

Abstract
C/EBP homologous protein (CHOP) is a stress-inducible nuclear protein that is crucial for the development of programmed cell death and regeneration; however, the regulation of its function has not been well characterized. Slbo, a Drosophila homolog of C/EBP (CCAAT/enhancer binding protein), was shown to be unstabilized by tribbles. Here, we identified TRB3 as a tribbles ortholog in humans, which associated with CHOP to suppress the CHOP-dependent transactivation. TRB3 is induced by various forms endoplasmic reticulum (ER) stress later than CHOP. Tunicamycin treatment enhanced the TRB3 promoter activity, while dominant-negative forms of CHOP suppressed the tunicamycin-induced activation. In addition, the tunicamycin response region in the TRB3 promoter contains amino-acid response elements overlapping the CHOP-binding site, and CHOP and ATF4 cooperated to activate this promoter activity. Knockdown of endogenous ATF4 or CHOP expression dramatically repressed tunicamycin-induced TRB3 induction. Furthermore, knockdown of TRB3 expression decreased ER stress-dependent cell death. These results indicate that TRB3 is a novel target of CHOP/ATF4 and downregulates its own induction by repression of CHOP/ATF4 functions, and that it is involved in CHOP-dependent cell death during ER stress.
Keywords: apoptosis, ATF4, CHOP, ER stress, TRB3
Introduction
CCAAT/enhancer binding proteins (C/EBPs) are a family of leucine zipper transcription factors that are critical for the regulation of various aspects of cellular differentiation and function in multiple tissues. This family consists of six members: C/EBPα, β (NF-IL6), γ (Ig/EBP), δ, ɛ and C/EBP homologous protein (CHOP)/growth arrest-DNA damage inducible 153 (GADD153) (Lekstrom-Himes and Xanthopoulos, 1998). The prototypic C/EBP consists of a transcriptional activation domain and a bZIP region for DNA binding and dimerization. All family members share strong homology in the carboxyl-terminal domain, which carries a basic DNA-binding domain and a leucine zipper motif.
CHOP was originally isolated as the gene induced in response to DNA-damaging agents; subsequently, it has been revealed that CHOP is induced by extracellular and endoplasmic reticulum (ER) stress (Fornace et al, 1989; Zinszner et al, 1998). From experiments on the overexpression of its protein and knockout mice, CHOP has been shown to act as an inducer of cell cycle arrest and apoptosis during ER stress (Barone et al, 1994; Matsumoto et al, 1996; Zinszner et al, 1998). At first, because the basic region (BR) of CHOP is less conserved than that of other C/EBP family proteins, CHOP was thought to lack DNA-binding activity. Actually, by forming heterodimers with other C/EBP proteins such as NF-IL6, CHOP inhibits their ability to bind DNA and their transcriptional activity (Ron and Habener, 1992). Interestingly, however, a CHOP-C/EBP heterodimer has been reported to bind to a unique DNA sequence different from classical C/EBP-binding sites and to act as a positive transactivator (Ubeda et al, 1996). In recent studies, several CHOP-inducible genes have been induced during ER stress via this CHOP-binding sequence (Wang et al, 1998).
ER stress responses are alterations in homeostasis following cellular stress, which prevent protein folding and cause misfolding or malfolding proteins to accumulate in the ER (Kaufman, 1999; Mori, 2000). Under such conditions, the homeostasis of protein folding in the ER is maintained by inter-organelle signaling from the ER to the nucleus, a process known as unfolded protein response (UPR). Thus, from yeast to humans, the transcription of genes encoding molecular chaperones and folding enzymes in the ER is induced in the nucleus in response to unfolding in the ER, and excessive or long-term accumulations of unfolding proteins in the ER result in the apoptosis of cells.
A large number of transcription factors undergo degradation via a ubiquitin–proteasome-dependent pathway (Hochstrasser, 1995; Pahl and Baeuerle, 1996). A genetic study on Drosophila revealed that Slbo, a Drosophila homolog of C/EBP, is specifically degraded by the ubiquitin–proteasome pathway (Rorth et al, 2000). As the degradation of Slbo is augmented by expression of tribbles, tribbles may be involved in the ubiquitin–proteasome pathway. In humans, we have previously reported that C/EBP family transcription factors, CHOP and Ig/EBP (C/EBPγ), are multiubiquitinated and subsequently degraded by proteasomes, but the molecular mechanism involved is still unclear (Hattori et al, 2003a). In an effort to clarify the mechanism by which C/EBP is degraded, we searched a database for the tribbles ortholog in humans, and identified TRB3 (tribbles-related protein 3), its gene having been cloned from HepG2 cells with function unknown. In this study, TRB3 was revealed to be a novel target of CHOP/ATF4 and downregulates its own induction by repression of CHOP/ATF4 functions, and also seemed to be involved in CHOP-dependent cell death as a second messenger during ER stress.
Results
Identification of a mammalian homolog of tribbles
To clarify the regulation of C/EBP degradation, we searched the GenBank database to determine the human ortholog of Drosophila tribbles. This search identified several mammalian genes displaying high levels of identity to tribbles and we determined the sequence of a full-length cDNA clone, a gene of unknown function that had been cloned from the human hepatoma cell line HepG2 (GenBank accession #AK026945). It was identical to human SKIP3 (GenBank accession #AK0250311), and very recently, Du et al (2003) identified its product (termed TRB3, tribbles-related protein 3) as a novel Akt-binding and -regulating protein (Du et al, 2003). A search of the GenBank database with the full coding sequence revealed that TRB3 is related to TRB1 or TRB2.
ER stress induces TRB3 expression
First, we analyzed the expression level of TRB3 mRNA by RT–PCR and Northern blotting. TRB3 mRNA was not expressed in steady-state 293 cells, human embryonic kidney cells, but was induced during ER stress by treatment with tunicamycin for 4–6 h (Figure 1A). In HepG2 cells, TRB3 mRNA was expressed in normal conditions and its expression was augmented 6 h after treatment (Figure 1B). TRB3 induction was also observed in tunicamycin-treated A375, HeLa and SH-SY5Y cells (see Supplementary Figure S1). The induction of TRB3 mRNA was late compared to that of CHOP mRNA. In addition, cycloheximide treatment blocked the expression of TRB3 mRNA, indicating that its induction required de novo protein synthesis (Figure 1A, lane 7). TRB3 mRNA was also induced by treatment with other ER stress inducers, such as MG132, a proteasome inhibitor, methanesulfonic acid methyl ester (MMS), a DNA alkylating agent and A23187, an ER Ca2+-ATPase inhibitor (Figure 1C). As shown in Figure 1D, this induction of TRB3 was observed at the protein level as well. In HepG2 cells, a small amount of TRB3 protein was detected, and its expression was upregulated by tunicamycin 8 h after treatment. TRB3 protein was not detected in steady-state 293 and A375 cells, but was detected 4–6 h after tunicamycin treatment (Figure 1D). At the protein level as well, CHOP induction usually preceded TRB3 induction.
Figure 1.
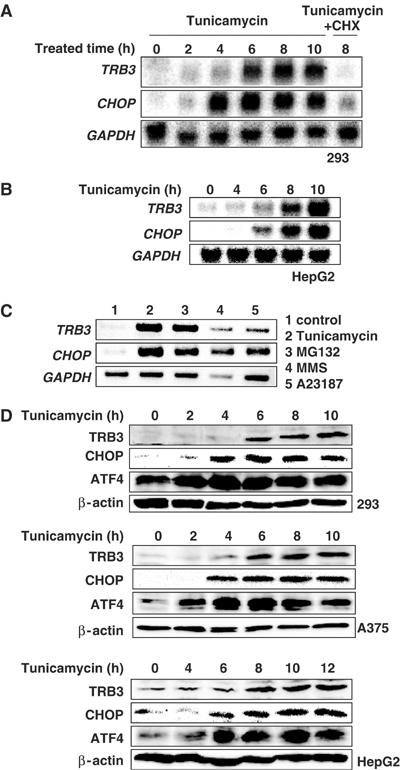
TRB3 is induced during ER stress. (A, B) 293 (A) or HepG2 cells (B) were treated with 2 μg/ml of tunicamycin in the presence or absence of 10 μg/ml of cycloheximide (CHX) for the indicated periods. Total RNA was prepared and analyzed by Northern blotting using each specific probe. (C) 293 cells were treated with 2 μg/ml of tunicamycin, 10 μM MG132, 100 μg/ml of MMS or 2 μM A23187 for 6 h. Each mRNA level in the cells was analyzed by RT–PCR using specific primers. TRB3, 32 cycles ; CHOP, 30 cycles ; GAPDH, 24 cycles. (D) 293 (top), A375 (middle) and HepG2 (bottom) cells were treated with 2 μg/ml of tunicamycin for the indicated periods. Cell lysate was prepared and equal amounts of protein were subjected to SDS–PAGE. Western blotting was performed with anti-human TRB3, anti-CHOP, anti-ATF4 or anti-β-actin antibodies.
TRB3 interacts with CHOP but does not promote CHOP degradation
In Drosophila, Slbo was reported to interact with tribbles to inhibit its function via protein degradation (Rorth et al, 2000). We first examined the physical interaction between TRB3 and CHOP. CHOP was co-immunoprecipitated with TRB3 in transfected 293 cells (Figure 2B, lane 4). To identify the domain of CHOP required for its interaction with TRB3, we analyzed the ability of several CHOP deletion mutants as shown in Figure 2A to co-immunoprecipitate with TRB3. CHOP interaction with TRB3 does not require either the BR or potential phosphorylation sites (Ser79,82), which are critical for DNA-binding and transactivation activity, respectively. Leucine zipper (LZ) domain, responsible for dimer formation, was not necessary for the binding with TRB3 either (see Supplementary Figure S2A). Full-length TRB3 strongly interacted with CHOP, CHOPΔN9 (amino acids (aa) 10–169) and CHOPΔ19–26 (aa 1–18, 27–169); in contrast, further truncation in the N-terminus of CHOP (CHOPΔN18 (aa 19–169) and CHOPΔN70 (aa 71–169)) abolished the interaction with TRB3 (Figure 2B, lane 5; see Supplementary Figure S2B). These results indicate that the region between aa 10 and 18 of CHOP is responsible for the interaction with TRB3 (Figure 2A). Next, to identify the domain of TRB3 required for its interaction with CHOP, we analyzed the possibility of CHOP co-immunoprecipitating with several TRB3 deletion mutants as shown in Figure 2C. TRB3ΔC (aa 1–282), TRB3Δ239–265 (aa 1–238, 266–358) and TRB3N179 (aa 1–179) interacted with CHOP as well as full-length TRB3, but TRB3ΔN (aa 128–358) and TRB3C179 (aa 180–358) have no or only faint interaction with CHOP (Figure 2D). These results indicate that the region between aa 1 and 127 of TRB3 is responsible for the interaction with CHOP (Figure 2C).
Figure 2.
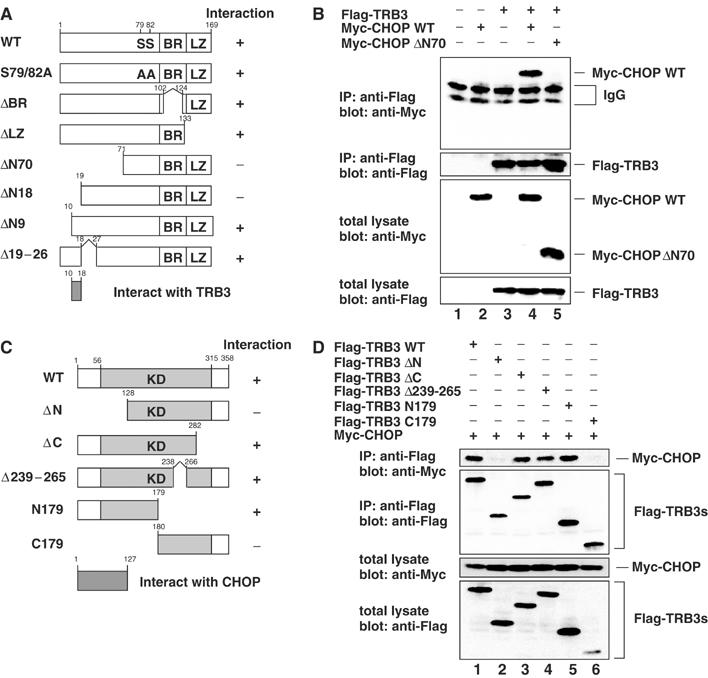
TRB3 interacts with CHOP in vivo. (A) Constructs of CHOP mutants. (B, D) 293 cells were transiently transfected with the indicated constructs. After 36 h, cells were treated with 10 μM MG132 for 12 h. The cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-Myc antibody. The expression level of each protein was assessed by the immunoblotting of cell lysates with anti-Flag or anti-Myc antibodies. (C) Deletion mutants of TRB3. KD: kinase-like domain.
Next, we examined whether TRB3 promotes the protein degradation of CHOP. Unexpectedly, the CHOP expression level and degradation rate were unchanged by coexpression with TRB3 or knockdown of endogenous TRB3 by expression of TRB3 siRNA (see Supplementary Figure S3). These results suggest that TRB3 interacts with CHOP; however, it does not promote degradation of CHOP.
TRB3 downregulates CHOP-dependent transcriptional activity
It has been shown that several CHOP-inducible genes have been induced during ER stress or mitochondrial stress via novel CHOP-binding sequence (Wang et al, 1998; Zhao et al, 2002) (Figure 3A). The region between aa 10 and 18 in CHOP is critical for the binding with TRB3 and also crucial for CHOP transcriptional activity (Figure 3B). Therefore, to explore the possibility that TRB3 affects the transcriptional activity of CHOP, we constructed a luciferase reporter gene containing four tandem repeats of the CHOP-binding site and performed a luciferase reporter assay. When CHOP alone or a low level of NF-IL6 alone was transiently expressed in A375 cells, the transcriptional activity was unaffected (Figure 3C). However, this activity was upregulated on coexpression of NF-IL6 and CHOP. Overexpression of TRB3 repressed the transcriptional activity stimulated by CHOP/NF-IL6 in a dose-dependent manner (Figure 3D). In addition, a high level of NF-IL6 expression alone also stimulated the transcriptional activity for these sites; however, TRB3 failed to suppress this activation (Figure 3C). These results suggest that TRB3 selectively inhibits the transcriptional activity of CHOP.
Figure 3.
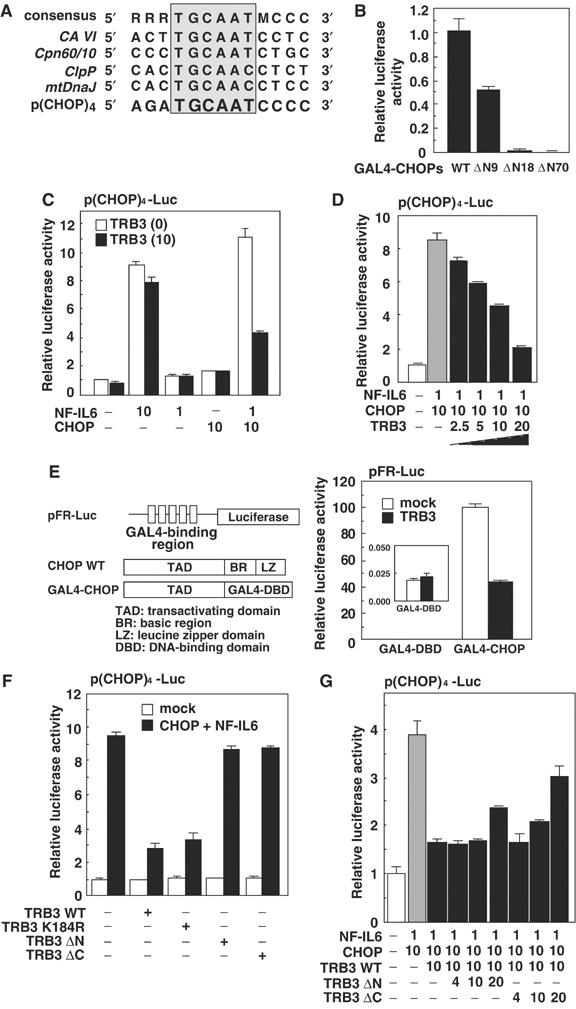
TRB3 represses CHOP transactivation. (A) Alignment of CHOP-responsive element in various genes. (B) 293 cells were transiently transfected with the expression plasmids for wild-type or deletion mutants of CHOP fused with GAL4-dbd, pCMV5-Gal4-CHOPs, pFR-luc and pCMV-β-gal. After 48 h, the luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (C, D) A375 cells were transiently transfected with p(CHOP)4-Luc, pCMV-β-gal and various combinations of the expression vectors for CHOP and NF-IL6 with or without TRB3. After 48 h, the luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (E) 293 cells were transiently transfected with the expression plasmids for Gal4-CHOP(WT) and/or TRB3, pFR-luc and pCMV-β-gal. After 48 h, the luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. The inset depicts the enlarged figure for the data of GAL4-dbd. (F, G) A375 cells were transiently transfected with p(CHOP)4-Luc, pCMV-β-gal and various combinations of the expression vectors for CHOP and NF-IL6 with or without wild type or deletion mutants of TRB3. After 48 h, the luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. Similar results were obtained in three independent experiments.
TRB3 interacted with CHOP via its transactivation domain (Figures 2A and 3B) but not DNA-binding or leucine zipper domains, and inhibited its transactivation activity. Consistent with the function of TRB3-binding region on CHOP, TRB3 did not interfere with the dimerization of CHOP with NF-IL6 nor with DNA binding of CHOP (see Supplementary Figure S4). In addition, TRB3 repressed even the transactivation activity of GAL4 fusion protein of CHOP (Figure 3E). These results suggest that TRB3 primarily inhibits CHOP transactivation, probably by inhibiting the modification of CHOP required for its transactivation or by interfering with the association of coactivator(s) or recruiting corepressor(s) to DNA.
To investigate the functional domain of TRB3 inhibiting the transactivating activity of CHOP, we examined the effect of several deletion and point mutants of TRB3 on this activity. Deletion of the N-terminal (Δ1–127, TRB3ΔN) or C-terminal region of TRB3 (Δ283–358, TRB3ΔC) abolished the inhibitory effect on the CHOP-dependent transactivation (Figure 3F). These results suggest that the functional domain of TRB3 for this inhibitory action exists in both the N-terminal region, necessary for the association with CHOP, and the C-terminal region. In addition, TRB3ΔC mutants strongly, and TRB3ΔN mutants slightly, interfered with CHOP-dependent transactivation in a dominant-negative manner (Figure 3G). Substitution of Lys184, a potential catalytic core in the kinase-like domain, with Arg (TRB3 K184R) resulted in almost the same inhibitory activity as wild-type TRB3, indicating that this region is not critical for the inhibitory action (Figure 3F).
CHOP overexpression activates TRB3 promoter activity
To study whether TRB3 mRNA expression induced by ER stress is regulated at the transcriptional level, we cloned the promoter region of human TRB3 (−1265 to +609), and constructed a luciferase reporter plasmid (pTRB3-Luc). As shown in Figure 4A, transient transfection experiments in 293 cells using this reporter gene confirmed that ER stress induced by tunicamycin caused the promoter activation. This activation was also observed in tunicamycin-treated HepG2 cells, and other ER stressors, thapsigargin and A23187, also stimulated the promoter (see Supplementary Figure S5). These results suggest that TRB3 expression is induced at the transcriptional level during ER stress.
Figure 4.
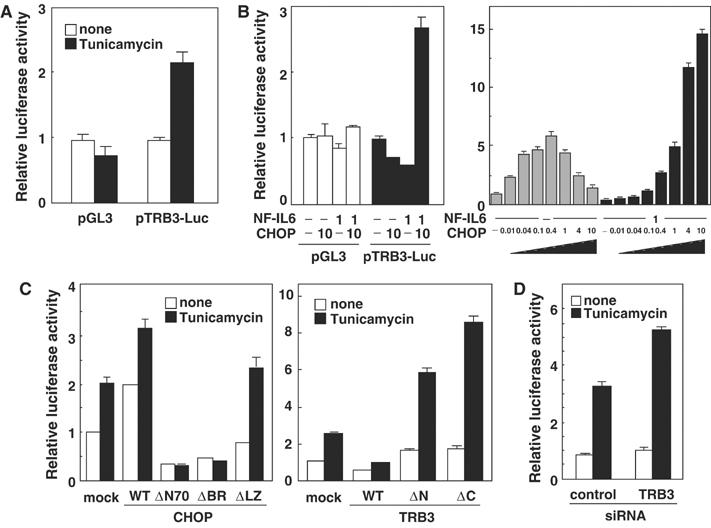
Overexpression of CHOP causes TRB3 induction. (A) 293 cells were transiently transfected with pTRB3-Luc and pCMV-β-gal. After 24 h, cells were left untreated or treated with 2 μg/ml of tunicamycin for 16 h. The luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (B) 293 cells were transiently transfected with pTRB3-Luc and pCMV-β-gal combination with expression vectors for CHOP and/or NF-IL6 for 48 h. The luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (C) 293 cells were transiently transfected with pTRB3-Luc and pCMV-β-gal in the presence of expression vectors for wild type or mutants of CHOP or TRB3. After 24 h, cells were left untreated or treated with 2 μg/ml of tunicamycin for 16 h. The luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (D) 293 cells were transiently transfected with pTRB3-Luc, pCMV-β-gal, control siRNA and/or TRB3 siRNA. After 36 h, cells were left untreated or treated with 2 μg/ml of tunicamycin for 16 h. The luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. Similar results were obtained in three independent experiments.
The expression of TRB3 mRNA during ER stress was blocked by cycloheximide treatment, and the induction of TRB3 was late compared to that of CHOP (Figure 1). In addition, TRB3 interacts with CHOP to downregulate its transactivation activity (Figures 2 and 3). We next investigated the effect of CHOP overexpression on TRB3 promoter activity, to explore the possibility that TRB3 expression is induced via CHOP expression. By cotransfection with CHOP and NF-IL6 expression plasmids, TRB3 promoter activity was enhanced (Figure 4B, left). This result was confirmed by Northern blotting experiments using cells transiently transfected with CHOP and NF-IL6 (see Supplementary Figure S6). In addition, a low to middle level of CHOP expression alone effectively activated TRB3 promoter activity; however, a high level of expression failed to activate it (Figure 4B, right). On the other hand, a high level of CHOP was able to activate TRB3 promoter activity when coexpressed with NF-IL6 (Figure 4B, right). These results suggest that ectopically expressed CHOP can activate TRB3 promoter activity via heterodimerization with endogenous proteins such as NF-IL6, but overexpressed CHOP cannot, due to a shortage of dimerization partners.
Overexpression of dominant-negative forms of CHOP, CHOPΔN70 or CHOPΔBR diminished the tunicamycin-induced TRB3 promoter activation, while that of CHOPΔLZ, which has no transactivation activity or a dominant-negative effect, did not affect or only slightly potentiated the activity (Figure 4C, left). Ectopic expression of TRB3 caused an inhibition of transcription, while dominant-negative forms of TRB3 (TRB3ΔN and TRB3ΔC) and TRB3 siRNA upregulated the tunicamycin-stimulated TRB3 expression (Figure 4C and D). These results strongly suggest that TRB3 is induced by ER stress via CHOP expression and suppresses CHOP transactivational activity via negative feedback.
Identification of ER stress response elements in TRB3 promoter
To clarify the regulation of TRB3 induction via CHOP expression, we identified the ER stress response region in the TRB3 promoter using luciferase reporter plasmids of its promoter in various lengths as shown in Figure 5A. The reporter genes containing the region +201 to +312 (pTRB3-Luc1, 2, 5, 6), but not the others, were activated by tunicamycin (Figure 5B, left). This result revealed that the tunicamycin-induced proteins activate the TRB3 promoter via this region.
Figure 5.
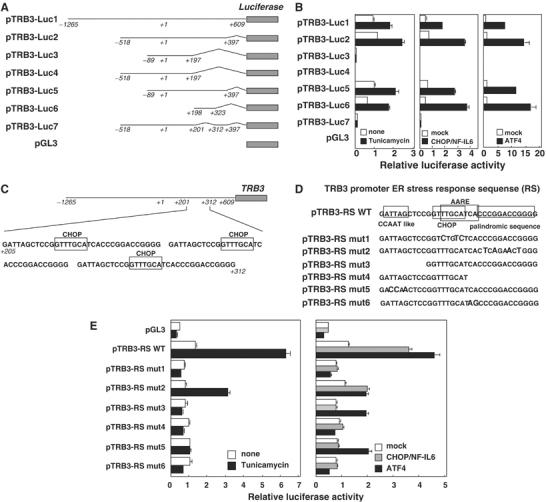
Mutagenesis analysis of the TRB3 promoter. (A) Constructs of TRB3 promoter reporter genes. Position +1 demonstrates the initiation site for the TRB3 transcription. (B) 293 cells were transiently transfected with pTRB3-Luc, pCMV-β-gal and the expression vectors for CHOP/NF-IL6 or ATF4. After 24 h, cells were left untreated or treated with 2 μg/ml of tunicamycin for 16 h. The luciferase activity in cell lysates was measured and was normalized with β-galactosidase activity. (C) The sequence of the ERSE in the TRB3 promoter. (D) Constructs of TRB3 promoter reporter genes. (E) HepG2 cells were transiently transfected with pTRB3-Luc and pCMV-β-gal, and after 24 h, treated with 2 μg/ml of tunicamycin for 16 h (left). 293 cells were transiently transfected with pTRB3-Luc, pCMV-β-gal and the indicated expression vectors for CHOP/NF-IL6 or ATF4. The luciferase activity in cell lysates was measured and normalized with β-galactosidase activity (right). The results in HepG2 cells were shown because the tunicamycin-induced activation level was higher than in 293 cells. Similar results were obtained in 293 cells as well. Similar results were obtained in three independent experiments.
As TRB3 mRNA expression induced by ER stress required de novo protein synthesis, we next examined the effects of overexpression of three transcription factors induced during ER stress, CHOP, ATF4 and XBP1, on TRB3 promoter activity. Not only CHOP/NF-IL6 but also ATF4 overexpression activated the TRB3 promoter via the region +201 to +312, which is similar to tunicamycin treatment; however, XBP1 overexpression had no effect (Figure 5B; see Supplementary Figure S7). These results suggest that ATF4 is able to activate the TRB3 promoter as well.
The ER stress response region in the TRB3 promoter, +201 to +312, contains three identical tandem repeats each consisting of 33 bp (Figure 5C). In addition, this region contains an amino-acid response element (AARE) overlapping a CHOP-binding site at the center, a CCAAT-like box on the left and a palindromic sequence on the right (Figure 5D). AARE has been discovered between nucleotides −313 and −295 in the CHOP promoter. The minimum core sequence (5′-ATTGCATCA-3′) is related to C/EBP- and ATF/CRE-binding sites and was described to bind in vitro ATF2/NF-IL6 or ATF4/NF-IL6 (Averous et al, 2003). This element is involved in the response to amino-acid starvation but not ER stress. On the other hand, ER stress response element (ERSE) was identified as a sequence of 19 nucleotides, the consensus of which is CCAAT-N9-CCAAG, in the CHOP and ER chaperone promoters (Yoshida et al, 2000). A mature form of ATF6 directly binds to the CCACG part of the ERSE in a manner dependent on the binding of NF-Y to the CCAAT part. To clarify whether these elements are involved in the tunicamycin-induced TRB3 promoter activation, we constructed the reporter gene containing one repeat of three tandem repeats sequence (pTRB3-RS) and its deletion and point mutants as shown in Figure 5D. As shown in Figure 5E (left), tunicamycin-induced ER stress did not cause the transactivation of the reporter gene containing mutations in the CHOP-binding site (pTRB3-RS mut1) or AARE not overlapping a CHOP-binding site (pTRB3-RS mut4, 6). Interestingly, the transactivation of mutants of the CCAAT-like box (pTRB3-RS mut3, 5) was not caused either. The mutation to a palindromic sequence (pTRB3-RS mut2) did not cause a significant decrease in tunicamycin-induced transactivation as compared with the wild type. These results revealed that the CHOP-binding sites, the AAREs and the CCAAT-like boxes, but not the palindromic sequences, are involved in the TRB3 promoter activation by tunicamycin-induced ER stress.
Next, we examined the effects of CHOP/NF-IL6 and ATF4 overexpression on these promoters (Figure 5E, right). CHOP/NF-IL6 overexpression demonstrated almost the same activation profile as that of tunicamycin treatment. On the other hand, ATF4 overexpression caused the transactivation of not only pTRB3-RS wt and mut2 but also pTRB3-RS mut3 and 5. Therefore, the TRB3 promoter activation by ATF4 overexpression is independent of the CCAAT-like box and not correlated with tunicamycin-induced activation. These results suggest that ATF4 expression alone is not enough for physiological TRB3 promoter activation and additional factors, such as CHOP, are necessary for this induction.
CHOP and ATF4 cooperate to activate TRB3 promoter activity
A consensus C/EBP-binding site consists of the palindromic decanucleotide ATTGCGCAAT and a consensus ATF/CRE box consists of the palindromic octanucleotide TGACGTCA (Montminy et al, 1986; Osada et al, 1996). Usually, the homodimers of members of the C/EBP family or those of the ATF/CREB family bind to the C/EBP-binding site or the ATF/CRE box, respectively (Bruhat et al, 2002). The AARE in the CHOP promoter and nutrient-sensing response element (NRSE) 1 (an AARE-related element) in the asparagine synthetase (AS) promoter (also termed C/EBP-ATF site) consist of one-half of each of the palindromic sequences that comprise an optimal C/EBP-binding site and an ATF/CRE box (Fawcett et al, 1999) (Figure 6A), and these sites were shown to bind C/EBP homodimers, ATF/CREB homodimers or C/EBP-ATF heterodimers. CHOP/NF-IL6 (heterodimers of the C/EBP family) or ATF4 (homodimers of the ATF/CREB family) caused transcriptional activation of the TRB3 promoter containing an AARE- and a CHOP-binding site. Therefore, we next examined whether C/EBP-ATF heterodimers can activate the TRB3 promoter as well. As ATF4 overexpression caused TRB3 promoter transcriptional activation, ATF4 is the strongest candidate for a component of the ATF part. As a component of C/EBP part, NF-IL6 is the partner of ATF4 for activation of the TRB3 promoter as well as the CHOP and AS promoters, but overexpression of NF-IL6 caused the potent repression of basal or ATF4-stimulated TRB3 promoter (Figure 6B, right). On the other hand, the coexpression of CHOP with ATF4 dramatically increased TRB3 promoter activation compared with the expression of ATF4 alone or CHOP/NF-IL6 (Figure 6B, left lane 5). In addition, this activation was stimulated by both CHOP and ATF4 in a dose-dependent manner (Figure 6C). This cooperative activation was not caused by the coexpression of a dominant-negative form of CHOP, CHOPΔBR or a dysfunctional CHOP, CHOPΔLZ, with ATF4 (Figure 6D). These results suggest that CHOP and ATF4 bind to the TRB3 promoter dependent on those DNA-binding regions and a precedent heterodimerization via the leucine zipper domains, and cooperate to activate this promoter.
Figure 6.
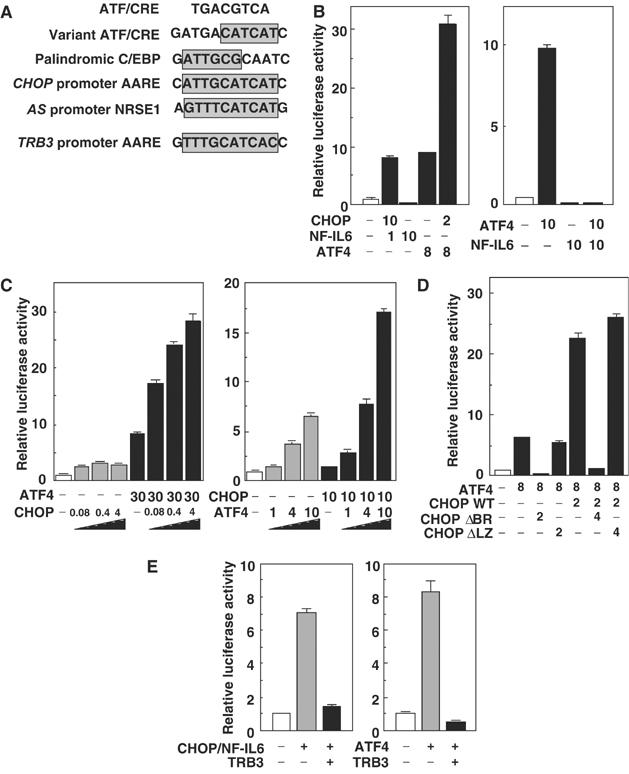
CHOP and ATF4 cooperate to activate the TRB3 promoter. (A) Alignment of the AARE in the TRB3 promoter with its related elements. (B–E) 293 cells were transiently transfected with pTRB3-Luc and pCMV-β-gal in combination with the indicated expression vectors for 48 h. The luciferase activity in cell lysates was measured and normalized with β-galactosidase activity. Similar results were obtained in three independent experiments.
Moreover, coexpression with TRB3 strongly suppressed the TRB3 promoter activation by ATF4 as well as CHOP (Figure 6E), indicating that TRB3 regulates its own expression via the repression of both CHOP and ATF4 transactivation activity in a double negative feedback loop.
Shutdown of ATF4–CHOP pathway represses TRB3 induction
The expression of DOCs (downstream of CHOP) during ER stress was not induced in CHOP knockout mouse embryonic fibroblasts (MEFs), and this suppression occurred in NF-IL6 knockout MEFs as well (Wang et al, 1998). From this observation, it is believed that NF-IL6 is a critical partner of CHOP for DOC induction during ER stress. In the expression of TRB3, CHOP/NF-IL6 and CHOP/ATF4 caused the critical activation of this promoter (Figures 3 and 6), and therefore we examined the effect of knockout/down of these genes on TRB3 induction. In NF-IL6 knockout MEFs, the expression of TRB3 mRNA was induced by tunicamycin treatment, comparable to normal MEFs (Figure 7A), while knockdown of ATF4 by the transfection of siRNA dramatically repressed TRB3 expression along with CHOP expression (Figure 7B). Furthermore, the knockdown of CHOP strongly repressed TRB3 expression 8 h after tunicamycin treatment (Figure 7C). These results suggest that CHOP causes TRB3 induction by cooperating with ATF4 at the early stage and is essential for maintenance of the induction at the later stage. These results also suggest that NF-IL6 is not essential for the induction of TRB3 as the partner of CHOP different from the induction of DOCs and the ATF4–CHOP pathway is critical for the TRB3 induction.
Figure 7.
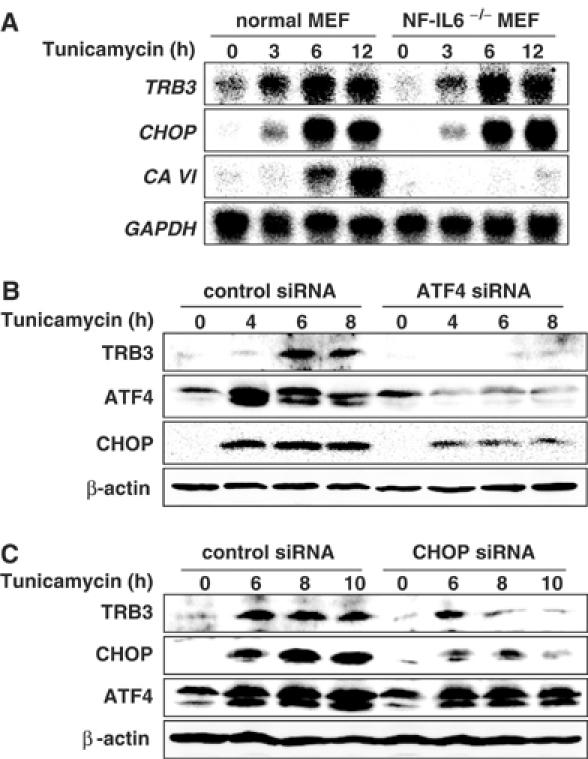
Knockdown of either ATF4 or CHOP repressed TRB3 induction. (A) Wild-type and NF-IL6−/− MEFs were treated with 2 μg/ml of tunicamycin for the indicated periods. Total RNA was prepared and analyzed by Northern blotting using indicated mouse cDNAs as probes. (B, C) 293 cells were transiently transfected with control siRNA, ATF4 siRNA or CHOP siRNA. After 48 h, cells were treated with 2 μg/ml of tunicamycin for the indicated periods. The cell lysates were analyzed by immunoblotting using anti-TRB3, anti-ATF4, anti-CHOP and anti-β-actin (bottom). Similar results were obtained in three independent experiments.
TRB3 is involved in cell death during ER stress
As CHOP-deficient mice are partially resistant to ER stress-dependent cell death (Zinszner et al, 1998; Oyadomari et al, 2002), CHOP can be related to the ER stress-induced apoptosis; however, the function of its target genes (DOCs) does not sufficiently account for the cell death. TRB3 negatively regulated CHOP-dependent transactivation, while TRB3 was also a target gene of CHOP. We speculated that TRB3 causes the ER stress-dependent apoptosis by a function different from the suppression of CHOP transactivation and accounts for CHOP-dependent cell death by a novel mechanism via TRB3 expression. As shown in Figure 8A, knockdown of TRB3 in 293 cells significantly caused resistance to the decrease in adherent cells by tunicamycin treatment. The TRB3-dependent cell death induced by tunicamycin was also observed in the HeLa cells (Figure 8B). In addition, ectopic TRB3 expression in 293 cells increased tunicamycin-dependent cell death, and this augmented cell death was inhibited by zVAD, a caspase inhibitor, indicating that it was apoptosis (Figure 8C). Condensation and fragmentation of nuclei in HeLa cells induced by tunicamycin were significantly decreased by knockdown of TRB3 (Figure 8D). These results suggest that TRB3 mediates ER stress-dependent apoptosis and could be a link between ER stress-induced CHOP and cell death.
Figure 8.
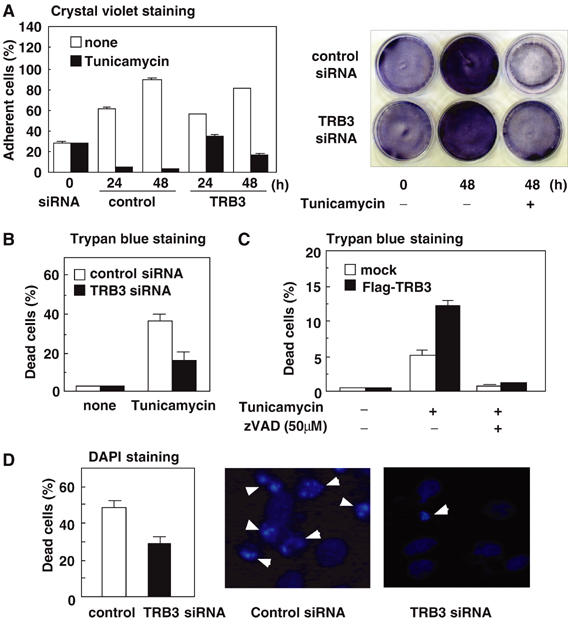
TRB3 induces ER stress-dependent cell death. (A) 293 cells were transiently transfected with control siRNA or TRB3 siRNA. After 48 h, cells were treated with 2 μg/ml of tunicamycin for the indicated periods. The percentage of adherent cells was measured by crystal violet staining. The right panel shows the stained cells treated with tunicamycin for 48 h. (B, C) HeLa cells (B) were transiently transfected with control siRNA or TRB3 siRNA. 293 cells (C) were transiently transfected with control vector or Flag-TRB3. After 48 h, cells were treated with 2 μg/ml of tunicamycin for 24 h or left untreated in the absence or presence of 50 μM of zVAD. The percentage of dead cells was measured by Trypan blue staining. (D) HeLa cells were transiently transfected with control siRNA or TRB3 siRNA. After 48 h, cells were treated with 2 μg/ml of tunicamycin for 24 h. Apoptotic cells were measured by staining with DAPI. Arrowheads in the right panel indicate apoptotic cells. Similar results were obtained in three independent experiments.
Discussion
CHOP, a member of the C/EBP family, has a dual role in the regulation of cellular gene expression: as an inhibitor of the binding of C/EBP to classical C/EBP target genes and as an activator of genes that have CHOP-C/EBP-binding sites. In this study, we identified a novel stress-inducible gene, TRB3, downstream of CHOP that is regulated by the latter part of CHOP. A luciferase assay using a reporter gene with the 5′-flanking region of TRB3 demonstrated that ER stress-dependent expression of TRB3 occurred at the transcriptional level. This region is approximately 1.9 kbp in length and consists of several putative CHOP/NF-IL6-binding elements. Consistent with this, ectopic expression of both CHOP and NF-IL6 stimulated promoter activation. In addition, a low to moderate level of CHOP expression alone activated TRB3 promoter activity. This activation would be caused via heterodimerization of CHOP with endogenous dimerization partners, probably ATF4 (a low level of ATF4 was detected in steady-state 293 cells). In this paper, we defined a novel CHOP-binding site consisting of two crucial sequences, an AARE and a CCAAT-like box, similar to NRSE1 and NRSE2. The authentic AARE is involved in the response to amino-acid starvation but not ER stress; however, NRSE1, which is an AARE-related element, is involved not only in the response to amino-acid starvation but also in the response to ER stress in the presence of NRSE2. In the TRB3 promoter, the presence of the second element, the CCAAT-like box, may be involved in the response to ER stress as well. Ectopic expression of CHOP/NF-IL6 promoted the transcriptional activity of a reporter gene containing four tandem repeats of the consensus CHOP-binding site (p(CHOP)4-Luc); however, the activation of this reporter gene was not caused by either a CHOP expression alone or treatment with tunicamycin (data not shown), indicating that an additional cis-regulatory element, such as CCAAT-like box, is required for the CHOP-dependent activation of TRB3 promoter and other promoters. NF-Y is one of the candidates to bind to the CCAAT-like box; however, the study using dominant-negative forms of NF-Y (Mantovani et al, 1994) suggested that NF-Y would not be related to this box and affect the transcriptional activity of the TRB3 promoter (see Supplementary Figure S8). In this paper, the proteins associated to the CCAAT-like boxes in the TRB3 promoter have not been identified. Identification of the proteins involved in the response to ER stress and amino-acid starvation and adaptor protein(s) is important for the elucidation of these stress responses.
In humans and mice, three orthologs of tribbles (TRB1, 2, 3) have so far been identified. Aspects of their expression are different. The expression levels of TRB3 mRNA and protein were quite low in untreated cells, and were induced by ER stress in several cells. In contrast, TRB1 and TRB2 mRNA was expressed in normal conditions and the expression level was not changed by treatment with tunicamycin in these cells (data not shown).
Here we demonstrate that TRB3 functions as a novel negative regulator of CHOP. The mechanism of the inhibitory action of TRB3 against CHOP is still unclear. The Drosophila tribbles has been shown to promote the degradation of slbo (Rorth et al, 2000); however, TRB3 did not promote the degradation of CHOP protein, and the degradation does not account for the inhibitory effect of TRB3 on transactivation activity. The TRB3-binding region in CHOP exists in the transactivation domain, indicating that binding may cause a block of the transactivation activity of CHOP.
The CHOP-binding region on TRB3 was in its N-terminal portion, and the deletion of this region sufficiently decreased its repression activity for CHOP (Figure 3F and Supplementary Figure S9). In addition, the deletion of carboxyl-terminal region or Akt-binding site (239–265) (Du et al, 2003) on TRB3 did not affect the binding with CHOP; however, these mutants lost CHOP repression ability like TRB3 ΔN (Figures 2D and 3F, and Supplementary Figure S9). These results suggest that the regulation of CHOP activity by TRB3 requires the association not only with CHOP but also with other molecule(s), for example, coactivator(s), corepressor(s) or other members of TRB family. Alternatively, TRB3/SKIP3 and its family contain the classic substrate-binding domains (C-terminal region) of a protein kinase, but lack the ATP-binding and kinase-activation domains (N-terminal region) (Bowers et al, 2003), so original kinase(s) or other modifying enzyme(s) cannot recruit to CHOP when CHOP binds to TRB3. TRB3 could be a novel type of endogenous kinase inhibitor, acting as a decoy kinase-like protein for CHOP, Akt or other substrates.
TRB3 functions as a negative regulator of ATF4, another bZIP protein that is critical for the induction of CHOP, as well. As TRB3/SKIP3 promotes the degradation of ATF4 (Bowers et al, 2003), this function may be explained by the ATF4 protein degradation.
Several genes are activated by ER or mitochondrial stress in a CHOP-dependent manner; however, the activation of these target genes does not account for CHOP-dependent cell growth inhibition. High levels of sustained ER stress can result in apoptosis, and some of the factors controlling this response, the activation of c-Jun N-terminal kinase (JNK) (Nishitoh et al, 2002), the activation of caspase-12 (Nakagawa et al, 2000) and transcriptional induction of CHOP (Zinszner et al, 1998), have recently been identified. During ER stress, CHOP is induced by a PERK–ATF4 pathway and/or IRE1/ATF6 pathway. The reports from the studies of overexpression or knockout of CHOP indicate that CHOP is related to ER stress-induced apoptosis; however, there is a missing link in the CHOP-mediated apoptosis-signaling pathway. We have indicated that TRB3 is a CHOP target gene and induces apoptosis during ER stress. Very recently, TRB3 has been shown to be a crucial factor for insulin resistance, and binds to Akt/PKB to inhibit its kinase activity (Du et al, 2003). Akt is known to maintain cell survival, by inhibiting apoptosis, and promote cell cycling (Brazil and Hemmings, 2001). On the other hand, it has recently been shown that CHOP-induced apoptosis is mediated by translocation of Bax from the cytosol to mitochondria (Gotoh et al, 2004). In addition, it has been described that the dephosphorylation of Akt is essential to the Bax conformational change and translocation to mitochondria of Bax (Pervin et al, 2003; Rathmell et al, 2003). Taken together, our observations and reports suggest that TRB3 could be the link between ER stress-induced CHOP and Akt dephosphorylation in inducing translocation of Bax to induce cell death.
Recently, there have been several reports that CHOP acts as an antiapoptotic factor (Southwood et al, 2002; Mayerhofer and Kodym, 2003). Southwood et al (2002) showed that oligodendrocytes but not kidney cells undergo apoptosis with greater frequency in Chop/rsh null mice than in controls. These data reveal a major divergence in the importance of CHOP as a crucial mediator of cell death for different cell types that conceivably could be related to the cell type-specific target genes of this transcription factor. A likely possibility is the pattering of CHOP with oligodendrocyte-specific bZIP transcription factor. Further studies should be carried out to resolve this question.
In a study using CHOP or NF-IL6 knockout MEFs, Wang et al have indicated that several CHOP target genes, DOC1 (stress-induced form of carbonic anhydrase VI), DOC4 and DOC6, are expressed dependent on the presence of NF-IL6 as well as CHOP. In this paper, by knockout/knockdown analysis, we have revealed that TRB3 expression during ER stress is independent of the presence of NF-IL6, but that the ATF4–CHOP pathway is crucial for this expression. From the promoter analysis, unlike CHOP-binding site, NF-IL6 alone negatively regulated the activation of the TRB3 promoter (Figures 3C and 6B). Overexpressed NF-IL6 supported the ectopic CHOP-dependent activation of TRB3 promoter probably because it worked as a dimerization partner of CHOP as shown in Figure 4B. However, tunicamycin-induced endogenous CHOP, whose expression level might be quite low, may choose ATF4 as a dimerization partner for TRB3 promoter activation, probably because the DNA-binding affinity of ATF4 to TRB3 promoter is higher than that of NF-IL6. ATF4 expression alone activated the TRB3 promoter; however, the promoter mutagenesis study revealed that this activation is not correlated with tunicamycin-induced activation. CHOP-induced TRB3 promoter activation is usually correlated with tunicamycin-induced activation, and TRB3 induction was late compared to CHOP induction. These findings suggest that ATF4 expression alone is not sufficient for TRB3 induction, and CHOP is essential for this as well. Indeed, CHOP and ATF4 cooperated to activate the TRB3 promoter, and the knockdown of CHOP significantly repressed the TRB3 expression. Here, we show that ATF4 is a novel partner of CHOP in the PERK–eIF2α pathway during ER stress.
CHOP is also induced by other stress signals such as oxidative stress, amino-acid deprivation and hypoxia. Therefore, it is possible that the CHOP–TRB3 pathway operates in response to these stresses as well. TRB3 is one of the targets of CHOP, and acts as a regulator of CHOP as well, suggesting that CHOP signaling is strictly regulated by TRB3 via a negative feedback mechanism. In terms of the physiological significance, TRB3 could be a sensor for ER stress-induced apoptosis. If the ER stress is transient and mild, the induced TRB3 blocks the CHOP and ATF4 function by binding to them. However, when potent and prolonged ER stress occurs, excess TRB3 will be produced and lead to apoptosis. TRB3 may be a potential therapeutic target for diseases with stress-dependent cell death, such as neurodegenerative diseases or type I diabetes.
Materials and methods
Reagents
RPMI 1640 medium, Dulbecco's modified Eagle's medium (DMEM), anti-β-actin monoclonal antibody (AC-15), anti-Flag monoclonal antibody (M2), tunicamycin and MMS were purchased from Sigma (St Louis, MO). Fetal bovine serum (FBS) was from HyClone (Logan, UT). Anti-Myc monoclonal antibody (9E10) was from Roche (Indianapolis, IN). Anti-GST monoclonal antibody (DG 122–2A7) was from Upstate Inc. (Lake Placid, NY). Anti-GADD153 monoclonal antibody (B-3) was from Santa Cruz (Santa Cruz, CA). A23187 was obtained from Calbiochem (La Jolla, CA); carbobenzoxyl-L-leucyl-L-leucyl-L-leucinal (MG132) and benzyloxycarbonyl-Val-Ala-Asp (OMe)-fluoromethylketone (zVAD) were obtained from Peptide Institute (Osaka, Japan). Cycloheximide was obtained from Nacalai Tesque (Kyoto, Japan).
Cell culture
The human melanoma cell line A375, the human embryonic kidney cell line 293 and the human hepatocellular carcinoma cell line HepG2 were cultured as described previously (Hattori et al, 2001, 2003a).
Construction of expression plasmids
TRB3 (GenBank accession #AK026945) was kindly provided by the human cDNA sequencing project of the New Energy and Industrial Technology Development Organization (NEDO) in Japan (Ota et al, 2004). pCMV5-Flag-TRB3, pCMV5-Flag-TRB3ΔN lacking aa 1–127, pCMV5-Flag-TRB3ΔC lacking aa 283–358, pCMV5-Flag-TRB3N179 lacking aa 180–358, pCMV5-Flag-TRB3C179 lacking aa 1–179 or pCMV5-Flag-TRB3Δ239–265 lacking aa 239–265, the region essential for the binding with Akt1 of human TRB3 were generated by PCR. pGEX-TRB3(1–50) was constructed by ligating pGEX-6P-1 (Amersham Bioscience, Little Chalfont, UK) with the TRB3 cDNA fragment (corresponding to aa 1–50). The plasmids pcDNA3.1-Myc-CHOP, pcDNA3.1-Myc-CHOPSer79,82Ala, pcDNA3.1-Myc-CHOPΔBR, pcDNA3.1-Myc-CHOPΔLZ and pCMV-Flag-NF-IL6 were constructed as described previously (Hattori et al, 2003a, 2003b). pcDNA3.1-Myc-CHOPΔN9, ΔN18, Δ19–26 and ΔN70 lacking the amino-terminus of human CHOP were generated by PCR. Gal4-CHOP(WT) was kindly provided by Dr D Ron (New York Univ.). pCMV5-Gal4-CHOPΔN9, ΔN18 and ΔN70, pcDNA3.1-NF-IL6 and pcDNA3.1-CHOP were generated by PCR. To obtain p(CHOP)4-Luc, four tandem repeats of the CHOP-binding element were amplified by PCR and ligated with pGL3 promoter (Promega, Madison, WI). pTRB3-Luc was generated by ligating the human TRB3 promoter region (−1265 to +609) with pGL3-basic. pcDNA3.1-Flag-ATF4, pcDNA3.1-Flag-XBP1, pcDNA3.1-Flag-NF-YA and pcDNA3.1-Flag-NF-YA DN, replacing Arg283, Gly284 and Glu285 with Ala, were generated by PCR. To obtain p(ERSE)2-Luc or p(UPRE)2-Luc, two tandem repeats of ERSE or two tandem repeats of the UPR response element, respectively, were amplified by PCR and ligated with pGL3 promoter. All constructs were verified by sequencing.
Reporter gene assays
Cells were transfected with luciferase reporter plasmids, and at 24 h post-transfection, treated for specific periods with ER stressors. Lysates were prepared and luciferase assays were performed according to the manufacturer's instructions (Promega). All experiments were performed a minimum of three times before calculating means and standard deviations.
RNA extraction, RT–PCR and Northern blot analysis
Total RNA was extracted from cells seeded in 60 mm plates. The RT–PCR and Northern blot analysis used were described previously (Matsumura et al, 2000). Human TRB3, CHOP, GAPDH, and mouse TRB3, CHOP-10, carbonic anhydrase VI, GAPDH-radiolabeled hybridization probes were generated using the individual cDNA fragments. Primers used for human TRB3 were 5′-ATGCGAGCCACCCCTCTAGC and 3′-CTAGCCATACAGAACCACTTC; for human CHOP were 5′-GCGTCTAGAATGGCAGCTGAGTCATTGCC and 3′-GCGTCTAGATCATGCTTGGTGCAGATTC; and for human GAPDH were 5′-TGAAGGTCGGAGTCAACGGATTTGGT and 3′-CATGTGGGCCATGAGGTCCACCAC.
Preparation of antiserum against human TRB3
TRB3 (1–50) protein fused with GST was prepared by transformation of JM109 with pGEX-TRB3 (1–50). This region of TRB3 (aa 1–50) has little homology with other human orthologs of tribbles, TRB1 or TRB2. Two female Japanese White rabbits (2–3 kg) were immunized with recombinant TRB3 (1–50) protein four times and bled to prepare the antiserum against human TRB3.
Immunoprecipitation and Western blot analysis
Cells were transiently transfected and treated as described in the figure legends. The cells were lysed in RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate and 1% Triton X-100) supplemented with protease inhibitors. The lysates were subjected to immunoprecipitation, and 1–2% of the lysate or co-immunoprecipitate was subjected to SDS–PAGE (12.5%), transferred onto PVDF membrane and probed with antibodies indicated in the figure legends. The immunoreactive proteins were visualized using ECL Western blotting detection reagents (Amersham Bioscience), and light emission was quantified with a LAS1000 lumino image analyzer (FUJI, Japan).
RNA interference
Double-stranded RNA duplexes corresponding to human TRB3 (5′-CGAGCUCGAAGUGGGCCCC-3′), human ATF4 (5′-GCCUAGGUCUCUUAGAUGA-3′) and human CHOP (5′-GCCUGGUAUGAGGACCUGC-3′) were designed. Control siRNA was used for the scramble II duplex (5′-GCGCGCUUUGUAGGAUUCG-3′). They were purchased from Dharmacon Inc. (Chicago, IL).
Transfection
A375 cells were transfected by a lipofection method using Effectene (Qiagen, Hilden, Germany) according to the manufacturer's instructions. 293 and HepG2 cells were transfected by the Chen–Okayama method (Chen and Okayama, 1987)
Cell proliferation assay and apoptosis assay
Cells were washed with PBS. Crystal violet was added to stain cells and the stained cells were lysed in 1% SDS and measured at an OD of 595. Cells were pelleted and washed with PBS. Trypan blue was added to cell pellets and stained cells were counted as dead.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Acknowledgments
We thank Dr Sumio Sugano, Dr Toshio Ota, Dr David Ron and Dr Shizuo Akira for generously providing expression plasmids and MEFs deficient in NF-IL6 and Dr Ryuichiro Sato for discussion. This work was supported in part by Grant-in-Aids for Scientific Research (B) from Japan Society for the Promotion of Science and Grant-in-Aids for Scientific Research on Priority Areas (C) from the Ministry of Education, Science, Sports and Culture.
References
- Averous J, Bruhat A, Carraro V, Thiel G, Fafournoux P (2003) Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem 279: 5288–5297 [DOI] [PubMed] [Google Scholar]
- Barone MV, Crozat A, Tabaee A, Philipson L, Ron D (1994) CHOP (GADD153) and its oncogenic variant, TLS-CHOP, have opposing effects on the induction of G1/S arrest. Genes Dev 8: 453–464 [DOI] [PubMed] [Google Scholar]
- Bowers AJ, Scully S, Boylan JF (2003) SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 22: 2823–2835 [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26: 657–664 [DOI] [PubMed] [Google Scholar]
- Bruhat A, Averous J, Carraro V, Zhong C, Reimold AM, Kilberg MS, Fafournoux P (2002) Differences in the molecular mechanisms involved in the transcriptional activation of the CHOP and Asparagine synthetase genes in response to amino acid deprivation or activation of the unfolded protein response. J Biol Chem 277: 48107–48114 [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7: 2745–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300: 1574–1577 [DOI] [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/camp-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 339: 135–141 [PMC free article] [PubMed] [Google Scholar]
- Fornace AJ, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, Holbrook NJ (1989) Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol Cell Biol 9: 4196–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, Mori M (2004) Hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ 11: 390–402 [DOI] [PubMed] [Google Scholar]
- Hattori T, Itoh S, Hayashi H, Chiba T, Takii T, Yoshizaki K, Onozaki K (2001) CHOP, a basic leucine zipper transcriptional factor, contributes to the antiproliferative effect of IL-1 on A375 human melanoma cells through augmenting transcription of IL-6. J Interferon Cytokine Res 21: 323–332 [DOI] [PubMed] [Google Scholar]
- Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K (2003a) C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene 22: 1273–1280 [DOI] [PubMed] [Google Scholar]
- Hattori T, Ohoka N, Hayashi H, Onozaki K (2003b) C/EBP homologous protein (CHOP) up-regulates IL-6 transcription by trapping negative regulating NF-IL6 isoform. FEBS lett 541: 33–39 [DOI] [PubMed] [Google Scholar]
- Hochstrasser M (1995) Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol 7: 215–223 [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13: 1211–1233 [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes J, Xanthopoulos KG (1998) Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem 273: 28545–28548 [DOI] [PubMed] [Google Scholar]
- Mantovani R, Li X, Pessara U, Hoojt van Huijsduijnen R, Benoist C, Mathis D (1994) Dominant negative analogs of NF-YA. J Biol Chem 269: 20340–20346 [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S (1996) Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS lett 395: 143–147 [DOI] [PubMed] [Google Scholar]
- Matsumura T, Ito A, Takii T, Hayashi H, Onozaki K (2000) Endotoxin and cytokine regulation of Toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res 20: 915–921 [DOI] [PubMed] [Google Scholar]
- Mayerhofer T, Kodym R (2003) Gadd153 restores resistance to radiation-induced apoptosis after thiol depletion. Biochem Biophys Res Commun 310: 115–120 [DOI] [PubMed] [Google Scholar]
- Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH (1986) Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci USA 83: 6682–6686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K (2000) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101: 451–454 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-specific apoptosis and cytotoxicity by amyloid-β. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada H, Yamamoto H, Nishihara T, Imagawa M (1996) DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. J Biol Chem 271: 3891–3896 [DOI] [PubMed] [Google Scholar]
- Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, Kimura K, Makita H, Sekine M, Obayashi M, Nishi T, Shibahara T, Tanaka T, Ishii S, Yamamoto J, Saito K, Kawai Y, Isono Y, Nakamura Y, Nagahari K, Murakami K, Yasuda T, Iwayanagi T, Wagatsuma M, Shiratori A, Sudo H, Hosoiri T, Kaku Y, Kodaira H, Kondo H, Sugawara M, Takahashi M, Kanda K, Yokoi T, Furuya T, Kikkawa E, Omura Y, Abe K, Kamihara K, Katsuta N, Sato K, Tanikawa M, Yamazaki M, Ninomiya K, Ishibashi T, Yamashita H, Murakawa K, Fujimori K, Tanai H, Kimata M, Watanabe M, Hiraoka S, Chiba Y, Ishida S, Ono Y, Takiguchi S, Watanabe S, Yosida M, Hotuta T, Kusano J, Kanehori K, Takahashi-Fujii A, Hara H, Tanase TO, Nomura Y, Togiya S, Komai F, Hara R, Takeuchi K, Arita M, Imose N, Musashino K, Yuuki H, Oshima A, Sasaki N, Aotsuka S, Yoshikawa Y, Matsunawa H, Ichihara T, Shiohata N, Sano S, Moriya S, Momiyama H, Satoh N, Takami S, Terashima Y, Suzuki O, Nakagawa S, Senoh A, Mizoguchi H, Goto Y, Shimizu F, Wakebe H, Hishigaki H, Watanabe T, Sugiyama A, Takemoto M, Kawakami B, Yamazaki M, Watanabe K, Kumagai A, Itakura S, Fukuzumi Y, Fujimori Y, Komiyama M, Tashiro H, Tanigami A, Fujiwara T, Ono T, Yamada K, Fujii Y, Ozaki K, Hirao M, Ohmori Y, Kawabata A, Hikiji T, Kobatake N, Inagaki H, Ikema Y, Okamoto S, Okitani R, Kawakami T, Noguchi S, Itoh T, Shigeta K, Senba T, Matsumura K, Nakajima Y, Mizuno T, Morinaga M, Sasaki M, Togashi T, Oyama M, Hata H, Watanabe M, Komatsu T, Mizushima-Sugano J, Satoh T, Shirai Y, Takahashi Y, Nakagawa K, Okumura K, Nagase T, Nomura N, Kikuchi H, Masuho Y, Yamashita R, Nakai K, Yada T, Nakamura Y, Ohara O, Isogai T, Sugano S (2004) Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet 36: 40–45 [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109: 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA (1996) Control of gene expression by proteolysis. Curr Opin Cell Biol 8: 340–347 [DOI] [PubMed] [Google Scholar]
- Pervin S, Singh R, Chaudhuri G (2003) Nitric-oxide-induced Bax integration into the mitochondrial membrane commits MDA-MB-468 cells to apoptosis: essential role of Akt. Cancer Res 63: 5470–5479 [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB (2003) Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23: 7315–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Habener JF (1992) CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev 6: 439–453 [DOI] [PubMed] [Google Scholar]
- Rorth P, Szabo K, Texido G (2000) The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mol Cell 6: 23–30 [DOI] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A (2002) The unfolded protein response modulates disease severity in Pelizaeus–Merzbacher disease. Neuron 36: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda M, Wang XZ, Zinszner H, Wu I, Habener JF, Ron D (1996) Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol Cell Biol 16: 1479–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D (1998) Identification of novel stress-induced genes downstream of chop. EMBO J 17: 3619–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 20: 6755–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ (2002) A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9