

Abstract
Animal models of human diseases that accurately recapitulate clinical pathology are indispensable for understanding molecular mechanisms and advancing preclinical studies. The Alzheimer's disease (AD) research community has historically used first‐generation transgenic (Tg) mouse models that overexpress proteins linked to familial AD (FAD), mutant amyloid precursor protein (APP), or APP and presenilin (PS). These mice exhibit AD pathology, but the overexpression paradigm may cause additional phenotypes unrelated to AD. Second‐generation mouse models contain humanized sequences and clinical mutations in the endogenous mouse App gene. These mice show Aβ accumulation without phenotypes related to overexpression but are not yet a clinical recapitulation of human AD. In this review, we evaluate different APP mouse models of AD, and review recent studies using the second‐generation mice. We advise AD researchers to consider the comparative strengths and limitations of each model against the scientific and therapeutic goal of a prospective preclinical study.
Keywords: Alzheimer's disease, amyloid precursor protein, amyloid β peptide, App knock‐in, APP transgenic
Subject Categories: Molecular Biology of Disease, Neuroscience
Modeling preclinical AD with APP mice
Alzheimer's disease (AD) is the most common neurodegenerative disease. In 2015, of the ~47 million individuals with dementia worldwide (Prince et al, 2016), AD accounted for 50–70% of these cases (Winblad et al, 2016). In the coming decades, global AD prevalence is projected to reach higher epidemic levels that will place a massive economic burden on society. Given the grim epidemiological forecast, scientifically based strategies to prevent AD are urgently needed, as evidenced by the fact that in the previous two decades more than 400 medication candidates failed to reach the clinic (Mangialasche et al, 2010). This large‐scale failure by the pharmaceutical industry and biomedical research community can be attributed to many factors, including inappropriate choice of mouse models, wrong timing for therapeutic interventions, over‐reliance on inappropriate assays for translational studies, or lack of precise biomarkers, most of which center on efficacy and reproducibility in preclinical studies of which animal models were a critical component. In this review, we focus on the important roles that genetically engineered mouse models have contributed to AD mechanistic studies and preclinical drug development. Recently, new mouse models have been introduced to the community, and we will summarize the construction, characteristics, and merits and demerits of current AD mouse models to facilitate understanding of these tools for the design of future studies. In this aim, we will be guided by the principle that AD is a disease that should ideally be prevented in preclinical stages where a potential interventional window of at least 20 years exists before dementia clinically manifests (Funato et al, 1998; Bateman et al, 2012).
Clinically, AD is characterized by early memory deficits followed by a decline in other cognitive functions (Scheltens et al, 2016; Winblad et al, 2016). The pathology of AD begins before overt cognitive symptoms and includes the accumulation of amyloid β peptide (Aβ) as extracellular plaques, hyper‐phosphorylated tau as intracellular neurofibrillary tangles (NFTs), and chronic neuroinflammation, followed by the loss of neuronal cells mainly in the cerebral cortex and hippocampus (Braak & Braak, 1991; Hyman et al, 2012). In parallel, a coordinated breakdown in vascular, astroglial, and oligodendrocytic responses demonstrates that AD is a systems disorder and the roles and interactions of different cell types in the decline of brain homeostasis and resultant dementia is a major research topic (De Strooper & Karran, 2016). Aβ pathology is initiated at least two decades before cortical tau pathology and the onset of clinical symptoms (Bateman et al, 2012; Maruyama et al, 2013). After disease onset, it is increasingly difficult to treat symptoms after postmitotic neurons start to degenerate (Fig 1) and finely tuned neuronal circuits and cognitive skills are not easily recovered at later stages. Thus, the development of accurate preclinical animal models of AD for studies of disease mechanisms and the development of medications for early prevention and treatment are considered vital research goals in accord with the global epidemiological status.
Figure 1. Cortical pathology, neurological symptoms, and APP mouse models of AD .
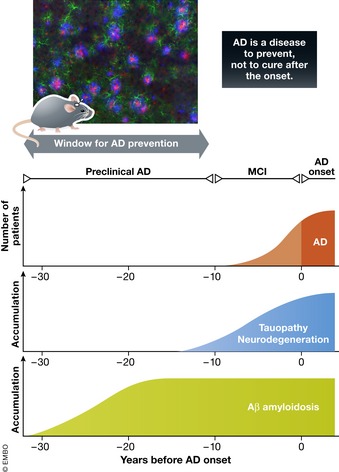
There are three neurological phases leading to the onset of AD and associated cortical pathology. The first phase is preclinical AD, where Aβ accumulates in cortex without neurological symptoms. The second phase is mild cognitive impairment (MCI), where tauopathy and neurodegeneration proceed with predementia symptoms. The third phase is AD, where neurodegeneration eliminates neurons and neuronal circuits in an irreversible manner with progressively serious symptoms of dementia. As models of preclinical AD, APP‐overexpressing mice or App knock‐in mice exhibit extensive Aβ pathology without tauopathy and neurodegeneration, for which there is a preventive window of approximately two decades. Modified from Ihara and Arai (2007). The pathology shown is from the cortex of a 9‐month‐old App NL‐G‐F/NL‐G‐F mouse. Blue: Aβ; red: microglia (Iba‐1); green: astrocyte (GFAP) (Saito et al, 2014).
Sporadic late‐onset AD (LOAD) accounts for more than 99% of all cases (Campion et al, 1999), and the ratio of LOAD patients to all AD patients continues to increase because aging is a primary risk factor aligned with aging of the world population. Early‐onset AD (EOAD), in contrast, is predominately familial and caused by mutations in genes that encode amyloid precursor protein (App), presenilin‐1 (PSEN1 or PS1) and presenilin‐2 (PS2). Proteolytic processing of APP by β‐secretase (β‐site APP cleaving enzyme 1 or BACE1) and γ‐secretase generates soluble Aβ fragments. γ‐Secretase is a protein complex composed of PS1 or PS2, nicastrin, Aph1 and presenilin enhancer 2 (PEN2). Most familial AD (FAD) mutations affect processivity of γ‐secretase resulting in the release of longer Aβ peptides and a shift in the relative ratios of the different peptides, including the Aβ42/Aβ40 ratio (Welander et al, 2009; Chávez‐Gutiérrez et al, 2012). Similarly, mutations in the App gene result in the production of longer Aβ peptides that aggregate more easily (Rosenberg et al, 2016). Interestingly, an App A673T mutation was claimed to reduce the risk of sporadic AD (SAD) and age‐related cognitive decline by decreasing the production of Aβ (Jonsson et al, 2012), although these findings require confirmation (Wang et al, 2015) and the mutation appears to affect the biophysical properties of Aβ peptides (Benilova et al, 2014; Maloney et al, 2014). The existence of familial mutations that directly affect Aβ production and influence AD risk is often cited as evidence that Aβ accumulation is central to AD pathogenesis (amyloid cascade hypothesis: Selkoe & Hardy, 2016).
In general, Aβ and tau pathology in sporadic and familial cases are morphologically similar, rationalizing the use of mouse models with genetically engineered FAD mutations for understanding SAD. However, the extent to which these models actually reproduce SAD remains unknown. A critical factor to consider in developing and using mouse APP models is the potential mechanism of Aβ accumulation. In FAD, Aβ deposition is primarily caused by the increased production of Aβ>40 except for intra‐Aβ sequence mutations that alter its structural properties (Selkoe & Hardy, 2016) or the Swedish mutation that increases all Aβ species by increasing cleavage at the β‐site. Whether the Iceland mutation App A673T is protective by decreasing Aβ production (Jonsson et al, 2012) remains controversial. However, because Aβ degradation declines with aging potentially due to a decrease in the major Aβ‐degrading enzyme neprilysin (Iwata et al, 2001, 2002; Hellström‐Lindahl et al, 2008) and because Aβ clearance is decreased in SAD patients (Mawuenyega et al, 2010), Aβ deposition in SAD is likely partially caused by an aging‐associated decrease in degradation/clearance of Aβ (Fig 2). In accord, increased production of Aβ or its decreased degradation/clearance might contribute to Aβ accumulation in AD (Saido & Iwata, 2006). Recent studies on the ubiquitin‐proteasome system and autophagy (Nilsson et al, 2013; Ciechanover & Kwon, 2015; Khaminets et al, 2016) point to the essential significance of protein degradation in many diseases including AD. While decreased Aβ degradation may be dominant in SAD, most APP mouse models have increased FAD‐like production. The selection of a mouse model for preclinical studies should consider this issue.
Figure 2. Aβ proteostasis determined by the balance of production and degradation.
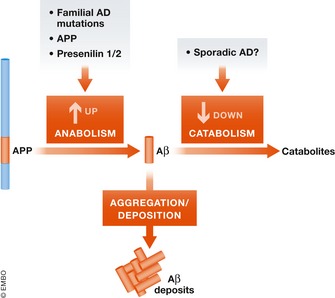
The balance of anabolism and catabolism determines the steady‐state quantity of a given protein in a biological system. In FAD, increased anabolism of pathogenic Aβ (Aβ42 and Aβ43) in cortex results in pathological deposition. In SAD, the causes of Aβ accumulation are not fully understood, but an aging‐associated decrease in catabolism is a candidate mechanism (Saido & Iwata, 2006; Hellström‐Lindahl et al, 2008).
Various AD mouse models (Onos et al, 2015; Puzzo et al, 2015; Drummond & Wisniewski, 2017) as well as other animal models including rats, non‐human primates, Drosophila, and Caenorhabditis elegans (Drummond & Wisniewski, 2017) have been recently reviewed. In this review, we will focus on genetically modified APP mouse models of AD as they are the most practical approach for in vivo screening and validation of preventive medications at this time (Zahs & Ashe, 2010). In the absence of gene manipulations, no small animal models exist at present that sufficiently or consistently mimic clinical disease pathology for experimental and preclinical studies of AD. Furthermore, we will focus on the preclinical stage when the time window for effective prevention and treatment is wider. Preclinical AD patients are cognitively normal (Bateman et al, 2012), and likewise, model mice in parallel preclinical stages having Aβ pathology without tauopathy and neurodegeneration should not exhibit strong cognitive impairment. However, at this early stage, pathological alterations in Aβ or tau are thought to initiate disease processes including synaptic dysfunction, local damage to spines and dendrites, and vascular pathology that are observed in AD mouse models and presumably in presymptomatic humans (Ashe & Zahs, 2010). There are other potential points for prevention at this stage, such as mechanism(s) by which Aβ amyloidosis affects tauopathy or by which TREM2 (Guerreiro et al, 2013; Jonsson & Stefansson, 2013) influences pathogenesis and these may also be studied in mouse models for Aβ pathology.
First‐generation mouse models
First, a note on terminology: The term “transgenic (Tg) mice” could be confusing because in a wide sense it means “genetically modified mice” and because it also means mice into the host genome of which transgene is inserted in single or multicopy number. We use the second definition for the Tg mice in this review because knock‐in and knockout mice are different from Tg mice in that they maintain the original murine genomic structure except for the introduced mutations.
Several groups generated Tg mice that overexpress APP with or without FAD mutations using various promoters (Table 1), such as platelet‐derived growth factor‐β (PDGF‐β), prion protein (PrP), and Thy1. Frequently used models include PDAPP (Games et al, 1995), Tg2576 (Hsiao et al, 1996), APP23 (Sturchler‐Pierrat et al, 1997), J20 (Mucke et al, 2000), and TgCRND8 (Chishti et al, 2001). The APP constructs differ among the lines: They include APP695, APP770, and minigenes. Some mice carry more than one mutation in the transgene, and the most commonly used mutation is the Swedish mutation (K670N/M671L; Citron et al, 1992), which causes the overproduction of total Aβ from APP. These mice exhibit extracellular Aβ deposits in the brain, which are reminiscent of plaques in human patients with some differences (refer to section “Limitations of first‐generation mouse models”). In addition, these mice develop cognitive dysfunction before the appearance of amyloid plaques in many cases. However, they are unable to recapitulate neurofibrillary tangle (NFT) formation or neuronal loss.
Table 1.
Comparison of current APP mouse models of AD
| Strain(s) | Genetic background | Promoter | Mutation(s) | General features | Potential disadvantages | Suitable applications | |
|---|---|---|---|---|---|---|---|
|
Single transgenic APP‐Tg |
PDAPP | C57B6 × DBA2 | PDGF‐β | APPV717F |
Moderate behavioral phenotype Neuronal loss in some models |
Random integration of a transgene Overexpression‐related artifacts: multiple APP fragments overproduced No perfect negative control mice Artificial expression pattern controlled by exogenous promoters No NFTs, a feature of preclinical AD Sudden death (unknown reason): Tg2576, APP23 Cognitive impairment often preceding Aβ accumulation Mixed genetic backgrounds in some cases |
Analysis of Aβ production, deposition, and Aβ‐associated neuroinflammation Drug development (targeting Aβ deposits and secretases) Analysis of behavior if caused by Aβ Identification of CSF biomarkers |
| Tg2576 | B6; SJL mixed background | hamster prion protein (PrP) | APPKM670/671NL | ||||
| APP23 | C57BL/6 | mouse Thy1 | APPKM670/671NL | ||||
| J20 | C57BL/6 | PDGF‐β | APPKM670/671NL,V717F | ||||
| TgCRND8 | Hybrid C3H/He‐C57BL/6 | hamster prion protein (PrP) | APPKM670/671NL,V717F | ||||
|
Double transgenic APP‐Tg × PSEN1‐Tg or KI |
APPPS1 | C57BL/6J | mouse Thy1 (APP, PS1) |
APPKM670/671NL
PS1I166P |
Moderate behavioral phenotype Aβ accumulation from early stage Neuronal loss in some models |
Random integration of a number of transgenes in some cases Overexpression‐related artifacts: multiple APP fragments overproduced No perfect negative control mice Artificial expression pattern controlled by exogenous promoters Multiple mutations (APP + PS1) Non‐specific ER stress may arise No NFTs, a feature of preclinical AD Mixed backgrounds in some cases Complicated crossbreeding in some cases |
Analysis of Aβ production, deposition, and Aβ‐associated neuroinflammation Drug development (targeting Aβ deposits and secretases) Analysis of behavior if caused by Aβ Identification of CSF biomarkers Analysis of cell death, in some cases |
| 5XFAD | (C57BL/6 × SJL)F1 and C57BL/6J | mouse Thy1.2 (APP, PS1) |
APPKM670/671NL,I716V,V717I
PS1M146L,L286V |
||||
| Triple transgenic | 3xTg‐AD | C57BL/6J |
mouse Thy1.2 (APP, Tau) endogenous (PS1) |
APPK670N,M671L
PS1M146V MAPTP301L |
Moderate to severe behavioral phenotype NFT formation Neuronal loss |
Random integration of transgenes Overexpression‐related artifacts: multiple APP fragments overproduced No perfect negative control mice Artificial expression pattern controlled by exogenous promoters Multiple mutations (APP + PS1 + FTDP‐17) where FTDP‐17 mutations are not causes of AD Mixed genetic backgrounds Complicated crossbreeding Cognitive impairment preceding Aβ accumulation |
Analysis of Aβ production, deposition, and Aβ‐associated neuroinflammation Drug development (targeting Aβ and tau) Analysis of behavior if caused by Aβ and tau Tau imaging Identification of CSF biomarkers Analysis of cell death |
| Single App knock‐in | NL‐F | C57BL/6J | endogenous APP | APPKM670/671NL,I716F |
Minor behavioral phenotype No overexpression of APP and byproducts except for CTFβ Endogenous App promoter‐driven gene expression Presence of relevant control mice (NL mice) Two lines for differential purposes NL‐F (wild‐type Aβ) NL‐G‐F (Arctic Aβ) : Aβ accumulation from early stage |
Multiple familial AD mutations of APP, the interaction between which has not been identified Unknown effects of Arctic mutation No severe behavioral phenotypes, a feature of preclinical AD No NFTs or neuronal loss, a feature of preclinical AD Genomic homozygosity in order to accelerate pathology and to remove murine endogenous Aβ (heterozygous mice accumulate Aβ, but take longer than homozygous mice.) Overproduction of CTFβ |
Analysis of Aβ production, deposition, and Aβ‐associated neuroinflammation Analysis of molecular pathways Analysis of neural network Omics analysis Reverse genetic analysis using the knockout and knock‐in mice Drug development (preventive) Additional gene manipulations (gene editing) Analysis of transcription and splicing of APP Identification of CSF and plasma biomarkers |
| NL‐G‐F | C57BL/6J | endogenous APP | APPKM670/671NL,E693G,I716F |
A side‐by‐side comparison of key factors to consider in selecting an APP mouse model for preclinical studies. Researchers should decide on a model depending on the specific scientific or therapeutic goal.
APP‐Tg mice recapitulate only a part of AD pathology, and efforts were made to combine them with other mutant mice to further reconstitute the remaining pathological hallmarks. PS1 is a constituent of the γ‐secretase complex that cleaves the C terminal fragment of APP generated by β‐secretase (CTF‐β) to produce Aβ (De Strooper et al, 1998). PS1 mutations cause the majority of FAD cases (Karch et al, 2014). The overexpression of mutant PS1 or a knock‐in pathogenic PSEN1 gene mutation alone did not induce Aβ pathology, presumably because the absolute amount of pathogenic longer Aβ such as Aβ42 and Aβ43 generated from mouse APP was insufficient (De Strooper et al, 1995). Alternately, mouse Aβ might have low amyloidogenic potential that might be caused by the existence of three different amino acids compared to human Aβ (Chui et al, 1999; Guo et al, 1999; Schmitz et al, 2004; Xu et al, 2015). However, the combination of these mice with human APP‐overexpressing Tg mice increased pathogenic Aβ production and conferred amyloidogenicity, which resulted in accelerated Aβ deposition, behavioral deficits, and neuronal loss. These combinations include Tg2576 and PS1M146L Tg (Holcomb et al, 1998), APPKM670/671NL Tg and PSA246E Tg (Borchelt et al, 1996, 1997), APP751KM670/671NL‐V717I Tg and PSM146L Tg (Schmitz et al, 2004), and APP KM670/671NL‐V717I and PSEN1 M233T/L235P knock‐in (Casas et al, 2004). Oakley et al (2006) generated 5XFAD mice carrying five FAD mutations in APP and PS1 transgenes (APPK670N/M671L/I716V/V717I Tg and PSEN1M146L/L286V Tg) driven by the Thy‐1 promoter. These mice exhibited cerebral Aβ pathology and gliosis as early as 2 months of age, synaptic degeneration and neuronal loss, and developed progressive cognitive deficits as early as 4–5 months. However, the 5XFAD mice also failed to develop NFTs despite their aggressive phenotypes and pathological changes (refer to section “Limitations of first‐generation mouse models”).
In efforts to replicate NFT pathology, crossbreeding of mutant Tau‐Tg mice with APP‐Tg mice enhanced tau pathology in the limbic system and olfactory cortex without affecting Aβ pathology (Tg2576 and JNPL3: Lewis et al, 2001; APP23 and JNPL3: Bolmont et al, 2007). Oddo et al (2003) generated a triple Tg model, 3xTg‐AD mice, which overexpress APPswe, and TauP301L transgenes on a PS1M146V knock‐in background. The mice exhibit neuropathology similar to AD patients, including the formation of Aβ plaques and NFTs, together with gliosis, synaptic damage, and memory deficits. However, the introduced mutations in the Mapt gene that encode tau protein are not causes of AD but rather of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP‐17). In addition, the overexpression of multiple genes causes an increased risk of artificial phenomena making it difficult to interpret the results. APP‐Tg mice crossbred with Mapt knockout mice exhibited improvements in memory deficits and survival in APP‐Tg mice, which suggests that tau may possibly confer Aβ toxicity (J20: Roberson et al, 2007; APP23: Ittner et al, 2010). Several combinations, such as APP‐Tg mice crossbred with BACE1 knockout mice (Ohno et al, 2004) or with apolipoprotein E4 (ApoE4) knock‐in mice (Fryer et al, 2005), may be more useful for specific applications.
Studies on first‐generation mouse models
APP‐ and APP/PS‐overexpressing mice exhibit key features of amyloid pathology that have allowed them to be applied in AD research. Although specific details of amyloid pathology such as plaque age of onset, size and regional distribution, and Aβ species content vary depending on the line, APP‐overexpressing mice recapitulate aspects of cerebral Aβ accumulation, including production and deposition of Aβ and associated neuroinflammation (microgliosis and astrogliosis). In some cases, downstream pathologic consequences of Aβ deposition in overexpressing mice, such as tau hyperphosphorylation, formation of dystrophic neurites, loss of synaptic markers, and the accumulation of BACE1 (Zhao et al, 2007), appear similar to those observed in AD. Other effects of Aβ deposition in overexpressing mice may also be relevant to AD. For example, 5XFAD mice exhibit neuron loss and memory deficits that are associated with amyloid pathology (Oakley et al, 2006). Importantly, BACE1 knockout abolishes Aβ deposition in 5XFAD mice and at the same time prevents both memory deficits and neuron loss in this and other mouse models (Ohno et al, 2007). Thus, cerebral Aβ accumulation is responsible for neuron loss and memory deficits in these lines, rather than transgene overexpression, although effects of β‐CTF overexpression cannot be ruled out.
APP‐overexpressing mice have also been useful in validating and assessing BACE1 and γ‐secretase inhibition as a therapeutic strategy for AD. BACE1 gene knockout abrogates cerebral Aβ accumulation in all APP‐ and APP/PS‐overexpressing mice tested to date (Luo et al, 2001, 2003; Ohno et al, 2004, 2007; Laird et al, 2005; McConlogue et al, 2007; Rabe et al, 2011), validating BACE1 as the major β‐secretase enzyme in the brain. Subsequently, overexpressing mice were used to screen small molecule inhibitors of BACE1, some of which could reduce Aβ levels in the brain and CSF by 90% or more. Some of which have advanced to clinical trials where they show similar Aβ lowering effects in human CSF (May et al, 2015; Neumann et al, 2015; Kennedy et al, 2016; Cebers et al, 2017). Another application of overexpressing mice translated successfully to humans is the preclinical testing of the anti‐Aβ antibody aducanumab. Plaque‐bearing Tg2576 mice that were chronically treated with aducanumab experienced a dose‐dependent reduction of cerebral Aβ levels by up to ~70% compared to vehicle (Sevigny et al, 2016). The mechanism of aducanumab‐mediated Aβ reduction appeared to involve binding to Aβ deposits in both human AD and aged Tg2576 mouse brains stimulating microglial phagocytosis of Aβ.
Criticisms of first‐generation mouse models have often focused on the failure of γ‐secretase‐based medications. However, mouse work (De Strooper et al, 1999; and many follow up studies in overexpressing models) had predicted almost all the side effects seen in human trials, long before phase III clinical trials were halted. Further work into the potential of tackling γ‐secretase in a safe manner is based on overexpressing mice (Weggen et al, 2001) and novel insights into more safe approaches continue (De Strooper, 2014), illustrating the utility of the overexpression paradigm for certain types of preclinical studies specifically targeting Aβ production and Aβ deposits, and possibly also for certain pathophysiologies associated with amyloid plaques such as neuroinflammation.
Limitations of first‐generation mouse models
APP undergoes sequential limited proteolysis catalyzed by proteases, collectively termed “secretases” (Fig 3A). In first‐generation transgenic mouse models, APP overexpression therefore results in the overproduction of various APP fragments in addition to Aβ (Fig 3). This makes it technically difficult to distinguish between the functional effects of additional Aβ and of other overproduced fragments. It is reasonable to assume that some of the phenotypes of the double and triple transgenic mutant mice might be of uncertain relevance to Alzheimer's disease. Box 1 summarizes the mutant APP or APP/PS‐overexpressing mouse models, including prospective ideas that may be experimentally validated to better consolidate the APP overexpression paradigm with human AD clinical pathogenesis (Huang & Mucke, 2012; Saito et al, 2014; Palop & Mucke, 2016).
Figure 3. Proteolytic processing of APP in wild‐type and Tg mice.
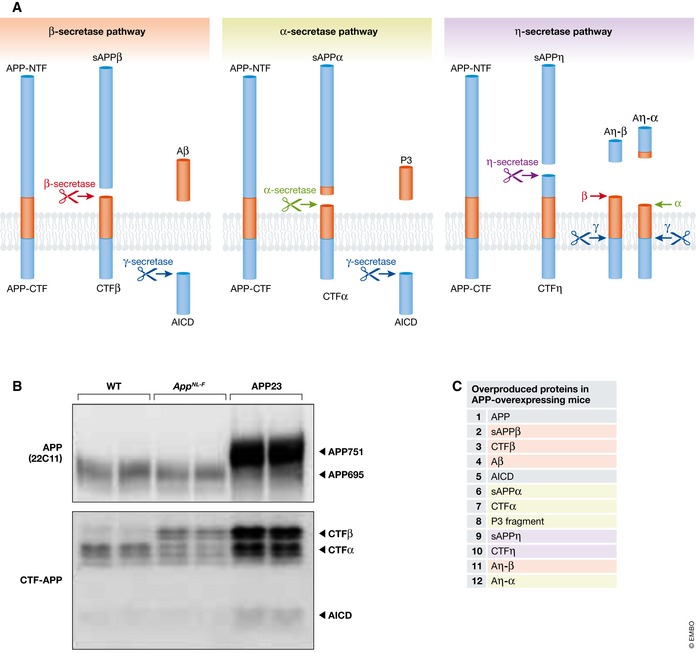
(A) APP processing by β‐secretase, α‐secretase, and η‐secretase pathways, respectively. (B) Western blot analysis of APP and APP‐derived fragments in wild‐type (WT), App NL‐F , and APP23 mice indicated that only APP23 mice produced APP and non‐Aβ APP fragments in substantial abundance. App NL‐F mice overproduce CTFβ; however, App NL mice produce the same amount without Aβ deposition (Saito et al, 2014), therefore serving as relevant negative controls. (C) Proteins that are overproduced in APP‐overexpressing mice.
Box 1: Limitations of mutant APP‐ and APP/PS‐overexpressing mouse models.
Transgene insertion may destroy endogenous gene loci (Kuro‐o et al, 1997; Verret et al, 2012; Saito et al, 2016).
Absence of non‐coding regions of the App gene precludes the analysis of splicing of APP mRNA and transcriptional regulation involving these gene regions (Nicolas et al, 2016).
Non‐matched negative controls due to variable transgene copy number and insertion site.
Overexpressed APP interacts unphysiologically with cellular proteins such as kinesin via JIP‐1 (Gunawardena & Goldstein, 2001; Chiba et al, 2014; Cassar & Kretzschmar, 2016; Laßek et al, 2016).
Overproduced non‐Aβ APP fragments may interact unphysiologically with cellular proteins (Chang & Suh, 2005; Mitani et al, 2012; Nicolas & Hassan, 2014; Kerridge et al, 2015; Nhan et al, 2015; Willem et al, 2015; Xia et al, 2016). See Fig 3.
Non‐specific ER stress may arise in APP/PS‐overexpressing mice (Barbero‐Camps et al, 2014; Chaudhari et al, 2014; Reinhardt et al, 2014; Borkham‐Kamphorst et al, 2016; Saito et al, 2016).
Aβ species may appear that are different from those found in clinical AD brain (Saido et al, 1995; Saito et al, 2016).
Atypical region specificity of Aβ pathology arises. Different Tg mice use different promoters to drive APP transgene expression that may affect Aβ in vivo propagation (Jucker & Walker, 2013). Höfling et al (2016) showed differences in expression level and brain regional patterning of exogenous APP among different APP‐Tg mouse lines.
Inconsistent drug effects occurred in some cases (Duggan & McCarthy, 2016; Ohno, 2016).
Crossbreeding with other mutant mice can generate additional artificial phenotype(s) (Higuchi et al, 2012; Saito et al, 2014). Refer to section “Limitations of first‐generation mouse models” for details.
Crossbreeding with particular mouse strains increase premature death in some lines (Tg2576 and TgCRND8; Carlson et al, 1997; Chishti et al, 2001).
In the absence of further validation in patient material or additional animal models in most cases, concerns remain with transgenic overexpression. APP overexpression for instance might induce behavioral abnormalities prior to Aβ pathology (Hsiao et al, 1996; Mucke et al, 2000), as Aβ pathology arises much earlier than overt disease onset in humans. Apart from potential overexpression artifacts, it has been difficult to standardize the phenotypes of the various models because of their construction with different promoters, transgene constructs, and mouse strains (Webster et al, 2014; Foley et al, 2015). In summary, further work is needed to reevaluate published results and to be aware that some phenotypes might be the result of APP‐ or APP/PS‐overexpression and not part of normal disease pathology. For instance, the extremely early lethality of calpastatin (CAST)‐deficient APP23 mice, half of which die within 10 weeks (Higuchi et al, 2012; Saito et al, 2014), clearly contradicts the chronic progressive nature of AD. Notably, intrinsic mouse–human differences cannot be ruled out as contributing factors as well (Espuny‐Camacho et al, 2017).
Second‐generation mouse models
To overcome intrinsic drawbacks of the APP overexpression paradigm, mouse models utilizing an App knock‐in strategy were generated to overproduce pathogenic Aβ such as Aβ42 without overexpressing APP. Single App knock‐in mouse models were generated in which the murine Aβ sequence was humanized by changing three amino acids that differ between mice and humans (G676R, F681Y, and H684R) and introduced two FAD mutations (KM670/671NL: Swedish and I716F: Beyreuther/Iberian mutations) into the endogenous mouse App gene (Saito et al, 2014). The identification of the Beyreuther/Iberian mutation using a phenylalanine scan (Lichtenthaler et al, 1999) opened up the possibility for a knock‐in strategy because this particular mutation increased the Aβ42/Aβ40 ratio by a factor of 30 in vitro. This mutation was subsequently identified as a cause for an aggressive form of FAD in Iberia (Guerreiro et al, 2010).
Mice that carry NL‐F mutations, denoted App NL‐F, exhibited increased Aβ42 production and a high Aβ42/Aβ40 ratio without alterations in the expression levels of APP or other fragments. The exception was that App knock‐in mice produced more CTF‐β and thus sAPPβ compared with wild‐type mice because of the Swedish mutation (Saito et al, 2014). Due to the increase in CTF‐β and concomitant decrease of CTF‐α in the App knock‐in mice, the total amount of CTF in App knock‐in mice remains the same as in wild‐type mice. To examine the effect of increased CTF‐β and sAPPβ in this case, App NL mice were generated that carried only the Swedish mutation and we confirmed that this amount of CTF‐β and sAPPβ exert no effects on the pathology or cognitive function of the mice (Saito et al, 2014; Masuda et al, 2016). The high levels of Aβ42 in App NL‐F mice led to pathological Aβ deposition in the cerebral cortex and hippocampus, which was accompanied by enhanced neuroinflammation, that is, infiltration of astrocytes and microglia that surround plaques from 6 months of age. Of particular note, the amyloid plaques in App NL‐F mice mainly consisted of pathogenic Aβ1/3pE‐42 (Saido et al, 1995) in a manner similar to the brains of AD patients, whereas the amyloid plaques in APP‐Tg mice were predominately composed of Aβ1–40 and were unphysiologically large, compared to those observed in App knock‐in mice and AD patients. A notable exception is the 5XFAD mice, which have amyloid deposits with an over twofold greater amount of Aβ1–42 as compared to Aβ1–40 (Oakley et al, 2006). Synaptic alterations in App NL‐F mice were also identified by the loss of presynaptic synaptophysin and postsynaptic PSD95 (Saito et al, 2014).
App NL‐F mice developed memory dysfunction at 18 months of age as detected by the Y‐maze test. In addition, Masuda et al (2016) analyzed the knock‐in mice using IntelliCage and determined that the App NL‐F mice exhibited various cognitive dysfunctions, including deficits in spatial memory and flexible learning, enhanced compulsive behavior, and reduced attention performance, depending on the age and pathology of the mice (Masuda et al, 2016). App knock‐in mice that harbor a third mutation, an E693G Arctic mutation (App NL‐G‐F), were also generated that makes Aβ more oligomerization/fibrillization‐prone (Cheng et al, 2007; Gessel et al, 2012), and these mice exhibited threefold faster and greater AD pathology and cognitive abnormalities compared with App NL‐F mice.
Reaume et al (1996) generated App knock‐in mice that harbored the Swedish mutation (K670N/M671L) with humanization of the murine Aβ sequences (App NLh/NLh). These mice overproduced human Aβ40 and Aβ42 without overexpressing APP; however, they failed to deposit Aβ in the brain at up to 22 months of age. This group subsequently crossbred their App knock‐in mice with mutant PSEN1 knock‐in mice (Flood et al, 2002; Malthankar‐Phatak et al, 2012), and the double knock‐in mice successfully exhibited Aβ pathology without depending on the overexpression paradigm. The double knock‐in mice, App NLh/NLh × PSEN P264L, exhibited less aggressive pathology compared with the double transgenic mice, likely because of the lower expression levels of APP and PS1. These mice are likely more difficult to use because of their double homozygous nature but have, for reasons that are unclear, not been used extensively by the community yet bear reassessment as experimental tools.
Li et al (2014) produced App knock‐in mouse models using multiple pathogenic mutations. The mice carried the Swedish (K670N/M671L), Dutch (E693Q), and London (V717I) mutations with the Aβ sequence humanized. The Dutch mutation causes intensive cerebral amyloid angiopathy (CAA) in humans, which results in brain hemorrhage and early mortality (Levy et al, 1990; Van Broeckhoven et al, 1990). Thus, this mutation is not a cause of FAD; however, its discovery inspired the first identification of an FAD mutation in the App gene (Goate et al, 1991; Hardy, 2017). These mice alone developed minimal Aβ deposits throughout life until the authors crossbred them with PSEN1 M146V knock‐in mice. The double knock‐in mice exhibited an age‐dependent deposition of Aβ not only in the parenchyma of the cerebral cortex but also the cerebral vasculature in a manner similar to human CAA pathology. Consistently, the double knock‐in mice without the Dutch mutation exhibited virtually no vascular pathology. They likely would not have had to introduce the PSEN knock‐in mice if they had used the Beyreuther/Iberian mutation instead of the London mutation in the mouse App gene. Nevertheless, the Dutch mutation‐harboring knock‐in mice can be considered to represent relevant models for CAA.
Studies on second‐generation mouse models
New studies suggest that a re‐examination of previous results obtained using first‐generation mouse models with their second‐generation counterparts is good practice. We previously reported that the activation of calpain, a calcium‐activated cysteine protease, is associated with Aβ plaque formation in the brains of AD patients and APP23 mice (Higuchi et al, 2012). Genetic ablation of calpastatin (CAST), a calpain‐specific inhibitor protein, exacerbated amyloid deposition, neuroinflammation, tau phosphorylation, and somato‐dendritic atrophy. Notably, when APP transgenic mice were crossed with CAST knockout mice, there was increased mortality (Higuchi et al, 2012) where half of the mice died in 10 weeks for unknown reason(s). In contrast, the double mutant App NL‐F mice crossbred with CAST knockout mice lived as long as wild‐type mice, indicating that the early lethality demonstrated in APP‐Tg crossbred with CAST knockout mice was inconsistent with the chronic nature of AD (Saito et al, 2014). Furthermore, Aβ was suggested to induce calpain‐dependent conversion of p35 to p25, a CDK5 activator, which may play an important role in AD pathogenesis (Seo et al, 2014). App NL‐F mice crossbred with CAST knockout mice, in which calpain is hyper activated, did not exhibit conversion of p35 to p25 despite the finding that calpastatin deficiency increases Aβ amyloidosis in the crossed mice (Saito et al, 2016). Thus, the conclusion that Aβ accumulation can cause p25 generation in neurons reported in 5XFAD mice may need to be revisited, as it might be caused by a non‐specific increase in calcium (Barbero‐Camps et al, 2014; Reinhardt et al, 2014) that might be unique to APP/PS1 double transgenic mice (Reinhardt et al, 2014), although further work is required to draw firm conclusions. A third result where differences between first and second‐generation mouse models are evident involved the reported down‐regulation of Nav1.1, a sodium channel expressed in PV‐positive interneurons in the APP‐Tg mouse line J20 and its resultant effect on epilepsy and AD phenotypes (Verret et al, 2012). In contrast, in App NL‐F mice, App NL‐F mice crossbred with CAST knockout mice, or APP23, down‐regulation of Nav1.1 was not observed (Saito et al, 2016) although it is possible that CAST deficiency makes calpain hyper activated (Higuchi et al, 2005; Takano et al, 2005). Hypofunction of Nav1.1 has been also observed in other mouse models such as Tg2576, TgCRND8, and BACE1 transgenic mice (Kim et al, 2007; Corbett et al, 2013; Hamm et al, 2017), and the effect of amyloid on Nav1.1 expression and its phenotypic consequences in AD mouse models should continue to be reviewed and validated in future studies.
Several basic findings using the second‐generation mouse models have advanced the basic biology of AD. Hama et al (2015) developed a new sorbitol‐based optical clearing method referred to as ScaleS that preserves the cellular structure of the tissue and proteins, including their immunochemical epitopes, enabling a 3D analysis of plaque deposition. App NL‐F mice treated with ScaleS allowed quantitative visualization of Aβ in an entire hemisphere, mapping of the 3D network of amyloid plaques in association with the vascular structure, and tracking of single plaques via successive light microscopy (LM) and electron microscopy (EM) observations. 3D images of microglial activation during amyloidosis of App NL‐F brains demonstrated that microglia association and active inflammation occur at an early stage of plaque formation. Such clearing methods combined with AD mouse models enable analysis of the degree of Aβ burden in larger brain volumes compared with conventional immunohistochemistry and are also applicable for the verification of immunotherapy (Sevigny et al, 2016) by visualizing therapeutic anti‐Aβ antibody binding to regional amyloid in situ.
One proposed mechanism for memory loss in AD is the destabilization of mushroom‐shaped postsynaptic spines, which may play an important role in memory storage. In accord, several reports indicate a reduction in mushroom spines in AD brain. In App NL‐F mice, Zhang et al (2015) demonstrated that hippocampal mushroom spines are lost and the STIM2 (stromal interaction molecule 2)‐nSOC (neuronal store‐operated calcium entry) pathway is altered as early as 3 months of age in a time‐dependent manner. The authors demonstrate the relationship between extracellular Aβ42 and spine loss concluding that Aβ42‐induced hyperactivation of mGluR5 and the subsequent overload of ER Ca2+ signaling likely represent the main cause for mushroom spine loss in App knock‐in mice. Moreover, an sSOC‐positive modulator NSN21778 recovered the reduction of mushroom spines and memory deficits via activation of transient receptor potential canonical 6 in App knock‐in mice (Zhang et al, 2016).
The orphan G protein (heterotrimeric guanine nucleotide‐binding protein)‐coupled receptor (GPCR) GPR3 is reported to regulate γ‐secretase activity and Aβ generation without affecting Notch receptor proteolysis (Thathiah et al, 2009). Recently, Huang et al (2015) demonstrated that genetic deletion of GPR3 reduced amyloid pathology in the brains of the App knock‐in models, as well as the APP‐Tg and APP/PS1‐Tg models. However, a reduction in the Aβ42/Aβ40 ratio following a genetic deficiency of GPR3 was detectable only in App NL‐F/NL‐F mice and not in the transgenic mice. In addition, they demonstrated that both the number and volume of amyloid plaques in App NL‐F/NL‐F mice crossbred with Gpr3 −/− mice were decreased compared with single App NL‐F/NL‐F (Gpr3 +/+) mice using 3D analysis with another clearing technique, CLARITY. These findings demonstrate that second‐generation mouse models can be used to evaluate the effect of new therapeutic targets on Aβ pathology.
BACE1 activity is up‐regulated in AD patients, after Aβ deposition, and in traumatic brain injury (Rossner et al, 2006). Kizuka et al (2015) hypothesized that bisecting N‐acetylglucosamine (GlcNAc) stabilizes BACE1 protein during oxidative stress, which results in an increase in Aβ generation. In a more recent paper, they demonstrated the up‐regulation of BACE1 protein and the level of bisecting GlcNAc in App NL‐G‐F/NL‐G‐F mouse brains, which was accompanied by an accumulation of oxidative damage (Kizuka et al, 2016).
Recently, the App knock‐in mice were used to refute the hypothesis that the new Alzheimer candidate gene PLD3 (Cruchaga et al, 2014) was involved in APP processing. Crossing of Pld3 deficient mice with App knock‐in mice demonstrated that there was no modulation of Aβ plaque or APP cleavage in these mice (Fazzari et al, 2017)
Limitations of second‐generation mouse models
Like single APP overexpression mice, the knock‐in mice do not exhibit tau pathology or neurodegeneration. This finding suggests that Aβ pathology may account, at least in part, for the cognitive dysfunction in AD via disturbances in neuronal activities because Zhang et al 2015 identified a reduction of mushroom spines, distinguishing spine structure at excitatory synapses, in the early stage in these mice. However, the Aβ‐induced memory failure alone might be insufficient to explain all symptoms of AD patients because tauopathy‐accompanying irreversible neurodegeneration has previously occurred at disease onset even in FAD‐mutation carriers (Bateman et al, 2012). Therefore, App knock‐in mice should be considered “models of preclinical AD”. A summary of features and limitations in App knock‐in mice is shown in Box 2.
Box 2: Limitations of single App knock‐in mouse model and potential solutions.
All the lines carry Swedish mutations (NL), which may exhibit different sensitivity to β‐secretase inhibitors from the wild‐type lines (KM). This is however easy to fix by converting NL to KM by gene editing, which cannot be applied to the APP‐overexpressing mice.
The Swedish mutation causes an increase of CTF‐β and concomitant reduction of CTF‐α. Consequently, total CTF levels (CTF‐β + CTF‐α) remain unchanged. If CTF‐β possesses particular biological or pathological functions, this may cause artifacts. Still, the amount of CTF‐β in single App knock‐in mice is much smaller than that in APP‐overexpressing mice.
The knock‐in mice possess two or three independent FAD mutations in the App gene. There is no evidence for an interaction between the Swedish mutation and Beyreuther/Iberian mutations, but the Arctic mutation increases CTFs (CTF‐β + CTF‐α) by 50% by unknown mechanisms (T Saito & TC Saido, unpublished; Cheng et al, 2004) and results in an unnatural Aβ conformation. It is important to perform delipidation pretreatment for the analysis of CTFs (Sato et al, 2003; Saito et al, 2014).
The negative control mice, App NL, accumulate no Aβ throughout life, but appear to induce minor microgliosis specifically in hippocampus and compulsive behavior (Masuda et al, 2016).
The knock‐in mice are used in a homozygous status, in order to achieve early pathology and to remove murine Aβ. It is advised to backcross them with the original line, B6/J, occasionally, to protect the mice from accumulating de novo mutations that could cause recessive defects.
APP may behave different from human APP because the App gene except for part of intron 15–17 is a murine sequence. For example, KPI domain‐containing APP variants are not expressed in mouse brain unlike in human brain (Saito et al, 2014). Therefore, App knock‐in mice may not be suitable for addressing the properties of KPI domain‐containing APP variants in endothelial cells, since these variants are expressed mostly in endothelial cells (Kitazume et al, 2010).
The absence of tauopathy and neurodegeneration in these mice, which live less than 3 years, may be simply a matter of AD time course because it requires more than two decades for Aβ amyloidosis to induce cortical tauopathy and neurodegeneration in humans (Bateman et al, 2012). To address these questions, further genetic manipulation to study the connection between Aβ pathology and tauopathy/neurodegeneration will be required. There are only three splice variants of tau (Mapt gene product) in adult mouse brain whereas there are six in humans. We therefore have generated human tau knock‐in mice, in which all the exons and introns of murine Mapt gene have been humanized (Saido et al, 2015). The mice are available to the research community from RIKEN. Other genes should not need to be humanized because overexpression of frontotemporal dementia with parkinsonism (FTDP) mutation‐carrying human tau is sufficient to reconstitute tauopathy composed of Neurofibrillary tangles (NFTs) and neurodegeneration (Lewis et al, 2000).
Biomarkers for the diagnosis and prognosis of preclinical AD will not only reduce the cost but also shorten the time necessary for drug development. App NL‐F and App NL‐G‐F mice are the only single knock‐in models that develop Aβ pathology and memory deficits. However, the presence of multiple mutations in the App gene, not observed in human patients, could in principle interact with each other in some cases that may not accurately represent clinical AD (Box 2). Thus, App NL‐F mice are suitable for analyzing the mechanisms that affect preclinical Aβ deposition compared with App NL‐G‐F that may be more useful for analyzing the mechanisms that alter downstream cascades.
Despite these potential drawbacks, App knock‐in mice may be useful as preclinical AD models for a number of purposes including (i) identification of biomarker(s) for preclinical AD, (ii) identification of molecules that evoke tauopathy in an Aβ pathology‐dependent manner, (iii) preclinical studies of preventive medicine(s), (iv) a platform for the generation of improved AD model(s) by crossbreeding with appropriate mutant mice (Table 1), and (v) to study the cellular phase of Alzheimer's disease (De Strooper & Karran, 2016), including the progressive response of vascular, astroglia, oligodendrocyte, and microglial cell populations upon amyloid stress. A distinct advantage is that expression from an endogenous promoter ensures that responses are not directed to cells that artifactually overexpress APP.
Future perspectives on AD mouse modeling
Previous first‐generation transgenic mouse models have made substantial contributions to our understanding of AD pathology. Many of these studies carried out with the best available mouse lines at the time have advanced the understanding of AD. The new second‐generation mouse lines solve some of the previous limitations and point the way to future third generation models. Until such next generation models become available, studies that investigate the interface of results from first‐ and second‐generation models will continue to reveal discrepancies and may in some cases indicate that findings from previous AD models may in part be a consequence of overexpression artifacts. Sorting out the clinically relevant phenotypes and mechanisms will require years of work with all the models but we urge AD researchers to remain vigilant and not to assume textbook status for any previous findings without extensive validation using the most appropriate mouse lines. We further emphasize that preclinical studies, including immunotherapy, may benefit from a re‐examination with new models to identify drug candidates for the preclinical prevention of earlier AD symptoms.
Species differences between rodents and humans in terms of neuroanatomy, genetics, and behaviors are also critical to control (Emes et al, 2003; Molnár & Clowry, 2012; Kaas, 2013; Nithianantharajah & Grant, 2013). Key molecules in AD such as Aβ, tau, and ApoE are different between mice and humans in their sequences, pathogenicity or number of isoforms expressed. In addition, immune systems in the brain also differ between mice and humans in certain aspects such as the proportion of microglial phenotypes, or the expression pattern of inflammation‐related genes (reviewed by Franco Bocanegra et al, 2017). The development of induced pluripotent stem cells (iPSCs) from AD patients can help to address species differences (Mungenast et al, 2016; Sullivan & Young‐Pearse, 2017). Recently, a novel chimeric AD mouse model was developed by transplanting human PSCs into AD mouse brain, showing pathological changes in tau and neurodegeneration in human neurons (Espuny‐Camacho et al, 2017). Furthermore, to leverage species differences for near‐clinical studies, we are generating non‐human primate models of AD (Okano et al, 2016) with more similarity to humans that could reduce species barriers and limit the time and cost of drug development.
AD research can benefit from modern views in the fields of immunology and cancer progression. Cancer stem cells arise in human bodies every day, but fail to develop into cancer in most cases because of cancer immunity (Yarchoan et al, 2017; Yeo & Angeli, 2017; Fig 4). We can draw an analogy between cancer immunity and AD protective mechanism(s) in the brain, which can maintain cognition essentially unaffected for more than two decades after the initial deposition of Aβ. If we could identify the molecules responsible for this protection, we should be able to facilitate studies of the major AD pathologies in mice by knocking out or modifying the corresponding genes. In the progressive systemic dysfunction that characterizes the cellular phase of AD, many types of cells are involved with complex feedback and feedforward responses at different disease stages.
Figure 4. Analogy between cancer immunity and protective mechanism in brain.
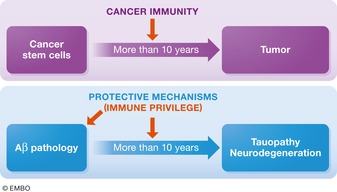
Analogy between cancer and AD. Because of cancer immunity mechanisms, it generally takes cancer stem cells more than a decade to develop pathological cancer. Human brains may possess similar protective mechanisms for AD, which can explain why it takes Aβ amyloidosis decades to induce neurodegeneration, whose identification may improve animal models and protective medications.
The emerging insight that AD is a multidimensional and multicellular process will require more integrated and complex forms of analysis (Tarasoff‐Conway et al, 2015; De Strooper & Karran, 2016). Knock‐in animals are most suitable to study these processes with target genes that are expressed in the correct cell types, with appropriate timing and amounts under the control of endogenous promoters. In addition, a number of common variants associated with LOAD have been identified in genes that participate in Aβ clearance or neuroinflammation in genome‐wide association studies (GWAS; Rosenberg et al, 2016; Naj et al, 2017). Effects of these variants can be precisely assessed by additional gene manipulations in App knock‐in mice. Both crossbreeding App knock‐in mice with other existing mutants and gene editing, which utilizes novel techniques, such as transcription activator‐like effector nuclease (TALEN) and clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 will reveal new mechanisms of Aβ pathology (Lee et al, 2016).
In conclusion, major steps to improve mouse models for AD are underway. While first‐generation models will remain important and relevant for AD research, the second‐generation mice solve some of the limitations of previous models for both basic and preclinical studies of AD. Standardization and sharing of disease models is essential for the objective interpretation of data, and the second‐generation mice can serve as one of the important baseline models for mechanisms of Aβ pathology obtained in different laboratories or under different conditions. However, every mouse model has limitations, and further side‐by‐side comparisons between App knock‐in mice, APP‐overexpressing mice, and other models and observations from human AD patients are required to move toward effective treatments. AD researchers can and should develop an ethic of sharing and comparing data and tools, carefully selecting the most suitable models for their purposes, and analyzing the data with an eye toward maximizing replication. To conquer AD as soon as possible, such collaboration ethics on a global scale will maximize the speed of drug development.
Acknowledgements
We thank Lennart Mucke, UCSF, for comments on the manuscript, Makoto Higuchi for sharing valuable information, and Charles Yokoyama, RIKEN, for editorial and editing support, respectively. This work was partially supported by a Grant‐in‐Aid for Young Scientists (B) and Scientific Research (B) from the Japan Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (S.H and T.S.); Special Postdoctoral Researchers Program from RIKEN (K.N. and S.H.); the Japan Science and Technology Agency Precursory Research for Embryonic Science and Technology (T.S.); the Strategic Research Program for Brain Sciences, Japan Agency for Medical Research and Development (AMED) (T.S.), and the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) project of AMED (T.C.S.); Aging Project of RIKEN (T.C.S.), the Swedish Research Council (P.N.), Alzheimerfonden (P.N.), Hållstens forskningsstiftelse (P.N.), and the National Institutes of Health (R01 AG022560 and R01 AG030142 to R.V.).
Conflict of interest
TCS and TS serve as the CEO and advisor, respectively, for RIKEN BIO Co. Ltd., which sublicenses animal models to for‐profit organizations, the profits from which are used for the identification of disease biomarkers.
The EMBO Journal (2017) 36: 2473–2487
Contributor Information
Hiroki Sasaguri, Email: hiroki.sasaguri@riken.jp.
Takaomi C Saido, Email: saido@brain.riken.jp.
References
- Ashe KH, Zahs KR (2010) Probing the biology of Alzheimer's disease in mice. Neuron 66: 631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero‐Camps E, Fernández A, Baulies A, Martinez L, Fernández‐Checa JC, Colell A (2014) Endoplasmic reticulum stress mediates amyloid β neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol 184: 2066–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 367: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Gallardo R, Ungureanu AA, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B (2014) The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid‐β (Aβ) aggregation. J Biol Chem 289: 30977–30989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Clavaguera F, Meyer‐Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M (2007) Induction of tau pathology by intracerebral infusion of amyloid‐β‐containing brain extract and by amyloid‐beta deposition in APP x Tau transgenic mice. Am J Pathol 171: 2012–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS (1996) Familial Alzheimer's disease‐linked presenilin 1 variants elevate Abeta1‐42/1‐40 ratio in vitro and in vivo. Neuron 17: 1005–1013 [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939–945 [DOI] [PubMed] [Google Scholar]
- Borkham‐Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL, Weiskirchen R (2016) CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress‐related apoptosis. Cell Signal 28: 34–42 [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1: 213–216 [DOI] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas‐Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget‐Darpoux F, Brice A, Frebourg T (1999) Early‐onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65: 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK (1997) Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet 6: 1951–1959 [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N‐terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165: 1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassar M, Kretzschmar D (2016) Analysis of amyloid precursor protein function in Drosophila melanogaster . Front Mol Neurosci 9: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebers G, Alexander RC, Haeberlein SB, Han D, Goldwater R, Ereshefsky L, Olsson T, Ye N, Rosen L, Russell M, Maltby J, Eketjäll S, Kugler AR (2017) AZD3293: pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer's disease. J Alzheimers Dis 55: 1039–1053 [DOI] [PubMed] [Google Scholar]
- Chang KA, Suh YH (2005) Pathophysiological roles of amyloidogenic carboxy‐terminal fragments of the beta‐amyloid precursor protein in Alzheimer's disease. J Pharmacol Sci 97: 461–471 [DOI] [PubMed] [Google Scholar]
- Chaudhari N, Talwar P, Parimisetty A, Lefebvre d'Hellencourt C, Ravanan P (2014) A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci 8: 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez‐Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B (2012) The mechanism of γ‐Secretase dysfunction in familial Alzheimer disease. EMBO J 31: 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Palop JJ, Esposito LA, Bien‐Ly N, Yan F, Mucke L (2004) Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med 10: 1190–1192 [DOI] [PubMed] [Google Scholar]
- Cheng IH, Scearce‐Levie K, Legleiter J, Palop JJ, Gerstein H, Bien‐Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L (2007) Accelerating amyloid‐beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 282: 23818–23828 [DOI] [PubMed] [Google Scholar]
- Chiba K, Araseki M, Nozawa K, Furukori K, Araki Y, Matsushima T, Nakaya T, Hata S, Saito Y, Uchida S, Okada Y, Nairn AC, Davis RJ, Yamamoto T, Kinjo M, Taru H, Suzuki T (2014) Quantitative analysis of APP axonal transport in neurons: role of JIP1 in enhanced APP anterograde transport. Mol Biol Cell 25: 3569–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA et al (2001) Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276: 21562–21570 [DOI] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T (1999) Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 5: 560–564 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47: e147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo‐Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the beta‐amyloid precursor protein in familial Alzheimer's disease increases beta‐protein production. Nature 360: 672–674 [DOI] [PubMed] [Google Scholar]
- Corbett BF, Leiser SC, Ling HP, Nagy R, Breysse N, Zhang X, Hazra A, Brown JT, Randall AD, Wood A, Pangalos MN, Reinhart PH, Chin J (2013) Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci 33: 7020–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, Jeng AT, Cooper B, Skorupa T, Carrell D, Levitch D, Hsu S, Choi J, Ryten M, UK Brain Expression Consortium (UKBEC) , Sassi C et al (2014) Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 505: 550–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG (1995) Production of intracellular amyloid‐containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J 14: 4932–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R (1999) A presenilin‐1‐dependent γ‐secretase‐like protease mediates release of Notch intracellular domain. Nature 398: 518–522 [DOI] [PubMed] [Google Scholar]
- De Strooper B (2014) Lessons from a failed γ‐secretase Alzheimer trial. Cell 159: 721–726 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164: 603–615 [DOI] [PubMed] [Google Scholar]
- Drummond E, Wisniewski T (2017) Alzheimer's disease: experimental models and reality. Acta Neuropathol 133: 155–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan SP, McCarthy JV (2016) Beyond γ‐secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell Signal 28: 1–11 [DOI] [PubMed] [Google Scholar]
- Emes RD, Goodstadt L, Winter EE, Ponting CP (2003) Comparison of the genomes of human and mouse lays the foundation of genome zoology. Hum Mol Genet 12: 701–709 [DOI] [PubMed] [Google Scholar]
- Espuny‐Camacho I, Arranz AM, Fiers M, Snellinx A, Ando K, Munck S, Bonnefont J, Lambot L, Corthout N, Omodho L, Vanden Eynden E, Radaelli E, Tesseur I, Wray S, Ebneth A, Hardy J, Leroy K, Brion JP, Vanderhaeghen P, De Strooper B (2017) Hallmarks of Alzheimer's disease in stem‐cell‐derived human neurons transplanted into mouse brain. Neuron 93: 1066–1081 [DOI] [PubMed] [Google Scholar]
- Fazzari P, Horre K, Arranz AM, Frigerio CS, Saito T, Saido TC, De Strooper B (2017) PLD3 gene and processing of APP. Nature 541: E1–E2 [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW (2002) FAD mutant PS‐1 gene‐targeted mice: increased Aβ42 and Aβ deposition without APP overproduction. Neurobiol Aging 23: 335–348 [DOI] [PubMed] [Google Scholar]
- Foley AM, Ammar ZM, Lee RH, Mitchell CS (2015) Systematic review of the relationship between amyloid‐β levels and measures of transgenic mouse cognitive deficit in Alzheimer's disease. J Alzheimers Dis 44: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco Bocanegra DK, Nicoll JAR, Boche D (2017) Innate immunity in Alzheimer's disease: the relevance of animal models? J Neural Transm https://doi.org/10.1007/s00702-017-1729-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005) Human apolipoprotein E4 alters the amyloid‐beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25: 2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Kusui K, Tamaoka A, Ishikawa K, Ohkoshi N, Namekata K, Okeda R, Ihara Y (1998) Quantitation of amyloid beta‐protein (A beta) in the cortex during aging and in Alzheimer's disease. Am J Pathol 152: 1633–1640 [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson‐Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliahparallel E, McConlogue L et al (1995) Alzheimer‐type neuropathology in transgenic mice overexpressing V717F beta‐amyloid precursor protein. Nature 373: 523–527 [DOI] [PubMed] [Google Scholar]
- Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT (2012) Familial Alzheimer's disease mutations differentially alter amyloid β‐protein oligomerization. ACS Chem Neurosci 3: 909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier‐Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak‐Vance M, Roses A, Williamson R, Rossor M, Owen M et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349: 704–706 [DOI] [PubMed] [Google Scholar]
- Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, Gómez‐Isla T, Hernández I, Lleó A, Machado A, Martínez‐Lage P, Masdeu J, Molina‐Porcel L, Molinuevo JL, Pastor P, Pérez‐Tur J et al (2010) Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging 31: 725–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila . Neuron 32: 389–401 [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP (1999) Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin‐1 mutant knock‐in mice. Nat Med 5: 101–106 [DOI] [PubMed] [Google Scholar]
- Hama H, Hioki H, Namiki K, Hoshida T, Kurokawa H, Ishidate F, Kaneko T, Akagi T, Saito T, Saido T, Miyawaki A (2015) ScaleS: an optical clearing palette for biological imaging. Nat Neurosci 18: 1518–1529 [DOI] [PubMed] [Google Scholar]
- Hamm V, Héraud C, Bott J‐B, Herbeaux K, Strittmatter C, Mathis C, Goutagny R (2017) Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer's disease. Sci Adv 3: e1601068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J (2017) The discovery of Alzheimer‐causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J 284: 1040–1044 [DOI] [PubMed] [Google Scholar]
- Hellström‐Lindahl E, Ravid R, Nordberg A (2008) Age‐dependent decline of neprilysin in Alzheimer's disease and normal brain: inverse correlation with A beta levels. Neurobiol Aging 29: 210–221 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC (2005) Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem 280: 15229–15237 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Iwata N, Matsuba Y, Takano J, Suemoto T, Maeda J, Ji B, Ono M, Staufenbiel M, Suhara T, Saido TC (2012) Mechanistic involvement of the calpain‐calpastatin system in Alzheimer neuropathology. FASEB J 26: 1204–1217 [DOI] [PubMed] [Google Scholar]
- Höfling C, Morawski M, Zeitschel U, Zanier ER, Moschke K, Serdaroglu A, Canneva F, von Hörsten S, De Simoni MG, Forloni G, Jäger C, Kremmer E, Roßner S, Lichtenthaler SF, Kuhn PH (2016) Differential transgene expression patterns in Alzheimer mouse models revealed by novel human amyloid precursor protein‐specific antibodies. Aging Cell 15: 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer‐type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97–100 [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102 [DOI] [PubMed] [Google Scholar]
- Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148: 1204–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Skwarek‐Maruszewska A, Horré K, Vandewyer E, Wolfs L, Snellinx A, Saito T, Radaelli E, Corthout N, Colombelli J, Lo AC, Van Aerschot L, Callaerts‐Vegh Z, Trabzuni D, Bossers K, Verhaagen J, Ryten M, Munck S, D'Hooge R, Swaab DF et al (2015) Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer's disease mouse models. Sci Transl Med 7: 309 [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y, Arai H (2007) Arutsuhaimaa‐byo ni naranai (Japanese). Tokyo, Japan: Asahi Books; [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J (2010) Dendritic function of tau mediates amyloid‐β toxicity in Alzheimer's disease mouse models. Cell 142: 387–397 [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC (2001) Metabolic regulation of brain Abeta by neprilysin. Science 292: 1550–1552 [DOI] [PubMed] [Google Scholar]
- Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC (2002) Region‐specific reduction of A beta‐degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res 70: 493–500 [DOI] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG et al (2012) A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 488: 96–99 [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson K (2013) TREM2 and neurodegenerative disease. N Engl J Med 369: 1568–1569 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas JH (2013) The evolution of brains from early mammals to humans. Wiley Interdiscip Rev Cogn Sci 4: 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Cruchaga C, Goate AM (2014) Alzheimer's disease genetics: from the bench to the clinic. Neuron 83: 11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, Jhee S, Kleijn HJ, Kuvelkar R, Li W, Mattson BA, Mei H, Palcza J, Scott JD, Tanen M, Troyer MD et al (2016) The BACE1 inhibitor verubecestat (MK‐8931) reduces CNS β‐amyloid in animal models and in Alzheimer's disease patients. Sci Transl Med 8: 363ra150 [DOI] [PubMed] [Google Scholar]
- Kerridge C, Kozlova DI, Nalivaeva NN, Turner AJ (2015) Hypoxia affects neprilysin expression through caspase activation and an APP intracellular domain‐dependent mechanism. Front Neurosci 9: 426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM (2007) BACE1 regulates voltage‐gated sodium channels and neuronal activity. Nat Cell Biol 9: 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Kato M, Yamaguchi Y, Honda T, Hashimoto Y, Wada Y, Saito T, Iwata N, Saido T, Taniguchi N (2010) J Biol Chem 285: 40097–40103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka Y, Kitazume S, Fujinawa R, Saito T, Iwata N, Saido TC, Nakano M, Yamaguchi Y, Hashimoto Y, Staufenbiel M, Hatsuta H, Murayama S, Manya H, Endo T, Taniguchi N (2015) An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer's disease. EMBO Mol Med 7: 175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka Y, Nakano M, Kitazume S, Saito T, Saido TC, Taniguchi N (2016) Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem J 473: 21–30 [DOI] [PubMed] [Google Scholar]
- Kuro‐o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki‐Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: 45–51 [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid‐beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25: 11693–11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laßek M, Weingarten J, Wegner M, Volknandt W (2016) The amyloid precursor protein‐A novel player within the molecular array of presynaptic nanomachines. Front Synaptic Neurosci 7: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HB, Sundberg BN, Sigafoos AN, Clark KJ (2016) Genome engineering with TALE and CRISPR systems in neuroscience. Front Genet 7: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez‐Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B (1990) Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248: 1124–1126 [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn‐Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402–405 [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487–1491 [DOI] [PubMed] [Google Scholar]
- Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, Pautler RG, Taffet GE, Zheng H (2014) Vascular and parenchymal amyloid pathology in an Alzheimer disease knock‐in mouse model: interplay with cerebral blood flow. Mol Neurodegener 9: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K (1999) Mechanism of the cleavage specificity of Alzheimer's disease gamma‐secretase identified by phenylalanine‐scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA 96: 3053–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu‐Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R (2001) Mice deficient in BACE1, the Alzheimer's β‐secretase, have normal phenotype and abolished beta‐amyloid generation. Nat Neurosci 4: 231–232 [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M (2003) BACE1 (β‐secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol Dis 14: 81–88 [DOI] [PubMed] [Google Scholar]
- Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, Atwal JK (2014) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289: 30990–31000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malthankar‐Phatak GH, Lin YG, Giovannone N, Siman R (2012) Amyloid deposition and advanced age fails to induce Alzheimer's type progression in a double knock‐in mouse model. Aging Dis 3: 141–155 [PMC free article] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer's disease: clinical trials and drug development. Lancet Neurol 9: 702–716 [DOI] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, Takashima A et al (2013) Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79: 1094–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Kobayashi Y, Kogo N, Saito T, Saido TC, Itohara S (2016) Cognitive deficits in single App knock‐in mouse models. Neurobiol Learn Mem 135: 73–82 [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 330: 1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Willis BA, Lowe SL, Dean RA, Monk SA, Cocke PJ, Audia JE, Boggs LN, Borders AR, Brier RA, Calligaro DO, Day TA, Ereshefsky L, Erickson JA, Gevorkyan H, Gonzales CR, James DE, Jhee SS, Komjathy SF, Li L et al (2015) The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J Neurosci 35: 1199–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson‐Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S (2007) Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem 282: 26326–26334 [DOI] [PubMed] [Google Scholar]
- Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, Ni K, Matsuoka N (2012) Differential effects between γ‐secretase inhibitors and modulators on cognitive function in amyloid precursor protein‐transgenic and nontransgenic mice. J Neurosci 32: 2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry G (2012) Cerebral cortical development in rodents and primates. Prog Brain Res 195: 45–70 [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson‐Wood K, McConlogue L (2000) High‐level neuronal expression of abeta 1‐42 in wild‐type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungenast AE, Siegert S, Tsai LH (2016) Modeling Alzheimer's disease with human induced pluripotent stem (iPS) cells. Mol Cell Neurosci 73: 13–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Schellenberg GD, for the Alzheimer's Disease Genetics Consortium (ADGC) (2017) Genomic variants, genes, and pathways of Alzheimer's disease: An overview. Am J Med Genet B Neuropsychiatr Genet 174: 5–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot‐Blomley M, Laue G, Beltz K, Vogg B, Schmid P, Frieauff W, Shimshek DR, Staufenbiel M, Jacobson LH (2015) A novel BACE inhibitor NB‐360 shows a superior pharmacological profile and robust reduction of amyloid‐β and neuroinflammation in APP transgenic mice. Mol Neurodegener 10: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhan HS, Chiang K, Koo EH (2015) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol 129: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M, Hassan BA (2014) Amyloid precursor protein and neural development. Development 141: 2543–2548 [DOI] [PubMed] [Google Scholar]
- Nicolas G, Wallon D, Goupil C, Richard AC, Pottier C, Dorval V, Sarov‐Rivière M, Riant F, Hervé D, Amouyel P, Guerchet M, Ndamba‐Bandzouzi B, Mbelesso P, Dartigues JF, Lambert JC, Preux PM, Frebourg T, Campion D, Hannequin D, Tournier‐Lasserve E et al (2016) Mutation in the 3'untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur J Hum Genet 24: 92–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC (2013) Aβ secretion and plaque formation depend on autophagy. Cell Rep 5: 61–69 [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Grant SG (2013) Cognitive components in mice and humans: combining genetics and touchscreens for medical translation. Neurobiol Learn Mem 105: 13–19 [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet‐Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal β‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39: 409–421 [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41: 27–33 [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R (2007) BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis 26: 134–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M (2016) Alzheimer's therapy targeting the β‐secretase enzyme BACE1: Benefits and potential limitations from the perspective of animal model studies. Brain Res Bull 126: 183–198 [DOI] [PubMed] [Google Scholar]
- Okano H, Sasaki E, Yamamori T, Iriki Shimogori T, Yamaguchi Y, Kasai K, Miyawaki A (2016) Brain/MINDS: a Japanese national brain project for marmoset neuroscience. Neuron 92: 582–590 [DOI] [PubMed] [Google Scholar]
- Onos KD, Sukoff Rizzo SJ, Howell GR, Sasner M (2015) Toward more predictive genetic mouse models of Alzheimer's disease. Brain Res Bull 122: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17: 777–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M, Comas‐Herrera A, Knapp M, Guerchet M, Karagiannidou M (2016) World Alzheimer Report 2016. Improving healthcare for people living with dementia. Coverage, quality and costs now and in the future. Alzheimer's Disease International. World Alzheimer's Report
- Puzzo D, Gulisano W, Palmeri A, Arancio O (2015) Rodent models for Alzheimer's disease drug discovery. Expert Opin Drug Discov 10: 703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe S, Reichwald J, Ammaturo D, De Strooper B, Saftig P, Neumann U, Staufenbiel M (2011) The Swedish APP mutation alters the effect of genetically reduced BACE1 expression on the APP processing. J Neurochem 119: 231–239 [DOI] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW (1996) Enhanced amyloidogenic processing of the β‐amyloid precursor protein in gene‐targeted mice bearing the Swedish familial Alzheimer's disease mutations and a “humanized” Aβ sequence. J Biol Chem 271: 23380–23388 [DOI] [PubMed] [Google Scholar]
- Reinhardt S, Schuck F, Grösgen S, Riemenschneider M, Hartmann T, Postina R, Grimm M, Endres K (2014) Unfolded protein response signaling by transcription factor XBP‐1 regulates ADAM10 and is affected in Alzheimer's disease. FASEB J 28: 978–997 [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid β‐induced deficits in an Alzheimer's disease mouse model. Science 316: 750–754 [DOI] [PubMed] [Google Scholar]
- Rosenberg RN, Lambracht‐Washington D, Yu G, Xia W (2016) Genomics of Alzheimer disease: a review. JAMA Neurol 73: 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner S, Sastre M, Bourne K, Lichtenthaler SF (2006) Transcriptional and translational regulation of BACE1 expression–implications for Alzheimer's disease. Prog Neurobiol 79: 95–111 [DOI] [PubMed] [Google Scholar]
- Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta‐amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 14: 457–466 [DOI] [PubMed] [Google Scholar]
- Saido TC, Iwata N (2006) Metabolism of amyloid β peptide and pathogenesis of Alzheimer's disease: towards presymptomatic diagnosis, prevention and therapy. Neurosci Res 54: 235–253 [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock‐in mouse models of Alzheimer's disease. Nat Neurosci 17: 661–663 [DOI] [PubMed] [Google Scholar]
- Saido T, Tsukakoshi K, Saito T (2015) ADPD5‐1906 Humanization of entire murine tau gene for a better model of AD. Neurodegener Dis 15(Suppl. 1): 22 [Google Scholar]
- Saito T, Matsuba Y, Yamazaki N, Saido TC (2016) Calpain activation in Alzheimer model mice is an artifact of APP and presenilin overexpression. J Neurosci 36: 9933–9936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, Maruyama H, Koo EH, Haass C, Takio K, Morishima‐Kawashima M, Ishiura S, Ihara Y (2003) Potential link between amyloid β‐protein 42 and C‐terminal fragment γ 49‐99 of β‐amyloid precursor protein. J Biol Chem 278: 24294–20301 [DOI] [PubMed] [Google Scholar]
- Scheltens P, Blennow K, Breteler MM, De Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer's disease. Lancet 388: 505–517 [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schäfer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA (2004) Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am J Pathol 164: 1495–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, Giusti‐Rodríguez P, Zhou Y, Rudenko A, Cho S, Ota KT, Park C, Patzke H, Madabhushi R, Pan L, Mungenast AE, Guan JS, Delalle I, Tsai LH (2014) Activity‐dependent p25 generation regulates synaptic plasticity and Aβ‐induced cognitive impairment. Cell 157: 486–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O'Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero‐Monzon O, Scannevin RH, Arnold HM, Engber T et al (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537: 50–56 [DOI] [PubMed] [Google Scholar]
- Sturchler‐Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease‐like pathology. Proc Natl Acad Sci USA 94: 13287–13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SE, Young‐Pearse TL (2017) Induced pluripotent stem cells as a discovery tool for Alzheimer's disease. Brain Res 1656: 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, Maki M, Saido TC (2005) Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem 280: 16175–16184 [DOI] [PubMed] [Google Scholar]
- Tarasoff‐Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ (2015) Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol 11: 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horré K, Vanbrabant M, Coun F, Baekelandt V, Delacourte A, Fischer DF, Pollet D, De Strooper B, Merchiers P (2009) The orphan G protein‐coupled receptor 3 modulates amyloid‐beta peptide generation in neurons. Science 323: 946–951 [DOI] [PubMed] [Google Scholar]
- Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter‐Van der Vlis M, Roos RA (1990) Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248: 1120–1122 [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ (2012) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149: 708–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LS, Naj AC, Graham RR, Crane PK, Kunkle BW, Cruchaga C, Murcia JD, Cannon‐Albright L, Baldwin CT, Zetterberg H et al (2015) Rarity of the Alzheimer disease‐protective APP A673T variant in the United States. JAMA Neurol 72: 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ (2014) Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet 5: 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez‐Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212–216 [DOI] [PubMed] [Google Scholar]
- Welander H, Frånberg J, Graff C, Sundström E, Winblad B, Tjernberg LO (2009) Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J Neurochem 110: 697–706 [DOI] [PubMed] [Google Scholar]
- Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LD, Moore S, Daria A, Hampel H, Müller V, Giudici C, Nuscher B, Wenninger‐Weinzierl A, Kremmer E, Heneka MT, Thal DR, Giedraitis V, Lannfelt L et al (2015) η‐Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526: 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Amouyel P, Andrieu S, Ballard C, Brayne C, Brodaty H, Cedazo‐Minguez A, Dubois B, Edvardsson D, Feldman H, Fratiglioni L, Frisoni GB, Gauthier S, Georges J, Graff C, Iqbal K, Jessen F, Johansson G, Jönsson L, Kivipelto M et al (2016) Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurol 15: 455–532 [DOI] [PubMed] [Google Scholar]
- Xia D, Gutmann JM, Götz J (2016) Mobility and subcellular localization of endogenous, gene‐edited Tau differs from that of over‐expressed human wild‐type and P301L mutant Tau. Sci Rep 6: 29074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Ran Y, Fromholt SE, Fu X, Yachnis A, Golde TE, Borchelt DR (2015) Murine Aβ over‐production produces diffuse and compact Alzheimer‐type amyloid deposits. Acta Neuropathol Commun 3: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Johnson BA III, Lutz ER, Laheru DA, Jaffee EM (2017) Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 17: 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo KP, Angeli V (2017) Bidirectional crosstalk between lymphatic endothelial cell and T cell and its implications in tumor immunity. Front Immunol 8: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs KR, Ashe KH (2010) ‘Too much good news’ ‐ are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer's disease? Trends Neurosci 33: 381–389 [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I (2015) Neuronal store‐operated calcium entry and mushroom spine loss in amyloid precursor protein knock‐in mouse model of Alzheimer's disease. J Neurosci 35: 13275–13286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Wu L, Pchitskaya E, Zakharova O, Fon Tacer K, Bezprozvanny I (2016) Store‐operated calcium channel complex in postsynaptic spines: A new therapeutic target for Alzheimer's disease treatment. J Neurosci 36: 11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R (2007) BACE1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci 27: 3639–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]