

Abstract
Objective:
To investigate the effect of disease-modifying treatment on short-term disability outcomes in secondary progressive multiple sclerosis (SPMS).
Methods:
Using MSBase, an international cohort study, we previously validated a highly accurate definition of SPMS. Here, we identified patients in MSBase who were either untreated or treated with a disease-modifying drug when meeting this definition. Propensity score matching was used to select subpopulations with comparable baseline characteristics. Disability outcomes were compared in paired, pairwise-censored analyses adjusted for treatment persistence, visit density, and relapse rates.
Results:
Of the 2,381 included patients, 1,378 patients were matchable (treated n = 689, untreated n = 689). Median pairwise-censored follow-up was 2.1 years (quartiles 1.2–3.8 years). No difference in the risk of 6-month sustained disability progression was observed between the groups (hazard ratio [HR] 0.9, 95% confidence interval [CI] 0.7–1.1, p = 0.27). We also did not find differences in any of the secondary endpoints: risk of reaching Expanded Disability Status Scale (EDSS) score ≥7 (HR 0.6, 95% CI 0.4–1.1, p = 0.10), sustained disability reduction (HR 1.0, 95% CI 0.8–1.3, p = 0.79), or change in disability burden (area under the EDSS-time curve, β = −0.05, p = 0.09). Secondary and sensitivity analyses confirmed the results.
Conclusions:
Our pooled analysis of the currently available disease-modifying agents used after conversion to SPMS suggests that, on average, these therapies have no substantial effect on relapse-unrelated disability outcomes measured by the EDSS up to 4 years.
Classification of evidence:
This study provides Class IV evidence that for patients with SPMS, disease-modifying treatment has no beneficial effect on short-term disability progression.
While substantial progress has been made in the development of effective treatments for relapsing-remitting multiple sclerosis (RRMS) in past decades, similar success has not been achieved in secondary progressive multiple sclerosis (SPMS).1,2 Currently, interferon (IFN) β-1b and mitoxantrone are the only anti-inflammatory agents that have been approved for SPMS treatment in Europe and the United States.3,4 However, a recent Cochrane review concluded that IFNβ is not useful in SPMS, and the use of mitoxantrone is limited by serious adverse events, namely cardiotoxicity and increased risk of leukemia.5,6 More recently, a randomized controlled trial of natalizumab in SPMS (A Clinical Study of the Efficacy of Natalizumab on Reducing Disability Progression in Participants With Secondary Progressive Multiple Sclerosis [ASCEND], NCT01416181) failed to show a statistically significant effect on confirmed disability progression.7 Newer oral drugs approved for RRMS such as fingolimod, dimethyl fumarate, or teriflunomide have not yet been studied.8 However, in the absence of other therapeutic options, these anti-inflammatory drugs are still being used in SPMS despite the weight of evidence not supporting this practice.
Because randomized controlled trials evaluating treatment outcomes in SPMS are associated with high cost, a feasible alternative strategy is to use existing longitudinal registries of clinical outcome data to examine potential therapeutic effects.9 MSBase is a large, international, observational cohort study of patients with MS, and we previously demonstrated its utility in the analysis of treatment outcomes using propensity matching to mitigate treatment indication bias.10–12 The aim of the present study was to investigate the effect of anti-inflammatory disease-modifying treatment used after conversion to SPMS on disability outcomes.
METHODS
Standard protocol approvals, registrations, and patient consents.
The MSBase cohort study (registered with World Health Organization International Clinical Trials Registry Platform, identifier ACTRN12605000455662) was approved by the Melbourne Health Human Research Ethics Committee and by the local ethics committees in all participating centers (or exemptions granted, according to applicable local laws and regulations). If required, written informed consent was obtained from enrolled patients in accordance with the Declaration of Helsinki.
Database and study population.
Longitudinal clinical data from 36,910 patients from 122 MS centers in 57 countries were extracted from the MSBase registry in January 2016. For this study, we selected patients with SPMS diagnosed retrospectively according to our previously validated definition.13 The minimal dataset comprised patient sex, year of birth, year of the first clinical presentation, MS course, treating center, and at least 2 clinical visits with recorded Expanded Disability Status Scale (EDSS) and functional system scores at least 6 months apart. Patients previously participating in randomized trials involving active agents or receiving alemtuzumab, cladribine, or autologous stem cell transplantation before baseline were excluded because we could not control for potential carryover effects of these treatments. The study baseline was defined as the time of the diagnosis of SPMS, regardless of the patients' treatment status, to avoid immortal-time bias.
The data quality assessment was conducted with a series of procedures to identify any invalid or inconsistent entries, as described elsewhere.14 The analyzed data were recorded as part of clinical practice, mostly at large tertiary MS centers. The usual data entry practice was real-time or near–real-time data entry (at the time of clinical visits). The MSBase protocol stipulates a required annual update of the minimum dataset, but patients with less frequent visits were not excluded from the analysis. Categorized results of brain MRI were reported by treating neurologists. The data entry portal was either the iMed patient record system or the MSBase online data entry system.
Study endpoints.
Our study was designed to provide class IV evidence to address the primary research question, which was the effect of disease-modifying treatment on the cumulative risk of disability progression after conversion to SPMS. Disability was scored by accredited scorers (Neurostatus certification was required at each center) using the EDSS, calculated on the basis of functional system and ambulation scores. Disability progression was defined as an EDSS score increase of 1 point (0.5 points if baseline EDSS score was ≥6) sustained for ≥6 months. Disability reduction was defined as decrease of EDSS score by 1 point (0.5 points if baseline EDSS score was ≥6.5) sustained for ≥6 months.15 The endpoint of EDSS score ≥7 was reached at the time a patient progressed to EDSS step 7 with confirmation over the next ≥6 months. A relapse was defined as occurrence of new symptoms or exacerbation of existing symptoms persisting for at least 24 hours, in the absence of concurrent illness or fever, and occurring at least 30 days after a previous relapse. Individual annualized relapse rate (ARR) was calculated as the annualized number of recorded relapses between baseline and a censoring event. Burden of disability over the follow-up period was quantified as the area under the EDSS-time curve (AUC) using the trapezium rule.11,16 MS duration was calculated from the first demyelinating event. The prospective on-study follow-up was defined as the time between the first and last available EDSS entries.
Matching and statistical analysis.
Matching and statistical analysis was conducted by the first author using R (version 3.1.2).17 Applying an intention-to-treat design, we allocated included patients to the treatment arm if they were receiving 1 of the following drugs on the day on which the diagnostic criteria for SPMS were first fulfilled: IFNβ-1a subcutaneously, IFNβ-1b subcutaneously, IFNβ-1a intramuscularly, glatiramer acetate, natalizumab, fingolimod, dimethyl fumarate, teriflunomide, mitoxantrone, or rituximab. Otherwise, they were assigned to the no-treatment arm. Patients were then matched on their propensity for receiving vs not receiving disease-modifying treatment with the MatchIt package.18 The propensity score was based on a multivariable logistic regression model with treatment allocation as the outcome variable and the demographic and clinical variables available to treating neurologists at the time of conversion to SPMS as the independent variables. These included sex, age, and disease duration at baseline, baseline EDSS, EDSS change leading to the diagnosis of SPMS, disability trajectory (i.e., the regression line projected over the EDSS/time points), number of relapses 12 months before baseline, and center. To adjust for residual imbalance between the treated and untreated groups, the observed postbaseline ARR was used as an additional independent variable for the final model. Patients were then matched in a 1:1 ratio with nearest-neighbor matching within a caliper of 0.1 SD of the propensity score, without replacement. The common on-treatment follow-up was determined as the shorter of the 2 individual follow-up periods for each matched patient pair (pairwise censoring) to control attrition bias.9,11 All subsequent analyses were then completed with paired models and were adjusted for treatment persistence during the follow-up and for visit density. The cumulative hazards of the confirmed disability progression or reduction events were evaluated with conditional proportional hazards models for both first event and recurrent events (with robust estimation of variance for the latter), with the frailty term indicating the matched pairs. For confirmed EDSS score ≥7, we applied a conditional proportional hazards model for time to single event, which was additionally adjusted for baseline EDSS. Proportionality of hazards was assessed with the Schoenfeld global test, and we used Kaplan-Meier plots to visualize the time-to-event data. After assessment of normality of data distribution, annualized changes in AUC were analyzed by a paired linear regression model.
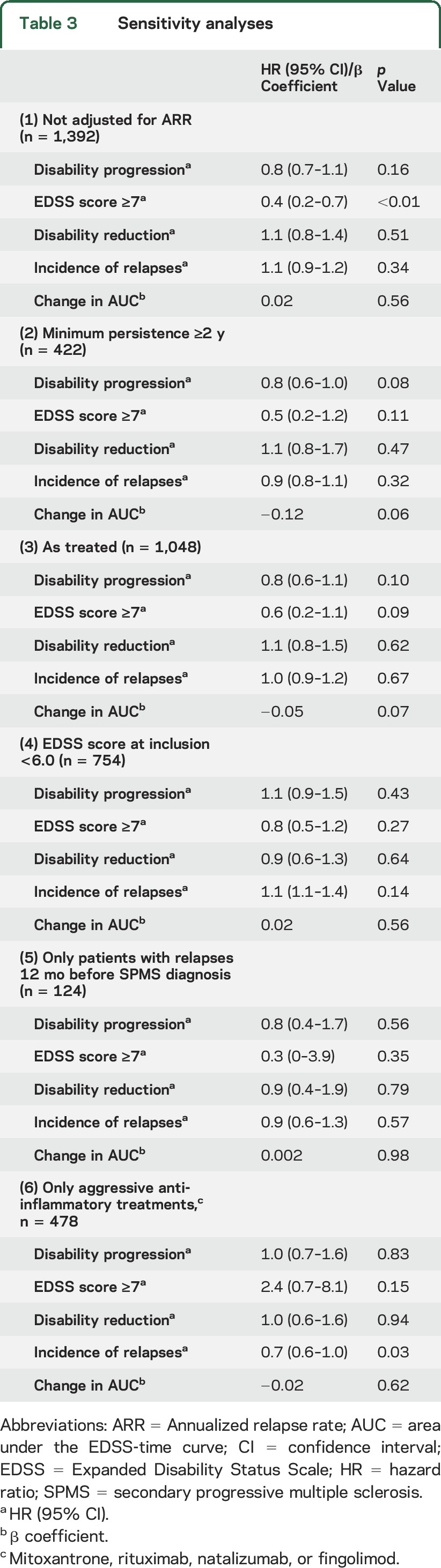
Six sensitivity analyses were carried out: (1) repeating the analyses without matching on observed postbaseline ARR, (2) assessing only patient pairs with treatment persistence ≥2 years, (3) an as-treated approach with pairwise censoring at the time of the change of the treatment status (i.e., discontinuing treatment in the treated group or starting treatment in the untreated group), (4) including only patients with a baseline EDSS score <6, (5) including only patients who experienced relapses in the year before baseline, and (6) including only patients treated with an aggressive anti-inflammatory agent (mitoxantrone, rituximab, natalizumab, or fingolimod).
A secondary analysis was performed to evaluate a potential for delayed treatment effect.19 The therapeutic time lag was estimated, depending on the baseline EDSS. For each matched pair, we subtracted 3 from the median EDSS step to obtain the number of years of the presumed lag. Only events that occurred after the calculated period were included in this analysis. An additional analysis was performed in which treatment status at conversion to SPMS was replaced by the proportion of follow-up time spent on disease-modifying agents in both the original model and the model for a therapeutic lag to evaluate the effect of overall treatment exposure.
Observed differences were considered significant at p ≤ 0.05. A post hoc power analysis was conducted to define the lower bounds of the minimum effect sizes at α = 0.05 detectable in the available data set at 1 − β = 0.9. Series of simulations (n = 200) were carried out for each of the statistical models using the observed distributions of the outcome variables. The minimum detectable effect sizes were 0.80 (hazard ratio [HR]) for confirmed disability progression, 0.56 (HR) for reaching confirmed EDSS score ≥7, 0.74 (HR) for confirmed disability reduction, 0.85 (HR) for relapses, and 0.07 EDSS-years for the change of AUC.
RESULTS
A total of 2,381 patients were included in the analysis (figure 1 and table e-1 at Neurology.org). Baseline characteristics are shown in table 1. Several demographic factors and markers of disease severity differed markedly between the unmatched patient groups. The logistic model, used to estimate propensity scores, showed that being untreated at the time of SPMS diagnosis was associated with lower ARR in the year before and after diagnosis of SPMS, older age, longer disease duration, faster disability increment, and center-specific practice (table e-2). The propensity-matching procedure retained 689 (50%) treated and 689 (69%) untreated patients, improving the overall match, as indicated by the decrease from 0.27 to 0.04 (by 92%) in the mean difference in propensity scores (figure e-1). This was reflected by the improved match in the individual determinants of treatment allocation, including age, disease duration, and number of relapses in the year before the diagnosis of SPMS (table 1).
Figure 1. CONSORT (Consolidated Standards of Reporting Trials) diagram of patient disposition (primary analysis).

DMT = disease-modifying therapy; EDSS = Expanded Disability Status Scale.
Table 1.
Patient characteristics at baseline
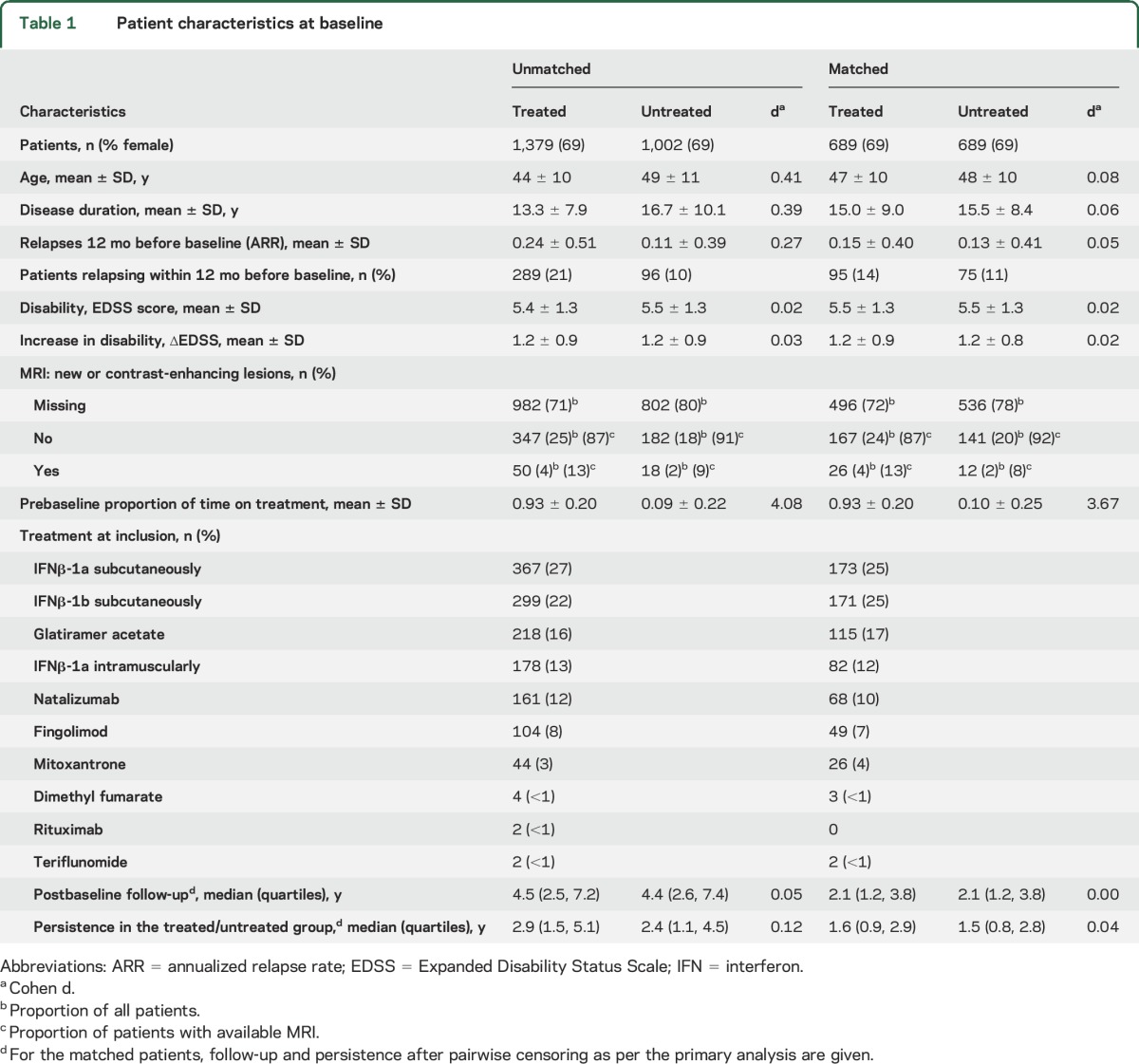
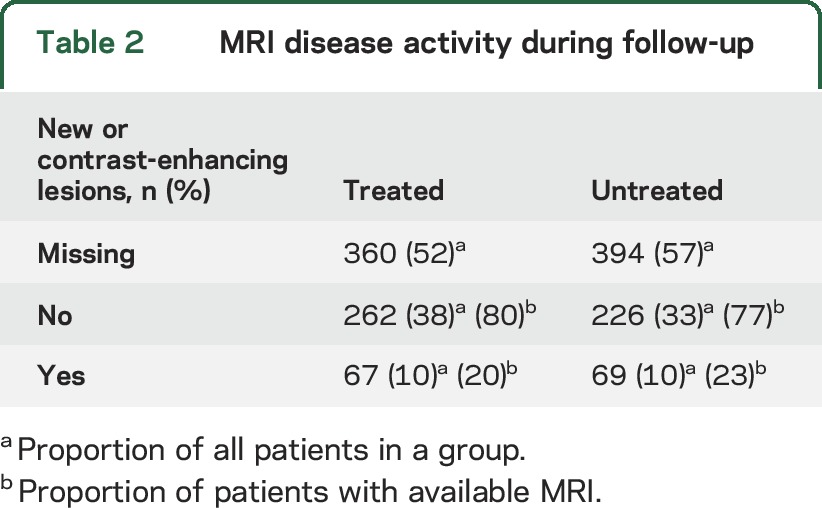
The results of the primary analysis are shown in figure 2. We did not observe any difference in the cumulative hazard of confirmed disability progression events (HR 0.9, 95% CI [confidence interval] 0.7–1.1, p = 0.27) or in the proportion of patients free from disability progression (HR 0.9, 95% CI 0.7–1.1, p = 0.19) between the treated and untreated groups at the time of SPMS conversion. There was no difference in the risk of reaching confirmed EDSS score ≥7 (HR 0.6, 95% CI 0.4–1.1, p = 0.10). The cumulative hazards of confirmed disability reduction also did not differ between the 2 groups (HR 1.0, 95% CI 0.8–1.3, p = 0.79), nor did the proportion of patients with sustained disability (HR 1.0, 95% CI 0.7–1.3, p = 0.96). We did not observe any difference in the change of AUC either (treated 0.07 [95% CI 0.03–0.11] vs untreated 0.12 [95% CI 0.08–0.16], p = 0.09). The matching process effectively adjusted the analysis for a potential effect of postbaseline relapses (HR 1.0, 95% CI 0.9–1.1, p = 0.80). In participants for whom sufficient MRI data were available (48% in the treated and 43% in the untreated group), the proportion of patients who showed new or gadolinium-enhancing lesions at any time during the follow-up was 20% in the treated and 23% in the untreated arm of the study (table 2).
Figure 2. Disability outcomes.

Proportion of patients free from disability progression (A), patients not reaching confirmed EDSS score ≥7 (B), and patients with sustained disability (i.e., no confirmed disability reduction) (C), and change in annualized area under the EDSS-time curve (D) among patients treated (DMT) or untreated (control) with an anti-inflammatory drug. Results of the paired matched analysis with pairwise censoring are shown. Error bars indicate 95% confidence interval (CI). DMT = disease-modifying therapy; HR = hazard ratio.
Table 2.
MRI disease activity during follow-up
The sensitivity analysis, which was not adjusted for postbaseline ARR (table 3), confirmed the outcomes of the primary analysis, except that the treated group showed a lower risk of reaching confirmed EDSS score ≥7 (HR 0.4, 95% CI 0.2–0.7, p < 0.01). Sensitivity analyses 2 through 5 (requiring a minimal treatment persistence of 2 years after the diagnosis of SPMS, using the as-treated approach, including only patients with an EDSS score <6 at baseline, and including only patients with relapses in the year before baseline) fully replicated the results of the primary analysis. The sensitivity analysis including patients treated with one of the more aggressive anti-inflammatory agents largely confirmed the primary analysis, but it also showed a lower risk of experiencing relapses in the treated group, despite being matched on postbaseline ARR (HR 0.7, 95% CI 0.6–1.0, p = 0.03).
Table 3.
Sensitivity analyses
The secondary analysis adjusted for a hypothesized treatment lag found no evidence for a delayed beneficial effect of disease-modifying treatment at the time of SPMS conversion on the cumulative hazard of disability progression events (HR 0.8, 95% CI 0.6–1.1, p = 0.28). When we replaced treatment status at conversion to SPMS in the primary analysis with the overall treatment exposure during the follow-up, there was no difference in the risk of EDSS progression associated with a greater proportion of time spent on treatment without (HR 0.8, 95% CI 0.6–1.0, p = 0.054) or with (HR 0.9, 95% CI 0.7–1.3, p = 0.67) adjustment for the therapeutic lag.
DISCUSSION
In this observational, propensity score–matched analysis of disability outcomes in patients with SPMS followed up for >4 years, we did not observe a therapeutic benefit of disease-modifying drugs that are currently used in RRMS on short-term progression of disability in patients meeting an objective definition of SPMS.
Of the 5 phase III randomized controlled trials investigating the effects of IFNβ compared to placebo in SPMS, only the European study of IFNβ-1b (placebo-controlled multicenter randomized trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis: European Study Group on Interferon Beta-1b in Secondary Progressive MS [EUSP]) showed a difference in time to confirmed progression in favor of IFNβ-1b.4 In contrast, the North American trial of IFNβ-1b and 3 randomized controlled trials using IFNβ-1a (Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS [SPECTRIMS], International MS Secondary Progressive Avonex Clinical Trial [IMPACT], and the Nordic trial) could not demonstrate a difference between the treated and untreated groups.20–23 In EUSP and SPECTRIMS, patients were younger and their disease duration was shorter than reported in other trials.6 This difference is of importance because a trend for better response was found for patients with prestudy relapses, younger age, and shorter disease duration in the European trial, which could explain the discordant results of that study.4 The Mitoxantrone in Multiple Sclerosis (MIMS) trial showed a beneficial effect of mitoxantrone in a mixed population of patients with worsening RRMS and SPMS, and the trial recruited patients with a markedly higher mean number of relapses in the year before inclusion (1.27–1.42) than our study (0.13–0.15).3 With a mean age at inclusion of 47 years, disease duration of 15 years, and EDSS score of 5.5, the core characteristics of our study population lay within the range of the North American, SPECTRIMS, IMPACT, and Nordic trials.20–23 In agreement with these studies, we could not demonstrate an effect of anti-inflammatory treatment on confirmed disability progression. There was also no significant difference in the risk of reaching an EDSS score ≥7, probability of disability reduction, or reduction in the overall disease burden (quantified as AUC).
Recently, a post hoc analysis of the SPECTRIMS trial and the study of glatiramer acetate in primary progressive MS (a multinational, multicenter, double-blind, placebo-controlled study to evaluate the efficacy, tolerability and safety of glatiramer acetate for injection in primary progressive multiple sclerosis patients [PROMiSe]) has suggested that an effect of immunomodulatory disease-modifying treatment on disability progression is potentially delayed in proportion to the preexisting disability.19,20,24 In our study, we did not identify a therapeutic lag; however, our median follow-up was 2.1 years, which has limited our ability to examine the treatment lag over an extended period of time. As demonstrated, our study was sufficiently powered to study the effect of therapy on disability progression independently of its effect on relapses.
In this study, we combined exposure to all disease-modifying drugs to maximize power. We also conducted a sensitivity analysis restricting therapy to highly aggressive therapies. While these drugs reduced the risk of relapses, they did not show a significant beneficial treatment effect on disability outcomes. The low number of patients exposed to individual drugs precluded us from drawing conclusions about the effect of single drug exposure vs no treatment. Therefore, we cannot rule out that 1 or more of these drugs, when studied in isolation, may demonstrate an effect on disability outcomes in SPMS. To clarify this point, further research is required.
Like other studies of treatment outcomes in SPMS, the limitation of our primary disability outcome is inherent in the limitation of the EDSS. The EDSS relies heavily on lower limb function, and its sensitivity to cognitive changes and upper limb function in more advanced MS is relatively low. It is also known to be less stable in the lower end of the EDSS spectrum.25 We have eliminated this variability by using only EDSS scores ≥4. To mitigate the known treatment indication bias, we used propensity score–based matching. Unlike randomization, propensity score–based matching does not eliminate unknown confounders. However, this is unlikely to have a substantial effect on our overall conclusions because sensitivity analyses with varying inclusion criteria, with varying definitions of follow-up, and without adjustment for postbaseline relapse activity confirmed the results of the primary analysis.
Because of the relative lack of MRI data, we were unable to match patients on MRI activity to analyze potential subgroup effects in patients with radiologically active disease or to study treatment effects on MRI outcomes. However, the occurrence of new T2 lesions or contrast-enhancing lesions on MRI, for the proportion of patients for whom it was recorded, was distributed evenly between the groups. Pairwise censoring was applied to control for attrition bias and to ensure validity of the patient match completed at baseline throughout the study. We also adjusted for reporting bias by taking into account the frequency of clinical follow-up. Finally, this study reports mainly the outcomes of platform injectable immunomodulatory therapies; only 21% of the treated patients received newer, more potent immunotherapies. On the other hand, our study used a validated definition of SPMS.13 This and the size of the analyzed multinational cohort argue for broad generalizability of our observations.26
In line with the outcomes of most of the previous trials, our present study shows that, on average, the currently available anti-inflammatory drugs do not prevent relapse-unrelated accumulation of disability in patients with established SPMS in the short term. However, our recent study showed that highly effective immunotherapy mitigates predominantly relapse-dependent disability accrual over 11 years in patients with moderately advanced or advanced relapse-onset MS (regardless of their SPMS status),27 a finding that is mirrored by our observation of difference in the risk of reaching EDSS step 7 in the sensitivity analysis unadjusted for postbaseline ARR. Moreover, the S1P receptor modulator siponimod has recently been reported to reduce disability progression in patients with SPMS.28 Therefore, separate evaluations of the newer, potentially more effective disease-modifying therapies in SPMS and studies of treatment efficacy in SPMS with high inflammatory profile are needed.
Supplementary Material
GLOSSARY
- ARR
annualized relapse rate
- ASCEND
A Clinical Study of the Efficacy of Natalizumab on Reducing Disability Progression in Participants With Secondary Progressive Multiple Sclerosis
- AUC
area under the EDSS-time curve
- CI
confidence interval
- EDSS
Expanded Disability Status Scale
- HR
hazard ratio
- IFN
interferon
- MIMS
Mitoxantrone in Multiple Sclerosis
- RRMS
relapsing-remitting multiple sclerosis
- SPECTRIMS
Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS
- SPMS
secondary progressive multiple sclerosis
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: MSBase Study Group, Raymond Hupperts, Ricardo Fernández Bolaños, Maria Edite Rio, Jose Antonio CabreraGomez, Freek Verheul, Mark Slee, Pamela McCombe, Javier Olascoaga, Maria Laura Saladino, Maria Pia Amato, Raed Alroughani, Edgardo Cristiano, Norma Deri, José Luis Sánchez Menoyo, Suzanne Hodgkinson, Shlomo Flechter, Fraser Moore, Thor Petersen, Radek Ampapa, Orla Gray, Olga Skibina, Tunde Csepan, Bhim Singhal, Leontien den Braber-Moerland, Carmen-Adella Sirbu, Steve Vucic, Walter Oleschko Arruda, Julie Prévost, Krisztian Kasa, Allan Kermode, Michael H. Barnett, Neil Shuey, Piroska Imre, Vera Daskalovska, and Norbert Vella
AUTHOR AFFILIATIONS
From the Department of Medicine (J.L., V.G.J., T.S., H.B., T.K.), University of Melbourne; Department of Neurology (J.L., V.G.J., H.B., T.K.), Royal Melbourne Hospital, Australia; Hospital Universitario Virgen Macarena (G. Izquierdo), Sevilla, Spain; Department of Biomedical and NeuroMotor Sciences (A.L.), Alma Mater Studiorum–Università di Bologna and IRCCS Istituto delle Scienze Neurologiche–“UOSI Riabilitazione Sclerosi Multipla,” Italy; Department of Neurology and Center of Clinical Neuroscience (E.H., D.H.), Charles University; First Faculty of Medicine and General University Hospital (E.H., D.H.), Prague, Czech Republic; Department of Basic Medical Sciences, Neuroscience and Sense Organs (M. Trojano), University of Bari, Italy; Hôpital Notre Dame (P.D., M.G., A.P.), Montreal; Neuro Rive-Sud (F. Grand'Maison), Hôpital Charles LeMoyne, Greenfield Park; Hôtel-Dieu de Lévis (P.G.), Quebec, Canada; Neurology Unit (E.P.), ASUR Marche, Ancona, Italy; Karadeniz Technical University (C.B.), Trabzon, Turkey; Nuovo Ospedale Civile S. Agostino/Estense (P.S., D.F.), Modeno, Italy; Department of Biomedic, Metabolic and Neurosciences (D.F.), University of Modena and Reggio Emilia, Italy; AORN San Giuseppe Moscati (D.S.), Avellino, Italy; Department of Neurology and John Hunter Hospital (J.L.-S.), School of Medicine and Public Health, University of Newcastle; Australia; Medical Faculty (M. Terzi), Mayis University, Samsun, Turkey; Cliniques Universitaires Saint-Luc (V.V.P.), Brussels, Belgium; Ospedali Riuniti di Salerno (G. Iuliano); Mondino National Neurological Institute of Pavia (R.B.), Italy; Hospital Germans Trias i Pujol (C.R.-T.), Barcelona, Spain; University of Parma (F. Granella), Italy; University Hospital San Carlos (C.O.-G.), Spain; and Department of Neurology (H.B.), Box Hill Hospital, Melbourne, Australia.
AUTHOR CONTRIBUTIONS
Johannes Lorscheider: study concept and design, analysis and interpretation of data, statistical analysis, drafting and revising the manuscript. Vilija G. Jokubaitis and Tim Spelman: analysis and interpretation of data, revising the manuscript. Guillermo Izquierdo, Alessandra Lugaresi, Eva Havrdova, Dana Horakova, Maria Trojano, Pierre Duquette, Marc Girard, Alexandre Prat, François Grand'Maison, Pierre Grammond, Eugenio Pucci, Cavit Boz, Patrizia Sola, Diana Ferraro, Daniele Spitaleri, Jeanette Lechner-Scott, Murat Terzi, Vincent Van Pesch, Gerardo Iuliano, Roberto Bergamaschi, Cristina Ramo-Tello, Franco Granella, and Celia Oreja-Guevara: analysis and interpretation of data, revising the manuscript, acquisition of data. Helmut Butzkueven and Tomas Kalincik: study concept and design, analysis and interpretation of data, revising the manuscript, acquisition of data, study supervision, obtaining funding.
STUDY FUNDING
This study was financially supported by Biogen (Fellowship in MS Registries Research), the National Health and Medical Research Council (practitioner fellowship 1,080,518; project grants 1,083,539 and 1,129,189; and Centre for Research Excellence 1,001,216), and the University of Melbourne (Faculty of Medicine, Dentistry and Health Sciences research fellowship). The MSBase Foundation is a not-for-profit organization that receives support from Merck, Biogen, Novartis, Bayer, Genzyme, Teva, and Sanofi-Aventis. The study was conducted separately and apart from the guidance of the sponsors.
DISCLOSURE
J. Lorscheider accepted conference travel support from Novartis and has received research support from Biogen. V. Jokubaitis received conference travel support from Merck and Novartis and honoraria from Novartis and Biogen. T. Spelman received compensation for travel from Biogen Idec. G. Izquierdo received speaking honoraria from Biogen-Idec, Novartis, Sanofi, Merck Serono, and Teva. A. Lugaresi is a Bayer, Biogen, Genzyme, and Merck Advisory Board Member. She received travel grants and honoraria from Bayer, Biogen, Merck, Novartis, Sanofi-Genzyme, Teva, and Associazione Italiana Sclerosi Multipla, and her institution received research grants from Bayer, Biogen, Merck, Novartis, Sanofi, Teva, and Fondazione Italiana Sclerosi Multipla. E. Havrdova received speaker honoraria and consultant fees from Biogen Idec, Merck, Novartis, Genzyme, and Teva, as well as support for research activities from the Czech Ministries of Education and Health (NT13237-4/2012, PRVOUK-P26/LF1/4), Biogen Idec, and Merck Serono. D. Horakova received speaker honoraria and consulting fees from Biogen Idec, Merck Teva, Genzyme, and Novartis, as well as support for research activities from the Czech Ministries of Education and Health (NT13237-4/2012, PRVOUK-P26/LF1/4) and Biogen Idec. M. Trojano received speaking honoraria from Biogen-Idec, Bayer-Schering, Sanofi Aventis, Merck-Serono, Teva, and Novartis and has received research grants from Biogen-Idec, Merck-Serono, and Novartis. P. Duquette has served on editorial boards and has been supported to attend meetings by EMDSerono, Biogen-Idec, Novartis, Genzyme, and TEVA Neuroscience. He holds grants from the Canadian Institutes of Health Research and the MS Society of Canada and has received funding for investigator-initiated trials from Biogen-Idec, Novartis, and Genzyme. M. Girard has received speaker honoraria and has been on advisory board for Novartis, Biogen Idec, Teva Neuroscience, EMD Serono, and Genzyme. He has received research grants from the Canadian Institutes of Health Research and the MS Society of Canada. A. Prat reports no disclosures relevant to the manuscript. F. Grand'Maison received honoraria or research funding from Biogen Idec, Genzyme, Novartis, Teva Neurosciences, Mitsubishi, and ONO Pharmaceuticals. P. Grammond is a Novartis, Teva Neuroscience, Biogen Idec, and Genzyme Advisory Board Member; is a consultant for Merck Serono; received payments for lectures from Merck Serono, Teva Neuroscience, and the Canadian MS society; and received grants for travel from Teva Neuroscience and Novartis. E. Pucci served on scientific advisory boards for Genzyme, Merck-Serono, and Biogen-Idec; he received honoraria, congress, and/or travel/accommodation expense compensations from Sanofi Aventis, Novartis, Biogen-Idec, Merck-Serono, Genzyme, Teva, and Associazione Marchigiana Sclerosi Multipla e altre malattie neurologiche. C. Boz received conference travel support from Biogen Idec, Novartis, Bayer-Schering, Merck-Serono, and Teva and has participated in clinical trials by Sanofi Aventis, Roche, and Novartis. P. Sola reports no disclosures relevant to the manuscript. D. Ferraro has received travel grants and/or speaking honoraria from Teva, Merck Serono, Genzyme, Biogen, and Novartis. D. Spitaleri received honoraria as a consultant on scientific advisory boards from Bayer-Schering, Novartis, and Sanofi-Aventis and compensation for travel from Novartis, Biogen Idec, Sanofi Aventis, Teva, and Merck-Serono. J. Lechner-Scott has accepted travel compensation from Novartis, Biogen, and Merck Serono. Her institution receives the honoraria for talks and advisory board commitment and clinic support from Bayer Health Care, Biogen Idec, CSL, Genzyme Sanofi, Merck Serono, and Novartis. M. Terzi received travel grants from Merck Serono, Novartis, Bayer-Schering, Merck-Serono, and Teva and has participated in clinical trials by Sanofi Aventis, Roche, and Novartis. V. Van Pesch has received travel grants from Biogen, Bayer Schering, Genzyme, Merck, Teva, and Novartis Pharma. His institution receives honoraria for consultancy and lectures from Biogen, Bayer Schering, Genzyme, Merck, Roche, Teva, and Novartis Pharma, as well as research grants from Novartis Pharma and Bayer Schering. G. Iuliano had travel/accommodations/meeting expenses funded by Bayer Schering, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis, and Teva. R. Bergamaschi has served on scientific advisory boards for Biogen Idec and Almirall; has received funding for travel and speaker honoraria from Sanofi-Aventis, Genzyme, Biogen Idec, Bayer Schering, Teva Neurosciences, Merck Serono, Almirall, and Novartis; and has received research support from Merck Serono, Biogen Idec, Teva Neurosciences, Bayer Schering, Novartis, Sanofi-Aventis. C. Ramo-Tello reports no disclosures relevant to the manuscript. F. Granella received research funding, consulting/advisory fees, or compensation for travel from Biogen-Idec, Novartis, Genzyme, Almirall, and Roche. C. Oreja-Guevara received honoraria as a consultant on scientific advisory boards from Biogen-Idec, Bayer-Schering, Merck-Serono, Teva, and Novartis and has participated in clinical trials/other research projects by Biogen-Idec, GSK, Teva, and Novartis. H. Butzkueven has served on scientific advisory boards for Biogen Idec, Novartis, and Sanofi-Aventis and has received conference travel support from Novartis, Biogen Idec, and Sanofi Aventis. He serves on steering committees for trials conducted by Biogen Idec and Novartis and has received research support from Merck Serono, Novartis, and Biogen Idec. T. Kalincik served on scientific advisory boards for Roche, Genzyme-Sanofi, Novartis, Merck, and Biogen; has received conference travel support and/or speaker honoraria from WebMD Global, Novartis, Biogen, Genzyme-Sanofi, Teva, BioCSL, and Merck; and has received research support from Biogen. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Miller AE, Rhoades RW. Treatment of relapsing-remitting multiple sclerosis: current approaches and unmet needs. Curr Opin Neurol 2012;25(suppl):S4–S10. [DOI] [PubMed] [Google Scholar]
- 2.Stuve O, Paul F. Progressive multiple sclerosis: desperately seeking remedy. Lancet Neurol 2013;12:840–841. [DOI] [PubMed] [Google Scholar]
- 3.Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002;360:2018–2025. [DOI] [PubMed] [Google Scholar]
- 4.Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis: European Study Group on interferon beta-1b in secondary progressive MS. Lancet 1998;352:1491–1497. [PubMed] [Google Scholar]
- 5.Martinelli Boneschi F, Vacchi L, Rovaris M, Capra R, Comi G. Mitoxantrone for multiple sclerosis. Cochrane Database Syst Rev 2013;5:CD002127. [DOI] [PubMed] [Google Scholar]
- 6.La Mantia L, Vacchi L, Di Pietrantonj C, et al. Interferon beta for secondary progressive multiple sclerosis. Cochrane Database Syst Rev 2012;1:CD005181. [DOI] [PubMed] [Google Scholar]
- 7.ClinicalTrials.gov. A Clinical Study of the Efficacy of Natalizumab on Reducing Disability Progression in Participants With Secondary Progressive Multiple Sclerosis (ASCEND in SPMS). Bethesda. Available at: https://clinicaltrials.gov/ct2/show/NCT01416181. [Google Scholar]
- 8.Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol 2015;14:208–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalincik T, Butzkueven H. Observational data: understanding the real MS world. Mult Scler 2016;22:1642–1648. [DOI] [PubMed] [Google Scholar]
- 10.He A, Spelman T, Jokubaitis V, et al. Comparison of switch to fingolimod or interferon beta/glatiramer acetate in active multiple sclerosis. JAMA Neurol 2015;72:405–413. [DOI] [PubMed] [Google Scholar]
- 11.Kalincik T, Horakova D, Spelman T, et al. Switch to natalizumab vs fingolimod in active relapsing-remitting multiple sclerosis. Ann Neurol 2015;77:425–435. [DOI] [PubMed] [Google Scholar]
- 12.Kalincik T, Jokubaitis V, Izquierdo G, et al. Comparative effectiveness of glatiramer acetate and interferon beta formulations in relapsing-remitting multiple sclerosis. Mult Scler 2015;21:1159–1171. [DOI] [PubMed] [Google Scholar]
- 13.Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain 2016;139:2395–2405. [DOI] [PubMed] [Google Scholar]
- 14.Kalincik T, Kuhle J, Pucci E, et al. Data quality evaluation for observational multiple sclerosis registries. Mult Scler 2017;23:647–655. [DOI] [PubMed] [Google Scholar]
- 15.Kalincik T, Cutter G, Spelman T, et al. Defining reliable disability outcomes in multiple sclerosis. Brain 2015;138:3287–3298. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Po ALW, Blumhardt LD. “Summary measure” statistic for assessing the outcome of treatment trials in relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry 1998;64:726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.R Core Development Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 18.Ho D, Imai K, King G, Stuart EA. MatchIt: nonparametric preprocessing for parametric causal inference. J Stat Softw 2011;42:1–28. [Google Scholar]
- 19.Sormani MP, Giovannoni G. Therapeutic lag: is treatment effect delayed in progressive MS? Mult Scler 2016;22:77. [DOI] [PubMed] [Google Scholar]
- 20.Panitch H, Miller A, Paty D, Weinshenker B. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology 2004;63:1788–1795. [DOI] [PubMed] [Google Scholar]
- 21.Randomized controlled trial of interferon- beta-1a in secondary progressive MS: clinical results. Neurology 2001;56:1496–1504. [DOI] [PubMed] [Google Scholar]
- 22.Cohen JA, Cutter GR, Fischer JS, et al. Benefit of interferon beta-1a on MSFC progression in secondary progressive MS. Neurology 2002;59:679–687. [DOI] [PubMed] [Google Scholar]
- 23.Andersen O, Elovaara I, Farkkila M, et al. Multicentre, randomised, double blind, placebo controlled, phase III study of weekly, low dose, subcutaneous interferon beta-1a in secondary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry 2004;75:706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolinsky J, Narayana P, O'Connor P, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann Neurol 2007;61:14–24. [DOI] [PubMed] [Google Scholar]
- 25.Amato MP, Portaccio E. Clinical outcome measures in multiple sclerosis. J Neurol Sci 2007;259:118–122. [DOI] [PubMed] [Google Scholar]
- 26.Koch MW, Cutter G, Stys PK, Yong VW, Metz LM. Treatment trials in progressive MS: current challenges and future directions. Nat Rev Neurol 2013;9:496–503. [DOI] [PubMed] [Google Scholar]
- 27.Lizak N, Lugaresi A, Alroughani R, et al. Highly active immunomodulatory therapy ameliorates accumulation of disability in moderately advanced and advanced multiple sclerosis. J Neurol Neurosurg Psychiatry 2017;88:196–203. [DOI] [PubMed] [Google Scholar]
- 28.Kappos L, Bar-Or A, Cree B, et al. Efficacy and safety of siponimod in secondary progressive multiple sclerosis: results of the placebo controlled, double-blind, phase III EXPAND study. ECTRIMS Congress, London, September 14–17, 2016:147077. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.