [Google Scholar]; b (BnO)2PO2H was first reported by:Zhang S.-Y.; He G.; Nack W. A.; Zhao Y.; Li Q.; Chen G. J. Am. Chem. Soc. 2013, 135, 2124–2127. 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]
- Hughes A. B.Amino Acids, Peptides and Proteins in Organic Chemistry, 1st ed.; Wiley-VCH: Weinheim, Germany, 2011; Vol. 4, Protection Reactions. [Google Scholar]
- Burke M. D.; Schreiber S. L. Angew. Chem., Int. Ed. 2004, 43, 46–58. 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Carter P. H.; Dinsmore C. J.; Barrow J. C.; Katz J. L.; Kung D. W. Tetrahedron Lett. 1997, 38, 4535–4538. 10.1016/S0040-4039(97)00972-6. [DOI] [Google Scholar]
- a Kong Too Lin P.; Kuksa V. A.; Maguire N. M. Synthesis 1998, 6, 859–866. 10.1055/s-1998-2073. [DOI] [Google Scholar]; b Matsumori N.; Sawada Y.; Murata M. J. Am. Chem. Soc. 2005, 127, 10667–10675. 10.1021/ja051597r. [DOI] [PubMed] [Google Scholar]
- Reeves J. T.; Tan Z.; Marsini M. A.; Han Z. S.; Xu Y.; Reeves D. C.; Lee H.; Lu B. Z.; Senanayake C. H. Adv. Synth. Catal. 2013, 355, 47–52. 10.1002/adsc.201200835. [DOI] [Google Scholar]
- Burrows J. N.; Duparc S.; Gutteridge W. E.; Hooft van Huijsduijnen R.; Kaszubska W.; Macintyre F.; Mazzuri S.; Möhrle J. J.; Wells T. N. C. Malar. J. 2017, 16, 26. 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters W.Chemotherapy and Drug Resistance in Malaria, 2nd ed.; Academic Press: London, 1987. [Google Scholar]
- Jensen M.; Mehlhorn H. Parasitol. Res. 2009, 105, 609–627. 10.1007/s00436-009-1524-8. [DOI] [PubMed] [Google Scholar]
- a Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A. M.; Kocken C. H.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. ACS Infect. Dis. 2016, 2, 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Maetani M.; Kato N.; Jabor V. A. P.; Calil F. A.; Nonato M. C.; Scherer C. A.; Schreiber S. L. ACS Med. Chem. Lett. 2017, 8, 438–442. 10.1021/acsmedchemlett.7b00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington S.; Nag P. P.; Michalska K.; Johnston S. E.; Jedrzejczak R. P.; Kaushik V. K.; Clatworthy A. E.; Siddiqi N.; McCarren P.; Bajrami B.; Maltseva N. I.; Combs S.; Fisher S. L.; Joachimiak A. J.; Schreiber S. L.; Hung D. T. Nat. Chem. Biol. 2017, 10.1038/nchembio.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.