

Abstract
Here we show that the adenovirus major late promoter produces a 31-nucleotide transcriptional start site small RNA (MLP-TSS-sRNA) that retains the 7-methylguanosine (m7G)-cap and is incorporated onto Ago2-containing RNA-induced silencing complexes (RISC) in human adenovirus-37 infected cells. RNA polymerase II CLIP (UV-cross linking immunoprecipitation) experiments suggest that the MLP-TSS-sRNA is produced by promoter proximal stalling/termination of RNA polymerase II transcription at the site of the small RNA 3′ end. The MLP-TSS-sRNA is highly stable in cells and functionally active, down-regulating complementary targets in a sequence and dose-dependent manner. The MLP-TSS-sRNA is transcribed from the opposite strand to the adenoviral DNA polymerase and preterminal protein mRNAs, two essential viral replication proteins. We show that the MLP-TSS-sRNA act in trans to reduce DNA polymerase and preterminal protein mRNA expression. As a consequence of this, the MLP-TSS-sRNA has an inhibitory effect on the efficiency of viral DNA replication. Collectively, our results suggest that this novel sRNA may serve a regulatory function controlling viral genome replication during a lytic and/or persistent adenovirus infection in its natural host.
Keywords: adenovirus, Ago2, m7G-cap, small RNA
INTRODUCTION
MicroRNAs (miRNAs) are abundant 22-nucleotide (nt) regulatory RNAs, typically derived from endogenous transcripts, with short hairpins, that modulate essential cellular processes at the post-transcriptional level in most eukaryotes (for review, see Filipowicz et al. 2008). Canonical miRNAs are produced from long primary miRNA transcripts by two RNaseIII-like cleavage events. First, the primary miRNA is processed by the Drosha/DGCR8 microprocessor complex in the nucleus into a 60- to 80-nt precursor miRNA hairpin that is exported to the cytoplasm. Here the Dicer enzyme cleaves the loop of the hairpin to produce an ∼22-base pair (bp) mature miRNA duplex (Kim et al. 2009). Based on the relative thermodynamic stability of the duplex ends (Khvorova et al. 2003; Schwarz et al. 2003), one or both of the strands are loaded onto one of four Ago proteins to form the RNA-induced silencing complex (RISC) (Yang and Lai 2011). The RISC complex functions through miRNA-directed base-pairing to target mRNA transcripts for post-transcriptional gene silencing (Wilson and Doudna 2013).
Human adenoviruses (HAds) encode for one or two virus-associated RNAs (VA RNAs), designated as VA RNAI and VA RNAII (for review, see Punga et al. 2013). The VA RNAs are ∼160-nt long noncoding RNAs, transcribed by RNA polymerase III, that fold into highly structured RNAs that show features similar to pre-miRNAs. The terminal stem of the VA RNAs from several HAdVs is processed by Dicer into small viral miRNAs, so-called mivaRNAs, that associate with Ago2-containing RISC (Andersson et al. 2005; Sano et al. 2006; Xu et al. 2007; Kamel et al. 2014). The HAdV-5 mivaRNAs function as miRNAs that regulate host cell gene expression by targeting complementary sequences in the 3′ UTR of an mRNA (Aparicio et al. 2010). However, the significance of these interactions for a lytic infection, at least in tissue culture cells, has been questioned (Kamel et al. 2013). Although most of the mivaRNA research has been done in the HAdV-5 model system, our work demonstrates that the mivaRNAs expressed in HAdV-11- and HAdV-37-infected cells are more abundant and more efficiently associated with Ago2-containing RISC (Kamel et al. 2014).
In addition to the classical miRNA biogenesis pathway, a variety of alternative noncanonical mechanisms to generate Ago-associated miRNAs have been reported (for review, see Yang and Lai 2011). For example, the mirtron pathway produces miRNAs by Dicer cleavage of spliced out introns (Okamura et al. 2007; Ruby et al. 2007; Flynt et al. 2010). Also, some miRNAs produced by the herpesvirus family of viruses are transcribed as tails to other noncoding RNAs and trimmed either by RNaseZ or the Integrator complex (Bogerd et al. 2010; Cazalla et al. 2011). Noncanonical miRNAs have also been reported from tRNA (Lee et al. 2009), small nucleolar RNAs (Ender et al. 2008), and endogenous short-hairpin RNAs (Babiarz et al. 2008). In addition, it has been demonstrated that the vertebrate miR-451 is generated directly by the Ago2 slicer activity rather than Dicer cleavage (Cheloufi et al. 2010; Cifuentes et al. 2010).
Here we present a novel mechanism to produce an Ago2-associated small RNA. Thus, we show that the promoter proximal region of the adenovirus major late promoter (MLP) produces transcriptional start site (TSS) small RNAs, which we termed MLP-TSS-sRNAs, that associates with Ago2 in HAd-infected cells. The MLP produces a major 31-nt MLP-TSS-sRNA with a 5′ end that coincides with the MLP +1 start site. The MLP-TSS-sRNAs were detected in all HAds analyzed (HAdV-4, HAdV-5, HAdV-11, and HAdV-37) at varying levels. The HAdV-37 MLP-TSS-sRNAs contains a 7-methylguanosine (m7G)-cap and are enriched in Ago2-containing RISC. The MLP-TSS-sRNAs were capable of inhibiting reporter gene expression and had a capacity to suppress viral DNA replication during a lytic virus infection. Further, we show that the HAdV-37 MLP-TSS-sRNAs, in contrast to the mivaRNAs, are produced by a distinct pathway not requiring the Exportin5 nuclear export receptor or the Dicer enzyme. Our data suggest that the MLP-TSS-sRNAs are generated by a novel mechanism where a short prematurely terminated RNA polymerase II (RNAPII) transcript becomes Ago2 associated.
RESULTS
Production of RISC-associated sRNAs from the HAdV-37 MLP transcriptional start site
In a recent study, we sequenced the pool of viral sRNAs from several adenovirus infections (Kamel et al. 2014), primarily to map the structure and abundance of the VA RNA-derived mivaRNAs expressed in the different serotypes. As expected, the vast majority of sRNAs mapped to the VA RNA region (Fig. 1B). However, at a closer examination of the data, we noted that a small fraction of the sRNAs enriched in RISC mapped to the MLP-TSS (Fig. 1C) with a major 31-nt species with a 5′ end at the classical MLP +1 cap site and a minor 34 nt species (20-fold lower) with a 5′ end located at position −3 relative to the MLP cap site. Since both MLP-TSS RNAs were enriched in immunopurified Flag-Ago2-containing RISC, we refer to them (+1 and −3 start site RNAs) as MLP-TSS-sRNAs. The same MLP-TSS-sRNAs were detected in all serotypes analyzed (HAdV-4, HAdV-5, HAdV-11, and HAdV-37) but were most highly expressed in HAdV-37-infected cells (Supplemental Table 1). Therefore, all subsequent experiments were done using this virus as the model virus.
FIGURE 1.

Production of RISC-associated sRNAs from the HAdV-37 MLP transcriptional start site. (A) Genomic organization of HAdV-37 showing the location of different viral transcription units. (B) Distribution of viral small RNA reads (shown as percentage of total mapped viral reads) across the HAdV-37 genome both in the cytoplasmic fraction (Blue) and F-Ago2-bound fraction (red). (C) Close-up view of the HAdV-37 MLP showing the mapped read coverage around the first leader (boxed) and the location of the MLP-TSS-sRNAs. (D) Size distribution of the MLP-TSS-sRNA (−3 on the left side and +1 on the right side) both in the cytoplasmic fraction (blue) and Ago2-associated fraction (red).
The HAdV-37 +1 and −3 MLP-TSS-sRNAs share a major 3′ end, located 9 nt upstream of the first leader 5′ splice site (Fig. 1C). The length distribution of the +1 TSS-sRNAs was predominantly of 31 nt with a trailing fraction generating a second peak of 35 nt in length and minor peaks of 24 and 32 (Fig. 1D). The −3 TSS-sRNAs had a major peak at 34 nt but a larger heterogeneity of both shorter and longer types (Fig. 1D). Collectively, our data show that the HAdV-37 MLP-TSS-sRNAs are the most abundant RISC-associated adenoviral sRNAs outside the VA region (Fig. 1B), accumulating to levels comparable to many of the most abundant cellular miRNAs expressed in HAdV-37-infected cells (Supplemental Fig. 1).
The 5′ end of the HAdV-37 MLP-TSS-sRNAs are modified
To characterize the MLP-TSS-sRNAs, we used carbodiimide-mediated cross-linking of RNA to a nylon membrane (Pall and Hamilton 2008). This method requires a free 5′ phosphate group for chemical binding to the membrane. We used this feature of the method to investigate whether the 5′ end of the MLT-TSS-sRNAs was modified. Since the 5′ ends of the large majority of these RNAs coincide with the well-characterized MLP cap site, we used tobacco acid pyrophosphatase (TAP) or RNA 5′ pyrophosphohydrolase (RppH) treatment to determine whether they contain a 5′ end modification that hinders cross-linking of the RNAs to the membrane. The TAP and RppH enzymes remove the m7G-cap from mRNAs leaving a 5′ monophosphate group (Lockard et al. 1980; Deana et al. 2008). As shown in Figure 2, TAP treatment of total cytoplasmic RNA from HAdV-37-infected cells increased the cross-linking efficiency of MLP-TSS-sRNA by 13- to 15-fold (lanes 3 and 4). In contrast, cross-linking of HAdV-37 VA RNAII and tRNA-lysine, which are RNA polymerase III products, or let-7b miRNA were not enhanced by TAP treatment (lanes 3 and 4). Similar results were obtained using RppH (Supplemental Fig. 2). Based on these findings we TAP treated the RNA before performing Northern blot analysis in all subsequent experiments. Interestingly, a series of longer transcripts was also enhanced after TAP or RppH treatment (Fig. 2, lane 4; Supplemental Fig. 2). These RNAs most likely correspond to promoter proximal preterminated MLP transcripts (see below). As expected from the expression profile of the MLP (Jansen-Durr et al. 1988) and the VA RNAs (Söderlund et al. 1976), the MLP-TSS-sRNAs start to accumulate toward the late phase of infection (12–18 h post infection [hpi]), whereas the full-length VA RNAII was detectable already at 6 hpi and the processed mivaRNAII at 12 hpi (Supplemental Fig. 3).
FIGURE 2.
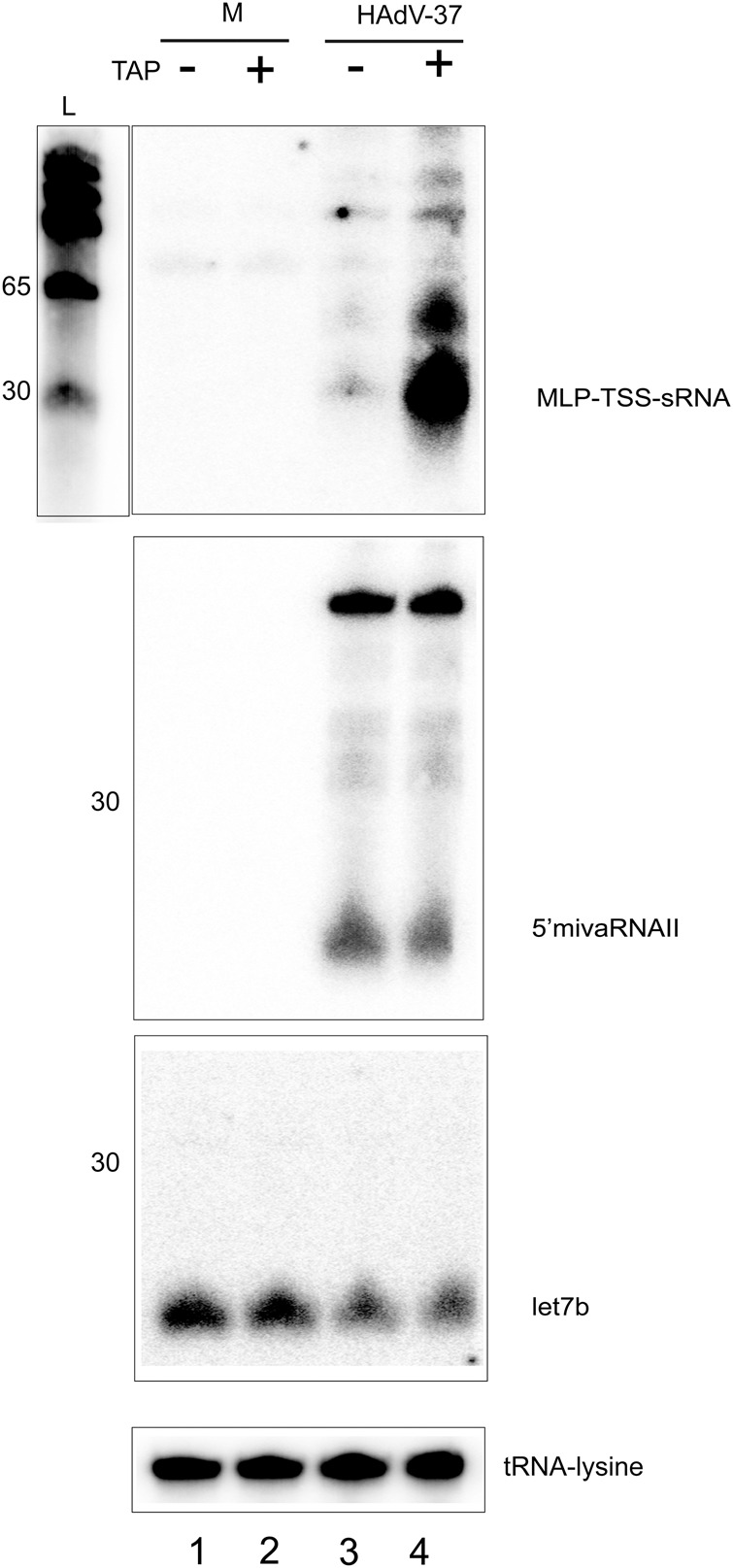
The 5′ end of the HAdV-37 MLP-TSS-sRNAs are modified and produced during the late phase of infection. Northern blot analysis of cytoplasmic RNA extracted from HAdV-37 (5 FFU per cell) and mock (M) infected cells. The presence of a modified 5′ end was probed using TAP treatment. The same membrane was sequentially probed for the MLP-TSS-sRNA, 5′ mivaRNAII, let7b, and tRNA lysine.
The HAdV-37 MLP-TSS-sRNAs are capped and enriched in Ago2-containing RISC
To determine whether the Ago2-associated MLP-TSS-sRNAs contain an m7G-cap we immunopurified Flag–Ago2 complexes from cytoplasmic extracts prepared from HAdV-37-infected 293-Flag–Ago2 cells at 24 hpi. As expected from our pervious results, the processed 5′ mivaRNAII was enriched in the Ago2–RISC (Fig. 3A, lane 4; Kamel et al. 2014). In agreement with our sRNA sequencing data the MLP-TSS-sRNA was also found in the Ago2–RISC (Fig. 3A, lane 4). As a control of specificity we show that the actin protein did not contaminate the Ago2–RISC fraction (Fig. 3A, lanes 3 and 4). The Ago2–RISC RNAs were purified by phenol extraction and subjected to a second round of immunoprecipitation using an anti-m7G-cap monoclonal antibody. As shown in Figure 3B, the MLP-TSS-sRNAs were recovered in the m7G immunoprecipitate (lane 4), whereas the 5′ mivaRNAII and the let7b miRNA, as expected, were not detectable in the m7G immunoprecipitate (lanes 3 and 4). Collectively, these results suggest that the MLP-TSS-sRNA retains the m7G-cap added during the transcription initiation process. Further, the TAP treatment of the cytoplasmic RNA fraction (Fig. 2) strongly argues that at least 95% of the MLP-TSS-sRNAs are capped.
FIGURE 3.

The HAdV-37 MLP-TSS-sRNAs are capped and enriched in Ago2-containing RISC. (A) Northern blot analysis of total cytoplasmic (Cyto) and Flag–Ago2-associated (F-Ago2-IP) RNA. The membrane was probed sequentially for MLP-TSS-sRNA and 5′ mivaRNAII. The lower panel is a Western blot for Flag–Ago2 and Actin in the cytoplasmic and Ago2-bound fractions. (B) Northern blot analysis demonstrating that the Ago2-associated MLP-TSS-sRNAs contain a 5′ cap structure. Ago2-associated RNAs were used for immunoprecipitation with an m7G-cap antibody. The membrane was sequentially probed for MLP-TSS-sRNA, 5′ mivaRNAII and let7b.
The HAdV-37 MLP-TSS-sRNAs are produced by a pathway distinct from the mivaRNAs
Previous studies have shown that VA RNA export requires Exportin5 (Yi et al. 2003; Lu and Cullen 2004) and that the mivaRNAs are produced by Dicer cleavage (Kamel et al. 2014). Considering the novel features of the MLP-TSS-sRNA, it was of interest to study whether the mivaRNAs and the MLP-TSS-sRNAs require the same cellular machinery for export and processing. To test this we used an siRNA approach to knock down candidate proteins. As shown in Supplemental Figure 4A, Dicer knockdown reduced 5′ mivaRNA processing, whereas MLP-TSS-sRNA production was unaffected (lanes 2 and 3). Also, knockdown of the Dis3 RNase in the nuclear exosome complex did not impair the maturation of the MLP-TSS-sRNA or the 5′ mivRNAII (lane 7). As expected from previous results (Yi et al. 2003), knockdown of Exportin5 reduced mivaRNA production (lane 6). In contrast, targeting Exportin5 did not impair export of the MLP-TSS-sRNA. To investigate whether MLP-TSS-sRNA production is dependent on the Ago2 slicer activity, like mir-451 (Cheloufi et al. 2010; Cifuentes et al. 2010), HEK293 cells were transfected with plasmids expressing Flag-epitope-tagged wild-type Ago2 or the catalytically dead Ago2 (D669A) mutant proteins (Cifuentes et al. 2010). Cells were infected with HAdV-37 and the sRNA content in the cytoplasmic Ago2 fractions analyzed at 24 hpi. The result shows that the Ago2 catalytic activity is not required for MLP-TSS-sRNA production (Supplemental Fig. 4C, lanes 9 and 10). The analysis further demonstrated that the MLP-TSS-sRNA also associates with Ago1, which lacks slicer activity.
The MLP-TSS-sRNA is an RNA polymerase II transcription termination product
In order to gain further insights into the biogenesis of the MLP-TSS-sRNA, we aimed to investigate the sequence of the potential precursor transcript(s) that could be the origin of MLP-TSS-sRNA. For this experiment, 293-Flag-Ago2 cells were transfected with three pGL4 reporter plasmids (Fig. 4A) for 24 h followed by total cytoplasmic RNA extraction and Northern blot analysis. Reporter plasmid UnMLT corresponds to the unspliced precursor RNA, and contains the MLP fused to the first 200 nt downstream from the transcriptional start site. Plasmid Trip contained the MLP fused to a cDNA copy of the spliced tripartite leader sequence, whereas reported plasmid RevTrip contained a cDNA copy of the tripartite leader where the second and third exons had been inserted in the reverse orientation. As shown in Figure 4B, all three reporter plasmids accumulated a small RNA ∼31 nt long. This result indicates the MLP-TSS-sRNAs can be produced from both the unspliced MLP precursor RNA and the spliced tripartite leader. Most interestingly, the MLP-TSS-sRNA was also produced in cells transfected with the plasmid containing a reversed copy of the tripartite leader exons 2 and 3. This plasmid contains the MLP-TSS-sRNA sequence plus only two extra nucleotides from the first leader exon (Fig. 1C). Collectively, these results argue that the MLP-TSS-sRNA is not processed from a precursor RNA requiring downstream sequences beyond 2 nt from the MLP-TSS-sRNA 3′ end. Alternatively, the MLP-TSS-sRNA could be generated as a direct product of RNAPII stalling/termination downstream from the MLP initiation site. To test this hypothesis, we determined whether we could detect an increased accumulation of RNAPII at the site of the MLP-TSS-sRNA 3′ end. For this experiment, 293Ago2 cells were infected with HAdV-37 or transfected with plasmid UnMLT. Following 24-h incubation, cells were UV-irradiated to halt further transcription, and RNAPII complexes were isolated by immunoprecipitation. The supernatant after RNAPII-IP was used to immunopurify Ago2 complexes, and the sRNA content in both fractions was analyzed by Northern blot. As shown in Figure 4C, a distinct band (lanes 4–6) of the size of the Ago2-associated MLP-TSS-sRNA (lane 8) appeared in the RNAPII precipitate, particularly in HAdV-37-infected cells (lane 5). The same band was also recovered from plasmid-transfected cells, although with a considerably lower efficiency (lane 6). The more pronounced site for RNAPII stalling in the transfected cells was a site located ∼60 nt downstream from the MLP-TSS. This sRNA can be seen in most experiments (lane 5 and Figs. 2, 4; Supplemental Figs. 1–5) and is also Ago2-RISC-associated (lanes 8 and 9). The specificity of the immunoprecipitations was confirmed by probing the same membrane for VA RNAII and tRNA lysine, which are RNA polymerase III products. The specificity of the immunoprecipitations was also confirmed by Western blotting of the indicated proteins (Fig. 4D). Collectively, these results are compatible with the hypothesis that the MLP-TSS-sRNA is an RNAPII transcript produced by RNA polymerase II stalling/termination shortly after initiation at the MLP.
FIGURE 4.

The HAdV-37 MLP-TSS-sRNAs are products of premature termination of RNA polymerase II transcription. (A) Schematic diagram of the different constructs used for the transfection assay. UnMLT contains the MLP fused to the first 200 nt downstream from the transcriptional start site, Trip contains the MLP fused to a cDNA copy of the spliced tripartite leader sequence, and RevTrip contains a cDNA copy of the tripartite leader where the second and third exons had been inserted in the reverse orientation. (B) Northern blot analysis of total cytoplasmic RNA isolated 24 h post transfection with the indicated plasmids. The same membrane was sequentially probed for the MLP-TSS-sRNA and tRNA lysine. (C) Northern blot analysis of total (Input), RNAPII-associated small RNA (RNAPII-IP), and Ago2-associated (Ago2-IP) RNA isolated from HAdV-37 infected 293-Flag-Ago2 cells at 30 hpi. The membrane was probed sequentially for the MLP-TSS-sRNA, 5′ mivaRNAII and tRNA lysine. (D) Western blot analysis of RNAPII, Ago2, and Actin in the fractions displayed in panel C.
The HAdV-37 MLP-TSS-sRNAs repress complementary targets
Importantly, the MLP-TSS-sRNA associates with the endogenous Ago2 protein in HAdV-37-infected ARPE-19 (Supplemental Fig. 5A). The specificity of this interaction was confirmed by the lack of tRNA lysine and the GAPDH protein in the Ago2 immunoprecipitate (Supplemental Fig. 5B).
To determine whether the MLP-TSS-sRNAs loaded onto RISC are functional, we tested the capacity of the complex to regulate expression of a firefly luciferase reporter construct with a complementary MLP-TSS-sRNA binding site in the 3′ UTR [pmirGlo(+), Fig. 5A]. For this experiment ARPE-19 cells were first infected with two concentrations of HAdV-37 followed by transfection of reporter plasmids pmirGlo, lacking an MLP-TSS-sRNA binding site, or pmirGlo(+) at 1 hpi. Cells were harvested at 24 hpi and the effect of the HAdV-37 infection on luciferase expression measured. As shown in Figure 5B, the relative luciferase expression was progressively reduced in pmirGlo(+)-infected cells, whereas luciferase expression in cells transfected with the pmirGlo reporter construct were unaffected. To further investigate the specificity of this interaction, we cloned the MLP-TSS-sRNA binding site in the reverse orientation, generating plasmid pmirGlo(Rev) (Fig. 5A). As shown in Figure 5C, an HAdV-37 infection significantly suppressed gene expression from pmirGlo(+) without having a negative effect on luciferase expression from reporter plasmids pmirGlo or pmirGlo(Rev). Taken together, these results suggest that during a HAdV-37 infection, RISC programmed with the MLP-TSS-sRNAs are functionally active and capable of suppressing reporter gene expression in a sequence-specific and dose-dependent manner.
FIGURE 5.

The HAdV-37 MLP-TSS-sRNAs repress complementary targets. (A) Schematic diagram of the different luciferase reporters used: pmirGlo lacks an MLP-TSS-sRNA binding site, pmirGlo(+) contains a complementary binding site, pmirGlo(Rev) has the binding site in reverse orientation, and pmirGlo(mut) has multiple mutations in the binding site. (B,C) ARPE-19 cells were infected with HAdV-37 or mock infected. Following a 1 h incubation, cells were transfected with the indicated reporter plasmids. At 23 h post transfection, total cell lysate was prepared and luciferase activity measured. The result from three independent experiments is shown in the graph. Error bars indicate the SD. (D) ARPE-19 cells were cotransfected with the in vitro–transcribed and capped MLP-TSS-sRNA or a control RNA (Ctrl) together with the indicated reporter plasmids. Luciferase expression was measured at 18 h post transfection. The mean from three independent experiments is shown in the graph. Error bars indicate the SD ([*] P < 0.05, [**] P < 0.01, [***] P < 0.001). (E) RNA secondary structure prediction for the 31-nt MLP-TSS-sRNA, in the upper panel, and a shorter sRNA version that lacks the last 12 nt (MLP-TSS-sRNA[trunc]) in the lower panel. (F) Northern blot analysis of total RNA isolated at the indicated time post transfection with 10 nM RNA oligonucleotides (either MLP-TSS-sRNA or MLP-TSS-sRNA[trunk]) in 293-Flag-Ago2 cells. The same membrane was sequentially probed for the MLP-TSS-sRNA and tRNA lysine. On the right side, the Northern blot of 0.01 pmol of each RNA oligonucleotide was probed for the MLP-TSS-sRNA. (G) HeLa cells were cotransfected with RNA oligonucleotides (either MLP-TSS-sRNA or MLP-TSS-sRNA[trunc]) together with pmirGlo or pmirGlo(+) plasmids. Normalized Luciferase expression of pmirGlo(+) relative to pmirGlo was measured at the indicated time points. The results are presented as the mean from three independent experiments. Error bars indicate the SD.
To confirm this observation we used a reductionist approach to minimize the potentially complex effects caused by the expression of the whole repertoire of viral proteins. For this experiment the m7G-capped MLP-TSS-sRNA was produced by in vitro transcription and cotransfected with luciferase reporter plasmids pmirGlo, pmirGlo(+), or pmirGlo(mut), where the MLP-TSS-sRNA binding site was mutated. As shown in Figure 5D, the MLP-TSS-sRNA was capable of down-regulating luciferase expression from the reporter containing a complementary binding site. Moreover, mutations in this binding site caused a complete loss of the inhibitory effects on luciferase expression. This experiment is significant because it suggests that the MLP-TSS-sRNA is capable of regulating gene expression in a sequence-specific manner also in the absence of other viral components.
The stability of the MLP-TSS-sRNA is dependent on a 3′ hairpin structure
Interestingly, the m7G-cap did not contribute significantly to the stability of the MLP-TSS-sRNA. Thus, the MLP-TSS-sRNA with a 5′ phosphate was as stable as the capped counterpart in transfected 293-Flag-Ago2 cells (Supplemental Fig. 5C). On the other hand, secondary structure prediction identified a potential short hairpin at the 3′ end of the MLP-TSS-sRNA (Fig. 5E). To investigate whether this hairpin was important as a stabilizing element, we disrupted this secondary structure by deleting the last 12 nt from the MLP-TSS-sRNA, generating MLP-TSS-sRNA(trunc). The full-length and truncated MLP-TSS-sRNA synthetic RNAs were transfected into 293Ago2 cells and small RNA stability measured in total RNA at 12, 24, 48, 72 h post transfection by Northern blot. As shown in Figure 5F, the full-length MLP-TSS-sRNA was highly stable during the course of the experiment (lanes 3–6) whereas the MLP-TSS-sRNA(trunc) was rapidly degraded and barely detectable at any of the time points assayed (lanes 8–11). Importantly, the input of synthetic sRNA was the same for the full-length and truncated MLP-TSS-sRNA (lanes 1 and 2). The low stability of MLP-TSS-sRNA(trunc) suggests that it should be inefficient as a suppressor of target mRNAs. As shown in Figure 5G, MLP-TSS-sRNA efficiently repressed luciferase expression during the 3-d experiment whereas MLP-TSS-sRNA(trunc) caused moderate repression at 24 h post transfection, a repression that was lost at the later time points. Collectively, these data suggest that the hairpin structure at the 3′ end of the MLP-TSS-sRNA is critical for stability and consequentially also important for the sRNA function.
The MLP-TSS-sRNA suppresses HAdV-37 DNA replication
The adenovirus E2B region encodes for two viral proteins that are essential for viral DNA replication: the preterminal protein (pTP), which is the primer protein needed to initiate viral DNA replication and the Adpol, which is the DNA polymerase copying the viral genome (Stillman et al. 1981; Hoeben and Uil 2013). Interestingly, the MLP-TSS-sRNA originates from the opposite strand to the adenovirus E2B region (Figs. 1A, 6A). Thus, the MLP-TSS-sRNA is perfectly complementary to the pTP and Adpol mRNAs and might therefore be functioning as a regulator repressing virus growth by targeting the expression of the E2B mRNAs. To determine whether the expression level of pTP and Adpol are key factors controlling subsequent steps in virus replication, like DNA replication and late protein synthesis, ARPE-19 cells were transfected with a synthetic MLP-TSS-sRNA or a scrambled sRNA for 4 h, followed by infection with HAdV-37. The transfection approach would mimic an overexpression of the MLP-TSS-sRNA. As shown in Figure 6B, transfection of the MLT-TSS-sRNA reduced expression of both Adpol and pTP mRNAs by more than 50%. As a consequence it also caused a decrease in the efficiency of viral DNA replication (Fig. 6C), which was accompanied by a reduction in viral capsid proteins synthesis (Fig. 6D).
FIGURE 6.

MLP-TSS-sRNA inhibits HAdV-37-replication. (A) Schematic drawing showing the position of the MLP-TSS-sRNA in relation to the E2B mRNAs. (B–D) Effect of synthetic MLP-TSS-sRNA overexpression on HAdV-37 growth. (B) ARPE-19 cells were transfected with a scrambled sRNA (Scr) or the MLP-TSS-sRNA (15 nM) for 4 h; after that, cells were infected with HAdV-37 (10 FFU per cell) for 12 h followed by measuring relative expression of both Adpol and pTP mRNAs by RT-qPCR. The results are presented as the mean from three independent experiments. Error bars indicate the SD ([**] P < 0.01, [*] P < 0.05). (C) The same transfection and infection protocol as in panel B was used but the infection extended to 24 hpi at which time point the viral DNA copy number was determined by qPCR. Results are presented as the mean from three independent experiments. Error bars indicate the SD ([*] P < 0.05). (D) ARPE-19 cells were transfected with the scrambled sRNA (Scr) or the MLP-TSS-sRNA at two different concentrations (15 and 50 nM) for 4 h, followed by infection with HAdV-37 (10 FFU per cell). At 24 hpi, viral late proteins were detected by Western blot analysis using a polyclonal anti-capsid antibody. (E–G) Effect of MLP-TSS-sRNA sponge constructs on HAdV-37 growth. (E) HeLa cells infected with HAdV-37 were transfected with pmirGlo(+), expressing a reporter mRNA containing an MLP-TSS-sRNA binding site, or control plasmid pmirGlo, lacking the binding site. Adpol and pTP mRNA expression was measured 24 hpi by RT-qPCR. The results are presented as the mean from three independent experiments. Error bars indicate the SD ([*] P < 0.05, [**] P < 0.01, [***] P < 0.001). (F) Three independent HeLa stable cell lines constitutively expressing the pmirGlo(+) or pmirGlo reporter mRNAs were infected with HAdV-37 and viral late protein expression detected by Western blot analysis at 24 hpi using a polyclonal anti-capsid antibody. (G) Same as in F with the exception that production of new virus particles was titrated in crude cell lysates prepared from the cells at 24 hpi. The results are presented as the mean from two independent experiments. Error bars indicate the SD ([*] P < 0.05, [**] P < 0.01).
Although these results suggest that the synthetic MLP-TSS-sRNA can target the Adpol and pTP mRNAs, we were curious to investigate whether the MLP-TSS-sRNA produced from the viral genome would exhibit a similar inhibitory effect. For this experiment we used the same plasmid transfection and virus infection approach as described in Figure 5. HeLa cells were infected with HAdV-37 followed by transfection with pmirGlo(+) or pmirGlo lacking a binding site (Fig. 5A). The rationale in this experiment was that the pmirGlo(+) would function as a MLP-TSS-sRNA sponge that would sequester the virus-produced MLP-TSS-sRNA and thereby alleviate the inhibitory effect of the sRNA on Adpol and pTP mRNA expression. As shown Figure 6E, transfection with pmirGlo(+) caused, indeed, a significant up-regulation of both Adpol, and pTP mRNA accumulation. To further confirm this result we generated stable HeLa cell lines constitutively expressing the pmirGlo(+) or pmirGlo luciferase reporter mRNAs. Three separate clones of respective plasmid-transfected cells were infected with HAdV-37 and late protein expression visualized by Western blot at 24 hpi. As shown in Figure 6F, HeLa(+) cells expressing the reporter mRNA with the MLP-TSS-sRNA sponge enhanced viral capsid protein expression considerably (lanes 5–7), compared to the HeLa(−) cells expressing the reporter mRNA with no binding site (lanes 2–4). The up-regulation of late protein synthesis was also accompanied by an increase of late viral mRNA expression, as illustrated by an enhanced accumulation of tripartite leader containing mRNAs (Fig. 6E). The enhanced late protein expression in the HeLa(+) cell lines (Fig. 6F) was also manifested in a three- to fourfold increase in new virus progeny formation (Fig. 6G). Taken together, these results suggest that the MLP-TSS-sRNAs indeed serve a regulatory function during virus multiplication.
DISCUSSION
Here we show, using a TAP-treatment small RNA sequencing approach, that in HAdV-37-infected cells large amounts of a novel Ago2-associated sRNA (MLP-TSS-sRNA) are produced. This sRNA shows several unique features not previously shown for Ago2-associed RNAs. For example, the MLP-TSS-sRNAs are unusually long—31 or 34 nt in length. The major 31-nt species has a 5′ end that coincides with the characterized MLP start site. Also, the MLP-TSS-sRNAs contain an m7G-cap at the 5′ end (Figs. 2, 3B; Supplemental Fig. 2). Based on available data (Fig. 2; Supplemental Fig. 2), we estimate that ∼95% of the MLP-TSS-sRNAs produced during a HAdV-37 infection carry the m7G-cap. Further, to our knowledge, the MLP-TSS-sRNAs represent the first Ago2-associated sRNA that does not require the microRNA machinery for processing and maturation (Supplemental Fig. 4). Interestingly, the MLP-TSS-sRNA also associates efficiently with Ago1 in HAdV-37-infected cells (Supplemental Fig. 4C). Our results indicate that this sRNA is produced by RNAPII stalling within the MLP first leader exon at a site coinciding with the MLP-TSS-sRNA 3′ end (Fig. 4C). Collectively, our data suggest a model where the MLP-TSS-sRNAs are produced through repeated cycles of RNAPII initiation of transcription, stalling, and premature termination. The production of promoter proximal TSS RNAs is not unique to the MLP. In fact, the majority of metazoan gene promoters appear to have high levels of RNAPII accumulation 20–60 bp downstream from the TSS (for review, see Liu et al. 2015). Many of these RNAPII represent paused polymerases that may resume productive elongation; others become preterminated, and the released short RNA most likely is degraded by the nuclear exosome (Kilchert et al. 2016). Our data suggest that a major reason that the MLP-TSS-sRNA accumulates to high levels during an infection may be that it contains a hairpin-like structure at the 3′ end of the sRNA that protects it from degradation (Fig. 5E–G). Small RNAs can be degraded by multiple pathways (Ha and Kim 2014; Kilchert et al. 2016). We note that our sRNA sequencing data show that the third largest population of MLP-TSS-sRNA is 24 nt long and accounts for ∼8% of the total MLP-TSS-sRNA population in RISC (Fig. 1D). This pool of small RNAs may represent processed products similar to miR-451, which becomes trimmed by poly(A)-specific ribonuclease PARN on the RISC complex (Yoda et al. 2013).
There have been several studies reporting the accumulation of capped TSS-sRNAs. However, no report has demonstrated an association of the capped TSS-sRNA with Ago2 RISC complexes. Rather, they appear to act as precursors for classical cellular microRNA processing (Zamudio et al. 2014) or piRNA generation (Gu et al. 2012), or associate with RNAPII to initiate viral transcription as shown for hepatitis D virus (Haussecker et al. 2008), or generated by the cap-snatching mechanism used by the influenza virus (Koppstein et al. 2015).
The crystal structure of an Ago2-miRNA complex shows that the 5′ end of the miRNA is anchored at the Ago2 MID domain, with extensive interactions between the 5′ residues including the 5′ monophosphate (Elkayam et al. 2012; Schirle and MacRae 2012). Further, several reports have demonstrated that a 5′ phosphate is required for double-stranded siRNA loading onto RISC (for examples, see Nykänen et al. 2001; Chiu and Rana 2002). These examples raise the question of how the capped MLP-TSS-sRNAs associate with Ago2. It has, in fact, been proposed that the bulkiness of the m7G cap structure might impede on capped sRNA association with the Ago2 MID domain (Haussecker et al. 2008; Xie et al. 2013). However, it has also been shown that a bulky 5′-flourescine group on the guide strand does not appear to impede on the silencing activity of an siRNA (Harborth et al. 2003). Our data suggest that the MLP-TSS-sRNA is produced as a stable single-stranded RNAPII stalled/preterminated transcript. Loading of single-stranded sRNAs onto RISC has previously been documented (for review, see Chak and Okamura 2014). Interestingly, the 5′ phosphate sensing mechanism operating for siRNA duplex loading onto RISC does not apply to single-stranded siRNA loading (Chen et al. 2008). In fact, single-stranded siRNA loading is more permissive compared to duplex loading, allowing that the siRNA 5′ end is modified without any negative effects on RISC activity. The failure to observe single-stranded siRNA loading in vivo appears to be due to a low stability of single-stranded siRNAs. However, chemically modified synthetic single-stranded siRNAs have been developed that are stable and enter an active RISC and induce RNAi also in cells (Lima et al. 2012; Yu et al. 2012). Although these examples show that there is some flexibility in sRNA RISC loading, it does not prove that RISC has an alternative structure that can accommodate a cap. Therefore, it remains to be established how the m7G-capped MLP-TSS-sRNA enters onto RISC. Final proof would most likely require determination of the structure of such a complex.
The VA RNAs are produced in massive amounts during virus infection and interfere with cellular miRNA production by suppressing pre-miRNA export (Lu and Cullen 2004) and Dicer cleavage (Andersson et al. 2005). Since the MLP-TSS-sRNAs use an alternative noncanonical route for production, the virus will maintain control over the miRNA pathway while being capable of producing other functional sRNAs.
Importantly, RISC programmed with the MLP-TSS-sRNAs is functional with a capacity to suppress reporter gene expression in a sequence-specific and dose-dependent manner (Fig. 5B–D). The MLP-TSS-sRNA originates from the antisense strand of the E2B mRNAs with a perfect complementary binding site within the Adpol gene and the 3′ UTR of the pTP mRNA (Figs. 1A, 6A). Both Adpol and pTP are essential DNA replication proteins. Further, the E2B mRNAs are among the scarcest mRNAs expressed during an adenovirus infection (Stillman et al. 1981), potentially through the MLP-TSS-sRNA targeting the pTP and Adpol mRNAs. Indeed, controlling pTP and Adpol mRNA accumulation (Fig. 6B) by transfecting a synthetic MLT-TSS-sRNA had a significant inhibitory effect on adenovirus genome replication (Fig. 6C) and late protein synthesis (Fig. 6D). However, it was central to determine whether the MLP-TSS-sRNAs produced from the virus act similarly as the synthetic mimic. By using an MLP-TSS-sRNA sponge assay, either in a transient transfection assay (Fig. 6E) or in HeLa stable cell lines (Fig. 6F), we demonstrated that sequestering the MLP-TSS-sRNA drastically increased E2B mRNA expression (Fig. 6E), late viral protein synthesis (Fig. 6F), and new virus progeny formation (Fig. 6G). Collectively, these results suggest that the MLP-TSS-sRNA has a function as a trans-acting viral regulator and is not simply produced as transcriptional noise.
It is well established that the activity of the MLP increases as a result of the increase in viral DNA copy number following the start of viral DNA replication (for review, see Akusjärvi 2008). In a hypothetical model, one would predict that synthesis of Adpol and pTP stimulates viral DNA replication. As a consequence, the activity of MLP increases, which results in an increase in MLP-TSS-sRNA production, which in turn reduces Adpol and pTP gene expression. Such a feedback mechanism may be important for the virus to control virus replication, particularly during the establishment and/or maintenance of persistent/latent infections where the capacity of the virus to multiply needs to be kept at a reduced level.
MATERIALS AND METHODS
Cell culture and virus infection
HEK-293, 293-Flag-Ago2, and HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) and A549 and ARPE-19 cells in DMEM/F12 medium (Invitrogen), supplemented with 10% fetal calf serum (FCS, Invitrogen), 1% penicillin/streptomycin (PEST) at 37°C in 7% CO2. Virus titers were measured as fluorescence forming units (FFU) (Philipson 1961) using a pan-hexon antibody (MAB8052, Millipore, 1:500 dilution). Infection of adherent cells was done as previously described (Kamel et al. 2014). HeLa stable cell lines constitutively expressing firefly reporter construct pmirGlo or pmirGlo(+) were made as previously described (Xu et al. 2007). For titration purposes crude cell lysate was prepared through five cycles of freezing (dry ice-ethanol) and thawing (37°C water bath).
Transient transfection and siRNA knockdown
Transfection of siRNA and plasmid DNA was done using the Jetprime (Polypus-transfection) transfection reagent according to the manufacturer's instructions. The sequence of the siRNAs used in this study is listed in Supplemental Table 2. A scrambled ON-TARGETplus Non-targeting siRNA pool (GE Dharmacon) was used as a negative control.
RNA immunoprecipitation
S15 cytoplasmic extract preparation and Flag/HA tagged Ago2 protein immunoprecipitation was done as previously described (Xu and Akusjärvi 2011). The endogenous Ago2 was immunoprecipitated using an anti-Ago2 antibody (Abcam, ab57113). Cell lysates were incubated with the antibody (2 µg Ab/mL lysate) on a rotating wheel overnight at 4°C. Antibody–Ago2 complexes were purified on Dynabeads Protein G magnetic beads (Thermo Fisher Scientific). The washing and RNA extraction were performed as previously described (Xu and Akusjärvi 2011). For RNAPII immunoprecipitation, 293-Flag-Ago2 cells were irradiated twice for 400 mJ/cm2 at 254 nm followed by protein extraction with RIPA buffer (150 mM NaCl, 50 mM HEPES [pH 7.4], 0.5% sodium deoxycholate, and 0.1% SDS, supplemented with 2 U DNase I/mL and protease inhibitors) and sonication, 10 cycles (30 sec on and 30 sec off). RNAPII complexes were immunoprecipitated using the 8WG16 antibody (ab817), which recognizes the RNAPII CTD repeat, using the same method as described above with the modification that the beads were washed four times in lysis buffer, followed by proteinase K digestion and phenol extraction and ethanol precipitation. Treatment of RNA with Cap-removing enzymes was performed using Tobacco acid Pyrophosphatase TAP (Epicenter) or RNA 5′ Pyrophosphohydrolase RppH (New England Biolabs) according to the manufacturer's instructions.
Capped RNA immunoprecipitation
RNA prepared from the RISC immunoprecipitation was incubated in cap binding buffer (100 mM Hepes-KOH [pH 7.0], 5 mM MgCl2, 5 mM KCl, 300 mM NaCl) together with 5 µg Anti-m3G-cap, m7G-cap Antibody (clone H20, Millipore) on a rotating wheel overnight at 4°C. The antibody-capped RNA complexes were purified on Dynabeads Protein G magnetic beads (Thermo Fisher Scientific) and washed three times in cap binding buffer, followed by RNA extraction as described below.
In vitro transcription and m7G capping of the MLP-TSS-sRNA
The template for the in vitro transcription was a double-stranded DNA oligonucleotide in which the forward strand contained a T7-promoter sequence. In vitro transcription was done using the TranscriptAid T7 High Yield Transcription Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. The RNA was purified by one round of phenol extraction and ethanol precipitation. Capping of the in vitro–transcribed RNA was done using the vaccine virus capping system (New England Biolabs), as described by the manufacturer. The final RNA used for transfection was purified by one round of phenol extraction and ethanol precipitation. The RNA was dissolved in sterile H2O and stored at −80°C.
Luciferase assay
Luciferase assay was performed with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Both firefly luciferase and renilla luciferase activity were measured on Infinite M200 luminometer (Tecan). Results shown are the normalized ratios of firefly to renilla and are presented as the mean from at least three biological replicates. Statistical analysis was performed using Prism6 (GraphPad Software) using a two-tailed unpaired t-test. The value of P < 0.05 was considered statistically significant.
Bioinformatics analysis
The cDNA library construction and small RNA sequencing has previously been described (Kamel et al. 2014). Sequencing reads were mapped to the human reference genome (GRCH37) and HAdV-37 genome (accession number DQ900900) using STAR aligner (Dobin et al. 2013). Visualization of mapped reads shown in Figure 1C was done using the Integrative Genomics Viewer (IGV) (Robinson et al. 2011). To characterize viral sRNA, reads were mapped to the corresponding adenovirus genome using BLASTN (no mismatch allowed). Read start site and viral sRNA read length was visualized using customized Perl scripts.
Total protein extraction
Cells were collected by low speed centrifugation and washed once with 1× PBS. The pellet was suspended in 180 µL of RIPA buffer (150 mM NaCl, 50 mM HEPES [pH 7.4], 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with 1 U/mL of Benzonase (EMD Millipore) and incubated for 1 h at 4°C. Cells were further disrupted by addition of 10% SDS and 1 M DTT to final concentrations of 1% and 100 mM, respectively, followed by a boiling step of 5 min (Holden and Horton 2009).
Total DNA/RNA extraction and quantitative real-time PCR
Genomic viral DNA was extracted using TRI Reagent (Sigma-Aldrich). To estimate viral DNA copy number, 50 ng DNA of each sample was amplified using VAII-specific primers and the copy number was calculated from a standard curve created from serial dilutions of puc19-VAII plasmid, which contain a single copy of HAdV-37 VAII gene, using the absolute quantification method (Whelan et al. 2003). RNA was extracted using TRI Reagent (Sigma-Aldrich) followed by cDNA synthesis using the SuperScript III Kit (Thermo Fisher Scientific). Relative expression of Adpol and pTP were normalized to HPRT1 and quantified using the ΔΔCt method (Schmittgen and Livak 2008). PCR was carried on the Applied Biosystems 7900 System (Life Technologies) using HOT FIREPol EvaGreen qPCR Supermix (Solis BioDyne). Statistical analysis was performed using Prism6 (GraphPad Software) using a two-tailed unpaired t-test. The value of P < 0.05 was considered statistically significant. The primers used in the qPCR are listed in Supplemental Table 2.
Northern and Western blot analysis
Northern and Western blots were performed as described in detail in Kamel et al. (2014). A list of oligonucleotide probes, details of the antibodies, dilutions, and the vendors used are shown in Supplemental Table 2.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anette Carlsson, Shady Younis, and Louise Andersson for excellent technical help, and Professor Göran Wadell and Niklas Arnberg for providing the HAdVs used in this study. This work was supported by the Swedish Cancer Society (grant number 130469) and the Swedish Research Council through a grant to the Uppsala RNA Research Centre (grant number 2006-5038-36531-16).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.061291.117.
REFERENCES
- Akusjärvi G. 2008. Temporal regulation of adenovirus major late alternative RNA splicing. Front Biosci 13: 5006–5015. [DOI] [PubMed] [Google Scholar]
- Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjärvi G. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol 79: 9556–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, Fortes P. 2010. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res 38: 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. 2008. Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, Dicer-dependent small RNAs. Genes Dev 22: 2773–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Karnowski HW, Cai X, Shin J, Pohlers M, Cullen BR. 2010. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Mol cell 37: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Xie M, Steitz JA. 2011. A primate herpesvirus uses the integrator complex to generate viral microRNAs. Mol Cell 43: 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chak LL, Okamura K. 2014. Argonaute-dependent small RNAs derived from single-stranded, non-structured precursors. Front Genet 5: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. 2010. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465: 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Weinmann L, Gaidatzis D, Pei Y, Zavolan M, Tuschl T, Meister G. 2008. Strand-specific 5′-O-methylation of siRNA duplexes controls guide strand selection and targeting specificity. RNA 14: 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Rana TM. 2002. RNAi in human cells: basic structural and functional features of small interfering RNA. Mol Cell 10: 549–561. [DOI] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. 2010. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328: 1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451: 355–358. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L. 2012. The structure of human argonaute-2 in complex with miR-20a. Cell 150: 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ender C, Krek A, Friedländer MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. 2008. A human snoRNA with microRNA-like functions. Mol Cell 32: 519–528. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. 2008. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114. [DOI] [PubMed] [Google Scholar]
- Flynt AS, Greimann JC, Chung WJ, Lima CD, Lai EC. 2010. MicroRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol Cell 38: 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Lee HC, Chaves D, Youngman EM, Pazour GJ, Conte D Jr, Mello CC. 2012. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell 151: 1488–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T. 2003. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev 13: 83–105. [DOI] [PubMed] [Google Scholar]
- Haussecker D, Cao D, Huang Y, Parameswaran P, Fire AZ, Kay MA. 2008. Capped small RNAs and MOV10 in human hepatitis delta virus replication. Nat Struct Mol Biol 15: 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeben RC, Uil TG. 2013. Adenovirus DNA replication. Cold Spring Harb Perspect Biol 5: a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden P, Horton WA. 2009. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes 2: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen-Durr P, Boeuf H, Kédinger C. 1988. Replication-induced stimulation of the major late promoter of adenovirus is correlated to the binding of a factor to sequences in the first intron. Nucleic Acids Res 16: 3771–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel W, Segerman B, Öberg D, Punga T, Akusjärvi G. 2013. The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res 41: 4802–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel W, Segerman B, Punga T, Akusjärvi G. 2014. Small RNA sequence analysis of adenovirus VA RNA-derived miRNAs reveals an unexpected serotype-specific difference in structure and abundance. PLoS One 9: e105746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. 2003. Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216. [DOI] [PubMed] [Google Scholar]
- Kilchert C, Wittmann S, Vasiljeva L. 2016. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol 17: 227–239. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. [DOI] [PubMed] [Google Scholar]
- Koppstein D, Ashour J, Bartel DP. 2015. Sequencing the cap-snatching repertoire of H1N1 influenza provides insight into the mechanism of viral transcription initiation. Nucleic Acids Res 43: 5052–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, Dutta A. 2009. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23: 2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WF, Prakash TP, Murray HM, Kinberger GA, Li W, Chappell AE, Li CS, Murray SF, Gaus H, Seth PP, et al. 2012. Single-stranded siRNAs activate RNAi in animals. Cell 150: 883–894. [DOI] [PubMed] [Google Scholar]
- Liu X, Kraus WL, Bai X. 2015. Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem Sci 40: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockard RE, Rieser L, Vournakis JN. 1980. Labeling of eukaryotic messenger RNA 5′ terminus with phosphorus-32: use of tobacco acid pyrophosphatase for removal of cap structures. Gene Amplif Anal 2: 229–251. [PubMed] [Google Scholar]
- Lu S, Cullen BR. 2004. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J Virol 78: 12868–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykänen A, Haley B, Zamore PD. 2001. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 107: 309–321. [DOI] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. 2007. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 130: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pall GS, Hamilton AJ. 2008. Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3: 1077–1084. [DOI] [PubMed] [Google Scholar]
- Philipson L. 1961. Adenovirus assay by the fluorescent cell-counting procedure. Virology 15: 263–268. [DOI] [PubMed] [Google Scholar]
- Punga T, Kamel W, Akusjärvi G. 2013. Old and new functions for the adenovirus virus-associated RNAs. Future Virol 8: 343–356. [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. 2007. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Kato Y, Taira K. 2006. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett 580: 1553–1564. [DOI] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ. 2012. The crystal structure of human Argonaute2. Science 336: 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208. [DOI] [PubMed] [Google Scholar]
- Söderlund H, Pettersson U, Vennström B, Philipson L, Mathews MB. 1976. A new species of virus-coded low molecular weight RNA from cells infected with adenovirus type 2. Cell 7: 585–593. [DOI] [PubMed] [Google Scholar]
- Stillman BW, Lewis JB, Chow LT, Mathews MB, Smart JE. 1981. Identification of the gene and mRNA for the adenovirus terminal protein precursor. Cell 23: 497–508. [DOI] [PubMed] [Google Scholar]
- Whelan JA, Russell NB, Whelan MA. 2003. A method for the absolute quantification of cDNA using real-time PCR. J Immunol Methods 278: 261–269. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA. 2013. Molecular mechanisms of RNA interference. Annu Rev Biophys 42: 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Li M, Vilborg A, Lee N, Shu MD, Yartseva V, Šestan N, Steitz JA. 2013. Mammalian 5′-capped microRNA precursors that generate a single microRNA. Cell 155: 1568–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Akusjärvi G. 2011. Characterization of RISC-associated adenoviral small RNAs. Methods Mol Biol 721: 183–198. [DOI] [PubMed] [Google Scholar]
- Xu N, Segerman B, Zhou X, Akusjärvi G. 2007. Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the rna-induced silencing complex and associate with polyribosomes. J Virol 81: 10540–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lai EC. 2011. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43: 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Qin Y, Macara IG, Cullen BR. 2003. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda M, Cifuentes D, Izumi N, Sakaguchi Y, Suzuki T, Giraldez AJ, Tomari Y. 2013. Poly(A)-specific ribonuclease mediates 3′-end trimming of Argonaute2-cleaved precursor microRNAs. Cell Rep 5: 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Pendergraff H, Liu J, Kordasiewicz HB, Cleveland DW, Swayze EE, Lima WF, Crooke ST, Prakash TP, Corey DR. 2012. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 150: 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio JR, Kelly TJ, Sharp PA. 2014. Argonaute-bound small RNAs from promoter-proximal RNA polymerase II. Cell 156: 920–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.