

Abstract
DNA base damage and non-coding apurinic/apyrimidinic (AP) sites are ubiquitous types of damage that must be efficiently repaired to prevent mutations. These damages can occur in both the nuclear and mitochondrial genomes. Base excision repair (BER) is the frontline pathway for identifying and excising damaged DNA bases in both of these cellular compartments. Recent advances demonstrate that BER does not operate as an isolated pathway but rather dynamically interacts with components of other DNA repair pathways to modulate and coordinate BER functions. We define the coordination and interaction between DNA repair pathways as pathway crosstalk. Numerous BER proteins are modified and regulated by post-translational modifications (PTMs), and PTMs could influence pathway crosstalk. Here, we present recent advances on BER/DNA repair pathway crosstalk describing specific examples and also highlight regulation of BER components through PTMs. We have organized and reported functional interactions and documented PTMs for BER proteins into a consolidated summary table. We further propose the concept of DNA repair hubs that coordinate DNA repair pathway crosstalk to identify central protein targets that could play a role in designing future drug targets.
Keywords: DNA Damage Base Excision Repair, BER DNA pathway crosstalk DNA repair hubs Post-translational modifications, PTMs
1. Introduction
DNA contained in both the nuclear and mitochondrial cellular compartments is subject to damage from multiple sources1–3. Diverse classes of DNA damage are caused by exogenous sources such as UV, ionizing radiation, alkylating agents, and heavy metals4–8. Endogenous sources of damage such as reactive oxygen species (ROS), are generated during normal metabolic functions as well as various cellular transactions2,9. Cells have therefore evolved numerous DNA repair and tolerance pathways to protect the genome from these types of damage.
Originally, each DNA repair pathway was analyzed in isolation to define repair of a specific subtype of DNA damage. For instance, base excision repair (BER) handles non-bulky DNA base damage, nucleotide excision repair (NER) manages bulky lesions, and homologous recombination (HR) repairs double strand breaks in S phase10. As each DNA repair pathway was characterized beyond the individual biochemical steps, it became apparent that coordination between DNA repair pathways is essential for proper cellular responses to DNA damage. We refer to such coordination as pathway crosstalk. In this context, pathway crosstalk occurs when components of one, biochemically distinct DNA repair pathway influence the repair of a substrate that is corrected by a different DNA repair pathway. For example, components of NER are indispensible for efficient repair of BER substrates through interactions with several N-glycosylases that initiate BER11–13. We focus on pathway crosstalk events primarily mediated through protein-protein interactions and extend the analysis of BER proteins to post-translational modifications (PTMs) that could affect BER activity and pathway crosstalk in response to DNA damage through multiple mechanisms. We describe several classical, as well as recently reported examples of pathway crosstalk, with an emphasis on how BER components are regulated in human cells.
Base excision repair is crucial for maintaining genome integrity through repair of non-bulky base damage in both the nuclear and mitochondrial cellular compartments1,3,9. As depicted in Figure 1, the N-glycosylase proteins initiate BER when they recognize and cleave a specific subset of DNA base damage leaving an apurinic/apyrimidinic (AP) site. Bifunctional N-glycosylases contain an AP lyase activity that further cleaves the DNA phosphodiester backbone resulting in single strand breaks. AP endonuclease (APEX1) performs end-cleaning duties following glycosylase AP lyase activity or cleaves the AP site following the action of a monofunctional N-glycosylase. Further processing by DNA polymerase β and subsequent ligation result in repair of the initial damage site9. Proper regulation and completion of each BER step is crucial as AP sites and single strand break intermediates created during the repair process are themselves types of DNA damage. Defining how DNA repair pathway proteins interact to ensure efficient completion of each BER intermediate step is therefore critical for understanding BER regulation within the context of genome stability.
Figure 1. Key human N-glycosylases and APEX1 interactions with components of other DNA repair pathways enhance BER activity.
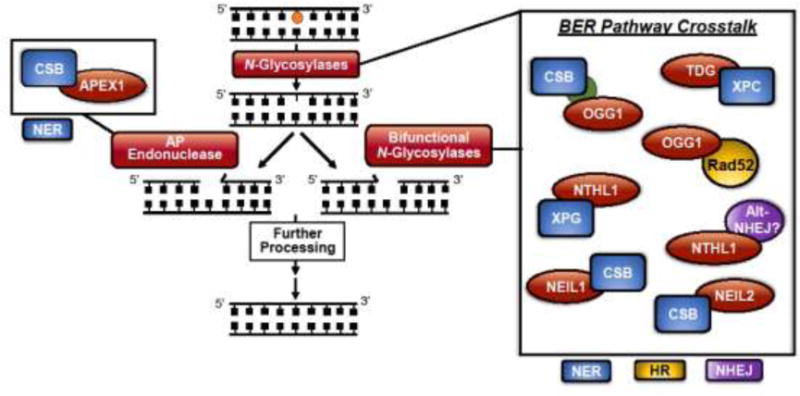
In the initiating steps of BER, a damage substrate is detected, and an N-glycosylase cleaves the damaged base, leaving an apurinic/apyrimidinic (AP) site. Bifunctional glycosylases cleave the phosphodiester backbone and create a single-strand break, while monofunctional glycosylases require APEX1-mediated cleavage of the phosphodiester backbone. Further processing results in repair of the initial damage site. Functional interactions with BER proteins in this review are depicted, with an emphasis on the initiating steps of BER. Pathway crosstalk of BER proteins (red) at the initiating steps of BER includes interactions with components of the NER (blue), HR (yellow), and alt-NHEJ (purple) pathways. We highlight recent advances that provide insight into BER functions, and the interactions displayed are discussed in this review.
Of note, a majority of BER crosstalk with other DNA repair pathways takes place at the initiating steps of BER. As BER glycosylases generate apurinic/apyrimidinic (AP) sites and/or strand breaks, which are themselves forms of DNA damage14,15, proper coordination and regulation of BER glycosylase proteins is important to ensure these intermediates do not accumulate. APEX1 (also known as APE1) also generates single strand breaks during BER and the coordinated handoff from APEX1 to downstream BER proteins must be properly regulated to avoid accumulation of BER intermediates.
One mechanism that could help coordinate DNA repair pathway regulation is PTMs of individual DNA repair proteins. Numerous examples of PTMs modulating cellular activities have been reported16–18. However, in only a small number of cases have sites of PTMs been defined and analyzed on BER proteins. Furthermore, the biological consequence of many of these BER protein PTMs have yet to be defined. General functions of PTMs, which may be relevant to BER regulation, include, but are not limited to modulating protein-protein interactions, pathway cascade signaling, cellular localization, conformational changes, and protein stability18,19. The research into BER protein PTM elucidation is expanding and several advances will be covered in this review. Table 1 summarizes documented PTM modifications on BER proteins together with documented BER protein interactions that aggregate data into a readily accessible resource.
Table 1. Human BER proteins undergo posttranslational modifications (PTMs).
A panel of BER proteins is represented with characterized PTMs and PTMs reported in the PHOSIDA database (www.PHOSIDA.com)69,70. PHOSIDA is a phosphorylation and acetylation database derived from experimental analysis of proteins from different species and conditions. Functional interactions characterized for human BER proteins are included to demonstrate the breadth of BER protein associations.
| BER Prote in | Activity | Level of Regulation | ||
|---|---|---|---|---|
| Post-translational Modifications (Literature) | PHOSIDA Post-translational Modifications (Human69’70) | Functional Interactions | ||
| UNG1/2 | Uracil DNA Glycosylase | Phosphorylation at T6 and T126110; Phosphorylation at S23, T60, S6486 | Phosphorylation at S12, S14, S23, S40, S54, S55, S58, S63, S64, S67; T31, T60, T51 Acetylation at K286, K5, K295 | Glycosylase activity increased by APEX1111; Associated with RPA88, XRCC1112, PCNA89, and PPM1D110 |
| SMUG1 | – | Phosphorylation at S241 | Glycosylase activity increased by APEX1113 | |
| MPG | 3-methyl adenine DNA Glycosylase | Acetylation114 | – | Association with hHR23115, XRCC1116, and ERα114 |
| NTHL1 | Endonuclease III DNA Glycosylase | Sumoylation85 | Phosphorylation at S71 | Glycosylase activity increased by APEX1117; Associated with XPG11, XRCC1116 and YB-1118 |
| NEIL1 | Endonuclease VIII DNA Glycosylase | – | – | Associated with FEN1119, XRCC1120, PCNA121, Pol δ121, CSB13, RPA122 and PARP1123 |
| NEIL2 | Acetylation K49 and K153124 | – | Interacts with p300124,125, XRCC1116, and YB-1126 | |
| NEIL3 | – | – | Associated with RPA127 | |
| OGG1 | 8-Oxoguanine DNA Glycosylase | Phosphorylation at S326128’129 Acetylation at K41 and K338130; Nitrosylated131 | – | Glycosylase activity increased by APEX1132; Associated with XRCC1133, 134, SIRT3135 |
| MUTYH | A-G Mismatch DNA Glycosylase | Phosphorylation at S524136,137 Ubiquitination90 | – | Associated with APEX1138, PCNA138, RPA138, MSH6139, and 91-1 complex140 |
| MBD4 | G-T Mismatch DNA Glycosylase | – | Phosphorylation at S112 | Associated with MLH1141, and HDAC1142, DMNT3B143 |
| TDG | Acetylation83 and Sumoylation151 | – | Glycosylase activity increased by APEX127. Associated with CBP/p30083, DMNT3A144, DMNT3B143, RXR/RAR145, SCR1146, p73α147, p63γ147, ERα148, RAD9149, XPC24, NEIL1/NEIL2150, SIRT177 | |
| APEX1 | AP Endonuclease | Acetylation at K27, K31, K32, K3598,152,153; Phosphorylation at T22396; Glutathionylation at C99103; Ubiquitination at K24, K25, K27154 | Phosphorylation at S128, K196 | Associated with UNG1111, SMUG113, NTHL1117, OGG1132, TDG133, MUTYH138, XRCC1155, TP53156, HSP70157, PCNA158, LIG1159, FEN1158, 159, WRN160 |
| APEX2 | – | – | Associated with PCNA161 | |
| XRCC1 | X-ray Repair Cross Complementing Group 1 | Phosphorylation162, 163 | Phosphorylation at Y211, Y515, T198, T257, T367, T440, T453, T519, T523; S199, S226, S229, S236, S241, S259, S266, S268, S357, S408, S409, S410, S416, S418, S421, S446, S447, S518, S525 | Associated with OGG1134, NTHL1116, NEIL1116, NEIL2116, MPG116, POLβ164–166, LIG3167–170, PARP1171, 172, PARP2172, UNG2112 PNK120, CK2162, TDP1173, ATPX174, PCNA175, and APEX1120 |
| PARP1 | Poly (ADP-ribose) Polymerase | Phosphorylation mapping176, S372/T373177, Y907178 Sumoylation at K486179, 180 and K203181; Acetylation at K498, K505, K508, K521, and K524182 Méthylation at K508183, K528184 Ribosylation mapping185 | Acetylation at K96, K104, K130, K547, K550, K599, K620 Phosphorylation at T94, S176, S178, S273, S464, S781, S863 | Enzyme activity increase by ROS186; Associated with LIG3179, 187, XRCC1172, 188, POL(3187, OGG1189, XPA190, BRCA1191, DNA-PKcs188, CHK1192, SIRT1193, SIRT6194, 195, CDK2196, TET1197, CTCF198, P21199, TMPRSS:ERG fusion200, and numerous other proteins201–203 |
| FEN1 | Flap Endonuclease | Phosphorylation at S187204 Sumoylation at K168205 Ubiquitination at K354205 Acetylation at K354, K375, K377, K380206 Méthylation at R192207 | Acetylation at K80, K267, K375 Phosphorylation at S197, S363, T195, T364 | Associated with PCNA89, 208, 209, WRN210, CDK1204, CDK2204, NEIL1119, LIG1211, p300212, APEX1158, FANCA213, APC214, MUS81215, 9-1-1 complex216, E2F1217 |
We also present a network map of specific DNA repair pathway protein interactions (Figure 2) to visualize central protein-protein interaction hubs. As more DNA repair pathway crosstalk interactions are elucidated, these hubs should lead to a better understanding of how DNA damage response systems are integrated and may be valuable in future drug design to target multiple DNA repair pathways at once for clinical applications.
Figure 2. Human base excision repair protein interactions with other DNA repair pathway components.
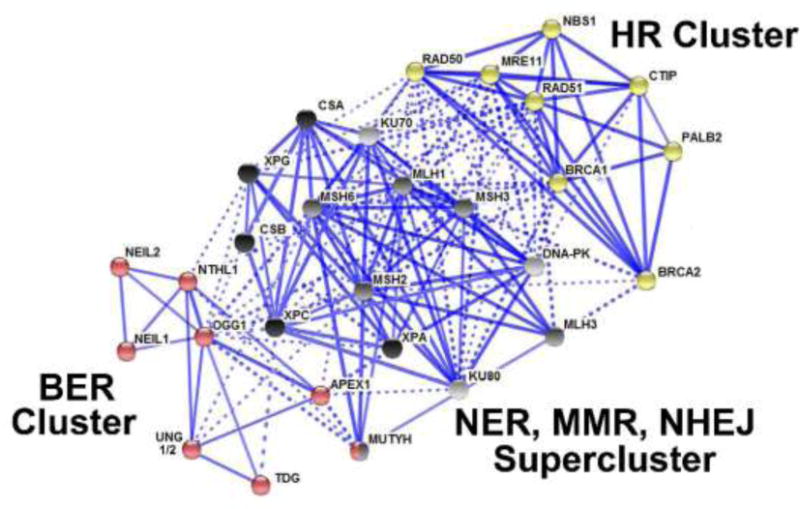
The STRING functional protein association network and Kmeans clustering reveal how DNA repair components interact68. A circle represents a node (protein), while edges (lines) indicate protein interactions. Solid edges denote interactions within the same cluster (pathway), while dashed lines are pathway crosstalk interactions that take place between proteins of different clusters. The line thickness is related to the strength of data that supports a particular interaction. Three clusters of interactions were generated and color-coded according to the canonical function of the protein- a BER cluster (red); a supercluster comprised of MMR (grey), NHEJ (light grey), NER (black), and an HR cluster (yellow). The MMR/NER/NHEJ supercluster was generated by the STRING algorithm during Kmeans clustering. The STRING map demonstrates that DNA repair pathways dynamically interact with each other as demonstrated by the dashed edges.
2. Nucleotide Excision Repair Crosstalk with BER Components
The canonical function of nucleotide excision repair (NER) is to eliminate bulky DNA damage, which can arise from exposure to UV radiation or certain chemical agents10. These lesions include UV-induced cyclobutane pyrimidine dimers, pyrimidine-pyrimidone photoproducts, and bulky chemical adducts20,21. NER also participates as a backup mechanism to base excision repair (BER) for the repair of certain oxidative induced DNA base damage22. Thus, the interplay between multiple NER and BER components is critical to ensure efficient BER processing of base damage. Several BER glycosylase-NER protein interactions have been characterized that impact the function of key BER glycosylase enzymes in processing of their respective DNA damage substrates. Several specific examples that illustrate this interplay are described here.
2.1 TDG Glycosylase
Well documented evidence that NER components can influence BER glycosylase activity comes from the analysis of the interaction between the BER thymine DNA glycosylase, TDG, and the NER XPC protein (Figure 1)23,24. In their respective pathways, TDG recognizes G:T mismatches in DNA and excises the mismatched thymine25. The XPC protein is involved in global genome NER (GG-NER)26. TDG is strongly product inhibited by the AP site that is produced in DNA following thymine cleavage and APEX1 helps displace TDG from these AP sites27. Previous work identified a physical interaction between TDG and XPC24. To assess whether XPC is an additional factor that contributes to displacement of TDG from AP sites, APEX1 and XPC were simultaneously added to DNA-bound TDG. Individually, APEX1 or XPC stimulated moderate TDG release from DNA. When present together, APEX1 and XPC resulted in a 6-fold increase in TDG release from DNA compared to control reactions24. These findings demonstrated that XPC is an additional component that triggers TDG release from DNA product. However, an exact mechanism for how XPC stimulates TDG release has yet to be defined. Furthermore, as XPC increased the ability of APEX1 to aid in TDG turnover, future studies are required to determine if XPC binds to and/or influences APEX1 activity.
2.2 NER components interact with BER substrates
Another example of BER and NER interplay includes two NER proteins, XPC and CSB. Both XPC and CSB localize to sites of oxidatively-induced DNA damage generated by laser (405 nm) excitation of a photosensitizer28. The primary product of this reaction is the BER substrate 8-oxoguanine (8-oxoG)28. Fluorescently tagged XPC and CSB were employed to track the localization and kinetics for both proteins within the nucleus. Upon DNA damage, XPC localized to sites of DNA damage exclusively in the nucleoplasm while CSB localized to sites of damage in both the nucleolus and the nucleoplasm28. As the nucleolus is a site of high transcriptional activity due to ribosomal DNA29, these results are in line with previous data assigning XPC to global genome NER and CSB to transcription coupled repair26. In contrast, downstream NER components such as the XPA and XPB proteins were not recruited to these sites of oxidative base damage28. This result indicates that the recruitment of XPC and CSB is independent of their respective NER functions and supports a role for XPC and CSB in influencing BER-mediated repair of oxidatively-induced DNA damage.
The NER protein, CSB, also influences binding and excision of the oxidative damage 8-oxoG by the BER glycosylase, OGG124,30–32. Despite a functional link to 8-oxoG repair, no direct interaction between the OGG1 and CSB proteins has been detected, suggesting that these proteins could function as part of a protein complex to ensure efficient BER function (Figure 1)33. By analyzing the kinetics of protein recruitment to DNA damage, Menoni et al. concluded that CSB is recruited to DNA damage prior to OGG128. Consistent with this model, there was no change detected in either XPC or CSB recruitment to damage in cells deficient for OGG1. Thus, OGG1 is not required for recruitment of CSB to sites of oxidative DNA damage28. XPC was recently described as a general DNA damage sensor independent of NER34. This role for XPC is supported both by the XPC link to TDG glycosylase and nucleoplasm localization of XPC to sites of DNA damage independent of other NER components28. How XPC may generally influence other BER glycosylases as a sensor for other BER substrates is unknown and will require further study.
2.3 NEIL Glycosylases
Another class of BER glycosylases that is modulated by CSB are the NEIL1 and NEIL2 glycosylases (Figure 1)12,13,35. While the NEIL glycosylases have substrate specificity that overlaps with other BER N-glycosylases, they are unique in their ability to excise oxidative DNA damage from single-stranded DNA that mimics a transcription bubble36. NEIL1 substrates include the 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) and 4,6-diamino-5-formamidopyrimidine (FapyA) open ring base damages13. As both FapyG and FapyA share an intermediate structure with 8-oxoguanine37, and as CSB impacts OGG1-mediated repair of 8-oxoguanine33, this led to the hypothesis that CSB could impact NEIL1 activity13. To address this question, levels of FapyG and FapyA damage were analyzed in the brain, liver, and kidneys of CSB−/− mice revealing that FapyA levels are increased in all three tissues relative to control mice. FapyG damage was also elevated in the brain and kidneys compared to control mouse tissues, providing evidence that CSB is required for efficient repair of NEIL1 substrates. Further analysis using NEIL1 in vitro incision assays revealed that CSB increases NEIL1-mediated incision activity up to 4-fold for FapyG, up to 2.5-fold for FapyA, and also stimulates NEIL1 AP lyase activity. The stimulation of NEIL1 by CSB was mapped to a region within the N-terminal domain of CSB (amino acids 2-341) and was independent of CSB ATPase activity. These results suggest that CSB does not mediate chromatin remodeling during repair of NEIL1 substrates. Immunoprecipitation experiments from HeLa lysates demonstrated that NEIL1 and CSB are present in the same protein complex although whether a direct interaction occurs between NEIL1 and CSB remains to be determined13. An interesting question is whether NEIL1 has a role in transcription-coupled repair of oxidatively-induced DNA base damage in light of the interaction with CSB and the ability of NEIL1 to initiate repair of single-stranded DNA.
Further work revealed that CSB also stimulates NEIL2 activity12,35. Immunoprecipitation experiments from HeLa cells revealed that CSB and NEIL2 are present in the same protein complex and that the protein-protein interaction is increased following exposure to the oxidizing agent menadione. Further analysis revealed a direct protein-protein interaction between CSB and NEIL212. Incision assays of a NEIL2 substrate revealed that CSB stimulates NEIL2-mediated incision of FapyA up to 4-fold in duplex DNA and up to 3-fold for 5-hydroxyuracil present in a bubble DNA structure. However, CSB did not affect NEIL2 binding to DNA damage, suggesting that CSB may play a role in NEIL2 release from the final DNA product. Collectively, these results demonstrate that repair of BER substrate from single-stranded DNA can be coordinated through interactions between BER and NER components.
2.4 NTHL1 Glycosylase
A crucial BER glycosylase, NTHL1, repairs a large subset of oxidized DNA bases including dihydrouracil and the replication and transcription blocking base damage, thymine glycol (Tg)11. However, in vitro reconstitution experiments showed that purified NTHL1 has poor incision activity on an oligonucleotide containing an NTHL1 damage substrate11. Based on findings that NER-deficient patient cells, which lack the XPG protein, also showed poor excision of Tg38, purified XPG was added into the reconstituted NTHL1 BER in vitro system. Surprisingly, the addition of XPG stimulated NTHL1 incision and release of DNA product for both Tg and dihydrouracil substrates. The addition of other NER components, XPA or XPC, had no such stimulatory effect on NTHL1 activity. To address whether XPG endonuclease activity is required to achieve this stimulation of NTHL1, two XPG protein variants (E791A and A792V) that have no XPG nuclease activity were employed11. Both variants could stimulate NTHL1-mediated base excision as effectively as wild type XPG, indicating that the XPG-dependent stimulatory effect on NTHL1 is independent of XPG nuclease function. Overall, this study demonstrates that XPG plays a critical role in NTHL1-mediated base excision of damage and release from DNA (Figure 1). Whether NTHL1 protein has an impact on XPG-mediated NER functions is not known.
2.5 AP Endonuclease (APEX1)
Platinum-based chemotherapeutics are used in the clinic as an effective treatment for multiple cancer types39–43. A common platinum therapeutic is cisplatin, which causes intra- and interstrand crosslinks primarily between guanine bases43,44. However, cisplatin has multiple negative side effects including peripheral neuropathy45,46, nephrotoxicity43, and an increase in cellular reactive oxygen species (ROS)2,47. NER is the main repair pathway to handle cisplatin adducts48,49 while BER initiates repair of the ROS-induced base damage49.
Previous studies showed that increased protein levels of the BER protein, APEX1 (also known as APE1), protect cells against cisplatin toxicity47,50. To assess the role of APEX1 in repair of cisplatin adducts, APEX1 levels were modulated in a neuronal tissue culture model exposed to cisplatin46. In this study, knockdown of APEX1 caused an increase in the level of unrepaired cisplatin adducts. Furthermore, cisplatin adduct repair was dependent on APEX1 endonuclease activity, and this repair activity was separate from APEX1 redox functions that are critical for transcriptional regulation47. Interestingly, when APEX1 was lost, an increase in the level of the NER protein, XPA, was detected. Whether this increase in XPA protein resulted from regulation at the protein or RNA level is unknown, and the exact mechanism of how APEX1 is involved in the removal of cisplatin adducts remains unclear. Thus, the level of cisplatin-DNA adducts, which are repaired by NER, increased in the absence of the BER protein, APEX1. Whether APEX1 involvement in cisplatin adduct repair is dependent on global genome NER (GG-NER) or transcription-coupled (TC-NER) NER is not known.
In support of a model where APEX1 plays a role in TC-NER of cisplatin adducts, the APEX1 and CSB proteins directly interact (Figure 1)51. CSB protein has been implicated in altering DNA conformation as well as chromatin remodeling during NER51,52. CSB−/− cells display hypersensitivity to reactive oxygen species (ROS)-generating agents, supporting a role for CSB in BER-mediated processing of oxidative DNA damage53–55. In fact, CSB also interacts with other BER proteins, including PARP1 and FEN151, in addition to APEX1. In vitro, APEX1 endonuclease activity is increased up to 4-fold on duplex DNA and up to 6-fold on single-stranded DNA by the interaction with CSB51. The larger stimulation for single-stranded DNA indicates that the CSB/APEX1 interaction might have a greater impact on damage present in transcriptionally active DNA compared to double-stranded DNA. Addition of ATP was not needed for the stimulation of APEX1 endonuclease activity by CSB, suggesting that increased APEX1 endonuclease activity is not due to CSB-mediated chromatin remodeling. While these results show that APEX1 has a role in repair of cisplatin adducts46, whether APEX1 has a reciprocal impact on CSB-mediated TC-NER activity is currently unknown.
3. Non-Homologous End Joining and Homologous Recombination Crosstalk with BER Components
Double strand breaks (DSBs) are the most deleterious class of DNA damage56, and cells have evolved multiple repair pathways to repair this damage. Homologous recombination (HR) repairs DSBs in the S and G2 phases of the cell cycle when a homologous sister chromatid is present57. Non-homologous end joining (NHEJ), while active in all phases of the cell cycle, primarily repairs DSBs in the G0 and G1 phases of the cell cycle. Importantly, BER can generate DSBs as a result of excision of closely opposed base damages or from single strand break intermediates if these intermediates are encountered by the replication or transcription machinery3. To avoid the accumulation of deleterious damage intermediates, BER must therefore be efficiently and precisely regulated to initiate and complete repair. In fact, the BER intermediates generated by the alkyladenine glycosylase induce more robust HR than the initial alkylation damage in vivo58. The impact of BER and DSB repair pathway cross regulation and how the cell cycle phase contributes to pathway crosstalk to maintain genome stability are areas that require further study. Recent examples that illustrate the interplay between BER and DSB repair pathways are described here.
3.1 Alternative Non-Homologous End Joining
End joining repair of DSBs can be prone to loss of genetic material as there is no template for extensive homology searching59,60. The NHEJ pathway is subdivided into two major sub-pathways that include classical NHEJ (c-NHEJ), which is dependent on the KU70/80 proteins, and alternative NHEJ (alt-NHEJ), which uses short stretches of end resection that result in microhomology often found at chromosomal translocations59. To elucidate which protein factors mediate alt-NHEJ, an RNAi library directed against DNA repair factors was screened using a fluorescent reporter assay for alt-NHEJ60. Interestingly, proteins from diverse DNA repair pathways were identified as top candidates required to perform alt-NHEJ. These proteins include the BER glycosylases, NTHL1 and UNG, the mismatch repair protein, MSH6, and the crosslink repair protein, FANCA. Subsequent analyses demonstrated that knockdown of the NTHL1 and UNG glycosylases significantly decreased alt-NHEJ. Furthermore, this result was specific for alt-NHEJ, as knockdown of either glycosylase did not have a significant impact on HR or single strand annealing events as determined by reporter assays. HR and alt-NHEJ are most active in the S and G2 phases of the cell cycle. Thus, accumulation of cells in G1 upon depletion of either NTHL1 or UNG could account for the apparent decrease in alt-NHEJ. However, NTHL1 knockdown resulted in an increase in the percent of G2 cells while UNG knockdown resulted in an increase of G1 phase cells. These distinct cell cycle changes suggest that, at least in the case of NTHL1, the cell cycle status does not account for the decrease observed in alt-NHEJ activity. This result implies that the NTHL1 glycosylase plays a role in promoting alt-NHEJ, perhaps while suppressing HR. Future experiments will need to pinpoint how BER and end joining repair mechanisms coordinate their activities to ensure efficient repair of DSBs.
3.2 Homologous Recombination
Another well-known NTHL1 partner is the NER protein, XPG (Figure 1). Recent work reveals that XPG is indispensible for HR recovery from collapsed replication forks61. Genomic instability can result from an inability to repair DSBs that result from these collapsed replication forks, which are normally repaired by HR62. In this study, loss of XPG led to DNA damage that resulted in genomic instability61. XPG is required for efficient loading of the Rad51 presynaptic filament by the BRCA2/PALB2 complex for HR following end resection61. NTHL1 is also upregulated in S phase63, presumably for BER glycosylase function. However, because NTHL1 appears to promote alt-NHEJ and is a binding partner for XPG, NTHL1 could modulate DSB repair pathway choice during S phase. Future experiments will be needed to assess the functional consequences of NTHL1 protein regulation for DSB repair.
In addition, recent results reveal a reciprocal effect between the BER protein, OGG1, and the HR protein, Rad5264. Rad52 is part of the Rad51 epistasis group that functions in presynaptic filament formation during HR64. Previous studies in budding yeast demonstrated that Rad52 aids in strand exchange by forming a bridge between RPA-coated single-stranded DNA and Rad5165,66. Curiously, yeast deficient in BER are further sensitized to oxidative damage when the RAD52 gene is disrupted, underscoring the importance of repair pathway crosstalk67. Studies determined that mammalian OGG1 and Rad52 proteins directly interact, and this interaction is increased in response to oxidative stress64. Furthermore, this interaction had reciprocal effects on the function of both the BER and HR pathways. Rad52 stimulates OGG1-mediated incision of 8-oxoG by up to 3-fold, and Rad52 promotes OGG1 release from DNA64. Conversely, OGG1 inhibits Rad52 single-strand annealing and strand exchange activity, while another glycosylase, UNG, has no such effect64. RNAi-mediated knockdown of Rad52 caused an increase in 8-oxoG and FapyG accumulation in genomic DNA64. Taken together, these results demonstrate that HR proteins can impact BER activity, and, conversely, a BER protein can influence HR function. These findings raise the question of whether BER glycosylases have reciprocal effects on the efficient function of the DNA repair pathways that interact with BER such as NER. Future research will be required to investigate BER protein regulation and understand how BER proteins influence the activities of other repair pathways.
Pathway Crosstalk Conclusions
To illustrate and examine DNA repair pathway crosstalk, we utilized the STRING protein-protein interaction network (www.string-db.org)68. Various types of protein interactions and databases are included in the STRING analysis. Visualization of the interaction map is straightforward; each protein is represented by a node (circle), while a protein interaction is represented by an edge (line) (Figure 2). We included a panel of DNA repair proteins from five repair pathways (BER, NER, MMR, NHEJ, and HR), which yielded three main clusters after Kmeans clustering analysis with a high confidence (0.700) minimum interaction score.
By examining the three different clusters, one can appreciate the breadth of coordination not only within a specific pathway but also between DNA repair pathways (Figure 2). For example, the red cluster highlights BER. Dashed lines indicate crosstalk with the black supercluster containing components of NER, MMR, and NHEJ. One could postulate that a BER interaction may, in fact, influence the HR pathway (yellow) by modulating a common interaction highlighted within the black supercluster. As DNA repair is a tightly regulated process, perturbations in pathway crosstalk may have untoward consequences for multiple DNA repair pathways. Therefore, understanding the nuances of regulation at the protein level by identifying central interaction hubs could be an effective approach to identifying new targets for therapeutic development, or for predicting how a patient may respond to existing chemotherapeutic options that target DNA repair.
To detect central interaction hubs, we propose a two-fold approach of assessing 1) the total number of edges that a node has, and 2) the number of dashed edges per node. In this way, interaction hubs for a specific pathway, or a hub that impacts the greatest number of interactions between pathways can be identified. For example, OGG1 has six solid edges to denote interactions with other BER components while nine dashed edges represents crosstalk with various components of the supercluster for a total of fifteen edges. Furthermore, many MMR proteins have interactions connected to HR. One could hypothesize that dysregulation at the protein level anywhere along this string of interactions could impact the functions of BER, MMR, and/or HR simultaneously. The same logic can be applied along any node-edge pathway.
Another striking observation is the large number of interactions that appear to be coordinated through the MMR proteins MLH1, MSH6, and MSH3 (Figure 2). These proteins emerge as central coordinators between certain BER proteins and HR. For example, an interesting case emerges with the MSH6 protein. MSH6 is strongly implicated in promoting alt-NHEJ60. From the STRING interaction network, one can see that MSH6 interacts with RAD51 as well as components of the BER pathway (Figure 2). Whether MSH6 can promote alt-NHEJ while suppressing HR through the interaction with RAD51 remains to be determined. Alternatively, whether MSH6 interaction with BER components aids in the suppression of HR has not been investigated. The fact that NTHL1 is also a top candidate for promoting alt-NHEJ, suggests that BER and MMR could potentially influence HR functions.
A distinct subset of BER proteins is coordinated with various components of NER. One example is the interaction between NTHL1 and XPG. As previously noted, recent studies demonstrate that XPG is indispensible for proper HR, and that this function is independent from the NER functions of XPG61. In turn, XPG is also crucial for catalytic turnover of the NTHL1 glycosylase11. Thus, protein dysregulation of any of these DNA repair components has the potential to affect more than one DNA repair pathway. For example, dysregulation of NTHL1 could ultimately affect the efficiency of HR through unregulated interactions with XPG. Examination at the protein level will provide a starting point for investigating the overall impact of DNA repair protein dysregulation and pathway crosstalk.
4. Post-translational Modifications Affecting Function and Protein Levels of BER Components
Many of the crosstalk examples described depend on DNA damage induced protein-protein interactions. One mechanism of regulating DNA repair is through reversible posttranslational modifications (PTMs) of repair proteins, which could directly impact protein activity or protein-protein interactions (Figure 3). While there are a limited number of PTMs annotated on BER proteins, even fewer of these PTMs have been assigned a biological function. However, recent studies have begun to elucidate how a small subset of PTMs regulates BER proteins. Table 1 lists PTMs that have been identified on individual BER proteins and includes results from proteome-wide mass spectrometry analyses to identify phosphorylated and acetylated peptides69,70. As many BER glycosylases are cell cycle regulated63,71–74, PTM modification by CDK/cyclin proteins could play a role in modulating BER function in a cell cycle-dependent manner. Some of the best-characterized examples of BER modifications are discussed below. We focus on results reported since our group last discussed this topic in 201075.
Figure 3. Maintenance of genomic integrity requires the integration of multiple factors for DNA repair in response to damage.
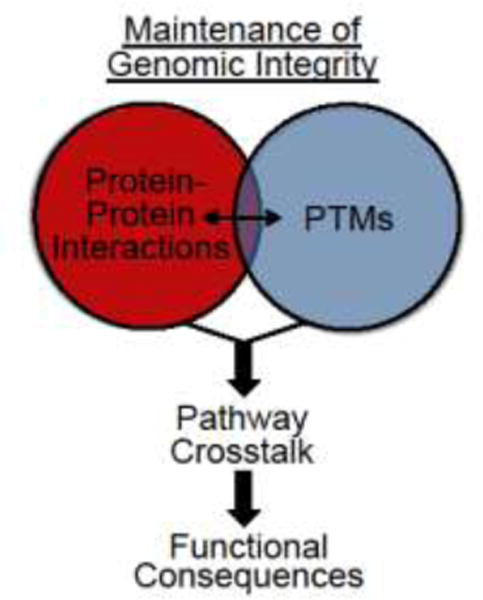
Mechanisms resulting in pathway crosstalk include the induction of functional protein-protein interactions (red) or PTMs (blue). A field that requires further analysis is how these individual areas intersect to result in pathway crosstalk, as illustrated by the overlap in the Venn diagram. Pathway crosstalk results in functional consequences that allow for a rapid and efficient DNA repair response to damage.
4.1 TDG Glycosylase
A classic and striking example of BER regulation by a PTM is SUMO modification of the G:T-mismatch glycosylase, TDG76. TDG has multiple functions which include BER glycosylase activity and transcription regulation, that are mediated by interactions with the CBP/p300 complex77. In BER, substrates for TDG include the G:T and G:U mismatched base pairs resulting from deamination of cytosine or 5-methylcytosine. TDG also functions in DNA demethylation through excision of 5-formylcytosine and 5-carboxylcytosine78. However, TDG is strongly product inhibited by the resulting AP site from TDG-mediated glycosylase activity, raising the question of how TDG is removed from DNA following TDG catalytic activity79,80. One mechanism of eliminating this AP product inhibition is through interaction with the NER protein, XPC, as discussed in the pathway crosstalk portion of this review24.
In addition to interaction with XPC and APEX1, TDG is modulated by a SUMO1 or SUMO2/3 modification, which regulates TDG turnover76. A structural approach was employed to probe the mechanism by which SUMO modification triggers TDG turnover. The crystal structures for unmodified TDG and TDG modified by SUMO1 and SUMO2/3 were resolved to provide insight into the impact of SUMO modification on TDG structure79,80. TDG modification by SUMO triggered TDG turnover, and did not influence the structure of the TDG core domain, which contains the glycosylase activity. Sumoylation occurs within the C-terminus of TDG, and the resulting C-terminal structural rearrangement involves non-covalent interactions with water. The conformational change causes protrusion of an α-helix that sterically clashes with the DNA phosphate backbone and causes release of TDG from the DNA. These results imply that TDG is only sumoylated following TDG glycosylase activity in order to activate release of TDG from product DNA.
In contrast to this model, another study suggested that TDG can be modified by SUMO as free protein not bound to DNA. Unbound TDG and TDG in complex with DNA containing a TDG substrate are sumoylated to approximately the same extent in vitro81. Furthermore, TDG bound to undamaged DNA and bound to DNA containing a TDG substrate is SUMO modified at similar rates81. TDG sumoylation does not influence TDG excision activity against 5-formylcytosine or 5-carboxylcytosine substrates78. These results confirm previous findings that TDG is sumoylated to relieve product inhibition. However, as unbound TDG can also be sumoylated, these results suggest that TDG sumoylation may have a distinct function in addition to relief of TDG product inhibition.
SUMO modification of TDG is regulated in a cell cycle-dependent manner82. When cells are synchronized in S phase, the steady state level of TDG decreases82. In contrast, in G2 phase and mitosis, both unmodified TDG and sumoylated TDG increase relative to other phases of the cell cycle82. Furthermore, TDG and TDG-SUMO protein fluctuation is dependent on proteasomal degradation, as treatment with the proteasome inhibitor, MG132, increases the steady state levels of both TDG and TDG-SUMO82. As the steady state level of TDG-SUMO is regulated in a cell-cycle dependent manner, and SUMO modification impacts TDG turnover, these results suggest that TDG-mediated repair is also cell cycle dependent.
Evidence that chemotherapeutic agents affect TDG modification comes from studies of TDG acetylation by CBP/p300. Acetylation of TDG weakens the interaction of TDG with APEX177. Thus, TDG acetylation may coordinate TDG BER glycosylase function and transcriptional activity83. A critical deacetylase, SIRT1, removes the acetyl group from TDG77. Upon de-acetylation, TDG displays increased excision activity of a G:T substrate. In fact, TDG de-acetylation stimulates TDG release from an AP site, demonstrating that together, XPC, APEX1, sumoylation, and acetylation all influence TDG release from DNA product. The opposite effect for TDG acetylation was observed in the case of excision of the chemotherapeutic–induced DNA base damage, 5-flurouracil (FU), from DNA. Acetylated TDG displayed enhanced FU:G excision activity, in contrast to slower excision for G:T nucleotide pairs. Therefore, acetylation of TDG results in opposing effects on substrate excision of a chemotherapeutic. Thus, TDG acetylation status within tumor cells could potentially impact clinical efficacy of 5-flurouracil. To date, TDG is the best characterized example of how the intersection of PTMs and protein interactions coordinate efficient BER repair of DNA damage.
4.2 NTHL1 Glycosylase
Sumoylation of the Saccharomyces cerevisae DNA N-glycosylase, Ntg1, is another PTM that was recently reported. The Ntg1 protein is a functional ortholog of the human NTHL1 glycosylase84,85. Recent work revealed that Ntg1 is sumoylated in response to both oxidative stress and the alkylating agent methyl methanesulfonate (MMS)84,85. Consistent with an evolutionarily conserved function of this modification, human NTHL1 is also SUMO modified in response to oxidative stress85. Sumoylated Ntg1 is enriched in the nuclear but not the mitochondrial cellular compartment84. Whether this SUMO modification mediates nuclear localization or occurs in the nucleus is not known. To understand the functional consequence of sumoylation of Ntg1, the sites of SUMO modification on Ntg1 were mapped, and an Ntg1 variant that could not be SUMO modified was created (Ntg1ΔSUMO)85. This variant provides one of the first tools to examine the functional consequences of SUMO modification of BER proteins in future studies.
To explore whether sumoylation of Ntg1 is required for a proper response to DNA damage, yeast with wild type or Ntg1ΔSUMO were reconstituted into BER/NER deficient cells, and these cells were treated with MMS. In the absence of MMS, there was no discernable difference in growth between strains expressing control or the Ntg1ΔSUMO variant85. Surprisingly, yeast expressing Ntg1ΔSUMO grew better than the control cells with wild type Ntg1 when challenged with MMS. However, four days post MMS exposure, yeast containing wild type NTG1 caught up to the growth of yeast expressing Ntg1ΔSUMO. This result implies that sumoylation of Ntg1 may play a role in DNA repair or coordinating a DNA damage response in response to DNA damage. Additional experiments defining the site(s) of human NTHL1 sumoylation, and how the phosphorylation at S71 (Table 1) influences NTHL1 activity will be a focus of future studies. Further work to understand how damage-dependent sumoylation of Ntg1 coordinates this growth phenotype will need to be performed to gain a fuller understanding of BER crosstalk with cell cycle progression.
4.3 UNG2 Glycosylase
The UNG glycosylases (UNG1 and UNG2) are the primary enzymes tasked with removing uracil from duplex DNA86. UNG1 is predominately localized to the mitochondria, while UNG2 is primarily responsible for removing uracil from nuclear DNA3,87. Uracil is mutagenic if left unrepaired and is particularly problematic once the cell undergoes replication. Therefore, understanding how the cell coordinates repair of uracil during S phase is critical to defining mechanisms that protect genome integrity. UNG2 binds to PCNA and RPA suggesting that UNG2 can process uracil in single stranded DNA during replication88,89. The steady-state level of UNG2 is regulated in S phase by phosphorylation of three residues, S23, T60, and S6486. Phosphorylation at these sites is mediated in a cell cycle-dependent manner by the CDK4/cyclinD complex86. UNG2 S64 is also modified in late S phase/early G2 phase by the CDK2/cyclinA and CDK1/cyclinB complexes86. Phosphorylation at S23 increases UNG2 association with both RPA and replicating chromatin, and also influences UNG2 catalytic turnover. Conversely, phosphorylation at T60 and S64 creates a phosphodegron triggering ubiquitination and proteolytic degradation in late S phase and early G286. Taken together these findings provide a model for how UNG2 is regulated and degraded during S phase in response to specific PTMs. As UNG2 also repairs uracil in all phases of the cell cycle, how additional PTMs regulate UNG2 activity in other phases of the cell cycle is a key question. In fact, the PHOSIDA database annotates multiple uncharacterized phosphorylation and acetylation sites within UNG1/2 (Table 1), which could influence the biological function of UNG1/2. Future studies will need to determine the biological function of these uncharacterized UNG PTMs.
4.4 MUTYH Glycosylase
MUTYH is a DNA N-glycosylase that excises adenine from A:8oxoG mispairs. Consistent with this function, deletion of the human MUTYH glycosylase is linked to colon cancer90–92. Like other BER components93, MUTYH is regulated by ubiquitination90. In vitro, MUTYH is modified by the E3 ligase, Mule, in conjunction with the E2 enzymes H5b, H5c, and H790. MUTYH ubiquitination occurs within a domain comprised of amino acids 475-535. Lysines in this region were systematically analyzed to abolish MUTYH ubiquitination and to explore the biological consequences of MUTYH ubiquitination. MUTYH that could not be ubiquitinated showed an increase in protein steady state levels and altered interaction with chromatin90. In cells deficient for the Mule E3 ligase, which have increased MUTYH levels, a decrease in the mutation frequency of the HPRT gene was observed as compared to control cells. Conversely, when Mule was overexpressed in cells, which have decreased MUTYH levels, an increase in HPRT gene mutations was detected. Therefore, the regulation of a BER glycosylase at the protein level influences mutation frequencies in mammalian cells. MUTYH protein regulation through specific PTMs may therefore serve as a regulatory paradigm for how other BER glycosylases could be influenced by PTMs that modulate steady state protein levels.
4.5 OGG1 Glycosylase
Another key player in repair of oxidative DNA damage is OGG1, which is primarily responsible for excision of 8-oxoguanine base damage94. A common variant of OGG1 present in the genome is the polymorphism coding for OGG1 S326C95. The presence of the S326C OGG1 variant predisposes carriers to multiple cancer types94,95, but how this variant responds to oxidative stress in vivo had not been defined. Recent work reveals that in response to physiological oxidative stress, the OGG1 S326C variant loses glycosylase activity94. Employing a prediction program for disulfide bridge formation, a potential for increased disulfide bond formation in the OGG1 S326C variant as compared to wild type OGG1 was identified94. One of the predicted inter- or intra-protein disulfide bridges includes amino acid 326. Thus, disulfide bridge formation and the resulting loss of glycosylase activity could impair OGG1 activity and cause increased mutation frequencies. This model for gain of a disulfide bridge could explain how the OGG1 S326C polymorphism predisposes to cancer. This study adds disulfide bridges to the list of functionally important modifications to BER protein variants.
4.6 AP Endonuclease (APEX1)
Another a key protein in BER, APEX1, contains multiple PTMs that affect both the endonuclease and transcription regulatory functions of the protein96. Documented PTMs detected in APEX1 include phosphorylation96,97, acetylation98–100, ubiquitination101, and S-nitrosylation (Table 1)96,102. However, S-glutathionylation also occurs in response to the altered redox state of the cell103. Glutathionylation, addition of a glutathione group, occurs at cysteine residues and three candidate cysteines are located in the redox responsive domain of APEX1103. As cysteine modifications include disulfide bridges, S-nitrosylation, and S-glutathionylation, understanding the competition between different PTMs and how they impact APEX1 function is crucial. APEX1 undergoes reversible glutathionylation in vitro103. Glutathionylation of APEX1 occurs on C99, and inhibits APEX1-mediated AP site cleavage by 90%. In fact, modified APEX1 does not form stable complexes with AP-DNA. HeLa cells exposed to mildly toxic doses of hydrogen peroxide also show APEX1 glutathionylation suggesting that this modification could be relevant in cells and potentially in vivo. Determining how the cellular redox environment impacts APEX1 modification in non-transformed cell lines will be important to define how PTMs of BER proteins influence genome stability.
5. Conclusions
As illustrated in Figure 1 and analyzed in Figure 2, functional interactions between proteins from different DNA repair pathways can influence efficient BER responses to DNA damage. Figure 3 highlights the mechanisms that can achieve pathway crosstalk, including protein-protein interactions and PTMs that coordinate function to ensure rapid and efficient repair of DNA damage. Individually, each of these sub-areas is a rapidly evolving field, but studies suggest that repair biological outcomes depend on the extent of overlap of each of these components (Figure 3). Comprehensive studies to uncover further mechanisms of regulation that achieve pathway crosstalk are warranted. What remains unclear is the extent to which BER proteins impact the function of other DNA repair pathways. Most research to date in the DNA repair field has focused on biochemical elucidation of pathways, identifying new disease predisposing single nucleotide polymorphisms (SNPs) in DNA repair genes, and transcriptional regulation of DNA repair genes. Although each of these areas has value in contributing to the depth and breadth of DNA repair knowledge, a relatively neglected area has been defining how diseases are influenced by dysregulation of DNA repair proteins at the protein level. For example, responses to chemotherapeutics are often influenced by the DNA repair protein status of a patient’s tumor cells104,105. Thus, there is a need to investigate DNA repair dysregulation at the protein level and to understand how DNA repair pathway crosstalk occurs in order to optimize treatment strategies. Furthermore, how efficient DNA repair is coordinated via protein-protein interactions and PTMs in order to influence repair activities requires future studies. As BER coordinates multiple interactions with other DNA repair pathways, we propose BER as a starting point for addressing how DNA repair can be dysregulated at the protein level, and how such dysregulation can influence other DNA repair pathways.
We also propose that by focusing on central interaction hubs (Figure 2), a directed effort at future drug design and DNA repair protein screening will open avenues previously unexplored for responses to chemotherapeutics. For instance, if BER and/or MMR proteins are dysregulated through altered interactions, PTMs, or steady-state protein levels to promote alt-NHEJ while suppressing HR, could this scenario impact clinical responses to chemotherapeutics? An analogous scenario is found in breast cancer patients with a germline mutation in the BRCA1/2 genes that results in inefficient HR function106,107. As a consequence of decreased HR function, cells are sensitive to PARP inhibitors as PARP1 and BRCA1 are synthetically lethal108,109. Conversely, if a patient does not have a BRCA1/2 mutation, but instead has suppressed HR as a consequence of dysregulated proteins in BER and/or MMR, this raises the issue of whether that patient would be sensitive to PARP inhibitors. Thus, patient tumors could also be screened for specific interactions or protein dysregulation as a potential biomarker for tumor responsiveness to chemotherapeutics. As studies reveal additional functional interactions within and among the various DNA repair pathways, the interaction networks displayed in Figure 2 will need to further evolve to accurately reflect this new information. As a consequence, pathway crosstalk through PTMs and protein-protein interactions may reveal a potential therapeutic avenue to sensitize a tumor previously thought to be unresponsive to certain treatment options. As cells are constantly “BERing” the burden of DNA damage, we must expand our knowledge of DNA repair in order to ultimately improve human health.
Acknowledgments
We would like to acknowledge the helpful discussions and guidance from Dr. Nicholas Seyfreid’s lab (Emory University) in preparing the STRING analysis for Figure 2. Support for this work was provided by NIH grant ESES011163 (PWD), Emory University School of Medicine (PWD), the Winship Cancer Institute of Emory University (PWD), and by NIH grant GM058728 (AHC).
Abbreviations
- BER
Base Excision Repair
- NER
Nucleotide Excision Repair
- MMR
Mismatch Repair
- HR
Homologous Recombination
- NHEJ
Non-homologous End Joining
- PTMs
Post-translational Modifications
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Conflicts of Interest
The Authors declare no conflicts of interest
References
- 1.Swartzlander DB, Griffiths LM, Lee J, Degtyareva NP, Doetsch PW, Corbett AH. Regulation of base excision repair: Ntg1 nuclear and mitochondrial dynamic localization in response to genotoxic stress. Nucleic Acids Res. 2010;38:3963–3974. doi: 10.1093/nar/gkq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS One. 2013;8:e81162. doi: 10.1371/journal.pone.0081162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015;43:10083–10101. doi: 10.1093/nar/gkv1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoeijmakers JH. Nucleotide excision repair. II: From yeast to mammals. Trends Genet. 1993;9:211–217. doi: 10.1016/0168-9525(93)90121-w. [DOI] [PubMed] [Google Scholar]
- 6.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA–repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ. Heavy metal toxicity and the environment. EXS. 2012;101:133–164. doi: 10.1007/978-3-7643-8340-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 9.Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klungland A, Hoss M, Gunz D, Constantinou A, Clarkson SG, Doetsch PW, Bolton PH, Wood RD, Lindahl T. Base excision repair of oxidative DNA damage activated by XPG protein. Mol Cell. 1999;3:33–42. doi: 10.1016/s1097-2765(00)80172-0. [DOI] [PubMed] [Google Scholar]
- 12.Aamann MD, Hvitby C, Popuri V, Muftuoglu M, Lemminger L, Skeby CK, Keijzers G, Ahn B, Bjoras M, Bohr VA, Stevnsner T. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech Ageing Dev. 2014;135:1–14. doi: 10.1016/j.mad.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J Biol Chem. 2009;284:9270–9279. doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase beta null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 15.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 16.Dery U, Masson JY. Twists and turns in the function of DNA damage signaling and repair proteins by post-translational modifications. DNA Repair (Amst) 2007;6:561–577. doi: 10.1016/j.dnarep.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 18.Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–672. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 20.DNA Damage [Internet] John Wiley & Sons Ltd; Chichester: 2014. Available from: http://www.els.net. [Google Scholar]
- 21.Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1:225–236. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 22.Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovtun IV, McMurray CT. Crosstalk of DNA glycosylases with pathways other than base excision repair. DNA Repair (Amst) 2007;6:517–529. doi: 10.1016/j.dnarep.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu Y, Iwai S, Hanaoka F, Sugasawa K. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 2003;22:164–173. doi: 10.1093/emboj/cdg016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardeland U, Bentele M, Jiricny J, Schar P. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J Biol Chem. 2000;275:33449–33456. doi: 10.1074/jbc.M005095200. [DOI] [PubMed] [Google Scholar]
- 26.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 27.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 28.Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J Cell Biol. 2012;199:1037–1046. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fusser M, Nesse GJ, Khobta A, Xia N, Li H, Klungland A, Epe B. Spontaneous mutagenesis in Csb(m/m)Ogg1(−)(/)(−) mice is attenuated by dietary resveratrol. Carcinogenesis. 2011;32:80–85. doi: 10.1093/carcin/bgq196. [DOI] [PubMed] [Google Scholar]
- 31.Khobta A, Kitsera N, Speckmann B, Epe B. 8-Oxoguanine DNA glycosylase (Ogg1) causes a transcriptional inactivation of damaged DNA in the absence of functional Cockayne syndrome B (Csb) protein. DNA Repair (Amst) 2009;8:309–317. doi: 10.1016/j.dnarep.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003;17:668–674. doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- 33.Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst) 2002;1:913–927. doi: 10.1016/s1568-7864(02)00116-7. [DOI] [PubMed] [Google Scholar]
- 34.Shell SM, Hawkins EK, Tsai MS, Hlaing AS, Rizzo CJ, Chazin WJ. Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair (Amst) 2013;12:947–953. doi: 10.1016/j.dnarep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aamann MD, Muftuoglu M, Bohr VA, Stevnsner T. Multiple interaction partners for Cockayne syndrome proteins: implications for genome and transcriptome maintenance. Mech Ageing Dev. 2013;134:212–224. doi: 10.1016/j.mad.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 37.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Cooper PK, Nouspikel T, Clarkson SG, Leadon SA. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science. 1997;275:990–993. doi: 10.1126/science.275.5302.990. [DOI] [PubMed] [Google Scholar]
- 39.Chovanec M, Hanna N, Cary KC, Einhorn L, Albany C. Management of stage I testicular germ cell tumours. Nat Rev Urol. 2016;13:663–673. doi: 10.1038/nrurol.2016.164. [DOI] [PubMed] [Google Scholar]
- 40.Group TIALCTC. Cisplatin-Based Adjuvant Chemotherapy in Patients with Completely Resected Non-Small-Cell Lung Cancer. New England Journal of Medicine. 2004;350:351–360. doi: 10.1056/NEJMoa031644. [DOI] [PubMed] [Google Scholar]
- 41.Rose PG, Bundy BN, Watkins EB, Thigpen JT, Deppe G, Maiman MA, Clarke-Pearson DL, Insalaco S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N Engl J Med. 1999;340:1144–1153. doi: 10.1056/NEJM199904153401502. [DOI] [PubMed] [Google Scholar]
- 42.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334:1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 43.Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 44.Fichtinger-Schepman AM, van der Veer JL, den Hartog JH, Lohman PH, Reedijk J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985;24:707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- 45.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9–17. doi: 10.1007/pl00007853. [DOI] [PubMed] [Google Scholar]
- 46.Kim HS, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, Lee SH. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat Res. 2015;779:96–104. doi: 10.1016/j.mrfmmm.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One. 2014;9:e106485. doi: 10.1371/journal.pone.0106485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Damia G, Imperatori L, Stefanini M, D’Incalci M. Sensitivity of CHO mutant cell lines with specific defects in nucleotide excision repair to different anti-cancer agents. Int J Cancer. 1996;66:779–783. doi: 10.1002/(SICI)1097-0215(19960611)66:6<779::AID-IJC12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 49.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Y, Guo C, Fishel ML, Wang ZY, Vasko MR, Kelley MR. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: use of APE1 small molecule inhibitors to delineate APE1 functions. DNA Repair (Amst) 2009;8:1273–1282. doi: 10.1016/j.dnarep.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Am J Hum Genet. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tuo J, Muftuoglu M, Chen C, Jaruga P, Selzer RR, Brosh RM, Jr, Rodriguez H, Dizdaroglu M, Bohr VA. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J Biol Chem. 2001;276:45772–45779. doi: 10.1074/jbc.M107888200. [DOI] [PubMed] [Google Scholar]
- 54.de Waard H, de Wit J, Gorgels TG, van den Aardweg G, Andressoo JO, Vermeij M, van Steeg H, Hoeijmakers JH, van der Horst GT. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair (Amst) 2003;2:13–25. doi: 10.1016/s1568-7864(02)00188-x. [DOI] [PubMed] [Google Scholar]
- 55.de Waard H, de Wit J, Andressoo JO, van Oostrom CT, Riis B, Weimann A, Poulsen HE, van Steeg H, Hoeijmakers JH, van der Horst GT. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol. 2004;24:7941–7948. doi: 10.1128/MCB.24.18.7941-7948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanaar R, Hoeijmakers JH, van Gent DC. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8:483–489. doi: 10.1016/s0962-8924(98)01383-x. [DOI] [PubMed] [Google Scholar]
- 57.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kiraly O, Gong G, Roytman MD, Yamada Y, Samson LD, Engelward BP. DNA glycosylase activity and cell proliferation are key factors in modulating homologous recombination in vivo. Carcinogenesis. 2014;35:2495–2502. doi: 10.1093/carcin/bgu177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11:e1004943. doi: 10.1371/journal.pgen.1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trego KS, Groesser T, Davalos AR, Parplys AC, Zhao W, Nelson MR, Hlaing A, Shih B, Rydberg B, Pluth JM, Tsai MS, Hoeijmakers JH, Sung P, Wiese C, Campisi J, Cooper PK. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol Cell. 2016;61:535–546. doi: 10.1016/j.molcel.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luna L, Bjoras M, Hoff E, Rognes T, Seeberg E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat Res. 2000;460:95–104. doi: 10.1016/s0921-8777(00)00015-x. [DOI] [PubMed] [Google Scholar]
- 64.de Souza-Pinto NC, Maynard S, Hashiguchi K, Hu J, Muftuoglu M, Bohr VA. The recombination protein RAD52 cooperates with the excision repair protein OGG1 for the repair of oxidative lesions in mammalian cells. Mol Cell Biol. 2009;29:4441–4454. doi: 10.1128/MCB.00265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 66.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci U S A. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2929–2935. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gnad F, Ren S, Cox J, Olsen JV, Macek B, Oroshi M, Mann M. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007;8:R250. doi: 10.1186/gb-2007-8-11-r250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boldogh I, Milligan D, Lee MS, Bassett H, Lloyd RS, McCullough AK. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001;29:2802–2809. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bouziane M, Miao F, Bates SE, Somsouk L, Sang BC, Denissenko M, O’Connor TR. Promoter structure and cell cycle dependent expression of the human methylpurine-DNA glycosylase gene. Mutat Res. 2000;461:15–29. doi: 10.1016/s0921-8777(00)00036-7. [DOI] [PubMed] [Google Scholar]
- 73.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Slupphaug G, Olsen LC, Helland D, Aasland R, Krokan HE. Cell cycle regulation and in vitro hybrid arrest analysis of the major human uracil-DNA glycosylase. Nucleic Acids Res. 1991;19:5131–5137. doi: 10.1093/nar/19.19.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daniel B, Swartzlander NCB, Anita H Corbett, Paul W Doetsch. Regulation of base excision repair in eukaryotes by dynamic localization strategies. Elsevier; 2012. [DOI] [PubMed] [Google Scholar]
- 76.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Madabushi A, Hwang BJ, Jin J, Lu AL. Histone deacetylase SIRT1 modulates and deacetylates DNA base excision repair enzyme thymine DNA glycosylase. Biochem J. 2013;456:89–98. doi: 10.1042/BJ20130670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McLaughlin D, Coey CT, Yang WC, Drohat AC, Matunis MJ. Characterizing Requirements for Small Ubiquitin-like Modifier (SUMO) Modification and Binding on Base Excision Repair Activity of Thymine-DNA Glycosylase in Vivo. J Biol Chem. 2016;291:9014–9024. doi: 10.1074/jbc.M115.706325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005;435:979–982. doi: 10.1038/nature03634. [DOI] [PubMed] [Google Scholar]
- 80.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Crystal structure of SUMO-3-modified thymine-DNA glycosylase. J Mol Biol. 2006;359:137–147. doi: 10.1016/j.jmb.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 81.Coey CT, Fitzgerald ME, Maiti A, Reiter KH, Guzzo CM, Matunis MJ, Drohat AC. E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex. J Biol Chem. 2014;289:15810–15819. doi: 10.1074/jbc.M114.572081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moriyama T, Fujimitsu Y, Yoshikai Y, Sasano T, Yamada K, Murakami M, Urano T, Sugasawa K, Saitoh H. SUMO-modification and elimination of the active DNA demethylation enzyme TDG in cultured human cells. Biochem Biophys Res Commun. 2014;447:419–424. doi: 10.1016/j.bbrc.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 83.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 84.Griffiths LM, Swartzlander D, Meadows KL, Wilkinson KD, Corbett AH, Doetsch PW. Dynamic compartmentalization of base excision repair proteins in response to nuclear and mitochondrial oxidative stress. Mol Cell Biol. 2009;29:794–807. doi: 10.1128/MCB.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Swartzlander DB, McPherson AJ, Powers HR, Limpose KL, Kuiper EG, Degtyareva NP, Corbett AH, Doetsch PW. Identification of SUMO modification sites in the base excision repair protein, Ntg1. DNA Repair (Amst) 2016;48:51–62. doi: 10.1016/j.dnarep.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hagen L, Kavli B, Sousa MM, Torseth K, Liabakk NB, Sundheim O, Pena-Diaz J, Otterlei M, Horning O, Jensen ON, Krokan HE, Slupphaug G. Cell cycle-specific UNG2 phosphorylations regulate protein turnover, activity and association with RPA. EMBO J. 2008;27:51–61. doi: 10.1038/sj.emboj.7601958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nagelhus TA, Haug T, Singh KK, Keshav KF, Skorpen F, Otterlei M, Bharati S, Lindmo T, Benichou S, Benarous R, Krokan HE. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J Biol Chem. 1997;272:6561–6566. doi: 10.1074/jbc.272.10.6561. [DOI] [PubMed] [Google Scholar]
- 89.Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res. 1996;24:2036–2043. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dorn J, Ferrari E, Imhof R, Ziegler N, Hubscher U. Regulation of human MutYH DNA glycosylase by the E3 ubiquitin ligase mule. J Biol Chem. 2014;289:7049–7058. doi: 10.1074/jbc.M113.536094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27:3975–3980. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 92.Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, Broderick P, Gallinger S, Croitoru EM, Jenkins MA, Win AK, Cleary SP, Koessler T, Pharoah PD, Kury S, Bezieau S, Buecher B, Ellis NA, Peterlongo P, Offit K, Aaltonen LA, Enholm S, Lindblom A, Zhou XL, Tomlinson IP, Moreno V, Blanco I, Capella G, Barnetson R, Porteous ME, Dunlop MG, Farrington SM. A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. Br J Cancer. 2010;103:1875–1884. doi: 10.1038/sj.bjc.6605966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV, Kessler BM, Sharma RA, McKenna WG, Dianov GL. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009;28:3207–3215. doi: 10.1038/emboj.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morreall J, Limpose K, Sheppard C, Kow YW, Werner E, Doetsch PW. Inactivation of a common OGG1 variant by TNF-alpha in mammalian cells. DNA Repair (Amst) 2015;26:15–22. doi: 10.1016/j.dnarep.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bravard A, Vacher M, Moritz E, Vaslin L, Hall J, Epe B, Radicella JP. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009;69:3642–3649. doi: 10.1158/0008-5472.CAN-08-3943. [DOI] [PubMed] [Google Scholar]
- 96.Busso CS, Lake MW, Izumi T. Posttranslational modification of mammalian AP endonuclease (APE1) Cell Mol Life Sci. 2010;67:3609–3620. doi: 10.1007/s00018-010-0487-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yacoub A, Kelley MR, Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–5459. [PubMed] [Google Scholar]
- 98.Lirussi L, Antoniali G, Vascotto C, D’Ambrosio C, Poletto M, Romanello M, Marasco D, Leone M, Quadrifoglio F, Bhakat KK, Scaloni A, Tell G. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell. 2012;23:4079–4096. doi: 10.1091/mbc.E12-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Poletto M, Di Loreto C, Marasco D, Poletto E, Puglisi F, Damante G, Tell G. Acetylation on critical lysine residues of Apurinic/apyrimidinic endonuclease 1 (APE1) in triple negative breast cancers. Biochem Biophys Res Commun. 2012;424:34–39. doi: 10.1016/j.bbrc.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 100.Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, Jung SB, Kim CS, Irani K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832–845. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Busso CS, Wedgeworth CM, Izumi T. Ubiquitination of human AP-endonuclease 1 (APE1) enhanced by T233E substitution and by CDK5. Nucleic Acids Res. 2011;39:8017–8028. doi: 10.1093/nar/gkr401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim YJ, Kim D, Illuzzi JL, Delaplane S, Su D, Bernier M, Gross ML, Georgiadis MM, Wilson DM., 3rd S-glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J Mol Biol. 2011;414:313–326. doi: 10.1016/j.jmb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 105.Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, Tursz T, Le Chevalier T, Soria JC, Investigators IB DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 106.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 109.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 110.Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 2004;15:621–634. doi: 10.1016/j.molcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 111.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:39926–39936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- 114.Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J Biol Chem. 2004;279:16875–16882. doi: 10.1074/jbc.M313155200. [DOI] [PubMed] [Google Scholar]
- 115.Miao F, Bouziane M, Dammann R, Masutani C, Hanaoka F, Pfeifer G, O’Connor TR. 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. J Biol Chem. 2000;275:28433–28438. doi: 10.1074/jbc.M001064200. [DOI] [PubMed] [Google Scholar]
- 116.Campalans A, Marsin S, Nakabeppu Y, O’Connor TR, Boiteux S, Radicella JP. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair (Amst) 2005;4:826–835. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 117.Marenstein DR, Chan MK, Altamirano A, Basu AK, Boorstein RJ, Cunningham RP, Teebor GW. Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J Biol Chem. 2003;278:9005–9012. doi: 10.1074/jbc.M212168200. [DOI] [PubMed] [Google Scholar]
- 118.Marenstein DR, Ocampo MT, Chan MK, Altamirano A, Basu AK, Boorstein RJ, Cunningham RP, Teebor GW. Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein B). Interaction between a base excision repair enzyme and a transcription factor. J Biol Chem. 2001;276:21242–21249. doi: 10.1074/jbc.M101594200. [DOI] [PubMed] [Google Scholar]
- 119.Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008;283:27028–27037. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 121.Hegde PM, Dutta A, Sengupta S, Mitra J, Adhikari S, Tomkinson AE, Li GM, Boldogh I, Hazra TK, Mitra S, Hegde ML. The C-terminal Domain (CTD) of Human DNA Glycosylase NEIL1 Is Required for Forming BERosome Repair Complex with DNA Replication Proteins at the Replicating Genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J Biol Chem. 2015;290:20919–20933. doi: 10.1074/jbc.M115.642918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Theriot CA, Hegde ML, Hazra TK, Mitra S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair (Amst) 2010;9:643–652. doi: 10.1016/j.dnarep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Noren Hooten N, Fitzpatrick M, Kompaniez K, Jacob KD, Moore BR, Nagle J, Barnes J, Lohani A, Evans MK. Coordination of DNA repair by NEIL1 and PARP-1: a possible link to aging. Aging (Albany NY) 2012;4:674–685. doi: 10.18632/aging.100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sengupta S, Mantha AK, Mitra S, Bhakat KK. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene. 2011;30:482–493. doi: 10.1038/onc.2010.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH, Hazra TK. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem. 2007;282:28474–28484. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morland I, Rolseth V, Luna L, Rognes T, Bjoras M, Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Luna L, Rolseth V, Hildrestrand GA, Otterlei M, Dantzer F, Bjoras M, Seeberg E. Dynamic relocalization of hOGG1 during the cell cycle is disrupted in cells harbouring the hOGG1-Cys326 polymorphic variant. Nucleic Acids Res. 2005;33:1813–1824. doi: 10.1093/nar/gki325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hu J, Imam SZ, Hashiguchi K, de Souza-Pinto NC, Bohr VA. Phosphorylation of human oxoguanine DNA glycosylase (alpha-OGG1) modulates its function. Nucleic Acids Res. 2005;33:3271–3282. doi: 10.1093/nar/gki636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jaiswal M, LaRusso NF, Nishioka N, Nakabeppu Y, Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 132.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 134.Marsin S, Vidal AE, Sossou M, Menissier-de Murcia J, Le Page F, Boiteux S, de Murcia G, Radicella JP. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J Biol Chem. 2003;278:44068–44074. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 135.Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS, Patel R, Wu H, Huber-Keener K, Yang JW, Liu D, Spratt TE, Yang JM. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013;4:e731. doi: 10.1038/cddis.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kundu S, Brinkmeyer MK, Eigenheer RA, David SS. Ser 524 is a phosphorylation site in MUTYH and Ser 524 mutations alter 8-oxoguanine (OG): a mismatch recognition. DNA Repair (Amst) 2010;9:1026–1037. doi: 10.1016/j.dnarep.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parker AR, O’Meally RN, Sahin F, Su GH, Racke FK, Nelson WG, DeWeese TL, Eshleman JR. Defective human MutY phosphorylation exists in colorectal cancer cell lines with wild-type MutY alleles. J Biol Chem. 2003;278:47937–47945. doi: 10.1074/jbc.M306598200. [DOI] [PubMed] [Google Scholar]
- 138.Parker A, Gu Y, Mahoney W, Lee SH, Singh KK, Lu AL. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J Biol Chem. 2001;276:5547–5555. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- 139.Gu Y, Parker A, Wilson TM, Bai H, Chang DY, Lu AL. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J Biol Chem. 2002;277:11135–11142. doi: 10.1074/jbc.M108618200. [DOI] [PubMed] [Google Scholar]
- 140.Luncsford PJ, Chang DY, Shi G, Bernstein J, Madabushi A, Patterson DN, Lu AL, Toth EA. A structural hinge in eukaryotic MutY homologues mediates catalytic activity and Rad9-Rad1-Hus1 checkpoint complex interactions. J Mol Biol. 2010;403:351–370. doi: 10.1016/j.jmb.2010.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005;25:4388–4396. doi: 10.1128/MCB.25.11.4388-4396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boland MJ, Christman JK. Characterization of Dnmt3b:thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase(s) and RNA. J Mol Biol. 2008;379:492–504. doi: 10.1016/j.jmb.2008.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li YQ, Zhou PZ, Zheng XD, Walsh CP, Xu GL. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Um S, Harbers M, Benecke A, Pierrat B, Losson R, Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1998;273:20728–20736. doi: 10.1074/jbc.273.33.20728. [DOI] [PubMed] [Google Scholar]
- 146.Lucey MJ, Chen D, Lopez-Garcia J, Hart SM, Phoenix F, Al-Jehani R, Alao JP, White R, Kindle KB, Losson R, Chambon P, Parker MG, Schar P, Heery DM, Buluwela L, Ali S. T:G mismatch-specific thymine-DNA glycosylase (TDG) as a coregulator of transcription interacts with SRC1 family members through a novel tyrosine repeat motif. Nucleic Acids Res. 2005;33:6393–6404. doi: 10.1093/nar/gki940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim EJ, Um SJ. Thymine-DNA glycosylase interacts with and functions as a coactivator of p53 family proteins. Biochem Biophys Res Commun. 2008;377:838–842. doi: 10.1016/j.bbrc.2008.10.058. [DOI] [PubMed] [Google Scholar]
- 148.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, Buluwela L, Coombes RC, Chambon P, Schar P, Ali S. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. 2003;278:38586–38592. doi: 10.1074/jbc.M304286200. [DOI] [PubMed] [Google Scholar]
- 149.Guan X, Madabushi A, Chang DY, Fitzgerald ME, Shi G, Drohat AC, Lu AL. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007;35:6207–6218. doi: 10.1093/nar/gkm678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schomacher L, Han D, Musheev MU, Arab K, Kienhofer S, von Seggern A, Niehrs C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat Struct Mol Biol. 2016;23:116–124. doi: 10.1038/nsmb.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Steinacher R, Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 152.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fantini D, Vascotto C, Marasco D, D’Ambrosio C, Romanello M, Vitagliano L, Pedone C, Poletto M, Cesaratto L, Quadrifoglio F, Scaloni A, Radicella JP, Tell G. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010;38:8239–8256. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009;28:1616–1625. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kenny MK, Mendez F, Sandigursky M, Kureekattil RP, Goldman JD, Franklin WA, Bases R. Heat shock protein 70 binds to human apurinic/apyrimidinic endonuclease and stimulates endonuclease activity at abasic sites. J Biol Chem. 2001;276:9532–9536. doi: 10.1074/jbc.M009297200. [DOI] [PubMed] [Google Scholar]
- 158.Dianova II, Bohr VA, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 159.Ranalli TA, Tom S, Bambara RA. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem. 2002;277:41715–41724. doi: 10.1074/jbc.M207207200. [DOI] [PubMed] [Google Scholar]
- 160.Ahn B, Harrigan JA, Indig FE, Wilson DM, 3rd, Bohr VA. Regulation of WRN helicase activity in human base excision repair. J Biol Chem. 2004;279:53465–53474. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- 161.Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, Sarno S, Meggio F, Pinna LA, Caldecott KW. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 163.Luo H, Chan DW, Yang T, Rodriguez M, Chen BP, Leng M, Mu JJ, Chen D, Songyang Z, Wang Y, Qin J. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol Cell Biol. 2004;24:8356–8365. doi: 10.1128/MCB.24.19.8356-8365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Marintchev A, Gryk MR, Mullen GP. Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray cross-complementing group 1, with DNA polymerase beta. Nucleic Acids Res. 2003;31:580–588. doi: 10.1093/nar/gkg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP. Domain specific interaction in the XRCC1-DNA polymerase beta complex. Nucleic Acids Res. 2000;28:2049–2059. doi: 10.1093/nar/28.10.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marintchev A, Mullen MA, Maciejewski MW, Pan B, Gryk MR, Mullen GP. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat Struct Biol. 1999;6:884–893. doi: 10.1038/12347. [DOI] [PubMed] [Google Scholar]
- 167.Wei YF, Robins P, Carter K, Caldecott K, Pappin DJ, Yu GL, Wang RP, Shell BK, Nash RA, Schar P, et al. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol Cell Biol. 1995;15:3206–3216. doi: 10.1128/mcb.15.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nash RA, Caldecott KW, Barnes DE, Lindahl T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry. 1997;36:5207–5211. doi: 10.1021/bi962281m. [DOI] [PubMed] [Google Scholar]
- 169.Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Caldecott KW, Tucker JD, Stanker LH, Thompson LH. Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 1995;23:4836–4843. doi: 10.1093/nar/23.23.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 173.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 174.Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia. Cell. 2003;112:7–10. doi: 10.1016/s0092-8674(02)01247-3. [DOI] [PubMed] [Google Scholar]
- 175.Fan J, Otterlei M, Wong HK, Tomkinson AE, Wilson DM., 3rd XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004;32:2193–2201. doi: 10.1093/nar/gkh556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gagne JP, Moreel X, Gagne P, Labelle Y, Droit A, Chevalier-Pare M, Bourassa S, McDonald D, Hendzel MJ, Prigent C, Poirier GG. Proteomic investigation of phosphorylation sites in poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase. J Proteome Res. 2009;8:1014–1029. doi: 10.1021/pr800810n. [DOI] [PubMed] [Google Scholar]
- 177.Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci U S A. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GRt, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol. 2003;23:5919–5927. doi: 10.1128/MCB.23.16.5919-5927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Messner S, Schuermann D, Altmeyer M, Kassner I, Schmidt D, Schar P, Muller S, Hottiger MO. Sumoylation of poly(ADP-ribose) polymerase 1 inhibits its acetylation and restrains transcriptional coactivator function. FASEB J. 2009;23:3978–3989. doi: 10.1096/fj.09-137695. [DOI] [PubMed] [Google Scholar]
- 181.Zilio N, Williamson CT, Eustermann S, Shah R, West SC, Neuhaus D, Ulrich HD. DNA-dependent SUMO modification of PARP-1. DNA Repair (Amst) 2013;12:761–773. doi: 10.1016/j.dnarep.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280:40450–40464. doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 183.Kassner I, Andersson A, Fey M, Tomas M, Ferrando-May E, Hottiger MO. SET7/9-dependent methylation of ARTD1 at K508 stimulates poly-ADP-ribose formation after oxidative stress. Open Biol. 2013;3:120173. doi: 10.1098/rsob.120173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Piao L, Kang D, Suzuki T, Masuda A, Dohmae N, Nakamura Y, Hamamoto R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia. 2014;16:257–264. doi: 10.1016/j.neo.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chapman JD, Gagne JP, Poirier GG, Goodlett DR. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J Proteome Res. 2013;12:1868–1880. doi: 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- 186.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 187.Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ying S, Chen Z, Medhurst AL, Neal JA, Bao Z, Mortusewicz O, McGouran J, Song X, Shen H, Hamdy FC, Kessler BM, Meek K, Helleday T. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016;76:1078–1088. doi: 10.1158/0008-5472.CAN-15-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Noren Hooten N, Kompaniez K, Barnes J, Lohani A, Evans MK. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1) J Biol Chem. 2011;286:44679–44690. doi: 10.1074/jbc.M111.255869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fischer JM, Popp O, Gebhard D, Veith S, Fischbach A, Beneke S, Leitenstorfer A, Bergemann J, Scheffner M, Ferrando-May E, Mangerich A, Burkle A. Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014;281:3625–3641. doi: 10.1111/febs.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, Brown M, Marto JA, Livingston DM. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014;4:1430–1447. doi: 10.1158/2159-8290.CD-13-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Min W, Bruhn C, Grigaravicius P, Zhou ZW, Li F, Kruger A, Siddeek B, Greulich KO, Popp O, Meisezahl C, Calkhoven CF, Burkle A, Xu X, Wang ZQ. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Commun. 2013;4:2993. doi: 10.1038/ncomms3993. [DOI] [PubMed] [Google Scholar]
- 193.Walko TD, 3rd, Di Caro V, Piganelli J, Billiar TR, Clark RS, Aneja RK. Poly(ADP-ribose) polymerase 1-sirtuin 1 functional interplay regulates LPS-mediated high mobility group box 1 secretion. Mol Med. 2015;20:612–624. doi: 10.2119/molmed.2014.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA, Gorbunova V, Seluanov A. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016;16:2641–2650. doi: 10.1016/j.celrep.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wright RH, Castellano G, Bonet J, Le Dily F, Font-Mateu J, Ballare C, Nacht AS, Soronellas D, Oliva B, Beato M. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 2012;26:1972–1983. doi: 10.1101/gad.193193.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ciccarone F, Valentini E, Zampieri M, Caiafa P. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget. 2015;6:24333–24347. doi: 10.18632/oncotarget.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhao H, Sifakis EG, Sumida N, Millan-Arino L, Scholz BA, Svensson JP, Chen X, Ronnegren AL, Mallet de Lima CD, Varnoosfaderani FS, Shi C, Loseva O, Yammine S, Israelsson M, Rathje LS, Nemeti B, Fredlund E, Helleday T, Imreh MP, Gondor A. PARP1- and CTCF-Mediated Interactions between Active and Repressed Chromatin at the Lamina Promote Oscillating Transcription. Mol Cell. 2015;59:984–997. doi: 10.1016/j.molcel.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 199.Cazzalini O, Dona F, Savio M, Tillhon M, Maccario C, Perucca P, Stivala LA, Scovassi AI, Prosperi E. p21CDKN1A participates in base excision repair by regulating the activity of poly(ADP-ribose) polymerase-1. DNA Repair (Amst) 2010;9:627–635. doi: 10.1016/j.dnarep.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 200.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, Palanisamy N, Siddiqui J, Yan W, Cao X, Mehra R, Sabolch A, Basrur V, Lonigro RJ, Yang J, Tomlins SA, Maher CA, Elenitoba-Johnson KS, Hussain M, Navone NM, Pienta KJ, Varambally S, Feng FY, Chinnaiyan AM. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xie S, Mortusewicz O, Ma HT, Herr P, Poon RY, Helleday T, Qian C. Timeless Interacts with PARP-1 to Promote Homologous Recombination Repair. Mol Cell. 2015;60:163–176. doi: 10.1016/j.molcel.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 202.Lu H, Wang X, Li T, Urvalek AM, Yu L, Li J, Zhu J, Lin Q, Peng X, Zhao J. Identification of poly (ADP-ribose) polymerase-1 (PARP-1) as a novel Kruppel-like factor 8-interacting and -regulating protein. J Biol Chem. 2011;286:20335–20344. doi: 10.1074/jbc.M110.215632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rodriguez MI, Gonzalez-Flores A, Dantzer F, Collard J, de Herreros AG, Oliver FJ. Poly(ADP-ribose)-dependent regulation of Snail1 protein stability. Oncogene. 2011;30:4365–4372. doi: 10.1038/onc.2011.153. [DOI] [PubMed] [Google Scholar]
- 204.Henneke G, Koundrioukoff S, Hubscher U. Phosphorylation of human Fen1 by cyclin-dependent kinase modulates its role in replication fork regulation. Oncogene. 2003;22:4301–4313. doi: 10.1038/sj.onc.1206606. [DOI] [PubMed] [Google Scholar]
- 205.Guo Z, Kanjanapangka J, Liu N, Liu S, Liu C, Wu Z, Wang Y, Loh T, Kowolik C, Jamsen J, Zhou M, Truong K, Chen Y, Zheng L, Shen B. Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol Cell. 2012;47:444–456. doi: 10.1016/j.molcel.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Friedrich-Heineken E, Henneke G, Ferrari E, Hubscher U. The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J Mol Biol. 2003;328:73–84. doi: 10.1016/s0022-2836(03)00270-5. [DOI] [PubMed] [Google Scholar]
- 207.Guo Z, Zheng L, Xu H, Dai H, Zhou M, Pascua MR, Chen QM, Shen B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat Chem Biol. 2010;6:766–773. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Warbrick E, Lane DP, Glover DM, Cox LS. Homologous regions of Fen1 and p21Cip1 compete for binding to the same site on PCNA: a potential mechanism to co-ordinate DNA replication and repair. Oncogene. 1997;14:2313–2321. doi: 10.1038/sj.onc.1201072. [DOI] [PubMed] [Google Scholar]
- 209.Li X, Li J, Harrington J, Lieber MR, Burgers PM. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem. 1995;270:22109–22112. doi: 10.1074/jbc.270.38.22109. [DOI] [PubMed] [Google Scholar]
- 210.Chung L, Onyango D, Guo Z, Jia P, Dai H, Liu S, Zhou M, Lin W, Pang I, Li H, Yuan YC, Huang Q, Zheng L, Lopes J, Nicolas A, Chai W, Raz D, Reckamp KL, Shen B. The FEN1 E359K germline mutation disrupts the FEN1-WRN interaction and FEN1 GEN activity, causing aneuploidy-associated cancers. Oncogene. 2015;34:902–911. doi: 10.1038/onc.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 212.Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell. 2001;7:1221–1231. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 213.Qian L, Yuan F, Rodriguez-Tello P, Padgaonkar S, Zhang Y. Human Fanconi anemia complementation group a protein stimulates the 5′ flap endonuclease activity of FEN1. PLoS One. 2013;8:e82666. doi: 10.1371/journal.pone.0082666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Jaiswal AS, Armas ML, Izumi T, Strauss PR, Narayan S. Adenomatous polyposis coli interacts with flap endonuclease 1 to block its nuclear entry and function. Neoplasia. 2012;14:495–508. doi: 10.1593/neo.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shin YK, Amangyeld T, Nguyen TA, Munashingha PR, Seo YS. Human MUS81 complexes stimulate flap endonuclease 1. FEBS J. 2012;279:2412–2430. doi: 10.1111/j.1742-4658.2012.08620.x. [DOI] [PubMed] [Google Scholar]
- 216.Querol-Audi J, Yan C, Xu X, Tsutakawa SE, Tsai MS, Tainer JA, Cooper PK, Nogales E, Ivanov I. Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability. Proc Natl Acad Sci U S A. 2012;109:8528–8533. doi: 10.1073/pnas.1121116109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Simbulan-Rosenthal CM, Rosenthal DS, Luo R, Samara R, Espinoza LA, Hassa PO, Hottiger MO, Smulson ME. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene. 2003;22:8460–8471. doi: 10.1038/sj.onc.1206897. [DOI] [PubMed] [Google Scholar]