

Abstract
How is noise in gene expression modulated? Do mechanisms of noise control impact genome organization? In yeast, the expression of one gene can affect that of a very close neighbor. As the effect is highly regionalized, we hypothesize that genes in different orientations will have differing degrees of coupled expression and, in turn, different noise levels. Divergently organized gene pairs, in particular those with bidirectional promoters, have close promoters, maximizing the likelihood that expression of one gene affects the neighbor. With more distant promoters, the same is less likely to hold for gene pairs in nondivergent orientation. Stochastic models suggest that coupled chromatin dynamics will typically result in low abundance-corrected noise (ACN). Transcription of noncoding RNA (ncRNA) from a bidirectional promoter, we thus hypothesize to be a noise-reduction, expression-priming, mechanism. The hypothesis correctly predicts that protein-coding genes with a bidirectional promoter, including those with a ncRNA partner, have lower ACN than other genes and divergent gene pairs uniquely have correlated ACN. Moreover, as predicted, ACN increases with the distance between promoters. The model also correctly predicts ncRNA transcripts to be often divergently transcribed from genes that a priori would be under selection for low noise (essential genes, protein complex genes) and that the latter genes should commonly reside in divergent orientation. Likewise, that genes with bidirectional promoters are rare subtelomerically, cluster together, and are enriched in essential gene clusters is expected and observed. We conclude that gene orientation and transcription of ncRNAs are candidate modulators of noise.
Keywords: non, coding transcripts, bidirectional promoter, expression noise, chromatin, chromatin remodeling
Introduction
In eukaryotic genomes, genes that are in close proximity tend to be expressed in a similar manner (Hurst et al. 2004; Michalak 2008). These can be large clusters (circa tens of genes), such as the clustering of highly expressed (Caron et al. 2001) or housekeeping (Lercher et al. 2002) genes in the human genome and clusters of coexpressed but functionally unrelated genes in the Drosophila genome (Spellman and Rubin 2002). In many instances, however, clusters are made up of just pairs of genes (see, e.g., Mijalski et al. 2005). In yeast, for example, there are many more neighboring pairs of highly co-expressed genes than expected by chance (Cho et al. 1998; Cohen et al. 2000).
In part, this coexpression of neighbors is thought to reflect the simultaneous opening and closing of chromatin domains, rendering all genes within a domain potentially accessible to or hidden from transcription factors (TFs) (Raj et al. 2006). This is supported by the finding that two transgenes inserted in tandem have simultaneous transcriptional bursting, whereas the same two when unlinked do not (Raj et al. 2006). Importantly, although tandem duplicates certainly underpin some coexpression of neighbors in some species (Lercher et al. 2003; Quijano et al. 2008), the high levels of coexpression of neighboring genes in yeast, the species we consider here, is not owing solely to tandem duplicates nor to similarity in TF usage of neighbors (Batada, Urrutia, et al. 2007). That the coexpression level reflects underlying nucleosome patterns (Batada et al. 2007) is consistent with the chromatin fluctuation hypothesis. The involvement of chromatin dynamics in defining expression clusters in other species is well described (Boutanaev et al. 2002; Lunyak et al. 2002; de Wit et al. 2008).
As might be expected were localized chromatin opening and shutting partially responsible for enabling coexpression, the patterns of coexpression of neighboring genes also reflects the proximity of the promoters of two genes. Gene pairs can come in one of three orientations: convergent (→←), co-oriented (→→ or ←←), or divergent (←→). High levels of coexpression are, as expected, relatively rare for genes in convergent orientation (Kruglyak and Tang 2000; Huynen and Snel 2003; Batada et al. 2007), their promoters being the most distant. At the other extreme, when two genes are in divergent orientation, they can share the same promoter (i.e., a bidirectional promoter). For brevity we refer to “bipromoter genes” to specify genes transcribed from the same bidirectional promoter. In Saccharomycescerevisiae, more than 60% of nonoverlapping divergent protein coding transcripts share the same promoter region (Xu et al. 2009). Such gene pairs can be (but aren’t necessarily [Trinklein et al. 2004]) very highly coexpressed, as observed in yeast (Kruglyak and Tang 2000; Batada et al. 2007) and Arabidopsis (Williams and Bowles 2004).
Through the fungi, divergent gene pairs are more conserved in orientation than convergent gene pairs (Hurst et al. 2002; Huynen and Snel 2003; Batada et al. 2007; Kensche et al. 2008). Likewise, in mammals, bidirectional gene organization tends to be both ancient and more conserved (Trinklein et al. 2004; Li et al. 2006). This may reflect selection to preserve the coexpression pattern of functionally coordinated gene pairs (Hurst et al. 2002; Huynen and Snel 2003; Poyatos and Hurst 2007). However, in yeast, only the top 2–5% of the most highly coexpressed genes pairs show evidence of similarity in gene ontology (GO) class, whereas the majority of neighboring gene pairs have moderate coexpression scores (0.1 < r < 0.4) and no similarity in GO class (Batada et al. 2007). Although a lack of similarity in GO class need not prove an absence of functional coupling, it is worth asking whether an alternative rationale for bipromoter architecture is imaginable. Here, we suggest that a bipromoter architecture will tend to reduce the noise levels of the genes concerned, noise being the variation between genetically identical cells in abundance of any given transcript or protein (Elowitz et al. 2002; Ozbudak et al. 2002; Blake et al. 2003; Kaern et al. 2005; Raj and van Oudenaarden 2008).
The Hypothesis
In the standard model outlined above, chromatin is opened and any gene within the open domain can be expressed if the relevant TFs are present. In this conceptualization, the genes don’t affect each other’s expression but, rather, are simply passive respondents to open or closed chromatin. Our hypothesis relies on logic importantly different to the standard model. We consider, that on a very local scale, the upregulation of one gene can directly affect the chromatin, and hence transcription, of its very close neighbor. As bipromoter genes are as close (in terms of promoter proximity) as is possible, this sort of transcriptional coupling should be most profound for such genes, with nonbipromoter divergent genes also potentially being affected. Mechanistically, one possibility is that, for divergent genes, the transcription or priming for transcription by PolII loading opens the chromatin for the focal gene; this then potentially affects the accessibility of the neighboring gene’s promoter to TFs, possibly by leaky chromatin opening.
Evidence for the transcription of one gene affecting the activity of neighbors (rather than simply being correlated) comes from analysis of the consequences of high expression levels in humans and yeast (Ebisuya et al. 2008). Upregulation of one gene causes a time-lagged increase in the expression of neighbors, sending a small ripple of transcription away from the focal gene. In yeast, the effect is highly localized, the ripple extending no further than 3 kb (Ebisuya et al. 2008). This spillover is at least in part owing to local relaxation of chromatin associated with the expression of the focal gene, as evidenced by changes in histone modifications (Ebisuya et al. 2008). That transcription affects chromatin status (Li et al. 2007) is similarly consistent with the above hypothesis.
Given this possibility, we suggest that a bipromoter gene pair can act almost as a block of self-reinforcing open chromatin, enabling expression when expression is needed. Such gene pairs should be low-noise genes, as they will be poised for expression when the relevant TFs appear and be much less prone to transcriptional bursting caused by stochastic opening and closing of chromatin (fig. 1). This is consistent with the finding that expression of an antisense transcript in the intergenic space near PHO5 in yeast can boost expression of the protein transcript on the opposite strand owing to the effects of chromatin status and promoter remodeling (Uhler et al. 2007).
FIG. 1.—
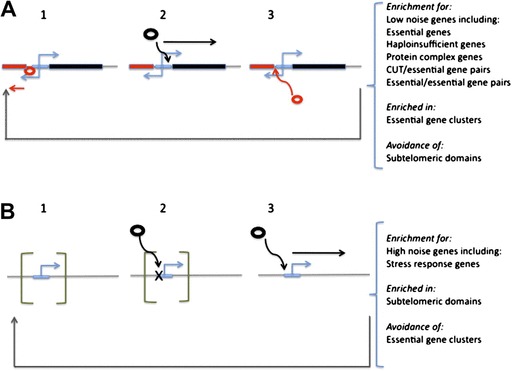
A simplified representation of the effect of bipromoter architecture on expression priming and noise. Consider a bipromoter architecture (A) or unidirectional architecture (B). With the bipromoter architecture, a transcript (red, with red TF and red gene) is made (stage 1). This keeps the chromatin open so that should the TF (black circle) for the gene on the opposite strand (black) become available transcription can occur (stage 2). This in turn makes it more likely that a TF for the red gene can have access should it appear (stage 3). This predisposes to avoidance of randomly spaced (in time) transcriptional bursting. Moreover, constant expression, for example, of a small ncRNA (red) (repetition of stage 1 or polymerase pausing) will prime the focal gene (black) for expression when expression is needed, assuming no interference between the two sets of TFs. By contrast, with unidirectional promoters (B), the promoter can be trapped in closed chromatin (shown as parentheses). When the relevant TF appears (stage 2) expression is hence not possible but instead must wait for the chromatin to open (stage 3). With random shutting of chromatin after expression (return to stage 1), bursting of gene expression is expected and hence typically high noise.
Assuming that coexpression dynamics of divergent genes are mediated in part through chromatin dynamics, then a coupling between gene orientation and noise accords with the finding that the central determinants of noise are chromatin control factors (Newman et al. 2006). Moreover, as expected, if genomic domains differ in the extent to which chromatin is open, noise of a transgene is dependent on the insertion site (Becskei et al. 2005). The notion that shared chromatin domains can modulate noise levels, has led to the suggestion that the genomic distribution of essential genes and chromatin control should coevolve, such that essential genes (by definition those sensitive to reductions in dose) end up clustered into domains with largely open chromatin, thereby ensuring low noise and expression when expression is needed (Batada and Hurst 2007). Our hypothesis is an extension, on a microscale, of similar logic.
Below we start by providing a simulation and analysis of a toy model to ask whether the logic described above is viable. We then test predictions of the hypothesis. Importantly, the hypothesis correctly predicts variation in noise levels as a function of gene orientation and distance between genes. It also correctly predicts which classes of gene should be disproportionately associated with which orientation and where in the genome they might be found. Perhaps most importantly, the hypothesis provides a functional explanation for the occurrence of noncoding RNA (ncRNA) driven off bidirectional promoters that is consistent with observation. We additionally show that the alternative hypothesis, that such ncRNA reflects spurious transcription, is inconsistent with observed data.
Materials and Methods
Data set
All yeast (S. cerevisiae) transcripts as observed by tiling arrays under three conditions (YPE, YPD, and YPGal) and their genomic coordinates were obtained from Xu et al. (2009). Two transcripts were considered as bipromoter transcripts if they share the same 5′ nucleosome free region (NFR), where NFR was defined as a nucleosome deplete region ≥80 bp, according to Xu et al. (2009). These transcripts were defined as divergent (←→), convergent (→←), or cooriented (→→ or ←←) by their coordinates in the genome. Essential genes in rich media were downloaded from the Web site of the Saccharomyces Genome Deletion Project (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). Both the yeast gene order (Version 2) and genome annotation information were taken from http://wolfe.gen.tcd.ie/ygob/. For more than 2,000 proteins, expression noise data in rich media were obtained from Newman et al. (2006). We used the distance to median noise level (DM_YEPD) in our analysis to get rid of the confounding influence of protein abundance. Genes whose promoter contains a TATA-box were derived from a large TATA-box gene enquiry experiment (Basehoar et al. 2004). Codon usage bias (FOP) was obtained from Drummond et al. (2006). The relationships between TFs and their target genes were derived from the yeast transcriptional regulatory network (Balaji et al. 2006). In total, 12,873 regulatory interactions were indentified in this network. Stress-related genes and growth-related genes were obtained from Wapinski et al. (2007) and coexpression level of adjacent gene pairs as previously reported (Batada et al. 2007). Haploinsufficent genes were taken from Deutschbauer et al. (2005), and genes with type I and type II promoters were obtained from Field et al. (2008). About 431 type I genes and 565 type II genes were included in our analysis. Protein complexes were gained from Wang et al. (2009).
Data Analysis
Transcripts that share the same 5′ NFR were described in Xu et al. (2009). The noise of each protein measured by Newman et al. (2006) was used to represent the noise of the transcript. In the comparison of the noise of proteins derived from divergent transcripts to the noise of proteins without divergent transcripts, transcripts with complex annotations were excluded (e.g., the annotation “other,” which means the transcript contains multiple open reading frames or is a mixture of noncoding and coding parts). In the calculation of the correlation between noise levels of protein pairs, transcripts that contain multiple annotation features, which means the transcript contains multiple open reading frames or is a mixture of noncoding and coding parts were excluded. In the calculation of the correlation between noise level and the distance between transcription start sites (TSSs), we used the mean noise level of the two proteins if the noise of both proteins had been measured. If one gene transcript shares its promoter with a noncoding transcript, the noise of this gene was chosen to represent the noise of the two transcripts in the calculation. We used the lawstat package in R to perform the Brunner–Munzel (B-M) test (Brunner and Munzel 2000; Hui et al. 2008).
Randomization Test of the Correlation between Noise Levels of Gene Pairs
Our model predicts that the expression noises of two divergent genes should be positively correlated due to the shared chromatin regulation, as chromatin regulation processes are responsible for much of the expression noise in yeast (Choi and Kim 2008, 2009). To check if there is a positive correlation between expression noise in divergent, convergent and cooriented gene pairs, and to obtain the significance level of any such correlation, we employed a randomization procedure. In this, we extract the noise level for each protein, orient the gene pairs by their strand location for divergent and convergent gene pairs, by their transcription order for cooriented gene pairs, calculate the spearman correlation level for this data, randomize one column of genes 10,000 times, and determine the correlation for each. The significance level of the observed correlation is (m + 1)/10,001, where m is the rank of the true correlation compared against the randomizations.
Randomization Test to Determine Whether Essential–Essential Gene Pairs Are More Likely to be Divergent Gene Pairs
The S. cerevisiae gene order was taken from the Yeast Gene Order Browser (http://wolfe.gen.tcd.ie/ygob/), Version 2. The procedure is as follows: 1) count the number of divergent essential gene pairs in the S. cerevisiae genome, 2) randomize the position of essential genes in each chromosome 1,000 times and calculate the number of divergent essential gene pairs for each, 3) the significance level of this number is (m + 1)/1,001, where m is the rank of the true number compared with the randomizations.
Method to Test the Density of Essential Genes in the Neighborhood of Different Gene Types
To calculate the density of essential genes surrounding essential bipromoter genes and essential nonbipromoter genes, a ±5 gene window was used to scan the yeast chromosomes (the S. cerevisiae gene order we used is from http://wolfe.gen.tcd.ie/ygob/, as described above). To avoid biases caused by the fact that essential genes tend to be in divergent gene pairs, the direct (+1 and −1) gene neighbors were excluded from the scan.
Results
Why Close Promoters Should Be Associated with Low Noise: Toy Model Simulation and Analysis
To consider our hypothesis more fully, we examine by simulation an extension of a standard telegraph model (i.e., chromatin is either open or shut), in which genes have some degree of coupling between them, measured by an independence parameter, i. For low values of i (i.e., dependent/coupled genes), expression of one gene is likely to affect the chromatin status of the other. There are two not mutually exclusive ways by which transcription of one gene might mediate such effects: either by reducing the probability that chromatin of the other promoter will shut, if open, or by increasing the probability of the chromatin opening if shut. We model both independently and consider a third model combining both.
Results from these simulations (see supplementary model 1; supplementary figs. 1–Supplementary Data, Supplementary Material online) confirm the expectation that coupled (low i) genes should generally be low-noise genes. For representative examples, see figure 2. Empirically, much of the between-gene variation in noise is accounted for by expression level, there being lower noise for more highly expressed genes (Raser and O'Shea 2004; Bar-Even et al. 2006; Newman et al. 2006; Yin et al. 2009). Even controlling for this, using an abundance-corrected noise (ACN) measure, there remains, however, much variation (Bar-Even et al. 2006; Newman et al. 2006). Our simulations also report that effects on noise of the coupling between neighboring genes has both dosage-dependent and, importantly, dosage-independent components (supplementary model 1.2; supplementary figs. 4 and Supplementary Data, Supplementary Material online), that is, that the effects aren’t simply a consequence of open chromatin, leading to higher expression, leading to lower noise (cf. Cook et al. 1998). We have further confirmed this through analytically solving a simpler Markov chain model (supplementary analysis 1, Supplementary Material online) and demonstrated that coupling between genes will typically reduce noise strength, an abundance-independent metric (Ozbudak et al. 2002) (supplementary fig. 6, Supplementary Material online). Simulations also reveal noise strength to be affected by coupling in the more complex simulation models (fig. 2; supplementary figs. 1.1, 2.1, 3.1, Supplementary Material online).
FIG. 2.—
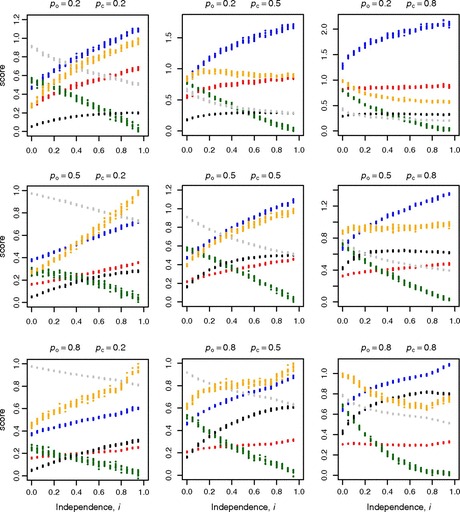
The relationship between the independence between two neighboring genes and various noise and coexpression parameters for nine values of pc (the probability of chromatin closing if open) and po (the probability of chromatin opening if closed) in simulations under the mixed model. Data: transcriptional noise, blue; protein noise, red; strength of protein noise, orange; coexpression, green; chromatin fluctuation, black; proportion of time chromatin open, gray. Other parameter values: N = 100, pt = 0.9, pd = 0.7. Noise strength is normalized to the highest value in any given plot so that the maximum value is unity.
Predictions
Merging the results of the general model with known biology, we can then make a series of predictions. As transcriptional spillover is stronger the closer two genes are (Ebisuya et al. 2008), we presume that bipromoter genes are the most strongly coupled, followed more generally by those in bidirectional orientation. More generally, we presume that interpromoter distance correlates positively with i. If so, we expect that bipromoter pairs will have relatively low ACN levels. Furthermore, as the transcriptional ripple effects are very local in yeast, we expect for bidirectional genes, both that the distance between their promoters should correlate with noise levels (high noise for distant promoters) and that genes within a pair should show correlated noise levels (supplementary model 1.3, Supplementary Material online). Given that prior analysis suggests that gene proximity is a predictor of noise levels (Newman et al. 2006), this hypothesis is worthy of further scrutiny.
Our hypothesis suggests a possible explanation for the transcription of short ncRNAs in proximity to genes, as observed in multiple taxa, yeast included. For example, mapping millions of short RNA reads generated from murine cells has revealed abundant short TSS–associated RNAs, many of which are antisense transcripts (Seila et al. 2008). Likewise in humans, depletion of the exonucleolytic RNA exosome reveals lots of highly unstable RNAs of promoter upstream transcripts (Preker et al. 2008). Similar RNAs are reported in chicken and Drosophila (Taft et al. 2009). Our hypothesis proposes that a protein-coding gene partnered with a ncRNA, driven off the same bidirectional promoter but from the opposite strand, will have low ACN, as the ncRNA poises the protein-coding gene ready for transcription, resisting stochastic chromatin shutting. This presents a functional and testable explanation for the transcription of ncRNA that is different from the null of spurious transcription (Brosius 2005). Pairs of ncRNAs separated by a bidirectional promoter are also possible (Yang et al. 2007) but, for want of data, are not considered.
We can also predict which sorts of genes might be in which pairwise architectures. Genes whose knockouts are not viable (“essential” genes) are those for which decreases in dose have, by definition, major fitness consequences. A priori they are thus expected to be under selection for low noise (Fraser et al. 2004), which is indeed observed (Fraser et al. 2004; Batada and Hurst 2007); as expected, haploinsufficient genes have yet lower noise levels (Batada and Hurst 2007). That essential genes tend to be highly expressed explains in part their low noise but even controlling for this they are “quiet” (Newman et al. 2006; Batada and Hurst 2007). Many features of essential genes can be explained as selection for low noise. For example, and counterintuitively for highly expressed genes, the mRNAs have short half lives (Pal et al. 2001), a feature that suppresses noise (Fraser et al. 2004). They tend not to be TATA controlled (a high-noise promoter) and reside clustered in genomic low noise/open chromatin domains (Batada and Hurst 2007). Experimental demonstration that essential genes are under selection for low noise (e.g., by showing decreased fitness owing to induced high noise of a usually low-noise essential gene) is, however, currently lacking (Raj and van Oudenaarden 2008). Assuming essential genes to be under selection for low noise, we expect them to adopt bipromoter architecture more commonly than expected by chance.
Similarly, genes whose proteins function in a complex are expected to have low noise (Fraser et al. 2004), as coordinated expression is likely to be important (Papp et al. 2003; Fraser et al. 2004). By contrast stress–response genes are thought to be under selection for high noise (Blake et al. 2006; Lopez-Maury et al. 2008) and should thus, in our hypothesis, avoid bipromoter architecture. Yeast subtelomeric domains are high-noise domains, so we also expect these to be depauperate in bipromoter genes and in ncRNAs driven off bipromoters. We test all these predictions and show them to be upheld and not explained by correlation to known covariates. We also consider whether modification of noise may be a more important determinant of gene orientation than coexpression. Several tests suggest this may indeed be the case.
Empirical Evidence
Evidence That Bipromoter Transcribed Genes Have Low Expression Noise.
We tested the hypothesis that bipromoter protein-coding genes have low protein noise with the help of recently published yeast whole-genome transcription data (Xu et al. 2009) to define gene orientation and presence of ncRNA. This we cross-referenced with high-resolution noise data for yeast grown on rich media, provided for over 2,000 protein-coding genes specified by Newman et al. (2006). In all, we analyzed 7,272 well-identified transcripts, of which 1,772 are noncoding transcripts (stable unannotated transcripts and cryptic unstable transcripts [cryptic unstable transcripts {CUTs}]). Among bidirectional transcripts with a mapped 5′ NFR, 61% of the unannotated transcripts and 48% of the protein-coding transcripts initiated bidirectionally from shared 5′ NFRs rather than initiating from their own promoters (Xu et al. 2009).
As we are not interested in the hypothesis that bipromoter architecture might modify noise through modification of abundance, we restrict analysis to ACN measures, as defined by Newman et al. (2006). We also repeated the analysis using residuals from a loess regression of noise against abundance and find no important differences (data not shown). Here after, when we refer to noise, we refer to ACN.
After removing the confounding transcript types (5′ NFR tandem transcript, 3′ NFR antisense transcript, and 3′ NFR tandem transcript) annotated by Xu et al. (2009), we find that protein-coding genes with a bipromoter structure, sharing their 5′ NFR either with a coding gene or with a noncoding gene, show significantly lower noise than the genes that do not have a bipromoter transcript structure (mean noise of bipromoter genes = 0.33 ± 0.11; of all nonbipromoter genes: 1.76 ± 0.15; B-M test P = 4.1 × 10−13, fig. 3). More generally, divergent genes (regardless of their NFR) have lower noise than those in alternative configurations (noise of nondivergent genes = 1.50 ± 0.18, mean noise of divergent genes = 0.88 ± 0.12, B-M test P = 0.0077). Consistent with the notion that divergently oriented genes are exceptional, convergent genes and cooriented genes have the same noise level (P = 0.68, B-M test). The above results are not explained by skewed TF usage or TATA control (supplementary results 1, Supplementary Material online). As expected, permanently open nucleosome architectures (type II promoters) do not show lower noise for bipromoter pairs (supplementary results 2, Supplementary Material online).
FIG. 3.—
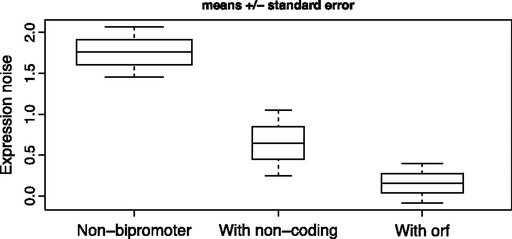
Genes that share a promoter (5′ NFR) with either a noncoding transcript or coding transcript (ORF) show lower abundance-corrected expression noise than genes without any bipromoter transcript. Number of genes that have noise value in each category: With noncoding: 216; with ORF: 537; other (genes that do not share 5′ NFR with other transcripts): 1,072. Box width indicates sample sizes. For the distribution of the data, see supplementary figure 8 (Supplementary Material online).
As expected, we find that bipromoter protein-coding genes have low noise both when they are partnered with a protein-coding gene (P = 5.0 × 10−14 compared with all other genes; B-M test), and when the partner is a ncRNA (P = 0.0030, fig. 3). Also as predicted, we find a significant correlation of the noise of two divergent transcripts but not for either convergent or cooriented gene pairs (supplementary results 3, Supplementary Material online). The mean noise level of the transcripts in divergent gene pairs is, as predicted, correlated with the distance between TSSs, a correlation not seen for convergent and cooriented pairs (Spearman rank correlation for divergent pairs r = 0.0936, P = 0.0055; for convergent pairs r = −0.0194, P = 0.49; for cooriented pairs r = −0.0282, P = 0.29; fig. 4). It may be the case also that for the most close divergent transcripts (less than 100 bp apart), noise is relatively high (fig. 4a). This suggests the possibility of some form of interference between sense and antisense transcription not considered in the toy models that we presented.
FIG. 4.—
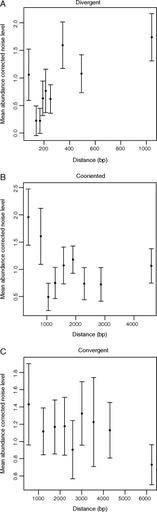
The relationship between the distance between TSSs and the mean noise level of the two genes as a function of gene orientation: (A) Divergent, (B) Cooriented, (C) Convergent. In each case, the data are split into bins with equal numbers in each bin, for a given orientation. Error bars indicate standard error of the mean. Only for the divergent genes is there a significant correlation between distance and noise.
Is Noise More Important than Coexpression?.
Classically, bipromoter genes are thought to be coexpressed genes. Indeed, in simulations, we find that coexpression is higher when genes are coupled (r = −0.86). In this regards too, our simulations are thus broadly consistent with observation. But can we then be confident that the function of bipromoter architecture is ever to reduce noise rather than to increase coexpression levels? The most highly coexpressed 2–5% of gene pairs tend to belong to the same functional class, are preserved as a pair over evolutionary time, and are enriched in divergent orientation (Batada et al. 2007). For these, coexpression is likely to be functionally relevant. However, several findings support the proposition that noise modification is relevant.
First, for observed data, there is no significant correlation between coexpression level and mean noise, neither for divergent gene pairs (r = −0.064, P = 0.424), convergent gene pairs (r = −0.1038, P = 0.152), nor cooriented gene pairs (r = −0.0672, P = 0.257). This suggests that low noise is not de facto a consequence of high coexpression (or vice versa). We do, nonetheless and as expected, find higher coexpression rates for divergent gene pairs (supplementary results 4, Supplementary Material online). Note too that in theory, when closure and opening rates are high we can recover negative coexpression scores for moderate levels of independence (supplementary fig. 7a and b, Supplementary Material online). The model is thus potentially compatible with a low but significant number of bipromoter pairs having negative coexpression, as observed in humans (Trinklein et al. 2004), without compromising low noise.
Second, we can consider a special class of ncRNA, these being cyptic unstable transcripts, CUTs. These are transcripts which, as their name suggests, are degraded as soon as they are produced (Neil et al. 2009). Unlike, for example, proteins in the same protein complex, there can thus be no advantage to coexpression per se, as copresence of the two products of transcription (a CUT and a protein) from the same bipromoter is possible only for a vanishingly small time if at all. Importantly then, we find that protein-coding genes partnered with CUTs through a bidirectional promoter have lower noise than other genes (P = 0.012) but no different from that of protein-coding genes partnered with protein-coding genes in a bipromoter architecture (P > 0.05). For these proteins, noise modification, or priming for expression, by the transcription of the CUT is more likely to be the focus of any selection.
A third line of evidence derives from examination of genes where a priori, we might know the fellow genes with which they might benefit from being coexpressed. The best candidates in this regard are proteins that belong to the same protein complex, that do indeed have high coexpression scores with fellow members (supplementary results 5, Supplementary Material online), as commonly observed (e.g., Papp et al. 2003; Fraser et al. 2004). Given the need for transcription when transcription is needed, as expected, complex-associated genes also have low noise (P = 7.3 × 10−7 B-M Test) as previously observed (Fraser et al. 2004). We are unaware that anyone has previously reported that, as we predict, genes specifying proteins in a complex tend to have bipromoter architecture more than expected by chance (P < 2.2 × 10−16, Fisher’s Exact Test), this being true after control for essentiality (P < 2.2 × 10−16, Fisher’s Exact Test). Importantly, although complex related genes both have low noise and are found more commonly in bipromoter architecture than expected by chance, we find no cases where two genes specifying proteins in the same complex are located in the same bipromoter pair. The bipromoter architecture thus does not drive the coexpression with the interacting protein but can drive the low noise.
Low Noise of Bipromoter Genes Predicts Gene and Genome Anatomy.
Above we showed that genes whose protein products function in a complex tend to have a bipromoter architecture more than expected by chance. Assuming that bipromoter architecture promotes low noise, as model and evidence supports, can we predict which other sorts of genes might prefer or avoid such an orientation? Similarly, as noise levels vary around the genome, can we understand variation in the distribution of different gene pair architectures?
Essential Genes Tend to be Low Noise with Bipromoter Architecture, Whereas the Opposite Is Seen for Stress Response Genes.
As predicted, essential genes are in bipromoter and divergent architecture more than expected by chance. Of 6,600 protein-coding genes in yeast, 2,627 are divergent with a partner protein-coding gene. Of these, 537 (20.4%) are essential, whereas only 577 (14.5%) of the 3,973 nondivergent genes are essential. There is thus enrichment of essential genes in the divergent class (P = 4.9 × 10−10, Fisher’s Exact test). There is a corresponding enrichment of essential genes in gene pairs with bipromoter architecture. Of 2,111 genes in bipromoter organization, 22% are essential, whereas only 649 of 4,489 (14.4%) nonbipromoter genes are essential (P = 5.9 × 10−13, Fisher’s Exact test). An analogous excess in divergent orientation has recently been reported in Drosophila (Yang and Yu 2009). Moreover, we see more bidirectional pairs of two essentials genes than expected by chance: there are 79 bidirectional essential gene pairs in yeast, this being more than ever found in 1,000 gene order randomizations (P < 0.001). Also as expected, haploinsufficent genes tend to be in bipromoter architecture more than expected (41% are bipromoter vs. 31% of all nonhaploinsufficient genes; P = 0.005).
For stress-related genes we see, as expected, the opposite pattern. Although those that are bipromoter have lower noise than stress-related genes in different configurations (mean noise for bipromoter stress genes: 1.59 ± 0.30, for nonbipromoter stress genes 3.63 ± 0.27, P = 1.6 × 10−8, B-M test), stress-related genes tend to avoid having a bipromoter architecture. Only 509 (24.1%) bipromoter genes are stress related, whereas 1,525 (34.0%) of the nonbipromoter genes are stress related (Fisher’s Exact test, P = 2.7 ×10−16). Similarly, stress genes tend not to be in divergent orientation (28% divergent, 32.5% nondivergent; P = 0.00024, Fisher’s Exact test).
Furthermore, if ncRNA is a mechanism of noise reduction, of the essential genes that are not bipromoter with another protein-coding gene, we expect to see more cases of antisense ncRNA associated with such genes than expected by chance. This we observe. Of 309 genes with an antisense CUT, 65 (21%) are essential genes, whereas only 624 (14.1%) of 4,441 genes without an antisense CUT are essential (P = 0.0014, Fisher’s Exact test).
If there are peculiar features of essential genes (e.g., short half-life, low usage of optimal codons), can we exclude the possibility that bipromoter genes have low noise just because of this enrichment for essential genes? Mean noise level of the 1,646 nonessential bipromoter genes is significantly lower than other nonessential genes (0.43 ± 0.12 vs. 1.83 ± 0.15, P = 5.5 × 10−12 in B-M test). This indicates that dispensability alone cannot account for the trends seen. That nonessential genes with bipromoter control have lower expression noise than essential genes (in all orientations) (P = 0.035) further suggests that dispensability cannot alone account for the low noise of bipromoter genes.
There must, however, be alternative methods to modulate noise. Notably, we find that the mean noise of bipromoter essential genes (with either an ncRNA or a protein-coding gene partner) is not significantly lower than the noise of nonbipromoter essential genes (0.18 ± 0.22 vs. 0.22 ± 0.26, P = 0.82 in B-M test; 0.03 ± 0.20 vs. 0.29 ± 0.30, P = 0.76 after removing type II genes). These results are then consistent with bipromoter architecture being a means to reduce noise, but, unsurprisingly, not the only mechanism.
What the other mechanisms might be is not immediately transparent. For example, although essential genes have a shorter mRNA half-life than nonessential genes (P = 2.8 × 10−16, B-M test), the mean mRNA half-life for bipromoter essential genes is no different to that of nonbipromoter essential genes (16.65 vs. 16.91, respectively: P = 0.25, B-M test). Increased usage of codons that specify abundant tRNAs is expected to enable fast translation and be associated with high noise. As expected, there is a positive correlation between the frequency of optimal codon usage (FOP) and expression noise in yeast (r = 0.107, P = 4.6 × 10−07, Spearman’s rank correlation). However, FOP of bipromoter essential genes does not differ from that of either essential nonbipromoter genes or essential nondivergent genes (P = 0.16 and 0.63, respectively, B-M tests).
Bipromoter Gene Pairs and CUTs Are Rare in Noisy Subtelomeric Domains
Does the fact that bipromoter gene pairs have low noise affect not only which sort of genes are found in this architecture but also where on chromosomes they are found? Previously, it was reported that essential genes and nonessential genes flanked by a high density of essential genes tend to have low noise (Batada and Hurst 2007). Could it be that nonbipromoter essential genes tend to reside in essential gene clusters, thus giving them low noise? Alternatively, might genes requiring low noise not only adopt bipromoter architecture but also aggregate into low-noise chromosomal domains?
Ignoring genes +1 and −1 from a focal essential gene (direct neighbors) and then asking about the number of essential genes in the flanking five genes on either side, we find that both bipromoter essential genes (P = 0.022) and bipromoter nonessential genes (P= 0.018) have more essential genes in their vicinity than expected by chance (table 1). Thus, bipromoter genes tend to be enriched in the vicinity of essential gene clusters, these having unusually low-noise levels (Batada and Hurst 2007). Clustering of bipromoter genes doesn’t, however, fully account for the low noise of genes in such domains. Examining nonbipromoter genes, those in essential gene clusters have lower noise than those not in clusters (P = 0.0007; controlling for essentiality, P = 0.01).
Table 1.
The Density of Essential Genes among the Ten Genes Flanking Focal Genes
| Bipromoter | Not Bipromoter | P Value | |
| Essential | 0.212 ± 0.006 | 0.195 ± 0.005 | 0.022 |
| Not essential | 0.188 ± 0.003 | 0.180 ± 0.003 | 0.018 |
| P value | 0.00089 | 0.010 |
Note.—Here, we ignore genes +1 and −1 of a focal gene (direct neighbors).
Yeast subtelomeric domains are high-noise domains and are depauperate in essential genes (Batada and Hurst 2007). From the logic that bipromoter architecture is a genomic device to minimize noise, we might expect that genes found in subtelomeric domains should be favored to be high-noise genes and hence not in a bipromoter architecture. In subtelomeric domains (20 kb from chromosome ends), 28 of 324 gene pairs (8.6%) are bipromoter; conversely, 2,083 of 6,276 (33%) nonsubtelomerics are bipromoter (P< 2.2 × 10−16, Fisher’s Exact test). However, as essential genes tend to be bipromoter and avoid subtelomeric domains, we may be seeing nothing more than the biased distribution of essential genes. Considering only nonessential genes, we see the same bias (8% subtelomeric nonessential genes in bipromoter architecture vs. 31% nonsubtelomeric, P < 2.2 × 10−16, Fisher’s Exact test). We similarly find that bipromoter CUT-associated genes are rare subtelomerically (1.2% subtelomeric genes have a bipromoter CUT compared with 4.8% otherwise, P = 0.001 Fisher’s Exact test; controlling for essentiality of the neighbor, P = 0.006). The high noise of subtelomeric genes and the avoidance of subtelomeric domains by bipromoter genes cannot explain the low noise of bipromoter genes, as they have low noise even compared with genes that are not subtelomeric (P < 10−11).
Discussion
We hypothesize that as transcription of one gene increases the probability of a neighbor being amenable to transcription, low noise of both is commonly expected. The promoters of divergent gene pairs, bipromoter gene pairs in particular, are very close. This maximizes the probability of chromatin nonindependence between the genes, and divergently transcribed genes are thus expected to be low-noise genes, even allowing for any effect of protein abundance. This model has striking predictive ability. As predicted, bipromoter genes are indeed low noise, and the noise is modulated by intergene distance and correlated across pairs. The model can predict biases both in which genes are or are not in bipromoter architecture (essential/complex genes and stress response genes, respectively) and which classes of gene should be more likely to have ncRNA in bipromoter architecture. Indeed, that our model can predict noise levels and skew in gene type associated with CUTs strengthens the view that noise control, independent of coexpression modulation, is a possible focus of selection. The model also predicts that bipromoter pairs should be rare subtelomerically as observed, such domains being high-noise domains.
Although the low noise associated with ncRNA is consistent with our model, can we exclude a simple null that envisages ncRNAs as spurious transcripts, a consequence of illegitimate TF activity in open chromatin (Brosius 2005)? This model most obviously predicts that ncRNA should be associated with highly expressed genes, these being in open chromatin. ncRNA is indeed associated with high expression levels (P = 0.00005). To be consistent with this result, note, the null makes the same presumption as our model, namely that the expression of one gene increases the likelihood for the expression of another very close by. The difference is that our model suggests that the expression of the ncRNA in bidirectional orientation enables priming for transcription of the protein-coding partner. Several results suggest that the null to be an incomplete explanation.
First, our model also predicts that ncRNA should be associated with high-expression levels. One component of this is because we expect ncRNA to be disproportionately associated with essential genes, and essential genes tend to have high-expression levels. By contrast, the null predicts ncRNA should be associated with high-expression levels regardless of dispensability. That nonessential genes associated with CUTs do not have higher expression levels than nonessential genes not associated with CUTs (P = 0.33) thus argues against the null. Second, that ncRNA from bipromoters is associated with low values of ACN of the partner protein-coding gene, indicates that abundance of the product is not per se the sole determinant of ncRNA presence/absence. Third, if spurious transcripts are simply a side consequence of neighboring gene expression, the properties of genes associated with ncRNA should not vary as regards their orientation to the neighbor. However, proteins with a cooriented ncRNA have higher expression noise than proteins with a ncRNA derived from a bidirectional promoter (bipromoter with ncRNA noise = 0.65, cooriented with ncRNA noise = 2.07, P = 0.036; B-M test) and higher noise levels than protein-coding genes which have a same strand protein-coding gene neighbor (P = 0.026). This difference, owing to the orientation, cannot be accounted for in terms of abundance differences as, not only is our noise measure abundance-corrected but also protein abundance of genes with an ncRNA is not a function of orientation (P = 0.9). Finally, our model predicts an excess of ncRNA associated with essential genes, which is both observed and cannot be accounted for solely by covariance with abundance (P = 0.018). That genes with cooriented ncRNA have higher ACN suggests that such ncRNAs may be a means to increase expression noise, a possibility we will not examine further.
These results suggest that gene orientation could be an important feature in the control of noise. They also suggest that, as with transcription at SER3 (Martens et al. 2004), it is the process of transcription, or priming for transcription, rather than the product of transcription, that can be important. Although the CUT associated with SER3 (a sense ncRNA) is associated with control of the expression of the downstream gene, our results suggest that transcription from the opposing strand is an effective mechanism for priming a focal sense strand gene for expression and hence for reduction in noise. The CUT transcript may well be unwanted, but it doesn’t follow that the making of the transcript is without functional relevance. This is also supported by the observation that upstream RNA PolII transcripts usually cannot be elongated effectively (Core et al. 2008; Seila et al. 2009). We might then also wonder how much expression in protein-coding genes from bidirectional promoters is to enable noise control rather than produce the protein product itself. Such a hypothesis could explain why many relatively highly coexpressed neighbors (0.4 > r > 0.1) in yeast have no functional (GO class) similarity (Batada et al. 2007).
These findings complement recent evidence that a substantial component of selection on gene arrangement within genomes may well be to modulate noise levels. In yeast, the clustering of essential genes may be also owing to such selection (see also Keller and Knop 2009). In bacteria colinearity, the tendency for genes to appear in the same order in the operon as the proteins are needed in a temporal fashion, appears also best explained by the consequences of selection on noise (Kovacs et al. 2009; Lovdok et al. 2009). Whether noise modulation mediated by changes in gene order/orientation is relevant in less compact genomes, such as those of mammals, is unknown.
Supplementary Material
Acknowledgments
We thank Claudia C. Weber, Lu Chen for helpful discussion and Araxi O. Urrutia for sharing her yeast coexpression data. We thank Pablo Emilio Verde for discussions on analytical stochastic models. G.Z.W. is supported by a Boehringer Ingelheim Fonds travel grant and by the visiting research scholar program of the University of Bath. L.D.H. is a Royal Society Wolfson Research Merit Award Holder. After the review process for this paper, the finding that low noise is associated with divergent orientation was published by another group (Woo and Li 2011).
References
- Balaji S, Babu MM, Iyer LM, Luscombe NM, Aravind L. Comprehensive analysis of combinatorial regulation using the transcriptional regulatory network of yeast. J Mol Biol. 2006;360:213–227. doi: 10.1016/j.jmb.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Bar-Even A, et al. Noise in protein expression scales with natural protein abundance. Nat Genet. 2006;38:636–643. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- Batada NN, Hurst LD. Evolution of chromosome organization driven by selection for reduced gene expression noise. Nat Genet. 2007;39:945–949. doi: 10.1038/ng2071. [DOI] [PubMed] [Google Scholar]
- Batada NN, Urrutia AO, Hurst LD. Chromatin remodelling is a major source of coexpression of linked genes in yeast. Trends Genet. 2007;23:480–484. doi: 10.1016/j.tig.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Becskei A, Kaufmann BB, van Oudenaarden A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nat Genet. 2005;37:937–944. doi: 10.1038/ng1616. [DOI] [PubMed] [Google Scholar]
- Blake WJ, et al. Phenotypic consequences of promoter-mediated transcriptional noise. Mol Cell. 2006;24:853–865. doi: 10.1016/j.molcel.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Blake WJ, Kaern M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Boutanaev AM, Kalmykova AI, Shevelyou YY, Nurminsky DI. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- Brosius J. Waste not, want not–transcript excess in multicellular eukaryotes. Trends Genet. 2005;21:287–288. doi: 10.1016/j.tig.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Brunner E, Munzel U. The nonparametric Behrens–Fisher problem: asymptotic theory and a small-sample approximation. Biometrical J. 2000;42:17–25. [Google Scholar]
- Caron H, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- Cho R, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. Epigenetic regulation and the variability of gene expression. Nat Genet. 2008;40:141–147. doi: 10.1038/ng.2007.58. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat Genet. 2009;41:498–503. doi: 10.1038/ng.319. [DOI] [PubMed] [Google Scholar]
- Cohen BA, Mitra RD, Hughes JD, Church GM. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- Cook DL, Gerber AN, Tapscott SJ. Modeling stochastic gene expression: implications for haploinsufficiency. Proc Natl Acad Sci U S A. 1998;95:15641–15646. doi: 10.1073/pnas.95.26.15641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Braunschweig U, Greil F, Bussemaker HJ, van Steensel B. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008;4:e1000045. doi: 10.1371/journal.pgen.1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- Ebisuya M, Yamamoto T, Nakajima M, Nishida E. Ripples from neighbouring transcription. Nat Cell Biol. 2008;10:1106–1113. doi: 10.1038/ncb1771. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Field Y, et al. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB. Noise minimization in eukaryotic gene expression. PLoS Biol. 2004;2:e137. doi: 10.1371/journal.pbio.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui W, Gel YR, Gastwirth JL. lawstat: an R package for law, public policy and biostatistics. J Stat Soft. 2008;28(3):1–26. [Google Scholar]
- Hurst LD, Pal C, Lercher MJ. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5:299–310. doi: 10.1038/nrg1319. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Williams EJ, Pal C. Natural selection promotes the conservation of linkage of co-expressed genes. Trends Genet. 2002;18:604–606. doi: 10.1016/s0168-9525(02)02813-5. [DOI] [PubMed] [Google Scholar]
- Huynen MA, Snel B. Exploiting the variations in the genomic associations of genes to predict pathways and reconstruct their evolution. In: Galperin MY, Koonin EV, editors. Frontiers in computational genomics. Norfolk (UK): Horizon Scientific Press; 2003. pp. 145–166. [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Knop M. Evolution of mutational robustness in the yeast genome: a link to essential genes and meiotic recombination hotspots. PLoS Genet. 2009;5:e1000533. doi: 10.1371/journal.pgen.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensche PR, Oti M, Dutilh BE, Huynen MA. Conservation of divergent transcription in fungi. Trends Genet. 2008;24:207–211. doi: 10.1016/j.tig.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Kovacs K, Hurst LD, Papp B. Stochasticity in protein levels drives colinearity of gene order in metabolic operons of Escherichia coli. PLoS Biol. 2009;7:e1000115. doi: 10.1371/journal.pbio.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak S, Tang H. Regulation of adjacent yeast genes. Trends Genet. 2000;16:109–111. doi: 10.1016/s0168-9525(99)01941-1. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Blumenthal T, Hurst LD. Coexpression of neighboring genes in Caenorhabditis elegans is mostly due to operons and duplicate genes. Genome Res. 2003;13:238–243. doi: 10.1101/gr.553803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher MJ, Urrutia AO, Hurst LD. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li YY, et al. Systematic analysis of head-to-head gene organization: evolutionary conservation and potential biological relevance. PLoS Comput Biol. 2006;2:e74. doi: 10.1371/journal.pcbi.0020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- Lovdok L, et al. Role of translational coupling in robustness of bacterial chemotaxis pathway. PLoS Biol. 2009;7:e1000171. doi: 10.1371/journal.pbio.1000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- Martens JA, Laprade L, Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature. 2004;429:571–574. doi: 10.1038/nature02538. [DOI] [PubMed] [Google Scholar]
- Michalak P. Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics. 2008;91:243–248. doi: 10.1016/j.ygeno.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Mijalski T, et al. Identification of coexpressed gene clusters in a comparative analysis of transcriptome and proteome in mouse tissues. Proc Natl Acad Sci U S A. 2005;102:8621–8626. doi: 10.1073/pnas.0407672102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H, et al. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- Newman JR, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet. 2002;31:69–73. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- Pal C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- Poyatos JF, Hurst LD. The determinants of gene order conservation in yeasts. Genome Biol. 2007;8:R233. doi: 10.1186/gb-2007-8-11-r233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- Quijano C, et al. Selective maintenance of Drosophila tandemly arranged duplicated genes during evolution. Genome Biol. 2008;9:R176. doi: 10.1186/gb-2008-9-12-r176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, et al. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Core LJ, Lis JT, Sharp PA. Divergent transcription: a new feature of active promoters. Cell Cycle. 2009;8:2557–2564. doi: 10.4161/cc.8.16.9305. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, et al. Tiny RNAs associated with transcription start sites in animals. Nat Genet. 2009;41:572–578. doi: 10.1038/ng.312. [DOI] [PubMed] [Google Scholar]
- Trinklein ND, et al. An abundance of bidirectional promoters in the human genome. Genome Res. 2004;14:62–66. doi: 10.1101/gr.1982804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler JP, Hertel C, Svejstrup JQ. A role for noncoding transcription in activation of the yeast PHO5 gene. Proc Natl Acad Sci U S A. 2007;104:8011–8016. doi: 10.1073/pnas.0702431104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. A complex-based reconstruction of the Saccharomyces cerevisiae interactome. Mol Cell Proteomics. 2009;8:1361–1381. doi: 10.1074/mcp.M800490-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- Williams EJB, Bowles DJ. Coexpression of neighboring genes in the genome of Arabidopsis thaliana. Genome Res. 2004;14:1060–1067. doi: 10.1101/gr.2131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo YH, Li WH. Gene clustering pattern, promoter architecture, and gene expression stability in eukaryotic genomes. Proc Natl Acad Sci U S A. 2011;108:3306–3311. doi: 10.1073/pnas.1100210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, et al. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yu J. A comparative analysis of divergently-paired genes (DPGs) among Drosophila and vertebrate genomes. BMC Evol Biol. 2009;9:55. doi: 10.1186/1471-2148-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MQ, Koehly LM, Elnitski LL. Comprehensive annotation of bidirectional promoters identifies co-regulation among breast and ovarian cancer genes. PLoS Comput Biol. 2007;3:e72. doi: 10.1371/journal.pcbi.0030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, et al. Dosage compensation on the active X chromosome minimizes transcriptional noise of X-linked genes in mammals. Genome Biol. 2009;10:R74. doi: 10.1186/gb-2009-10-7-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.