

Abstract
The ABCs of PXE
Pseudoxanthoma elasticum (PXE) is a genetic disorder caused by mutations in ABCC6 that is characterized by calcium deposition outside of the skeletal system, specifically in the blood vessels, skin, and eyes. Using patient-derived fibroblasts and genetic knockout mouse models, Ziegler et al. demonstrate that ABCC6 mutant cells are osteogenic and that loss of ABCC6 reduces pyrophosphate, an inhibitor of calcification. In mice, ectopic calcification was seen only when ABCC6 was deleted jointly from the liver and from Wnt1+ cells, suggesting systemic and local contributions to the phenotype. Treating mice and cells with a tissue-nonspecific alkaline phosphatase (TNAP) inhibitor prevented pyrophosphate degradation and ectopic calcification progression.
Biallelic mutations in ABCC6 cause pseudoxanthoma elasticum (PXE), a disease characterized by calcification in the skin, eyes, and blood vessels. The function of ATP-binding cassette C6 (ABCC6) and the pathogenesis of PXE remain unclear. We used mouse models and patient fibroblasts to demonstrate genetic interaction and shared biochemical and cellular mechanisms underlying ectopic calcification in PXE and related disorders caused by defined perturbations in extracellular adenosine 5′-triphosphate catabolism. Under osteogenic culture conditions, ABCC6 mutant cells calcified, suggesting a provoked cell-autonomous defect. Using a conditional Abcc6 knockout mouse model, we excluded the prevailing pathogenic hypothesis that singularly invokes failure of hepatic secretion of an endocrine inhibitor of calcification. Instead, deficiency of Abcc6 in both local and distant cells was necessary to achieve the early onset and penetrant ectopic calcification observed upon constitutive gene targeting. ABCC6 mutant cells additionally had increased expression and activity of tissue-nonspecific alkaline phosphatase (TNAP), an enzyme that degrades pyrophosphate, a major inhibitor of calcification. A selective and orally bioavailable TNAP inhibitor prevented calcification in ABCC6 mutant cells in vitro and attenuated both the development and progression of calcification in Abcc6−/− mice in vivo, without the deleterious effects on bone associated with other proposed treatment strategies.
INTRODUCTION
Three human diseases—generalized arterial calcification of infancy (GACI), calcification of joints and arteries (CALJA), and pseudoxanthoma elasticum (PXE)—are characterized by debilitating ectopic calcification. The current understanding of the pathogenesis of GACI and CALJA suggests that aberrations in the extracellular adenosine 5′-triphosphate (ATP) catabolic pathway cause ectopic calcification; it is unclear whether a similar mechanism also applies to PXE. GACI [Online Mendelian Inheritance in Man (OMIM) #20800], the most serious of these disorders, often presents by 3 months of age with myocardial infarction secondary to occlusive coronary artery calcification (1). Patients also have extensive medial calcification of their medium-sized and large arteries, predisposing to strokes and heart failure (2). GACI generally results from biallelic loss-of-function mutations in ENPP1, which encodes an extracellular ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) that converts ATP into adenosine 5′-monophosphate (AMP) and pyrophosphate (PPi) (1), a potent inhibitor of calcification in vitro (3) and in vivo (4). Loss of ENPP1 activity results in decreased quantity of PPi both locally and systemically, and GACI patients reportedly have low plasma (5) and urinary (6) PPi concentrations.
CALJA (OMIM #211800) is an adult-onset disorder of medial arterial and joint calcification. Patients typically present in their third decade of life with lower extremity claudication due to calcification of the femoral, popliteal, and dorsalis pedis arteries, in addition to extra-articular joint calcification. CALJA is caused by biallelic loss-of-function mutations in NT5E, which encodes CD73, an ecto-5′-nucleotidase that participates in ATP metabolism by degrading AMP to adenosine and inorganic phosphate (7). Although calcification in GACI appears directly related to PPi deficiency, dysfunctional CD73 has been linked to increased PPi degradation by tissue-nonspecific alkaline phosphatase (TNAP). Increased TNAP activity in CALJA is believed to be secondary to reduced amounts of extracellular adenosine and consequent impairment of intracellular adenosine receptor signaling that inhibits TNAP expression (7). Thus, in both GACI and CALJA, ectopic calcification appears to be related to reduced extracellular concentration of the calcification inhibitor PPi.
In contrast to our understanding of GACI and CALJA, the mechanism of PXE (OMIM #264800), an autosomal recessive disorder of elastic fiber calcification, remains largely unknown. PXE patients exhibit calcification of elastic fibers in the skin, eyes, and arterial wall, resulting in characteristic papular lesions and skin laxity at flexure regions, fragmentation of Bruch’s membrane underlying the retina leading to central vision loss, and medial arterial calcification causing peripheral vascular insufficiency. PXE typically results from mutations in ABCC6, which encodes a presumptive ATP-dependent exporter (8, 9). Markedly, rare patients with biallelic mutations in ABCC6 develop GACI (OMIM #614473) instead of PXE, without compelling evidence for a genotype-phenotype correlation (4).
The function of ATP-binding cassette C6 (ABCC6) remains unclear. ABCC6 is a member of the multidrug resistance protein family with demonstrated transporter activity (10), but its endogenous substrate is unknown. The ABCC6 protein has very low expression in the peripheral cells directly affected in PXE, such as dermal fibroblasts and vascular smooth muscle cells (11, 12), but strong expression in the liver and, to a lesser extent, kidney. The prevailing mechanistic hypothesis suggests that hepatocellular ABCC6 exports an endocrine inhibitor of calcification that acts at distant target sites (13–16) and that failure of this event is sufficient to cause the systemic manifestations of PXE; only circumstantial evidence exists for this pathogenic model. Lack of understanding of disease pathogenesis has resulted in limited treatment options for PXE. Here, we attempt to unravel the mechanisms underlying PXE to better understand the pathways involved in ectopic calcification and conceive new therapeutic approaches.
RESULTS
Crossing Abcc6 to Enpp1 and Nt5e mutant mice reveals genetic interaction
Because of the observed locus heterogeneity within the cohort of patients manifesting GACI and the clinical overlap among PXE, GACI, and CALJA, our initial hypothesis was that ABCC6 functions within the extracellular ATP metabolism pathway. To test this, we generated all possible genetic allele combinations by crossing Abcc6 mutant mice to Enpp1- or Nt5e-deficient mice. Micro–computerized tomography (micro-CT) was used to quantify the extent of calcification of the fibrous capsule surrounding the mouse vibrissae (whiskers on the muzzle; fig. S1), an early marker of ectopic calcification (17). By 15 weeks of age, Abcc6−/− mice showed only moderate calcification, whereas Abcc6−/− mice with one mutated Enpp1 allele showed worsening of the phenotype (Fig. 1, A and B). Enpp1−/− mice showed very aggressive calcification, with no further accentuation upon deleting Abcc6 alleles. There was significant interaction between Abcc6 and Enpp1 [two-way analysis of variance (ANOVA): Abcc6 effect, P = 2.2 × 10−16; Enpp1 effect, P = 2.2 × 10−16; interaction effect, P = 2.2 × 10−16]. The fact that the calcification phenotype is saturated upon full loss of Enpp1 function, with no added effect of targeting Abcc6 alleles, is consistent with a model where Enpp1 functions upstream of Abcc6.
Fig. 1.

Crossing Abcc6 to Enpp1 or Nt5e mutant mice reveals genetic interaction.
Abcc6 mutant mice were crossed to Enpp1 or Nt5e mutant mice to generate all possible genetic allele combinations. (A and C) Micro-CT scans of the muzzle to evaluate the extent of vibrissae fibrous capsule calcification were obtained at 15 weeks of age. Representative coronal z-stacked images of the mouse muzzle with the nasal bones and sinuses midline (indicated by white asterisk) and the pathological calcification seen as radiodense lesions (indicated by the yellow arrow) in the surrounding soft tissue. (B and D) Quantification of ectopic calcification from micro-CT images. A two-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. Two-way ANOVA: (B) Abcc6 effect, P = 2.2 × 10−16; Enpp1 effect, P = 2.2 × 10−16; interaction effect, P = 2.2 × 10−16; (D) Abcc6 effect, P = 2.2 × 10−16; Nt5e effect, P = 1.2 × 10−4; interaction effect, P = 1.1 × 10−3. P values of post hoc comparisons are indicated in the figure.
Crosses between Abcc6- and Nt5e-deficient mice also revealed evidence for genetic interaction (Fig. 1, C and D). By 15 weeks of age, Nt5e−/− mice with or without one null Abcc6 allele showed no evidence of calcification, and deleting one Nt5e allele in Abcc6−/− mice did not exacerbate calcification. However, Nt5e−/− mice with two null Abcc6 alleles showed calcification that was more severe than that observed in Abcc6−/− mice. This interaction was statistically significant (two-way ANOVA: Abcc6 effect, P = 2.2 × 10−16; Nt5e effect, P = 1.2 × 10−4; interaction effect, P = 1.1 × 10−3). When aged to 1 year, Nt5e−/− mice showed mild calcification that was exacerbated by homozygous loss of Abcc6, again documenting genetic interaction (fig. S2, A and B; two-way ANOVA: Abcc6 effect, P = 2.2 × 10−16; Nt5e effect, P = 1.7 × 10−13; interaction effect, P = 5.8 × 10−7). In this set of crosses, the observation that maximal phenotypic severity is only observed upon complete loss of function for both Abcc6 and Nt5e suggests that they work in combination rather than in tandem. Together, these findings suggest that PXE is caused by defects in the same pathway as GACI and CALJA; a parsimonious model places ABCC6 acting downstream of ENPP1 and in parallel with CD73, but more complex scenarios cannot be excluded.
Patient fibroblasts with biallelic ABCC6 mutations can calcify in vitro and have alterations in the extracellular ATP catabolic pathway
To further probe whether metabolic defects observed in GACI and CALJA might also underlie the calcification phenotype in PXE, we generated primary fibroblast cell lines from patients with confirmed biallelic loss-of-function mutations in ABCC6 (ABCC6Mut/Mut), ENPP1 (ENPP1Mut/Mut), or NT5E (NT5EMut/Mut). The disease-causing mutations were missense, nonsense, frameshift, or deletion (table S1). In contrast to control fibroblasts, ABCC6Mut/Mut cell lines, when cultured to confluency and then stimulated with osteogenic media for 21 days, exhibited fully penetrant but variably severe calcification, as assessed by Alizarin red staining (Fig. 2, A and B; one-tailed Student’s t test, P = 0.045). As previously reported, the same stimulation with osteogenic media is needed to elicit calcification in ENPP1Mut/Mut and NT5EMut/Mut fibroblasts (7). These data demonstrate a cell-autonomous predisposition in ABCC6Mut/Mut cells that requires exogenous provocation for phenotypic expression and validate the use of these cells for further biochemical analysis of the functional consequences of ABCC6 mutations in vitro.
Fig. 2.

Evidence for a provoked cell-autonomous defect and alterations in enzymes integral to the extracellular catabolism of ATP in ABCC6 mutant cells.
(A) Primary dermal fibroblasts derived from patients with biallelic mutations in ABCC6 (ABCC6Mut/Mut) calcify in vitro when stimulated with osteogenic media, as indicated by alizarin red staining. Representative images demonstrating the spectrum of calcification are shown. (B) Quantification of the alizarin red staining was determined by colorimetric analysis. One-tailed Student’s t test was performed (P = 0.045). (C to F) Quantification of enzyme activity and gene expression for ENPP1 (ENPP1) and CD73 (NT5E) in primary dermal fibroblasts derived from patients with biallelic mutations in ABCC6, ENPP1, or NT5E. A one-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. One-way ANOVA: (C) P = 0.001; (D) P = 0.016; (E) P = 3.51 × 10−5; (F) P = 0.038. P values of post hoc comparisons are indicated in the figure.
We measured the steady-state enzymatic activity of ENPP1 and CD73, and their respective gene expression, in confluent-cultured ABCC6Mut/Mut, ENPP1Mut/Mut, and NT5EMut/Mut fibroblasts. As expected, ENPP1Mut/Mut cells had negligible ENPP1 activity. ABCC6Mut/Mut and NT5EMut/Mut cells had increased ENPP1 enzymatic activity compared to controls (Fig. 2C; one-way ANOVA, P = 0.001). In addition, there was a marked increase in ENPP1 mRNA expression in ABCC6Mut/Mut cells compared to controls (Fig. 2D; one-way ANOVA, P = 0.016). ENPP1 mRNA expression was also elevated in ENPP1Mut/Mut cells (all containing at least one missense allele), but the mutated protein lacked function, as predicted. These data are consistent with the ordered biochemical pathway inferred from our genetic interaction data and suggest that mutations in genes encoding for proteins distal to ENPP1 lead to compensatory up-regulation of ENPP1, with a predicted increase in PPi production. This compensation is impossible with biallelic loss-of-function mutations in ENPP1, perhaps reconciling the particular severity of the GACI phenotype.
As expected, NT5EMut/Mut fibroblasts had no measurable CD73 enzymatic activity. Both ABCC6Mut/Mut and ENPP1Mut/Mut cells exhibited decreased CD73 activity compared to controls (Fig. 2E; one-way ANOVA, P = 3.51 × 10−5). Quantity of NT5E mRNA in ABCC6Mut/Mut and ENPP1Mut/Mut cells was similar to that in control cells but decreased in NT5EMut/Mut cell lines, all of which have biallelic mutations creating a premature termination codon expected to elicit nonsense-mediated mRNA decay (Fig. 2F; one-way ANOVA, P = 0.038). These data suggest that reduced production and/or bioavailability of substrate (AMP) limits CD73 activity but not expression. This demonstration of metabolic cross-talk among ENPP1, CD73, and ABCC6 further validates the conclusion that ABCC6 contributes to extracellular ATP metabolism.
Liver-specific deletion of Abcc6 does not recapitulate the Abcc6−/− phenotype implicating both local and systemic factors in ectopic calcification
Previous reports demonstrating high expression of ABCC6 in the liver and low expression of ABCC6 in disease-affected tissues (11) have advanced a liver-centric mechanistic hypothesis for PXE (14): that peripheral tissue calcification reflects failed liver secretion of an endocrine inhibitor of calcification. In contrast, our in vitro findings suggest that a cell-autonomous perturbation of extracellular ATP metabolism in ABCC6Mut/Mut fibroblasts is sufficient to predispose to calcification but that an exogenous trigger is required for phenotypic expression. To explore this issue in vivo, we generated a mouse carrying a conditional Abcc6 allele (Abcc6flox/flox) that was subsequently crossed to a number of different lines that express Cre recombinase in a cell type– or tissue-specific manner.
As evidenced by micro-CT, global ablation of Abcc6 with cytomegalovirus (CMV)–Cre resulted in calcification of the fibrous capsule surrounding the muzzle vibrissae at 20 weeks of age, fully recapitulating the phenotype of Abcc6−/− mice (Fig. 3, A and B; one-way ANOVA, P = 2.2 × 10−16). Contrary to the proposed liver-centric model, however, efficient liver-specific deletion of Abcc6 using albumin-Cre (Abcc6flox/flox; Alb-Cre) failed to induce any calcification at 20 weeks (Fig. 3A). We verified efficient deletion of Abcc6 in hepatocytes by breeding the Alb-Cre mice to a RosamTmG mouse line; all cells that recombine change expression from membrane Tomato (mT; red fluorescence) to green fluorescent protein (mG; green fluorescence). All hepatocytes in the RosamTmG; Alb-Cre mice showed green fluorescence (Fig. 3C).
Fig. 3.

Liver-specific deletion of Abcc6 does not phenocopy constitutive ablation of Abcc6 in mice.
Micro-CT scans of the muzzle to evaluate the extent of vibrissae fibrous capsule calcification were obtained at 20 weeks (A) and 1 year (D) of age. (B and E) Quantification of ectopic calcification from micro-CT images. A one-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. One-way ANOVA: (B) P = 2.2 × 10−16; (E) P = 4.68 × 10−11. P values of post hoc comparisons are indicated in the figure. (C) Visualization and confirmation of Cre-targeted tissues and cell types using the RosamTmG reporter mouse line. All cells that are successfully recombined transition from expression of membrane Tomato (mT; red fluorescence) to green fluorescent protein (mG; green fluorescence). Representative images are shown.
Cell type– or tissue-specific ablation of Abcc6 using Cre drivers specific for vascular smooth muscle (SM22α-Cre), vascular endothelium (VE-cadherin–Cre), skeletal muscle (Pax7-Cre), renal tubular cells (Cdh16-Cre), pericytes (Pdgfrβ-Cre), adipocytes (Fabp4-Cre), and bone marrow (CD45-Cre) also failed to induce calcification at 20 weeks of age (table S2). Curiously, 1 of 22 mice with Wnt1-Cre mediated ablation of Abcc6 in the neural crest, including local cells surrounding the vibrissae, showed mild calcification of the fibrous capsule at 20 weeks of age (Fig. 3, A and B, and table S2). Upon breeding the Wnt1-Cre mouse line to a RosamTmG reporter, we discovered mosaic recombination within the liver; at 20 weeks of age, green fluorescence (indicating recombination) was observed in about 12% of hepatocytes (Fig. 3C).
When aged to 1 year, Abcc6flox/flox; Alb-Cre and Abcc6flox/flox; Wnt1-Cre mice showed reduced penetrance and variable expressivity of calcification of the vibrissae fibrous capsule compared to Abcc6flox/flox; CMV-Cre mice (Fig. 3, D and E, and table S2; one-way ANOVA, P = 4.68 × 10−11). Deleting Abcc6 in all other cell types and tissues tested did not result in calcification at 1 year of age (table S2). These data suggest that the loss of Abcc6 expression in the liver sensitizes to calcification but that this event in isolation is insufficient to achieve the threshold loss of function needed for highly penetrant and severe phenotypic expression. The efficiency of the tissue-specific Abcc6 ablation was confirmed by measuring Abcc6 expression at 1 year of age. Like Abcc6−/− mice, Abcc6flox/flox; CMV-Cre mice had no measureable expression of Abcc6 in the liver or kidney when compared to normal expression in control mice [fig. S3, A (one-way ANOVA, P = 2.2 × 10−16) and B (one-way ANOVA, P = 2.2 × 10−16)]. Abcc6flox/flox; Alb-Cre mice had no Abcc6 expression in the liver (fig. S3A), though normal expression in the kidney (fig. S3B). Abcc6flox/flox; Wnt1-Cre mice had a substantial (fivefold), yet incomplete, reduction of Abcc6 expression in the liver (fig. S3A) but not in the kidney (fig. S3B).
Next, we deleted Abcc6 in a combinatorial manner in an attempt to recapitulate the robust calcification seen at 20 weeks in Abcc6−/− and Abcc6flox/flox; CMV-Cre mice. Deleting Abcc6 in all organs caudal to the heart and lungs (Abcc6flox/flox; Alb-Cre; Cdx1-Cre), including the liver and kidney, did not result in calcification at 20 weeks of age, providing strong evidence against a pathogenic hypothesis that singularly invokes an endocrine mechanism (Fig. 4, A and B). Knocking out Abcc6 in the liver and all skeletal muscle cells, including those resident in the muzzle (Abcc6flox/flox; Alb-Cre; Pax7-Cre), also did not induce calcification. Targeting Abcc6 in the liver and in Wnt1+ cells, including local cells in the fibrous capsule surrounding the vibrissae (Abcc6flox/flox; Alb-Cre; Wnt1-Cre), induced calcification at 20 weeks, albeit with reduced penetrance (5 of 13 mice calcified). All Cre-line combinations were also bred to a RosamTmG mouse line to confirm efficient targeting of the cell and/or tissue type (Fig. 4C).
Fig. 4.

Evidence that both local and systemic defects in ATP metabolism are needed to promote PXE-associated ectopic calcification in mice.
(A) Micro-CT scans of the muzzle demonstrating ectopic calcification at 20 weeks of age upon deletion of Abcc6 in both the liver and local Wnt1+ cells in the fibrous capsule, albeit with reduced penetrance compared to constitutive targeting (Fig. 3, A and B). (B) Quantification of ectopic calcification from micro-CT images. (C) Visualization and confirmation of Cre-targeted tissues and cell types using the RosamTmG reporter mouse line. Representative images are shown.
Although circulating PPi amount appears decreased in Abcc6−/− mice and PXE patients (18), it remains uncertain whether decreased plasma PPi is the primary determinant of disease. To explore whether deleting Abcc6 in a combinatorial method was further decreasing circulating PPi concentrations in an additive manner that correlated with the severity of ectopic calcification, we measured plasma PPi concentrations in different cell- and tissue-specific Abcc6 knockout mouse models at 1 year of age and simultaneously quantified vibrissae fibrous capsule calcification via micro-CT (Fig. 5). As expected, constitutive deletion of Abcc6 (Abcc6flox/flox; CMV-Cre) resulted in a robust calcification phenotype (Fig. 5, A and B; one-way ANOVA, P = 1.09 × 10−5), along with plasma PPi concentrations that were significantly below those observed in control animals (Abcc6flox/flox) (Fig. 5C; one-way ANOVA, P = 0.0087). However, two knockout combinations (Abcc6flox/flox; Alb-Cre and Abcc6flox/flox; Alb-Cre; Cdx1-Cre) exhibited much milder vibrissae calcification compared with the Abcc6flox/flox; CMV-Cre mice (Fig. 5, A and B) and yet showed comparably reduced concentrations of circulating PPi (Fig. 5C). The same knockout combinations showed calcification equivalent to that observed in Abcc6flox/flox; Wnt1-Cre mice, despite normal circulating PPi concentrations in the latter. These data document poor correlation between the severity of calcification and the amount of circulating PPi.
Fig. 5.

Circulating PPi concentration does not correlate with severity of calcification phenotype in mice.
(A) Micro-CT scans of the muzzle to evaluate the extent of vibrissae fibrous capsule calcification were obtained at 1 year of age. (B) Quantification of ectopic calcification from micro-CT images. (C) Quantification of plasma PPi concentration. (B and C) A one-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. One-way ANOVA: (B) P = 1.09 × 10−5; (C) P = 0.0087. P values of post hoc comparisons are indicated in the figure.
Of all the cell type– or tissue-specific Cre drivers used in this study, only the use of Wnt1-Cre was associated with recombination within the fibrous capsule of the vibrissae (Fig. 3C). Furthermore, the increase in disease penetrance in Abcc6flox/flox; Alb-Cre; Wnt1-Cre mice compared to Abcc6flox/flox; Alb-Cre animals cannot plausibly relate to enhanced liver recombination. Together, these data suggest cooperation between local and systemic events in the initiation and/or progression of calcification in PXE.
TNAP inhibition prevents in vitro calcification in cell lines with biallelic ABCC6 mutations under osteogenic conditions
With evidence that local cells contribute to PXE pathogenesis, ABCC6Mut/Mut patient fibroblasts emerged as a viable model for investigating potential therapies. We explored whether the calcification was related to increased TNAP activity, because it is known that TNAP is a major regulator of in vitro (19) and in vivo calcification (20–22). When stimulated with osteogenic media for 5 days, ABCC6Mut/Mut cells had increased TNAP enzymatic activity compared to controls (fig. S4A; two-way ANOVA: genotype effect, P = 0.010; treatment effect, P = 0.0029; interaction effect, P = 0.039). Expression of ALPL, the gene encoding TNAP, was concordantly increased (fig. S4B; two-way ANOVA: genotype effect, P = 0.012; treatment effect, P = 0.048; interaction effect, P = 0.18).
Arylsulfonamides are potent and selective inhibitors of TNAP (23). SBI-425, an arylsulfonamide derivative with optimized pharmacokinetic properties, effectively inhibits TNAP in vivo (21). Treatment of ABCC6Mut/Mut cells under osteogenic conditions with SBI-425 prevented in vitro calcification, whereas mutant cells treated with vehicle proceeded to calcify (fig. S4, C and D; two-way ANOVA: genotype effect, P = 0.029; treatment effect, P = 0.028; interaction effect, P = 0.029). These data demonstrate that in vitro calcification of ABCC6Mut/Mut cells is TNAP-dependent, suggesting a potential therapeutic target for PXE.
TNAP inhibition attenuates both the development and progression of calcification in a PXE mouse model
Our demonstration of excessive TNAP expression and activity in ABCC6Mut/Mut cells prompted a treatment trial in 6-week-old Abcc6−/− mice with the TNAP inhibitor SBI-425 (30 mg/kg per day), the non-nitrogenous bisphosphonate etidronate (240 mg/kg per day), which inhibits calcium and phosphate precipitation at high concentrations, or control food for 14 weeks. Micro-CT scans at 20 weeks of age revealed significant and equivalent attenuation of the calcification phenotype in both SBI-425– and etidronate-treated Abcc6−/− mice (Fig. 6, A and B; two-way ANOVA: genotype effect, P = 1.2 × 10−12; treatment effect, P = 3.7 × 10−5; interaction effect, P = 2.1 × 10−5). Mice were treated before the onset of any sign of calcification, as evidenced by micro-CT at 6 weeks of age, although microscopic nidi of calcification cannot be excluded. The therapeutic effect of both SBI-425 and etidronate was substantiated by measuring the calcium phosphate precipitate in muzzle tissue (Fig. 6C; one-way ANOVA, P = 8.4 × 10−4). Serum samples taken from treated mice demonstrated that SBI-425 robustly inhibited plasma TNAP activity, whereas etidronate and control treatments did not, as expected (Fig. 6D). Because there was no effect of genotype (two-way ANOVA: genotype effect, P = 0.64; treatment effect, P = 8.7 × 10−12; interaction effect, P = 0.92), genotype was collapsed to evaluate for differences across treatment groups (one-way ANOVA, P = 1.5 × 10−13). Whereas Abcc6−/− mice had decreased circulating PPi concentrations compared to control mice, SBI-425 did not significantly increase PPi concentrations, potentially highlighting the contribution of local events to the pathogenesis of PXE (fig. S5; two-way ANOVA: genotype effect, P = 0.0017; treatment effect, P = 0.24; interaction effect, P = 0.72).
Fig. 6.

TNAP inhibition attenuates calcification in a PXE mouse model.
(A and B) Micro-CT scans revealed significant attenuation of the calcification phenotype in both SBI-425 and etidronate-treated Abcc6−/− mice. Control mice did not calcify. A two-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. Two-way ANOVA: genotype effect, P = 1.2 × 10−12; treatment effect, P = 3.7 × 10−5; interaction effect, P = 2.1 × 10−5. P values of post hoc comparisons are indicated in the figure. (C) Micro-CT results were validated by dissolving the muzzle tissue and quantifying the calcium phosphate precipitate. OD575, optical density at 575 nm. (D) Quantification of TNAP activity from plasma from mice treated with SBI-425 or etidronate. (C and D) A one-way ANOVA with Tukey’s honest significance difference post hoc analysis was performed. One-way ANOVA: (C) P = 8.4 × 10−4; (D) P = 1.5 × 10−13. P values of post hoc comparisons are indicated in the figure.
Femora were evaluated for bone microarchitecture, mineralization, and mechanical properties at the conclusion of the treatment trial. Imaging of the femora showed significant effects of sex and treatment, but not genotype, across all trabecular bone parameters. It has been well established that trabecular architecture differs between male and female C57BL/6 mice, in an age-related manner, starting between 2 and 6 months of age (24). SBI-425 treatment did not result in any changes to bone microarchitecture. In contrast, etidronate treatment resulted in increased trabecular bone volume (fig. S6, A and E; three-way ANOVA: gender effect, P = 4.16 × 10−12; treatment effect, P = 8.06 × 10−8; genotype effect, P = 0.49) and increased number of trabeculae (fig. S6, B and E; three-way ANOVA: gender effect, P = 1.08 × 10−13; treatment effect, P = 4.7 × 10−10; genotype effect, P = 0.48), with decreased intertrabecular space (fig. S6, C and E; three-way ANOVA: gender effect, P = 2.52 × 10−12; treatment effect, P = 5.59 × 10−5; genotype effect, P = 0.77) in the distal femur in both male and female mice. Etidronate-treated male, but not female, mice also had significantly increased trabecular thickness compared to vehicle-treated mice (fig. S6, D and E; three-way ANOVA: gender effect, P = 7.2 × 10−9; treatment effect, P = 0.0050; genotype effect, P = 0.62). Trichrome staining of the undecalcified distal femora showed no abnormalities of bone mineralization in SBI-425 mice, but as previously reported, increased accumulation of osteoid was apparent in etidronate-treated animals (fig. S6F) (25, 26). Neither of the treatment arms altered the cortical bone of the femoral diaphysis, as quantified by micro-CT (table S3) and mechanical testing (table S4).
To assess for therapeutic potential of SBI-425 in mice with established calcification, we aged Abcc6−/− animals to 20 weeks and then treated them with either vehicle or SBI-425 (30 mg/kg per day) for 16 weeks. Whereas vehicle-treated Abcc6−/− mice showed progressive muzzle calcification, SBI-425–treated animals did not (fig. S7; one-way ANOVA, P = 0.050). These data suggest that although TNAP inhibition does not reverse existing calcification in this experimental context, it can prevent progression of the phenotype. The potential for chronic treatment to achieve therapeutic tissue remodeling in patients remains to be determined.
DISCUSSION
Here, both genetic and metabolic analyses provide compelling evidence that ABCC6 acts in concert with ENPP1 and CD73 to regulate extracellular PPi, a major physiologic inhibitor of calcification. This work suggests that ENPP1 is required for generation of PPi, whereas ABCC6 and CD73 cooperate downstream to inhibit TNAP expression and activity and maintain normal PPi amount (Fig. 7). This interactive network reconciles the pronounced clinical severity of GACI and may provide insight into the phenotypic diversity associated with ABCC6 deficiency, ranging from early-onset GACI to late-onset PXE. Perturbation of this pathway limits the bioavailability of PPi, implying the potential for broad therapeutic relevance of TNAP inhibitors.
Fig. 7.
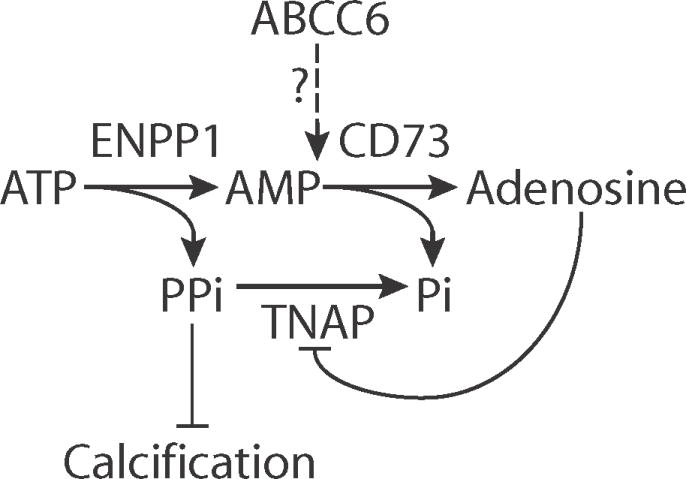
Proposed involvement of ABCC6 in extracellular ATP metabolism and the suppression of ectopic calcification.
ENPP1 metabolizes ATP into AMP and PPi, whereas CD73 further degrades AMP into adenosine and inorganic phosphate (Pi). Adenosine can bind to its cell surface receptor to repress ALPL, the gene encoding TNAP. TNAP degrades PPi into Pi and is a primary distal regulator of PPi, a major negative inhibitor of calcification. Our data suggest that ABCC6 is integral to the extracellular ATP metabolism pathway and likely works downstream of ENPP1 and in tandem with CD73 to maintain low TNAP amount and prevent pathological calcification.
We demonstrate that ABCC6Mut/Mut cells have the intrinsic capacity to calcify in vitro and have altered activity of enzymes involved in ATP catabolism—specifically, increased ENPP1 and TNAP and decreased CD73 enzymatic activities. Consistent with our findings, ABCC6Mut/Mut dermal fibroblasts were previously shown to be morphologically and biochemically distinct from controls (27–29) and to have a tendency for matrix mineralization (30). Although our data concur with previous studies demonstrating that ABCC6Mut/Mut cells have higher gene expression and activity of TNAP (30, 31), we showed that ABCC6Mut/Mut cells had increased (rather than decreased) ENPP1 enzymatic activity and mRNA expression. These discrepancies might arise from the differences in experimental design—we measured ENPP1 enzymatic activity and mRNA after 5 days in culture, whereas previous reports assayed after 21 days in culture; compensatory mechanisms might be at play (30).
It has been proposed that the altered behavior of PXE fibroblasts in culture manifests memory for an in vivo imbalance of a circulating factor (13). However, there exists additional evidence that inherent, cell-autonomous defects are operative in PXE. For example, abcc6-deficient zebrafish show ectopic calcification in the vicinity of osteoblasts that normally express Abcc6 (32). A reconciling view might invoke a role for ABCC6 in determining the amount of a circulating factor that regulates calcification and a local sensitization to its perturbation.
Previous work had shown that fibroblasts from GACI patients deficient for ENPP1 activity can also calcify in vitro, yet overexpression of ENPP1 in some, but not all locations in enpp1-deficient zebrafish, could attenuate calcification at distant target sites (33). Together, these data are consistent with the concept that, although necessary, systemic perturbations may not be sufficient to elicit disease in ectopic calcification disorders. These considerations highlight the relative importance of in vivo models to interrogate disease pathogenesis.
Jiang and Uitto (14) and Jiang et al. (15) have postulated that PXE is specifically caused by a defect in liver secretion of an endocrine inhibitor of calcification at distant target sites. Parabiosis between Abcc6−/− and control mice showed attenuation of the calcification phenotype in the mutant animals, when compared to parabiosis between knockouts (16). However, if PXE is solely driven by a deficiency of a systemic factor that equilibrates in the circulation, the wild-type partner in a wild-type–to–Abcc6−/− pairing (established before the onset of calcification) should show the same phenotype as the knockout mouse; this was not observed. Hence, isolated deficiency of a circulating metabolite appears insufficient to initiate PXE-associated calcification, indicating that another factor, perhaps imposed by a local cell, contributes to PXE pathogenesis. Consistent with this, we found that deletion of Abcc6 in both the liver and local cells in the fibrous capsule surrounding the vibrissae was required to phenocopy the early-onset calcification seen upon constitutive gene ablation; the ongoing observation of reduced penetrance heralds additional complexity regarding the critical threshold level and spatial distribution of ABCC6 function.
The observation of decreased circulating PPi in PXE mice and patients (18) has led to the suggestion that PPi is the protective endocrine factor that is missing in PXE; its deficiency in the extracellular space could result from impaired transport of ATP out of cells, with consequent reduction in AMP and PPi production from ATP catabolism (18). Overexpression of ABCC6 in human embryonic kidney–293 cells resulted in increased extracellular monophosphates, including AMP, a finding that is thought most consistent with enhanced ATP secretion. However, whereas increased extracellular ATP was observed after concomitant treatment with an ENPP1 inhibitor, there was also a generalized increase in triphosphates, diphosphates, and monophosphates, suggesting promiscuous effects in this experimental system (18). In our studies, there was poor correlation between plasma PPi concentration and the extent of vibrissae fibrous capsule calcification in mouse models. Although these data do not exclude a contribution of low circulating PPi, they suggest other determinants of disease pathogenesis.
Although our data have not revealed the precise function of ABCC6, they provide evidence that ABCC6 acts downstream of ENPP1, a view inconsistent with the hypothesis that ABCC6 is primarily involved in ATP transport. In addition, the fact that the classical GACI phenotype is more severe than later-onset PXE also suggests that ENPP1 functions upstream of ABCC6. If ABCC6 were needed to export ATP for extracellular processing by ENPP1, then it would be expected that PXE would routinely have a phenotype as severe as, or more severe than, GACI; this is not observed. Our data are consistent with a model in which ATP is degraded, at least in part, by intracellular ENPP1 to AMP and PPi, with ABCC6 potentially serving to secrete AMP for subsequent extracellular processing by CD73 to adenosine, which is then presumed to act through cell surface adenosine receptors to inhibit TNAP expression (Fig. 7) (7). Although this remains to be formally tested, it is notable that previous reports have described intracellular ENPP1 activity, and there is no described AMP exporter (34–36). In theory, ABCC6 could also contribute to the secretion of PPi, although this is not directly inferred from the data, and there is already an established plasma membrane PPi transporter, ANK (37). Finally, the model proposed by Jansen et al. (18) that ABCC6 acts upstream of ENPP1 and transports ATP could be partially consistent with our genetic data if there is an alternative but limited source of extracellular ATP. It remains notable, however, that such model would not reconcile our findings, suggesting that ABCC6 and CD73 do not act sequentially but rather show cooperative function.
Because of the elusive pathological mechanism underlying PXE, therapeutic targets have been limited. Oral bisphosphonates, which directly disrupt calcium and phosphate precipitation and hence deposition, have been proposed as a treatment strategy for PXE (38). Although the first-generation bisphosphonate etidronate (100 μg/kg, intraperitoneally administered twice a week) failed to prevent calcification in Enpp1−/− mice (39), it effectively attenuated calcification in Abcc6−/− mice when used at a high dose (240 mg/kg per day, orally) (40). Nevertheless, etidronate is currently being tested in the treatment of patients with GACI (2) and CALJA (ClinicalTrials.gov ID: NCT01585402). One drawback is that etidronate results in detrimental changes to bone microarchitecture in mice (40) and in acquired hypophosphatemia with severe skeletal mineralization defects in GACI patients (41). Our findings suggest that the TNAP inhibitor SBI-425 does not have these side effects.
Demonstration that TNAP inhibition attenuates both the development and progression of calcification in both in vitro and in vivo models of PXE helps further establish TNAP as a disease mediator and an attractive therapeutic target in calcification disorders, including PXE, GACI, and CALJA. This finding might also shed light on disease mechanism. The prevailing view that PXE relates to the liver’s inability to secrete ATP predicts that there is a profound impairment in the generation of extracellular PPi. In this scenario, TNAP inhibition and consequent prevention of PPi degradation would not be effective. Here, we provide both genetic and biochemical studies that suggest a role for ABCC6 distal to the degradation of ATP to AMP and PPi by ENPP1. This offers the first rationale that TNAP inhibition would be effective. Given the lack of apparent toxicity, TNAP inhibition might also be considered for other disorders of ectopic calcification including common conditions, such as aortic valve calcification (42, 43) and chronic kidney disease–associated vascular calcification (44, 45), in which decreased PPi has also been documented.
Despite this progress, a number of limitations should be considered. First, although our study draws attention to the potential importance of local events in PXE pathogenesis, the precise nature of microenvironmental alterations remains speculative, and the ability to robustly monitor relevant metabolites is subject to both practical and technical limitations in the absence of specific information regarding the physiologic ABCC6 cargo. Second, this study uses the early-onset and highly penetrant vibrissae fibrous capsule calcification phenotype as a surrogate for tissue calcification events with relevance to patients with PXE, such as those in the eye and vasculature. Although there has been no documentation that vibrissae fibrous capsule calcification is somehow specialized in this regard, the broader relevance of findings made in this context remains assumed and will need to be documented. Finally, as always, observations made using model systems allow the generation of hypotheses that will require validation in patients with PXE.
MATERIALS AND METHODS
Study design
The purpose of this study was to elucidate the pathophysiological mechanisms underlying PXE and the function of ABCC6 in an effort to rationally design treatment strategies for this rare disease patient population. To explore the functional relationships among Abcc6, Enpp1, and Nt5e, we generated double-mutant mice and evaluated vibrissae fibrous capsule calcification via micro-CT. Because we saw strong evidence for genetic interaction, we further explored the role of extracellular ATP metabolism in primary fibroblasts derived from PXE, GACI, and CALJA patients. We found evidence for a provoked cell-autonomous defect and tested the relevance of these findings in vivo by generating a conditional Abcc6 knockout mouse model. The ability to recapitulate pathogenic events in vitro also allowed us to use ABCC6Mut/Mut cell lines to explore possible therapeutic targets. When provoked under osteogenic conditions, ABCC6Mut/Mut demonstrated TNAP-dependent in vitro calcification. To extend these findings in vivo, we treated our Abcc6−/− mice with a TNAP inhibitor. Efficacy and potential negative effects of therapy were evaluated using micro-CT, quantification of calcium phosphate deposition, serum collection, and studies of bone microarchitecture, histology, and mechanical strength.
Sample sizes were determined on the basis of statistical considerations and on pilot experiments that indicated the number of mice per group needed to generate statistical significance. For human cell lines experiments, our sample size was limited by the number of skin biopsy samples we were able to collect from patients with these very rare conditions. Both male and female mice and cell lines obtained from males and females were evaluated in this study. Mice were randomly assigned to treatment groups; sex was equally distributed among the groups. All experiments were performed blindly to genotype and/or treatment. No outliers were excluded. The number of biological replicates in each group is specified in the figures. Individual-level data are included in table S5. Detailed materials and methods are provided in the Supplementary Materials and Methods.
Statistical analysis
All statistical analyses and graphs were generated in RStudio. One-way, two-way, or three-way ANOVAs with Tukey’s honest significance difference post hoc analysis were used to assess for major effects and determine whether there were differences between multiple groups. One-tailed Student’s t test was used to analyze between two groups. All significant pairwise comparisons are shown, and P values are indicated in the figures. No outliers were excluded. α ≤ 0.05 was considered statistically significant. Data obtained from the intercrossing of Enpp1-, Nt5e-, and Abcc6-deficient mice were log-transformed before analysis to normalize the data. For all graphs, the lower and upper margins of each box define the 25th and 75th percentiles, respectively; the internal line defines the median, and the whiskers define the range. Values outside 1.5 times the interquartile distance are shown as open circles, whereas values outside 2 times the interquartile distance are shown as filled circles. The number of biological replicates in each group is specified.
Supplementary Material
Acknowledgments
We thank G. Green, D. Jacob, P. Shah, and Y. Wang for expert technical assistance with micro-CT imaging; J. D. Beckett, Y. Chen, G. S. Knipp, G. Rykiel, and C. Schiefer for micro-CT image analysis; K. Hsu for Sanger sequencing; and K. E. Johnson for mouse genotyping.
Funding: This work was supported by the Howard Hughes Medical Institute, the Johns Hopkins University School of Medicine Medical Scientist Training Program (T32 GM007309), the Predoctoral Training Program in Human Genetics (T32 GM007814), and the Intramural Research Program of the National Human Genome Research Institute. Instrumentation funded by NIH (S10OD016374) was also used.
Footnotes
Author contributions: E.G.M, W.A.G., and H.C.D. aided in experimental design and interpretation of the data. C.R.F. obtained patient samples and provided valuable guidance and clinical expertise. R.C.R. and R.E.T. analyzed mouse bone microarchitecture and integrity, respectively. E.Y.C. and L.M. provided patient samples. C.-T.M. and E.S. performed the plasma alkaline phosphatase assay. A.B.P. and J.L.M. provided SBI-425 and advice for drug dosage. S.G.Z. generated mouse models and performed all other experiments and analyses. S.G.Z., W.A.G., and H.C.D. wrote the paper.
Competing interests: S.G.Z, A.B.P, J.L.M., W.A.G., and H.C.D. are inventors on a patent application entitled “Methods of treating PXE with TNAP inhibitors” (no. PCT/US2015/52967), jointly filed on 29 September 2015 by the Johns Hopkins University School of Medicine, NIH, and the Sanford Burnham Prebys Medical Discovery Institute. All other authors declare that they have no competing interests.
www.sciencetranslationalmedicine.org/cgi/content/full/9/393/eaal1669/DC1
Materials and Methods
Fig. S1. Location of calcification in the fibrous capsule surrounding the vibrissae.
Fig. S2. Demonstration of genetic interaction between Abcc6 and Nt5e mice when aged to 1 year.
Fig. S3. Demonstration of efficient liver-specific deletion of Abcc6 in mice.
Fig. S4. Primary dermal fibroblasts derived from patients with biallelic mutations in ABCC6 show TNAP-dependent in vitro calcification.
Fig. S5. TNAP inhibition does not alter circulating PPi concentration in mice.
Fig. S6. TNAP inhibition had no negative effects on bone microarchitecture or mineralization in a PXE mouse model.
Fig. S7. TNAP inhibition prevents progression of established calcification in a PXE mouse model.
Table S1. List of patient mutations in ABCC6, ENPP1, and NT5E.
Table S2. Number of calcified mice at 20 weeks and 1 year of age.
Table S3. Cortical bone microarchitecture.
Table S4. Cortical bone strength.
Table S5. Individual-level data.
REFERENCES AND NOTES
- 1.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nürnberg P. Mutations in ENPP1 are associated with “idiopathic” infantile arterial calcification. Nat Genet. 2003;34:379– 381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira C, Ziegler S, Gahl W. Generalized arterial calcification of infancy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong C-T, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews. University of Washington; 2014. www.ncbi.nlm.nih.gov/books/NBK253403/ [PubMed] [Google Scholar]
- 3.Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O’Neill WC. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J Am Soc Nephrol. 2004;15:1392– 1401. doi: 10.1097/01.asn.0000128955.83129.9c. [DOI] [PubMed] [Google Scholar]
- 4.Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier MF, Chassaing N, Roche O, Feenstra I, Loechner K, Deshpande C, Garber SJ, Chikarmane R, Steinmann B, Shahinyan T, Martorell L, Davies J, Smith WE, Kahler SG, McCulloch M, Wraige E, Loidi L, Höhne W, Martin L, Hadj-Rabia S, Terkeltaub R, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90:25– 39. doi: 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stuart AG. Idiopathic arterial calcification of infancy and pyrophosphate deficiency. J Pediatr. 1993;123:170– 171. doi: 10.1016/s0022-3476(05)81575-6. [DOI] [PubMed] [Google Scholar]
- 6.Rutsch F, Schauerte P, Kalhoff H, Petrarulo M, August C, Diekmann L. Low levels of urinary inorganic pyrophosphate indicating systemic pyrophosphate deficiency in a boy with idiopathic infantile arterial calcification. Acta Pediatr. 2000;89:1265– 1266. doi: 10.1080/080352500750027682. [DOI] [PubMed] [Google Scholar]
- 7.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432– 442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergen AAB, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, ten Brink JB, de Jong PTVM. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228– 231. doi: 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- 9.Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali-Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, de Paepe A, Boyd CD. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223– 227. doi: 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- 10.Iliás A, Urbán Z, Seidl TL, Le Saux O, Sinkó E, Boyd CD, Sarkadi B, Váradi A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6) J Biol Chem. 2002;277:16860– 16867. doi: 10.1074/jbc.M110918200. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzaki Y, Nakano A, Jiang QJ, Pulkkinen L, Uitto J. Tissue-specific expression of the ABCC6 gene. J Invest Dermatol. 2005;125:900– 905. doi: 10.1111/j.0022-202X.2005.23897.x. [DOI] [PubMed] [Google Scholar]
- 12.Pomozi V, Le Saux O, Brampton C, Apana A, Iliás A, Szeri F, Martin L, Monostory K, Paku S, Sarkadi B, Szakács G, Váradi A. ABCC6 is a basolateral plasma membrane protein. Circ Res. 2013;112:e148– e151. doi: 10.1161/CIRCRESAHA.111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Saux O, Bunda S, VanWart CM, Douet V, Got L, Martin L, Hinek A. Serum factors from pseudoxanthoma elasticum patients alter elastic fiber formation in vitro. J Invest Dermatol. 2006;126:1497– 1505. doi: 10.1038/sj.jid.5700201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Q, Uitto J. Pseudoxanthoma elasticum: A metabolic disease? J Invest Dermatol. 2006;126:1440– 1441. doi: 10.1038/sj.jid.5700267. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Q, Endo M, Dibra F, Wang K, Uitto J. Pseudoxanthoma elasticum is a metabolic disease. J Invest Dermatol. 2009;129:348– 354. doi: 10.1038/jid.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre AE, Horwitz EM, Uitto J. Parabiotic heterogenetic pairing of Abcc6 − / − / Rag1 − / − mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol. 2010;176:1855– 1862. doi: 10.2353/ajpath.2010.090983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klement JF, Matsuzaki Y, Jiang QJ, Terlizzi J, Choi HY, Fujimoto N, Li K, Pulkkinen L, Birk DE, Sundberg JP, Uitto J. Targeted ablation of the Abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. 2005;25:8299– 8310. doi: 10.1128/MCB.25.18.8299-8310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansen RS, Küçükosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IEM, Bergen AAB, Gorgels TGMF, Borst P, van de Wetering K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci USA. 2013;110:20206– 20211. doi: 10.1073/pnas.1319582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narisawa S, Harmey D, Yadav MC, O’Neill WC, Hoylaerts MF, Millán JL. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res. 2007;22:1700– 1710. doi: 10.1359/jbmr.070714. [DOI] [PubMed] [Google Scholar]
- 20.Millán JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int. 2016;98:398– 416. doi: 10.1007/s00223-015-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Chhea TN, Sergienko EA, Kapoor K, Jackson MR, Hoylaerts MF, Pinkerton AB, O’Neill WC, Millán JL. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res. 2015;30:824– 836. doi: 10.1002/jbmr.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savinov AY, Salehi M, Yadav MC, Radichev I, Millán JL, Savinova OV. Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc. 2015;4:e002499. doi: 10.1161/JAHA.115.002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, Smith LH, O’Neill WC, Millán JL, Cosford NDP. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP) J Med Chem. 2009;52:6919– 6925. doi: 10.1021/jm900383s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22:1197– 1207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- 25.Schenk R, Merz WA, Mühlbauer R, Russell RG, Fleisch H. Effect of ethane-1-hydroxy-1, 1-diphosphonate (EHDP) and dichloromethylene diphosphonate (Cl 2 MDP) on the calcification and resorption of cartilage and bone in the tibial epiphysis and metaphysis of rats. Calcif Tissue Res. 1973;11:196– 214. doi: 10.1007/BF02547219. [DOI] [PubMed] [Google Scholar]
- 26.Price PA, Faus SA, Williamson MK. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler Thromb Vasc Biol. 2001;21:817– 824. doi: 10.1161/01.atv.21.5.817. [DOI] [PubMed] [Google Scholar]
- 27.Tiozzo Costa R, Baccarani Contri MR, Cingi MR, Pasquali Ronchetti I, Salvini R, Rindi S, De Luca G. Pseudoxanthoma elasticum (PXE): Ultrastructural and biochemical study on proteoglycan and proteoglycan-associated material produced by skin fibroblasts in vitro. Coll Relat Res. 1988;8:49– 64. doi: 10.1016/s0174-173x(88)80035-9. [DOI] [PubMed] [Google Scholar]
- 28.Quaglino D, Jr, Boraldi F, Barbieri D, Croce A, Tiozzo R, Pasquali Ronchetti I. Abnormal phenotype of in vitro dermal fibroblasts from patients with Pseudoxanthoma elasticum (PXE) Biochim Biophys Acta. 2000;1501:51– 62. doi: 10.1016/s0925-4439(00)00007-7. [DOI] [PubMed] [Google Scholar]
- 29.Gheduzzi D, Boraldi F, Annovi G, DeVincenzi CP, Schurgers LJ, Vermeer C, Quaglino D, Ronchetti IP. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab Invest. 2007;87:998– 1008. doi: 10.1038/labinvest.3700667. [DOI] [PubMed] [Google Scholar]
- 30.Dabisch-Ruthe M, Kuzaj P, Götting C, Knabbe C, Hendig D. Pyrophosphates as a major inhibitor of matrix calcification in pseudoxanthoma elasticum. J Dermatol Sci. 2014;75:109– 120. doi: 10.1016/j.jdermsci.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Boraldi F, Annovi G, Vermeer C, Schurgers LJ, Trenti T, Tiozzo R, Guerra D, Quaglino D. Matrix Gla protein and alkaline phosphatase are differently modulated in human dermal fibroblasts from PXE patients and controls. J Invest Dermatol. 2013;133:946– 954. doi: 10.1038/jid.2012.460. [DOI] [PubMed] [Google Scholar]
- 32.Mackay EW, Apschner A, Schulte-Merker S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development. 2015;142:1095– 1101. doi: 10.1242/dev.113811. [DOI] [PubMed] [Google Scholar]
- 33.Apschner A, Huitema LFA, Ponsioen B, Peterson-Maduro J, Schulte-Merker S. Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of infancy (GACI) and pseudoxanthoma elasticum (PXE) Dis Model Mech. 2014;7:811– 822. doi: 10.1242/dmm.015693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson K, Vaingankar S, Chen Y, Moffa A, Goldring MB, Sano K, Jin-Hua P, Sali A, Goding J, Terkeltaub R. Differential mechanisms of inorganic pyrophosphate production by plasma cell membrane glycoprotein-1 and B10 in chondrocytes. Arthritis Rheum. 1999;42:1986– 1997. doi: 10.1002/1529-0131(199909)42:9<1986::AID-ANR26>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 35.Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43:1560– 1570. doi: 10.1002/1529-0131(200007)43:7<1560::AID-ANR21>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 36.Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1– C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]
- 37.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank :An integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199– 1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drake MT, Clarke BL, Khosla S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032– 1045. doi: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huesa C, Staines KA, Millán JL, MacRae VE. Effects of etidronate on the Enpp1 − / − mouse model of generalized arterial calcification of infancy. Int J Mol Med. 2015;36:159– 165. doi: 10.3892/ijmm.2015.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q, Sundberg JP, Levine MA, Terry SF, Uitto J. The effects of bisphosphonates on ectopic soft tissue mineralization caused by mutations in the ABCC6 gene. Cell Cycle. 2015;14:1082– 1089. doi: 10.1080/15384101.2015.1007809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, Kiffer-Moreira T, Sheen C, Millán JL, Ericson KL, Whyte MP. Severe skeletal toxicity from protracted etidronate therapy for generalized arterial calcification of infancy. J Bone Miner Res. 2013;28:419– 430. doi: 10.1002/jbmr.1752. [DOI] [PubMed] [Google Scholar]
- 42.Rattazzi M, Bertacco E, Iop L, D’Andrea S, Puato M, Buso G, Causin V, Gerosa G, Faggin E, Pauletto P. Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: Implications for aortic valve calcification. Atherosclerosis. 2014;237:568– 576. doi: 10.1016/j.atherosclerosis.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 43.Rathan S, Yoganathan AP, O’Neill CW. The role of inorganic pyrophosphate in aortic valve calcification. J Heart Valve Dis. 2014;23:387– 394. [PMC free article] [PubMed] [Google Scholar]
- 44.Lomashvili KA, Khawandi W, O’Neill WC. Reduced plasma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol. 2005;16:2495– 2500. doi: 10.1681/ASN.2004080694. [DOI] [PubMed] [Google Scholar]
- 45.O’Neill WC, Sigrist MK, McIntyre CW. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol Dial Transplant. 2010;25:187– 191. doi: 10.1093/ndt/gfp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeCorre Y, LeSaux O, Froeliger F, Libouban H, Kauffenstein G, Willoteaux S, Leftheriotis G, Martin L. Quantification of the calcification phenotype of Abcc6- deficient mice with microcomputed tomography. Am J Pathol. 2012;180:2208– 2213. doi: 10.1016/j.ajpath.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berndt A, Li Q, Potter CS, Liang Y, Silva KA, Kennedy V, Uitto J, Sundberg JP. A single-nucleotide polymorphism in the Abcc6 gene associates with connective tissue mineralization in mice similar to targeted models for pseudoxanthoma elasticum. J Invest Dermatol. 2013;133:833– 836. doi: 10.1038/jid.2012.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) BioTechniques. 2000;29:52– 54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 49.Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Váradi A, Plomp A, Bergen AA, Oude Elferink RPJ, Borst P, van de Wetering K. ABCC6 – mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation — Brief report. Arterioscler Thromb Vasc Biol. 2014;34:1985– 1989. doi: 10.1161/ATVBAHA.114.304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sergienko EA, Sun Q, Ma CT. A method for direct assessment of tissue-nonspecific alkaline phosphatase (TNAP) inhibitors in blood samples. Methods Mol Biol. 2013;1053:103– 113. doi: 10.1007/978-1-62703-562-0_6. [DOI] [PubMed] [Google Scholar]
- 51.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468– 1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.