

Abstract
Protein S-sulfinylation (R-SO2−) and S-sulfonylation (R-SO3−) are irreversible oxidative post-translational modifications of cysteine residues. Greater than 5% of cysteines are reported to occupy these higher oxidation states, which effectively inactivate the corresponding thiols and alter the electronic and physical properties of modified proteins. Such higher oxidation states are reached after excessive exposure to cellular oxidants, and accumulate across different disease states. Despite widespread and functionally relevant cysteine oxidation across the proteome, there are currently no robust methods to profile higher order cysteine oxidation. Traditional data-dependent liquid chromatography/tandem mass spectrometry (LC/MS/MS) methods generally miss low occupancy modifications in complex analyses. Here, we present a data-independent acquisition (DIA) LC/MS-based approach, leveraging the high IR absorbance of sulfoxides at 10.6 μm, for selective dissociation and discovery of S-sulfonated peptides. Across peptide standards and protein digests, we demonstrate selective infrared multiphoton dissociation (IRMPD) of S-sulfonated peptides in the background of unmodified peptides. This selective DIA IRMPD LC/MS-based approach allows identification and annotation of S-sulfonated peptides across complex mixtures while providing sufficient sequence information to localize the modification site.
TOC image
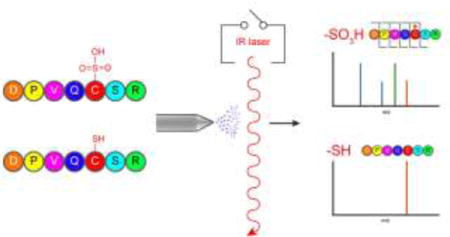
Originally thought to be merely by-products of cellular processes, reactive oxygen species such as hydrogen peroxide have emerged as important second messengers in cellular signaling.1–3 In proteins, the amino acid cysteine is particularly susceptible to redox chemistry and can be successively oxidized from a thiol (RSH) to sulfenic (RSOH), sulfinic (RSO2H), or sulfonic acid (RSO3H). Misregulation of these post-translational modifications (PTMs) has been associated with hypertension, cancer, aging, and neurodegenerative diseases.4–7 Thus, developing analytical techniques to profile these modifications is imperative.
Bottom up liquid chromatography/tandem mass spectrometry (LC/MS/MS) has become the leading technique for protein PTM analysis.8–12 Proteins are typically digested with trypsin and, when available, enrichment strategies for a particular modification are employed followed by LC separation of tryptic peptides online with MS/MS analysis. The eluting peptides are detected in MS1 mode, the observed ions are then sequentially isolated and dissociated, typically via collision induced dissociation (CID). The resulting product ions are searched against a genome-derived database or peptide MS/MS library to identify the isolated precursor ions and to locate any modifications. This “data-dependent analysis” (DDA) workflow has found widespread use due to the increasing availability of fast scanning, sensitive instruments for interrogating moderately abundant peptides and the existence of robust bioinformatics approaches.13–17 In more complex samples, however, this workflow’s dependence on semistochastic isolation of individual peptides prohibits the interrogation of low abundance species.18–20 Mann and co-workers suggested that a standard DDA experiment only analyzes ~16% of the eluting peptide ions.21 This low sampling rate is particularly concerning in PTM analysis, as the occupancy of any modification at a given site is often low.10
To partially overcome the low concentration of these modifications, a number of chemical enrichment techniques have been developed.10,22 For oxidative-based cysteine modifications specifically, the recent development of chemical proteomics methods has greatly advanced our understanding of redox regulation.7,23–25 The most widely used approach involves immediate alkylation of free cysteine residues, followed by the reduction and subsequent capture of any newly reduced cysteines. These methods are valuable for identifying proteins with redox-based modifications and have led to the discovery of many redox-regulated proteins and pathways.23,24,26–30 Reduction-switch methods, however, only capture reversible modifications and cannot access more advanced oxidation PTMs such as RSO2H and RSO3H. To enrich the latter largely unreactive modifications, RSO2H-specific probes and affinity-based techniques have been developed.25,32,33
A complementary approach to the aforementioned enrichment strategies is to eliminate the need for precursor-ion isolation by leveraging a physical (rather than chemical) attribute unique to modified peptides. Previous studies have shown that irradiation of all eluting peptides with 10.6 μm IR photons from a continuous wave (CW) CO2 laser selectively activates and dissociates phosphorylated peptides, enabling their differentiation from non-phosphorylated peptides.34–38 Other reports have demonstrated the use of gas-phase IR spectroscopic signatures as a means to identify the presence of phosphorylation, nitrosation, and oxidative-based PTMs.39–41 Despite the fact that sulfoxides absorb at a similar wavelength as phosphates, the application of infrared multiphoton dissociation (IRMPD) to sulfoxide-containing peptides has been limited.42–45 In fact, this methodology has, to our knowledge, not previously been applied to peptides containing cysteine-based oxidative modifications. Furthermore, we are not aware of any studies that have investigated the selectivity of this wavelength for the differentiation of sulfoxide-containing species.
Here, we demonstrate the preferential activation of S-sulfonated peptides by leveraging the strong S-O bond IR absorbance at 10.6 μm. This selective activation enables the discrimination of oxidized peptides in a data independent manner; thus permitting the facile identification of cysteine sulfonic acid-containing peptides in complex mixtures.
EXPERIMENTAL METHODS
Materials
Dithiothreitol, optima LC/MS grade formic acid, acetonitrile, and water were all purchased from Fisher Scientific (Fairlawn, NJ). Laminin β-1 Chain (925–933) (CDPGYIGSR), Substance P (RPKPQQFFGLM-NH2), Angiotensin I (DRVYIHPFHL), and ACTH 1–10 (SYSMEHFRWG) were all procured from BaChem (Bubendorf, Switzerland). Leucine enkephalin (YGGFL), 30% hydrogen peroxide, oxidized Insulin chain A (GIVEQC[SO3H]C[SO3H]ASVC[SO3H]SLYQLENYC[SO3H]N), and Insulin chain B (FVNQHLC[SO3H]GSHLVEALYLVC[SO3H]GERGFFYTPKA) were obtained from Sigma Aldrich (St. Louis, MO). The phosphorylated peptides (TSTEPQpYQPENL) and neuropeptide F (KRSpYEEHIP) were purchased from Millipore (Billerica, MA) and GenScript (Piscataway, NJ), respectively. Sequencing grade trypsin and urea were acquired from Promega (Madison, WI).
Protein Expression and Purification
DJ-1 and AhpC cDNAs were amplified from 293T cDNA, cloned into pET45b, a bacterial 6-His expression vector, and transformed into BL21 E. coli. Bacteria were grown in LB media at 37 °C to an OD600 of 0.6 and induced with 0.4 mM IPTG for four additional hours at 37 °C. Following sonication and lysozyme treatment, the lysate was incubated with Talon resin (Clontech) and loaded on a gravity column. After washing, the purified recombinant protein was eluted with imidazole and dialyzed into PBS buffer, typically yielding 10–15 mg/L of protein.
Oxidation Reactions
Oxidation of proteins and peptides was performed with performic acid. Performic acid was generated by mixing 4.5 mL formic acid with 0.5 mL 30% hydrogen peroxide and incubating the mixture at room temperature for 30 min as previously described by Kinumi et. al.46 The performic acid solution was then cooled on ice for 30 min. Simultaneously, 10 μL of formic acid was added to 20 μL of each protein or peptide stock solution and also placed on ice. Once cooled, 90 μL of performic acid was added to each protein or peptide stock solution and allowed to incubate on ice for 4 hours. The oxidized solutions were then diluted with 200 μL cool water, flash frozen on liquid nitrogen, and lyophilized overnight using a Labconco FreeZone 2.5 plus freeze drier (Kansas City, MO). The peptides were subsequently dissolved in water and proteins were dissolved in 10 mM triethylamine acetate.
Proteolytic Digestion
Proteolytic digestion of both oxidized and native proteins was performed with trypsin. Prior to digestion both oxidized and native proteins were subjected to denaturation and reduction with 2 mM urea and 1 mM DTT for 30 min at 60 °C. In order to effectively digest each protein, an overnight incubation at 37 °C at a trypsin:substrate ratio of 1:25 was used. The reaction was then diluted five-fold into water and immediately analyzed.
HPLC Separation
Separations of peptide standards and protein digests were conducted using an Agilent 1100 HPLC system (Agilent, Wilmington, DE) equipped with a Luna C18 column (25 cm × 1 mm, 5 μm particle size; Phenomenex, Torrance, CA). A two-step gradient was used to achieve separation of oxidized and unoxidized peptide standards. Acetonitrile with 0.1% formic acid was increased from 0.1 to 15 % over 10 minutes followed by a shallow gradient of 15 to 38% acetonitrile over 40 minutes. Proteolytic peptides were eluted using a linear gradient of acetonitrile containing 0.1% formic acid, increasing from 5% to 45% acetonitrile over 64 min at a flow rate of 100 μL/min. In both experiments, the eluent was directly introduced into the mass spectrometer.
Mass Spectrometry
All experiments were performed on either a 7 T Bruker SolariX (Billerica, MA) quadrupole-FT-ICR- mass spectrometer equipped with a Synrad Firestar 25 W, 10.6 μm infrared laser (Mukilteo, WA) or on the National High Magnetic Field Laboratory’s custom built FT-ICR instrument47 based on a 9.4 T Oxford Instruments 8” bore magnet (Oxney Mead, UK) equipped with a Synrad Firestar 25 W 10.6 μm infrared laser fitted with a 2.5× beam-expander. These instruments were both fitted with electrospray ionization (ESI) sources. For the SolariX LC/MS experiments, the capillary voltage, nebulizing gas flow rate, drying gas flow rate, and drying gas temperature were set to 4,900 V, 2.5 L/min, 5 L/min, and 220 °C respectively. For direct infusion experiments, the nebulizing gas and drying gas flow rates were set to 0.9 L/min and 2.5 L/min, respectively. For all experiments, a 0.25 s external accumulation time and a resolution of 512k were used. For the NHMFL instrument, the electrospray voltage and tube lens were set to 2,500 and 350 V, respectively.
For IRMPD experiments, both instruments utilized a BaF2 window for photon transmission. Irradiation times ranged from 0.12 to 0.5 s with laser powers varying from 10 to 23 W. Data analysis was performed manually using either Bruker Data Analysis 4.0 or Predator Analysis 4.1.8. All assignments were made with less than ±10 ppm error.
RESULTS AND DISCUSSION
Selective Dissociation of Cysteine Sulfonic Acid
To examine the ability of IR irradiation to selectively induce the dissociation of S-sulfonated peptides, we first examined the model peptide Laminin β-1 Chain (925–933, CDPGYIGSR). Oxidized Laminin β-1 Chain (925–933) was produced via performic acid oxidation and the modification site was confirmed with CID MS/MS (Figure S1 in the Supporting Information). The doubly protonated ions of both oxidized and unoxidized Laminin β-1 Chain were quadrupole isolated and subjected to an IR pulse of 500 ms at 23 W (using a x2.5 beam expander). The abundance of the unoxidized peptide was not affected by this IR pulse, while the oxidized Laminin β was extensively fragmented upon IR irradiation (Figure 1 and Figure S2 in the Supporting Information). The product ions generated from the oxidized peptide, particularly the y7 fragment lacking the modification and the b5* fragment containing the modification, enable the confirmation of the PTM to the first two N-terminal residues of the peptide.
Figure 1.
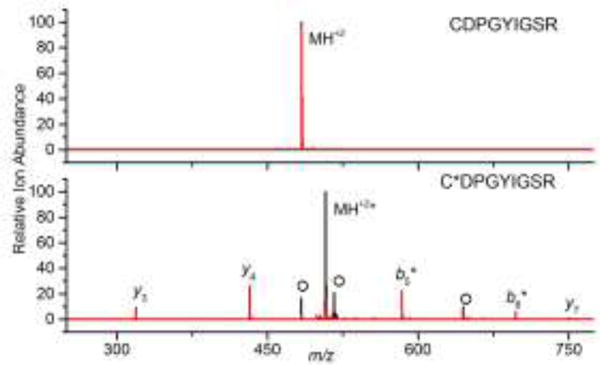
Overlaid mass spectra of isolated unoxidized (top) and oxidized (bottom) Laminin β (CDPGYIGSR and C[SO3]DPGYIGSR, respectively) before (black) and after (red) IR irradiation at 10.6 μm for 0.5 s at 23 W (using a x2.5 beam expander). Product ions that contain the sulfonic acid are designated with *. O denotes additional oxidation products.
The drastically different behavior we observe between the oxidized and unoxidized Laminin β peptides is not due to changes in critical energy as both ions require similar voltages (~7 V) to dissociate in CID (data not shown). This observation is in agreement with previous IRMPD work on phosphopeptides, demonstrating that selectivity is due to differences in photoabsorptivity and not critical energies.34,–38 Our findings are also consistent with previous CID work demonstrating that, under mobile-proton conditions, S-sulfonylation has little influence on critical energies.48 By contrast, it has been reported that this modification can enhance fragmentation under charge-remote conditions for both ESI and fast atom bombardment-generated peptide ions.48,49 However, we focus on multiply charged standard peptides with one or less arginines, and on tryptic peptide ions.
Because reduced thiols are typically alkylated in standard proteomics workflows (e.g., data below), we alkylated unoxidized Laminin β to mimic a typical proteomics experiment. Similar to the unoxidized peptide, the alkylated peptide was not affected by IR irradiation (Figure S3 in the Supporting Information).
Optimization and Characterization of Selective Infrared Multiphoton Dissociation
To further understand the observed differences in photoabsorptivity between oxidized and unoxidized peptides, a set of six standard peptides; oxidized Laminin β, methionine-oxidized ACTH 1–10, ACTH 1–10, Angiotensin I, oxidized Insulin chain A, and oxidized Insulin chain B were irradiated for 150 ms with increasing amounts of laser power. At the lowest power all peptide ions responded similarly, displaying only minor changes in abundance primarily due to the increased time trapped in the ICR cell (Figure 2). As the laser power approaches 10 W, however, the three S-sulfonylated peptides dissociate. Insulin A displays the highest sensitivity to IR irradiation, which could potentially be due to the presence of four S-sulfonylated cysteine residues. Interestingly, Insulin B and oxidized Laminin β demonstrate similar sensitivities despite possessing differing numbers of S-sulfonylated cysteine residues. This similarity could be explained by the fact that Laminin β is more labile than Insulin B requiring only ~7 V of collision energy compared to 17 V for Insulin B. In contrast to these S-sulfonylated peptides, the unmodified and methionine-oxidized peptides demonstrated no significant changes in ion abundance upon IR irradiation around 10 W. As the laser power increased past 15 W, both methionine-oxidized and unmodified ACTH began to dissociate. Angiotensin I remained intact throughout the experiment.
Figure 2.

Relative abundance of oxidized Laminin β, methionine-oxidized ACTH 1–10, ACTH 1–10, Angiotensin I, oxidized Insulin chain A, and oxidized Insulin chain B following IR irradiation of various laser powers for 0.15 s.
In addition to the six peptides included in Figure 2, a handful of phosphorylated and other unmodified peptides were irradiated. Consistent with previous work, the phosphorylated peptides dissociated readily upon the onset of IR irradiation (Table S1 in the Supporting Information).34–38 The ion abundance change for unmodified peptides fluctuated from 0 to as much as 40 % (Table S1 and S2 in the Supporting Information). Further experimentation identified the source of these fluctuations as the additional 150 ms spent trapped in the ICR cell, normal instrumental variability, and small amounts of fragmentation (not shown). Ion abundance fluctuations for unmodified peptides are consistently of a lower extent as compared with changes due to modification-induced IR sensitivity, in agreement with previous work.34 To maximize the difference in response between unmodified and S-sulfonylated peptides, all further experiments involved irradiation for 150 ms with 15 W laser power.
Selective IRMPD of Peptide Standards on an LC Time Scale
The ability to selectively induce the dissociation of S-sulfonated peptides through the use of IRMPD could enable the discovery of modified peptides in complex mixtures without individually isolating and fragmenting each eluting ion. Additionally, the generation of fragment ions enables the identification of discovered oxidized peptides and localization of the modification.
In order to examine the feasibility of this approach for differentiating modified peptides from unmodified ones on LC time scales, a mixture of the six standard peptides (Figure 2) was prepared in water at approximately equimolar (5 μM) concentrations. This sample was injected, separated via LC, and directly introduced into the mass spectrometer. The laser was fired during alternating scans, for an LC compatible 150 ms (15 W), allowing both MS and IRMPD spectra to be collected in the same LC run. Figure 3A displays the extracted ion chromatograms for each peptide with and without IR irradiation. The differences in peak areas indicate how sensitive the eluting peptide ions are to IR irradiation. The abundances of Angiotensin I (# 4, Figure 3A), ACTH1-10 (#3, Figure 3A), and methionine-oxidized ACTH 1–10 (#2, Figure 3A) are not affected by IR irradiation at this wavelength. This impotence is further demonstrated by overlaid mass spectra before and after IR irradiation for Angiotensin 1 (Figure 3C), ACTH 1–10, and methionine-oxidized ACTH 1–10 (Figure S4 in the Supporting Information). Importantly, the absence of fragmentation for oxidized ACTH 1–10 suggests that the presence of an oxidized methionine residue alone is not sufficient to enhance activation upon exposure to 10.6 μm IR light; congruent with studies that have shown oxidized methionine residues absorb light at approximately 9.5 μm, unless ion structure promotes S=O bond elongation.40,50
Figure 3.
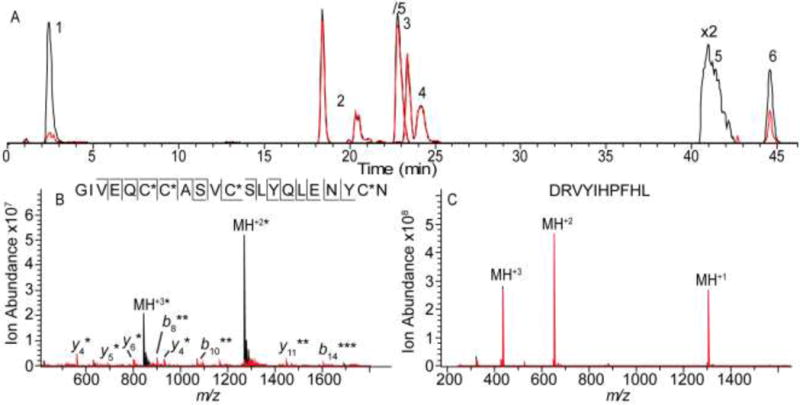
A) Extracted ion chromatograms for 1) oxidized Laminin β, 2) methionine-oxidized ACTH 1–10, 3) ACTH 1–10, 4) Angiotensin I, 5) oxidized Insulin chain A, and 6) oxidized Insulin chain B before (black) and after (red) IR irradiation (15 W for 0.15 s). Representative mass spectra for B) Insulin chain A (peak 5) and C) Angiotensin I (peak 4). The spectra following IR irradiation (red) are overlaid on the spectra acquired without irradiation (black). Product ions that retained the sulfonic acid are designated with *.
By contrast, the sulfonic acid-containing oxidized Laminin β (#1, Figure 3A), oxidized Insulin chain B (#5, Figure 3A), and oxidized Insulin chain A (#6, Figure 3A) all show dramatic abundance decreases upon IR irradiation with the corresponding peak areas dropping by 90, 99, and 51 %, respectively. Mass spectra for oxidized Insulin chain A (Figure 3B), Insulin chain B, and Laminin β (Figure S4 in the Supporting Information) illustrate the extent of fragmentation for these peptides. In each case, the resulting product ions enable confident peptide assignment and often provide site-specific localization of each oxidation site (Figure 3B and Figure S4 in the Supporting Information). For insulin chain A the product ions enable the localization of all four sulfonic acid modifications (Figure 3B).
Application of IRMPD to Digests of Oxidized Proteins
As illustrated above, IR irradiation induces significantly different behavior for peptides containing cysteine sulfonic acid compared with unmodified peptides. To further explore this selectivity, we probed a tryptic digest of oxidized protein deglycase DJ-1, associated with oxidative signaling and protection.51,52 Figure 4A shows the extracted ion chromatograms of each identified DJ-1 peptide with and without IR irradiation. The majority of eluting peptides were found to fluctuate in abundance as described above but did not significantly dissociate upon IR irradiation. Upon further investigation, these signals corresponded to unmodified or methionine-oxidized peptides (Table S2 in the Supporting Information). The DJ-1 peptide 13–27 (Figure 4, Peak 1, GAEEM[O2]ETVIPVDVM[O2]R) is an extreme example of these fluctuations, with the ion abundance dropping approximately 35% upon irradiation (Figure S5 A in the Supporting Information). Several eluting peptides did fragment significantly upon the onset of IR irradiation (Figure 4A Peaks 2–6). For peaks 2, 4, 5, and 6 the mass of the precursor ion and the resulting product ions identified these peaks as corresponding to oxidized DJ-1 peptides 49–62 (Figure 4B, DVVIC[SO3]PDASLEDAK), 100–115 (Figure 4C, GLIAAIC[SO3]AGPTALLAH), 100–122 (GLIAAIC[SO3]AGPTALLAHEIGFGSK), and 33–48 (VTVAGLAGKDPVQC[SO3]SR), respectively, and enabled the assignment of oxidation to all three cysteine residues in the protein.
Figure 4.
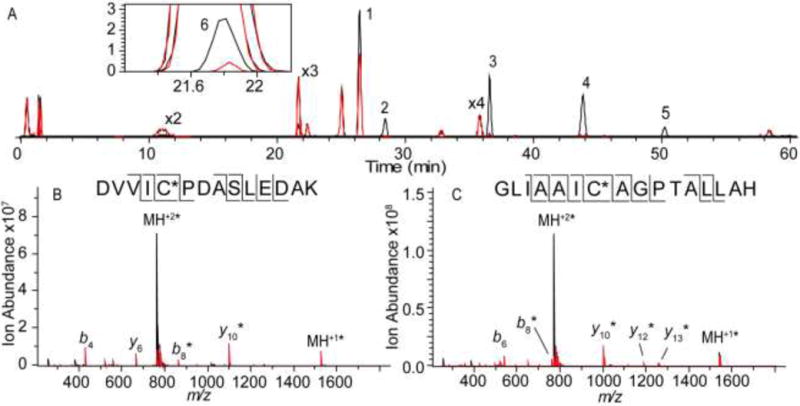
A) Extracted ion chromatograms for DJ-1 tryptic peptides before (black) and after (red) IR irradiation (15 W for 0.15 s). DJ-1 peptides 13–27 (Peak 1, GAEEM[O2]ETVIPVDVM[O2]R), 49–62 (peak 2, DVVIC[SO3]PDASLEDAK), 64–89 (peak 3, EGPYDVVVLPGGNLGAQNLSESAAVK), 100–115 (peak 4, GLIAAIC[SO3)AGPTALLAH), 100–122 (peak 5, GLIAAIC[SO3]AGPTALLAHEIGFGSK), and 33–48 (peak 6, VTVAGLAGKDPVQC[SO3]SR) are indicated on the chromatogram. The inset expands on the EIC for DJ-1 peptide 33–48. Mass spectra for B) DJ-1 peptide 49–62 (peak 2) and C) DJ-1 peptide 100–115 (peak 4) are also shown. The spectra following IR irradiation (red) are overlaid with spectra obtained without irradiation (black). Product ions that retained the sulfonic acid are designated with *.
Interestingly, Figure 4 peak 3 was identified as the unmodified peptide 64–89 (EGPYDVVVLPGGNLGAQNLSESAAVK). Upon further examination, it was found that a shorter version of this peptide, DJ-1 peptide 64–81 (Table S2 in the Supporting Information, EGPYDVVVLPGGNLGAQN), also fragmented significantly upon IR irradiation, suggesting that this sequence is somehow sensitive to IR irradiation. The unexpected facile fragmentation of these peptides may be explained by the combination of two factors: 1) the presence of multiple proline residues, which introduce gas phase labile peptide bonds into the peptide53 and 2) the N-terminal glutamic acid effect.54,55 The conversion of N-terminal glutamic acid to pyroglutamate and the subsequent loss of water requires very low energy to proceed.54 This cyclization results in the conversion of the N-terminal amine into an imide, thus reducing its proton affinity. Because the N-terminus is likely protonated in tryptic peptides, the reduction in proton affinity results in more facile mobile proton-induced dissociation throughout the peptide at significantly lower energies than in peptides lacking N-terminal glutamic acid.54 The presence of product ions prior to IR activation, the observed loss of water from all observed b-type product ions (Figure S5 B in the Supporting Information), and the fact that the related peptide 63–89 (KEGPYDVVVLPGGNLGAQNLSESAAVK, Table S2 in the Supporting Information) does not fragment supports this hypothesis. While ring strain likely prohibits this process in the oxidized DJ-1 peptide 49–62 (DVVIC[SO3]PDASLEDAK), we examined an unoxidized digest and subjected the unoxidized peptide analogue to IR irradiation to ensure that the peptide was not fragmenting due to a similar effect. IR irradiation failed to induce significant fragmentation in the unoxidized peptide (Table S2 and Figure S6 in the Supporting Information), thus confirming selective detection of the S-sulfonated peptide.
To supplement the DJ-1 analysis, we oxidized, digested, and probed the redox regulation-associated protein alkyl hydroperoxide reductase C (AhpC), which endogenously undergoes terminal oxidation upon excessive cellular oxidative load.56,57 As with DJ-1, we were able to selectively dissociate and differentiate S-sulfonated peptides from unmodified peptides with the exception of one peptide with an N-terminal glutamic acid (Table S2 in the Supporting Information). In total, we have examined the effect of IR irradiation on 74 peptides. Figure 5 summarizes the extent of fragmentation observed for all peptides and illustrates the significant difference in abundance distributions between modified and unmodified peptides. Overall, this bimodal distribution enables the differentiation of S-sulfonated peptides from unmodified peptides, allowing the facile discovery of these modifications.
Figure 5.
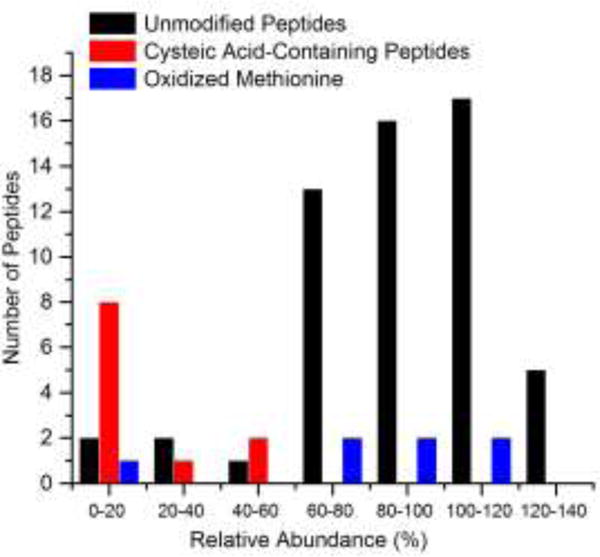
Peptides divided into bins by percent abundance remaining after IR irradiation.
CONCLUSIONS
We have examined the feasibility of using IR irradiation to screen for cysteine sulfonic acid in complex peptide mixtures. By applying this technique to peptide standards, we demonstrated that IR irradiation induces significantly higher extent of dissociation in S-sulfonated peptides compared with unmodified peptides. Utilizing this difference in IR absorbance, we applied IR irradiation without precursor ion isolation (i.e., data independent acquisition) to all eluting peptides from DJ-1 and AhpC digests. This selective IRMPD-based technique was able to induce significant fragmentation in oxidized peptides, while leaving unmodified peptides more intact. This differential behavior enabled the rapid assignment of modified peptides. Additionally, IRMPD-generated product ions often allowed the localization and assignment of the modification to a single residue. The ability to rapidly identify and sequence modified peptides in a single LC/MS run is a potentially powerful approach for the analysis of oxidative PTMs in cellular systems.
Supplementary Material
Acknowledgments
We thank Greg Blakney, Logan Krajewski and Lissa Anderson (NHMFL) for technical assistance. The NHMFL User Facility is funded by NSF (DMR-11-57490) and the State of Florida. The presented research was funded by the National Institutes of Health, grants DP2 GM114848 (to B.R.M.) and R01 GM107148 (to K.H.), and the University of Michigan.
Footnotes
Supporting Information
Additional example MS/MS spectra and raw abundance data can be found in the supplemental information. These items can be found online free of charge at http://pubs.acs.org/.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Antelmann H, Helmann JD. Antioxid Redox Signal. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC. Antioxid Redox Signal. 2009;11:2985–3011. doi: 10.1089/ars.2009.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veal EA, Day AM, Morgan BA. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Circ Res. 2013;112:382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyström T, Yang J, Molin M. Genes Dev. 2012;26:2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas JA, Mallis RJ. Exp Gerontol. 2001;36:1519–1526. doi: 10.1016/s0531-5565(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 7.Gu L, Robinson RAS. Proteomics - Clin Appl. 2016;10:1159–1177. doi: 10.1002/prca.201600015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witze ES, Old WM, Resing KA, Ahn NG. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 9.Mann M, Jensen ON. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 10.Olsen JV, Mann M. Mol Cell Proteomics. 2013;12:3444–3452. doi: 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner E, Luche S, Penna L, Chevallet M, Van Dorsselaer A, Leize-Wagner E, Rabilloud T. Biochem J. 2002;366:777–785. doi: 10.1042/BJ20020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eyers CE, Gaskell SJ. Wiley Encyclopedia of Chemical Biology. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2008. pp. 1–33. [Google Scholar]
- 13.Gu L, Evans AR, Robinson RAS. J Am Soc Mass Spectrom. 2015;26:615–630. doi: 10.1007/s13361-014-1059-9. [DOI] [PubMed] [Google Scholar]
- 14.Gu L, Robinson RA. Analyst. 2016;141:3904–3915. doi: 10.1039/c6an00417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin J, Meredith GE, Chen L, Zhou Y, Xu J, Shie FS, Lockhart P, Zhang J. Mol Brain Res. 2005;134:119–138. doi: 10.1016/j.molbrainres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Bateman NW, Goulding SP, Shulman NJ, Gadok AK, Szumlinski KK, MacCoss MJ, Wu CC. Mol Cell Proteomics. 2014;13:329–338. doi: 10.1074/mcp.M112.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porter CJ, Bereman MS. Anal Bioanal Chem. 2015;407:6627–6635. doi: 10.1007/s00216-015-8819-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Law KP, Lim YP. Expert Rev Proteomics. 2013;10:551–566. doi: 10.1586/14789450.2013.858022. [DOI] [PubMed] [Google Scholar]
- 19.Bauer M, Ahrné E, Baron AP, Glatter T, Fava LL, Santamaria A, Nigg EA, Schmidt A. J Proteome Res. 2014;13:5973–5988. doi: 10.1021/pr500860c. [DOI] [PubMed] [Google Scholar]
- 20.Mayne J, Ning Z, Zhang X, Starr AE, Chen R, Deeke S, Chiang C-K, Xu B, Wen M, Cheng K, Seebun D, Star A, Moore JI, Figeys D. Anal Chem. 2016;88:95–121. doi: 10.1021/acs.analchem.5b04230. [DOI] [PubMed] [Google Scholar]
- 21.Michalski A, Cox J, Mann M. J Proteome Res. 2011;10:1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Jensen ON. Proteomics. 2009;9:4632–4641. doi: 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couvertier SM, Zhou Y, Weerapana E. Biochim Biophys Acta. 2014;1844:2315–2330. doi: 10.1016/j.bbapap.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 24.Leonard SE, Carroll KS. Curr Opin Chem Biol. 2011;15:88–102. doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 25.Majmudar JD, Konopko AM, Labby KJ, Tom CTMB, Crellin JE, Prakash A, Martin BR. J Am Chem Soc. 2016;138:1852–1859. doi: 10.1021/jacs.5b06806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones DP, Go YM. Curr Opin Chem Biol. 2011;15:103–112. doi: 10.1016/j.cbpa.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Murray CI, Van Eyk JE. Circ Cardiovasc Genet. 2012;5:591–601. doi: 10.1161/CIRCGENETICS.111.961425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeon S, Kim YJ, Kim ST, Moon W, Chae Y, Kang M, Chung MY, Lee H, Hong MS, Chung JH, Joh TH, Lee H, Park H. J Proteomics. 2008;8:4822–4832. doi: 10.1002/pmic.200700955. [DOI] [PubMed] [Google Scholar]
- 29.De Iuliis A, Grigoletto J, Recchia A, Giusti P, Arslan P. Clin Chim Acta. 2005;357:202–209. doi: 10.1016/j.cccn.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 30.Licker V, Kövari E, Hochstrasser DF, Burkhard PR. J Proteomics. 2009;73:10–29. doi: 10.1016/j.jprot.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 31.Klomsiri C, Karplus PA, Poole LB. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang YC, Huang CN, Lin CH, Chang HC, Wu CC. Proteomics. 2010;10:2961–2971. doi: 10.1002/pmic.200900850. [DOI] [PubMed] [Google Scholar]
- 33.Lo Conte M, Lin J, Wilson MA, Carroll KS. ACS Chem Biol. 2015;10:1825–1830. doi: 10.1021/acschembio.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowe MC, Brodbelt JS. Anal Chem. 2005;77:5726–5734. doi: 10.1021/ac0509410. [DOI] [PubMed] [Google Scholar]
- 35.Crowe MC, Brodbelt JS. J Am Soc Mass Spectrom. 2004;15:1581–1592. doi: 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 36.Flora JW, Muddiman DC. Anal Chem. 2001;73:3305–3311. doi: 10.1021/ac010333u. [DOI] [PubMed] [Google Scholar]
- 37.Flora JW, Muddiman DC. J Am Chem Soc. 2002;124:6546–6547. doi: 10.1021/ja0261170. [DOI] [PubMed] [Google Scholar]
- 38.Flora JW, Muddiman DC. J Am Soc Mass Spectrom. 2004;15:121–127. doi: 10.1016/j.jasms.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Lanucara F, Chiavarino B, Crestoni ME, Scuderi D, Sinha RK, Maître P, Fornarini S. Int J Mass Spectrom. 2012;330–332:160–167. [Google Scholar]
- 40.Ravi J, Hills AE, Cerasoli E, Rakowska PD, Ryadnov MG. Eur Biophys J. 2011;40:339–345. doi: 10.1007/s00249-010-0656-1. [DOI] [PubMed] [Google Scholar]
- 41.Correia CF, Clavaguera C, Erlekam U, Scuderi D, Ohanessian G. Chem Phys Chem. 2008;9:2564–2573. doi: 10.1002/cphc.200800469. [DOI] [PubMed] [Google Scholar]
- 42.Scuderi D, Ignasiak MT, Serfaty X, de Oliveira P, Houée Levin C. Phys Chem Chem Phys. 2015;17:25998–26007. doi: 10.1039/c5cp03223g. [DOI] [PubMed] [Google Scholar]
- 43.Paciotti R, Coletti C, Re N, Scuderi D, Chiavarino B, Fornarini S, Crestoni ME. Phys Chem Chem Phys. 2015;17:25891–25904. doi: 10.1039/c5cp01409c. [DOI] [PubMed] [Google Scholar]
- 44.Wilson JJ, Brodbelt JS. Anal Chem. 2006;78:6855–6862. doi: 10.1021/ac060760d. [DOI] [PubMed] [Google Scholar]
- 45.Keough T, Youngquist RS, Lacey MP. Proc Natl Acad Sci U S A. 1999;96:7131–7136. doi: 10.1073/pnas.96.13.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinumi T, Shimomae Y, Arakawa R, Tatsu Y, Shigeri Y, Yumoto N, Niki E. J Mass Spectrom. 2006;41:103–112. doi: 10.1002/jms.973. [DOI] [PubMed] [Google Scholar]
- 47.Kaiser NK, Quinn JP, Blakney GT, Hendrickson CL, Marshall AG. J Am Soc Mass Spectrom. 2011;22:1343–1351. doi: 10.1007/s13361-011-0141-9. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Vivekananda S, Men L, Zhang Q. J Am Soc Mass Spectrom. 2004;15:697–702. doi: 10.1016/j.jasms.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Burlet O, Yang CY, Gaskell SJ. J Am Soc Mass Spectrom. 1992;3:337–344. doi: 10.1016/1044-0305(92)87061-3. [DOI] [PubMed] [Google Scholar]
- 50.Ignasiak M, Scuderi D, de Oliveira P, Pedzinski T, Rayah Y, Houée Levin C. Chem Phys Lett. 2011;502:29–36. [Google Scholar]
- 51.Wilson MA. Antioxid Redox Signal. 2011;15:111–122. doi: 10.1089/ars.2010.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canet-Avilés RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaisar T, Urban J. J Mass Spectrom. 1996;31:1185–1187. doi: 10.1002/(SICI)1096-9888(199610)31:10<1185::AID-JMS396>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 54.Godugu B, Neta P, Simón-Manso Y, Stein SE. J Am Soc Mass Spectrom. 2010;21:1169–1176. doi: 10.1016/j.jasms.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 55.Khatun J, Ramkissoon K, Giddings MC. Anal Chem. 2007;79:3032–3040. doi: 10.1021/ac061455v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biteau B, Labarre J, Toledano MB. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 57.Poole LB. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.