

Abstract
Benzenoids in principle represent attractive and abundant starting materials for the preparation of substituted cyclohexanes; however, the synthetic tools available for overcoming the considerable aromatic energies inherent to these building blocks limit the available product types. In this paper, we demonstrate access to heretofore unknown heterotricyclic structures by leveraging oxidative dearomatization of 2-hydroxymethyl phenols with concurrent N-hydroxycarbamate dehydrogenation using a common oxidant. The pairwise-generated, mutually reactive species then participate in a second stage acylnitroso Diels–Alder cycloaddition. The reaction chemistry of the derived [2.2.2]-oxazabicycles, bearing four orthogonal functional groups and three stereogenic centers, is shown to yield considerable diversity in downstream products. The methodology allows for the expeditious synthesis of a functionalized intermediate bearing structural and stereochemical features in common with the complex alkaloid tetrodotoxin.
The latent functionality embedded within the benzene nucleus offers opportunities to chemists seeking to deploy the parent aromatic core structure as a point of departure for synthesizing chiral, functionalized cyclohexanes. The key challenge thwarting the realization of this potential is the design not only of transformations that overcome the formidable aromatic stabilization energy associated with benzenoids1–7 but also a suitable second stage reaction that takes advantage of the functionality, often of tenuous stability, revealed in the first stage. The work described here uses the cyclohexane substructure of the complex natural product tetrodotoxin (TTX, 1, Scheme 1a) as an inspiration to develop a new oxidative dearomatization cascade sequence for the de novo creation of heterotricyclic compounds presenting four orthogonal functional groups. We demonstrate that considerable product diversity is available from this new platform.
Scheme 1.

Synthetic Strategy for Tetrodotoxin and Proposed Pairwise Oxidation/Cycloaddition Cascade
Tetrodotoxin (1), a potent neurotoxin first isolated from puffer fish, has garnered significant attention since its structure was first elucidated in 1964 due to its unique biological properties and densely functionalized structure (Scheme 1a).8 (−)-TTX inhibits voltage gated sodium ion channels and is consequently a leading molecular probe for further study in this area.9 Additionally, (−)-TTX has been under study as a treatment for chemotherapy-induced neuropathic pain.10 Since the first synthesis of (±)-TTX by Kishi in 1972, there have been five total syntheses, each enabled by the development of creative tactics to access the densely functionalized core.11–19 “Tetrodamine” (2, Scheme 1a) is a common conceptual target in tetrodotoxin syntheses.11–18 The C6–C7–C8–C8a stereotetrad comprises a challenging functional group constellation. The C7/C8 syn diol is suggestive of a cis alkene difunctionalization; direct dihydroxylation of a suitably substituted benzene has been suggested as a potential entry point for tetrodotoxin20 and OsO4-catalyzed alkene functionalization was realized en route to 1.18 Perhaps more nettlesome is the C6/C8a syn-1,4-amino-cyclohexanol where both heteroatoms reside at fully substituted stereogenic centers (Scheme 1a).
In considering retrosynthetic approaches, the presence of a C7/C8 alkene and antithetic reconnection of the C6/C8a amino alcohol (red arrows) would create a retron for an acylnitroso Diels–Alder cycloaddition.21 We envisioned oxidative remodeling of an achiral benzenoid could be triggered by Adler oxidation of the corresponding salicyl alcohol 4.22 Trapping the resulting spiroepoxydienone 6 with a concurrently generated dienophile 5 might trigger a second stage hetero-Diels–Alder cycloaddition (Scheme 1b). An enabling feature of the envisioned strategy is that acyl nitroso 5 and epoxydienone 6 could in principle be prepared by oxidation of N-hydroxycarbamate 323 and salicyl alcohol 4, respectively, with a common oxidant.24 Development of this methodology would allow for the rapid preparation of rigid, bicyclic products with four orthogonal functional groups from simple, aromatic feedstock while potentially allowing for an expeditious, flexible synthesis of the tetrodamine substructure and ultimately (±)-TTX and its structural congeners.25
We began by investigating the proposed acylnitroso Diels–Alder cycloaddition. The known dimerization of spiroepoxydienones was a concern;26 therefore, spiroepoxydienone 7, a compound we observed to be resistant to dimerization, was chosen as a model substrate. Moreover, dienone 7 bears an analogous substitution pattern to an idealized substrate for elaboration to TTX. Treating spiroepoxydienone 7 with N-hydroxycarbamate 8 under Cu(II)-catalyzed aerobic oxidation conditions28 afforded a 1.5:1 mixture of the desired Diels–Alder cycloadduct 9 (5.4:1 dr) and salicyl alcohol 12 (Table 1a, entry 1). Formation of alcohol 12 only occurred in the presence of 8, suggesting the in situ reduction of 7 could not be prevented under these reaction conditions. Stoichiometric quantities of tetra-n-butylammonium periodate (nBu4NIO4) in CH2Cl2 afforded cycloadduct 9 (2.3:1 dr) in low conversion without any observable formation of 12 (entry 2). Repeating the reaction in CDCl3 at 45 °C showed trace conversion to 9, but full consumption of 8 after 1 h. On the basis of these observations, we speculated decomposition of the transient acylnitroso species, which has a lifetime on the order of 1 ms at infinite dilution,23 was likely the source of the poor reactivity.
Table 1.
Optimization of the Sequential and One-Pot Adler Oxidation/Acylnitroso Cycloaddition Protocol
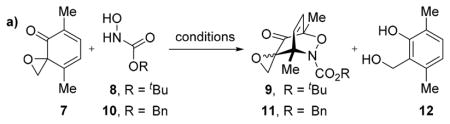
| ||||||
|---|---|---|---|---|---|---|
| entry | R = | oxidant | solvent | T(°C) | conv. of 7 | dr (yield) |
| 1a,b | tBu | CuCl2 L, O2 |
MeOH | rt | 85% (1.5:1 9/12) | 5.3:1 |
| 2c | tBu | nBu4NIO4 | CH2Cl2 | rt-35 | 30% | 2.2:1 |
| 3c,d | tBu | nBu4NIO4 | CDCl3 | 45 | 57% | 3.2:1 |
| 4e | tBu | nBu4NIO4 | CDCl3 | 45 | 58% | 2.1:1 |
| 5e | tBu | nBu4NIO4 | CHCl3f | 45 | 100% (1.4:1 9/12) | 2.3:1 |
| 6g | Bn | nBu4NIO4 | CDCl3 | 45 | 68% | 2.7:1 (12%)h |
| 7g | Bn | nBu4NIO4 | CDCl3 | 30 | 87% | 4.0:1 (51%)h |
| 8g | Bn | nBu4NIO4 | CDCl3 | rt | 91% | 4.3:1 (74%)h |
| |||||
|---|---|---|---|---|---|
| entry | oxidant | solvent | catalyst | conv. of 13 | product |
| 1 | NaIO4 | MeOH/water | — | 100% | 13i |
| 2 | NaIO4 | THF/water | — | 100% | 13i |
| 3 | nBu4NIO4 | MeOH/water | — | 100% | 13i |
| 4 | nBu4NIO4 | THF/water | — | 100% | 13i |
| 5 | nBu4NIO4 | THF | — | 20% | 14/15(10:1) |
| 6 | NaIO4 | CDCl3/water | BnEt3NClj | 100% (60%)k | 14/15(10:1) |
| 7 | NaIO4 | CH2Cl2/water | BnEt3NClj | 100% (76%)k | 14/15(20:1) |
CuCl2 (10 mol %), L = 2-ethyloxazoline (20 mol %).
1.5 equiv 8.
1.5 equiv 8, 1.5 equiv nBu4NIO4.
2.0 additional equiv of nBu4NIO4 and 8 (2 h addition) were added.
2.0 equiv 8, 2.0 equiv nBu4NIO4.
0.75% EtOH as stabilizer.
2.0 equiv 10, 2.0 equiv nBu4NIO4.
Isolated yields.
Initial full conversion to 15 was observed followed by reduction back to 13.
10 mol %.
Isolated yields in parentheses.
Accordingly, we proposed the reactivity could be enhanced by maintaining an excess of spiroepoxydienone 7 relative to the acylnitroso species; therefore, an additional 2 equiv of nBu4NIO4 was added followed by the dropwise addition of 8 (2 equiv) over 2 h, affording cycloadduct 9 (3.2:1 dr) in 57% conversion (entry 3). When the reaction was conducted in CHCl3 (entry 5), phenol 12 was observed in similar quantities relative to the Cu(II)-catalyzed aerobic oxidation conditions (entry 1). The variance in reactivities observed between CHCl3 and its deuterated counterpart are likely the result of reduction of 7 promoted by ethanol, typically present in CHCl3 as a stabilizer. Indeed, a similar effect was observed when methanol was used as the solvent (entry 1). In light of these observations, we avoided these and similar reaction media in subsequent experiments. Further optimization showed increased conversion of 7 using Cbz analog 10, but cycloadduct 11 was isolated in only 12% yield (entries 5, 6). Repeating the reaction at cooler temperatures resulted in increased conversions and isolated yields of 11 (entries 7, 8). The diminished yields at elevated temperatures indicated possible thermal instability of the cycloadducts. Specifically, retro Diels–Alder cycloaddition followed by incomplete recombination and decomposition of the acylnitroso species was hypothesized. This hypothesis was later confirmed via a crossover experiment (vide infra).
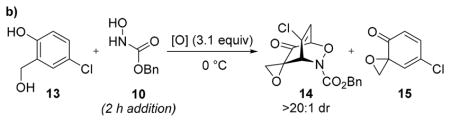
To explore the viability of a one-pot Adler oxidation/acylnitroso cycloaddition sequence, we added a solution of 10 over 2 h to a mixture of salicyl alcohol 13 and NaIO4 in methanol/water (Table 1b, entry 1). Full conversion of phenol 13 to spiroepoxydienone 15 was observed after 15 min, but complete reduction of 15 back to 13 was observed after 1 h. Screening nBu4NIO4 as the oxidant and removing methanol as a solvent returned identical results (entries 2–4). Excluding water from the reaction afforded cycloadduct 14 (>20:1 dr), albeit in low conversion (entry 5). These results indicated that excess water was promoting the Adler oxidation while simultaneously promoting the reduction of intermediate spiroepoxydienone in the presence of the acylnitroso species in water-miscible solvents. These results collectively suggested that biphasic reactions under phase-transfer catalysis conditions might be efficacious (entry 6, 7). Indeed, using CH2Cl2 as the cosolvent afforded 14 in excellent yield with complete consumption of 13 (entry 7).
We next probed the scope of the corresponding one-pot procedure (Table 2). Electron-rich and electron-poor salicyl alcohols29–31 were tolerated, affording the derived cycloadducts in good yield. For most substrates, the cycloadduct is initially formed with >20:1 dr; however, cycloadducts 11 and 19 were isolated as a mixture of equilibrating diastereomers (determined by chemical derivatization and selective 1D NOESY NMR analysis; see the Supporting Information (SI) for details).
Table 2.
Scope of the One-Pot Oxidative Dearomatization/Acylnitroso Cycloaddition Cascade

|
Equilibrated dr.
Thermal isomerization occurs ≥40 °C.
Isomerization is observed after standing at rt (see SI).
An X-ray diffraction study of bicyclic dihydrooxazine 9 revealed a regiochemical preference for the illustrated isomer, which matched that needed for extrapolation to TTX,27 and acylnitroso approach from the methylene face of the epoxide of 7. The observed facial selectivity was surprising as C═C dienophile approach from the oxygen face of spiroepoxydienones is well documented.26 By contrast, an X-ray diffraction study of dihydrooxazine 14 revealed acylnitroso approach from the oxygen face of the epoxide (see SI). We hypothesize acylnitroso approach from the oxygen face of the epoxide is kinetically favored whereas the “methylene endo” product is the more thermodynamically stable diastereomer. This hypothesis is additionally supported by the diminished pre-equilibration diastereoselectivities observed for cycloadducts 11 and 19 (see SI), the slow isomerization of cycloadduct 22 at rt, and the thermally promoted isomerization of cycloadduct 16 (40–65 °C for 50 h). A crossover experiment between 19 and Boc–NO confirmed a retro-Diels–Alder/recombination sequence, providing a likely pathway for isomerization of the dihydroxazine cycloadducts (see the SI for details of the crossover experiment and thermal isomerization).
The reactivity of cycloadduct 16 was explored in a number of orthogonal transformations that independently probed each functional group (Scheme 2). Reduction of the ketone afforded alcohol 24 in good yield while ring-opening of the epoxide was achieved by treating ketone 16 with bromodimethylsulfonium bromide,32 yielding bromohydrin 25. Gram-scale dihydroxylation of 16 gave diol 26 with high diastereoselection and matched the stereochemistry needed for elaboration toward tetrodamine 2. Subsequent one-pot oxazinane reduction33 and epoxide ring-opening34 afforded cyclohexanone 27 in good yield. Oxidative rearrangement of 16 provided fused lactone 28 in 50% yield.35 Reductive fragmentation36 of epoxyoxime 29 afforded primary alcohol 30 and thence the crystalline bis(p-nitrobenzoate) 31. Corey–Chaykovsky epoxidation37 afforded bis-epoxide 32 (relative stereochemistry assigned by NOESY analysis, see SI).
Scheme 2. Chemoselective Reactions of Heterocycloadducts*.

*Conditions: aNaBH4 (0.33 equiv), THF, 0 °C; bBr2 (2.0 equiv), Me2S (2.5 equiv), CH2Cl2, 0 °C to rt; cOsO4 (1 mol %), NMO (6.0 equiv), THF/acetone/H2O (5:5:1), 0 °C to rt; di. Pd/C (10 wt %), AcOH (5.0 equiv), MeOH, rt, ii. HCl in 1,4-dioxane (4 N, 25.0 equiv), rt; emCPBA (1.5 equiv), NaHCO3 (2.0 equiv) CH2Cl2, rt; fHONH2· HCl (1.5 equiv), NaOAc (2.0 equiv), THF, rt; gNaBH4 (2.0 equiv), anhydrous MeOH, 0 °C; hp-O2NC6H4COCl (2.2 equiv), TEA (3 equiv), DMAP (10 mol %), anhydrous THF, rt; iNaH (60 wt %, 1.1 equiv), Me3SO·I (1.1 equiv), anhydrous THF, 0 °C.
Potential extrapolation of this new oxidative dearomatization/cycloaddition cascade reaction to the synthesis of (±)-TTX would require deployment of a more functionalized phenolic starting material. To that end, we prepared salicyl alcohol 33 in five steps from commercial 2,3,6-trimethyl phenol. Without interference from the distal benzylic alcohol, the desired cycloadduct 34 was prepared with excellent facial selectivity through the action of both the sequential and one-pot Adler oxidation/ANDA procedures on salicyl alcohol 33 (Scheme 3). The relative stereochemistry of 34 was confirmed via X-ray crystallographic analysis (see SI). Subsequent dihydroxylation proceeded on a 3 g scale with excellent diastereoselectivity to afford diol 35, which underwent reductive N–O cleavage on gram scale to yield lactol 36.33 The favorable display of functionality in the latter structure vis-à-vis that required for tetrodamine (2) provides flexibility in exploring reactions that could ultimately facilitate an efficient synthesis of 1 and its congeners.
Scheme 3. Application of the Oxidation/Cycloaddition Cascade Procedure towards the TTX Core Structure*.

*Conditions: ai. NaIO4, MeOH/H2O (2:1), ii. BnO2CNHOH (2 equiv, 2 h addition), BnEt3NCl (20 mol %), NaIO4 (2.1 equiv), CH2Cl2/H2O (1.3:1), 0 °C; bNaIO4 (5.1 equiv), BnEt3NCl (20 mol %), CH2Cl2/H2O (1.3:1), rt then BnO2CNHOH (4 equiv, 3 h addition), 0 °C; cOsO4 (1 mol %), NMO (5.0 equiv), THF/acetone/H2O (5:5:1), rt; dH2, Pd/C (10 wt %), AcOH (5 equiv), MeOH/THF (3:2).
In summary, we have developed a one-pot oxidative dearomatization/acylnitroso Diels–Alder cycloaddition cascade sequence for the rapid preparation of rigid [2.2.2]-bicyclic products bearing four orthogonal functional groups from simple, aromatic feedstocks. The synthetic utility of these new cycloadducts was demonstrated through an array of chemoselective transformations. Additionally, we have applied this methodology toward the synthesis of an advanced intermediate that may have relevance to a projected synthesis of tetrodotoxin.
Supplementary Material
Acknowledgments
The project described was supported by Award R35 GM118055 from the National Institute of General Medical Sciences. R.J.S. acknowledges an NSF Graduate Research Fellowship. X-ray crystallography was performed by Dr. Peter White and Blane Zavesky (UNC). We thank Dr. Roger Sommer (North Carolina State University) for helpful discussions regarding the X-ray structures.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07745.
Experimental procedures, characterization, spectral data (PDF)
Crystallographic data for 9 (CIF)
Crystallographic data for 14 (CIF)
Crystallographic data for 31 (CIF)
Crystallographic data for 34 (CIF)
Crystallographic data for 35 (CIF)
References
- 1.(a) Magdziak D, Meek SJ, Pettus TRR. Chem Rev. 2004;104:1383. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]; (b) Pouységu L, Deffieux D, Quideau S. Tetrahedron. 2010;66:2235. [Google Scholar]
- 2.Rabideau PW, Marcinow Z. Org React. 2004;42:1. [Google Scholar]
- 3.(a) Pape AR, Kaliappan KP, Kündig EP. Chem Rev. 2000;100:2917. doi: 10.1021/cr9902852. [DOI] [PubMed] [Google Scholar]; (b) Chordia MD, Harman WD. J Am Chem Soc. 2000;122:2725. [Google Scholar]
- 4.Hudlicky T, Reed JW. Synlett. 2009;2009:685. [Google Scholar]
- 5.Uyanik M, Yasui T, Ishihara K. Angew Chem, Int Ed. 2013;52:9215. doi: 10.1002/anie.201303559. [DOI] [PubMed] [Google Scholar]
- 6.Uyanik M, Sasakura N, Mizuno M, Ishihara K. ACS Catal. 2017;7:872. [Google Scholar]
- 7.Roche SP, Porco JA. Angew Chem, Int Ed. 2011;50:4068. doi: 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodward RB. Pure Appl Chem. 1964;9:49. [Google Scholar]
- 9.Moczydlowski EG. Toxicon. 2013;63:165. doi: 10.1016/j.toxicon.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 10.Nieto FR, Cobos EJ, Tejada MÁ, Sánchez-Fernández C, González-Cano R, Cendán CM. Mar Drugs. 2012;10:281. doi: 10.3390/md10020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kishi Y, Nakatsubo F, Aratani M, Goto T, Inoue S, Kakoi H, Sugiura S. Tetrahedron Lett. 1970;11:5127. doi: 10.1016/s0040-4039(00)96956-9. [DOI] [PubMed] [Google Scholar]
- 12.Kishi Y, Nakatsubo F, Aratani M, Goto T, Inoue S, Kakoi H. Tetrahedron Lett. 1970;11:5129. doi: 10.1016/s0040-4039(00)96957-0. [DOI] [PubMed] [Google Scholar]
- 13.Kishi Y, Aratani M, Fukuyama T, Nakatsubo F, Goto T, Inoue S, Tanino H, Sugiura S, Kakoi H. J Am Chem Soc. 1972;94:9217. doi: 10.1021/ja00781a038. [DOI] [PubMed] [Google Scholar]
- 14.Kishi Y, Fukuyama T, Aratani M, Nakatsubo F, Goto T, Inoue S, Tanino H, Sugiura S, Kakoi H. J Am Chem Soc. 1972;94:9219. doi: 10.1021/ja00781a039. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ohyabu N, Nishikawa T, Isobe M. J Am Chem Soc. 2003;125:8798. doi: 10.1021/ja0342998. [DOI] [PubMed] [Google Scholar]; (b) Nishikawa T, Urabe D, Isobe M. Angew Chem, Int Ed. 2004;43:4782. doi: 10.1002/anie.200460293. [DOI] [PubMed] [Google Scholar]
- 16.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- 17.Akai S, Seki H, Sugita N, Kogure T, Nishizawa N, Suzuki K, Nakamura Y, Kajihara Y, Yoshimura J, Sato K-i. Bull Chem Soc Jpn. 2010;83:279. [Google Scholar]
- 18.Maehara T, Motoyama K, Toma T, Yokoshima S, Fukuyama T. Angew Chem, Int Ed. 2017;56:1549. doi: 10.1002/anie.201611574. [DOI] [PubMed] [Google Scholar]
- 19.(a) Koert U. Angew Chem, Int Ed. 2004;43:5572. doi: 10.1002/anie.200461097. [DOI] [PubMed] [Google Scholar]; (b) Chau J, Ciufolini MA. Mar Drugs. 2011;9:2046. doi: 10.3390/md9102046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mendelsohn BA, Ciufolini MA. Org Lett. 2009;11:4736. doi: 10.1021/ol901914u. [DOI] [PubMed] [Google Scholar]
- 20.Trant JF, Froese J, Hudlicky T. Tetrahedron: Asymmetry. 2013;24:184. [Google Scholar]
- 21.(a) Bodnar BS, Miller MJ. Angew Chem, Int Ed. 2011;50:5630. doi: 10.1002/anie.201005764. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto Y, Yamamoto H. Eur J Org Chem. 2006;2006:2031. [Google Scholar]; (c) Lu PH, Yang CS, Devendar B, Liao CC. Org Lett. 2010;12:2642. doi: 10.1021/ol100840n. [DOI] [PubMed] [Google Scholar]
- 22.(a) Adler E, Brasen S, Miyake H. Acta Chem Scand. 1971;25:2055. [Google Scholar]; (b) Becker HD, Bremholt T, Adler E. Tetrahedron Lett. 1972;13:4205. [Google Scholar]
- 23.(a) Cohen AD, Zeng BB, King SB, Toscano JP. J Am Chem Soc. 2003;125:1444. doi: 10.1021/ja028978e. [DOI] [PubMed] [Google Scholar]; (b) Kirby GW. Chem Soc Rev. 1977;6:1. [Google Scholar]
- 24.Wee AG, Slobodian J, Fernández-Rodríguez MA, Aguilar E. e-EROS Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2006. Sodium Periodate. [Online] http://onlinelibrary.wiley.com/doi/10.1002/047084289X.rs095.pub2/full. [Google Scholar]
- 25.Kao CY, Yeoh PN, Goldfinger MD, Fuhrman FA, Mosher HS. J Pharmacol Exp Ther. 1981;217:416. [PubMed] [Google Scholar]
- 26.Singh V. Acc Chem Res. 1999;32:324. [Google Scholar]
- 27.CCDC 1549797 (9), CCDC 1548692 (14), CCDC 1478943 (31), CCDC 1548690 (34) and CCDC 1548691 (35) contain the supplementary crystallographic data for this paper. This data can be obtained free of charge from the Cambridge Crystallographic Date Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 28.(a) Chaiyaveij D, Cleary L, Batsanov AS, Marder TB, Shea KJ, Whiting A. Org Lett. 2011;13:3442. doi: 10.1021/ol201188d. [DOI] [PubMed] [Google Scholar]; (b) Frazier CP, Bugarin A, Engelking JR, Read de Alaniz J. Org Lett. 2012;14:3620. doi: 10.1021/ol301414k. [DOI] [PubMed] [Google Scholar]
- 29.Li HJ, Wu YY, Wu QX, Wang R, Dai CY, Shen ZL, Xie CL, Wu YC. Org Biomol Chem. 2014;12:3100. doi: 10.1039/c4ob00228h. [DOI] [PubMed] [Google Scholar]
- 30.Belyanin ML, Filimonov VD, Krasnov EA. Russ J Appl Chem. 2001;74:103. [Google Scholar]
- 31.(a) Hansen TV, Skattebøl L. Org Synth. 2005;82:64. [Google Scholar]; (b) Yang D, Wong MK, Cheung KK, Chan EWC, Xie Y. Tetrahedron Lett. 1997;38:6865. [Google Scholar]
- 32.Das B, Krishnaiah M, Venkateswarlu K. Tetrahedron Lett. 2006;47:4457. [Google Scholar]
- 33.Bollans L, Bacsa J, Iggo JA, Morris GA, Stachulski AV. Org Biomol Chem. 2009;7:4531. doi: 10.1039/b912963d. [DOI] [PubMed] [Google Scholar]
- 34.Singh V, Praveena GD, Karki K, Mobin SM. J Org Chem. 2007;72:2058. doi: 10.1021/jo062416m. [DOI] [PubMed] [Google Scholar]
- 35.Meinwald J, Frauenglass E. J Am Chem Soc. 1960;82:5235. [Google Scholar]
- 36.Dresler R, Uzarewicz A. Polish J Chem. 2000;74:1581. [Google Scholar]
- 37.Corey EJ, Chaykovsky M. J Am Chem Soc. 1965;87:1353. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.