

Abstract
Objective:
To define molecular mechanisms underlying the clinical spectrum of epilepsy and movement disorder in individuals with de novo mutations in the GNAO1 gene.
Methods:
We identified all GNAO1 mutations reported in individuals with epilepsy (early infantile epileptiform encephalopathy 17) or movement disorders through April 2016; 15 de novo mutant alleles from 25 individuals were introduced into the Gαo subunit by site-directed mutagenesis in a mammalian expression plasmid. We assessed protein expression and function in vitro in HEK-293T cells by Western blot and determined functional Gαo-dependent cyclic adenosine monophosphate (cAMP) inhibition with a coexpressed α2A adrenergic receptor.
Results:
Of the 15 clinical GNAO1 mutations studied, 9 show reduced expression and loss of function (LOF; <90% maximal inhibition). Six other mutations show variable levels of expression but exhibit normal or even gain-of-function (GOF) behavior, as demonstrated by significantly lower EC50 values for α2A adrenergic receptor–mediated inhibition of cAMP. The GNAO1 LOF mutations are associated with epileptic encephalopathy while GOF mutants (such as G42R, G203R, and E246K) or normally functioning mutants (R209) were found in patients with movement disorders with or without seizures.
Conclusions:
Both LOF and GOF mutations in Gαo (encoded by GNAO1) are associated with neurologic pathophysiology. There appears to be a strong predictive correlation between the in vitro biochemical phenotype and the clinical pattern of epilepsy vs movement disorder.
Epilepsy is one of the most common neurologic disorders in the United States.1 Severe early-onset seizures can result in epileptic encephalopathy.2 There are at least 52 different gene mutations that cause early infantile epileptiform encephalopathy (EIEE).3 Mutations in the same genes also cause other neurodevelopmental abnormalities.4,5 A key challenge in genetic epilepsies has been understanding the genotype–phenotype relationships of causal genes; this may require biochemical analysis.6
Mutations in the heterotrimeric G protein Gαo (GNAO1 gene) cause an autosomal dominant epileptiform encephalopathy (EIEE17, OMIM: 615473).7 In this original article, all 4 mutations were characterized as having loss of function (LOF).7 More recently, an extended spectrum of GNAO1 encephalopathies was identified in which individuals had movement disorders but minimal to no seizures.8,9 Here, we use GNAO1 encephalopathy to describe the entire clinical spectrum of individuals with pathologic GNAO1 mutations. A genotype–phenotype correlation was also recently noted10; certain mutations (e.g., E246K and several R209 alleles) were found specifically in children with hypotonia, developmental delay, and chorea but no epilepsy. However, the mechanistic basis for this genotype–phenotype correlation remains unknown.
GNAO1 encodes the α subunit of Go, a heterotrimeric G protein (consisting of α and βγ subunits) that is highly abundant in the CNS, comprising about 1% of brain membrane protein.11 Go mediates signals from a wide range of inhibitory receptors including GABAB, α2A adrenergic, adenosine A1, and dopamine D2 receptors. A canonical function of Gαi/o family proteins is inhibition of cyclic adenosine monophosphate (cAMP).12 The identification of GOF mutations in ADCY5 (which encodes adenylate cyclase 5, the enzyme that produces cAMP) in dyskinesia and chorea patients directly links reduced cAMP to involuntary movement disorders.13–17 This is consistent with GOF behavior of Gαo.
Here we assessed expression and function of human mutations in GNAO1.7–9,18–27 Our biochemical analysis identified both LOF and gain-of-function (GOF) behaviors; the latter are associated with movement disorders while the former are primarily found in individuals with epileptiform encephalopathies. This mechanistic insight has important implications for therapies of GNAO1 encephalopathies.
METHODS
The detailed sources of reagents and antibodies, and the methods for mutagenesis, cell culture, transfections, Western blot, and cAMP assays, are described in the e-Methods at Neurology.org. The primer sets used in this study are listed in table e-1.
RESULTS
Most pathogenic GNAO1 mutations cause reduced Gαo protein expression.
To evaluate 15 mutations in GNAO1 (figures 1A and e-1) that were previously identified in patients with epilepsy or other neurodevelopmental disorders,7–9,18–27 we performed Western blots in HEK-293T cells transiently transfected with each mutant. The majority of mutants (12) showed significantly lower protein levels than wild-type (WT) Gαo, whereas 3 separate Arg209 mutant alleles showed essentially normal expression, as did the previously described GOF mutant G184S28 (figure 1, B–E).
Figure 1. Location and protein expression levels of human GNAO1 mutations related to epileptic encephalopathy.

(A) Location of 15 mutations (G40R, G42R, D174G, T191_F197del, L199P, G203R, R209C, R209G, R209H, A227V, Y231C, E246K, N270H, F275S, and I279N) mapped on the Gαo amino acid sequence. (B, C) Representative Western blots of Gαo protein expression from HEK293T cells transiently transfected with each Gαo mutant. (D, E) Quantification of relative protein levels of each Gαo mutant compared to wild-type Gαo. Graphs are the result of 3 independent experiments and data are presented as mean ± SEM. **p < 0.01, ****p < 0.0001 using 1-way analysis of variance with Bonferroni post hoc test for pairwise comparison.
Validation of an in vitro assay to assess function of GNAO1 mutations.
We used inhibition of forskolin-stimulated cAMP levels as a functional readout to allow efficient quantification of Gαo effects with full concentration curves for agonist-mediated signaling. We cotransfected Gαo plasmids with α2A adrenergic receptor (α2A AR) cDNA.29 Robust inhibition of cAMP by the α2 adrenergic agonist UK14,304 depends on both the transfected receptor and the Gαo protein (figure e-2, A and B). A pertussis toxin (PTX)–insensitive Gαo (C351G)30 was used to create all mutant constructs and WT, enabling inactivation of endogenous Gi/o proteins using PTX.
When PTX eliminates the inhibitory signaling through Gi/o, α2A AR couples weakly to Gs and stimulates adenylate cyclase (AC),31 resulting in increased cAMP levels after PTX treatment in the absence of a transfected Gαo (figure e-2B). Consequently, the fractional inhibition of AC by Gαo mutants was assessed as the decrease from the high control level of cAMP (PTX but no Gαo—0%) to the low level with WT Gαo (PTX and PTXi Gαo—100%). This is termed normalized % inhibition. PTX treatment did not alter the ability of the PTX-insensitive Gαo to mediate α2A AR-stimulated cAMP inhibition (figure e-2C). Hence, this is a good system to study functional consequences of Gαo mutations.
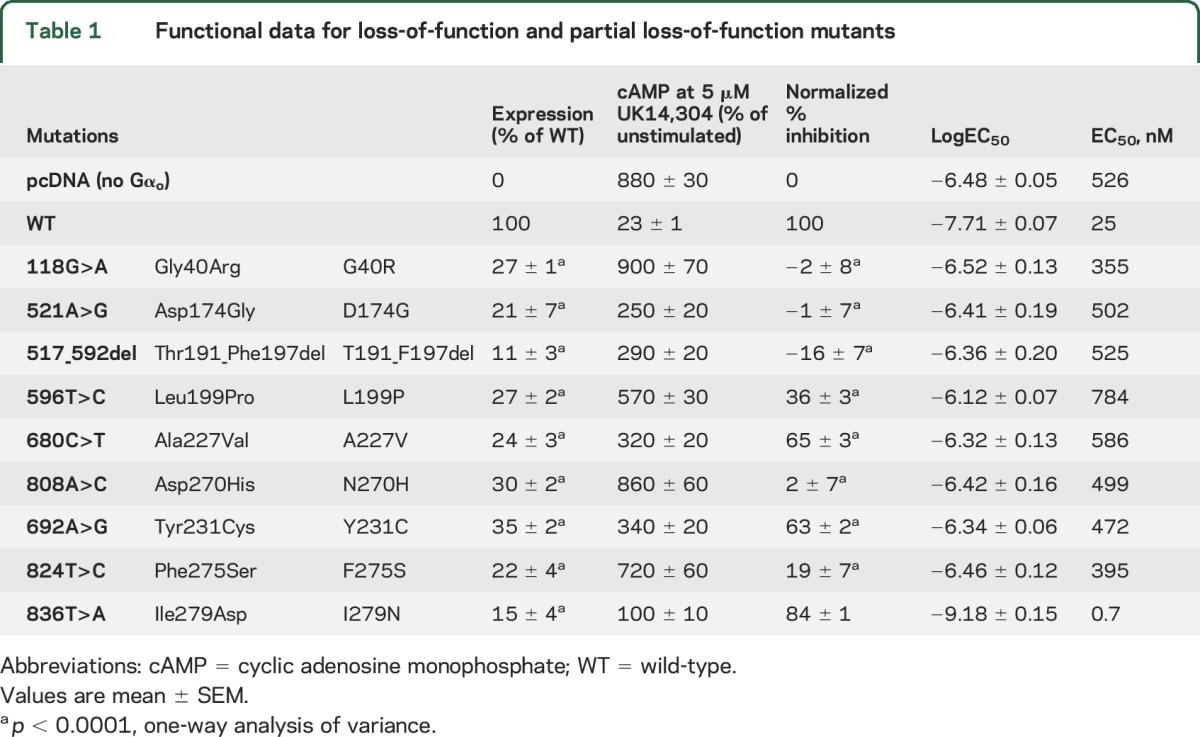
Nine GNAO1 mutations result in LOF or partial LOF.
Six mutants showed essentially complete LOF with normalized inhibition below 40% (figure 2A and table 1). All of these mutants also showed low expression levels (11%–35% of control; figure 1). Three mutants (A227V, Y231C, and I279N; figure 2, A and E, and table 1) had intermediate effects and were classified as partial LOF (PLOF) mutants. The I279N mutation showed a modestly reduced maximal inhibition of cAMP levels (figure 2E and table 1). This mutant also produced a very low EC50 value (0.7 nM vs 25 nM for WT Gαo), which might explain the discrepancy between the low expression levels (15% of WT) while maintaining good maximal inhibition in the cAMP inhibition assay. Based on the maximum inhibition below 90% of control, however, we classified this mutation as PLOF (table 1).
Figure 2. Effect on α2A adrenergic receptor (α2A AR)–mediated cyclic adenosine monophosphate (cAMP) inhibition by GNAO1 mutants.

(A–C) Dose–response curves of representative GNAO1 mutants. (A) Dose–response curves of loss-of-function (LOF) and partial loss-of-function (PLOF) mutants (G40R, L199P, N270H, F275S, A227V, Y231C) show changes in cAMP production in response to the adenylate cyclase activator forskolin and α2 AR agonist UK14,304, compared to the positive control (wild-type [WT]) and negative control (pcDNA). (B) Dose–response curves of functioning Gαo mutants show changes in cAMP production in response to the α2 AR agonist UK14,304. All dose–response curves are shown in comparison with WT and G184S. (C) G42R displays a biphasic dose–response curve with cAMP inhibition at low concentrations (gain of function [GOF]), followed by enhancement of cAMP levels at higher concentrations of UK14,304. (D) Quantification of EC50 of functioning Gαo mutants. G42R, G203R, and E246K exhibit significantly increased potency for α2A AR–mediated cAMP inhibition similar to the known GOF mutation G184S. *p < 0.05, **p < 0.01, ***p < 0.001 using paired t test between WT and each mutant separately (figure e-4). (E) Percentage of maximum inhibition (n = 5) was normalized to pcDNA (0%; resulting in activation of cAMP) and WT (100%). ****p < 0.0001 using 1-way analysis of variance with Bonferroni post hoc test for pairwise comparison. Note that the maximum inhibition of G42R was calculated at UK14,304 of 15.8 nM. NF = normal function; PTX = pertussis toxin.
Table 1.
Functional data for loss-of-function and partial loss-of-function mutants
The dominant nature of the clinical picture in the GNAO1 encephalopathies raised the question of whether the LOF mutations are actually dominant negative mutations that interfere with the function of the remaining normal Gαo protein expressed in heterozygous individuals. However, in coexpression studies of WT and mutant Gαo at plasmid ratios of 1:1 or 1:2, there was no evidence of a dominant negative action (figure e-3). This suggests that the effect of LOF mutations is through a haploinsufficiency mechanism rather than a dominant negative one.
Six GNAO1 mutations result in GOF or normal function.
Unexpectedly, a significant number of pathologic GNAO1 mutants showed essentially normal or even GOF behavior (figure 2, B–E, and table 2). As a benchmark for GOF behavior, we used our previously described RGS-insensitive G184S mutant,28,29,32,33 which shows a mild seizure phenotype in mouse models.32 It produced a small increase in the maximum inhibition of forskolin-stimulated cAMP levels in response to UK14,304 (table 2). More importantly, it had a significantly more potent response to the α2AR agonist UK14,304 (figures 2D and e-4A). This represents a 2- to 3-fold increase in signal strength at low agonist concentrations. Three of the human pathologic GNAO1 mutants also showed GOF behavior by this criterion. The G203R, E246K, and G42R mutants produced robust inhibition of cAMP with significantly lower EC50 values for the α2AR agonist UK14,304 (figures 2D and e-4, and table 2). G203R and E246K showed normal inhibition with modest decreases in EC50 (table 2 and figures 2, D and E, and e-4). This is similar to the effect on EC50 seen for the bona fide GOF mutant G184S. The G42R mutant showed the lowest EC50 of any of the mutants (figure e-4), at least 50-fold lower than the WT protein (figure 2, C and D). However, the inhibition mediated by G42R is followed by activation of cAMP with increasing concentrations of UK14,304 (figure 2C). The calculated normalized % inhibition for the G42R mutant is essentially identical to that of WT Gαo, which combined with its very high potency for agonist-mediated inhibition suggests GOF behavior.
Table 2.
Functional data for normal and gain-of-function mutants
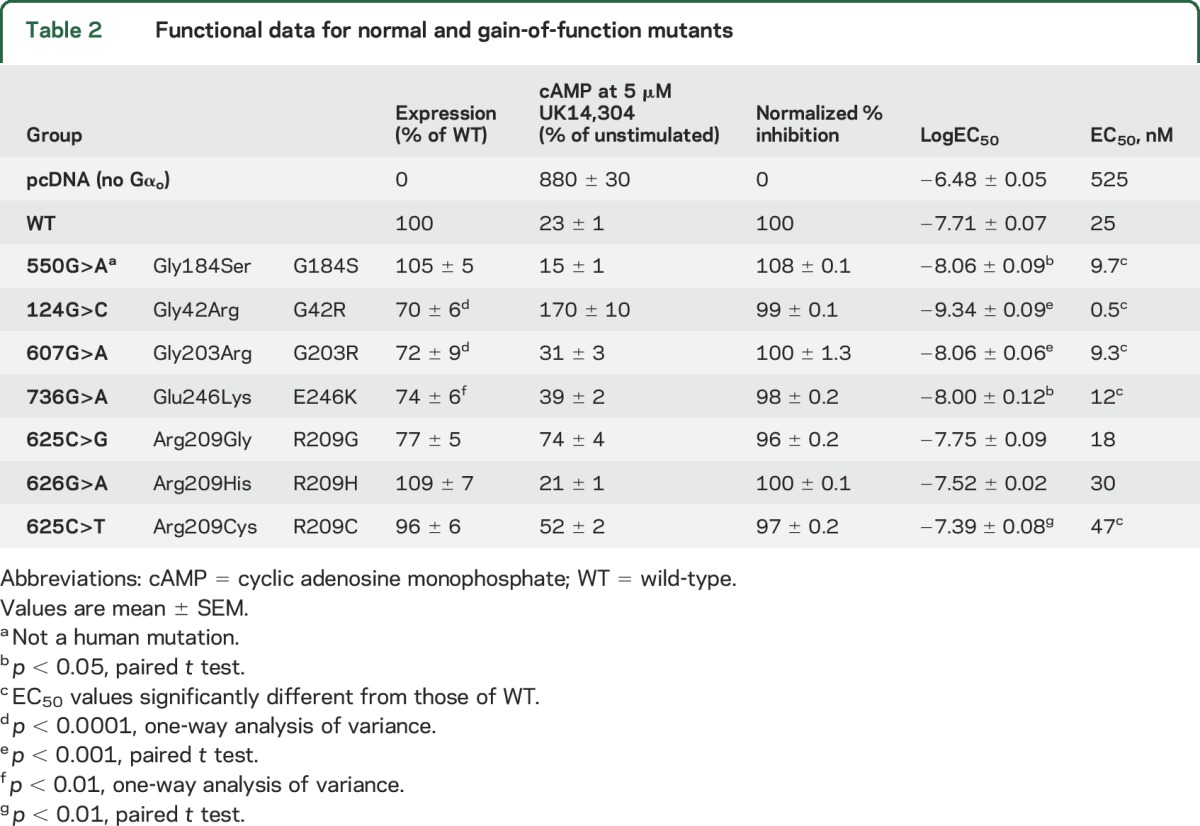
Three other patient-derived mutations (R209G, R209H, and R209C) showed almost completely normal function (NF) and nearly normal expression levels and are designated as NF mutants (figures 2, B, D, and E, and e-4, and table 2). The EC50 values for R209G and R209H mutant were not significantly different from WT, while the value for R209C was modestly but significantly higher (table 2 and figures 2D and e-4).
Clinical correlation with biochemical behavior of mutant GNAO1 alleles.
To address genotype–phenotype correlations for GNAO1 encephalopathy, we reviewed the case reports of all 25 individuals who had GNAO1 mutations that had been reported by April 2016. They have a range of clinical patterns, which extend from early severe epileptic encephalopathy with prominent tonic seizure activity to individuals with a dominant choreoathetotic movement disorder with virtually no evidence of seizures. There are also individuals,18,21 including one of the original 4 cases7 (patient 13—G203R; table 3), who had multiple seizures but also showed prominent choreoathetosis.
Table 3.
Correlation between cyclic adenosine monophosphate (cAMP) inhibition and clinical diagnosis
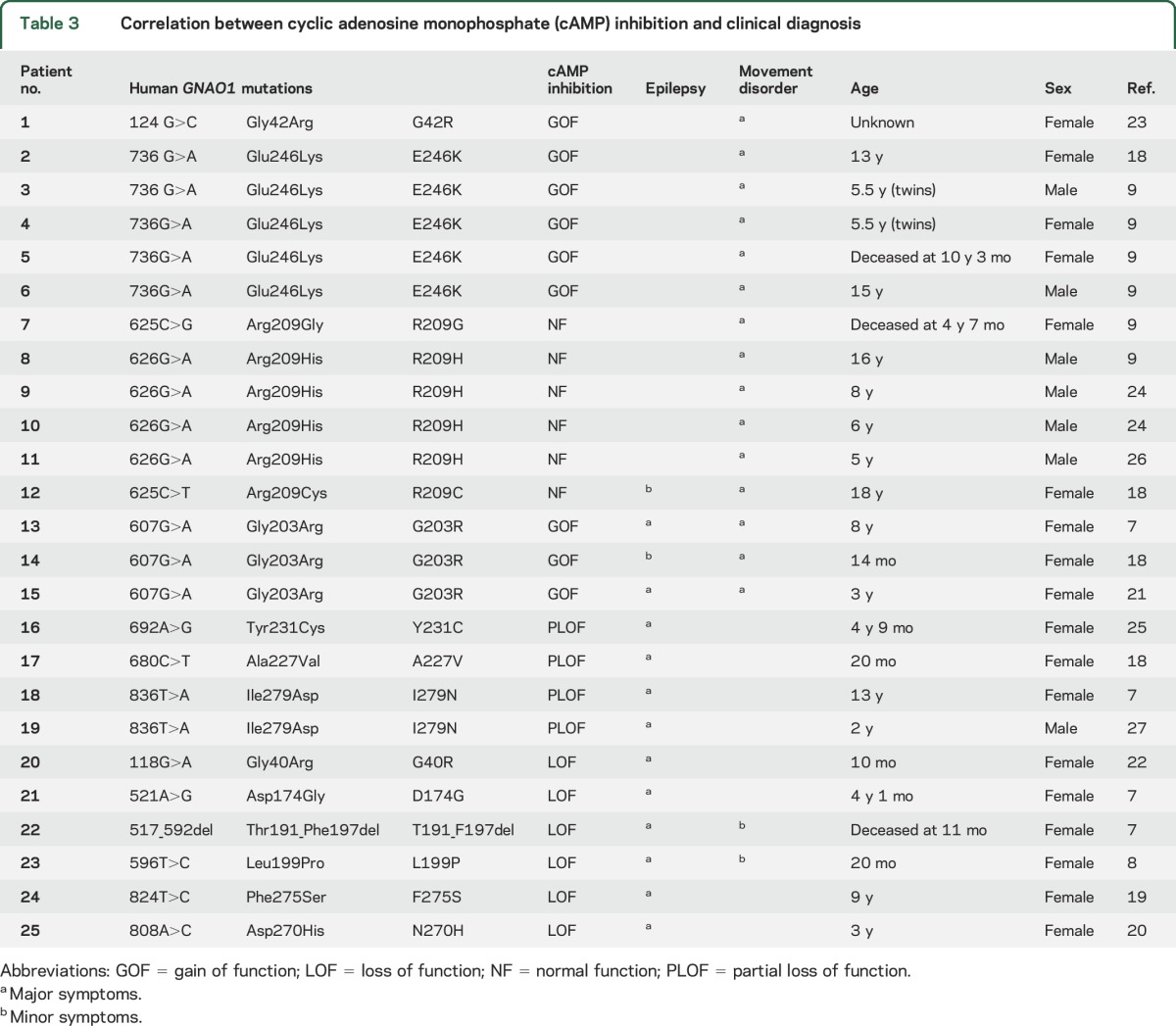
In 2016, a clinical report9 described a series of 6 patients with GNAO1 mutations and a pronounced movement disorder, virtually without seizures. They had global developmental delay and hypotonia from infancy and all developed chorea by ages 4–11 years. In the majority of cases it was intractable, leading to death in 2 cases. Four patients carried the E246K allele, which we have found to be a GOF mutation. The other 2 mutations found in this group (R209G and R209C) exhibited essentially normal function in our cAMP inhibition measurements. There are several other reports8,9,18–27 of GNAO1 mutations in individuals with a predominant movement disorder with or without seizures. This distinction of clinical patterns based on certain mutant alleles in GNAO1 encephalopathy patients was noted very recently10 but without information about biochemical mechanisms. Table 3 summarizes the Gαo biochemical function from the present report and its relation to seizure disorder or movement disorder in literature reports for these mutations. GOF and NF mutations are nearly always found when movement disorder is the predominant feature of the clinical pattern. Mutations that have pure LOF or PLOF biochemical phenotypes are seen in individuals with epileptic encephalopathy without pronounced choreoathetosis. A number of patients exhibit both seizures and movement disorder. We have indicated in table 3 with major symptoms or minor symptoms which of these features is predominant or less so. Further studies will be needed based on new cases or additional mutations, but there does appear to be a clear pattern emerging about a genotype–phenotype correlation that is driven by a GOF/LOF difference in mutant GNAO1 alleles.
Location of mutations linked to GNAO1 encephalopathies in the Gαo protein.
To investigate the structural basis for the effects of mutations in Gαo, the mutations were modeled onto the published crystal structure of Gαo in complex with RGS16 (PDB: 3C7K; figure e-5). The locations of mutations within the Gαo structure segregated according to their function. The GOF mutations are all near G184S and close to the ribose and phosphate moieties of the bound GDP. The LOF mutants are more broadly scattered throughout the GTPase domain and may destabilize protein folding or stability consistent with their markedly reduced expression levels. The PLOF mutations are clustered in the GTPase domain but away from the bound GDP. This striking structure–function correlation may facilitate prediction of the function of new mutations but ultimately a rigorous biochemical analysis will provide definitive understanding of function.
DISCUSSION
The concept of a GNAO1 encephalopathy has developed based on the identification of at least 15 different mutations in the GNAO1 gene7–9,18–27 associated with various combinations of epilepsy, developmental delay, hypotonia, and choreoathetotic movement disorders. Our study demonstrates GOF as well as LOF mutations in GNAO1 and describes a clear correlation between biochemical and clinical characteristics. The existence of these unexpected GOF mutations has important therapeutic implications. Specifically, one might expect that different approaches to therapy would be needed for different mutations (i.e., agonists for LOF and antagonists for GOF mutants).
We chose inhibition of cAMP production as the functional readout to assess the Gαo mutants because of the robust measurements permitting complete agonist concentration response studies. This was critical to our findings since the GOF mutants were detected primarily through their ability to increase signals at low agonist concentrations (figure 2 and table 2). Our previously studied GOF mutant (G184S), which is insensitive to the inhibitory influence of RGS proteins, shows such a left shift of agonist concentration response curves in vitro28,34 and in vivo29,35 and also has a mild seizure phenotype in a mouse model.32 One might argue that cAMP is not the best choice of functional measures for epilepsy since N-type Ca++ channels or the synaptic release mechanism proteins are critical for regulation of neurotransmitter release. However, the apparent correlation of clinical patterns with the biochemical behavior in our cAMP assay suggests that function assessed in this way is relevant to functionality in humans. The clear pathologic effect of the R209 mutations (with at least 3 individuals carrying distinct alleles), however, does raise the question of why a protein with normal expression and function would cause pathology. It is possible that the R209 mutations have a selective loss of one of the other functional outputs while retaining a normal ability to inhibit AC. Alternatively, there may be selective alterations in expression or localization in neurons that are not accurately reflected in our HEK-293T cell studies of cAMP regulation. A full understanding of the causal mechanisms in GNAO1 encephalopathies requires additional studies of these mutant Gαo proteins in neurons and with different functional readouts.
The locations of mutations in the protein structure may partially explain their functional influences. All functioning mutants (NF and GOF) are located around the RGS binding domain, while most of the LOF or PLOF mutants are near the GDP binding region. Two exceptions are D174G and T191_F197del. D174 forms a salt bridge with R162 and mutations in this position may disrupt this interaction. T191_F197del truncates 2 β sheets as well as their linking region, which would be expected to decrease protein stability of Gαo.
A dominant genetic effect from GOF mutants is not unusual but the fact that the LOF mutations result in a severe autosomal dominant disorder is a bit surprising. We have ruled out a biochemical dominant negative mechanism of these mutations, at least for cAMP regulation, suggesting a haploinsufficiency mechanism. In mice, homozygous Gαo−/− knockouts exhibit seizures as well as hyperactive turning behavior.36 We did not, however, observe spontaneous seizures or an increased sensitivity to pentylenetetrazol kindling in heterozygous Gαo+/− knockouts.32 This suggests that humans are more susceptible to haploinsufficiency of Gαo than are mice. In contrast, we observed enhanced kindling sensitivity and reduced survival in our Gnao1+/G184S knock-in mouse model possibly due to seizures.32 Furthermore, these mice display early neonatal lethality32 of unclear mechanism, which may be similar to the hypotonia seen in human patients carrying GOF mutations. We do not know whether the abnormalities in these mice are due to brain developmental abnormalities or acute signaling effects. Further studies are needed to better understand this and to determine whether our Gαo+/G184S mutant mouse might represent a useful preclinical model for individuals with GNAO1 GOF mutations.
To date, all characterized GNAO1 mutants have been reported as LOF mutations. The G203R mutant in the original article7 was reported as a LOF mutant for regulation of N-type Ca++ channels. Similarly, a G42R mutation in Gαi1 was reported as a LOF mutant based on biochemical studies.37 The unique approach that we have taken with detailed cAMP dose–response studies in a mammalian cell model permitted our recognition of the GOF mechanisms (e.g., the Gαo G42R mutation). The patient with the G203R mutation, which we found to have GOF for cAMP inhibition, had a very different clinical pattern than the other 3 patients in the original study.7 She had a much later onset of disease (7 months) as well as developmental delay and severe chorea with only localized seizures.7 A similar clinical pattern was observed in 2 more recently described patients with this same mutation.18,21 All patients carrying the GOF mutations identified here appear distinct from the strict EIEE pattern (table 3). In comparison, patients with LOF or PLOF mutations were diagnosed with either Ohtahara syndrome (Y231C, I279N, D174G, T191_F197del) or EIEE (A227V, L199P, N270H, F275S). Thus GOF and LOF mutations in Gαo appear to result in different disease mechanisms likely requiring different therapeutic approaches.
It has remained challenging to convert knowledge about genetic epilepsy mutations into therapies. The GNAO1 encephalopathies may be different because of the eminently druggable nature of the receptors that drive Gαo signaling pathways. A critical question then becomes which receptors might be involved. Interestingly, activation of many Gi/o-coupled receptors is associated with suppression of seizures. Adenosine A1 receptors may play a role in the efficacy of the ketogenic diet38 and agonists at group II metabotropic glutamate receptors are anticonvulsant in various models.39 The opposite situation is also seen; GABABR agonists exacerbate absence seizures while GABABR antagonists suppress them.40 Identifying which receptors or downstream signaling effectors of Gαo contribute to mechanisms of encephalopathy from LOF or GOF GNAO1 mutations could therefore reveal potential targets for novel anticonvulsant drug development.
We have identified distinct biochemical mechanisms of pathogenic human GNAO1 mutations that may improve the understanding of the heterogeneous clinical spectrum of GNAO1-associated epilepsy and movement disorders. Furthermore, these results also carry significant implications for personalized therapeutics in GNAO1 encephalopathies.
Supplementary Material
GLOSSARY
- α2A AR
α2A adrenergic receptor
- AC
adenylate cyclase
- cAMP
cyclic adenosine monophosphate
- EIEE
early infantile epileptiform encephalopathy
- GOF
gain of function
- LOF
loss of function
- NF
normal function
- PLOF
partial loss of function
- PTX
pertussis toxin
- WT
wild-type
Footnotes
Supplemental data at Neurology.org
Editorial, page 754
AUTHOR CONTRIBUTIONS
Huijie Feng: research design, acquisition, analysis, and interpretation of data, manuscript writing. Benita Sjögren: research design, acquisition, analysis, and interpretation of data, critical revision of manuscript for intellectual content. Behirda Karaj: acquisition of data. Vincent Shaw: interpretation of data. Aysegul Gezer: acquisition of data. Richard R. Neubig: research design, interpretation of data, critical revision of manuscript for intellectual content.
STUDY FUNDING
Study funded by the NIH (R01 GM039561 and T32 GM092715).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kobau R, Ly ZM, Helmers S, Thurman D. Epilepsy in adults and access to care: United States. MMWR Morb Mortal Wkly Rep 2012;61:909–931. [PubMed] [Google Scholar]
- 2.Capovilla G, Wolf P, Beccaria F, Avanzini G. The history of the concept of epileptic encephalopathy. Epilepsia 2013;54(suppl 8):2–5. [DOI] [PubMed] [Google Scholar]
- 3.McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 2016;15:304–316. [DOI] [PubMed] [Google Scholar]
- 4.Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr 2003;15:567–571. [DOI] [PubMed] [Google Scholar]
- 5.Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557. [DOI] [PubMed] [Google Scholar]
- 6.Noebels J. Pathway-driven discovery of epilepsy genes. Nat Neurosci 2015;18:344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura K, Kodera H, Akita T, et al. De novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013;93:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcé-Grau A, Dalton J, López-Pisón J, et al. GNAO1 encephalopathy: further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J Rare Dis 2016;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ananth AL, Robichaux-Viehoever A, Kim YM, et al. Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr Neurol 2016;59:81–84. [DOI] [PubMed] [Google Scholar]
- 10.Menke LA, Engelen M, Alders M, Odekerken VJ, Baas F, Cobben JM. Recurrent GNAO1 mutations associated with developmental delay and a movement disorder. J Child Neurol 2016;31:1598–1607. [DOI] [PubMed] [Google Scholar]
- 11.Sternweis PC, Robishaw JD. Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J Biol Chem 1984;259:13806–13813. [PubMed] [Google Scholar]
- 12.Ghahremani MH, Cheng P, Lembo PM, Albert PR. Distinct roles for Galphai2, Galphai3, and Gbeta gamma in modulation of forskolin- or Gs-mediated cAMP accumulation and calcium mobilization by dopamine D2S receptors. J Biol Chem 1999;274:9238–9245. [DOI] [PubMed] [Google Scholar]
- 13.Raskind WH, Friedman JR, Roze E, Meneret A, Chen DH, Bird TD. ADCY5-related dyskinesia: comments on characteristic manifestations and variant-associated severity. Mov Disord 2017;32:305–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carapito R, Paul N, Untrau M, et al. A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov Disord 2015;30:423–427. [DOI] [PubMed] [Google Scholar]
- 15.Chang FC, Westenberger A, Dale RC, et al. Phenotypic insights into ADCY5-associated disease. Mov Disord 2016;31:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mencacci NE, Erro R, Wiethoff S, et al. ADCY5 mutations are another cause of benign hereditary chorea. Neurology 2015;85:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YZ, Friedman JR, Chen DH, et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol. 2014;75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitsu H, Fukai R, Ben-Zeev B, et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet 2016;24:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium E-R, Project EPG, Consortium EK. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietel T. Genotype and phenotype in GNAO1-mutation: case report of an unusual course of a childhood epilepsy. In: Haack TB, Schlüter G, Cordes I, Trollmann R, Bast T, editors. Neuropediatrics. New York: Thieme; 2016:P01–P17. [Google Scholar]
- 22.Law CY, Chang ST, Cho SY, et al. Clinical whole-exome sequencing reveals a novel missense pathogenic variant of GNAO1 in a patient with infantile-onset epilepsy. Clin Chim Acta 2015;451:292–296. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 2015;17:774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulkarni N, Tang S, Bhardwaj R, Bernes S, Grebe TA. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol 2016;31:211–214. [DOI] [PubMed] [Google Scholar]
- 25.Talvik I. Clinical phenotype of de novo GNAO1 mutation: case report and review of literature. Child Neurol Open 2015;2:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhamija R. GNAO1-Associated movement disorder. In: Mink JW, Shah BB, Goodkin HP, editors. Movement Disorders Clinical Practice. Hoboken: Wiley Online Library; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epi4K Consortium. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet 2016;99:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu Y, Zhong H, Nanamori M, et al. RGS-insensitive G-protein mutations to study the role of endogenous RGS proteins. Methods Enzymol 2004;389:229–243. [DOI] [PubMed] [Google Scholar]
- 29.Goldenstein BL, Nelson BW, Xu K, et al. Regulator of G protein signaling protein suppression of Galphao protein-mediated alpha2A adrenergic receptor inhibition of mouse hippocampal CA3 epileptiform activity. Mol Pharmacol 2009;75:1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeda SR, Jeong SW. Use of RGS-insensitive Galpha subunits to study endogenous RGS protein action on G-protein modulation of N-type calcium channels in sympathetic neurons. Methods Enzymol 2004;389:170–189. [DOI] [PubMed] [Google Scholar]
- 31.Wade SM, Lim WK, Lan KL, Chung DA, Nanamori M, Neubig RR. G(i) activator region of alpha(2A)-adrenergic receptors: distinct basic residues mediate G(i) versus G(s) activation. Mol Pharmacol 1999;56:1005–1013. [DOI] [PubMed] [Google Scholar]
- 32.Kehrl JM, Sahaya K, Dalton HM, et al. Gain-of-function mutation in Gnao1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm Genome 2014;25:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lan KL, Sarvazyan NA, Taussig R, et al. A point mutation in Galphao and Galphai1 blocks interaction with regulator of G protein signaling proteins. J Biol Chem 1998;273:12794–12797. [DOI] [PubMed] [Google Scholar]
- 34.Clark MJ, Harrison C, Zhong H, Neubig RR, Traynor JR. Endogenous RGS protein action modulates mu-opioid signaling through Galphao: effects on adenylyl cyclase, extracellular signal-regulated kinases, and intracellular calcium pathways. J Biol Chem 2003;278:9418–9425. [DOI] [PubMed] [Google Scholar]
- 35.Lamberts JT, Smith CE, Li MH, Ingram SL, Neubig RR, Traynor JR. Differential control of opioid antinociception to thermal stimuli in a knock-in mouse expressing regulator of G-protein signaling-insensitive Galphao protein. J Neurosci 2013;33:4369–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang M, Gold MS, Boulay G, et al. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA 1998;95:3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosch DE, Willard FS, Ramanujam R, et al. A P-loop mutation in Gα subunits prevents transition to the active state: implications for G-protein signaling in fungal pathogenesis. PLoS Pathog 2012;8:e1002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masino SA, Li T, Theofilas P, et al. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J Clin Invest 2011;121:2679–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalby NO, Thomsen C. Modulation of seizure activity in mice by metabotropic glutamate receptor ligands. J Pharmacol Exp Ther 1996;276:516–522. [PubMed] [Google Scholar]
- 40.Han HA, Cortez MA, Snead OC III. GABAB receptor and absence epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies, 4th ed. Bethesda: National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.