

Abstract
The characterization of RNA–protein complexes (RNPs) is a difficult but increasingly important problem in modern biology. By combining the compact RNA Mango aptamer with a fluorogenic thiazole orange desthiobiotin (TO1-Dtb or TO3-Dtb) ligand, we have created an RNA tagging system that simplifies the purification and subsequent characterization of endogenous RNPs. Mango-tagged RNP complexes can be immobilized on a streptavidin solid support and recovered in their native state by the addition of free biotin. Furthermore, Mango-based RNP purification can be adapted to different scales of RNP isolation ranging from pull-down assays to the isolation of large amounts of biochemically defined cellular RNPs. We have incorporated the Mango aptamer into the S. cerevisiae U1 small nuclear RNA (snRNA), shown that the Mango-snRNA is functional in cells, and used the aptamer to pull down a U1 snRNA-associated protein. To demonstrate large-scale isolation of RNPs, we purified and characterized bacterial RNA polymerase holoenzyme (HE) in complex with a Mango-containing 6S RNA. We were able to use the combination of a red-shifted TO3-Dtb ligand and eGFP-tagged HE to follow the binding and release of the 6S RNA by two-color native gel analysis as well as by single-molecule fluorescence cross-correlation spectroscopy. Together these experiments demonstrate how the Mango aptamer in conjunction with simple derivatives of its flurophore ligands enables the purification and characterization of endogenous cellular RNPs in vitro.
Keywords: RNA, Mango, fluorophore, desthiobiotin, purification, RNP pull-down, TO1, TO3
INTRODUCTION
RNPs play essential roles in gene expression and regulation in all domains of life (Wang and Chang 2011; Cech and Steitz 2014), yet many RNPs remain poorly characterized due to the challenges inherent in their purification and subsequent biochemical characterization. While protein-based tags can be used for both purification (Lichty et al. 2005) and fluorescent labeling of RNPs (Cranfill et al. 2016), few options exist that simultaneously facilitate affinity-based native purification and fluorescent labeling of the RNA components of an RNP complex (Panchapakesan et al. 2015). Small RNA aptamer tags that are easy to insert into an RNA of interest and that bind to derivatizable fluorophore tags offer a potential solution to this problem. To be effective, such an approach requires high-affinity aptamers able to bind their fluorophore ligands so that the fluorescent aptamer/fluorophore complex can also serve as a purification handle. A key problem in this respect has been the difficulty in isolating aptamer fluorophore–ligand pairs that have sufficiently high binding affinity and brightness to serve as a dual use RNA tag.
The RNA Mango aptamer binds with nanomolar affinity to thiazole orange (TO) derivatives such as TO1-Biotin and can increase their fluorescence by ∼1000-fold (Dolgosheina et al. 2014). Therefore, it appeared to be an ideal aptamer/fluorophore system for native RNP purification and fluorescent characterization. Our interest in this system was increased by the recent finding that the Mango aptamer consists of a compact 19-nt fluorophore binding G-quadruplex domain that is physically connected to an arbitrary stem via a novel GAA^A tetraloop-like motif ([^] indicates fluorophore quadruplex binding domain insertion site, see Fig. 1A and Trachman et al. 2017). Since many biological RNAs contain GNRA tetraloops that are isosteric with a GAAA loop (Uhlenbeck 1990), we reasoned that the Mango tag might be biologically well-tolerated if it was used to replace preexisting stem–loops whose sequences or structures are not biologically essential.
FIGURE 1.

Native purification of RNP complexes using the fluorescent, high-affinity Mango tool set. (A) RNA Mango aptamer core quadruplex and the GAA^A tetraloop-like connecting motif sequence (in brown, [^] indicates site of core quadruplex insertion). N and N′ are complementary base pairs of arbitrary sequence forming a RNA duplex (Trachman et al. 2017). (B) Structures of Dtb-derivatized TO1 (n = 1) or TO3 (n = 3) fluorophores. (C) General schematic for purification of native RNP complexes using Mango.
To demonstrate the potential of the Mango system to enrich and fluorescently characterize native RNP complexes from both eukaryotic and prokaryotic cell lysates, we synthesized two derivatives of TO1-Biotin, TO1-Dtb, and the significantly red-shifted TO3-Dtb (Fig. 1B) and used these ligands to analyze RNPs at different purification scales. Insertion of a single Mango tag into stem–loop VII of the S. cerevisiae (yeast) U1 small nuclear RNA (snRNA) maintained yeast viability and enabled the single step pull-down of the U1 snRNP using TO1-Dtb under native conditions. Tagging a release-defective form of the bacterial 6S RNA (Oviedo Ovando et al. 2014) with Mango (6SRDM) allowed the purification of the 6SRDM:RNA polymerase complex in two steps: the second of which involved the use of TO1-Dtb fluorescence to track RNP elution. Using Mango-tagged 6S RNA, eGFP-tagged RNA polymerase, and TO3-Dtb, we could track the binding and release of the 6S RNA from the RNP (Wassarman and Storz 2000; Wassarman and Saecker 2006; Panchapakesan and Unrau 2012) by an electrophoretic mobility shift assay (EMSA). Finally, the tight binding of TO3-Dtb to the Mango-tagged 6S RNA also enables single-molecule fluorescent cross-correlation spectroscopy of this complex. Thus in both eukaryotic and prokaryotic contexts we demonstrate the utility of Mango for pull-down, purification, and biophysical characterization of RNPs.
RESULTS AND DISCUSSION
Mango tag insertion into yeast U1 snRNA maintains biological viability
To study the utility of RNA Mango for eukaryotic RNP pull-down experiments, we inserted the Mango motif into the stem–loop VII of the 568-nt Saccharomyces cerevisiae (yeast) U1 sRNA (U1M). The U1 snRNA is the highly conserved RNA component of the U1 snRNP that initially recognizes 5′ splice sites of introns during spliceosome assembly (Seraphin and Rosbash 1989) and is essential for eukaryotic cell growth. This U1M gene, which replaces the 5-nt loop of the U1 stem–loop VII with the Mango GAAA tetraloop-like adapter and core sequence (Fig. 1A; Supplemental Fig. 1), also replaced the wild-type (WT) U1 snRNA allele in a U1 deletion (snr19Δ strain) and was able to maintain yeast viability. The presence of the U1M gene and the absence of the WT allele was confirmed by PCR and DNA sequencing (Supplemental Fig. 2). The survival of U1M yeast construct demonstrates that Mango can be incorporated into a noncoding RNA essential for eukaryotic cell proliferation while maintaining biological function.
RNP pull-down using Mango and TO1-Dtb
In order to enable native purification and characterization of RNPs, we synthesized TO1-Desthiobiotin (TO1-Dtb, ex/em: 260, 510/535 nm) and the significantly red-shifted TO3-Desthiobiotin (TO3-Dtb, ex/em: 260, 637/658 nm). Desthiobiotin, having fivefold less affinity to streptavidin than biotin (Magalhães et al. 2011), can be displaced from streptavidin by the addition of free biotin (Hirsch et al. 2002). TO1-Dtb (Trachman et al. 2017) and TO3-Dtb bind to the Mango tag with nanomolar affinity (Supplemental Fig. 3), similar to what has been reported previously for other TO1 derivatives (Dolgosheina et al. 2014). Using the TO1-Dtb ligand, we developed a protocol for the native purification of RNP complexes using the Mango tag (Fig. 1C). Native extract containing the Mango-tagged RNP is first bound to a TO1-Dtb saturated streptavidin solid support. Binding can be performed either at RT or at 4°C, depending upon the stability of the RNA:protein complex. The solid support is then washed to remove nonspecific components of the extract with the stringency of washing being dependent upon the stability of the complex under study. After washing, the complexes are eluted by the addition of excess free biotin (see Materials and Methods).
We used Mango together with TO1-Dtb to pull down the U1 snRNP complex in nondenaturing conditions. The U1M snRNA was significantly enriched as judged by in vitro solution hybridization (Fig. 2A) and primer extension (Supplemental Fig. 4). A dual-labeled yeast strain containing both U1M and a fast SNAP-tagged U1 snRNP protein (Snp1-SNAPf) was also prepared in order to easily detect copurification of snRNA and protein components of the U1 snRNP. As expected, the U1M snRNA was enriched only in Mango-tagged strains while another noncoding snRNA (U2) was not, indicating that enrichment was specific for the Mango tag (Fig. 2B). The Snp1-SNAPf tag could only be detected in extract prepared from cells containing both the U1M and Snp1-SNAPf, again suggesting the specific enrichment of the U1 snRNP complex (Fig. 2C). Thus, not only is U1M viable in yeast, but the Mango aptamer/TO1-Dtb system facilitates the pull-down of the U1 snRNP complex from yeast cell extract. It is worthwhile to note that the Mango system enables U1M pull-down of an RNP present in only a few hundred molecules per cell and at nanomolar concentrations in whole-cell extract (Riedel et al. 1986), suggesting that Mango can be used to pull down and characterize low-abundance RNPs.
FIGURE 2.

Native purification of yeast U1 snRNP complex using RNA Mango. (A) Solution hybridization analysis of snRNAs present in native extract (NE) from U1M tagged yeast and following Mango batch purification using TO1-Dtb (Biotin Eluate). The probed snRNAs are labeled on the right and correspond to the number labels above each lane. U1M migrates as a doublet following RNA deproteinization and denaturation, but as a single band upon Mango-based purification. (B) Primer extension analysis to detect U1M or U2 snRNAs following TO1-Dtb bead-based purification. (C) SDS-PAGE analysis of Snp1-SNAPf following Mango purification.
Two-step Mango tag enrichment of the 6SRDM:HE RNP complex
While the high affinity of the Mango aptamer to its TO1-Dtb ligand facilitates pull-down type experiments, we wondered whether the intrinsic fluorescent properties of the biotin-eluted RNP complex could be exploited for larger-scale RNP purification. We tagged a release-defective mutant of the 6S RNA (R9-33) (Oviedo Ovando et al. 2014) with Mango (6SRDM) (Supplemental Fig. 5) and overexpressed 6SRDM in E. coli cells. Cell lysate from the 6SRDM expressing bacteria was then incubated with TO1-Dtb derivatized streptavidin agarose beads as before, washed, and the desthiobiotin-bound material eluted by addition of free biotin. Analysis of the recovered RNA fraction showed considerable enrichment in an RNA band of the expected size (Supplemental Fig. 6). LC–MS/MS analysis revealed that this single-step purification yielded the four core RNA polymerase proteins as the highest ranked polypeptides, consistent with enrichment of intact HE (Table 1; Supplemental Table 1). As this biotin-eluted RNP complex should still be complexed to TO1-Dtb, we reasoned that the Mango-based fluorescence of this complex could be used to follow the further purification of the complex by size-exclusion chromatography (SEC). Three TO1-Dtb dependent fluorescent peaks were observed by SEC (Fig. 3). Loading in vitro synthesized 6SRDM and free TO1-Dtb controls into the column revealed that the second and third peaks corresponded to 6SRDM:TO1-Dtb and free TO1-Dtb, while the earliest-eluting peak was consistent with the 6SRDM:HE RNP complex. MS analysis of this peak yielded all five protein components of the HE as high-ranking hits (Table 1; Supplemental Table 2). Thus, by using the binding and fluorescent properties of TO1-Dtb, we could rapidly enrich in bacterial RNA polymerase using the Mango-tagged 6S regulatory RNA.
TABLE 1.
Mass spectrometry analysis of the proteins enriched by 6SRDM-based purification

FIGURE 3.
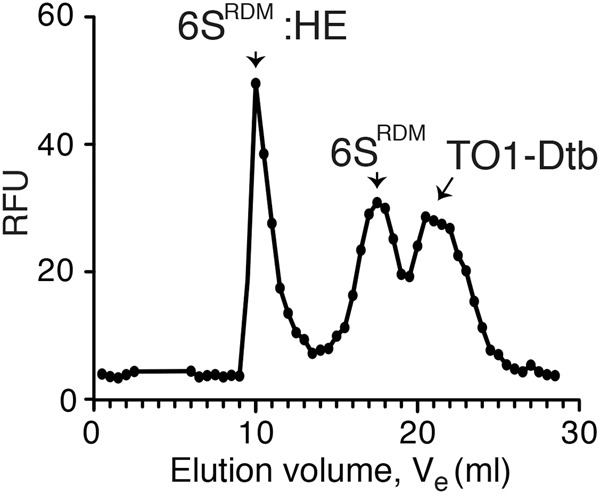
SEC purification of 6SM:HE complex from E. coli cells. The SEC elution profile represents the RNA Mango:TO1-Dtb fluorescence observed in each fraction. The 6SRDM:HE peak fraction corresponds to the sample analyzed by LC–MS/MS in Table 1.
Unexpectedly, two additional proteins were significantly enriched together with the core components of HE: GroEL and thioredoxin (Table 1). Throughout the purification of the 6SRDM:HE complex we were able to detect GroEL by LC–MS/MS, suggesting that a significant fraction of the 6SRDM:HE RNP complex may be associated with GroEL. We also found that thioredoxin was significantly enriched after SEC, suggesting that it may also make stable interactions with the 6SRDM:HE complex. To explore whether this was potentially an artifactual result of the Mango-based purification, we compared our results with an equivalent LC–MS/MS analysis of RNAP obtained from a commercial source. The GroEL and thioredoxin proteins were also present as high scoring MS hits in the commercial RNAP (Table 1; Supplemental Table 3), supporting the idea that they copurify with RNAP even in the absence of the 6S RNA.
Two-color fluorescence gel-shift analysis of 6S RNP assembly and disassembly using Mango
Next, we tested the functionality of the Mango system for multiwavelength fluorescence imaging. We reasoned that since the Mango:TO3-Dtb complex (Ex/Em 637/658 nm, Fig. 1B) and enhanced GFP (eGFP, Ex/Em: 488/509 nm) are spectrally distinct, this pair could be used to dual-label RNP complexes. We combined the TO3-Dtb ligand with a wild-type 6S RNA tagged with RNA Mango (6SM) (Dolgosheina et al. 2014) and prepared partially enriched HE tagged on the β′ subunit with eGFP (eGFP-HE) (Bratton et al. 2011). We mixed eGFP-HE with the 6SM:TO3-Dtb complex and analyzed the assembly and pRNA-dependent release of 6SM from the HE complex by native gel-shift analysis (Wassarman and Saecker 2006). As little as 10 pmol of the 6SM:HE complex could be clearly detected by dual-color fluorescence imaging. Upon the addition of partially purified eGFP-HE, 6SM formed a single, dual-labeled RNP complex (Fig. 4A). As expected, the addition of NTPs and MgCl2 triggered release of 6SM RNA from eGFP-HE and produced a faster mobility, fluorescent 6SM:pRNA band. As the release of the 6S RNA is rapid (∼30 sec) and still only partially understood (Panchapakesan et al. 2015), the ability to dual label the 6S RNA:HE complex promises to be beneficial for the further characterization of the 6S RNA release process.
FIGURE 4.
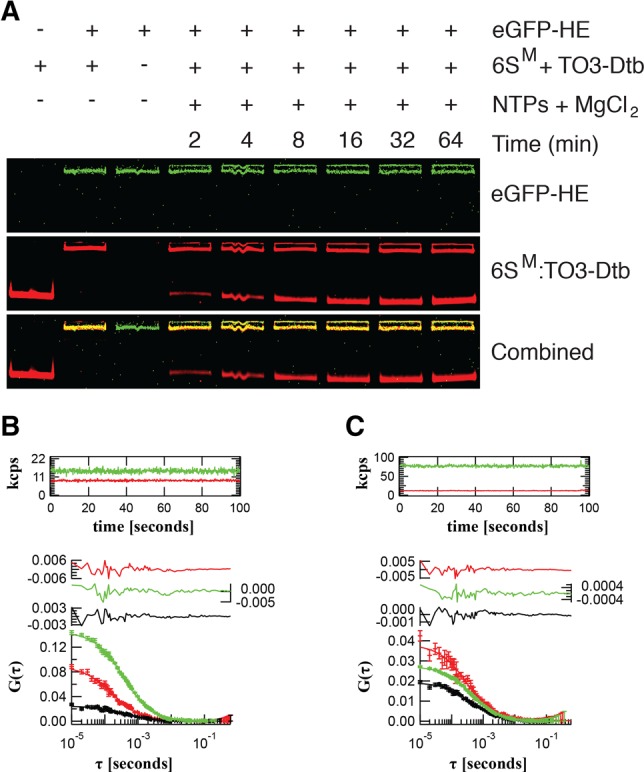
Multiwavelength fluorescence characterization of the 6SM:HE complex. (A) EMSA assay of the 6SM:eGFP-HE complex visualized using eGFP-HE (green, top) and 6SM:TO3-Dtb (red, middle) fluorescence. The bottom panel is the composite of the green and red images. Product RNA (pRNA) release was induced by the addition of NTPs and MgCl2. (B) Fluorescence auto- and cross-correlation spectroscopy of 6SM:TO3-Dtb (red) in the presence of 25.4 ± 0.3 nM free eGFP-HE (green) and 1% 6SM:eGFP-HE complex (yellow, cross-correlation amplitude). (C) 6SM:TO3-Dtb (red) in the presence of 127.5 ± 0.9 nM (right) free eGFP-HE (green) and 20% 6SM:eGFP-HE complex (yellow, cross-correlation amplitude). Fluorescence intensity traces in thousands of photon counts per second (kcps, both red and green channels) are shown in the top panels. Residuals are shown in the middle panels. Correlation functions are shown in the bottom panels.
Single-molecule fluorescence cross-correlation spectroscopy analysis of the 6S RNP
Finally, we set out to test the effectiveness of Mango for single-molecule analysis of dual-labeled RNPs. Fluorescence cross-correlation spectroscopy (FCCS) is a single-molecule approach that can be used to study biomolecular interactions in real-time (Bacia and Schwille 2007). Further, this can be combined with two-photon excitation to minimize photobleaching (Zipfel et al. 2003). We carried out a two-photon FCCS analysis of RNPs containing in vitro synthesized 6SM bound to the TO3-Dtb ligand and reconstituted with partially purified eGFP-HE. Analysis of the two-photon fluorescence excitation spectrum for 6SM:TO3-Dtb identified a peak near ∼840 nm (Supplemental Fig. 7). At this two-photon wavelength, we were able to determine the diffusion coefficient and molecular brightness of 6SM:TO3-Dtb (D6SM = 140 ± 8 µm2 sec−1) and eGFP-HE (DHE = 58 ± 2 µm2 sec−1) in isolation (Supplemental Fig. 8; Supplemental Table 5). Next, we added increasing amounts of eGFP-HE (green channel) solution to the 6SM:TO3 (red channel) solution. As expected, we observed cross-correlation (Kim et al. 2005) between the fluorescent signals in the red and green channels upon the addition of holoenzyme (Fig. 4B,C), indicating the detection of single molecular complexes containing both the eGFP and TO3 fluorophores. Further, the abundance of these dual-labeled complexes, likely representing 6SM:HE complex, increased in proportion to the amount of eGFP-HE added (see Materials and Methods and Supplemental Information for further details). These results demonstrate the ability of the RNA Mango fluorescent system to be used as a fluorescent partner with existing fluorescent proteins to perform two-photon FCCS.
Conclusions
We have developed a multifunctional Mango fluorophore system for the purification and biochemical analysis of cellular RNA and RNPs. The variety of applications demonstrated here are made possible due to the unique combination of the high affinity between TO derivatives and the Mango aptamer, the large fluorescence enhancement in TO that occurs upon binding to RNA Mango, and the small size and biological compatibility of the aptamer. It is likely that many RNAs in addition to the U1 snRNA and the 6S RNA studied here contain modifiable stem–loop structures or will permit insertion of RNA Mango-containing stem–loop sequences. As comprehensive libraries of eGFP-tagged proteins already exist for a range of organisms (Huh et al. 2003; Kitagawa et al. 2006; Buszczak et al. 2007), the Mango system should allow the rapid and systematic study of RNPs via the utilization of RNA-based pull-downs and RNA tagged, two-color fluorescent analysis.
MATERIALS AND METHODS
Synthesis of desthiobiotin derivatives of thiazole orange acetate
TO1 PEG4 desthiobiotin (TO1-Dtb) and TO3 PEG4 desthiobiotin (TO3-Dtb) were synthesized using TO1-acetate and TO3-acetate, respectively, together with EZ link amine PEG4 desthiobiotin (Thermo Fisher) as precursors following an established protocol (Dolgosheina et al. 2014; Trachman et al. 2017). Product identity was confirmed by electrospray ionization–mass spectrometry in the positive mode. Expected mass (in Da) for TO1-Dtb: C40H55N6O7S+: 763.3847, obtained mass: 763.3855, and expected mass for TO3-Dtb: C42H57N6O7S+: 789.4003, obtained mass: 789.3999.
Preparation of TO1-Dtb derivatized streptavidin agarose affinity resin
In all experiments, streptavidin agarose beads (Invitrogen) were used for batch purification in either 1.7 mL or 5 mL tubes, as required, and solutions containing the beads were mixed using a rotator. The beads (400 µL) were washed twice with 0.1 M NaOH and 0.05 M NaCl (Buffer A) as per the manufacturer's protocol and three times with 15 mM HEPES pH 7.5, 90 mM KCl (Buffer B). TO1-Dtb was added (22.5 nmol) in 400 µL Buffer B and incubated with the washed beads for 15 min at RT. The beads were then washed once with 400 µL Buffer B to remove any unbound dye.
Yeast strain and extract preparation
The yeast U1M strain was prepared from the BJ2168 derivative yAAH0441 (Mat a, prc1-407, prb1-1122, pep4-3, leu2, trp1, ura3-52, his3::loxP gal2 snr19::loxP [pMKU1-7 URA CEN]), a kind gift of Dr. Magda Konarska. The U1M gene along with 500 bp of upstream and 723 bp of downstream genomic DNA sequence was constructed by Genewiz, subcloned into the BamHI and NotI sites of the pRS413 plasmid (HIS3 CEN6), and sequenced. The U1M plasmid was then transformed into yAAH0441. Transformants were selected on dropout media, and single colonies were streaked onto medium containing 5-fluoroorotic acid (5-FOA, 1 mg/mL) to select for loss of the WT U1 URA3-marked plasmid. The presence of the U1-Mango gene and the absence of the WT allele in the resulting strain (yAAH1204) were confirmed by using PCR to amplify the U1 gene and DNA sequencing (Supplemental Fig. 2). A double-tagged strain containing both U1M and a SNAP-tagged Snp1 protein (yAAH1362) was created by homologous recombination to insert the SNAPf gene and a downstream hygromycin selection marker as previously described (Hoskins et al. 2011). Transformants were confirmed by PCR and by labeling of Snp1-SNAPf with fluorescent SNAP ligands. Yeast whole-cell extracts were prepared from strains yAAH1204 and yAAH1362 as previously described using the liquid N2 method and a ball mill (Hoskins et al. 2011).
SNAP labeling of yeast whole-cell extract
Yeast whole-cell extracts (500 µL) were first fluorescently tagged with the SNAP-Surface 549 tag (NEB) by incubating the extracts with 1 U/µL Murine RNase inhibitor (NEB), Protease Inhibitors (cOmplete mini at the recommended concentration, EDTA Free, Sigma), 5 mM DTT together with 5 µM SNAP-Surface 549 tag in Buffer B at RT for 30 min.
Pull-down of U1M-associated snRNPs
Yeast whole-cell extracts (500 µL of extract either with or without SNAP label) were added to 200 µL of TO1-Dtb saturated agarose beads and incubated at 4°C for 1 h in a rotator. The beads were washed twice each with 5 mL Buffer B for 15 min at 4°C. The beads were washed once more with buffer B at 30°C for 5 min. The snRNP was then eluted by the addition of 200 µL Buffer B supplemented with 20 mM biotin (Sigma-Aldrich) at 37°C for 30 min. For visualizing the fluorescent labeled SNAPf tag, 100 µL of the biotin eluate was concentrated in a speed vac to 10 µL and loaded directly onto a 10% SDS PAGE gel. The gel was imaged using a Typhoon scanner using 532 nm excitation and 580 nm emission filter settings and with PMT kept at 1000 V. The remaining 100 µL of the biotin eluate was phenol extracted once, chloroform extracted twice, and ethanol precipitated using 2 µg glycogen. The pellet was resuspended in 5.1 µL of water and analyzed by RT-primer extension assay.
RNA analysis by RT-primer extension and solution hybridization
Either 1 µL of the precipitated RNA or 1 µg of total RNA isolated from WT yeast extract was added to the corresponding 5′ [32P]-labeled primers (Supplemental Table 4) and the reverse transcription reaction was then carried out using the Maxima Reverse Transcriptase (Thermo Fisher Scientific) at 45°C for 1 h. Reactions were then loaded onto an 8% (19:1 acrylamide:bisacrylamide) denaturing polyacrylamide gel and run at 500 V at RT. For solution hybridization, 5′ [32P]-labeled primers (Supplemental Table 4) were added directly to the precipitated RNAs and incubated for 15 min at RT prior to loading onto a 5% (37.5:1 acrylamide:bisacrylamide) native polyacrylamide gel and electrophoresis at 4°C in 1× TBE at 350 V.
Construction of the p6SRDM_T7 plasmid
The 6SRDM plasmid was created as previously described (Dolgosheina et al. 2014) but with point mutations corresponding to the R9-33 construct (Oviedo Ovando et al. 2014). The 6SRDM is flanked by a T7 promoter and lac operator at its 5′ end and an intrinsic terminator at its 3′ end and was cloned into the pEcoli-Cterm-6xHN (Clontech) plasmid between the SgrAI and ClaI sites. Refer to Supplemental Figure 5 for sequences.
Expression of the 6SRDM RNA in E. coli and extract preparation
The 6SRDM RNA was expressed in E. coli BL21 (DE3) cells transformed with the p6SRDM_T7 plasmid (Supplemental Fig. 5). E. coli cells transformed with p6SRDM_T7 were grown overnight at 37°C in a shaking incubator in Luria-Bertani (LB) media containing 100 µg/mL ampicillin. A portion of the overnight culture (10 mL) was then used to inoculate 1 L of LB media containing 100 µg/mL ampicillin in a 4 L Erlenmeyer flask. The cells were then grown at 37°C in a shaking incubator at 250 rpm until the OD600 reached ∼1, at which point IPTG was added to a final concentration of 50 µM to induce 6SRDM RNA transcription. After 40 min of induction, cells were harvested by centrifugation at 3800g for 10 min at 4°C and resuspended in 20 mL native extract buffer (20 mM Tris pH 8, 150 mM KCl, 1 mM MgCl2, and 1 mM DTT). The resuspended cells were then lysed by two passages through a French press prechilled to 4°C and at 1000 psi. The lysate was centrifuged at 12,000g for 10 min at 4°C to remove cell debris to yield native extract (NE). This extract was then frozen in liquid N2 and stored at −80°C.
Purification of 6S RNARDM RNP complexes
For the purification of 6SRDM associated RNPs, 3.8 mL of E. coli native extract, was made up to 5 mL final volume by the addition of 1 mL of 5× Buffer B and supplemented with final concentrations of Heparin at 75 µg/mL and DTT at 0.75 mM. This solution was mixed with the TO1-Dtb derivatized agarose beads for 15 min while incubating at RT. The beads were washed twice with WB (15 mM HEPES pH 7.5, 90 mM KCl, 75 µg/mL Heparin and 0.75 mM DTT) at RT for 2 min each prior to elution with 400 µL of WB containing 20 mM free biotin at 37°C for 20 min (called Biotin Eluate).
6SRDM RNP complex fractionation by size-exclusion chromatography (SEC)
This biotin eluate (380 µL) was loaded onto a manually packed Superdex 200 Prep Grade (GE Healthcare) column (30 × 1 cm) and eluted with Buffer B using a flow rate of 0.1 mL/min. Fractions (500 µL) were collected and TO1-Dtb fluorescence was measured using a Spectramax M5 fluorescent plate reader (Ex/Em 495/535 nm, PMT at Medium) reading from the bottom of a 96-well plate (Greiner Bio-one).
Mass spectrometry analysis of 6SRDM RNP complexes and commercial RNAP
Samples were analyzed at the University of British Columbia Proteomics Core Facility. LC–MS/MS analysis used a quadrupole–time-of-flight mass spectrometer (Impact II; Bruker Daltonics) coupled to an Easy nano LC 1000 HPLC precolumn (Thermo Fisher Scientific). Analysis of mass spectrometry data was performed using MaxQuant 1.5.1.0. The peptide search was performed against a database comprised of the protein sequences from E. coli K12. Commercial holoenzyme was from Epicentre.
Partial purification of eGFP-HE
E. coli eGFP-HE was partially purified from E. coli strain RL1314 which was a kind gift from Dr. Robert Landick, UW Madison (Bratton et al. 2011). The partial purification was adapted from the protocol of Burgess and Jendrisak (1975). Briefly, 1 L of RL1314 E. coli cells at OD ∼1 were harvested and resuspended in 8 mL of 50 mM Tris pH 8, 5% (v/v) glycerol, 2 mM EDTA, 0.1 mM DTT, and 233 mM NaCl along with protease inhibitors (cOmplete mini at the recommended concentration, EDTA Free, Sigma) and lysed through a French press (prechilled to 4°C) at 1000 psi. The crude lysate was diluted with 8 mL of TGED Buffer (10 mM Tris pH 8, 5% v/v glycerol, 0.1 mM EDTA, and 0.1 mM DTT) supplemented with 0.2 M NaCl and was centrifuged at 12,000g for 45 min to remove cell debris. To this cleared lysate, 0.175 mL of 10% v/v Polymin P at pH 8 was added to 100 mL of the cleared lysate and stirred for 5 min, before pelleting at 4300g for 15 min. The resulting pellet was washed once for 10 min with 10 mL TGED + 0.5 M NaCl and repelleted at 4300g for 15 min. The washed pellet was once again resuspended in 10 mL TGED + 1 M NaCl and stirred for 10 min and the insoluble proteins were removed by spinning at 4300g for 15 min. The resulting material was precipitated with 50% saturated ammonium sulfate (35 g/100 mL) and resuspended in 500 µL dialysis buffer (TGED + 0.1 M NaCl but with 50% w/v glycerol) and was dialyzed overnight against 1 L of dialysis buffer. All the above steps were done either at 4°C or on ice.
Two-color gel-imaging analysis of the Mango and eGFP-tagged 6S:RNAP complex
In vitro synthesized 6SM RNA (1 µM) was added to 2 µM TO3-Dtb and one-tenth of the reaction volume of the partially purified eGFP-HE in 15 mM HEPES pH 7.5, 90 mM KCl, 0.75 mM DTT and 75 µg/mL heparin. This solution was then incubated at 37°C for 30 min to form bound complex. To induce pRNA synthesis-dependent 6SM release, 500 µM NTPs and 5 mM MgCl2 were then added and incubation continued at 37°C. Samples were loaded into a 5% (37.5:1 acrylamide: bisacrylamide) native polyacrylamide gel using 15 mM HEPES pH 7.5 and 90 mM KCl as both the gel and running buffer. Electrophoresis was carried out for 90 min at 4°C at 100 V. The gel was then imaged using a Typhoon Trio+ Variable Mode Imager (GE LifeSciences) using a 488 nm laser and 526 BP filter for eGFP-HE visualization and a 644 nm laser and 670 BP filter for imaging TO3-Dtb:6SM complex. PMT settings were kept at 1000 V and 600 V for eGFP and Mango/TO3-Dtb, respectively. Image data were analyzed and a composite image was made using ImageQuant TV v8.1.0.0 (GE LifeSciences).
Two-photon spectrum and cross-correlation experiments sample conditions
The two-photon spectrum of 6SM:TO3-Dtb was obtained by measuring fluorescence intensity in photon/s over a range of two-photon excitation wavelengths. The solution contained 1 µM in vitro transcribed 6SM, 1 µM TO3-Dtb, 15 mM HEPES pH 7.5, 90 mM KCl, 0.75 mM DTT, and 75 µg/mL heparin. The wavelength was first set to 800 nm, and then tuned to higher wavelengths at a constant rate up to 900 nm. During this tuning, data were acquired in the red channel at a rate of 100 Hz.
Fluorescence cross-correlation spectroscopy was performed by adding increasing amounts of RNAP-eGFP extract to a fixed amount of 6SM and TO3-Dtb. The starting mixture (128 µL) contained 1 µM in vitro transcribed 6SM, 1 µM TO3-Dtb, 15 mM HEPES pH 7.5, 90 mM KCl, 0.75 mM DTT, and 75 µg/mL heparin. To this was added 12.5 µL of partially purified E. coli eGFP-HE (see above), which was prediluted fivefold in 15 mM HEPES pH 7.5, 90 mM KCl, 0.75 mM DTT, and 75 µg/mL heparin. The sample was mixed and cross-correlation spectroscopy was performed. This was repeated for five sequential 12.5 µL additions, and two final 62.5 µL additions of the prediluted eGFP-HE. The molecular brightness of the 6SM:TO3-Dtb alone was determined by correlation spectroscopy on the mixture prior to adding any eGFP-HE. The molecular brightness of eGFP-HE alone was determined by adding 12.5 µL to 128 µL water and performing fluorescence correlation spectroscopy. A mixture containing 6SM in 15 mM HEPES pH 7.5, 90 mM KCl, 0.75 mM DTT, and 75 µg/mL heparin, without TO3-Dtb, was measured and showed no fluorescence above background.
Fluorescence cross-correlation spectroscopy was performed on an ISS Alba microscope controlled using ISS Vista Vision 4.0 Software. Two-photon laser excitation was achieved using a Titanium Sapphire laser (Spectra Physics, Tsunami) mode locked at a median wavelength of 840 nm and a laser power of 45 mW unless otherwise specified. Light was collected using two PerkinElmer SPCM-ARQ Avalanche Photodiodes. Laser light was filtered using a 780 nm short pass filter (Semrock) and split using a 561-nm-long pass dichroic filter (Semrock) before directing the fluorescence emission into the detectors. The excitation lasers were focused onto the sample using a Nikon 60× water immersion objective with a 1.2 numerical aperture. Data were acquired at 100 MHz for 100 sec using a 16-bit ISS FCS PCI card to transfer data between the photodiodes and computer. Each measurement was repeated three times, correlated and fit independently using Craig Markwardt's MPFIT library in IDL 8.3 (Exellis) using custom written software.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Sen, Dr. Audas, and the members of the Unrau Laboratory (Sunny Jeng, Razvan Cojocaru, and Amir Abdolahzadeh) for critical reading of the manuscript. We would also like to thank Robert Landick for providing us with eGFP-tagged HE containing the E. coli strain. This work was supported by a National Science and Engineering Research Council Operating grant (grant number RGPIN238948) to P.J.U.; National Institutes of Health (grant numbers R00 GM086471, R01 GM112735); Shaw Scientist and Beckman Young Investigator awards; startup funding from the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation (WARF) and the Department of Biochemistry to A.A.H. We also acknowledge partial funding from the National Institute of General Medical Sciences of the National Institutes of Health (grant number R15GM123446) as well as a Research Corporation and the Gordon and Betty Moore Foundation Grant (grant number GBMF5263.10) to M.L.F.; the National Science Foundation (grant number MCB-1413664) to E.J.H.; and startup funding from Boise State University to E.J.H. and M.L.F. Funding for open access charge was provided by the National Science and Engineering Research Council.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062166.117.
REFERENCES
- Bacia K, Schwille P. 2007. Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat Protoc 2: 2842–2856. [DOI] [PubMed] [Google Scholar]
- Bratton BP, Mooney RA, Weisshaar JC. 2011. Spatial distribution and diffusive motion of RNA polymerase in live Escherichia coli. J Bacteriol 193: 5138–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RR, Jendrisak JJ. 1975. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry 14: 4634–4638. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J, Planck J, Owen S, Skora AD, Nystul TG, Ohlstein B, Allen A, et al. 2007. The Carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics 175: 1505–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Steitz JA. 2014. The noncoding RNA revolution—trashing old rules to forge new ones. Cell 157: 77–94. [DOI] [PubMed] [Google Scholar]
- Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, Kremers G-J, Davidson MW, Ustione A, Piston DW. 2016. Quantitative assessment of fluorescent proteins. Nat Methods 13: 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgosheina EV, Jeng SCY, Panchapakesan SSS, Cojocaru R, Chen PSK, Wilson PD, Hawkins N, Wiggins PA, Unrau PJ. 2014. RNA Mango aptamer-fluorophore: a bright, high affinity, complex for RNA labeling and tracking. ACS Chem Biol 9: 2412–2420. [DOI] [PubMed] [Google Scholar]
- Hirsch JD, Eslamizar L, Filanoski BJ, Malekzadeh N, Haugland RP, Beechem JM, Haugland RP. 2002. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: uses for protein labeling, detection, and isolation. Anal Biochem 308: 343–357. [DOI] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ. 2011. Ordered and dynamic assembly of single spliceosomes. Science 331: 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686–691. [DOI] [PubMed] [Google Scholar]
- Kim SA, Heinze KG, Bacia K, Waxham MN, Schwille P. 2005. Two-photon cross-correlation analysis of intracellular reactions with variable stoichiometry. Biophys J 88: 4319–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2006. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12: 291–299. [DOI] [PubMed] [Google Scholar]
- Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. 2005. Comparison of affinity tags for protein purification. Protein Expr Purif 41: 98–105. [DOI] [PubMed] [Google Scholar]
- Magalhães MLB, Czekster CM, Guan R, Malashkevich VN, Almo SC, Levy M. 2011. Evolved streptavidin mutants reveal key role of loop residue in high-affinity binding. Protein Sci 20: 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviedo Ovando M, Shephard L, Unrau PJ. 2014. In vitro characterization of 6S RNA release-defective mutants uncovers features of pRNA-dependent release from RNA polymerase in E. coli. RNA 20: 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchapakesan SSS, Unrau PJ. 2012. E. coli 6S RNA release from RNA polymerase requires σ70 ejection by scrunching and is orchestrated by a conserved RNA hairpin. RNA 18: 2251–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchapakesan SSS, Jeng SCY, Unrau PJ. 2015. RNA complex purification using high-affinity fluorescent RNA aptamer tags. Ann N Y Acad Sci 1341: 149–155. [DOI] [PubMed] [Google Scholar]
- Riedel N, Wise JA, Swerdlow H, Mak A, Guthrie C. 1986. Small nuclear RNAs from Saccharomyces cerevisiae: unexpected diversity in abundance, size, and molecular complexity. Proc Natl Acad Sci 83: 8097–8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraphin B, Rosbash M. 1989. Identification of functional U1 snRNA-pre-mRNA complexes committed to spliceosome assembly and splicing. Cell 59: 349–358. [DOI] [PubMed] [Google Scholar]
- Trachman RJ III, Demeshkina NA, Lau MWL, Panchapakesan SSS, Jeng SCY, Unrau PJ, Ferré-D'Amaré AR. 2017. Structural basis for high-affinity thiazole orange binding and fluorescence activation by a small in vitro selected RNA. Nat Chem Biol 13: 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenbeck OC. 1990. Tetraloops and RNA folding. Nature 346: 613–614. [DOI] [PubMed] [Google Scholar]
- Wang KC, Chang HY. 2011. Molecular mechanisms of long noncoding RNAs. Mol Cell 43: 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Saecker RM. 2006. Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science 314: 1601–1603. [DOI] [PubMed] [Google Scholar]
- Wassarman KM, Storz G. 2000. 6S RNA regulates E. coli RNA polymerase activity. Cell 101: 613–623. [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW. 2003. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21: 1369–1377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.