

Abstract
Background
Disrupting sleep during development leads to lasting deficits in chordates and arthropods. To address lasting impacts of sleep deprivation in Caenorhabditis elegans, we established a nonlethal deprivation protocol.
Results
Deprivation triggered protective insulin-like signaling and two unfolded protein responses (UPRs): the mitochondrial (UPRmt) and the endoplasmic reticulum (UPRER) responses. While the latter is known to be triggered by sleep deprivation in rodent and insect brains, the former was not strongly associated with sleep deprivation previously. We show that deprivation results in a feeding defect when the UPRmt is deficient and in UPRER-dependent germ cell apoptosis. In addition, when the UPRER is deficient, deprivation causes excess twitching in vulval muscles, mirroring a trend caused by loss of egg-laying command neurons.
Conclusions
These data show that nonlethal deprivation of C. elegans sleep causes proteotoxic stress. Unless mitigated, distinct types of deprivation-induced proteotoxicity can lead to anatomically and genetically separable lasting defects. The relative importance of different UPRs post-deprivation likely reflects functional, developmental, and genetic differences between the respective tissues and circuits.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0407-1) contains supplementary material, which is available to authorized users.
Keywords: Sleep, C. elegans, Lethargus, Worm sleep, Unfolded protein response, Proteotoxicity, Proteostasis, Mitochondrial UPR, Endoplasmic reticulum UPR
Background
Disrupting mammalian sleep during development correlates with negative effects on physical, cognitive, and social health, suggesting that sleep is important for appropriate development [1–3]. Nonlethal sleep deprivation also causes lasting neurological and behavioral deficits in Drosophila melanogaster [4]. However, a mechanistic grasp of why inadequate sleep during development is particularly deleterious is lacking.
Caenorhabditis elegans exhibits developmentally timed sleep during lethargus, a 2- to 3-h-long period at the termination of each larval stage [5–7]. Similar to mammalian sleep, lethargus is characterized by locomotion and feeding quiescence, sensory gating, a typical posture, rebound sleep, and deeply conserved regulation [6, 8–15].
Severe sleep deprivation activates DAF-16/FoxO, the C. elegans Forkhead box O (FoxO) transcription factor. FoxOs function broadly in regulating metabolism, lifespan, and responses to environmental stressors [16–24]. Nuclear translocation of DAF-16/FoxO is inhibited by the insulin/insulin-like growth factor signaling (IIS) pathway [17, 18]. In response to prolonged and continuous sleep deprivation, DAF-16 translocates to the nucleus to mitigate or delay lethality [10]. DAF-16 is also required for rebound sleep following much weaker disruptions [11].
A particular proteotoxic outcome of sleep deprivation is endoplasmic reticulum (ER) stress. In all species examined, including rodents and flies, expression of the ER chaperone immunoglobulin binding protein (BiP/Grp78) from the heat shock 70 protein family is upregulated upon sleep deprivation [25–30]. The C. elegans homolog of mammalian BiP is HSP-4 [31–33]. BiP/HSP-4 upregulation requires the action of the ribonuclease inositol-requiring protein-1 (IRE-1), a key receptor for sensing misfolded ER proteins [32–37]. IRE-1 signaling activates the XBP-1 transcription factor, which changes the expression of BiP and other genes in the deeply conserved unfolded protein response (UPRER) pathway [25, 28, 30, 38–41].
Prolonged wakefulness increases daily energy expenditure [42–44], for instance, in the brain [45, 46]. Consequently, energy production by the mitochondrial oxidative phosphorylation system is upregulated [25, 30, 47–50]. Nevertheless, the roles of the mitochondrial UPR (UPRmt) following sleep deprivation are largely unknown, although one study found that sleep deprivation induces mitochondrial chaperones (to a lesser degree than BiP) in rat cerebral cortexes [30].
Upon mitochondrial stress, expression of ubl-5, encoding a ubiquitin-like protein, is upregulated, and UBL-5 plays a key role in activating dedicated chaperones and proteases of the UPRmt [51–54]. In C. elegans, chemically induced mitochondrial stress upregulates the mitochondrion-specific chaperones HSP-6 and HSP-60 (from the Hsp70 and Hsp10/16 superfamilies) [51, 55].
Tractable model organisms have been prominently used to study responses to environmental stressors, such as oxidation or heat [56, 57]. In contrast, C. elegans sleep deprivation was minimally explored, and its lasting impacts, other than lethality, were never characterized. Here we established an automated approach to inflicting severe yet nonlethal deprivation of developmentally timed sleep in C. elegans. We found that worm sleep deprivation inflicts both mitochondrial and ER stress, as indicated by the triggering of the UPRmt and the UPRER. When the UPRs were genetically impeded, lasting defects in feeding, fecundity, and egg-laying physiology were detected. Moreover, different UPRs protected different tissues from the impacts of deprivation.
To assay feeding, we measured the pumping motion of the pharynx, a neuromuscular organ that takes in bacterial food, expels excess liquid, and passes food to the intestine [58, 59]. Maintaining these functions requires speed and regularity and is energetically costly. The pharyngeal nervous system consists of 20 neurons, is isolated from the rest of the animal by the basal lamina, and can operate independently [60, 61]. The rate of pumping depends on feeding history, quality of food, and endogenous serotonin levels [62–65]. In addition, pumping can be stimulated with exogenous serotonin [66, 67]. We found that sleep deprivation-induced mitochondrial stress impacts pharyngeal neurons and slows pumping.
In contrast, sleep deprivation-induced ER stress resulted in germ cell apoptosis and abnormal activity in the egg-laying circuit. The key determinant of C. elegans brood size is the number of available sperm [68]. Germ cell apoptosis can be triggered to protect sperm against DNA damage or environmental stressors that are not directly genotoxic. In both cases, highly conserved core apoptotic genes are strictly required for the initiation of programmed cell death [69–73]. One of these is CED-3, a cysteine-aspartate protease essential for execution of apoptosis [74–79]. Conveniently, this process can be visualized: the transmembrane receptor CED-1 mediates engulfment of early apoptotic corpses by surrounding sheath cells [80, 81]. Thus, the translational ced-1::gfp reporter is used to indicate the occurrence of germ cell apoptosis [82].
In addition, the egg-laying circuit exhibited a post-deprivation defect similar to the outcome of genetically ablating an egg-laying command neuron [83]. Opposite to the case of the feeding circuit, the UPRER (but not the UPRmt) mitigated the impact of deprivation in the egg-laying circuit. Collectively, these findings implicate two UPRs and insulin/insulin-like growth factor signaling in mitigating the impacts of disrupting worm sleep. They show that developmentally timed sleep is a vulnerable period: external stimuli that are benign outside of lethargus are proteotoxic when administered during lethargus. Adequate sleep promotes normal functions in tissues differing in developmental dynamics and physiological activity, and distinct UPRs mitigate different impacts of nonlethal deprivation.
Results
Substantial deprivation of lethargus quiescence can be automatically inflicted
Forced locomotion inflicted during C. elegans sleep by manually delivering harsh touch was previously shown to be lethal [6, 10]. Considerably more gentle mechanical vibrations can transiently force motion. We previously identified rebound sleep when vibrations were applied infrequently [11] and found that worms desensitized to vibrations delivered too frequently. In response to a 3-min on/off cycle of vibrations, we measured a 50% reduction in mean quiescence (Fig. 1a, b). This indicated that, in contrast to previously published conditions, the 3-min on/off cycle robustly overwhelmed the capacity of the worms to compensate for excess motion. We further found that this disruption affected wild-type animals and daf-16 mutants similarly.
Fig. 1.
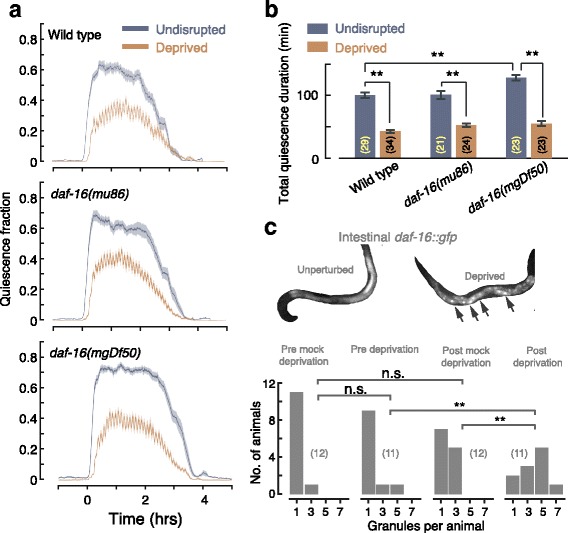
A periodic mechanical stimulus can partially and nonlethally reduce lethargus quiescence and induce translocation of DAF-16. a The fraction of quiescence measured during L4 lethargus of undisrupted (gray) and deprived (orange) wild-type animals and daf-16 mutants. Locomotion was forced using a square wave of mechanical vibrations with a 6-min period and 50% duty cycle. N = 30 animals, shaded areas depict mean ± standard error of the mean (SEM). b The total time in which quiescence was observed integrated over the data presented in a. Error bars depict mean ± SEM. c DAF-16::GFP fluorescence in the intestine before and after 1 h of sleep deprivation. Top: examples of green fluorescent protein (GFP) fluorescence in unperturbed and partially deprived animals. Arrowheads point to bright particles indicating concentration of DAF-16::GFP. Bottom: Histograms of the number of bright particles per animal identified under each set of conditions. Sample sizes are denoted in parentheses. Single and double asterisks denote significant differences with p < 0.05 and p < 0.01, respectively
Manually forced locomotion during worm sleep drives translocation of DAF-16 to nuclei of intestinal and body wall muscle cells [10]. We asked whether our 3-min on/off disruptions would induce similar translocation of DAF-16. To address this, we exposed animals expressing fluorescently labeled DAF-16 to 1 h of the vibration stimuli during the first half of the fourth lethargus stage (L4 lethargus). In agreement with the manual (lethal) deprivation protocol, we observed nuclear localization in intestinal cells (Fig. 1c). Translocation was not observed following a mock perturbation protocol, where vibrations were not applied during an equivalent 1-h period. In our hands, a clear translocation response was not observed in body wall muscles.
In contrast to the consequences of continuous manual deprivation [10], we did not observe any molting defects or lethality following our deprivation conditions. Possibly, this was a consequence of not depriving the animals of quiescence for a continuous period that exceeded 3 min. Collectively, these data demonstrate the ability to automatically and severely disrupt quiescence to a stressful yet nonlethal degree.
Considering these results, the experiments described throughout the manuscript employ three types of deprivation conditions: 1 h of disruptions for acute responses in individual animals expressing fluorescent markers (“mock” animals were loaded to identical observation chambers), 4 h of disruptions for gene expression assays in small groups of tightly synchronized animals, and 12 h of disruptions for lasting effects of deprivation assayed in large groups of animals. To control for nonspecific effects of the prolonged stimulation period, we compared sleep-deprived animals to those exposed to vibrations outside of lethargus. These groups were labeled “control” (see Methods section for details). In the latter two protocols (4 h and 12 h), sleep was disrupted for no more than the 3-h duration of lethargus. To identify effects of vibrations that are nonspecific to lethargus, we assayed a subset of strains without exposing them to any vibrations. These groups were labeled “unperturbed,” and they consistently exhibited similar phenotypes to those of the “control” animals. Thus, nonspecific effects of vibrations were found to be minor and genetically separable from impacts of sleep deprivation.
Two unfolded protein responses are triggered by nonlethal deprivation of C. elegans sleep
DAF-16/FoxO is associated with a broad spectrum of stress responses, and ER stress in particular was detected in previously examined sleep-deprived animals [25, 38–40]. To address whether the UPRER was triggered by worm sleep deprivation, we used a transcriptional reporter for HSP-4/BiP expression, hsp-4p∷gfp (zcIs4) [33, 51]. Animals expressing this indicator were subjected to a 1-h deprivation protocol. During L4 lethargus, hsp-4 was notably expressed in the epithelial seam of undisrupted animals (Fig. 2a). This array of hypodermal stem cells, termed seam cells, regulate hypodermal/cuticle formation and transform to their adult fate at the time of the fourth molt [7, 84–86]. We observed that hsp-4 expression in seam cells coincided with the generation of the alae — the adult cuticular ridges.
Fig. 2.
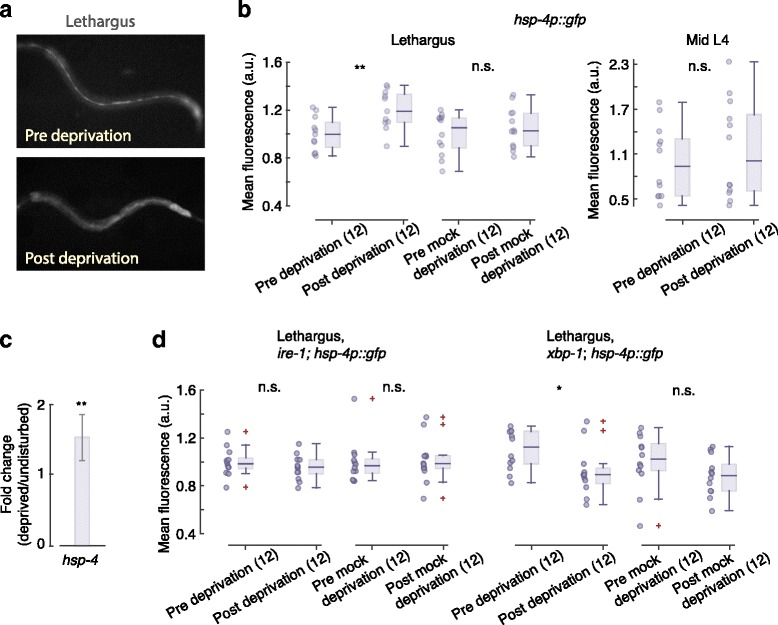
Worm sleep deprivation triggers the UPRER. a Example of pre- (top) and post- (bottom) deprivation fluorescence of the hsp-4p::gfp reporter. Prior to deprivation the reporter was prominently observed in the seam cells. b Fluorescence of the hsp-4p::gfp reporter before and after deprivation, mock deprivation, and stimulation of mid-L4 larvae. c Quantification of hsp-4 expression using real-time PCR. Error bars depict 99% confidence intervals (1 biological replicate, 20 animals per sample). d Fluorescence of the hsp-4p::gfp reporter in mutants where the function of the UPRER genes ire-1 and xbp-1 was lost. On these mutant backgrounds sleep deprivation did not upregulate the expression of hsp-4. In all box plots, horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Crosses denote outliers. Single and double asterisks denote significant differences with p < 0.05 and p < 0.01, respectively
Neither mock deprivation nor 1 h of vibration stimuli prior to lethargus affected hsp-4 expression. In contrast, sleep-deprived animals significantly upregulated the expression of hsp-4 (Fig. 2b). Consistently, hsp-4 expression remained elevated after a 4-h period of disruptions, as indicated by quantitative PCR (Fig 2c). Upregulation of hsp-4 requires essential components of the UPRER including IRE-1 and XBP-1 [33, 34, 87]. Consistent with induction of ER stress by environmental stressors or genetic perturbations, sleep deprivation failed to upregulate hsp-4 expression on ire-1 or xbp-1 mutant backgrounds (Fig. 2d).
The roles of the UPRmt following sleep deprivation are less understood, although evidence of mitochondrial stress was detected in sleep-deprived rodents [30]. Therefore, we similarly used fluorescent indicators to assay the activation of the UPRmt [51–53]. The ubl-5 translational reporter of the UPRmt expresses broadly at low levels and brightly in the posterior bulb of the pharynx, the posterior of the intestine, and the anterior edge of the intestine near the pharyngeal-intestinal valve. Following 1 h of sleep deprivation, we observed a small but significant upregulation of intestinal Publ-5::ubl-5::gfp expression. No increase in reporter fluorescence was observed following the mock protocol or when vibrations were applied at the mid-L4 larval stage (Fig. 3a).
Fig. 3.
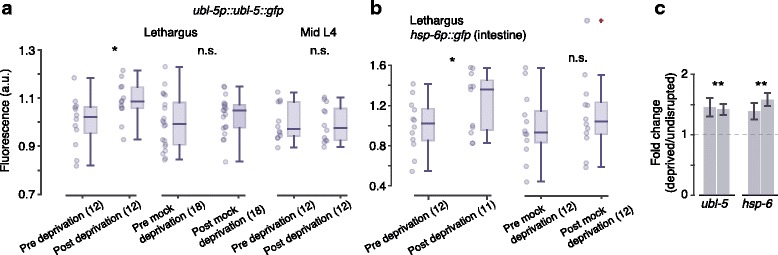
Worm sleep deprivation triggers the UPRmt. a Intestinal fluorescence of the ubl-5p::ubl-5::gfp UPRmt reporter before and after deprivation, mock deprivation, and stimulation of mid-L4 larvae. b Intestinal fluorescence of the mitochondrial chaperone hsp-6p::gfp reporter after deprivation and mock deprivation. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Single and double asterisks denote significant differences with p < 0.05 and p < 0.01, respectively. c Relative expression of ubl-5 and hsp-6 in deprived as compared to undisrupted wild-type animals (2 biological replicates, 20 animals per sample). The mechanical stimulus was applied for 4 h (which included L4 lethargus), and RNA was prepared immediately after this period. Error bars depict mean ± SEM
We similarly assayed two additional indicators of the UPRmt: the hsp-6 and hsp-60 transcriptional reporters. Expression of hsp-6 was broad and most clearly visible in the intestine. Strong expression of hsp-6 or an accumulation of the reporter led to bright staining of a posterior segment of the intestine. Sleep deprivation did not affect the posterior bright patch, but expression in the rest of the intestine was upregulated after the 1-h deprivation protocol. Mock deprivation did not affect hsp-6 expression (Fig. 3b). Real-time PCR was used to assay the relative expression of these genes after a 4-h period of administering the disruptive stimuli that included L4 lethargus. Consistently, we observed elevated expression of ubl-5 and hsp-6 in deprived animals (Fig. 3c). In our hands, we could not detect upregulation of hsp-60 post-deprivation (Additional file 1: Figure S1). Combined, these results show that nonlethal deprivation of worm sleep is proteotoxic and induces both mitochondrial and ER stress. Comparable stimulation outside of lethargus did not trigger these UPRs, indicating that the period of sleep is particularly vulnerable.
The UPRmt plays a role in mitigating effects of nonlethal sleep deprivation on pumping
Pharyngeal pumping for the purpose of feeding is an energetically demanding behavior that can readily be quantified [65]. To identify lasting effects of sleep deprivation on pumping, each animal was assayed for 60 min at a food concentration corresponding to an optical density (OD600) = 2.5 of the bacterial suspension, where pumping activity was intermediate (Additional file 2: Figure S2). Since control and unperturbed animals (as defined above) exhibited nearly identical feeding behaviors, deprivation conditions were typically compared to stimulation outside of lethargus (control).
Pharyngeal pumping can be adequately described as bursts of rapid pumping interspersed with pauses [65]. Similar to a previous study, we measured the mean instantaneous pumping rate and the duty ratio of rapid pumping [88]. These summary statistics did not reveal significant differences between deprived and control wild-type animals. However, sleep deprivation reduced pumping in daf-16 mutants, and the native promoter rescue of DAF-16 function restored the wild-type phenotype (Fig. 4a and Additional file 3: Figure S3). These results suggest that worm sleep deprivation can negatively impact feeding in a DAF-16-dependent manner.
Fig. 4.
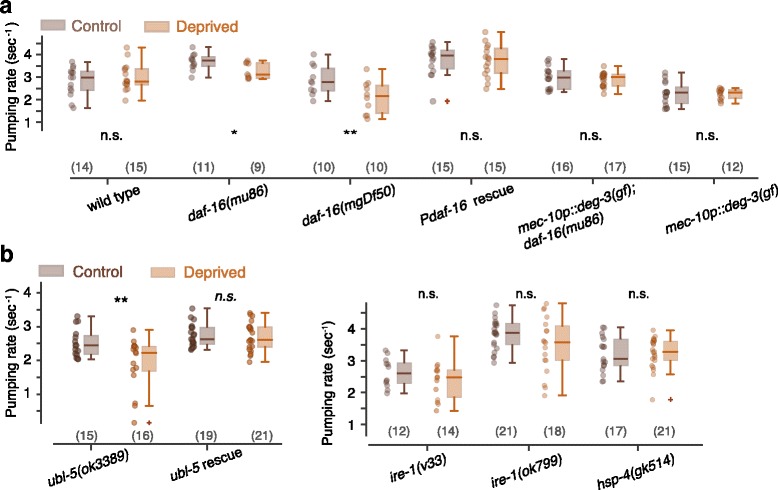
Mitigating mitochondrial (but not ER) stress is required for maintaining normal post-deprivation pumping rates. a Box plots of average pumping rates for control (i.e., stimulated outside of lethargus) and deprived animals obtained with the optical tracking method. Loss of DAF-16 or mec-10 expressing neurons confers a broad defect in stress responses or the loss of gentle mechanosensation, respectively. b Average pumping rates of animals deficient in the UPRmt (left) and the UPRER (right). Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses; asterisks and double asterisks denote significant differences (p < 0.05 and p < 0.01, respectively)
To control for potential biomechanical impacts of the vibrations, we assayed worms expressing deg-3(u662), a degeneration-causing constitutively active nicotinic acetylcholine receptor (nAChR) channel subunit, in touch neurons expressing the mec-10 gene [89–93]. These transgenics did not respond to vibrations (Additional file 4: Figure S4). In the absence of mec-10 expressing neurons, vibration stimuli during lethargus did not affect subsequent pumping on both wild-type and daf-16 mutant backgrounds (Fig. 4a and Additional file 3: Figure S3). These data indicate that mechanosensation by mec-10 expressing neurons and the ensuing loss of quiescence are required for pumping fatigue following sleep deprivation.
To test whether the UPRmt plays a role in mitigating consequences of nonlethal sleep deprivation, we examined feeding in ubl-5 mutants. Deprived ubl-5 mutants exhibited a decrease in pumping rate as compared to the control group. This pumping defect was rescued by expressing the Publ-5::ubl-5::gfp translational reporter (Fig. 4b and Additional file 3: Figure S3). In contrast, UPRER defective worms did not exhibit pumping fatigue: animals carrying two ire-1 putative null alleles and hsp-4 mutants did not significantly change their mean pumping rate post-deprivation (Fig. 4b). A mild change in the duty ratio of deprived ire-1 mutants may indicate a weak contribution of the UPRER maintaining post-deprivation pumping (Additional file 3: Figure S3). These results demonstrate that ER proteotoxic stress, unlike mitochondrial stress, does not play a major role in mitigating pumping fatigue following sleep deprivation.
Complementarily, electropharyngeograms (EPGs) enabled us to precisely time contractions and relaxations of the pharyngeal corpus and terminal bulb. We therefore used EPGs to measure durations of individual pumps and to confirm our optical measurements of pumping rates (Additional file 5: Figure S5, Additional file 6: Figure S6, and Additional file 7: Figure S7). We found that repeated mechanical stimuli extended the duration of individual pumps. However, the extension of single pumps was not specific to sleep deprivation; deprived animals and controls exposed to the mechanical stimuli before and after lethargus exhibited similar pump durations. The extended duration of individual pumps in ubl-5 mutants was comparable to that of wild-type animals and daf-16 mutants (Additional file 6: Figure S6). Thus, we could differentiate between deprivation-related and nonspecific impacts of mechanical stimuli, and we did not identify a role for the UPRmt in mitigating the nonspecific effect. Combined, our results indicate that the UPRmt (but not the UPRER) mitigates lasting effects of sleep deprivation on pharyngeal pumping.
Nonlethal sleep deprivation impacts pharyngeal pumping by affecting regulatory neurons
The pharynx is isolated from the rest of the animal and can exhibit pumping tens of minutes after it has been dissected out [60, 61]. Pumping defects induced by sleep deprivation can thus originate from pharyngeal regulatory neurons or pharyngeal muscles. We note that our mechanical stimuli do not noticeably affect the buccal plug, a cap of extracellular material that prevents food from entering the pharynx during lethargus [7], and they do not induce pumping. Consequently, the stimuli do not activate the pharyngeal muscles during lethargus, and “wear and tear” damage caused by anachronistic muscle activation is unlikely.
Serotonin or 5-hydroxytryptamine (5-HT) robustly activates rapid pumping through the action of the neuronally expressed SER-7/5-HT7 receptor. Several labs have shown that 5-HT-induced rapid pumping is abolished in ser-7 null mutants [64, 66, 67, 88]. We similarly activated pharyngeal neurons with 10 mM 5-HT instead of food and assayed pumping fatigue. We found that 5-HT-induced pumping was rapid in undisrupted and deprived daf-16(mgDf50) and ubl-5 mutants (Fig. 5a).
Fig. 5.
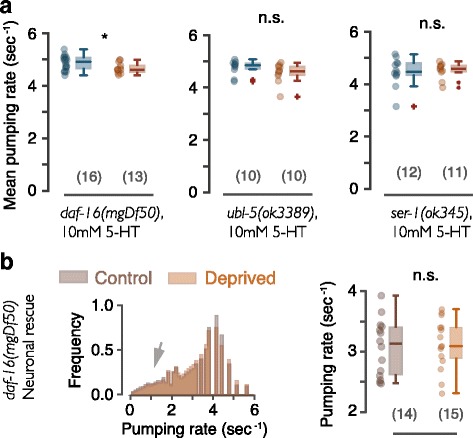
A neuronal deficiency underlies the post-deprivation slowdown of feeding. a Box plots of pumping rates for 5-HT-triggered pumping in undisrupted and deprived daf-16(mgDf50), ubl-5, and ser-1 mutants. b Left: distributions of pumping rates for deprived and control daf-16 mutants where neuronal function of DAF-16 was restored. Arrow points to the indistinguishable left tails of the distributions, indicating similar statistics of sporadic pumping. Right: mean pumping rates under these conditions. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses; asterisks and double asterisks denote significant differences (p < 0.05 and p < 0.01, respectively)
An additional serotonin receptor, SER-1/5-HT2, is expressed in pharyngeal muscles [94] but is not required for rapid pumping, whether induced by food or by 5-HT [64, 95]. Consistently, mutants carrying the putative null allele ser-1(ok345) and treated with 5-HT exhibited high pumping rates and no pumping fatigue (Fig. 5a). The absence and presence of rapid 5-HT-induced pumping in ser-7 and ser-1 mutants, respectively, indicates that 5-HT acts through activating pharyngeal neurons. The ability of deprived daf-16 and ubl-5 mutants to pump rapidly suggests that their deficits, exhibited in the presence of food, are the result of regulation rather than a biomechanical defect. In addition, neuronal rescue of DAF-16 function abolished the post-deprivation phenotype (Fig. 5b). Combined, these data indicate that rapid pumping is mechanically possible even in sleep-deprived mutants upon activation of pharyngeal neurons. Thus, sleep deprivation likely leads to lasting deficits in the neural circuit regulating pumping.
Nonlethal deprivation results in UPRER-dependent reduction in brood size
Sleep deprivation was recently implicated in affecting fertility in rodents [96, 97]. To address whether nonlethal sleep deprivation impacts C. elegans fecundity, we compared brood sizes of deprived and control animals using the 12-h protocol. We found that brood size in the control group was indistinguishable from that of undisrupted animals. However, sleep deprivation reduced wild-type brood size by 10% (Fig. 6a). The negative impact of nonlethal deprivation was exacerbated in daf-16(mu86) mutants, where brood size was reduced by 29% (Fig. 6b). In daf-16(mgDf50) null mutants, where brood size was overall lower, we observed an 18% reduction (Fig. 6c). When the function of DAF-16 was restored by driving expression with its native promoter, brood size was not reduced following deprivation (Fig. 6d).
Fig. 6.
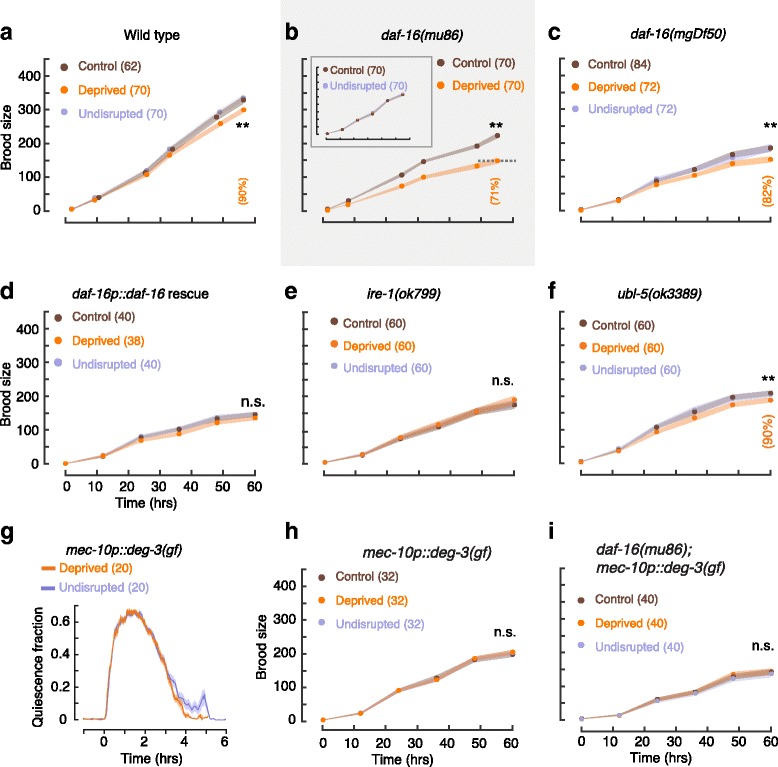
Worm sleep deprivation reduces brood size. a Brood sizes of wild-type animals during the first 3 days after L4 lethargus (t = 0 is 10–12 h after the fourth molt). Prior to the assay, animals were exposed to the 12-h deprivation protocol. The control group was stimulated ouside of lethargus, and the undisrupted group was not stimulated (see Methods). The dotted line depicts the brood size of deprived animals at the latest time point (see also Fig. 7a). Inset: undisrupted and control animals were indistinguishable. b, c The same as a for daf-16(mu86) and daf-16(mgDf50) mutants. d The same as a for daf-16(mgDf50) mutants where expression of daf-16 cDNA was driven by the daf-16 native promoter. e, f The same as a for UPRER deficient ire-1 mutants and UPRmt deficient ubl-5 mutants. The wild-type phenotype was exhibited by ubl-5 mutants, but fecundity was not reduced in ire-1 mutants. g Quiescence fraction during L4 lethargus of animals deficient in touch sensation (mec-10p::deg-3(gf)), whose responses to vibrations were mostly or entirely abolished (see also Additional file 4: Figure S4). h, i The same as a for transgenic animals deficient for touch sensation on wild-type or daf-16 mutant backgrounds. Shaded areas depict mean ± SEM, numbers of animals assayed are noted in parentheses, and double asterisks denote a significant difference in brood size at t = 60 h (p < 0.01)
To address the possibility of a floor effect in the daf-16(mgDf50) animals, we increased the brood size of daf-16(mgDf50) mutants through male mating [68] and observed a 24% reduction in brood size when both hermaphrodites and males were deprived. Disrupting sleep of either hermaphrodites or males resulted in intermediate phenotypes (Additional file 8: Figure S8A).
Since germ cell apoptosis can be enhanced by pharmacologically or genetically induced ER stress [98], we hypothesized that the UPRER may mediate an adverse impact of sleep deprivation on fecundity. To test this, we compared ire-1 mutants deficient in the UPRER to wild-type animals, daf-16 mutants, and UPRmt deficient ubl-5 mutants. In contrast to the other genotypes, where deprivation reduced fecundity, brood size was not reduced in the absence of IRE-1. Rather, deprived, control, and unperturbed ire-1 mutants were indistinguishable (Fig. 6e). Similarly, fecundity was impervious to sleep deprivation when ASI neurons, required for UPRER-mediated germ cell apoptosis, were genetically ablated (Additional file 8: Figure S8B) [98, 99]. The wild-type phenotype exhibited by ubl-5 mutants served as a negative control and indicated that the UPRmt did not mediate the impact of deprivation on brood size (Fig. 6f).
To control for potential biomechanical effects of the vibrations, we assayed deg-3(u662) transgenics. Failure to respond to vibrations abolished the reduction in brood size in the presence of the stimuli (Fig. 6h, i). Forced locomotion also did not appreciably increase the number of retained eggs (Additional file 9: Figure S9). Overall, these results suggest that nonlethal sleep deprivation in C. elegans negatively impacts brood size and that the UPRER and IIS play distinct roles in this process. Together with the relevance of the UPRmt to pumping fatigue, these data demonstrate distinct UPRs for mitigating impacts of sleep deprivation in different tissues.
Germ cell apoptosis causes the brood size reduction following nonlethal deprivation
The requirement of the UPRER for the reduction in brood size suggested that sleep deprivation may trigger germ cell apoptosis [98]. To test this, we assayed mutants lacking core apoptotic machinery caspase, quantified a fluorescent indicator of germ cell apoptosis, and counted fluorescently labeled germ cells. Animals carrying the putative null allele ced-3(n1286) were crossed to daf-16(mu86) mutants, which exhibited the most pronounced effect on fecundity. In both ced-3 single and daf-16; ced-3 double mutants the post-deprivation reduction in brood size was eliminated; i.e., the egg-laying dynamics of deprived, control, and unperturbed animals were indistinguishable (Fig. 7a). Moreover, brood sizes of daf-16; ced-3 double mutants were identical to those of undisrupted daf-16(mu86) single mutants. This suggested that the ced-3 mutation did not affect fecundity independently of sleep deprivation on a daf-16 background.
Fig. 7.
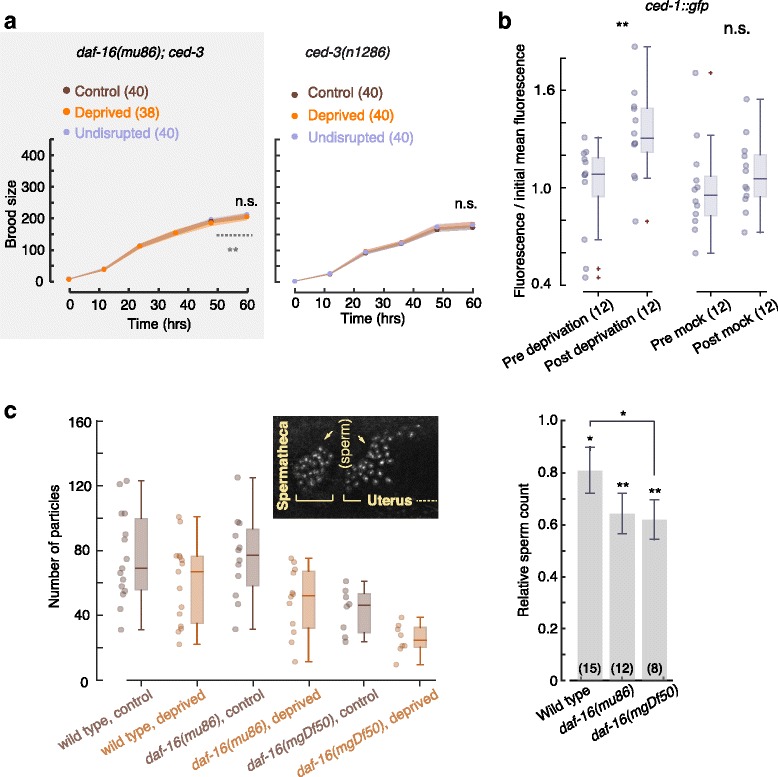
Worm sleep deprivation causes germ cell apoptosis. a Brood sizes of daf-16(mu86); ced-3(n1286) double mutants during the first 3 days after L4 lethargus (t = 0 is 10–12 h after the fourth molt). Deprivation failed to induce a reduction in brood size. The dotted line depicts the brood size of deprived daf-16(mu86) mutants at t = 60 h from Fig. 6a. b Box plots of ced-1::gfp fluorescence after deprivation (left) or mock deprivation (right), normalized by the mean pre-treatment fluorescence. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are denoted in parentheses; double asterisks depict a significant difference (p < 0.01). c Left: the number of sperm detected in a single gonad per animal. Horizontal lines, boxes, and bars depict means, 95% confidence intervals, and standard deviations, respectively. Inset: a confocal image of adult hermaphrodite sperm nuclei, specifically labeled by GFP-histone fusion driven by the Pcomp-1 promoter. Right: the ratio between the sperm count of deprived and control animals. Error bars depict mean ± SEM. Sample sizes are denoted in parentheses; single and double asterisks depict significant differences (between the mean and 1 or between genotypes) with p < 0.05 or p < 0.01, respectively
The ced-1::gfp translational reporter is an established indicator of germ cell apoptosis [82]. Animals expressing this reporter were subjected to the 1-h deprivation protocol. Deprivation resulted in a significant increase in ced-1::gfp fluorescence, suggesting that germ cell corpses were actively being disposed of. In contrast, the mock deprivation group, i.e., worms not exposed to vibration stimuli, did not exhibit a change in reporter fluorescence during the equivalent period (Fig. 7b).
Next, we counted fluorescently labeled sperm in self-fertilized deprived and control hermaphrodites as previously described [100, 101]. We observed reductions in sperm count following forced locomotion during L4 lethargus as compared to the control groups in wild-type and the two daf-16 strains (Fig. 7c). The reduced sperm counts were sufficient to explain the corresponding reductions in brood sizes. The reductions in daf-16(mgDf50) and daf-16(mu86) mutants were similar, perhaps due to the variability of the measurement, the 3-day assay having fallen short of revealing the full extent of the brood size deficiency, or further loss of sperm during adulthood. Direct indication of germ cell corpse engulfment mediated by CED-1, the requirement of the caspase CED-3, and the reduced sperm count provide three consistent lines of evidence. Together, they show that sleep deprivation induces germ cell apoptosis, consistent with the role of the UPRER in brood size reduction.
The UPRER (but not the UPRmt) mitigates the effects of nonlethal sleep deprivation on activity in the egg-laying circuit
The absence of hermaphrodite-specific neurons (HSNs) suppresses egg laying and doubles the frequency of calcium transients in vulval muscles (vms) [83, 102]. As germ cell apoptosis does not preclude an independent impact of deprivation on the egg-laying circuit, we asked whether sleep deprivation can cause abnormally high calcium activity in the vms. To address this, we assayed calcium dynamics indicative of vm twitching using a ratiometric reporter: co-expression of the calcium indicator GCaMP5 and the red fluorescent protein mCherry in the vms [83, 103] (Fig. 8a, b).
Fig. 8.
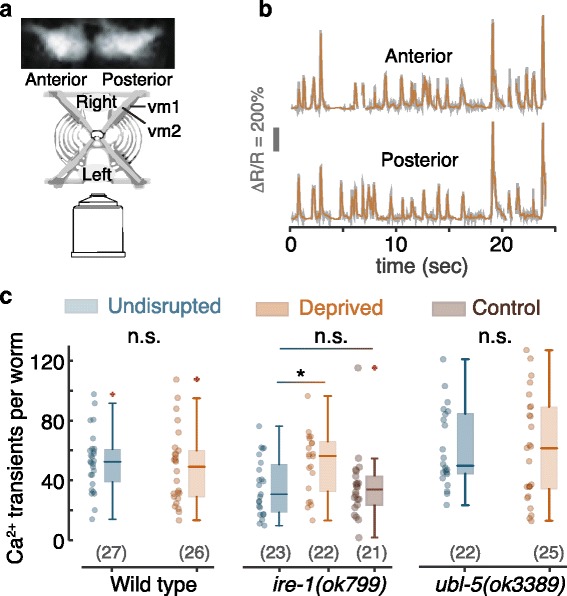
Worm sleep deprivation results in excess twitching of vulval muscles. a A schematic representation of the imaging setup. Top: fluorescently labeled anterior and posterior vulval muscle groups. Worms crawl on their left or right side such that their dorsoventral axis is parallel to the imaging plane. b Example traces of the ratio of GCaMP5 to mCherry fluorescence from anterior and posterior vms. c The total number of calcium transients in vms of undisrupted and sleep-deprived worms. An average increase of 30% in the number of vm twitches was detected in ire-1 (UPRER deficient) mutants. No significant changes were detected in wild-type animals and ubl-5 (UPRmt deficient) mutants. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses, and the asterisk denotes a significant difference (p < 0.02)
Opposite to the pharyngeal circuit, the egg-laying circuit was affected by ER but not mitochondrial stress. Neither wild-type animals nor UPRmt deficient ubl-5 mutants exhibited abnormal post-deprivation activity. When UPRER deficient ire-1 mutants were deprived, their mean number of vm twitches increased by 30%, mirroring the trend reported for HSN deficient animals (Fig. 8c).
Physiological activity in the egg-laying circuit is coupled with body posture and locomotion during brief periods around egg-laying events [83, 103, 104]. Therefore, we could not rule out the possibility that differences in locomotion over long timescales may also correlate with activity in the egg-laying circuit. Specifically, we asked whether a potential effect of deprivation on locomotion may indirectly cause the vm twitching phenotype observed in ire-1 mutants. To address this, we assayed the effect of sleep deprivation on mean velocity and measured the correlations between mean velocity and physiological activity (twitches) in the vms.
In all genotypes assayed, the mean velocity did not vary significantly between undisrupted and deprived animals (Additional file 10: Figure S10). While correlations between the mean velocity and vm twitching were found in wild-type, UPRER deficient, and UPRmt deficient worms, they were weakest in ire-1 mutants (Additional file 11: Figure S11). Moreover, the differences between mean or median velocities of deprived and nondeprived worms were insufficient to account for the 30% increase in vm twitching. Thus, excess vm twitching was not an indirect consequence of elevated locomotion activity in our assays.
Interestingly, the correlations between activity in the egg-laying circuit and locomotion were stronger in deprived wild-type animals and ubl-5 mutants as compared to their respective undisrupted groups. This trend was reversed in ire-1 mutants (Additional file 11: Figure S11). This observation may indicate that elevated coordination between distinct behaviors during stress may require secreted proteins, such as neuropeptides, whose function depends on processing in the ER [105]. These results support the conclusion that sleep deprivation causes ER stress which, if not mitigated, impacts the egg-laying circuit. Taken together, our data demonstrate that distinct types of proteotoxic stress caused by nonlethal deprivation of worm sleep adversely affected different circuits.
Discussion
The cognitive, physiological, and behavioral changes resulting from deprivation of human sleep can be subtle and elude superficial phenotyping. Detecting them requires functional imaging and/or proper design of the task being assayed, a clear definition of the sleep deprivation conditions, and careful measurements. Interpreting such results involves consideration of details such as differences in vulnerability to deprivation between individuals and whether the task was monotonous or complex [106–109]. Similarly, wild-type rodents and insects do not typically exhibit gross defects or substantial damage to brain cells following nonlethal deprivation protocols [4, 110–112]. This may be due to protective responses, activated by sleep deprivation, with the capacity to effectively prevent or repair the resulting damage.
To address the lasting impact of sleep deprivation in C. elegans, we established an experimental method enabling severe reduction in quiescence with no lethality or molting defects. Our periodic stimulus allowed for some quiescent behavior to take place throughout lethargus. Possibly, not forcing locomotion for extended continuous periods was key to avoiding a lethal outcome. In contrast, previous work assessed the impact of total sleep deprivation, i.e., consecutive forced movement for 30 min, which resulted in lethal molting defects. The impact on molting was interpreted to indicate a defect in metabolic regulation during lethargus, as the loss of DAF-16 sensitized the animals to this effect [10]. Lasting defects in surviving animals were not previously assayed.
The complete lack of lethality despite a loss of 50% of quiescence during lethargus suggests that quiescence, in and of itself, is an imperfect measure of the quality and restorative benefits of developmentally timed sleep in C. elegans. If homeostatic compensation can affect the quality of sleep [13], periodically allowing for rebound could confer greater restoration per unit time as compared to uninterrupted sleep. Testing this hypothesis will be key for understanding lethargus and may promote our understanding of additional quiescent states.
Identifying protective responses to worm sleep deprivation may clarify ancient functions of sleep. Cellular damage and repair can vary in molecular detail and occur at various rates. A comparison between tissues with different functions, developmental states, and metabolic demands may resolve requirements for a rest period due to accumulating damage. We found that deprivation triggers the UPRmt, which protects feeding behavior in sleep-deprived worms. In contrast to the UPRER, the UPRmt was not strongly associated with sleep deprivation previously. This may be partly due to a focus on sleep-related changes in gene expression in the brain [25, 27, 28, 38–40, 113–116]. One study reported that the mitochondrial chaperones Hsp60 and glucose-regulated protein 75 (Grp75, from the Hsp70 superfamily) were upregulated in the cerebral cortex of rats after sleep deprivation, although not as much as BiP [30].
Broad transcriptional responses to sleep deprivation were reported in mouse livers, lungs, and hearts [117, 118]. These studies demonstrate that the molecular consequences of disrupting sleep are not limited to the brain and that sleep contributes to normal function in a manner that may vary between different tissues or organs. For instance, the pharynx of C. elegans is a highly active organ, required to repeatedly generate powerful contractions. Neurons that regulate pumping may face a distinct cellular metabolic challenge and therefore may particularly benefit from the UPRmt post-deprivation.
Nonlethal sleep deprivation upregulates the expression of BiP in rodents and flies, thus indicating the activation of the UPRER [25, 38–40]. We have shown a similar upregulation of HSP-4/BiP in response to disrupting developmentally timed worm sleep. Furthermore, loss of function of the misfolded ER protein receptor IRE-1 affected both fecundity and the egg-laying circuit post-deprivation. These findings in C. elegans show that the activation of the UPRER is a deeply conserved response to sleep deprivation [15]. It remains to be determined whether the complementing set of phenotypes exhibited by the pharyngeal and the egg-laying circuits, whose function depended on the UPRmt and the UPRER, respectively, is associated with differences in their developmental states, energy expenditure, or additional factors.
Finally, we characterized outcomes of nonlethal sleep deprivation with respect to feeding, fecundity, and egg-laying physiology. Consistent with previous findings, our deprivation protocol triggered translocation of DAF-16/FoxO into intestinal cell nuclei [10] and implicated it in mitigating lasting adverse effects of deprivation. Three lines of evidence indicated that fecundity was reduced due to germ cell apoptosis: a dependence of the effect on CED-3 and IRE-1, a low sperm count in sleep-deprived worms, and the engulfment of early germ cell corpses by surrounding sheath cells as visualized by the ced-1 reporter [74, 75, 81, 98]. Interestingly, sperm quality was recently shown to be negatively impacted by disruptions to sleep in rodents [96, 97].
Conclusions
Hypotheses explaining the core functions of sleep include the notion that its utility may differ across species [119]. The synaptic homeostasis hypothesis proposes that continuous learning during wakefulness is bound to saturate synaptic connections. Therefore, renormalization of net synaptic strength during sleep is required to restore homeostasis [120, 121]. Other suggestions focus on “wear and tear” in physiologically active neurons during wakefulness due to accumulation of protein fragments, unfolded proteins, or other molecular stressors [112, 122–125]. On a larger scale, metabolite clearance from the brain can increase during mammalian sleep [126]. In addition, disrupting sleep is linked to abnormal glucose metabolism and appetite regulation. These findings suggest that sleep is key to normal metabolic and hormonal processes outside the brain [127, 128].
This work describes multiple pathways by which sleep deprivation can upset cellular proteostasis, likely by creating unfavorable biochemical conditions. Specific characteristics of physiological activity and development could affect the balance between accumulation and relief of allostatic load. To the best of our knowledge, how this balance might scale with different types of metabolic loads has not been systematically studied, let alone connected to sleep. Protection of post-deprivation pharyngeal function by the UPRmt is consistent with the notions that sleep reduces cellular metabolic stress and that highly active organs may invoke distinct responses in this context. Implicating the UPRs in mitigating consequences of worm sleep loss indicates that these responses are deeply conserved.
Methods
Strains
Wild-type, transgenic, and mutant C. elegans strains were cultivated with OP50 bacteria according to standard protocols at 20 °C. The following strains were used: N2 Bristol (wild type), CF1038 daf-16(mu86), GR1307 daf-16(mgDf50), NQ116 muIs211 [pNL213(ges-1p::GFP::daf-16) + rol-6(su1006)] (gift from D. Raizen), NQ441 daf-16(mgDf50); qnIs45[Pdaf-16:GFP::daf-16; Pmyo-2:mCherry] (gift from D. Raizen at the University of Pennsylvania), SJ4151 zcIs19 [ubl-5p::ubl-5::gfp], SJ4100 zcIs13 [hsp-6p::gfp], SJ4058 zcIs9 [hsp-60p::GFP + lin-15(+)], VC2564 ubl-5(ok3389), SJ4200 zcIs41 [ubl-5p::3xmyc-His tag::ubl-5 + myo-3p::gfp]; SJ4151 zcIs19 [ubl-5p::ubl-5::gfp], NQ128 muEx169[unc-119p::GFP::daf-16 + rol-6(su1006)] (gift from D. Raizen at the University of Pennsylvania), DA184 ser-1(ok345), Is[Pmec-10::deg-3(u662)] (gift from M. Treinin at the Hebrew University of Jerusalem), MT3002 ced-3(n1286), MD701 bcIs39 [lim-7p::ced-1::GFP + lin-15(+)], SJ4005 zcIs4 [hsp-4p::gfp], RE666 ire-1(v33), SJ17 xbp-1(zc12), SJ30 ire-1(zc14) II; zcIs4 V, SJ17 xbp-1(zc12) III; zcIs4 V, RB925 ire-1(ok799), VC1099 hsp-4(gk514)II, UX564 jnSi118[Pcomp-1::GFP::H2B::3’comp-1; Cb-unc-119(+)]; him-5(ok1896) (gift from G. Stanfield at the University of Utah), LX1938 egl-1(n986dm)V; vsIs164 X; lite-1(ce314) X; lin-15(n765ts) X (gift from K. Collins at the University of Miami), LX1918 vsIs164 X; lite-1(ce314) X; lin-15(n765ts) X (gift from K. Collins at the University of Miami), IV205 ueEx122 [str-3::TeTx::GFP; elt-2::sl2GFP] (gift from S. Chalasani at the Salk Institute for Biological Studies).
Severe nonlethal deprivation protocol
Motion and quiescence were identified using the image difference method as described in [129]. To disrupt quiescence during lethargus, mid-L4 animals were transferred to 60-mm nematode growth media (NGM) plates containing 10 mL of medium and seeded with 50 μL of OP50 culture that was grown overnight at 37 °C. Vibrations (1 kHz) were delivered using the mechanical clamps described in [11]. In contrast to the brief and well-spaced disruptions described previously, the stimuli described here were composed of 3 min of vibrations interspersed with 3 min of “quiet” intervals, i.e., a period of 6 min and a duty cycle of 50%.
Synchronization was performed in two steps. Gravid adults were allowed to lay eggs on a fresh plate for 2 h. Of the resulting larvae, mid and early L4s were manually picked for the deprived and control groups, respectively. Manually picking the L4 larvae reduced the variability of the timing of lethargus onset to 2 h.
Assays described throughout the manuscript employ one of three deprivation protocols: (1) fluorescent markers were assayed in individual worms during the first half of lethargus following a 1-h period of 3-min on/off disruptions. Control animals were either not stimulated at all (“mock”) or stimulated for an hour prior to lethargus. (2) Gene expression was assayed in small groups of tightly synchronized animals. Deprived groups were exposed to 3-min on/off disruptions for a period of 4 h that included lethargus, and control groups were exposed to 4 h of vibrations outside of lethargus. (3) Lasting defects were assayed in large groups of animals following a 12-h period of 3-min on/off stimulation. The stimulation period was initiated prior to lethargus and terminated at the young adult stage. Control animals were exposed to vibrations 5 h before lethargus, not stimulated for 5 h that included lethargus, and stimulated again for 5 h after lethargus. In cases when animals that were never stimulated were assayed, they were labeled “unperturbed” to differentiate them from the standard “control” groups.
Optical measurements of pharyngeal pumping
Post-stimulus (see above) young adults were picked into liquid NGM and loaded into a WormSpa microfluidic device [65, 130]. An Escherichia coli OP50 overnight culture, concentration-adjusted in NGM to OD600 = 2.5 (an intermediate food concentration), was flown through the device at a constant rate throughout the assay. After 1 h of acclimation in the device, the animals were imaged for an additional 1 h at a magnification of 10× and 62.5 frames per second using a Basler acA1920-25um complementary metal-oxide-semiconductor (CMOS) camera mounted on Celestron 44104 microscopes [131]. Pumping events were identified using a custom Python script which aligned and subtracted consecutive images and calculated the entropy of the difference image. A pumping event resulted in a characteristic spike in this entropy [65, 132].
Electropharyngeograms (EPGs)
Worms were loaded to a NemaMetrix ScreenChip System microfluidic device (NemaMetrix, Inc., Eugene, OR, USA) on a standard dissection microscope and allowed to acclimate for at least 30 min before recording. The device was filled with either bacterial food (OD600 = 2.5) or a solution of 10 mM 5-HT in NGM buffer. Immediately before the onset of the measurement, the output tube was disconnected to reduce electromagnetic noise. The pumping frequency was measured as described in the ScreenChip User Guide, and each recording lasted 5–10 min (measurements that terminated prematurely were discarded).
Imaging of green fluorescent protein (GFP) reporters
The 1-h deprivation protocol consisted of 3-min-long vibration pulses interspersed with 3-min-long pauses, starting during the first 30 min of L4 lethargus and lasting a total of 1 h. Animals expressing a fluorescent reporter were exposed to these mechanical stimuli in artificial dirt microfluidic devices placed inside a standard petri dish. They were imaged in the device immediately before and after the 1-h period of disruption. Imaging was performed at a magnification of 20× (0.5 numerical aperture, NA) using a Nikon Eclipse Ti microscope (Nikon Inc., Melville, NY, USA) and an Andor iXon X3 EMCCD camera (Andor, Belfast, UK). Fluorescence intensity was determined using custom Python scripts.
Calcium imaging in freely behaving animals
To reduce background fluorescence, calcium imaging was performed in artificial dirt microfluidic devices [133]. Animals co-expressing GCaMP5 and mCherry in their vms were mounted in the presence of bacterial food on an epi-fluorescence Nikon Eclipse Ti inverted microscope. Each worm was imaged at a magnification of 20× (0.5 NA) and a frame rate of 6 frames per second. Images were captured with an Andor iXon X3 EMCCD camera. A Dual View (DV2) two-channel system was used for simultaneous imaging of the red and green channels (Photometrix, Tucson, AZ, USA). Each animal was tracked manually and continuously imaged for a total period of 30 min. Calcium transients were analyzed using custom Matlab scripts (The Mathworks Inc., Natick, MA, USA).
Brood size
Brood size was counted by visual inspection: 10 h after mechanical stimulation ended, animals were transferred to individual 60-mm NGM plates seeded with a 50-μL drop of OP50 (two animals per plate). Plates were scored in the morning and evening of the following 3 days. To avoid the accumulation of hatched offspring, animals were transferred to new plates prior to the evening counts each day. For male mating, two males and two hermaphrodites were transferred to each plate.
Sperm counting
Adult Pcomp-1::GFP::H2B::3’comp-1 animals 24 h post-L4 lethargus were sealed into individual “artificial dirt” chambers filled with NGM and 10 μM levamisole. Confocal images of fluorescently labeled sperm were obtained using a Zeiss LSM 880 microscope with a Plan-Apochromat 40×/1.4 oil differential interference contrast (DIC) objective. Image stacks were analyzed using the FARSIGHT Nucleus Editor (http://www.farsight-toolkit.org/wiki/NucleusEditor). We note that when the fluorescent marker was crossed to a daf-16 mutant background, it was not confined to the nuclei. However, individual sperm cells were still detectable. We observed this in all daf-16(mgDf60) mutants and 25% of the daf-16(mu86) mutants.
Real-time PCR
The lmn-1 gene, encoding the C. elegans nuclear laminin, was chosen as the endogenous control gene. Primers (except for lmn-1, Shaham lab, Rockefeller University, private communication) were designed using Wormbase.org and the National Center for Biotechnology Information (NCBI) Primer-Basic Local Alignment Search Tool (BLAST) software. They were tested for specificity using NCBI BLAST and by agarose gel electrophoresis (using genomic DNA) and purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). The primers used for lmn-1, hsp-4, ubl-5, and hsp-6 were TCGAGGCGGAAAAGGCTC (Fwd), GCTCCAGCGAGTTCTCTCTC (Rev), GCCGACAAGGAAAAACTCGG (Fwd), GTGGGGTTGGGTTGGGAAA (Rev), ACAAACTGGAACACGATGGGA (Fwd), TCCCTCGTGAATCTCGTAATCC (Rev), AAGAACTCTGGAGGTGACGC (Fwd), and ACGTTGGGGGTTTCTAAAGAT (Rev), respectively. Real-time quantitative PCR amplifications were performed using 25 μL of QuantiTect SYBR Green Real-Time PCR master mix (QIAGEN, Hilden, Germany), 2 μL of diluted reverse transcription product (2 ng/reaction), 1.5 μL each of forward and reverse primer, and 20 μL of DNase/RNase-free water in a total volume of 50 μL. Amplification was carried out in an AB 7900 HT Real-Time PCR cycler (Applied Biosystems, Foster City, CA, USA) with initial polymerase activation at 95 °C for 15 min, followed by cycles of 94 °C for 15 s denaturation, 57 °C for 30 s for primer-specific annealing, and 72 °C for 30 s for extension. A melting curve analysis was carried out (60–95 °C) to verify the specificity of amplicons, i.e., the absence of primer dimers and nonspecific products. Each assay included six technical replicates and a no-template control for every primer pair.
Statistical analysis
Pairwise comparisons of data represented in bar or box plot were done using the Student’s t test. In the case of multiple comparisons, significance was calculated using a one-way analysis of variance (ANOVA) test and the Bonferroni post hoc correction. Distributions represented by histograms were compared using the k-sample Anderson-Darling test and the Bonferroni post hoc correction for multiple comparisons (when applicable).
For each figure panel, the following p values are listed in order of the positions of asterisks and not significant (n.s.) labels (top to bottom and then left to right). Fig. 1b: 1.1 × 10–3, 6.5 × 10–15, 2.2 × 10–15, 9.5 × 10–7. Fig. 1c: 0.59, 0.84, 0.002, 0.001. Fig. 2b: 0.003, 0.41, 0.63. Fig. 2c: 4.9 × 10–5. Fig. 2d: 0.36, 0.98, 0.033, 0.164. Fig. 3a: 0.016, 0.11, 0.34. Fig. 3b: 0.04, 0.22. Fig. 3c: 0.003, 0.001, 0.005, 0.002. Fig. 4a: 0.21, 0.048, 0.010, 0.50, 0.26, 0.46. Fig. 4b: 0.006, 0.40, 0.142, 0.074, 0.42. Fig. 5a: 0.018, 0.095, 0.38. Fig. 5b: 0.50. Fig. 6a: 4.8 × 10–3 . Fig. 6b: 5.4 × 10–14. Fig. 6c: 6.1 × 10–3. Fig. 6d: 0.39. Fig. 6e: 0.33. Fig. 6f: 0.007. Fig. 6h: 0.19. Fig. 6i: 0.48. Fig. 7a: 0.46. 0.18. Fig. 7b: 0.009, 0.44. Fig. 7c: 0.050, 0.043, 0.009, 0.008. Fig. 8c: 0.38, 0.017, 0.79. Additional file 1: Figure S1: 0.43. Additional file 3: Figure S3A: 0.37, 0.44, 0.003, 0.50, 0.34, 0.41. Additional file 3: Figure S3B: 0.048, 0.028, 0.40, 0.070, 0.27, 0.50. Additional file 5: Figure S5: 0.16, 0.050, 0.010. Additional file 6: Figure S6B: 0.035, 0.042, 0.021. Additional file 6: Figure S6C: 0.002. Additional file 6: Figure S6D: 0.061, 0.005, 0.28. Additional file 7: Figure S7 (OD600 = 2.5): 0.15, 0.37, 0.22, 0.34, 0.092, 0.36, 0.30, 0.30. Additional file 7: Figure S7 (10 mM 5-HT): 0.039, 0.043, 0.22, 0.47, 0.27, 0.30. Additional file 8: Figure S8A: 2.5 × 10–4, 0.82. Additional file 8: Figure S8B: 0.77. Additional file 10: Figure S10: 0.10, 0.34, 0.26, 0.29.
Additional files
Fluorescence of the hsp-60p::GFP fluorescent reporter before and after deprivation. In our hands, elevated expression of the reporter was not observed after 1 h of disrupting worm sleep. (PDF 368 kb)
Pumping rates increase as a function of food availability. The average pumping rate of wild-type animals at different concentrations of ambient bacterial food (as measured by optical density, OD 600). N = 10 animals per condition. Error bars depict mean ± standard error of the mean (SEM). (PDF 318 kb)
Duty ratios of rapid pumping of deprived and control worms. (A) Box plots of duty ratios for control and deprived animals. Continuous pumping was defined as a period in which the delay between pumps did not exceed 500 ms. (B) Same as (A) for UPR mutants and the neuronal rescue of daf-16. (PDF 1224 kb)
Genetic ablation of the mec-10 expressing touch neurons abolishes responses to the vibration stimuli. Locomotion in response to 1-kHz vibrations was quantified as the fraction of the body area the animal traversed per second. Wild-type animals (left) responded robustly to the stimulus, while touch-insensitive mec-10p::deg-3(gf) transgenics (right) did not exhibit a detectable response. N = 20 animals from each genotype were assayed, and shaded areas depict mean ± SEM. (PDF 468 kb)
EPG measurements of pumping fatigue are consistent with the results of the optical tracking method. Instantaneous pumping rates were calculated as 1/(duration between consecutive contraction peaks). Mean (per animal) rates for wild-type animals, daf-16(mu86) mutants, and ubl-5(ok3389) mutants reproduced the phenotypes detected by optical tracking. (PDF 480 kb)
The duration of an individual pumping motion is extended by exposure to mechanical vibrations irrespective of the timing of the stimuli. (A) Top: a sample EPG trace of a wild-type animal in the presence of food at OD600 = 2.5 concentration. Peaks correspond to corpus and terminal bulb contraction. Troughs correspond to corpus relaxation. Bottom: average contraction and relaxation EPG traces for undisrupted and deprived wild-type animals. Distributions of pump durations are shown for undisrupted, deprived, and control (exposed to vibrations outside of lethargus) animals. The outline of the distribution for undisrupted animals was duplicated as a guide to the eye. (B) The mean (per animal) pump durations and amplitudes of EPG peaks and troughs for wild-type animals (top) and daf-16(mu86) mutants (bottom). (C) Same as (A, B) for UPRmt deficient ubl-5 mutants. (D) Same as (A, B) for mutants treated with 10 mM 5-HT instead of food. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses; asterisks and double asterisks denote significant differences (p < 0.05 and p < 0.01, respectively). (PDF 774 kb)
EPG characteristics of individual pumping motions. Mean (per animal) EPG peak and trough amplitudes for animals presented with food or 10 mM 5-HT. Vibration stimuli during or outside lethargus did not typically affect these amplitudes. Sample sizes are noted in parentheses, and asterisks denote significant differences (p < 0.05). (PDF 1006 kb)
(A) Brood size can be reduced by sleep depriving either males or hermaphrodites. The numbers of egg laid by daf-16(mgDf50) mutants that mated with males. Either the males, the hermaphrodites, or both were sleep deprived. (B) ASI neurons are required for sleep deprivation to impact fecundity. Brood size was not reduced by deprivation when ASI neurons were genetically ablated using tetanus toxin (in contrast to phenotypes shown in Fig. 6a–c, f). (PDF 176 kb)
Nonlethal sleep deprivation does not increase egg retention. The numbers of fertilized eggs retained in the uterus of wild-type animals and daf-16 mutants 24 h and 48 h after L4 lethargus. Deprived animals were exposed to the stimulus before, during, and after L4 lethargus. Control animals were exposed to the stimulus before and after L4 lethargus. (PDF 381 kb)
Mean velocities are not affected by deprivation. No significant differences were found between mean velocities (averaged over the 30 min of the assay) of undisrupted or deprived wild-type animals, ire-1 mutants, or ubl-5 mutants. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses. (PDF 564 kb)
Long-term mean velocities with vm twitching. Mean velocities and vm twitching were significantly correlated in wild-type animals, ubl-5 mutants, and undisrupted ire-1 mutants. Notably, these correlations were stronger in sleep-deprived wild-type animals and ubl-5 mutants as compared to undisrupted worms of the corresponding genotype. However, correlations did not increase in ire-1 mutants, suggesting that secreted proteins may be required for deprivation-induced enhancement of coordination between vm activity and locomotion. (PDF 481 kb)
Acknowledgements
We thank D. Raizen, K. Collins, M. Treinin, G. Stanfield, and S. Chalasani for reagents. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the National Science Foundation (NSF, IOS 1256989 to DB) and the Howard Hughes Medical Institute International (HHMI) Student Research fellowship to MS.
Funding
Funding was provided by the NSF (IOS 1256989, to DB).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
Authors’ contributions
JS, MS, and DB conceptualized the study; JS, MS, IM, and DB developed the methodology; JS, MS, and IM were responsible for the investigation; JS, MS, and DB for the software; JS, MS, IM, and DB for the formal Analysis; and JS, MS, and DB for the visualization. DB wrote the original draft, and JS, MS, IM, and DB reviewed and edited the manuscript. DB was responsible for funding acquisition and supervision. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0407-1) contains supplementary material, which is available to authorized users.
References
- 1.Halbower AC, Degaonkar M, Barker PB, Earley CJ, Marcus CL, Smith PL, Prahme MC, Mahone EM. Childhood obstructive sleep apnea associates with neuropsychological deficits and neuronal brain injury. PLoS Med. 2006;3:1391–402. doi: 10.1371/journal.pmed.0030301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jan JE, Reiter RJ, Bax MCO, Ribary U, Freeman RD, Wasdell MB. Long-term sleep disturbances in children: a cause of neuronal loss. Eur J Paediatr Neurol. 2010;14:380–90. doi: 10.1016/j.ejpn.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Owens J. Insufficient sleep in adolescents and young adults: an update on causes and consequences. Pediatrics. 2014;134:e921–32. doi: 10.1542/peds.2014-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kayser MS, Yue Z, Sehgal A. A critical period of sleep for development of courtship circuitry and behavior in Drosophila. Science. 2014;344:269–74. doi: 10.1126/science.1250553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byerly L, Cassada RC, Russell RL. The life cycle of the nematode Caenorhabditis elegans. Dev Biol. 1976;51:23–33. doi: 10.1016/0012-1606(76)90119-6. [DOI] [PubMed] [Google Scholar]
- 6.Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You Y-J, Sundaram MV, Pack AI. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–72. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- 7.Singh RN, Sulston JE. Some observations on moulting in Caenorhabditis Elegans. Nematologica. 1978;24:63–71. doi: 10.1163/187529278X00074. [DOI] [Google Scholar]
- 8.Iwanir S, Tramm N, Nagy S, Wright C, Ish D, Biron D. The microarchitecture of C. elegans behavior during lethargus: homeostatic bout dynamics, a typical body posture, and regulation by a central neuron. Sleep. 2013;36:385–95. doi: 10.5665/sleep.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz J, Spies J-P, Bringmann H. Reduced muscle contraction and a relaxed posture during sleep-like Lethargus. Worm. 2012;1:0–2. [DOI] [PMC free article] [PubMed]
- 10.Driver RJ, Lamb AL, Wyner AJ, Raizen DM. DAF-16/FOXO regulates homeostasis of essential sleep-like behavior during larval transitions in C. elegans. Curr Biol. 2013;23:501–6. doi: 10.1016/j.cub.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagy S, Tramm N, Sanders J, Iwanir S, Shirley IA, Levine E, Biron D. Homeostasis in C. elegans sleep is characterized by two behaviorally and genetically distinct mechanisms. Elife. 2014;3:1–21. doi: 10.7554/eLife.04380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JY, Sternberg PW. Multilevel modulation of a sensory motor circuit during C. elegans sleep and arousal. Cell. 2014;156:249–60. doi: 10.1016/j.cell.2013.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh K, Chao MY, Somers GA, Komatsu H, Corkins ME, Larkins-Ford J, Tucey T, Dionne HM, Walsh MB, Beaumont EK, Hart DP, Lockery SR, Hart AC. C. elegans Notch signaling regulates adult chemosensory response and larval molting quiescence. Curr Biol. 2011;21:825–34. doi: 10.1016/j.cub.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tramm N, Oppenheimer N, Nagy S, Efrati E, Biron D. Why do sleeping nematodes adopt a hockey-stick-like posture? PLoS One. 2014;9:e101162. doi: 10.1371/journal.pone.0101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh K, Ju JY, Walsh MB, DiIorio MA, Hart AC. Deep conservation of genes required for both Drosphila melanogaster and Caenorhabditis elegans sleep includes a role for dopaminergic signaling. Sleep. 2014;37(9):1439–51. doi: 10.5665/sleep.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–80. doi: 10.1016/S0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 17.Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–45. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 18.Lee RYN, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–7. doi: 10.1016/S0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- 19.Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science. 2003;300(5627):1921. doi: 10.1126/science.1080147. [DOI] [PubMed] [Google Scholar]
- 20.Miyata S, Begun J, Troemel ER, Ausubel FM. DAF-16-dependent suppression of immunity during reproduction in Caenorhabditis elegans. Genetics. 2008;178:903–18. doi: 10.1534/genetics.107.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, Emanuel BS, Rovera G, Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 22.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–9. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 23.Kaestner KH, Knöchel W, Martínez DE, Kno W, Martı DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–6. [PubMed] [Google Scholar]
- 24.Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–51. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 25.Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science. 2000;287:1834–7. doi: 10.1126/science.287.5459.1834. [DOI] [PubMed] [Google Scholar]
- 26.Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature. 2002;417:287–91. doi: 10.1038/417287a. [DOI] [PubMed] [Google Scholar]
- 27.Cirelli C. Cellular consequences of sleep deprivation in the brain. Sleep Med Rev. 2006;10:307–21. doi: 10.1016/j.smrv.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Naidoo N. Cellular stress/the unfolded protein response: relevance to sleep and sleep disorders. Philadelphia: Elsevier Ltd; 2009. pp. 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naidoo N. Roles of endoplasmic reticulum and energetic stress in disturbed sleep. Neuromolecular Med. 2012;14:213–9. doi: 10.1007/s12017-012-8179-9. [DOI] [PubMed] [Google Scholar]
- 30.Cirelli C, Tononi G. Gene expression in the brain across the sleep-waking cycle. Brain Res. 2000;885:303–21. doi: 10.1016/S0006-8993(00)03008-0. [DOI] [PubMed] [Google Scholar]
- 31.Heschl MFP, Baillie DL. The HSP70 multigene family of Caenorhabditis elegans. Comp Biochem Physiol B. 1990;96(4):633–7. doi: 10.1016/0305-0491(90)90206-9. [DOI] [PubMed] [Google Scholar]
- 32.Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/S0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 33.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 34.Urano F, Calfon M, Yoneda T, Yun C, Kiraly M, Clark SG, Ron D. A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J Cell Biol. 2002;158:639–46. doi: 10.1083/jcb.200203086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2 Suppl 1):S102–9. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 36.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146(6):743–50. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 37.Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–47. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terao A, Steininger TL, Hyder K, Apte-Deshpande A, Ding J, Rishipathak D, Davis RW, Heller HC, Kilduff TS. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience. 2003;116:187–200. doi: 10.1016/S0306-4522(02)00695-4. [DOI] [PubMed] [Google Scholar]
- 39.Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/S0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- 40.Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92:1150–7. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- 41.Naidoo N, Casiano V, Cater J, Zimmerman J, Pack AI. A role for the molecular chaperone protein BiP/GRP78 in Drosophila sleep homeostasis. Sleep. 2007;30:557–65. doi: 10.1093/sleep/30.5.557. [DOI] [PubMed] [Google Scholar]
- 42.Everson CA, Gilliland MA, Kushida CA, Pilcher JJ, Fang VS, Refetoff S, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: IX. Recovery. Sleep. 1988;12:60–7. [PubMed] [Google Scholar]
- 43.Jung CM, Melanson EL, Frydendall EJ, Perreault L, Eckel RH, Wright KP. Energy expenditure during sleep, sleep deprivation and sleep following sleep deprivation in adult humans. J Physiol. 2011;589(Pt 1):235–44. doi: 10.1113/jphysiol.2010.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Sleep and brain energy levels: ATP changes during sleep. J Neurosci. 2010;30:9007–16. doi: 10.1523/JNEUROSCI.1423-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steriade M. Impact of network activities on neuronal properties in corticothalamic systems. J Neurophysiol. 2001;86:1–39. doi: 10.1152/jn.2001.86.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Timofeev I, Grenier F, Steriade M. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc Natl Acad Sci U S A. 2001;98:1924–9. doi: 10.1073/pnas.98.4.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–93. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 48.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase—development of the energy sensor concept. J Physiol. 2006;574(Pt 1):7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jing M, Ismail-Beigi F. Role of 5’-AMP-activated protein kinase in stimulation of glucose transport in response to inhibition of oxidative phosphorylation. Am J Physiol Cell Physiol. 2006;290:C484–91. doi: 10.1152/ajpcell.00321.2005. [DOI] [PubMed] [Google Scholar]
- 50.Nikonova EV, Naidoo N, Zhang L, Romer M, Cater JR, Scharf MT, Galante RJ, Pack AI. Changes in components of energy regulation in mouse cortex with increases in wakefulness. Sleep. 2010;33:889–900. doi: 10.1093/sleep/33.7.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;2004(Pt 18):4055–66. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 52.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–39. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–80. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 54.Haynes CM, Ron D. The mitochondrial UPR — protecting organelle protein homeostasis. J Cell Sci. 2010;123(Pt 22):3849–55. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 55.Kimura K, Tanaka N, Nakamura N, Takano S, Ohkuma S. Knockdown of mitochondrial heat shock protein 70 promotes progeria-like phenotypes in Caenorhabditis elegans. J Biol Chem. 2007;282:5910–8. doi: 10.1074/jbc.M609025200. [DOI] [PubMed] [Google Scholar]
- 56.Marques FC, Volovik Y, Cohen E. The roles of cellular and organismal aging in the development of late-onset maladies. 2015. [DOI] [PubMed]
- 57.Mardones P, Martınez G, Hetz C. Control of systemic proteostasis by the nervous system. Trends Cell Biol. 2015;25:1–10. doi: 10.1016/j.tcb.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Avery L, Horvitz H. Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron. 1989;3:473–85. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- 59.Avery L. The genetics of feeding in Caenorhabditis elegans. Genetics. 1993;133:897–917. doi: 10.1093/genetics/133.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albertson DG, Thomson JN. The pharynx of Caenorhabditis elegans. Philos Trans R Soc London B. 1976;275:299–325. doi: 10.1098/rstb.1976.0085. [DOI] [PubMed] [Google Scholar]
- 61.Raizen DM, Lee RY, Avery L. Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics. 1995;141:1365–82. doi: 10.1093/genetics/141.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shtonda BB, Avery L. Dietary choice behavior in Caenorhabditis elegans. J Exp Biol. 2006;209:89–102. doi: 10.1242/jeb.01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song B, Faumont S, Lockery S, Avery L. Recognition of familiar food activates feeding via an endocrine serotonin signal. Elife. 2013;2:e00329. doi: 10.7554/eLife.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hobson RJ, Hapiak VM, Xiao H, Buehrer KL, Komuniecki PR, Komuniecki RW. SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics. 2006;172:159–69. doi: 10.1534/genetics.105.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scholz M, Lynch DJ, Lee KS, Levine E, Biron D. A scalable method for automatically measuring pharyngeal pumping in C. elegans. J Neurosci Methods. 2016;274:172–8. doi: 10.1016/j.jneumeth.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 66.Hobson RJ, Geng J, Gray AD, Komuniecki RW. SER-7b, a constitutively active Galphas coupled 5-HT7-like receptor expressed in the Caenorhabditis elegans M4 pharyngeal motorneuron. J Neurochem. 2003;87:22–9. doi: 10.1046/j.1471-4159.2003.01967.x. [DOI] [PubMed] [Google Scholar]
- 67.Song B, Avery L. Serotonin activates overall feeding by activating two separate neural pathways in Caenorhabditis elegans. J Neurosci. 2012;32:1920–31. doi: 10.1523/JNEUROSCI.2064-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ward S, Carrel JS. Fertilization and sperm competition in the nematode Caenorhabditis elegans. Dev Biol. 1979;73:304–21. doi: 10.1016/0012-1606(79)90069-1. [DOI] [PubMed] [Google Scholar]
- 69.Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO. A conserved checkpoint pathway mediates DNA damage—induced apoptosis and cell cycle arrest in C. elegans. Mol Cell. 2000;5:435–43. doi: 10.1016/S1097-2765(00)80438-4. [DOI] [PubMed] [Google Scholar]
- 70.Aballay A, Ausubel FM. Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc Natl Acad Sci U S A. 2001;98:2735–9. doi: 10.1073/pnas.041613098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alpi A, Pasierbek P, Gartner A, Loidl J. Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma. 2003;112:6–16. doi: 10.1007/s00412-003-0237-5. [DOI] [PubMed] [Google Scholar]
- 72.Salinas LS, Maldonado E, Navarro RE. Stress-induced germ cell apoptosis by a p53 independent pathway in Caenorhabditis elegans. Cell Death Differ. 2006;13:2129–39. doi: 10.1038/sj.cdd.4401976. [DOI] [PubMed] [Google Scholar]
- 73.Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A. Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell. 2008;15:87–97. doi: 10.1016/j.devcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 75.Yuan J, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-H. [DOI] [PubMed] [Google Scholar]
- 76.Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 1996;10:1073–83. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- 77.Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–9. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 78.Conradt B, Horvitz HR. The C. elegans Protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–29. doi: 10.1016/S0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 79.Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7:97–108. doi: 10.1038/nrm1836. [DOI] [PubMed] [Google Scholar]
- 80.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/S0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 81.Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann R, Hengartner M, Schedl T, Gartner A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell. 2005;120:357–68. doi: 10.1016/j.cell.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 82.Gartner A, Boag PR, Blackwell TK. Germline survival and apoptosis. WormBook 2008;Sep 4:1–20. doi:10.1895/wormbook.1.145.1. [DOI] [PMC free article] [PubMed]
- 83.Collins KM, Bode A, Fernandez RW, Tanis JE, Brewer J, Creamer MS, Koelle MR. Activity of the C. elegans egg-laying behavior circuit is controlled by competing activation and feedback inhibition. Elife. 2016;5:1–24. doi: 10.7554/eLife.21126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57. doi: 10.1016/0092-8674(89)90171-2. [DOI] [PubMed] [Google Scholar]
- 85.Liu ZC, Ambros V. Heterochronic genes control the stage-specific initiation and expression of the dauer larva developmental program in Caenorhabditis elegans. Genes Dev. 1989;3(12 B):2039–49. doi: 10.1101/gad.3.12b.2039. [DOI] [PubMed] [Google Scholar]
- 86.Thein MC, McCormack G, Winter AD, Johnstone IL, Shoemaker CB, Page AP. Caenorhabditis elegans exoskeleton collagen COL-19: an adult-specific marker for collagen modification and assembly, and the analysis of organismal morphology. Dev Dyn. 2003;226:523–39. doi: 10.1002/dvdy.10259. [DOI] [PubMed] [Google Scholar]
- 87.Hou NS, Gutschmidt A, Choi DY, Pather K, Shi X, Watts JL, Hoppe T, Taubert S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci U S A. 2014;111:E2271–80. doi: 10.1073/pnas.1318262111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee KS, Iwanir S, Kopito RB, Scholz M, Calarco JA, Biron D, Levine E. Serotonin-dependent kinetics of feeding bursts underlie a graded response to food availability in C. elegans. Nat Commun. 2017;8:14221. doi: 10.1038/ncomms14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Albeg A, Smith CJ, Chatzigeorgiou M, Feitelson DG, Hall DH, Schafer WR, Miller DM, Treinin M. C. elegans multi-dendritic sensory neurons: morphology and function. Mol Cell Neurosci. 2011;46:308–17. doi: 10.1016/j.mcn.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–70. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- 91.Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron. 1995;14:871–7. doi: 10.1016/0896-6273(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 92.Treinin M, Gillo B, Liebman L, Chalfie M. Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc Natl Acad Sci U S A. 1998;95:15492–5. doi: 10.1073/pnas.95.26.15492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sanders J, Nagy S, Fetterman G, Wright C, Treinin M, Biron D. The Caenorhabditis elegans interneuron ALA is (also) a high-threshold mechanosensor. BMC Neurosci. 2013;14:156. doi: 10.1186/1471-2202-14-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsalik EL, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O. LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev Biol. 2003;263:81–102. doi: 10.1016/S0012-1606(03)00447-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dernovici S, Starc T, Dent JA, Ribeiro P. The serotonin receptor SER-1 (5HT2ce) contributes to the regulation of locomotion in Caenorhabditis elegans. Dev Neurobiol. 2007;67:189–204. doi: 10.1002/dneu.20340. [DOI] [PubMed] [Google Scholar]
- 96.Alvarenga TA, Hirotsu C, Mazaro-Costa R, Tufik S, Andersen ML. Impairment of male reproductive function after sleep deprivation. Fertil Steril. 2015;103:1355–1362. doi: 10.1016/j.fertnstert.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 97.Torres M, Laguna-Barraza R, Dalmases M, Calle A, Pericuesta E, Montserrat JM, Navajas D, Gutierrez-Adan A, Farré R. Male fertility is reduced by chronic intermittent hypoxia mimicking sleep apnea in mice. Sleep. 2014;37:1757–65. doi: 10.5665/sleep.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levi-Ferber M, Salzberg Y, Safra M, Haviv-Chesner A, Bülow HE, Henis-Korenblit S. It’s all in your mind: determining germ cell fate by neuronal IRE-1 in C. elegans. PLoS Genet. 2014;10:e1004747. doi: 10.1371/journal.pgen.1004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calhoun AJ, Tong A, Pokala N, Fitzpatrick JAJ, Sharpee TO, Chalasani SH. Neural mechanisms for evaluating environmental variability in Caenorhabditis elegans. Neuron. 2015;86:428–41. doi: 10.1016/j.neuron.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeiser E, Frokjaer-Jensen C, Jorgensen E, Ahringer J. MosSCI and gateway compatible plasmid toolkit for constitutive and inducible expression of transgenes in the C. elegans germline. PLoS One. 2011;6(5):e20082. doi: 10.1371/journal.pone.0020082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hansen JM, Chavez DR, Stanfield GM. COMP-1 promotes competitive advantage of nematode sperm. Elife. 2015;2015:1–26. doi: 10.7554/eLife.05423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–47. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Collins KM, Koelle MR. Postsynaptic ERG potassium channels limit muscle excitability to allow distinct egg-laying behavior states in Caenorhabditis elegans. J Neurosci. 2013;33:761–75. doi: 10.1523/JNEUROSCI.3896-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Waggoner LE, Zhou GT, Schafer RW, Schafer WR. Control of alternative behavioral states by serotonin in Caenorhabditis elegans. Neuron. 1998;21:203–14. doi: 10.1016/S0896-6273(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 105.Strand FL. Neuropeptides: General characteristics and neuropharmaceutical potential in treating CNS disorders. Prog Drug Res. 2003;61:1–37. doi: 10.1007/978-3-0348-8049-7_1. [DOI] [PubMed] [Google Scholar]
- 106.Pilcher JJ, Huffcutt AI. Effects of sleep deprivation on performance: a meta-analysis. Sleep. 1996;19:318–26. doi: 10.1093/sleep/19.4.318. [DOI] [PubMed] [Google Scholar]
- 107.Harrison Y, Home JA. The impact of sleep deprivation on decision making: a review. J Exp Psychol Appl. 2000;6:236–49. doi: 10.1037/1076-898X.6.3.236. [DOI] [PubMed] [Google Scholar]
- 108.Alhola P, Polo-Kantola P. Sleep deprivation: impact on cognitive performance. Neuropsychiatr Dis Treat. 2007;3(5):553–67. [PMC free article] [PubMed] [Google Scholar]
- 109.Chee MWL, Chuah LYM. Functional neuroimaging insights into how sleep and sleep deprivation affect memory and cognition. Curr Opin Neurol. 2008;21:417–23. doi: 10.1097/WCO.0b013e3283052cf7. [DOI] [PubMed] [Google Scholar]
- 110.Cirelli C, Shaw PJ, Rechtschaffen A, Tononi G. No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res. 1999;840:184–93. doi: 10.1016/S0006-8993(99)01768-0. [DOI] [PubMed] [Google Scholar]
- 111.Gopalakrishnan A, Ji LL, Cirelli C. Sleep deprivation and cellular responses to oxidative stress. Sleep. 2004;27:27–35. doi: 10.1093/sleep/27.1.27. [DOI] [PubMed] [Google Scholar]
- 112.Vyazovskiy VV, Harris KD. Sleep and the single neuron: the role of global slow oscillations in individual cell rest. Nat Rev Neurosci. 2013;14:443–51. doi: 10.1038/nrn3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zimmerman JE, Rizzo W, Shockley KR, Raizen DM, Naidoo N, Mackiewicz M, Churchill GA, Pack AI. Multiple mechanisms limit the duration of wakefulness in Drosophila brain. Physiol Genomics. 2006;27:337–50. doi: 10.1152/physiolgenomics.00030.2006. [DOI] [PubMed] [Google Scholar]
- 114.Cirelli C, LaVaute TM, Tononi G. Sleep and wakefulness modulate gene expression in Drosophila. J Neurochem. 2005;94:1411–9. doi: 10.1111/j.1471-4159.2005.03291.x. [DOI] [PubMed] [Google Scholar]
- 115.Wang H, Liu Y, Briesemann M, Yan J. Computational analysis of gene regulation in animal sleep deprivation. Physiol Genomics. 2010;42:427–36. doi: 10.1152/physiolgenomics.00205.2009. [DOI] [PubMed] [Google Scholar]
- 116.Jones S, Pfister-Genskow M, Benca RM, Cirelli C. Molecular correlates of sleep and wakefulness in the brain of the white-crowned sparrow. J Neurochem. 2008;105:46–62. doi: 10.1111/j.1471-4159.2007.05089.x. [DOI] [PubMed] [Google Scholar]
- 117.Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O’Hara BF, Franken P, Tafti M. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–5. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anafi RC, Pellegrino R, Shockley KR, Romer M, Tufik S, Pack AI. Sleep is not just for the brain: transcriptional responses to sleep in peripheral tissues. BMC Genomics. 2013;14:362. doi: 10.1186/1471-2164-14-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siegel JM. Clues to the functions of mammalian sleep. Nature. 2005;437:1264–71. doi: 10.1038/nature04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 121.Tononi G, Cirelli C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron. 2014;81:12–34. doi: 10.1016/j.neuron.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Varshavsky A. Augmented generation of protein fragments during wakefulness as the molecular cause of sleep: a hypothesis. Protein Sci. 2012;21:1634–61. doi: 10.1002/pro.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kang J-E, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-B dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience. 2008;156:71–80. doi: 10.1016/j.neuroscience.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–25. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 126.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–178. doi: 10.1016/j.smrv.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reutrakul S, Van Cauter E. Interactions between sleep, circadian function, and glucose metabolism: implications for risk and severity of diabetes. Ann N Y Acad Sci. 2014;1311:151–73. doi: 10.1111/nyas.12355. [DOI] [PubMed] [Google Scholar]
- 129.Nagy S, Raizen DM, Biron D. Measurements of behavioral quiescence in Caenorhabditis elegans. Methods. 2014;68:500–7. doi: 10.1016/j.ymeth.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kopito RB, Levine E. Durable spatiotemporal surveillance of Caenorhabditis elegans response to environmental cues. Lab Chip. 2014;14:764–70. doi: 10.1039/C3LC51061A. [DOI] [PubMed] [Google Scholar]
- 131.Lynch D, Biron D. A primer on prototyping. Methods Mol Biol. 2015;1327:231–40. doi: 10.1007/978-1-4939-2842-2_17. [DOI] [PubMed] [Google Scholar]
- 132.Jing G, Siong C, Rajan D. Foreground motion detection by difference-based spatial temporal entropy image. TENCON 2004. 2004 IEEE Region 10 Conference, vol. A, p. 379–82.
- 133.Lockery SR, Lawton KJ, Doll JC, Faumont S, Coulthard SM, Thiele TR, Chronis N, McCormick KE, Goodman MB, Pruitt BL. Artificial dirt: microfluidic substrates for nematode neurobiology and behavior. J Neurophysiol. 2008;99:3136–43. doi: 10.1152/jn.91327.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fluorescence of the hsp-60p::GFP fluorescent reporter before and after deprivation. In our hands, elevated expression of the reporter was not observed after 1 h of disrupting worm sleep. (PDF 368 kb)
Pumping rates increase as a function of food availability. The average pumping rate of wild-type animals at different concentrations of ambient bacterial food (as measured by optical density, OD 600). N = 10 animals per condition. Error bars depict mean ± standard error of the mean (SEM). (PDF 318 kb)
Duty ratios of rapid pumping of deprived and control worms. (A) Box plots of duty ratios for control and deprived animals. Continuous pumping was defined as a period in which the delay between pumps did not exceed 500 ms. (B) Same as (A) for UPR mutants and the neuronal rescue of daf-16. (PDF 1224 kb)
Genetic ablation of the mec-10 expressing touch neurons abolishes responses to the vibration stimuli. Locomotion in response to 1-kHz vibrations was quantified as the fraction of the body area the animal traversed per second. Wild-type animals (left) responded robustly to the stimulus, while touch-insensitive mec-10p::deg-3(gf) transgenics (right) did not exhibit a detectable response. N = 20 animals from each genotype were assayed, and shaded areas depict mean ± SEM. (PDF 468 kb)
EPG measurements of pumping fatigue are consistent with the results of the optical tracking method. Instantaneous pumping rates were calculated as 1/(duration between consecutive contraction peaks). Mean (per animal) rates for wild-type animals, daf-16(mu86) mutants, and ubl-5(ok3389) mutants reproduced the phenotypes detected by optical tracking. (PDF 480 kb)
The duration of an individual pumping motion is extended by exposure to mechanical vibrations irrespective of the timing of the stimuli. (A) Top: a sample EPG trace of a wild-type animal in the presence of food at OD600 = 2.5 concentration. Peaks correspond to corpus and terminal bulb contraction. Troughs correspond to corpus relaxation. Bottom: average contraction and relaxation EPG traces for undisrupted and deprived wild-type animals. Distributions of pump durations are shown for undisrupted, deprived, and control (exposed to vibrations outside of lethargus) animals. The outline of the distribution for undisrupted animals was duplicated as a guide to the eye. (B) The mean (per animal) pump durations and amplitudes of EPG peaks and troughs for wild-type animals (top) and daf-16(mu86) mutants (bottom). (C) Same as (A, B) for UPRmt deficient ubl-5 mutants. (D) Same as (A, B) for mutants treated with 10 mM 5-HT instead of food. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses; asterisks and double asterisks denote significant differences (p < 0.05 and p < 0.01, respectively). (PDF 774 kb)
EPG characteristics of individual pumping motions. Mean (per animal) EPG peak and trough amplitudes for animals presented with food or 10 mM 5-HT. Vibration stimuli during or outside lethargus did not typically affect these amplitudes. Sample sizes are noted in parentheses, and asterisks denote significant differences (p < 0.05). (PDF 1006 kb)
(A) Brood size can be reduced by sleep depriving either males or hermaphrodites. The numbers of egg laid by daf-16(mgDf50) mutants that mated with males. Either the males, the hermaphrodites, or both were sleep deprived. (B) ASI neurons are required for sleep deprivation to impact fecundity. Brood size was not reduced by deprivation when ASI neurons were genetically ablated using tetanus toxin (in contrast to phenotypes shown in Fig. 6a–c, f). (PDF 176 kb)
Nonlethal sleep deprivation does not increase egg retention. The numbers of fertilized eggs retained in the uterus of wild-type animals and daf-16 mutants 24 h and 48 h after L4 lethargus. Deprived animals were exposed to the stimulus before, during, and after L4 lethargus. Control animals were exposed to the stimulus before and after L4 lethargus. (PDF 381 kb)
Mean velocities are not affected by deprivation. No significant differences were found between mean velocities (averaged over the 30 min of the assay) of undisrupted or deprived wild-type animals, ire-1 mutants, or ubl-5 mutants. Horizontal lines, boxes, and bars depict medians, 1st and 3rd quartiles, and 5th and 95th percentiles, respectively. Sample sizes are noted in parentheses. (PDF 564 kb)
Long-term mean velocities with vm twitching. Mean velocities and vm twitching were significantly correlated in wild-type animals, ubl-5 mutants, and undisrupted ire-1 mutants. Notably, these correlations were stronger in sleep-deprived wild-type animals and ubl-5 mutants as compared to undisrupted worms of the corresponding genotype. However, correlations did not increase in ire-1 mutants, suggesting that secreted proteins may be required for deprivation-induced enhancement of coordination between vm activity and locomotion. (PDF 481 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files.