

Abstract
Daunorubicin is extensively used in chemotherapy for diverse types of cancer. Over the years, evidence has suggested that the mechanisms by which daunorubicin causes cytotoxic effects are also associated with interactions at the membrane level. The aim of the present work was to study the interplay between daunorubicin and mimetic membrane models composed of different ratios of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), sphingomyelin (SM) and cholesterol (Chol). Several biophysical parameters were assessed using liposomes as mimetic model membranes. Thereby, the ability of daunorubicin to partition into lipid bilayers, its apparent location within the membrane and its effect on membrane fluidity were investigated. The results showed that daunorubicin has higher affinity for lipid bilayers composed of DMPC, followed by DMPC : SM, DMPC : Chol and lastly by DMPC : SM : Chol. The addition of SM or Chol into DMPC membranes not only increases the complexity of the model membrane but also decreases its fluidity, which, in turn, reduces the amount of anticancer drug that can partition into these mimetic models. Fluorescence quenching studies suggest a broad distribution of the drug across the bilayer thickness, with a preferential location in the phospholipid tails. The gathered data support that daunorubicin permeates all types of membranes to different degrees, interacts with phospholipids through electrostatic and hydrophobic bonds and causes alterations in the biophysical properties of the bilayers, namely in membrane fluidity. In fact, a decrease in membrane fluidity can be observed in the acyl region of the phospholipids. Ultimately, such outcomes can be correlated with daunorubicin's biological action, where membrane structure and lipid composition have an important role. In fact, the results indicate that the intercalation of daunorubicin between the phospholipids can also take place in rigid domains, such as rafts that are known to be involved in different receptor processes, which are important for cellular function.
Keywords: drug–membrane interactions, daunorubicin, liposomes, partition, location, fluidity
1. Introduction
Daunorubicin [1] was the first anthracycline antibiotic to be used in cancer treatment. Despite its extensive clinical application for the treatment of acute lymphoblastic or myeloblastic leukaemias [2], a major problem with daunorubicin administration is the dose-dependent toxicity in healthy tissues, especially cardiotoxicity [2]. Daunorubicin continues to be widely studied because its mechanisms of action are still a matter of substantial controversy [2]. Nevertheless, it is thought that daunorubicin exhibits cytotoxic activity mainly through intercalation with DNA; alteration of topoisomerase II, which, in turn, inhibits DNA and RNA synthesis and leads to apoptosis; disturbance of membrane fluidity and ion transport; and production of oxygen radicals [3].
In addition to intracellular targets, strong evidence suggests that the cell membrane can itself be a target for this type of agent [4]. In fact, it has been demonstrated that anthracyclines, without entering the cell, can exert their cytotoxic effect only through their interplay with the membrane [5]. In 1995, it was shown that daunorubicin reduced the formation of the non-lamellar hexagonal (HII) phase of liposomes composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and stabilized the bilayer structure just by interacting with membrane phospholipids [6]. One repercussion of such an interaction is an alteration of various cellular events involved in signal transduction, in which these lipid phases are involved [6]. The gathered data highlight the role of the cell membrane and reveal that membrane lipids have a fundamental function in signal transduction that can be influenced by xenobiotics, such as anthracyclines.
The transport of daunorubicin across biological membranes, an important factor that can influence the drug's pharmacokinetics properties, is determined by the balance between lipophilicity and hydrophilicity and by its charge state [7]. The determination of the drug's partition coefficient, which gives information regarding the lipophilicity of a compound, can help to infer information about the process of passive diffusion. However, it is also relevant to take into account the drug's degree of ionization, because this can affect the ability of the compound to move through the bilayer and can differ depending on the medium pH, which is more acidic in the case of cancer cells. Several studies describe the transport of daunorubicin through cell membranes as a process of passive diffusion [8,9]. However, such an explanation only takes into consideration the non-ionized form of the drug. Since most of the daunorubicin molecules are charged at physiological pH, it seems unlikely that most of them cross the cell by simple diffusion. In this context, Regev et al. [10] studied the transport of daunorubicin across a membrane and demonstrated that it occurs by a passive ‘flip-flop’ mechanism, rather than by diffusion down a continuous concentration gradient.
Although the biological action of daunorubicin has been broadly described, the biophysical mechanisms that depend on the interactions with membranes are far from being completely understood. In 1996, Gallois et al. [11] demonstrated that the incorporation of daunorubicin molecules within the bilayers was independent of their charge, but the amounts of neutral and positively charged drug forms embedded were dependent on the membrane's electrostatic parameters. In 1998, the same authors [12] investigated the interplay of daunorubicin with large unilamellar vesicles (LUVs) composed of phosphatidylcholine, phosphatidic acid and cholesterol (Chol), and showed that, due to its lipophilicity and chemical nature, daunorubicin binds to the membrane through both electrostatic and hydrophobic interactions. Also, Escriba et al. [13] described that different lipid contents can determine how daunorubicin binds to membranes. While the presence of phosphatidylserine greatly increased both the drug's affinity and stoichiometry of binding to model lipid membranes, the presence of Chol caused a reverse effect. A strong affinity of daunorubicin to cardiolipin has also been described [14]. As a consequence of electrostatic interactions, a complex between both molecules is formed and this is thought to be involved in the cardiotoxicity of daunorubicin [14]. Finally, it was reported that daunorubicin-triggered cell death is mediated by a signalling pathway that starts with the early production of a sphingomyelin (SM)-derived ceramide [15], which emphasizes the importance of the cell membrane's lipid content for this anticancer drug's activity. Recent studies have also shown that daunorubicin induces apoptosis via signalling through plasma membrane-lipid rafts, where the death receptor pathway is involved [16,17]. The process is initiated by the activation of a sphingomyelinase, which in turn causes the hydrolysis of SM and generates ceramide. It was later found [16,17] that ceramide is responsible for triggering different signalling pathways, among which a stress-activated protein kinase cascade plays a crucial role.
More details on the effects of daunorubicin and other anthracyclines with mimetic model membranes, such as liposomes and monolayers, can be found in a recent review [18].
The different structural, biophysical and chemical aspects of the membrane can define whether and how the cell will react to the drug and whether the drug will cross this boundary. On the other hand, the drug's properties can also modulate its penetration, conformation and/or location within membranes [19]. Simultaneously, drugs can initiate membrane adjustments that can vary from changes in lipid conformation and surface charge to alterations in the fluidity and, consequently, in cell function [7]. For these reasons, drug–membrane interactions become important to understand the mechanisms of multi-drug resistance or the development of undesirable side effects [13]. Such knowledge is crucial, especially for anthracyclines, because these drawbacks are the reason why their therapeutic index needs to be improved. Additionally, due to the huge variety in the composition and structure of membranes, these interactions can significantly affect the drug's activity and toxicity [19]. For that reason, the goal of the current work is to elucidate whether and how the features of daunorubicin can affect the biophysical characteristics of biological membranes and what is the importance of the lipid composition and membrane structure in the drug's pharmacokinetic and pharmacodynamic properties.
Since biological membranes are too complex to accurately assess biophysical parameters, liposomes were used as membrane model systems. The main lipid components of biological membranes are glycerophospholipids, sphingolipids and sterols and, for this reason, they were chosen to develop the model systems. The mimetic models were developed in order to simulate several biological environments that daunorubicin can encounter. Accordingly, the purpose was to study the role of different types of membrane organization and fluidity in daunorubicin interactions, using LUVs constituted of different molar ratios of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), SM and Chol. The choice of lipids can be explained as follows: (i) phosphatidylcholines are among the most abundant phospholipids in natural plasma membranes [20] and were, therefore, included in the mimetic systems as the dominant constituent; (ii) SM, the major sphingolipid found in membranes, plays several roles in biological membranes and is involved in many cellular functions [21], and (iii) sterols are the main non-polar lipids of cell membranes, Chol being the most abundant and one of the most important regulators of lipid organization [22], and thus it was added in order to mimic cellular targets enriched in Chol domains. The presence of Chol, known to regulate membrane fluidity and permeability, is crucial for the preservation of the membrane's organization and, consequently, contributes to the cell membrane's physiology and function [23]. In addition, Chol and SM form liquid-ordered domains in a more fluid matrix, the so-called lipids rafts, which are fundamental for membrane organization and where several proteins and other biomolecules are embedded [24]. Furthermore, in a pharmacological context, it is crucial to know whether a drug partitions into specific lipid domains and whether this partition can have implications in specific functional effects.
2. Material and methods
2.1. Reagents
Daunorubicin was obtained from Biovision (Milpitas, CA, USA). The lipids 1DMPC, SM (milk, bovine) and Chol (ovine) were supplied by Avanti Polar Lipids, Inc. (Alabaster, AL, USA). The probes 1,6-diphenyl-1,3,5-hexatriene (DPH) and 1-(4-trimethylammoniophenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH) were purchased from Molecular Probes (Invitrogen Corporation, Carlsbad, CA, USA). N-(2-Hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). All reagents were used without further purification. Drug solutions were prepared with HEPES buffer (10 mM, pH 7.4). The buffer was prepared with double deionized water (conductivity inferior to 0.1 µS cm−1) and ionic strength was adjusted with NaCl (I = 0.1 M).
2.2. Preparation of liposomes
Liposomes were prepared according to an adapted classic thin film hydration method [25]. The molar ratio mixture of the lipids (DMPC, DMPC : SM [8 : 2], DMPC : Chol [8 : 2] and DMPC : SM : Chol [7 : 1.5 : 1.5]) was dissolved in chloroform/methanol (3 : 2 v/v) and the organic solvents were evaporated under a stream of nitrogen in a rotary evaporator (Buchi R-200; Sigma-Aldrich Corp., Buchs, Switzerland), equipped with a thermostatic bath (Buchi R-490; Sigma-Aldric Corp., Buchs, Switzerland). The resultant dried lipid film was dispersed in HEPES (pH 7.4) and the mixture was vortexed to form multilamellar vesicles (MLVs). To obtain LUVs the MLVs were extruded 10 times (extruder supplied by Lipex Biomembranes Inc., Vancouver, Canada) through polycarbonate filters with a pore diameter of 100 nm (Nucleopore) at 60°C (temperature above the main phase transition temperature of the lipids).
For fluorescence studies, the fluorescence probe (DPH or TMA-DPH) was co-dissolved with the lipid in the organic solvents mixture to give a probe/lipid molar ratio of 1 : 300 in order to prevent changes in the membrane's structure. The labelled LUVs were similarly obtained by evaporation of the organic solvents, followed by hydration of the dried lipid film and extrusion of the MLVs.
2.3. Hydrodynamic diameter and zeta potential characterization by dynamic light scattering and electrophoretic light scattering
The hydrodynamic diameter and zeta potential were assessed using a Brookhaven™ BI-MAS and Zeta-Pals (Brookhaven Instruments, Holtsville, NY, USA). Both the size and zeta potential distribution of the extruded liposomes, with and without drug, were determined at pH 7.4 (HEPES buffer) at 37 ± 0.1°C. Lipid concentration was kept constant at 500 µM and daunorubicin concentrations ranged from 0 to 75 µM. The hydrodynamic diameter and zeta potential values obtained were calculated from at least three independent assays.
2.4. Determination of partition coefficients by derivative UV–Vis spectrophotometry
The partition coefficient (Kp) of daunorubicin in the different model membranes (LUVs) was assessed by derivative UV–Vis spectrophotometry. A set of samples was prepared by the addition of anticancer drug HEPES-buffered solutions with a fixed drug concentration (40 µM) and increasing concentrations of lipid solution (in the range of 0–1500 µM). The corresponding reference solutions were prepared identically but without daunorubicin. All samples and references were incubated for 30 min at 37°C. The absorption spectra of all solutions were recorded at 37°C using a multidetection microplate reader (Synergy™ HT; BioTek Instruments Inc., Winooski, VT, USA). Mathematical treatment of the results obtained was performed using a developed routine, the Kp calculator [26], where each reference spectrum is subtracted from the corresponding sample spectrum to eliminate scattering from the vesicles, and its second- and third-derivative spectra are determined to enhance the ability to detect minor spectral features and to improve the band resolution. The Kp values are then calculated using a nonlinear regression method that fits equation (2.1) to the experimental data (DT versus [L]),
| 2.1 |
where, DT is the second- or third-derivative intensity (D = dnAbs/dλn) obtained from the absorbance values of the total amount of daunorubicin, the daunorubicin distributed on the lipid membrane phase (Dm) and the daunorubicin distributed in the aqueous phase (Dw). [L] represents the lipid concentration (in M) and Vm is the lipid molar volume. The Vm values for the mimetic systems studied (DMPC, DMPC : SM, DMPC : Chol and DMPC : SM : Chol) were 0.663, 0.682, 0.596 and 0.633 l mol−1, respectively. The obtained Vm values were calculated using the specific volume of each lipid and taking into account the different lipid molar ratio used for each model membrane. The Kp values obtained are adimensional and presented as the mean ± standard deviation calculated from at least three independent assays.
2.5. Drug location studies performed by fluorescence quenching
The location of daunorubicin within the model systems was assessed by fluorescence studies that included steady-state and time-resolved measurements, according to an already described method [27]. These experiments were conducted by incubating the anticancer drug with DPH- and TMA-DPH-labelled liposomes. The samples containing a fixed concentration of lipid (500 µM) and increasing concentrations of daunorubicin (0–75 µM) were incubated at 37°C for 30 min in the dark. The fluorescence measurements were carried out at 37°C, at excitation and emission wavelengths defined at 357 nm and 429 nm, and 359 nm and 429 nm, for DPH and TMA-DPH, respectively. Fluorescence steady-state measurements were performed in a Jasco FP-6500 spectrofluorometer (Jasco, Great Dunmow, UK) equipped with a constant temperature cell holder and all data were recorded in a 1 cm path length cuvette. The fluorescence intensity values obtained were corrected for inner filter effects (quencher absorbance) at the excitation wavelength [28]. The fluorescence time-resolved measurements were done using a Fluorolog Tau-3 Lifetime system. The modulation frequencies were acquired between 10 and 200 MHz, with an integration time of 10 s. The fluorescence emission was detected with a 90° scattering geometry. All measurements were made using Ludox as a reference standard (τ = 0.00 ns).
2.6. Membrane fluidity studies performed by fluorescence anisotropy
The influence of daunorubicin on the membrane's fluidity, in the different model membranes developed, was assessed by fluorescence anisotropy studies. The samples were prepared with a fixed concentration of lipid (500 µM), labelled with DPH or TMA-DPH, and increasing concentrations of daunorubicin (0, 40 and 75 µM), and incubated for 30 min at 37°C in the dark. The steady-state fluorescence anisotropy measurements (rs) were recorded between 10°C and 60°C at intervals of 2°C. The experiments were performed in the Jasco FP-6500 spectrofluorometer used for the location studies with polarizers inserted (excitation/emission wavelengths were set as described for each fluorescent probe) and equipped with a Peltier temperature controller. The results obtained are presented as the mean ± standard deviation calculated from at least three independent assays.
2.7. Statistical analysis
Statistical analysis was performed using IBM® SPSS® Statistics software (v. 20.0.0.0; IBM, Armonk, NY, USA). The measurements were repeated at least three times and the data were expressed as the mean ± standard deviation. Data were statistically analysed through the one-way analysis of variance (ANOVA) method and differences between groups were compared by Bonferroni's and Tukey's post hoc tests in which p < 0.05 was considered statistically significant.
3. Results and discussion
3.1. Hydrodynamic diameter and zeta potential characterization
The different mimetic systems were analysed regarding their hydrodynamic radius and surface charge in the absence and presence of daunorubicin and the results obtained are depicted in table 1 for all model membranes studied. The hydrodynamic diameter measurements demonstrated that the different model systems present an average diameter between 100 and 110 nm independently of their composition. With these sizes the surface curvature effects can be neglected at the molecular level [29], as in biological membranes. However, it should be noted that daunorubicin influenced the liposome hydrodynamic diameter in a concentration-dependent way, indicating that the compound interacts with the different types of membranes. The anticancer drug had a greater effect in the hydrodynamic diameter of models containing Chol.
Table 1.
Characterization of the liposomes' hydrodynamic diameter and zeta potential with increasing daunorubicin concentration. Studies conducted at pH 7.4 and 37°C.a
| model | hydrodynamic diameter (nm) |
zeta potential (mV) |
||||||
|---|---|---|---|---|---|---|---|---|
| 0 µM | 5 µM | 40 µM | 75 µM | 0 µM | 5 µM | 40 µM | 75 µM | |
| DMPC | 113 ± 2 | 114 ± 2 | 113 ± 3 | 115 ± 1 | −14 ± 2 | 4 ± 1* | 15 ± 1* | 20 ± 1* |
| DMPC : SM | 100 ± 5 | 104 ± 6 | 117 ± 5* | 120 ± 6* | −18 ± 1 | −6 ± 1* | 17 ± 2* | 19 ± 1* |
| DMPC : Chol | 102 ± 3 | 118 ± 2* | 123 ± 1* | 120 ± 4* | −8 ± 1 | −4 ± 2* | 7 ± 1* | 12 ± 1* |
| DMPC : SM : Chol | 107 ± 4 | 117 ± 5* | 119 ± 5* | 122 ± 5* | −16 ± 1 | −7 ± 1* | 8 ± 1* | 11 ± 1* |
aAll values represent the mean ± s.d. (n = 3).
*p < 0.05, statistically different from the corresponding sample without daunorubicin.
Regarding zeta potential values, the model membranes presented an overall negative surface charge that significantly altered in the presence of the anticancer drug, as can be seen in table 1. This outcome can be explained by the fact that daunorubicin molecules are positively charged at physiological pH, interacting with the negative moieties of phospholipids by electrostatic interactions.
Overall, the results point towards an interplay of daunorubicin with model membranes driven through both electrostatic interactions and hydrogen bonds.
3.2. Daunorubicin partition coefficients
Drug–membrane interaction is strongly influenced by drug lipophilicity, which can affect the drug's pharmacodynamics and pharmacokinetics [30]. In this context, the assessment of the partition coefficient (Kp) can be crucial to understanding and explaining a drug's pharmacological activity [31]. The daunorubicin partition coefficient in the several model membranes was determined by derivative UV–Vis spectrophotometry [25,32,33]. This technique, based on the fact that the drug's spectral characteristics (λmax) change when it permeates from the aqueous to the lipid medium, allows quantification of the distribution of the drug in each phase. Furthermore, it also provides a better resolution of the overlapped bands and eliminates the lipid light scattering interference by the use of the derivative method.
The electronic supplementary material, figure S1a, presents, as an example, the third-derivative absorption spectra of daunorubicin with increasing lipid concentration, calculated to improve the resolution of the spectra and to eliminate the interference caused by the vesicles scattering. From this spectrum, it is possible to observe a slight shift in the λmax, which indicates that the drug partitions from the aqueous to the lipid phase of the liposomes. The best fit of equation (2.1) to the third-derivative data was achieved at 546 nm and is shown in the electronic supplementary material, figure S1c.
The experimental daunorubicin partition coefficients obtained for each mimetic system (expressed as Kp and log D) along with the theoretical octanol/water partition coefficient (expressed as log D), predicted using Marvin sketch calculator software (ChemAxon), are depicted in table 2.
Table 2.
Partition coefficients (expressed as Kp and log D) of daunorubicin for the different model membranes at pH 7.4 and 37°C.a
| model | Kp | experimental log D | theoretical log D |
|---|---|---|---|
| DMPC | 3363 ± 41* | 3.53 ± 0.01* | 0.91 |
| DMPC : SM | 2983 ± 63* | 3.47 ± 0.01* | |
| DMPC : Chol | 1413 ± 82* | 3.15 ± 0.04* | |
| DMPC : SM : Chol | 869 ± 57* | 2.94 ± 0.03* |
aAll values represent the mean ± s.d. (n = 3).
*p < 0.05, statistically different from the other models.
The experimental results (table 2) revealed that daunorubicin presents a significant partition into the several mimetic models. When comparing the experimental results (experimental log P) and the values predicted for the octanol/water system (theoretical log D), it is possible to observe a significant variation between them. Such inconsistency is related to the differences between these techniques and can be explained by the fact that the octanol–water system does not take into account several crucial characteristics of biological membranes. Indeed, membranes are constituted by amphiphilic phospholipids that can establish electrostatic and hydrophobic interactions. Furthermore, the compound's partition is highly dependent on the lipid composition of the mimetic models studied, emphasizing the fact that, by using liposomes, it is possible to obtain more realistic information about daunorubicin lipophilicity and, therefore, its in vivo membrane partition.
On the other hand, the pKa values (9.17) calculated using Marvin sketch calculator software predict that nearly 80% of daunorubicin molecules are in the cationic form at pH 7.4, which means that the drug–membrane interaction should be mainly driven by electrostatic interactions with the polar headgroups of the phospholipids. For that reason, the theoretical log D value is much smaller than the experimental log D because it mainly reflects the hydrophobic interactions of the small amount of molecules that are in the neutral form at physiological pH. Hence, daunorubicin interplays with membranes through: electrostatic forces of the amino sugar moiety (positively charged) and the lipid phosphate groups (negatively charged); and hydrophobic interactions between the dihydroanthraquinone residue and the lipid fatty acid chains.
Comparing the results acquired for the different mimetic systems it is clear that the drug concentration between the membrane and aqueous medium is different from model to model, depending on the lipid composition and biophysical characteristics, particularly membrane fluidity (see §3.4). Therefore, daunorubicin presents a higher partition for the model composed by DMPC, followed by DMPC : SM, DMPC : Chol and lastly by DMPC : SM : Chol. The addition of SM or Chol into DMPC membranes increases the complexity of the system but also decreases its fluidity, which, in turn, reduces the amount of anticancer drug that can partition into these mimetic models. Ultimately, the presence of SM together with Chol confers on the membrane an increased organization and lipid packing (see §3.4) and, consequently, hinders daunorubicin partition into the lipid membrane. Nevertheless, a small partition can be observed for this model, probably due to the drug's planar structure that allows it to intercalate between the phospholipids of the bilayer, just as it does with the DNA strands, even in more rigid membranes where the lipid–lipid interactions are stronger.
The drug's membrane concentration (Kp) values demonstrated that daunorubicin permeates the several models to different degrees. Nevertheless, its partition appears to be mainly driven by electrostatic (which can be measured using the liposome/water system, contrary to the octanol/water method) and hydrophobic interactions in all models. Such outcomes are in agreement with previous studies [11,34–36] and evidence the fact that the membrane partition of specific molecules is dependent on the lipid composition and can help to explain its biodistribution. Furthermore, the transport of daunorubicin across membranes probably occurs by initial electrostatic binding to phospholipids of the mimetic systems, followed by a ‘flip-flop’ mechanism within the bilayer, as demonstrated by Regev et al. [10].
3.3. Drug location studies using different fluorescent probes
The daunorubicin location within the bilayer was also evaluated by fluorescence quenching of two probes [37]. The quenching efficiency of daunorubicin in the several models was determined by adding increasing drug concentrations to liposomes labelled with DPH and TMA-DPH. It is only possible to obtain this information because the fluorophore position in the membrane of each fluorescent probe (TMA-DPH and DHP) is well defined and documented. While DPH is deeply incorporated in the hydrophobic regions of the lipid bilayer [38], TMA-DPH is reported to be anchored in the polar head groups region of phospholipids due to its charged group [39].
The drug's efficiency to quench the fluorophore is associated with its proximity to the probe. In this sense, because the fluorophore is inserted in the membrane, only the drug that partitions into the lipid phase will be able to act as a quencher. In order to determine the daunorubicin quenching efficiency it is important to calculate its membrane concentration ([Q]m), which is given by equation (3.1):
| 3.1 |
where αm is the volume fraction of the membrane phase (αm = Vm/VT; Vm and VT represent the volumes of the membrane and water phase, respectively).
Daunorubicin molecules were effectively able to quench all probes in all mimetic membranes. The wavelength corresponding to the absorption band of the probes did not change with daunorubicin presence and no additional band was observed at a longer wavelength. As such, the quenching of fluorescence was analysed by the modified Stern–Volmer equation as follows:
| 3.2 |
where I and I0 are the steady-state fluorescence intensities with and without the quencher (daunorubicin), respectively; KSV is the Stern–Volmer constant; [Q]m is the membrane daunorubicin concentration; and τ0 and τ are the fluorophore lifetime in the absence and presence of daunorubicin, respectively. The molecular contact between the probe (fluorophore) and the drug (quencher) can be due to different molecular interactions and can be observed by the decrease in fluorescence intensity (fluorescence quenching) as represented in the electronic supplementary material, figure S2.
The quenching process can result from collisional encounters between the fluorophore and quencher, and is therefore called collisional or dynamic quenching, or it can be due to the formation of a complex, which is called static quenching [37]. These two types of quenching can be distinguished by lifetime measurements. This happens because while complexed fluorophores are non-fluorescent, and the observed fluorescence is just from the uncomplexed fluorophores, the uncomplexed fraction is unperturbed and hence its lifetime is about the same as τ0. In this context, for static quenching τ0/τ = 1, while for dynamic quenching I0/I = τ0/τ [37]. Accordingly, besides the determination of the fluorescence emission intensity, fluorescence lifetime measurements were also performed.
In many cases, the fluorescent probe can be quenched both by collisions and by formation of a complex with the same quencher. Such events result in an upward curvature of the Stern–Volmer plots, a positive deviation that can be seen in the electronic supplementary material, figure S2. Accordingly, the dynamic portion (KD) of the observed quenching is determined by lifetime measurements, through the following equation [37]:
| 3.3 |
By knowing the dynamic component, the static contribution may be calculated by linearization of the following equation [37]:
| 3.4 |
This modified form of the Stern–Volmer equation is second order in [Q]m, which accounts for the upward curvature observed when both static and dynamic quenching occur for the same fluorophore.
In the electronic supplementary material, figure S2, the Stern−Volmer plots of I0/I − 1 and τ0/τ − 1 as a function of the daunorubicin concentration in the membrane are shown. From these plots, it is possible to conclude that both collisional and static quenching processes exist, which means that daunorubicin must diffuse to the fluorophore during the lifetime of the excited state but it also might form a non-fluorescent complex with the probe.
According to the previous description of the quenching behaviour, the quenching parameters values, namely the dynamic constant (KD), the static constant (KS) and the Stern–Volmer constant (KSV), obtained from this technique are shown in table 3.
Table 3.
Fluorescence quenching parameters (KD, dynamic constant; KS, static constant; KSV, Stern–Volmer constant) calculated from the quenching of TMA-DPH and DPH by daunorubicin in the different mimetic models at pH 7.4 and 37°C.a
| model | probe | KD (M−1) | KS (M−1) | KSV (M−1) |
|---|---|---|---|---|
| DMPC | DPH | 17 ± 1* | 81 ± 2* | 98 ± 3* |
| TMA-DPH | 34 ± 2* | 12 ± 1* | 45 ± 1* | |
| DMPC : SM | DPH | 41 ± 2* | 38 ± 4* | 80 ± 1* |
| TMA-DPH | 28 ± 2* | 13 ± 2* | 42 ± 2* | |
| DMPC : Chol | DPH | 8 ± 1 | 11 ± 1* | 19 ± 1* |
| TMA-DPH | 7 ± 1 | ∼0* | 7 ± 1* | |
| DMPC : SM : Chol | DPH | 31 ± 1* | 51 ± 1* | 82 ± 2* |
| TMA-DPH | 20 ± 1* | 29 ± 1* | 49 ± 1* |
aAll values represent the mean ± s.d. (n = 3).
*p < 0.05, statistically different from the other probe, for each biophysical parameter.
From table 3, it can be seen that the quenching process results from dynamic and static interactions, except for the model composed by DMPC : Chol, where it results only from dynamic interactions. Analysis of the obtained data leads to the conclusion that all probes were quenched by daunorubicin but with different quenching efficiency depending on the type of mimetic model used. Daunorubicin induced a reduction in the light emitted by both probes (DPH and TMA-DPH) incorporated into liposomes in a concentration-dependent manner, indicating a broad distribution of the drug across the bilayer thickness.
Nevertheless, the decrease in the probe's fluorescence was more pronounced for DPH than for TMA-DPH in all the model systems, which translates into higher values of KSV. This fact suggests that the drug is buried inside the lipid bilayer but also near the phospholipid head. Once more, it appears that two types of interactions coexist and are responsible for the distribution of daunorubicin at the membrane level: electrostatic interactions between the positive group of the anticancer drug and the negative pole of the phospholipid headgroup, and hydrophobic interactions between the drug's dihydroanthraquinone residue and the lipid fatty acid chains. Furthermore, the membrane location of daunorubicin can be explained by the fact that being an amphiphilic molecule it permeates the lipid bilayer (quenches DPH and TMA-DPH), which is in turn related to the partition coefficients determined for this compound in the different model systems.
3.4. Effect of daunorubicin on membrane fluidity
Alterations in membrane fluidity can severely affect the cell's functional properties and also activate apoptotic pathways which ultimately result in cell death. Since anticancer drugs interact with membranes in order to reach their intracellular target, it is crucial to understand how these compounds can induce perturbation in the physicochemical state of the phospholipid bilayer.
Membrane fluidity studies were performed by steady-state anisotropy [40], which shows the microenvironment around the fluorescent probe once it conditions the degree of rotational motion of the probe [41].
The effect of temperature on the DPH and TMA-DPH fluorescence anisotropy of the different model membranes without daunorubicin is presented in figure 1. For all model systems developed, the anisotropy values decrease with increasing temperature to a greater extent in the acyl chain region (shown by the probe DPH) than in the phospholipid head region (shown by the TMA-DPH probe). It is well documented that the membrane is characterized by a fluidity gradient from the aqueous interface to the bilayer interior, where the acyl chain end presents increased disorder [42]. Furthermore, from the graphical analysis shown in figure 1, one can observe that, while the main phase transition increases the cooperativity of the process, fluidity decreases with increasing membrane-lipid composition and complexity, for both probes. Evidence suggests that the presence of SM and Chol in membranes promotes the formation and maintenance of specific domains in the liquid-ordered phase that are more condensed and organized, which results in higher anisotropy values [43].
Figure 1.
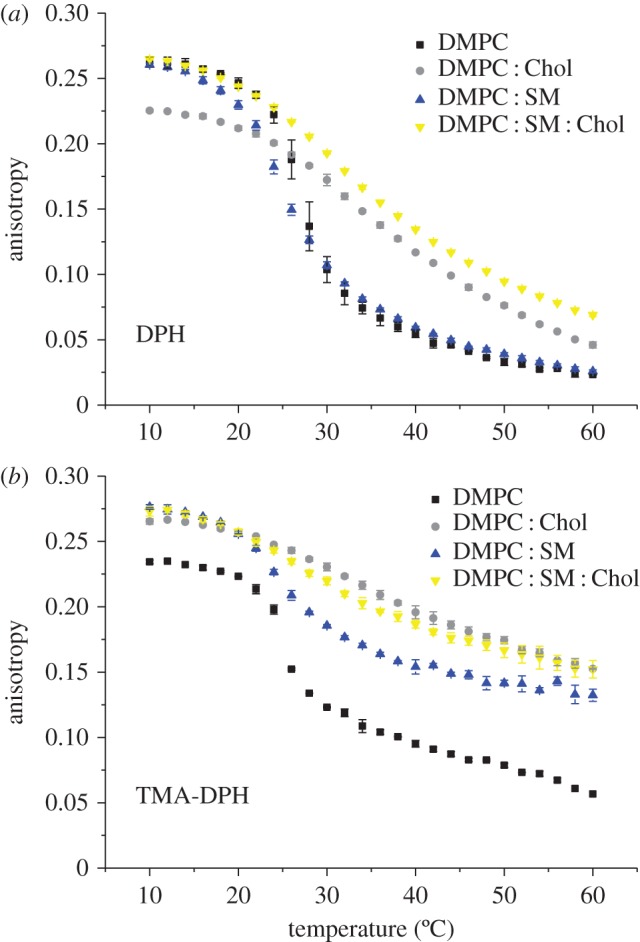
Steady-state anisotropy of (a) DPH and (b) TMA-DPH as a function of temperature in each mimetic model, at pH 7.4. Results present the mean of at least three independent assays. (Online version in colour.)
Our results clearly demonstrate a decrease in fluidity when cholesterol is added to the DMPC : SM bilayer. In addition, although there is no evidence that there is in fact phase separation in the chosen mixtures, the purpose of the present work was to mimic the liquid-ordered (Lo) phase. In fact, for that reason, a saturated phospholipid (DMPC) was chosen over a typical lipid containing unsaturated bonds (such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), which is more biologically relevant). In the latter case, phase separation would occur due to the hydrophobic mismatch between Chol and the unsaturated lipid component, and both the Lo and liquid disordered (Ld) phases would be present in the bilayer, contrary to the goal of the current investigation. Furthermore, there is evidence to demonstrate that although cholesterol can recognize and interact preferentially with SM in a laterally phase-separated system, it cannot do so when in a miscible bilayer system composed of, for example, palmitoylsphingomyelin (PSM) and DMPC [44] as intended in this work.
Through the use of DMPC : SM and DMPC : SM : Chol model membranes, which attempt to represent the Ld and Lo states, respectively, it is thus possible to perform a comparison of daunorubicin's behaviour in each lipid phase in a separate manner. If a model system known to phase separate were used, it would otherwise be very difficult, with the techniques employed, to discriminate the role of each lipid phase in the daunorubicin–membrane interaction and whether the drug interplays preferentially with a certain membrane state.
The influence of daunorubicin on the DPH and TMA-DPH fluorescence anisotropy as a function of temperature for each model system studied is shown in figures 2 and 3, respectively. Overall, the results demonstrate that the anticancer drug influences the bilayer fluidity in a way that depends on the mimetic model membrane studied and the location of the probe used (i.e. the type of microenvironment). Therefore, while a decrease in membrane fluidity takes place in the acyl region, almost no significant change in membrane fluidity near the phospholipid head groups is observed. This can be explained by the fact that, although daunorubicin is positively charged at pH 7.4 and it can establish electrostatic interactions between the phospholipid headgroups, its backbone is deeply buried in the membrane. For this reason, the major alterations in membrane fluidity are along the acyl chains as revealed by the DPH results.
Figure 2.

(a–d) Steady-state anisotropy of DPH as a function of temperature in each mimetic model, in the absence (square), and in the presence of daunorubicin (DAN) 40 µM (circle) and 75 µM (triangle), at pH 7.4. Results present the mean of at least three independent assays. (Online version in colour.)
Figure 3.

(a–d) Steady-state anisotropy of TMA-DPH as a function of temperature in each mimetic model, in the absence (square), and in the presence of daunorubicin (DAN) 40 µM (circle) and 75 µM (triangle), at pH 7.4. Results present the mean of at least three independent assays. (Online version in colour.)
On the other hand, incorporation of daunorubicin into the membrane due to the hydrophobic interactions is easier when the phospholipid bilayer is less packed and the interplay between the acyl chains of the lipid molecules is not so strong, as in the case of DMPC. As a consequence, a more pronounced effect in DMPC anisotropy can be observed in the presence of daunorubicin than in the other membranes that contain SM and Chol. It can also be verified that Chol presence impairs the compound's permeation in the membrane and therefore, only small changes in fluidity can be observed, which ultimately are in agreement with drug partition studies at 37°C. Therefore, these outcomes allow one to infer that the anticancer drug leads to an ordering effect of the bilayer, which is manifested by the decrease in fluidity, except for membranes containing Chol.
Daunorubicin is a molecule that possesses an aromatic ring wherein its planar structure resembles the structure of cholesterol. Evidence suggests that there is a competition between Chol and drugs presenting an identical planar structure [45]. The membrane fluidity studies clearly demonstrate such an event and a bigger influence of daunorubicin along the membrane is achieved in the systems composed of DMPC and DMPC : SM. In this sense, daunorubicin interaction with these models is facilitated probably because there is no competition between the drug and Chol for a place within the bilayer. Furthermore, it is possible that the interactions between daunorubicin and Chol involve the formation of hydrogen bonds between the protonated  group of daunorubicin and the OH group of Chol, as already mentioned, and which is more prone to happen due to steric effects than with, for instance, the negatively charged moieties of sphingomyelin. In fact, measurements show that the size of liposomes containing Chol increased in the presence of daunorubicin, which is probably associated with daunorubicin adsorption at the surface. For that reason, only small changes in the membrane fluidity near the phospholipids heads can be observed.
group of daunorubicin and the OH group of Chol, as already mentioned, and which is more prone to happen due to steric effects than with, for instance, the negatively charged moieties of sphingomyelin. In fact, measurements show that the size of liposomes containing Chol increased in the presence of daunorubicin, which is probably associated with daunorubicin adsorption at the surface. For that reason, only small changes in the membrane fluidity near the phospholipids heads can be observed.
These findings are, once again, consistent with the previous results of partition and location, where it has been observed that the anticancer drug establishes electrostatic interactions with the negative phosphate group, hydrophobic interactions with the hydrocarbon chains of the phospholipids and can also form H bonds with Chol. On the other hand, the presence of Chol in bilayers leads to a decrease in the overall effects caused by daunorubicin in membrane fluidity. This fact is especially pronounced in the model containing SM, which is a more rigid and compact model due to its lipid composition, where the anticancer drug cannot permeate more than in the other mimetic systems.
Albeit to a smaller degree than with simpler model membranes, daunorubicin partitions in bilayers that contain SM and Chol, which can be correlated with its biological action. Lipid rafts are small, heterogeneous, highly dynamic platforms for signalling molecules that modulate various cellular functions, including cell survival [46]. These particular structures within the membrane are enriched in sterol- and sphingolipids [47] and play important roles in the activation of signalling pathways induced by several anticancer drugs [48]. In fact, evidence reveals that such regions are implicated in drug-induced apoptosis [49]. It has been shown that different anticancer drugs can produce changes in the lipid content of membrane rafts, which compromises their structure and integrity and, consequently, impairs cell growth and triggers cellular death [48]. It has also been demonstrated that some cancer cell lines contain more lipid rafts than their normal counterparts and, when depleted of cholesterol, are more sensitive to induced cell death [46]. Furthermore, resistance to chemotherapy exhibited by cancer cells is a major clinical problem that leads to therapy failure [50]. This phenomenon has been associated with the presence of molecular ‘pumps’ in tumour cell membranes, such as p-glycoprotein (Pgp), which actively transports the anticancer drugs to the exterior of the cell [51]. Pgp appears to be associated with specific membrane microdomains (lipid rafts), where its activity and function can be regulated by changes in the membrane-lipid environment [52,53]. It has also been evidenced by several structure–activity studies that hydrophobicity and the presence of planar aromatic regions, which occurs in the case of daunorubicin, favour the drug–Pgp interaction [54]. In addition, it has also been reported that compounds able to increase membrane fluidity cause an inhibition of Pgp function [55]. Nevertheless, our results showed that daunorubicin is not capable of producing a significant alteration in membrane fluidity in models that include Chol. Since it has also been demonstrated that Pgp seems to be associated with the microdomains referred to above and that the drug's binding sites are via the lipid membrane [52], it is possible to correlate the results of this study with the resistance problem associated with daunorubicin administration.
The present work also highlights the importance and contribution of biological membranes in the pharmacological activity of anticancer drugs, because this is where several events associated with the compounds' cytotoxicity take place, such as apoptosis activation through signalling platforms (lipid rafts).
4. Conclusion
The interplay between daunorubicin and membranes composed of different lipid contents is expressed as modifications in the overall biophysical properties of the membrane. In fact, anticancer drugs showed a high partition with membranes, which seems to be greatly driven by electrostatic interactions. Furthermore, the use of two fluorescent probes with different locations within the membrane allowed us to confirm that daunorubicin is placed within phospholipids and is positioned between the polar region (where it establishes electrostatic interactions with the negative phosphate group) and the membrane core (where it establishes hydrophobic interactions with the acyl chain tails). Because of its location, the compound induces a decrease in membrane fluidity in the hydrophobic tails of the phospholipids, in bilayers where cholesterol is not present.
These interactions are intrinsically dependent on the lipid composition and the membrane physical state and structure, which can favour or restrain the drug's mechanism of action and/or its side effects. Moreover, such characteristics are key factors in influencing anticancer drug partition, the location of the drug within the membrane and, ultimately, membrane fluidity. Indeed, the differences in drug incorporation within cell membranes and the alteration in membrane fluidity caused by daunorubicin reveal that the plasma membrane is a crucial place for bioaccumulation of an anticancer drug and its action, and also for its cytotoxicity and chemoresistance.
The combination of the interplay between daunorubicin and the membrane assessed in this work and the results of previous studies leads to the idea that, when designing and developing new drugs, the different membrane characteristics should be taken into account. In fact, the results of our study demonstrate that the different properties of a cell membrane can modulate daunorubicin behaviour. For that reason, membrane-lipid therapy, which considers plasma membranes as targets for drugs, emerges as a promising way of treating cancer. This approach might help the future development of drugs with increased specificity and, therefore, higher efficiency against cancer.
Supplementary Material
Acknowledgements
The authors thank Manuela Barros for administrative and technical support, and the anonymous reviewers who helped to improve the manuscript.
Data accessibility
All data needed to evaluate the conclusions are present in the paper itself and in the electronic supplementary material or are available upon request to the authors.
Authors' contributions
A.C.A., C.N., S.R.: conceived and designed the experiments; A.C.A., M.H., D.R.: performed the experimental work; A.C.A., C.N., D.R., S.R.: analysed the experimental data; A.C.A., C.N.: wrote the paper. All authors reviewed the manuscript.
Competing interests
We declare we have no competing interests.
Funding
A.C.A. thanks the Foundation for Science and Technology (FCT) for the fellowship (SFRH/BD/82443/2011). C.N. thanks FCT for the Investigator Grant (IF/00293/2015). This work received financial support from EU (FEDER funds POCI/01/0145/FEDER/007728) and national funds (FCT/MEC, Fundação para a Ciência e a Tecnologia and Ministério da Educação e Ciência) under the Partnership Agreement PT2020 UID/MULTI/04378/2013.
References
- 1.Grein A, Spalla C, Canevazz G, Dimarco A. 1963. Descrizione e classificazione di un attinomicete (Streptomyces peucetius sp. nova) produttore di una sostanza ad attivita antitumorale—la daunomicina. Giorn. Microbiol. 11, 109–118. [Google Scholar]
- 2.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. 2004. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229. ( 10.1124/pr.56.2.6) [DOI] [PubMed] [Google Scholar]
- 3.Gewirtz DA. 1999. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 57, 727–741. ( 10.1016/S0006-2952(98)00307-4) [DOI] [PubMed] [Google Scholar]
- 4.Escriba PV. 2006. Membrane-lipid therapy: a new approach in molecular medicine. Trends Mol. Med. 12, 34–43. ( 10.1016/j.molmed.2005.11.004) [DOI] [PubMed] [Google Scholar]
- 5.Tritton TR, Yee G. 1982. The anticancer agent adriamycin can be actively cyto-toxic without entering cells. Science 217, 248–250. ( 10.1126/science.7089561) [DOI] [PubMed] [Google Scholar]
- 6.Escriba PV, Sastre M, Garciasevilla JA. 1995. Disruption of cellular signaling pathways by daunomycin through destabilization of nonlamellar membrane structures. Proc. Natl Acad. Sci. USA 92, 7595–7599. ( 10.1073/pnas.92.16.7595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucio M, Lima JLFC, Reis S. 2010. Drug-membrane interactions: significance for medicinal chemistry. Curr. Med. Chem. 17, 1795–1809. ( 10.2174/092986710791111233) [DOI] [PubMed] [Google Scholar]
- 8.Siegfried JM, Burke TG, Tritton TR. 1985. Cellular transport of anthracyclines by passive diffusion. Implications for drug resistance. Biochem. Pharmacol. 34, 593–598. ( 10.1016/0006-2952(85)90251-5) [DOI] [PubMed] [Google Scholar]
- 9.Willingham MC, Cornwell MM, Cardarelli CO, Gottesman MM, Pastan I. 1986. Single cell analysis of daunomycin uptake and efflux in multidrug-resistant and -sensitive KB cells: effects of verapamil and other drugs. Cancer Res. 46, 5941–5946. [PubMed] [Google Scholar]
- 10.Regev R, Yeheskely-Hayon D, Katzir H, Eytan GD. 2005. Transport of anthracyclines and mitoxantrone across membranes by a flip-flop mechanism. Biochem. Pharmacol. 70, 161–169. ( 10.1016/j.bcp.2005.03.032) [DOI] [PubMed] [Google Scholar]
- 11.Gallois L, Fiallo M, Laigle A, Priebe W, Garnier-Suillerot A. 1996. The overall partitioning of anthracyclines into phosphatidyl-containing model membranes depends neither on the drug charge nor the presence of anionic phospholipids. Eur. J. Biochem. 241, 879–887. ( 10.1111/j.1432-1033.1996.00879.x) [DOI] [PubMed] [Google Scholar]
- 12.Gallois L, Fiallo M, Garnier-Suillerot A. 1998. Comparison of the interaction of doxorubicin, daunorubicin, idarubicin and idarubicinol with large unilamellar vesicles—circular dichroism study. Biochim. Biophys. Acta Biomembr. 1370, 31–40. ( 10.1016/S0005-2736(97)00241-1) [DOI] [PubMed] [Google Scholar]
- 13.Escriba PV, Ferrermontiel AV, Ferragut JA, Gonzalezros JM. 1990. Role of membrane lipids in the interaction of daunomycin with plasma membranes from tumor cells—implications in drug-resistance phenomena. Biochemistry 29, 7275–7282. ( 10.1021/Bi00483a017) [DOI] [PubMed] [Google Scholar]
- 14.Schwartz HS, Kanter PM. 1979. Chemical interactions of cardiolipin with daunorubicin and other intercalating agents. Eur. J. Cancer 15, 923–928. ( 10.1016/0014-2964(79)90235-4) [DOI] [PubMed] [Google Scholar]
- 15.Jaffrezou JP, Levade T, Bettaieb A, Andrieu N, Bezombes C, Maestre N, Vermeersch S, Rousse A, Laurent G. 1996. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. Embo. J. 15, 2417–2424. [PMC free article] [PubMed] [Google Scholar]
- 16.Grazide S, Maestre N, Veldman RJ, Bezombes C, Maddens S, Levade T, Laurent G, Jaffrezou JP. 2002. Ara-C- and daunorubicin-induced recruitment of Lyn in sphingomyelinase-enriched membrane rafts. FASEB. J. 16, 1685–1687. ( 10.1096/fj.01-0794fje) [DOI] [PubMed] [Google Scholar]
- 17.Dimanche-Boitrel M-T, Meurette O, Rebillard A, Lacour S. 2005. Role of early plasma membrane events in chemotherapy-induced cell death. Drug Resist. Updat. 8, 5–14. ( 10.1016/j.drup.2005.02.003) [DOI] [PubMed] [Google Scholar]
- 18.Alves AC, Ribeiro D, Nunes C, Reis S. 2016. Biophysics in cancer: the relevance of drug-membrane interaction studies. Biochim. Biophys. Acta Biomembr. 1858, 2231–2244. ( 10.1016/j.bbamem.2016.06.025) [DOI] [PubMed] [Google Scholar]
- 19.Seydel JK, Wiese M. 2002. Drug-membrane interactions: analysis, drug distribution, modeling. New York, NY: Wiley-VCH Verlag GmbH & Co. [Google Scholar]
- 20.Escriba PV, et al. 2008. Membranes: a meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 12, 829–875. ( 10.1111/j.1582-4934.2008.00281.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slotte JP. 2013. Biological functions of sphingomyelins. Prog. Lipid Res. 52, 424–437. ( 10.1016/j.plipres.2013.05.001) [DOI] [PubMed] [Google Scholar]
- 22.Maxfield FR, Tabas I. 2005. Role of cholesterol and lipid organization in disease. Nature 438, 612–621. ( 10.1038/nature04399) [DOI] [PubMed] [Google Scholar]
- 23.Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. 2002. Cholesterol interactions with phospholipids in membranes. Prog. Lipid. Res. 41, 66–97. ( 10.1016/S0163-7827(01)00020-0) [DOI] [PubMed] [Google Scholar]
- 24.Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327, 46–50. ( 10.1126/science.1174621) [DOI] [PubMed] [Google Scholar]
- 25.Nunes C, Lopes D, Pinheiro M, Pereira-Leite C, Reis S. 2013. In vitro assessment of NSAIDs-membrane interactions: significance for pharmacological actions. Pharm. Res. 30, 2097–2107. ( 10.1007/s11095-013-1066-8) [DOI] [PubMed] [Google Scholar]
- 26.Magalhaes LM, Nunes C, Lucio M, Segundo MA, Reis S, Lima JL. 2010. High-throughput microplate assay for the determination of drug partition coefficients. Nat. Protoc. 5, 1823–1830. ( 10.1038/nprot.2010.137) [DOI] [PubMed] [Google Scholar]
- 27.Brittes J, Lucio M, Nunes C, Lima JL, Reis S. 2010. Effects of resveratrol on membrane biophysical properties: relevance for its pharmacological effects. Chem. Phys. Lipids 163, 747–754. ( 10.1016/j.chemphyslip.2010.07.004) [DOI] [PubMed] [Google Scholar]
- 28.Coutinho A, Prieto M. 1993. Ribonuclease T1 and alcohol dehydrogenase fluorescence quenching by acrylamide: a laboratory experiment for undergraduate students. J. Chem. Educ. 70, 425 ( 10.1021/ed070p425) [DOI] [Google Scholar]
- 29.Okamura E, Wakai C, Matubayasi N, Sugiura Y, Nakahara M. 2004. Limited slowdown of endocrine-disruptor diffusion in confined fluid lipid membranes. Phys. Rev. Lett. 93, 248101 ( 10.1103/PhysRevLett.93.248101) [DOI] [PubMed] [Google Scholar]
- 30.Marcelino J, Lima JL, Reis S, Matos C. 2007. Assessing the effects of surfactants on the physical properties of liposome membranes. Chem. Phys. Lipids 146, 94–103. ( 10.1016/j.chemphyslip.2006.12.008) [DOI] [PubMed] [Google Scholar]
- 31.Martinez MN, Amidon GL. 2002. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J. Clin. Pharmacol. 42, 620–643. ( 10.1177/00970002042006005) [DOI] [PubMed] [Google Scholar]
- 32.Pereira-Leite C, Nunes C, Lima JL, Reis S, Lucio M. 2012. Interaction of celecoxib with membranes: the role of membrane biophysics on its therapeutic and toxic effects. J. Phys. Chem. B 116, 13 608–13 617. ( 10.1021/jp304037v) [DOI] [PubMed] [Google Scholar]
- 33.Nunes C, Brezesinski G, Lopes D, Lima JL, Reis S, Lucio M. 2011. Lipid-drug interaction: biophysical effects of tolmetin on membrane mimetic systems of different dimensionality. J. Phys. Chem. B 115, 12 615–12 623. ( 10.1021/jp206013z) [DOI] [PubMed] [Google Scholar]
- 34.Dupou-Cezanne L, Sautereau AM, Tocanne JF. 1989. Localization of adriamycin in model and natural membranes. Influence of lipid molecular packing. Eur. J. Biochem. 181, 695–702. ( 10.1111/j.1432-1033.1989.tb14779.x) [DOI] [PubMed] [Google Scholar]
- 35.Calzolai L, Gaggelli E, Maccotta A, Valensin G. 1996. Interaction of daunomycin with dipalmitoylphosphatidylcholine model membranes. A 1H NMR study. J. Magn. Reson. B 112, 228–235. ( 10.1006/jmrb.1996.0135) [DOI] [PubMed] [Google Scholar]
- 36.Banuelos S, Arrondo JL, Canaves JM, Ferragut JA, Muga A. 1993. The interaction of daunomycin with model membranes. Effect of the lipid physical state and the lipid composition. Eur. J. Biochem. 213, 1269–1275. ( 10.1111/j.1432-1033.1993.tb17878.x) [DOI] [PubMed] [Google Scholar]
- 37.Lakowicz JR. 2006. Quenching of fluorescence. In Principles of fluorescence spectroscopy, 3rd edn, pp. 278–327. Berlin, Germany: Springer. [Google Scholar]
- 38.Kaiser RD, London E. 1998. Location of diphenylhexatriene (DPH) and its derivatives within membranes: comparison of different fluorescence quenching analyses of membrane depth. Biochemistry 37, 8180–8190. ( 10.1021/bi980064a) [DOI] [PubMed] [Google Scholar]
- 39.Illinger D, Duportail G, Mely Y, Poirel-Morales N, Gerard D, Kuhry JG. 1995. A comparison of the fluorescence properties of TMA-DPH as a probe for plasma membrane and for endocytic membrane. Biochim. Biophys. Acta 1239, 58–66. ( 10.1016/0005-2736(95)00135-P) [DOI] [PubMed] [Google Scholar]
- 40.Lakowicz JR. 2006. Fluorescence anisotropy. In Principles of fluorescence spectroscopy, 3rd edn, pp. 366–375. Berlin, Germany: Springer. [Google Scholar]
- 41.Lucio M, Ferreira H, Lima JL, Reis S. 2006. Interactions between oxicams and membrane bilayers: an explanation for their different COX selectivity. Med. Chem. 2, 447–456. ( 10.2174/157340606778250199) [DOI] [PubMed] [Google Scholar]
- 42.Collins JM, Dominey RN, Grogan WM. 1990. Shape of the fluidity gradient in the plasma membrane of living HeLa cells. J. Lipid Res. 31, 261–270. [PubMed] [Google Scholar]
- 43.Barenholz Y. 2004. Sphingomyelin and cholesterol: from membrane biophysics and rafts to potential medical applications. Subcell. Biochem. 37, 167–215. ( 10.1007/978-1-4757-5806-1_5) [DOI] [PubMed] [Google Scholar]
- 44.Calhoun WI, Shipley GG. 1979. Sphingomyelin–lecithin bilayers and their interaction with cholesterol. Biochemistry 18, 1717–1722. ( 10.1021/bi00576a013) [DOI] [PubMed] [Google Scholar]
- 45.Tessier C, Nuss P, Staneva G, Wolf C. 2008. Modification of membrane heterogeneity by antipsychotic drugs: an X-ray diffraction comparative study. J. Colloid Interface Sci. 320, 469–475. ( 10.1016/j.jcis.2008.01.034) [DOI] [PubMed] [Google Scholar]
- 46.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. 2006. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 168, 1107–1118. ( 10.2353/ajpath.2006.050959) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patra SK. 2008. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim. Biophys. Acta Rev. Cancer 1785, 182–206. ( 10.1016/j.bbcan.2007.11.002) [DOI] [PubMed] [Google Scholar]
- 48.George KS, Wu S. 2012. Lipid raft: a floating island of death or survival. Toxicol. Appl. Pharmacol. 259, 311–319. ( 10.1016/j.taap.2012.01.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bezombes C, Laurent G, Jaffrezou JP. 2003. Implication of raft microdomains in drug induced apoptosis. Curr. Med. Chem. Anticancer Agents 3, 263–270. ( 10.2174/1568011033482413) [DOI] [PubMed] [Google Scholar]
- 50.Koehn FE. 2013. Natural products and cancer drug discovery. Berlin, Germany: Springer. [Google Scholar]
- 51. 2000 Cancer multidrug resistance. Nat. Biotech. 18 , IT18–IT20. ( ) [DOI] [PubMed]
- 52.Modok S, Heyward C, Callaghan R. 2004. P-glycoprotein retains function when reconstituted into a sphingolipid- and cholesterol-rich environment. J. Lipid Res. 45, 1910–1918. ( 10.1194/jlr.M400220-JLR200) [DOI] [PubMed] [Google Scholar]
- 53.dos Santos SM, Weber CC, Franke C, Muller WE, Eckert GP. 2011. Erratum to: Cholesterol: coupling between membrane microenvironment and ABC transporter activity (vol. 354, pg 216, 2007). Biochem. Biophys. Res. Commun. 408, 193 ( 10.1016/j.bbrc.2011.03.111) [DOI] [PubMed] [Google Scholar]
- 54.Seelig A, Landwojtowicz E. 2000. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 12, 31–40. ( 10.1016/S0928-0987(00)00177-9) [DOI] [PubMed] [Google Scholar]
- 55.Orlowski S, Martin S, Escargueil A. 2006. P-glycoprotein and ‘lipid rafts’: some ambiguous mutual relationships (floating on them, building them or meeting them by chance?) Cell. Mol. Life Sci. 63, 1038–1059. ( 10.1007/s00018-005-5554-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions are present in the paper itself and in the electronic supplementary material or are available upon request to the authors.