

Ewing sarcoma is rare in adults, and there are no dedicated clinical trials in the adult population. This article reviews the results of therapy with vincristine, ifosfamide, and doxorubicin in the multidisciplinary treatment of adults with Ewing sarcoma.
Keywords: Ewing sarcoma, Adults, Ifosfamide, Doxorubicin, Vincristine
Abstract
Background.
There are no clinical trials specifically addressing chemotherapy for adults with Ewing sarcoma (ES). Five‐year event‐free survival (EFS) of adults on pediatric studies of ES (44%–47%) is worse than that of children treated with the same therapy (69%). The object of this study was to review the results of therapy with vincristine, ifosfamide, and doxorubicin (VID) in the multidisciplinary treatment of adults with ES at our institution.
Materials and Methods.
Charts for adults treated for ES from 1995 to 2011 were retrospectively reviewed. Clinician‐reported radiographic tumor response, type of local therapy, pathologic response, and survival data were collected.
Results.
Seventy‐one patients were identified who received VID as initial therapy. The median age was 25 (range: 16–64). Forty‐two patients (59%) presented with a localized disease and 29 patients (41%) presented with a distant metastasis. Of all patients treated with VID, 83.6% showed a radiological response. Patients who presented with a localized disease had a 5‐year overall survival (OS) of 68% (median not reached), compared with 10.3% (median: 1.9 years) in those who presented with distant metastases. Five‐year EFS was 67%. The nine patients with a pelvic primary tumor had inferior 5‐year OS (42%) to the 33 with primary tumors at other sites (75%). The 5‐year OS of those who had greater than or equal to 95% necrosis after neoadjuvant VID (n = 20; 5‐year OS: 84%) was superior to those who had less than 95% necrosis (n = 13; 5‐year OS: 53%).
Conclusion.
In adults with primary ES, VID combined with an adjuvant strategy based on post‐treatment percent necrosis has favorable outcomes compared with historical adult controls.
Implications for Practice.
Ewing sarcoma (ES) is a rare tumor in adults, and there are no dedicated clinical trials in the adult population. Most therapy is modeled after the published pediatric studies, although the small numbers of adult patients included on those studies did significantly worse than the children. We modeled our treatment on other adult sarcomas and reviewed the charts of 71 adult patients with ES treated with vincristine, ifosfamide, and doxorubicin (VID). In adults with primary ES, VID combined with an adjuvant strategy based on post‐treatment percent necrosis has favorable outcomes compared with historical adult controls.
Introduction
For nearly 20 years, vincristine (2 mg/m2 to a maximum of 2 mg), doxorubicin (75 mg/m2), and cyclophosphamide (1.2 g/m2) (VDC), alternating with ifosfamide (9 g/m2) and etoposide (500 mg/m2) (IE) for 14 cycles, has been the standard of care for upfront therapy in pediatric patients with localized Ewing sarcoma (ES) [1]. Patients older than 18 years old had worse outcomes compared with children in the initial VDC‐IE studies, with 5‐year event‐free survival (EFS) of 44%–47% compared with 69% in the younger population [2], [3]. Although these studies were not powered to detect whether dose‐dense therapy every 2 weeks offers benefit to adult patients, adult patients do not appear to have a survival benefit to the extent that children do in response to dose‐dense chemotherapy, and the preferred treatment regimens for this age group vary by institution. In the U.S., clinicians affiliated with large cancer centers that have significant experience treating ES patients frequently use current phase 3 Children's Oncology Group (COG) protocol, whereas our European counterparts have traditionally favored vincristine (1.5 mg/m2), ifosfamide (9 g/m2), doxorubicin (60 mg/m2), and etoposide (150 mg/m2/day on days 1–3) (VIDE) per the EURO‐Ewing99 protocol [4].
Standard treatment in adults with ES at our institution since 1995 has been initial therapy with vincristine 2 mg on day one, doxorubicin 25 mg/m2 continuous intravenous infusion for three days, and ifosfamide 10 g/m2 divided into four to five daily doses administered over 2–3 hours each day (VID) for six cycles in the neoadjuvant setting rather than alternating cyclophosphamide and ifosfamide. Similar to strategies employed in the recent European studies (e.g., EURO‐EWING99) [5], our institution's treatment approach has been customized to take neoadjuvant drug response into account. For patients with localized disease who undergo surgical resection, adjuvant therapy is tailored based upon percent necrosis on pathology and by provider preference, with most patients receiving adjuvant high‐dose ifosfamide and etoposide.
Our practice is to start treatment with VID with the goal of inducing rapid tumor regression. This allows for a higher initial exposure to doxorubicin and ifosfamide than would otherwise have been provided when VDC and IE are alternated. Additionally, this approach allows physicians to more accurately ascribe treatment outcomes to specific chemotherapies so that when patients respond or progress, we have a clearer understanding of the drugs their tumor is sensitive or resistant to. The latest COG protocols use seven (or more) drugs, significantly confounding decisions regarding which agents may still retain activity in patients with disease recurrence. We assessed VID as initial therapy for adult ES patients—with tailored adjuvant therapy based on response to VID for a goal of 14 total cycles of chemotherapy—and present our initial findings of the long‐term clinical results.
Methods
With an institutional review board‐approved protocol, we reviewed the charts of adult ES patients treated with VID as initial therapy at our institution between 1995 and 2011. Cases were identified through our tumor registry and confirmed with our sarcoma pathology database. Patients were excluded if they received any other form of initial front‐line therapy. Variables collected included patient demographics, site of primary tumor, extent of disease at initial presentation, date of diagnosis, date of start of chemotherapy, date and type of local therapy, percent necrosis at the time of surgery as assessed by a sarcoma pathologist, date of relapse or progression, and vital status. Descriptive statistics were obtained and analyzed with SPSS (statistical software, IBM SPSS Statistics Version 22, Armonk, NY, https://www.ibm.com/). Due to the retrospective nature of this analysis, the lack of consistency with primary imaging modality, and availability of primary images, the best radiographic response was determined by clinician assessment from the electronic medical record. When images were available, the assessment in the medical record was confirmed by review of the images per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 [6]. Survival was calculated using the Kaplan‐Meier method and statistical analysis used log rank tests and Cox models. Overall survival (OS) was calculated from the date of diagnosis until the date of the last assessment or death. EFS was calculated from the date of diagnosis until the date of event (e.g., progression, death, secondary malignancy).
Results
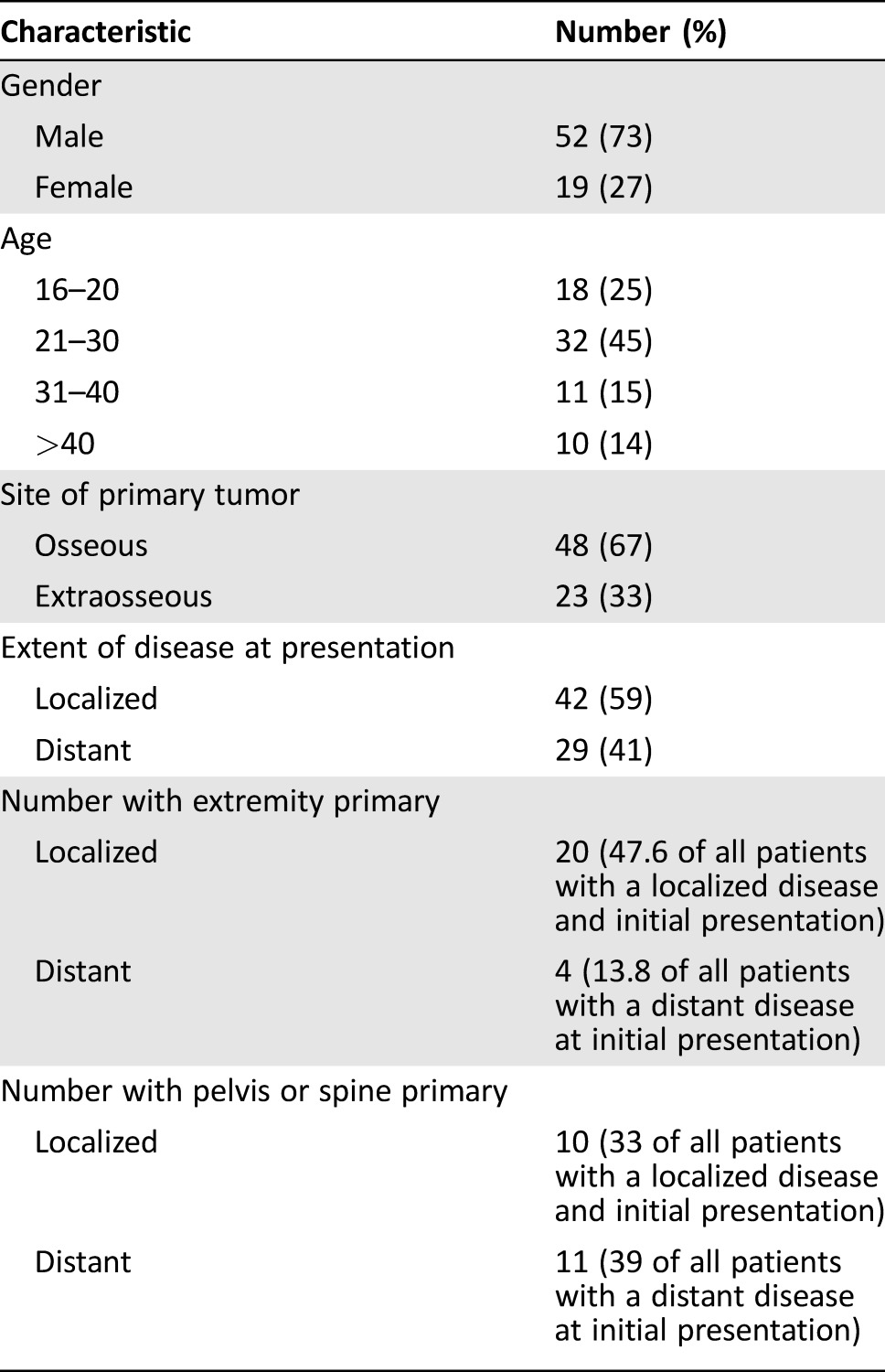
Seventy‐one patients treated with VID‐based therapy were identified from a total of 107 chemotherapy‐naive patients who presented to our institution during the study period (Fig. 1). The median age was 25 (range: 16–64). Nineteen (27%) were female and 52 (73%) were male. Forty‐eight (67%) had skeletal primary disease and 23 (33%) had extra‐skeletal primary disease. Forty‐two (59%) presented with a localized disease and 29 (41%) presented with a distant metastasis. Nearly half (47.6%) of the patients presenting a localized disease had an extremity‐based primary tumor, and 13.8% of the patients presenting with a distant disease at the initial presentation had an extremity‐based primary tumor (Table 1).
Figure 1.

Flow chart of patient treatment characteristics.
Abbreviations: ES, Ewing sarcoma; VID, vincristine, ifosfamide, and doxorubicin.
Table 1. Baseline patient characteristics.
Radiographic response was assessed as a physician‐reported outcome in the electronic medical record, which was then confirmed if images were available in our system. Of all patients with radiographic assessment (n = 63), 83.6% had either a partial or complete response to VID (Table 2).
Table 2. Radiographic clinician‐assessed best response.
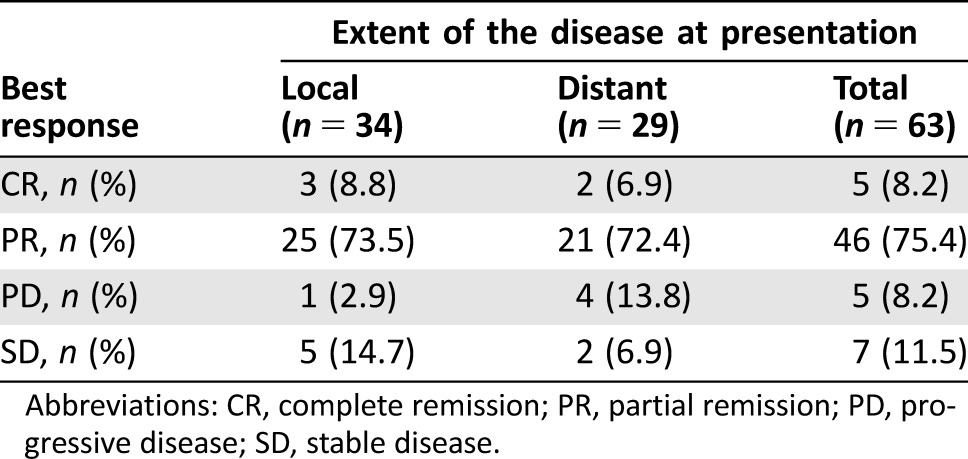
Abbreviations: CR, complete remission; PR, partial remission; PD, progressive disease; SD, stable disease.
Patients who presented with a local disease had a 5‐year OS of 68% (median OS not reached), compared with 10.3% (median OS: 1.9 years) in those who presented with a distant disease (Table 3; Fig. 2A; log rank p < .001). Two (4.7%) patients presented with local disease‐developed metastases while on initial therapy, one of whom did not have local treatment. Following neoadjuvant VID chemotherapy, 29 (69%) underwent surgery alone, 5 (12%) had radiation therapy (RT) alone, and 7 (17%) underwent both surgery and RT, with all but one receiving neoadjuvant radiation after VID and one receiving adjuvant radiation. All patients with more than scattered viable tumor cells on surgical pathology, defined as less than 95% necrosis (Fig. 3A, 3B), underwent adjuvant chemotherapy, including high‐dose ifosfamide, IE, or irinotecan‐temozolomide with or without vincristine (Table 4). Six patients out of 21 with greater than or equal to 95% necrosis (85% of all patients eligible for adjuvant chemotherapy on this study) did not receive adjuvant chemotherapy (Fig. 3C, 3D). None of the six relapsed; five of the six patients were alive at the last follow‐up; the sixth died from a secondary leukemia. Five‐year EFS in patients presenting with local disease was 67%, with most (86%) events occurring within the first year of treatment (Fig. 2B). When analyzed by percent necrosis at the time of surgery, there was improvement in 5‐year OS in those who had greater than or equal to 95% percent necrosis (n = 21; 5‐year OS: 90%) compared with those who had less than 95% necrosis (n = 13; 5‐year OS 53%) (Fig. 2C; log rank p = .036). The nine patients with a pelvic primary tumor had inferior 5‐year OS (42%) to the 33 with primary tumors at other sites (75%) in keeping with prior observations and the finding in Pretz et al. [7] (Fig. 2D; log rank p = .041). There was no difference in survival between patients below 30 years old versus those 30 years or older (p = .85), or between patients with primary bone versus primary extraskeletal tumors (p =.45) (supplemental online Fig. 1).
Table 3. Overall survival by patient characteristics.
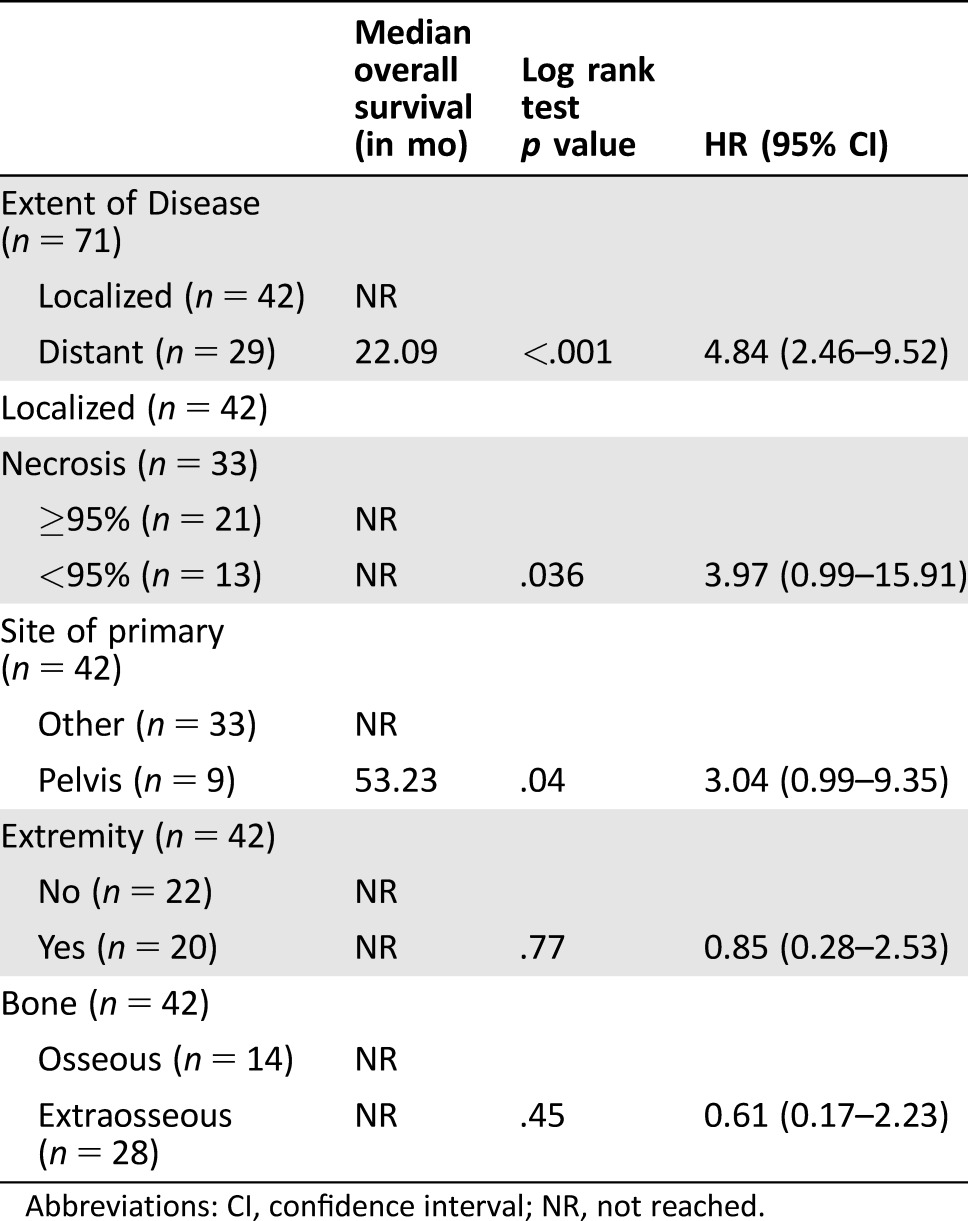
Abbreviations: CI, confidence interval; NR, not reached.
Figure 2.

Cumulative survival. (A): Overall survival (OS) of Ewing sarcoma patients receiving vincristine, ifosfamide, and doxorubicin (VID) for initial therapy stratified by the extent of disease (localized n = 42 vs. metastatic n = 29) at initial presentation. (B): Event‐free survival of patients presenting with a localized disease. (C): OS of patients presenting with localized disease stratified by percent necrosis after initial treatment with VID (≥95% necrosis n = 21 vs. <95% necrosis n = 13). (D): OS of patients presenting with a localized disease stratified by the site of primary (pelvic n = 9 vs. other n = 33).
Abbreviation: Cum, cumulative.
Figure 3.

Examples of Ewing sarcoma (ES) histological responses. (A): Low power shows tumor has viable areas (darker blue) as well as areas of necrosis and hyalinization. The treatment effect was estimated to be 75% (H&E, 10x). (B): Poor responder at high power reveals viable ES (small round blue tumor). (C): Hypocellular hyalinized tissue with thin‐walled vessels, hemosiderin and scattered histiocytes. No residual tumor cells seen (H&E, 100x). (D): Extensive necrosis. No viable tumor cells seen (H&E, 100x).
Table 4. Adjuvant treatments in patients with localized disease.
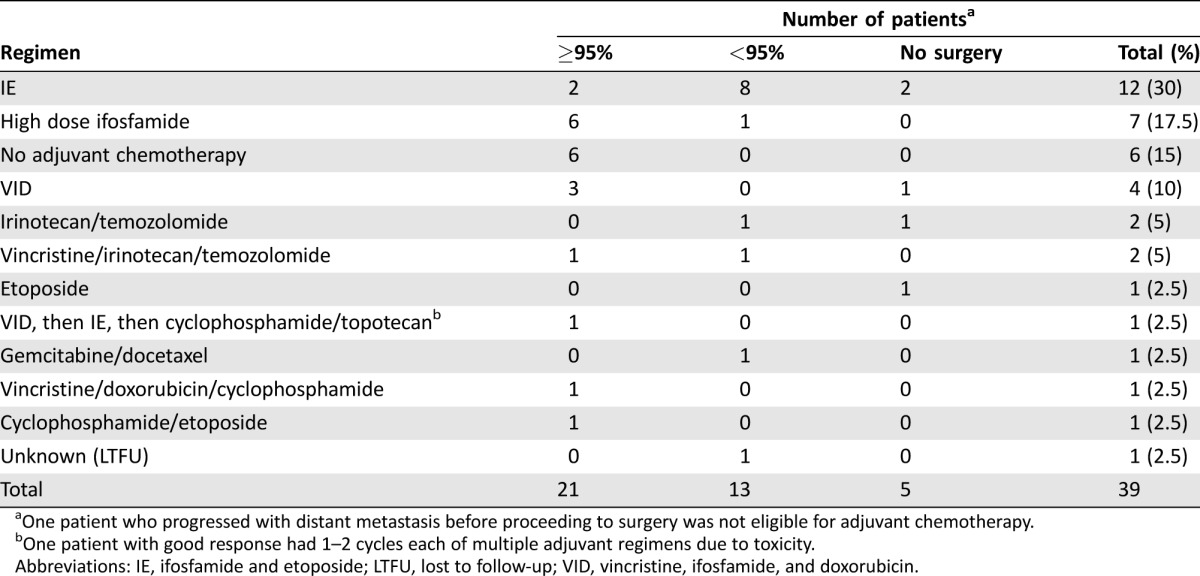
One patient who progressed with distant metastasis before proceeding to surgery was not eligible for adjuvant chemotherapy.
One patient with good response had 1–2 cycles each of multiple adjuvant regimens due to toxicity.
Abbreviations: IE, ifosfamide and etoposide; LTFU, lost to follow‐up; VID, vincristine, ifosfamide, and doxorubicin.
Additional subgroup analysis by adjuvant chemotherapy regimen in patients who were eligible for adjuvant chemotherapy demonstrated that no obvious differences were seen between ifosfamide‐based, antitopoisomerase1‐based, other adjuvant systemic therapy, or no adjuvant therapy in patients with good histological response (supplemental online Fig. 2). No statistics were performed on subgroup analysis by chemotherapy regimen due to the small numbers of patients in each group. Similarly, there was no difference in disease‐free survival between patients who received etoposide in the adjuvant setting compared with those who did not (supplemental online Fig. 3). Similarly, statistical analysis and definitive conclusions regarding the potential beneficial role for etoposide are limited by sample size in this study.
Discussion
Adults with ES do not do as well as children treated with the same regimen. We hypothesized that upfront dosing of VID would increase radiographic and pathological response rates compared with alternating therapy by maximizing upfront anthracycline and ifosfamide exposure while permitting later addition of dose‐intensified ifosfamide, usually together with etoposide, for poor responders. In the neoadjuvant setting, alternating dose‐dense cycles of VDC and IE as used in Womer et al. [2] results in cumulative doses of vincristine (6 mg/m2 to a maximum of 6 mg), doxorubicin (225 mg/m2), cyclophosphamide (3.6 mg/m2), ifosfamide (27 g/m2), and etoposide (1500 mg/m2). In contrast, six cycles of neoadjuvant VID deliver vincristine (12 mg), doxorubicin (450 mg/m2) and ifosfamide (60 g/m2). By accelerating the dose of VID in upfront treatment and adding intensified ifosfamide later, we attempted to improve the responses compared with alternating therapy. Although not formally assessed in this retrospective review, VID in our experience is tolerated with approximately the same profile as standard doxorubicin and ifosfamide for soft tissue sarcoma [8].
Our outcome with VID when combined with an adjuvant strategy based on post‐treatment percent necrosis compares favorably with that of historical adult controls treated with VDC‐IE in large trials (5‐year EFS 67% compared with 44%–47%) and in a similar retrospective cohort [9]. Though never shown to improve survival, minimizing the number of agents a patient is exposed to prior to surgery allows for tailoring subsequent regimens based on response to the initial therapy. Acknowledging that this is a single‐institution, retrospective study and the associated limitation of including only patients able to come to our institution, we believe that the results presented here validate the approach that we have used.
It could be argued that the omission of etoposide, a known effective drug, from our regimen, might lead to an inferior result. Our data, however, do not support such an argument. Our rate of good response compares favorably with that seen with etoposide‐containing regimens. Etoposide could not have helped the survival of our good responders. The fact that none of the six patients with greater than or equal to 95% necrosis who did not receive additional chemotherapy relapsed throws into question the practice of prolonged postoperative adjuvant chemotherapy with any agent(s) for patients with good response to preoperative chemotherapy. This question might be addressed in a subsequent randomized study. Most of the patients with a poor response did receive etoposide and derived marginal benefit (supplemental online Fig. 3).
A large prospective randomized trial would be needed to confirm or refute the benefits of our approach over the conventional VDC‐IE regimen; however, that comparison is neither cost‐effective nor feasible because the current chemotherapy backbone recommended by COG to treat patients with localized ES has integrated additional chemotherapies (e.g., topotecan). The protocols used for those presenting with metastatic disease have also evolved and now include biologically targeted therapies aimed at the insulin‐like growth factor receptor 1 or downstream signaling proteins. Given these practicalities, a sensible approach would compare our treatment outcomes to those obtained by other major cancer centers that treated a similar adult cohort of ES patients with VDC‐IE, such as the cohort described in Pretz et al. [7]. The favorable results in that report suggest that neither chemotherapy regimen is clearly superior and that the favorable results may be influenced, at least partially, by treatment by medical oncologists at centers with sarcoma expertise.
In our cohort, as in others, patients with metastatic disease do poorly universally, as do almost half of the patients with subpar neoadjuvant responses. Patients with pelvic primary tumors also did poorly. Alternative treatment strategies are urgently needed for these patient populations. Insulin‐like growth factor‐targeted drugs had promising initial results in ES models [10], [11]. Unfortunately, only a small percentage of patients seemed to derive benefit from these agents, with response rates ranging from 3%–16% [12], [13], [14], [15], [16], [17]. The addition of inhibitors of the mamallian target of rapamycin yielded a clinical benefit rate of 29%, with a median duration of benefit for more than a year [18], [19]. In spite of these results, to date none of these agents have achieved approval for use in ES. Transcription factor‐targeting drugs such as trabectedin or lurbinectedin [20], [21] and YK‐4‐279 [22] have also demonstrated promising preclinical activity, and clinical trials have just recently been initiated to assess their antineoplastic activity in ES patients. Some of the drugs in development have been shown to synergize with vinca‐alkaloids [23], and the addition of new drugs may further abrogate the number of cytotoxics needed.
The strategy of combining six or more cytotoxic chemotherapies together is unlikely to further elevate the 5‐year survival rate of ES patients. As additional agents are entering the clinical arena, prospectively one may consider the addition of biologically targeted therapies to a VID‐based backbone while removing other less effective chemotherapies such as cyclophosphamide. Topoisomerase II inhibition is still provided by maximizing upfront exposure to doxorubicin when giving VID and can be continued with the later addition of etoposide in the treatment course in the adjuvant setting, or it can be added for poor responders as a regimen similar to VIDE as given in other studies [5]. Though impractical in patients for reasons outlined above, one approach to determine the best modern‐day chemotherapy regimen would be to model polyagent chemotherapy using human patient‐derived tumor explants, which presently maintain the highest correlation of drug response to their human tumor counterparts. Ideally, as novel drug candidates are considered for incorporation into the multidrug ES regimen, one could rapidly elucidate if synergy exists when combined with a prescribed chemotherapy standard.
A complementary research approach could employ an emerging body “‐omic” data coming from a longitudinal study of patients’ tumors, pre‐ and post‐chemotherapy treatment, to determine how each patient's tumor responded at the genomic, proteomic, or epigenetic level to neoadjuvant chemotherapy. This information would, at its core, prove insightful by directly validating whether our current polyagent chemotherapy approach—whether this is VID or VDC‐IE—achieves its intended purpose, which is to eradicate tumor cells that would have otherwise survived individual therapies. Toward that end, while our study determined that poor tumor necrosis is linked to poor survival, an important question yet to be answered is how to use cutting‐edge technologies in the clinic to realize the expected clinical potential of precision‐based medicine.
Conclusion
A VID‐based chemotherapy backbone has been our institution's preferred method for neoadjuvant treatment of adults that present with primary ES. When combined with tailored adjuvant therapy based upon post‐treatment pathologic response, this approach yields favorable outcomes as compared with historical controls of adults treated with VDC‐IE. Further research would be required to determine if the improved survival observed in our study can be attributed to higher dosages of upfront doxorubicin and ifosfamide, or, instead, to extrinsic factors inherent in our clinical practice caring for ES patients. Given the intractable poor survival rate of those who present with metastatic disease at diagnosis, major clinical advances will hinge upon our ability to safely integrate drugs with novel mechanisms of action into a VID‐based chemotherapy backbone.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
Michael J. Wagner is supported by NIH grant T32 CA009666.
Footnotes
See the companion paper, “Localized Adult Ewing Sarcoma: Favorable Outcomes with Alternating Vincristine, Doxorubicin, Cyclophosphamide, and Ifosfamide, Etoposide (VDC/IE)‐Based Multimodality Therapy” by Michael J. Wagner, Vanceswaran Gopalakrishnan, Vinod Ravi et al., on page 1265 of this issue.
Author Contributions
Conception/design: Robert S. Benjamin, Michael J. Wagner, Dejka Araujo, Anthony P. Conley, Neeta Somaiah, Shreyaskumar R. Patel, Alexander Lazar
Provision of study material or patients: Robert S. Benjamin, Dejka Araujo, Joseph A. Ludwig, Ravin Ratan, J. Andrew Livingston, Vinod Ravi, Maria A. Zarzour, Anthony P. Conley, Neeta Somaiah, Shreyaskumar R. Patel, Alexander Lazar
Collection and/or assembly of data: Michael J. Wagner, Robert S. Benjamin, Vancheswaran Gopalakrishnan, J. Andrew Livingston, Vinod Ravi, Anthony P. Conley, Neeta Somaiah, Shreyaskumar R. Patel, Alexander Lazar
Data analysis and interpretation: Michael J. Wagner, Robert S. Benjamin, Vancheswaran Gopalakrishnan, Vinod Ravi, Anthony P. Conley, Neeta Somaiah, Shreyaskumar R. Patel, Alexander Lazar
Manuscript writing: Michael J. Wagner, Robert S. Benjamin, Vancheswaran Gopalakrishnan, Joseph A. Ludwig, Anthony P. Conley, Neeta Somaiah, Shreyaskumar R. Patel, Alexander Lazar
Final approval of manuscript: Robert S. Benjamin, Michael J. Wagner, Vancheswaran Gopalakrishnan, Vinod Ravi, J. Andrew Livingston, Dejka Araujo, Anthony P. Conley, Neeta Somaiah, Maria A. Zarzour, Ravin Ratan, Wei‐Lien Wang, Shreyaskumar R. Patel, Alexander Lazar, Joseph A. Ludwig
Disclosures
Vancheswaran Gopalakrishnan: UT MD Anderson Cancer Center (IP); Anthony P. Conley: Nektar, Novartis (C/A); Shreyaskumar R. Patel: Novartis, Janssen, Bayer, Eli Lilly & Co., CytRx, Eisai, EMD‐Serono (C/A), Janssen, Eisai, Morphotek (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.National Comprehensive Cancer Network. Soft Tissue Sarcomas (1.2015). Available at http://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf. Accessed September 27, 2015.
- 2. Womer RB, West DC, Krailo MD et al. Randomized controlled trial of interval‐compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children's Oncology Group. J Clin Oncol 2012;30:4148–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grier HE, Krailo MD, Tarbell NJ et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694–701. [DOI] [PubMed] [Google Scholar]
- 4. Juergens C, Weston C, Lewis I et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO‐E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 2006;47:22–29. [DOI] [PubMed] [Google Scholar]
- 5. Ladenstein R, Potschger U, Le Deley MC et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro‐EWING 99 trial. J Clin Oncol 2010;8:3284–3291. [DOI] [PubMed] [Google Scholar]
- 6. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 7. Pretz JL, Barysauskas CM, George S et al. Localized adult Ewing sarcoma: Favorable outcomes with alternating vincristine, doxorubicin, cyclophosphamide, and ifosfamide, etoposide (VDC/IE)‐based multimodality therapy. The Oncologist 2017;22:1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Judson I, Verweij J, Gelderblom H et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first‐line treatment of advanced or metastatic soft‐tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol 2014;15:415–423. [DOI] [PubMed] [Google Scholar]
- 9. Ahmed SK, Robinson SI, Okuno SH et al. Adult Ewing sarcoma: Survival and local control outcomes in 102 patients with localized disease . Sarcoma 10.1155/2013/681425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wagner MJ, Maki RG. Type 1 insulin‐like growth factor receptor targeted therapies in pediatric cancer. Front Oncol 2013;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scotlandi K, Benini S, Nanni P et al. Blockage of insulin‐like growth factor‐I receptor inhibits the growth of Ewing's sarcoma in athymic mice. Cancer Res 1998;58:4127–4131. [PubMed] [Google Scholar]
- 12. Pappo AS, Patel SR, Crowley J et al. R1507, a monoclonal antibody to the insulin‐like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 2011;29:4541–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Juergens H, Daw NC, Geoerger B et al. Preliminary efficacy of the anti‐insulin‐like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 2011;29:4534–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malempati S, Weigel B, Ingle AM et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: A report from the Children's Oncology Group. J Clin Oncol 2012;30:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tap WD, Demetri G, Barnette P et al. Phase II study of ganitumab, a fully human anti‐type‐1 insulin‐like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 2012;30:1849–1856. [DOI] [PubMed] [Google Scholar]
- 16. Tolcher AW, Sarantopoulos J, Patnaik A et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin‐like growth factor receptor 1. J Clin Oncol 2009;27:5800–5807. [DOI] [PubMed] [Google Scholar]
- 17. Olmos D, Postel‐Vinay S, Molife LR et al. Safety, pharmacokinetics, and preliminary activity of the anti‐IGF‐1R antibody figitumumab (CP‐751,871) in patients with sarcoma and Ewing's sarcoma: A phase 1 expansion cohort study. Lancet Oncol 2010;11:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Naing A, LoRusso P, Fu SQ et al. Insulin growth factor‐receptor (IGF‐1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 2012;18:2625–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartz GK, Tap WD, Qin LX et al. Cixutumumab and temsirolimus for patients with bone and soft‐tissue sarcoma: A multicentre, open‐label, phase 2 trial. Lancet Oncol 2013;14:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ordonez JL, Amaral AT, Carcaboso AM et al. The PARP inhibitor olaparib enhances the sensitivity of ewing sarcoma to trabectedin. Oncotarget 2015;6:18875–18890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amaral AT, Ordonez JL, Otero‐Motta AP et al. Innovative therapies in Ewing Sarcoma. Adv Anat Pathol 2014;21:44–62. [DOI] [PubMed] [Google Scholar]
- 22. Lamhamedi‐Cherradi SE, Menegaz BA, Ramamoorthy V et al. An oral formulation of YK‐4‐279: Preclinical efficacy and acquired resistance patterns in Ewing sarcoma. Mol Cancer Ther 2015;14:1591–1604. [DOI] [PubMed] [Google Scholar]
- 23. Zöllner SK, Commins R, Hong SH et al. Abstract 485: EWS‐FLI1 targeted small molecule YK‐4‐279 synergizes with vinca alkaloids through double hit to mitotic machinery. Cancer Res 2015;75:485. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.