

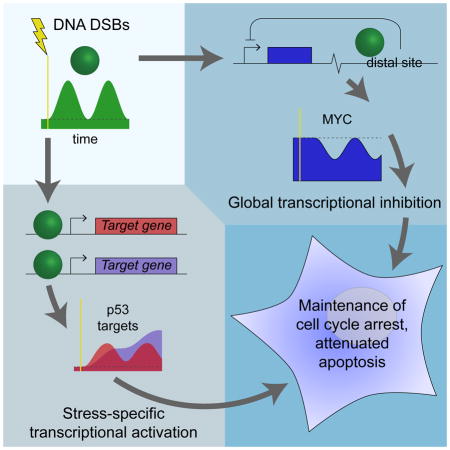
Summary
In response to stresses, cells often halt normal cellular processes; yet stress-specific pathways must bypass such inhibition to generate effective responses. We investigated how cells redistribute global transcriptional activity in response to DNA damage. We show that oscillatory increase of p53 levels in response to double-strand breaks drives counter-oscillatory decrease of MYC levels. Using RNA-seq of newly synthesized transcripts, we found that p53-mediated reduction of MYC suppressed general transcription, with the most highly expressed transcripts reduced to a greater extent. In contrast, upregulation of p53 targets was relatively unaffected by MYC suppression. Reducing MYC during the DNA damage response was important for cell fate regulation, as counteracting MYC repression reduced cell cycle arrest and elevated apoptosis. Our study shows that “global inhibition with specific activation” of transcriptional pathways is important for the proper response to DNA damage, and this mechanism may be a general principle used in many stress responses.
Graphical abstract
Introduction
During times of stress, it may be beneficial for cells to transiently halt normal processes to mount an appropriate stress response; paradoxically, effecting the response may require the use of the same basic cellular processes. For example, when misfolded proteins accumulate in the endoplasmic reticulum, cells activate the unfolded protein response, in which global protein synthesis is suppressed through signaling via PERK and eIF2α (Hetz et al., 2015; Walter and Ron, 2011). Meanwhile, transcripts related to protein folding, amino acid metabolism, and other processes important for alleviation of unfolded protein stress bypass the general inhibition through selective translation (Hetz et al., 2015; Walter and Ron, 2011). Thus, resources are diverted toward the production of stress response mediators while general protein production is reduced. Does a similar mechanism exist to redistribute transcriptional resources during times of stress?
A key regulator in the response to many forms of cellular stress, including different types of DNA damage, is the transcription factor p53 (Levine and Oren, 2009). Upon activation, p53 upregulates many genes to mediate multiple stress responses, including apoptosis, cell cycle arrest, and senescence (Riley et al., 2008). Different stresses give rise to different p53 dynamics, i.e., temporal patterns of rising and falling p53 levels (Batchelor et al., 2011). In response to DNA double-strand breaks (DSBs), a particularly damaging form of stress, p53 levels rise and fall in pulses of fixed amplitude and duration with a period of ~5.5 h (Lahav et al., 2004). These oscillatory dynamics are necessary for proper p53 function, as abolishing them pharmacologically with an inhibitor of p53 degradation changes cell fate (Purvis et al., 2012).
In a recent study of gene expression dynamics during the DSB response, we observed that many genes are upregulated in a series of pulses in a p53-dependent manner (Porter et al., 2016). Strikingly, we found that the expression dynamics of one particular gene, MYC, followed an opposite pattern: as p53 levels rose, MYC mRNA levels fell, and vice versa (Porter et al., 2016). While MYC has been observed to be repressed at least indirectly in a p53-dependent manner (Ho et al., 2005; Levy et al., 1993; Sachdeva et al., 2009), the mechanism for the regulation and the impact of the expression dynamics on cell fate remain poorly understood.
The proto-oncogene MYC codes for the transcription factor c-Myc, or MYC, which regulates numerous targets involved in a wide range of cellular processes. While MYC has been shown to regulate particular target genes, including a core “Myc signature” broadly associated with increasing cellular biomass (Ji et al., 2011), the full set of targets regulated by MYC has been difficult to define consistently (Levens, 2013). Recent work has led to a more unifying principle of MYC action, the “amplifier model,” in which MYC does not simply target specific genes but universally amplifies transcription of all expressed genes (Lin et al., 2012; Nie et al., 2012). This model explains the diverse functions of MYC upregulation in the context of cellular proliferation; however, the implications of the model for MYC activity during cellular stress responses have not been determined.
Based on the amplifier model, we hypothesized that MYC may act coordinately with p53 to redistribute the transcriptome during the DSB response. Here, we show that MYC dynamics are tightly, but inversely, coupled to p53 dynamics following DNA damage – as p53 accumulates, MYC levels are reduced. To investigate the role of this inverse regulation of p53 and MYC in the DSB response, we developed a system to exogenously control MYC expression. Using this system, we performed RNA-seq of newly synthesized transcripts to determine how the transcriptome is redistributed during the response to DNA damage and how maintaining MYC above its basal level alters the redistribution. We found that p53-mediated reduction of MYC downregulates transcription of most actively transcribed genes, especially highly expressed genes. In contrast, we found that activation of most p53 target genes is unaffected by alteration of MYC levels.
We further show that maintaining MYC above its basal level changes cell fate in the DNA damage response. Specifically, we show that bypassing MYC repression leads to reduced cell cycle arrest and elevated apoptosis. Based on gene set enrichment analysis of our transcriptome profiling, we found that the changes in cell fate occur potentially through the combined alteration of expression of numerous genes rather than select transcripts. Taken together, our findings show that p53-dependent reduction of MYC diverts cellular resources toward stress-response pathways and away from other pathways whose continued activation in the presence of DNA damage is detrimental to proper cell homeostasis. This mechanism of “global inhibition with specific activation” provides a potentially general principle of transcriptional control, analogous to translational control during the unfolded protein response, that may be broadly applicable to a range of cellular stress responses.
Results
Levels of MYC mRNA and MYC protein pulse opposite p53 during the DNA DSB response
To identify coordinated regulation of p53 and MYC during the DNA damage response, we first measured the dynamics of p53 and MYC expression. In response to DNA DSBs, p53 levels can increase in a series of pulses with a relatively fixed period of ~5.5 h (Geva-Zatorsky et al., 2006; Lahav et al., 2004) (Figure 1A). Oscillatory p53 accumulation generates oscillatory dynamics in the expression of p53 target genes with short mRNA half-lives, such as MDM2 (Porter et al., 2016; Purvis et al., 2012) (Figure 1B). In turn, oscillatory mRNA expression dynamics can generate oscillatory protein expression dynamics, as has been observed for Mdm2 (Geva-Zatorsky et al., 2006; Lahav et al., 2004; Porter et al., 2016) (Figure 1C). Whereas the oscillations are initially well-synchronized among cells, variation in pulse frequency between individual cells results in the appearance of pulse amplitude dampening in population-level measurements of mRNA and protein abundances as time progresses (Lahav et al., 2004). We observed that MYC mRNA levels were repressed in a series of pulses with the same frequency as that of upregulated p53 target genes (Porter et al., 2016) (Figure 1B, D). Among genes previously analyzed (Porter et al., 2016), such oscillations of reduced expression were unique to MYC.
Figure 1.

Dynamics of key regulators during the DNA damage response. Levels of (A) p53 protein (n = 5), (C) Mdm2 protein (n = 3), and (E) MYC protein (n = 5) were measured by western blotting in MCF-7 cells treated with NCS. Levels of (B) MDM2 mRNA (n = 4) and (D) MYC mRNA (n = 4) were measured by qPCR in MCF-7 p53-Venus cells treated with NCS, as described previously (Porter et al., 2016). Levels of (F) p53 protein (n = 3) and (H) MYC protein (n = 3) were measured by western blotting in p53-knockdown cells (MCF-7 sh-p53) treated with NCS. Levels of (G) MYC mRNA (n = 4) were measured by qPCR in MCF-7 sh-p53 cells treated with NCS, as described previously (Porter et al., 2016). Measurements in (F–H) are normalized to those in untreated control cells (MCF-7 pSuper) (Brummelkamp et al., 2002). Gray backgrounds denote early response prior to p53-driven response. Data are represented as mean ± SEM.
To determine whether similar oscillatory dynamics occur for MYC protein, we measured MYC levels by western blotting in MCF-7 cells treated with neocarzinostatin (NCS) to induce DNA double-strand breaks. After a ~2-h-long early response, MYC protein indeed pulsed opposite p53 (Figure 1E), similar to the dynamics of MYC mRNA (Figure 1D). As expected, the dynamics of MYC protein lagged those of the mRNA by ~1 h (Figure 1D–E), which in turn lagged those of p53 protein by another ~1 h (Figure 1A). The time lags were consistent with delays observed for other p53 target genes: for example, Mdm2 protein pulsed with p53 after a ~2-h early response, lagging the dynamics of its mRNA by ~1 h and those of p53 by ~2 h (Figure 1B–C). To verify that the pulsatile dynamics of MYC protein were dependent on p53, we measured MYC mRNA levels and MYC protein levels in MCF-7 sh-p53 cells, in which p53 expression is knocked down by a constitutively expressed shRNA (Figure 1F) (Brummelkamp et al., 2002). As expected, neither MYC mRNA nor MYC protein displayed pulsatile dynamics when p53 was knocked down (Figure 1G–H). Thus, p53 represses MYC in a pulsatile manner during the DNA DSB response.
p53 represses MYC by binding to a distal repressive element
To quantify the impact of reduction of MYC levels on the transcriptome during the DSB response, we required a method to alter MYC expression independent of p53 regulation. We first sought to identify the mechanism for p53-mediated repression of MYC during the DNA DSB response. Our observation that knocking down p53 abrogated MYC mRNA pulsing during the DSB response (compare Figure 1A, D with Figure 1F–G) implied that p53 regulates MYC mRNA at the level of transcription, processing, or degradation in this context. Two microRNAs, miR-34a and miR-145, have been reported to be p53 targets and to negatively regulate MYC expression (Christoffersen et al., 2009; Sachdeva et al., 2009). To determine whether either of these microRNAs is responsible for p53-mediated MYC repression in the DSB response, we transfected MCF-7 cells with anti-miR oligonucleotides for 24 h, then treated cells with NCS for 4 h and measured MYC protein levels by western blotting. No significant differences were observed between cells treated with anti-miRs and control cells, with or without NCS treatment (Figure S1A), suggesting that the two miRNAs are not mediators of MYC reduction in this context.
Next we determined whether p53 binds directly to the MYC promoter to regulate MYC transcription. We performed ChIP-seq on MCF-7 cells treated with NCS for 3 h to identify peaks of p53 binding near the MYC locus. Contrary to previous reports (Ho et al., 2005), we found no evidence of p53 binding directly upstream of MYC (Figure 2A).
Figure 2.

Mechanism of p53-mediated repression of MYC. (A) ChIP-seq reads for p53 binding near MYC in MCF-7 cells treated with NCS for 3 h (top track) and untreated MCF-7 cells (second track) are shown along with p53 binding peaks (red) and their scores as called by MACS. Read counts are normalized to total reads for each treatment. Peaks shown are the intersection of those identified in n = 2 biological replicates. All peaks in a 1-Mb-wide window centered on MYC are shown. Untreated cells had no p53 binding peaks in this window. RefSeq annotated genes are shown (third track); enhancers and repressive element (fourth track) were identified by Fulco et al. (2016). H3K27ac ChIP-seq reads (bottom track) are from the ENCODE project (Dunham et al., 2012). (B) Close-up view of the largest p53 binding peak shown in (A). p53 consensus binding motifs RRRCWWGYYY (El-Deiry et al., 1992) are shown in addition to data in (A). (C) Region of chromosome 8 excised by CRISPR/Cas9 to generate MCF-7 ΔMYCp53RE cell line. (D) Levels of MYC protein were measured by western blotting in NCS-treated MCF-7 ΔMYCp53RE cells. Measurements are normalized to those in untreated MCF-7 CRISPR-control cells. Data are represented as mean ± SEM (n = 3). (E) Nucleosomes in the region of chromosome 8 shown in (B) in MCF-7 and MCF-7 sh-p53 cells, treated with NCS for 3 h or not treated. Nucleosomes were identified by DANPOS from ATAC-seq data. RefSeq annotated genes are shown (bottom track). (F) Nucleosomes around the MYC promoter. RefSeq annotated genes are shown along with alternate MYC promoters P1 and P2 (bottom track). See also Figure S1.
p53 has been observed to repress genes by interfering with distal enhancers (Li et al., 2012); thus, we examined our ChIP-seq data to identify locations of p53 binding in the 1-Mb-wide window centered on the MYC locus. We identified three locations at which p53 was bound downstream of MYC (Figure 2A); binding at each of these regions increased in response to NCS treatment (Figure S1B–E). One peak of p53 binding, located ~50 kb downstream of MYC, stood out as being much larger than the other two (Figure 2B). To determine whether this binding site was involved in p53-mediated MYC repression, we used CRISPR/Cas9 to excise a 585-bp region of genomic DNA that included the location of highest p53 binding (Figure 2C). In cells lacking this region, MYC protein did not pulse after NCS treatment (Figure 2D) and its dynamics resembled those found in MCF-7 sh-p53 cells (Figure 1H), suggesting that p53 binding at this site is necessary for p53-mediated MYC repression.
The most plausible mechanism for p53 repressing MYC via this binding site is through the action of an enhancer. Several lines of evidence support this explanation: first, this p53 binding peak overlapped with a region recently identified as having a repressive effect on MYC expression by a CRISPRi screen for regulatory elements (Figure 2A–B, “r1”) (Fulco et al., 2016). Second, the region around this peak was identified as a super-enhancer of MYC as determined by H3K27ac ChIP-seq in several cell lines (Zhang et al., 2015). Data available from the ENCODE project support this conclusion in MCF-7 cells as well (Figure 2A) (Dunham et al., 2012) (GEO: GSM945854 and GSM945859). Third, we observed p53 binding at this location even though the pair of p53 consensus binding motifs there (El-Deiry et al., 1992) is separated by 143 bp (Figure 2B). Generally, p53 is not effectively recruited by split binding motifs (Chang et al., 2014; Tonelli et al., 2017); when p53 binds at a site distal to a gene without an unsplit pair of binding motifs, it tends to co-localize with chromatin marks characteristic of active enhancers (Tonelli et al., 2017).
Since p53 appeared to repress MYC at the level of transcription, we next sought to determine whether chromatin remodeling plays a role in this process. To assess the effect of the DSB response, and p53 in particular, on chromatin accessibility and nucleosome positioning, we performed ATAC-seq on MCF-7 cells treated with NCS for 3 h, comparing the results with those obtained from untreated MCF-7 cells and similarly treated MCF-7 sh-p53 cells. In the region of p53 binding located ~50 kb downstream of MYC, chromatin accessibility as measured by ATAC-seq fragment density increased with NCS treatment (Figure S1F), corresponding to the removal of a single nucleosome (Figure 2E). These results are consistent with increased accessibility of a distal regulatory element at this locus that is dependent on DNA damage and p53 expression. Strikingly, at the MYC promoter region there is a p53-dependent decrease in chromatin accessibility in response to DSBs (Figure S1G), even in the absence of a direct p53 binding site near the region. Whereas both the P1 and P2 promoters of MYC (Levens, 2008) are relatively free of nucleosomes in MCF-7 sh-p53 (Figure 2F), both P1 and P2 are occluded in cells expressing WT levels of p53, with an increase of nucleosomal occlusion in response to DNA damage (Figure 2F).
Taken together, our data combined with previous observations suggest MYC repression during the DNA DSB response results from nucleosomal occlusion of both the P1 and P2 promoters and is dependent on p53 binding to a downstream enhancer within a MYC super-enhancer region.
Reducing MYC levels during the DNA damage response redistributes the transcriptome
Given that we observed p53-mediated repression of MYC occurring at the level of transcription, we developed a system to bypass the repression by controlling MYC expression through an inducible, exogenous promoter. We constructed two clonal cell lines from parental MCF-7 cells: one cell line in which GFP-tagged MYC (Nie et al., 2012) was expressed from a doxycycline-inducible promoter (“MYC-GFP”), and a control cell line in which GFP alone was expressed from the same promoter (“GFP”) (Figure 3A). As previously determined, the MYC-GFP construct is fully functional in terms of MYC activity (Nie et al., 2012) and could therefore be used to probe functional effects of MYC expression. We treated both cell lines with doxycycline for 18 h to induce expression of MYC-GFP or GFP using a concentration of inducer that resulted in expression of MYC-GFP near endogenous MYC levels (Figure 3B). We then treated cells with NCS to induce DNA DSBs. In the MYC-GFP cells, this treatment maintained total MYC levels (the sum of MYC and MYC-GFP) at or slightly above the basal MYC expression level throughout the DSB response (Figure 3B), as desired.
Figure 3.

A cell system to control MYC expression. (A) MCF-7 variants with doxycycline-inducible constructs, including schematics of desired MYC expression in cell lines. (B) MYC dynamics in MYC-GFP cells during the DSB response. Levels of MYC protein were measured by western blotting in MYC-GFP cells treated with NCS; bands for endogenous MYC and MYC-GFP were quantified separately. Gray backgrounds denote early response prior to p53-driven response. Data are represented as mean ± SEM (n = 3).
Having developed a method to maintain MYC above its basal expression level, we next sought to determine functional impacts of p53-mediated reduction of MYC levels. Since MYC amplifies expression of all active genes (Lin et al., 2012; Nie et al., 2012), we predicted that reducing MYC levels would have pleiotropic, transcriptome-wide effects during the DNA DSB response. Specifically, we expected that most actively transcribed genes would be transcribed at a lower rate, while genes required for the DSB response, including most genes regulated by p53, would undergo increased transcription. To test this hypothesis, we used RNA-seq to measure the transcripts synthesized within a one-hour interval under four conditions: in either the GFP control cell line or the MYC-GFP cell line, and in either the absence of NCS treatment or 2.5–3.5 h after NCS treatment (around the first peak of p53 expression and before the first trough of MYC expression) (Figure 1A, E). For each condition, we labeled the RNA synthesized during the one-hour interval with the synthetic uridine analog 5-ethynyl uridine. Since MYC alteration changes the total amount of mRNA in cells, we needed to control for size differences in cells with different amounts of MYC (Lovén et al., 2012); therefore, we developed a labeled spike-in mRNA control. We grew fission yeast in the presence of 5-ethynyl uridine, then spiked yeast RNA into our human RNA samples proportional to the number of human cells counted. After conjugating labeled RNA with biotin and isolating it by binding to streptavidin beads, we performed RNA-seq on the isolated labeled RNA, normalizing counts of human transcripts to counts of highly expressed yeast transcripts. Samples from the two cell lines and two treatment conditions could be distinguished using principal component analysis, in which the first two principal components could be readily interpreted as relating to DNA damage and regulation of MYC expression, respectively (Figure S2A).
Quantifying transcripts produced during the time between when p53 levels are high and when MYC levels are reduced enabled us to estimate transcriptome-wide transcription rates. Strikingly, the majority of transcripts in GFP control cells (75.7%) were transcribed at a lower level during the time interval 2.5–3.5 h after NCS treatment than in a one-hour time interval without NCS treatment (Figure 4A). This repression was much less pronounced in MYC-GFP cells, in which MYC was held above its basal level during the DSB response: only 56.0% of transcripts were transcribed at a lower level than in GFP cells without NCS treatment (Figure 4B). While not all changes in transcription of individual genes were statistically significant, the general downward trend in transcription of particular genes during the DSB response (p < 10−16, Wilcoxon signed-rank test) and general reversal of this trend with MYC expressed above its basal level (p < 10−16, Wilcoxon signed-rank test) are consistent with a significant MYC-mediated effect on global transcription.
Figure 4.

Redistribution of the transcriptome during the DNA DSB response. (A–B) Fold-changes in mean production of all cellular transcripts 2.5–3.5 h after NCS treatment in GFP cells (A) and MYC-GFP cells (B) compared with untreated GFP cells. Transcripts with at least 5 reads in each replicate (n = 3) of each cell line/treatment condition are included. Transcripts are ordered by fold-change in mean production. (C–D) Fold-changes in mean production of p53 target transcripts, as identified by Allen et al. (2014), 2.5–3.5 h after NCS treatment in GFP cells (C) and MYC-GFP cells (D) compared with untreated GFP cells. (E) Foldchanges in mean transcription of select p53 target genes and control genes 2.5–3.5 h after NCS treatment in MYC-GFP and GFP cells, compared with untreated GFP cells, as measured by RNA-seq. Data are represented as mean ± SEM (n = 3 for each condition). (F) Density plot of fold-changes in mean production of cellular transcripts in untreated MYC-GFP cells relative to untreated GFP cells as a function of mean production in untreated GFP cells. Spearman correlation coefficient between expression in untreated GFP cells and fold-change due to MYC above its basal level is ρ = 0.155. (G) Density plot of fold-changes in mean production of cellular transcripts in NCS-treated GFP cells relative to untreated GFP cells as a function of mean production in untreated GFP cells. Spearman correlation coefficient between expression in untreated GFP cells and fold-change due to NCS is ρ = −0.151. For (F–G), transcripts of length ≥ 1500 bases with at least 5 reads in each replicate (n = 3) of each cell line/treatment condition are included. Pink lines represent median fold-change in each bin, calculated for bins with at least 40 transcripts. See also Figure S2 and Table S1.
In contrast to the lower level of transcription of most genes in GFP control cells, most p53 target genes had elevated transcription 2.5–3.5 h after NCS treatment compared with untreated cells, as expected (Figure 4C, examples in Figure 4E). Maintaining MYC above its basal level did not significantly change transcription of p53 targets during the DSB response (p > 0.05, Wilcoxon signed-rank test; Figure 4D, examples in Figure 4E). Still, many of the genes that showed elevated transcription during the DSB response with or without elevated MYC were not p53 targets (Figure S2B–C). To better understand the minority set of genes with upregulated transcription during the DSB response, we performed gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005), comparing NCS-treated GFP cells with GFP cells not treated with NCS. Predictably, most gene sets tested were enriched in cells not treated with NCS. The most highly enriched gene sets in NCS-treated cells (Table S1) involved responses to ionizing radiation and other forms of DNA damage, the p53 pathway, and the NF-κB pathway, which is also activated during the DSB response (Habraken and Piette, 2006) (examples in Figure S2D–F).
It has been observed that MYC amplifies expression of more highly transcribed genes to a greater degree than genes transcribed at a lower level (Lin et al., 2012; Nie et al., 2012). Consistent with this observation, we found that untreated MYC-GFP cells have higher transcription of most genes compared with untreated GFP cells, and the magnitude of this effect is greater for more highly transcribed genes (Figure 4F). Since MYC has a stronger amplifying effect on more highly transcribed genes, we predicted that highly transcribed genes would be downregulated to a greater degree during the DSB response compared with genes transcribed at a lower level. Indeed, we found this to be the case (Figure 4G). Taken together, these data indicate that transcription is redistributed in cells during the DSB response, with upregulation of a relatively small number of targets focused on genes involved in the stress response and simultaneous MYC-dependent downregulation of the remainder of the transcriptome, especially those genes most highly expressed.
To better understand how lowering MYC levels during the DSB response affects transcription of individual genes, we compared the fold-change in production of different transcripts during the DSB response in MYC-GFP cells to that in GFP cells (Figure 5A). In general, the behavior of transcripts was qualitatively consistent during the DSB response with or without MYC maintained above its basal level: the majority of transcripts downregulated during the DSB response in GFP cells were also downregulated, or at least upregulated to a lesser degree, during the DSB response in MYC-GFP cells. Transcripts could be divided into four groups (Figure 5A). Group I contained genes whose transcription was increased during the DSB response and increased further by MYC; this group included many canonical p53 targets. Group II contained transcripts that were repressed during the DSB response and upregulated by MYC. This group corresponded to more than half (54%) of expressed transcripts. Groups III and IV, corresponding to genes downregulated by MYC during the DSB response, together contained ~27% of transcripts. Genes in Groups III and IV were potentially subject to more complex regulation; for example, they could have been repressed by a factor whose expression was upregulated by MYC. Overall, these data reveal general trends that transcription of genes decreases during the DNA DSB response and increases with additional MYC.
Figure 5.

Effects of MYC levels on the transcriptome during the DNA DSB response. (A) Density plot of fold-changes in mean transcription in NCS-treated MYC-GFP cells (n = 3) relative to untreated GFP cells (n = 3) vs. fold-changes in mean transcription in NCS-treated GFP cells (n = 3) relative to untreated GFP cells (n = 3). Transcripts are divided into Groups I–IV based on their fold-change due to the DSB response and their fold-change due to MYC held above its basal level during the DSB response. (B–C) Examples of apoptosis- and cell cycle-related gene sets found to be significantly enriched in NCS-treated MYC-GFP cells over NCS-treated GFP cells. Normalized enrichment scores (NES), false discovery rate q-values, and nominal p-values are shown. See also Figure S3 and Table S2.
Reducing MYC levels during the DNA damage response impacts cell fate
In light of the broad transcriptional impact of MYC reduction during the DSB response, we next sought to determine its cell-physiological impacts by identifying the pathways most sensitive to changes in MYC following DNA damage. We performed GSEA to compare transcripts produced 2.5–3.5 h after NCS treatment in MYC-GFP cells against those in GFP cells (Groups I and II vs. Groups III and IV, Figure 5A). Predictably, most of the gene sets tested are enriched in MYC-GFP cells. Among the gene sets with the highest enrichment scores and false discovery rate q < 0.2 (Table S2) we identified two major categories: 1.) apoptosis and cell death; and 2.) cell cycle, proliferation, and division (examples in Figure 5B–C, Figure S3).
To determine how reduction in MYC levels during the DSB response affects cell cycle regulation, we treated MYC-GFP and GFP cells with NCS for different lengths of time, then measured the distribution of cells through the cell cycle using flow cytometry. Among populations of MYC-GFP cells not treated with NCS, more cells were found in S phase, and fewer were found in G2/M phase, compared with populations of GFP cells not treated with NCS (Figure 6A). After 24 h of NCS treatment, both MYC-GFP and GFP populations had significantly more cells in G2/M phase and fewer cells in S phase than populations not treated with NCS, indicating that cell cycle arrest mechanisms were active (Figure 6A–B). After 48 h of NCS treatment, both MYC-GFP and GFP populations still had many more cells in G2/M phase and fewer cells in S phase than cells not treated with NCS, but MYC-GFP populations had significantly more cells in S phase and significantly fewer cells in G2/M phase than GFP populations (Figure 6C). Thus, preventing MYC reduction during the DSB response resulted in cell cycle redistribution, which suggests that lowering MYC levels is necessary for maintaining proper cell cycle checkpoints during the DNA DSB response.
Figure 6.

MYC-mediated effects on cell fate during the DNA DSB response. (A–C) Cell cycle distributions of MYC-GFP and GFP cells that were (A) not treated with NCS, (B) treated with NCS for 24 h, or (C) treated with NCS for 48 h. Data are represented as mean ± SEM (n = 3). (D) Annexin V (AV) and propidium iodide (PI) staining showing the percentage of cells in early apoptosis (high AV, low PI) and late apoptosis (high AV, high PI) in RPE1 MYC-GFP and RPE1 GFP cells after the indicated treatments. Data are represented as mean ± SEM (n = 4). Asterisks (*) denote statistically significant differences between MYC-GFP and GFP cells or between RPE1 MYC-GFP and RPE1 GFP cells (p < 0.05, two-sample T test).
To determine whether MYC reduction during the DSB response affects the activation of apoptosis, we assessed cell viability by annexin V and PI staining. Since MCF-7 cells do not express caspase-3 (Jänicke et al., 1998), we used hTERT-RPE1 immortalized retinal pigmented epithelium cells as a more physiologically relevant model for apoptosis. As with the MCF-7 cells, we developed clonal hTERT-RPE1 cells in which either GFP-tagged MYC (RPE1 MYC-GFP) or GFP alone (RPE1 GFP) was expressed from a doxycycline-inducible promoter. After 48 h of NCS treatment, RPE1 MYC-GFP cells had a significant increase in apoptosis compared with RPE1 GFP cells (Figure 6D). Thus, counteracting MYC repression increases apoptosis during the DSB response, which suggests that p53-mediated reduction of MYC functions to suppress apoptosis during the stress response.
Discussion
Our results show that MYC, when reduced in expression in a p53-dependent manner during the DNA DSB response, reduces transcription of most expressed genes while a smaller subset of genes can bypass the general transcriptional inhibition to increase in expression. This MYC reduction functions to maintain the physiologically observed cell cycle profile during the DSB stress response while suppressing the activation of apoptosis (Figure 7). p53 represses MYC by binding to a downstream enhancer region within a MYC super-enhancer and is required for nucleosomal occlusion of the MYC P1 and P2 promoters that occurs during the DNA DSB response, providing a mechanism for generating the strong coupling between p53 and MYC dynamics required to mediate the transcriptomic and cell fate effects.
Figure 7.

During the DSB response, p53 acts through MYC along with its other targets, suppressing transcription of most genes while selectively activating genes involved in the response. This redistribution of the transcriptome guides cell fate decisions.
Our data support a model of “global inhibition with specific activation” that may be a common feature of cellular stress responses. Mechanisms of repressing global gene expression in response to other forms of DNA damage have previously been observed in mammalian cells. In cells treated with UV radiation (which leads to the formation of thymine dimers and exposure of tracts of single-stranded DNA) or hydrogen peroxide (which generates DNA-damaging reactive oxygen species), transcription initiation by RNA polymerase II is inhibited, leading to global transcriptional repression (Heine et al., 2008; Proietti-De-Santis et al., 2006; Rockx et al., 2000). Moreover, p53 can repress transcription of ribosomal genes, thus downregulating translation globally (Budde and Grummt, 1999; Zhai and Comai, 2000). The mechanism described here of lowering MYC levels to repress transcription differs notably from these other mechanisms in its selectivity: most transcription is reduced (Figure 4A) while a subset of genes, including those known to be important for the DNA DSB response, is upregulated (Figure 4C, Figure S2D–F, Table S1). This selectivity resembles that seen in the unfolded protein response, in which global protein synthesis is suppressed while transcripts involved in alleviation of unfolded protein stress are selectively translated, bypassing the general inhibition (Hetz et al., 2015; Walter and Ron, 2011). Similar arrangements of global inhibition with specific activation of gene expression, albeit by distinct mechanisms, can be found in other organisms as well, suggesting convergent evolution of a broadly useful principle in biological regulation. For example, alternative sigma factors in bacteria activate particular sets of genes during stress conditions; these compete with the housekeeping sigma factor, which is involved in general transcription (Browning and Busby, 2016). In bacteria transferred from nutrient-depleted to nutrient-rich medium, enzymes that metabolize the carbon source present in the medium are selectively transcribed, while most promoters are not active; normal promoter activity is restored upon transition to normal growth (Madar et al., 2013).
One of the physiological implications of MYC repression during the DSB response, and its concomitant redistribution of transcription, is a redistribution of the cell cycle. Consistent with previous reports (Ho et al., 2005; Levy et al., 1993; Sheen and Dickson, 2002), in this study we found that raising the level of MYC during the DSB response yielded more cells in S phase and fewer cells in G2/M phase. Thus, it can be inferred that reducing MYC levels yields fewer cells in S phase and more in G2/M, particularly after a long DSB response (Figure 6C). These data suggest that a lower level of MYC promotes both G1 and G2/M cell cycle arrest: G2/M arrest raises the number of cells in G2 and M phases while decreasing the number of cells that proceed into G1; meanwhile, G1 arrest maintains cells in G1 and prevents progression into S phase, leading to the observed phenotype. Cell cycle arrest is an important feature of the DNA DSB response, as it prevents damaged cells from replicating, and MYC repression evidently promotes this. The mechanism by which it does so is less clear. It has been shown that MYC can repress expression of CDKN1A, which encodes p21, a key effector of G1 arrest (Seoane et al., 2002); however, we did not observe a significantly different level of CDKN1A transcription in MYC-GFP cells compared to GFP cells (Figure 4E). As determined from our study, the broad influence of MYC reduction on the transcriptome, as indicated by the numerous gene sets significantly upregulated upon maintenance of MYC above basal levels (Table S2, Figure 5B–C, Figure S3), suggests that it enhances cell cycle arrest not through the specific activity of any one arrest mediator but rather through a combination of many small effects across the transcriptome. Thus, the physiological impact on the cell cycle may arise as an emergent property of a larger-scale network of interactions.
Our results further indicate that MYC repression during the DSB response reduces the number of cells undergoing apoptosis. Consistent with previous studies (Hoffman and Liebermann, 2008; Maclean et al., 2003; Phesse et al., 2014; Seoane et al., 2002), we here found that cells with higher-than-normal MYC levels undergo more apoptosis than control cells, particularly after a long DSB response (Figure 6D). This result suggests that MYC reduction limits apoptosis. In the DSB response, this could function to defer a terminal and irreversible cell fate decision until sufficient time has elapsed for the cell to exhaust its possibilities for DNA repair. While the exact mechanism by which MYC affects apoptosis is not certain, the simplest explanation, based on our findings and MYC’s known activity as a broad regulator of transcription, is that this phenomenon arises due to a combination of small effects from a multitude of downregulated genes rather than a single gene or pathway (Figure S3). It has been proposed that increased MYC levels tilt the balance of cell fate toward apoptosis by repressing CDKN1A transcription (Seoane et al., 2002). This is not a complete explanation, however, as (1) increased MYC levels also promoted apoptosis, albeit not as much, in cells lacking CDKN1A in the same study, and (2) we did not observe a significant change in CDKN1A transcription in cells with elevated MYC. It has also been observed that increased MYC sensitizes mouse embryonic fibroblasts to DSB-induced apoptosis by suppressing Bcl-XL, a negative regulator of apoptosis (Maclean et al., 2003). We did not find significant differences in transcription of BCL2L1, the gene coding for the human ortholog of murine Bcl-XL (data not shown); any MYC-mediated effect on BCL2L1 expression would need to be post-transcriptional. In light of our results, future wide-scale analysis should aid in dissecting the mechanistic connections between MYC levels and the regulation of the numerous pro- and anti-apoptotic factors during the DSB response.
Our ChIP-seq measurements and CRISPR/Cas9-mediated deletion suggest that p53 represses MYC expression during the DSB response via a downstream enhancer element. In principle, two other genomic features close to the p53 binding site identified in this study could be responsible for p53-mediated repression of MYC transcription; however, neither is well supported by the available evidence. A microRNA, miR-1204, is transcribed just 3′ of the location we identified (Figure 6B); however, MYC is not among miR-1204’s predicted targets (Agarwal et al., 2015). Also present at this location is PVT1, a gene believed to produce a long non-coding RNA. While PVT1 has been proposed to regulate MYC through several mechanisms, it is, on the whole, poorly understood (Cui et al., 2016), and additional studies will be required to disentangle potential interactions between p53 and the PVT1 locus in the fine-tuning of MYC expression. Based on our ATAC-seq data, a more likely mechanism is that p53 binding to the distal element induces changes in chromatin structure, leading to the occlusion of both the P1 and P2 MYC promoters and resulting in reduction in MYC transcription. Future work will be required to understand additional changes in chromatin structure and histone modification that may also contribute to the repression. Interestingly, the p53 binding site downstream of MYC identified in this study has been shown to be a locus for genomic instability in diverse types of cancer, including medulloblastomas (Northcott et al., 2012), multiple myelomas (Nagoshi et al., 2012), small cell lung cancer (Iwakawa et al., 2013), and gastric cancer (Kim et al., 2014), suggesting a potentially important role for this regulatory site in preventing malignancies.
MYC has a relatively unstable mRNA and protein, which makes it a particularly effective regulator of rapid cellular responses. Its mRNA half-life of ~10 minutes (Dani et al., 1984) and protein half-life of ~20 minutes (Wall et al., 2008) are among the shortest of any gene product in human cells. Such rapid turnover enables MYC levels to be quickly reduced upon inhibition of synthesis. Numerous signaling pathways alter MYC expression at the transcriptional, post-transcriptional, and protein regulatory levels (Levens, 2013). In particular, degradation of MYC results from the activity of many E3 ubiquitin ligases (Farrell and Sears, 2014). It is likely that other signaling stimuli and environmental stresses can reduce MYC expression in a manner similar to that observed in the DNA DSB response, yielding a rapid reduction in general transcription due to MYC’s function as a transcriptional amplifier while simultaneously enabling stimulus- or stress-specific transcriptional responses to bypass the general inhibition. Future studies will help to determine the extent to which the mechanism of “global inhibition with specific activation” functions in other stress response pathways, as well as to identify how such responses are altered in pathological conditions with MYC amplification.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Eric Batchelor (batchelore@mail.nih.gov).
Experimental Model and Subject Details
Plasmids
A doxycycline-inducible pTRIPZ-derived vector (GE Dharmacon, RHS4750) was used to generate a system for inducible MYC-GFP and GFP expression. The vector was digested with restriction enzymes BspE1 (NEB, R0540) and MluI (NEB, R0198) and the large fragment was ligated with AgeI- (NEB, R0552) and MluI-digested PCR products obtained from amplification of plasmid p-c-Myc-pd4-EGFP-N1 (Nie et al., 2012) using the primers 5′-TCCGTCCGGAGCCACCATGCCCCTCAACGTTAG-3′ and 5′-ACTACACGCGTAAGATACATTGATATGAGT-3′ (for MYC-GFP) or 5′-GATCCACCGGTCGCCACC-3′ and 5′-ACTACACGCGTAAGATACATTGATATGAGT-3′ (for GFP). To reduce the size of the plasmids to aid lentiviral packaging, each plasmid was next digested with restriction enzyme SfaAI (ThermoFisher, ER2091) and digested with PscI (ThermoFisher, ER1871). The larger fragment was ligated with a PscI- and SfaAIdigested PCR product obtained from amplification of pTRIPZ using the primers 5′-TTTGACCTTGACATGCT- 3′ and 5′-ACTTATGCGATCGCGCCCCAGCTGGTTCT-3′, resulting in plasmids pTRIPZ-del89-c-Myc-d4EGFP and pTRIPZ-del89-d4EGFP for MYC-GFP and GFP expression, respectively.
A separate smaller doxycycline-inducible MYC-GFP expression plasmid was constructed for integration into hTERT-RPE1 cells. The MYC-GFP coding sequence was amplified from pTRIPZ-del89-c-Myc-d4EGFP using the primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCATGCCCCTCAACGTTAGCTTC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCTACACATTGATCCTAGCAG-3′. Gateway cloning (ThermoFisher, 12537023) was used to generate a donor vector by integrating the PCR product into pDONR221 (ThermoFisher, 12536017) via a BP cloning reaction. The subsequent DONR vector was used in an LR cloning reaction with the destination vector pInducer20 (Meerbrey et al., 2011) to generate the expression plasmid pInducer20-c-Myc-d4EGFP.
Packaging of the plasmids pTRIPZ-del89-c-Myc-d4EGFP and pTRIPZ-del89-d4EGFP into lentiviral vectors was performed using a Trans-Lentiviral Packaging Kit (ThermoFisher, TLP4641) following the manufacturer’s protocol and grown in 293T cells (ATCC CRL-11268). Packaging of plasmid pInducer20-c-Myc-d4EGFP into lentiviruses was performed by transient transfection of 10-cm plates of 293T cells with 12 μg of the plasmid construct, 9 μg of psPAX2 (Addgene #12260), 3.6 μg of MD2.VSVG (Addgene #12259) and 49.2 μL of Lipofectamine 2000 (ThermoFisher, 11668019) in 11.5 mL of Opti-MEM I (ThermoFisher, 31985070). 16 h after transfection, medium was changed to 10 mL DMEM + 10% heat inactivated FBS. Virus was collected and pooled 40 and 64 h after transfection and filtered through 0.45-μm PVDF filters. Virus was concentrated and resuspended into 2 mL of DMEM:F12 medium (ATCC, 30-2006) by ultracentrifugation at 23,000 rpm for 2.5 hours at 4°C.
Human cell lines and culture
MCF-7 breast carcinoma cells were cultured in RPMI containing 10% fetal bovine serum (FBS), 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, and 250 ng/mL amphotericin B. MCF-7 p53-Venus cells (Batchelor et al., 2008) were cultured in base medium plus 400 μg/mL neomycin. MCF-7 sh-p53 and MCF-7 pSuper cells (Brummelkamp et al., 2002) were cultured in base medium plus 0.125 μg/mL puromycin.
For CRISPR/Cas9-mediated deletion of the genomic region at which p53 binds downstream of the MYC locus, we first developed sgRNA expression plasmids targeting sites flanking the region of interest. To target the region upstream of the p53 binding site, we used oligonucleotide pairs with the sequences 5′-CACCGTGCAAGGATTTAGGGACAAG and 5′-AAACCTTGTCCCTAAATCCTTGCAC; to target downstream, we used oligonucleotides with the sequences 5′-CACCGTGTACCCATTGAATTCCCAG and 5′-AAACCTGGGAATTCAATGGGTACAC. Oligonucleotide pairs were phosphorylated by T4 polynucleotide kinase (NEB, M0201S) for 30 min at 37°C and annealed to each other by heating to 95°C for 5 min, then slowly cooling to 25°C at a rate of 5°C/min. pLentiCRISPRv2 (Addgene, 52961) was digested with Esp3I (ThermoFisher, FD0454) and dephosphorylated with FastAP (ThermoFisher, EF0654) for 30 min at 37°C. The 13-kb fragment of the digested plasmid was isolated by agarose gel electrophoresis and purified using a QiaQuick Gel Extraction Kit (Qiagen, 28704). The upstream- and downstream-targeting annealed oligonucleotides were ligated into the digested pLentiCRISPRv2 fragment using Quick T4 DNA Ligase (NEB, M2200S) following the manufacturer’s protocol to yield plasmids pLentiCRISPR-ΔMYCp53RE-sg2 and pLentiCRISPR-MYCΔp53RE-sg7, respectively. 1.25 μg of pLentiCRISPR-ΔMYCp53RE-sg2 and pLentiCRISPR-MYCΔp53RE-sg7 each were combined and transfected into MCF-7 cells using Lipofectamine 3000 (ThermoFisher, L3000001) following the manufacturer’s protocol. To generate a control cell line, 2.5 μg of pLentiCRISPRv2 was transfected into MCF-7. Two days after transfection, stable cells were selected using growth medium containing 0.5 μg/ml puromycin. Clonal cells of each stable line were grown from single cells to generate the cell lines MCF-7 ΔMYCp53RE and MCF-7 CRISPR-control, respectively. Deletion of the genomic region chr8:128,807,770-128,808,354 (hg19 coordinates) from MCF-7 ΔMYCp53RE was verified by sequencing. MCF-7 ΔMYCp53RE and MCF-7 CRISPR-control cells were cultured in base medium plus 0.5 μg/mL puromycin.
hTERT-RPE1 retina pigmented epithelial cells (ATCC, CRL-4000) were cultured in DMEM:F12 medium (ATCC, 30-2006) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, 250 ng/mL amphotericin B, and 0.01 mg/mL hygromycin B. To generate doxycycline-inducible GFP and MYC-GFP cell lines, MCF-7 cells were infected with lentivirus in which pTRIPZ-del89-c-Myc-d4EGFP or pTRIPZ-del89-d4EGFP had been packaged, and hTERT-RPE1 cells were infected with lentivirus in which pInducer20-c-Myc-d4EGFP or pTRIPZ-del89-d4EGFP had been packaged. Infection was performed for 5 h in the presence of 8 μg/ml protamine sulfate and 10 μM HEPES. Stable cells were selected by growth in the appropriate base medium supplemented with puromycin at a concentration of 0.5 μg/mL for MCF-7 cells or 8 μg/mL for hTERT-RPE1 cells starting 2 days after lentiviral infection. Clonal cell lines were grown from single stable cells and used for all subsequent studies. MCF-7 GFP and MCF-7 MYC-GFP clonal cells were cultured in RPMI containing 10% tetracycline-free FBS (Clontech, 631106), 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, 250 ng/mL amphotericin B, and 0.5 μg/mL puromycin. hTERT-RPE1 GFP and hTERT-RPE1 MYC-GFP clonal cells were cultured in DMEM:F12 containing 10% tetracycline-free FBS, 100 U/mL penicillin G, 100 μg/mL streptomycin sulfate, 250 ng/mL amphotericin B, 0.01 mg/ml hygromycin B, and 8 μg/mL puromycin.
All cell lines were cultured at 37°C and 5% CO2.
Yeast cell lines and culture
S. pombe cells expressing human equilibrative nucleoside transporter 1 (hENT1) and thymidine kinase (tk) (h+ leu1-32::[hENT1 leu1+] his7-366::[hsv-tk his7+] ura4-D18 ade6-M210) (Hodson et al., 2003) were grown at 30°C in EMM containing 225 mg/L of each of adenine, histidine, lysine, and uracil.
Method Details
Western blotting
1.4×105 cells were plated on a 35-mm dish, or 4×105 cells were plated on a 6-cm dish, 2 days prior to treatment. For cell lines containing doxycycline-inducible genes, expression was induced with 150 ng/mL doxycycline 18 h prior to treatment. For experiments with anti-miRs, cells were transfected with 100 nM of miR inhibitors (IDT) targeting the sequences UCGUUAAUCGGCUAUAAUACGC (control), UGGCAGUGUCUUAGCUGGUUGU (miR-34a), or GUCCAGUUUUCCCAGGAAUCCCU (miR-145) using Lipofectamine 3000 reagent (ThermoFisher, L3000001) following the manufacturer’s suggested protocol 24 h prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks. At indicated times following treatment, cells were harvested by scraping and frozen in a dry ice-ethanol bath. Cells were lysed in M-PER Mammalian Protein Extraction Reagent (ThermoFisher, 78501) according to manufacturer’s protocol, and protein samples were quantified by Bradford assay. Equal masses of protein were separated by electrophoresis on 4–20% gradient gels (Bio-Rad, 567–1095 and 456–1096) and blotted onto Immobilon-P PVDF membranes (Millipore, IPVH00010). Membranes were incubated with the following primary antibodies: mouse monoclonal anti-p53 DO-1 (Santa Cruz Biotechnology, sc-126), rabbit monoclonal anti-β-actin 13E5 (Cell Signaling Technology, 4970), mouse monoclonal anti-Mdm2 SMP14 (Santa Cruz, sc-965), rabbit monoclonal anti-c-Myc Y69 (Abcam, ab32072), and mouse monoclonal anti-β-actin AC-74 (Sigma, A5316). Membranes were incubated with the following secondary antibodies: donkey anti-mouse 680RD (LI-COR, 926-68072) and donkey anti-rabbit 800CW (LI-COR, 926-32213), or donkey anti-rabbit 680RD (LI-COR, 926-68073) and donkey anti-mouse 800CW (LI-COR, 926-32212). Membranes were then scanned with a LI-COR Odyssey system (LI-COR).
mRNA expression measurements
mRNA levels in cell populations were measured as described previously (Porter et al., 2016). Specifically, 4×105 MCF-7 p53-Venus cells or 8×105 MCF-7 sh-p53 cells were plated on a 6-cm dish 2 days prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks. At indicated times following treatment, cells were harvested by scraping and frozen in a dry ice-ethanol bath. RNA was extracted using RNeasy (Qiagen, 74134) and QIAshredder (Qiagen, 79654) kits. For each sample, RNA concentration was determined using a UV spectrophotometer, and 604.8 ng of RNA was used to generate complementary DNA in a 20 μL reaction using a High Capacity cDNA Reverse Transcription Kit (ThermoFisher, 4374966). Gene expression was then measured in duplicate for each sample by qPCR using a 96.96 Dynamic Array on a BioMark™ system (Fluidigm) according to the manufacturer’s instructions (Fluidigm, 100-4109 B1). Samples and assays were subjected to thermal mixing (70°C for 40 min, 60°C for 30 s), hot start (95°C for 1 min), and 35 cycles of PCR (96°C for 5 s, 64°C for 20 s), followed by melt curve acquisition (64–95°C with 0.5°C resolution).
ChIP-seq
MCF-7 cells were plated on 15-cm dishes. After 24 h, cells were treated for 3 h with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks; “untreated” cells were given fresh medium. ChIP samples were prepared as described by Barski et al. (2007) with minor changes. Briefly, 4×107 cells were cross-linked with 1% formaldehyde for 10 min. Cross-linking was stopped by the addition of glycine to 125 mM final concentration, and cells were washed twice with PBS. Cells were then harvested by scraping, and the pellet was washed once with PBS + 0.5% BSA and resuspended in RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 200 mM NaCl) supplemented with cOmplete™ protease inhibitor tablet (Roche, 11697498001) to a final concentration of 5×106 cells/mL. Samples were sonicated for 15 min with 20-sec pulses, 40-sec resting, using the Q700 Sonicator (QSONICA) to produce chromatin fragments of 300 bp on average.
After clarification by centrifugation, sonicated extracts (other than input DNA) were subjected to immunoprecipitation. 4 μg of mouse monoclonal anti-p53 DO-1 X (Santa Cruz Biotechnology, sc-126 X) was mixed with 40 μL of Dynabeads Protein A (ThermoFisher, 10002D) and incubated at 4°C for 6 h with rotation. Chromatin from 5×106 cells was added to the Protein A-antibody complexes and incubated overnight at 4°C with rotation. Immunoprecipitates were washed twice with RIPA buffer, twice with RIPA buffer + 300 mM NaCl, twice with LiCl buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate), and twice with TE buffer. The beads were then resuspended in TE buffer + 0.25% SDS + 500 μg/mL proteinase K (New England Biolabs, P8102S) and incubated overnight at 65°C. After elution, the DNA was recovered from the eluate by phenol chloroform extraction followed by ethanol precipitation in the presence of 20 μg of glycogen (Roche, 10901393001) and dissolved in TE buffer.
Samples were quantified using a Quant-iT™ PicoGreen™ dsDNA Assay Kit (ThermoFisher, P11496), and fragment sizes were assessed using a Bioanalyzer (Agilent) with a High Sensitivity DNA Kit (Agilent, 5067-4626). Libraries were then prepared using TruSeq ChIP Library Preparation Kits (Illumina, IP-202-1012 and IP-202-1024) by the CCR Sequencing Facility (Frederick, MD) according to the manufacturer’s instructions. Briefly, DNA was end-repaired, and 3′ ends were adenylated to prepare for adapter ligation. After ligation of adapters, ligation products were purified and size-selected for fragments of length 250–300 bp. Fragments with adapters were enriched by 18 cycles of PCR, then pooled for sequencing. Single-end sequencing of pooled samples was performed by the CCR Sequencing Facility (Frederick, MD) using an Illumina NextSeq 500 system.
ATAC-seq
6.4×105 MCF-7 or 1.0×106 MCF-7 sh-p53 cells were plated on 6-cm dishes one day prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks; “untreated” cells were given fresh medium. Cells were harvested 3 h after treatment following the protocol of Buenrostro et al. (2015), with modifications, as follows. Cells were incubated for 5 min with 0.25% trypsin at 37°C and 5% CO2. Fresh medium was then added to the trypsin in a 3:1 ratio, and cells were centrifuged at 500x g for 5 min at 4°C. Cells were resuspended in 1.0 mL of cold PBS, then counted using a Millipore Scepter 2.0 handheld cell counter (Millipore). 150,000 cells were removed and centrifuged at 500x g for 5 min at 4°C. These cells were then resuspended in 150 μL of lysis buffer (10 mM Tris-HCl, pH 7.5; 10 mM NaCl; 3 mM MgCl2; 0.1% IGEPAL CA-630), incubated on ice for 5 min, and centrifuged at 500x g for 10 min at 4°C. The nuclear pellet was resuspended in 60 μL of resuspension buffer consisting of 3 parts water and 1 part 2x TD Reaction Buffer from the Nextera DNA Library Preparation Kit (Illumina, FC-121-1030). 10 μL of resuspended nuclei (representing 25,000 cells) was removed and combined with 10 μL of 2x TD Reaction Buffer and 5 μL of TDE1 transposase from the same kit, as suggested by Takaku et al. (2016). This mixture was incubated at 37°C for 30 min, gently shaking, then purified with a Qiagen MinElute PCR Purification Kit (Qiagen, 28004). Purified DNA was amplified and barcoded by 8 cycles of PCR using NEBNext® High-Fidelity 2X PCR Master Mix (New England BioLabs, M0541) and the barcoding primers used by Buenrostro et al. (2013); amplified fragments were then purified again with the MinElute kit. Size selection was performed by gel electrophoresis, selecting fragments of length 100–600 bp. Size-selected fragments were then quantified by qPCR, amplified by 4–7 additional cycles of PCR with the NEBNext master mix, and purified again using the MinElute kit. Paired-end sequencing of fragments was performed by the CCR Sequencing Facility (Frederick, MD) using an Illumina NextSeq 500 system.
RNA-seq
Because MYC is a universal amplifier of expressed genes (Lin et al., 2012; Nie et al., 2012), experiments measuring the effects of changes in MYC levels must be designed to account for changes in global gene expression. In particular, one cannot normalize gene expression measurements to those of housekeeping genes, whose expression can be expected to change with MYC levels. A better method involves counting the cells from which RNA is being harvested, adding external (spike-in) reference RNA in proportion to cell count, and normalizing gene expression measurements to those of the reference RNA (Lovén et al., 2012). We grew fission yeast (Schizosaccharomyces pombe) in the presence of EU, then used the EU-labeled yeast RNA as the spike-in reference RNA for gene expression normalization, as described below.
Generation of EU-labeled RNA spike-ins
S. pombe cells expressing human equilibrative nucleoside transporter 1 (hENT1) and thymidine kinase (tk) (Hodson et al., 2003) were grown overnight in medium supplemented with 0.5 mM EU (Jena Bioscience, CLK-N002-10). Cells were harvested during logarithmic phase, and RNA was extracted using TRIzol® LS Reagent (ThermoFisher, 10296010) according to manufacturer’s standard protocol. RNA concentration was determined using a UV spectrophotometer.
To estimate the fraction of yeast RNA labeled with EU, labeled RNA was isolated from total RNA using a Click-iT® Nascent RNA Capture Kit (ThermoFisher, C10365). Specifically, 1.0 μg of total RNA was used in a click reaction in which EU-labeled RNA was biotinylated according to manufacturer’s protocol. 100 ng of biotinylated RNA was bound to streptavidin-coated magnetic beads to isolate EU-labeled RNA according to manufacturer’s protocol. EU-labeled RNA was then reverse-transcribed in a 20-μL reaction with random hexamers using a High Capacity cDNA Reverse Transcription Kit (ThermoFisher, 4374966), following manufacturer’s protocol and shaking at 1000 rpm during thermal cycling. For comparison, 100 ng of total biotinylated RNA was reverse-transcribed under the same conditions. Expression of act1+, adh1+, and tdh1+ was measured in each sample by qPCR; the labeled fraction was estimated as the geometric mean of the labeled fractions for each gene.
Cell treatment and RNA isolation
4×105 MCF-7 GFP or MCF-7 MYC-GFP cells were plated on 6-cm dishes 2 days prior to treatment. Expression of GFP or MYC-GFP was induced with 150 ng/mL doxycycline 18 h prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks or with fresh medium containing 150 ng/mL doxycycline as a control. After 2.5 h, medium was replaced with fresh medium containing 0.5 mM EU and 150 ng/mL doxycycline.
After 1 h of labeling with EU (3.5 h after initial treatment), for each cell line and treatment, three plates of cells were incubated for 5 min with 0.25% trypsin at 37°C and 5% CO2. After trypsinization, cells in each plate were counted using a Millipore Scepter 2.0 handheld cell counter (Millipore). Then, for each cell line and treatment, three more plates of cells were lysed directly and RNA harvested using Qiagen QIAshredder and RNeasy Mini Plus kits. EU-labeled RNA from S. pombe was added to each cell lysate at 150 ng of total RNA per million MCF-7 cells, where the number of cells was taken to be the average count of the three replicates of that cell line and treatment. This amount of RNA was determined so that yeast RNA would constitute ~2% of the total EU-labeled RNA in each sample. Total RNA concentration was determined using a UV spectrophotometer.
Sequencing library preparation
For each sample, labeled RNA was isolated from total RNA using a Click-iT® Nascent RNA Capture Kit (ThermoFisher, C10365). Specifically, 5.0 μg of total RNA was used in a click reaction in which EU-labeled RNA was biotinylated according to manufacturer’s protocol. 1.0 μg of biotinylated RNA was bound to streptavidin-coated magnetic beads to isolate EU-labeled RNA according to manufacturer’s protocol. EU-labeled RNA was then used as a template for first-strand cDNA synthesis with a SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing (Clontech, 634889), following manufacturer’s protocol and shaking at 1000 rpm during thermal cycling. Second-strand cDNA synthesis and amplification (6 cycles of PCR) were then performed with the same kit, and amplified cDNA was purified using Agencourt AMPure XP beads (Beckman Coulter, A63880). Concentration and size distribution of amplified cDNA were assessed using a Bioanalyzer (Agilent) with a High Sensitivity DNA Kit (Agilent, 5067-4626). For each sample, 136.45 pg of amplified cDNA was fragmented, tagged with adapter sequences, and amplified using a Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1024) according to manufacturer’s protocol. DNA libraries were then purified again using Agencourt AMPure XP beads, and fragment size distribution was assessed using a Bioanalyzer. DNA concentration was measured using a Quant-iT™ High-Sensitivity dsDNA Assay Kit (ThermoFisher, Q33120) on a microplate reader, and equimolar amounts of DNA were pooled for sequencing. Paired-end sequencing was performed by the CCR Sequencing Facility (Frederick, MD) using an Illumina HiSeq® 2500 system.
Flow cytometry
For cell cycle profiling, 1.4×105 MCF-7 cell lines containing doxycycline-inducible genes were plated on a 35-mm dish 2 days prior to treatment. MYC-GFP or GFP expression was induced with 150 ng/mL doxycycline 18 h prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks. At indicated times following treatment, cells were harvested by trypsinization and then vortexed for 30 s to break apart cell aggregates. Cells were centrifuged at room temperature, 400x g for 4 min. After aspiration of the supernatant, cells were resuspended in 1 mL of phosphate-buffered saline (PBS) supplemented with 1% tetracycline-free FBS and 5 μL of 2000 U/mL DNase (New England Biolabs, M0303) and vortexed for 30 s. Cells were pelleted by centrifugation in a swinging-bucket rotor at 200x g for 5 min at 4 °C. After aspiration of the supernatant, cells were resuspended in 300 μL of ice-cold PBS and strained through a 40 μm nylon mesh. Cells were fixed by dropwise addition of 700 μL of ice-cold 100% ethanol while constantly vortexing the cell suspension. After storage at −20 °C for 24 h, cells were centrifuged in a swinging bucket rotor at 200x g for 5 min at 4 °C. The supernatant was aspirated and cell pellets were resuspended in 1 mL of PBS at 4 C, and centrifuged again at 200x g for 5 min at 4 °C. After aspirating the supernatant, cells were resuspended in 500 μL PBS supplemented with 10 μg/mL propidium iodide (Sigma, P4864) and 100 μg/mL RNase A (ThermoFisher, EN0531). Cells were stored in the dark for 30 min at room temperature. Cells were strained through a 40-μm nylon mesh, and flow cytometry was performed on a BD FACSCanto (BD Biosciences). Single cells were isolated based on forward scatter and side scatter measurements, and DNA content was measured as a function of propidium iodide fluorescence using a 488-nm blue laser and 585/42 filter.
For cell death profiling, 1.2×105 hTERT-RPE1 cells containing doxycycline-inducible genes were plated on a 35-mm dish 2 days prior to treatment. Cells were treated with 400 ng/mL neocarzinostatin (Sigma, N9162) to induce DNA double-strand breaks. At indicated times following treatment, cells were harvested by trypsinization, centrifuged at 400x g for 4 min, washed once with PBS at 4 °C, and then centrifuged again at 400x g for 4 min. Following aspiration of the supernatant, cells were stained with propidium iodide and APC-annexinV using the APC Annexin V Apoptosis Detection Kit with PI (BioLegend, 640932) following the manufacturer’s protocol. Cells were strained through a 40-μm nylon mesh, and flow cytometry was performed on a BD FACSCanto (BD Biosciences). Single cells were isolated based on forward scatter and side scatter measurements, and APC-Annexin V and propidium iodide fluorescences were detected using 633-nm red laser and 660/20 nm filter and a 488-nm blue laser and 585/42 filter, respectively.
Quantification and Statistical Analysis
Western blotting
Western blots were quantified using LI-COR Image Studio v3.1 software (LI-COR). After subtracting background, quantified bands were normalized to quantified β-actin band in the same lane on the same membrane. n, discussed in figure legends, represents biological replicates of a given experiment; error bars represent SEM.
mRNA expression measurements
qPCR measurements were processed using Fluidigm Real-Time PCR Analysis v4.0.1 software. For each gene, samples with a melt peak more than 0.5°C from the median of melt peaks were discarded. Relative expression values were computed from Ct values assuming a PCR efficiency of 100%. Two technical replicate measurements of gene expression were normalized to measurements of GAPDH expression and averaged. n, discussed in figure legends, represents biological replicates of a given experiment; error bars represent SEM.
ChIP-seq
After trimming bases on both ends with quality scores below 20, reads were aligned to the human genome (hg19) using Bowtie v2.2.5 (Langmead and Salzberg, 2012) with default parameters. ChIP-seq peaks were called using MACS v2.1.1.20160309 (Zhang et al., 2008) with false discovery rate cutoff q < 0.05. For each sample, input DNA from untreated MCF-7 cells (not subjected to immunoprecipitation) sequenced on the same lane as the sample was used as the control. Model building by MACS was bypassed in favor of a fixed 200-bp extension size (--nomodel, --extsize 200). Peaks shown in this paper are the intersection of those identified in two biological replicates.
ATAC-seq
Paired-end reads were trimmed using Trimmomatic v0.36 (Bolger et al., 2014) to remove Nextera adapter sequences and bases on the 3′ end with quality scores below 30. Trimmed reads were aligned to the human genome (hg19) using Bowtie v2.2.9 (Langmead and Salzberg, 2012) with parameter -X 2000 to ensure that long fragments were able to align. Properly paired alignments in a 20-Mb region of chromosome 8 (chr8:120000000-140000000) were extracted, and duplicate fragments were removed from this set using Sambamba (Tarasov et al., 2015). To identify nucleosome positions, fragments were processed as described by Buenrostro et al. (2013): fragments of length 180–247 bp were considered to be one nucleosome, and fragments of length 315–473 bp were considered to be two nucleosomes and split into two fragments. Nucleosome positions were identified using DANPOS v2.2.2 (Chen et al., 2013) with parameters -p 1, -a 1, -jd 20, and --clonalcut 0. Fragments shorter than 100 bp were used as the background signal (parameter -b). Read counts in DANPOS were normalized to the number of properly paired reads aligning to the aforementioned 20-Mb region of chromosome 8 with duplicates removed (parameter -c).
RNA-seq
RNA-seq data was processed using Partek Flow (Partek, St. Louis, MO). After trimming bases on both ends with quality scores below 20, reads were aligned to the human transcriptome (hg19, RefSeq Transcripts 2016-05-02) using TopHat v2.1.0 (Kim et al., 2013) with parameters --mate-inner-dist 82, -- mate-std-dev 134. Unaligned reads were then aligned to the S. pombe transcriptome (ASM294v2) using TopHat v2.1.0 with the same parameters.
A “pombe score” representing the amount of S. pombe RNA in each sample, and thus proportional to the number of cells in each sample, was calculated as follows: From the reads mapped to the S. pombe transcriptome, reads with the same start position, reads that mapped to more than one location, and unpaired reads were removed. The remaining reads were quantified using the Partek E/M algorithm. For each transcript, the number of reads mapping to that transcript was divided by the transcript length to yield RPK (reads per kilobase of transcript). From the top 50 expressed transcripts as measured by average RPK over all 12 samples, the transcript with the lowest average correlation with all the others across all 12 samples was removed; this procedure was performed iteratively until a set of 10 highly correlated transcripts was reached. This served to identify transcripts that collectively best represented the quantity of spike-in RNA in each sample. The pombe score for each sample was then calculated as the geometric mean of the expression (RPK) of those 10 genes in that sample.
Reads mapped to the human transcriptome (not filtered) were quantified using the Partek E/M algorithm. For each transcript, the number of reads quantified to that transcript was divided by the transcript length to yield RPK, then divided by the pombe score for the sample to normalize to the number of cells. This quantity, which approximates the number of transcripts per cell, is reported throughout this paper.
Principal component analysis was performed on the logarithm of normalized transcript counts, considering only genes with at least 5 reads in each sample. n, discussed in figure legends, represents biological replicates of a given experiment; error bars represent SEM.
Gene set enrichment analysis
Gene set enrichment analysis was performed using GSEA v2.2.3 (Mootha et al., 2003; Subramanian et al., 2005). Data from n = 3 biological replicates of each cell line/treatment combination were analyzed. Hallmark gene sets (H), curated gene sets (C2), motif gene sets (C3), computational gene sets (C4), GO gene sets (C5), and oncogenic signatures (C6) were obtained from the Molecular Signatures Database v5.2; gene sets containing between 15 and 500 genes were tested for enrichment in one cell line/treatment combination over the other. Genes were ranked by signal-to-noise ratio; 1000 random gene set permutations were used to assess statistical significance of enrichment scores. Default GSEA parameters were used for all other settings.
Flow cytometry
Cell cycle distributions for the number of cells in G1, S, and G2/M phases were determined by fitting flow cytometry histograms using ModFit LT software v4.1 (Verity). For cell cycle distributions and cell death assays, n, discussed in figure legends, represents biological replicates of a given experiment; error bars represent SEM.
Data and Software Availability
Data Resources
Raw and processed data from the ChIP-seq experiment described in this paper have been deposited in the NCBI Gene Expression Omnibus (GEO) database with accession code GSE101737. Data from the ATAC-seq experiment described in this paper have been deposited in the GEO database with accession code GSE101736. Data from RNA-seq of newly synthesized transcripts have been deposited in the GEO database with accession code GSE101738.
Supplementary Material
Gene set enrichment analysis comparing GFP cells treated with NCS with GFP cells not treated with NCS. Gene sets with false discovery rate q < 0.2 and enrichment score > 0 (enriched in NCS-treated GFP cells over non-NCS-treated GFP cells) are shown.
Gene set enrichment analysis comparing MYC-GFP cells with GFP cells during the DSB response. Gene sets with false discovery rate q < 0.2 and enrichment score > 0 (enriched in NCS-treated MYC-GFP cells over NCS-treated GFP cells) are shown.
Acknowledgments
We thank Jie Liu, Toren Finkel, Ji Luo, Ying Zheng, Liqiang Xi, Mark Raffeld, and all members of the Batchelor lab and the Levens lab for their help through technical assistance and many useful discussions. We also thank Qizong Lao, Brian Larsen, Sohyoung Kim, and Saori Fujiwara for their generous help with ATAC-seq. S. pombe were kindly provided by Julie Cooper. We thank Charles Fulco for providing data from CRISPRi experiments in the vicinity of the MYC locus. RNA-Seq and ChIP-Seq sequencing were performed by the Center for Cancer Research Sequencing Facility. We thank the ENCODE Consortium and the Peggy Farnham lab (USC) for data from ChIP-seq of H3K27ac in MCF-7 cells (GEO: GSM945854 and GSM945859). This work used computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
The authors declare no conflicts of interest.
Footnotes
Author contributions
Conceptualization, D.L. and E.B.; Methodology, J.R.P., B.E.F., D.M.K., Z.N., and J.L.; Investigation, J.R.P., B.E.F., L.B., J.C.L., D.M.K., W.S.K., and E.B.; Formal Analysis, J.R.P., J.M.S., and E.B.; Writing – Original Draft, J.R.P. and E.B.; Writing – Review & Editing, J.R.P., L.B., J.C.L., J.M.S., D.L., and E.B.; Supervision, J.M.S., D.L., and E.B.; Funding Acquisition, J.M.S., D.L., and E.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife. 2014;3:e02200. doi: 10.7554/eLife.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30:277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor E, Loewer A, Mock C, Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7 doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning DF, Busby SJW. Local and global regulation of transcription initiation in bacteria. Nat Rev Microbiol. 2016;14:638–650. doi: 10.1038/nrmicro.2016.103. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A System for Stable Expression of Short Interfering RNAs in Mammalian Cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Budde A, Grummt I. p53 represses ribosomal gene transcription. Oncogene. 1999;18:1119–1124. doi: 10.1038/sj.onc.1202402. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current Protocols Molecular Biology. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GS, Chen XA, Park B, Rhee HS, Li P, Han KH, Mishra T, Chan-Salis KY, Li Y, Hardison RC, et al. A Comprehensive and High-Resolution Genome-wide Response of p53 to Stress. Cell Rep. 2014;8:514–527. doi: 10.1016/j.celrep.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2009;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- Cui M, You L, Ren X, Zhao W, Liao Q, Zhao Y. Long non-coding RNA PVT1 and cancer. Biochem Biophys Res Commun. 2016;471:10–14. doi: 10.1016/j.bbrc.2015.12.101. [DOI] [PubMed] [Google Scholar]
- Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P. Extreme instability of myc mRNA in normal and transformed human cells. Proc Natl Acad Sci USA. 1984;81:7046–7050. doi: 10.1073/pnas.81.22.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Farrell AS, Sears RC. MYC Degradation. CSH Perspect Med. 2014;4:a014365. doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Rosenfeld N, Itzkovitz S, Milo R, Sigal A, Dekel E, Yarnitzky T, Liron Y, Polak P, Lahav G, et al. Oscillations and variability in the p53 system. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken Y, Piette J. NF-kappaB activation by double-strand breaks. Biochem Pharmacol. 2006;72:1132–1141. doi: 10.1016/j.bcp.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Heine GF, Horwitz AA, Parvin JD. Multiple Mechanisms Contribute to Inhibit Transcription in Response to DNA Damage. J Biol Chem. 2008;283:9555–9561. doi: 10.1074/jbc.M707700200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JSL, Ma W, Mao DYL, Benchimol S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol Cell Biol. 2005;25:7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson JA, Bailis JM, Forsburg SL. Efficient labeling of fission yeast Schizosaccharomyces pombe with thymidine and BUdR. Nucleic Acids Res. 2003;31:e134. doi: 10.1093/nar/gng134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–6472. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- Iwakawa R, Takenaka M, Kohno T, Shimada Y, Totoki Y, Shibata T, Tsuta K, Nishikawa R, Noguchi M, Sato-Otsubo A, et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes & Cancer. 2013;52:802–816. doi: 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 Is Required for DNA Fragmentation and Morphological Changes Associated with Apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al. Cell-Type Independent MYC Target Genes Reveal a Primordial Signature Involved in Biomass Accumulation. PLoSONE. 2011;6:e26057. doi: 10.1371/journal.pone.0026057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Cho GA, Han SW, Shin JY, Jeong EG, Song SH, Lee WC, Lee KH, Bang D, Seo JS, et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene. 2014;33:5434–5441. doi: 10.1038/onc.2013.490. [DOI] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levens D. How the c-myc Promoter Works and Why It Sometimes Does Not. J Natl Cancer Inst Monogr. 2008;2008:41–43. doi: 10.1093/jncimonographs/lgn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levens D. Cellular MYCro Economics: Balancing MYC Function with MYC Expression. CSH Perspect Med. 2013;3:a014233. doi: 10.1101/cshperspect.a014233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy N, Yonish-Rouach E, Oren M, Kimchi A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: induction of cell cycle exit and cooperativity with c-myc suppression. Mol Cell Biol. 1993;13:7942–7952. doi: 10.1128/mcb.13.12.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. Revisiting Global Gene Expression Analysis. Cell. 2012;151:476–482. doi: 10.1016/j.cell.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc Augments Gamma Irradiation-Induced Apoptosis by Suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar D, Dekel E, Bren A, Zimmer A, Porat Z, Alon U. Promoter activity dynamics in the lag phase of Escherichia coli. BMC Syst Biol. 2013;7:136. doi: 10.1186/1752-0509-7-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci USA. 2011;108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrrale M, Laurila E, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nagoshi H, Taki T, Hanamura I, Nitta M, Otsuki T, Nishida K, Okuda K, Sakamoto N, Kobayashi S, Yamamoto-Sugitani M, et al. Frequent PVT1 Rearrangement and Novel Chimeric Genes PVT1-NBEA and PVT1-WWOX Occur in Multiple Myeloma with 8q24 Abnormality. Cancer Res. 2012;72:4954–4962. doi: 10.1158/0008-5472.CAN-12-0213. [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJH, Peacock J, Garzia L, Sorana Morrissy A, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phesse TJ, Myant KB, Cole AM, Ridgway RA, Pearson H, Muncan V, van den Brink GR, Vousden KH, Sears R, Vassilev LT, et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014;21:956–966. doi: 10.1038/cdd.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JR, Fisher BE, Batchelor E. p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst. 2016;2:272–282. doi: 10.1016/j.cels.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti-De-Santis L, Drané P, Egly JM. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBOJ. 2006;25:1915–1923. doi: 10.1038/sj.emboj.7601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis JE, Karhohs KW, Mock C, Batchelor E, Loewer A, Lahav G. p53 dynamics control cell fate. Science. 2012;336:1440–1444. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rockx DAP, Mason R, van Hoffen A, Barton MC, Citterio E, Bregman DB, van Zeeland AA, Vrieling H, Mullenders LHF. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci USA. 2000;97:10503–10508. doi: 10.1073/pnas.180169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massagué J. Myc suppression of the p21-Cip1 Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- Sheen JH, Dickson RB. Overexpression of c-Myc Alters G1/S Arrest following Ionizing Radiation. Mol Cell Biol. 2002;22:1819–1833. doi: 10.1128/MCB.22.6.1819-1833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaku M, Grimm SA, Shimbo T, Perera L, Menafra R, Stunnenberg HG, Archer TK, Machida S, Kurumizaka H, Wade PA. GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol. 2016;17 doi: 10.1186/s13059-016-0897-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Morelli MJ, Sabò A, Verrecchia A, Rotta L, Capra T, Bianchi S, Campaner S, Amati B. Genome-wide analysis of p53-regulated transcription in Myc-driven lymphomas. Oncogene. 2017;36:2921–2929. doi: 10.1038/onc.2016.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M, Poortinga G, Hannan KM, Pearson RB, Hannan RD, McArthur GA. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood. 2008;112:2305–2317. doi: 10.1182/blood-2007-09-111856. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Zhai W, Comai L. Repression of RNA Polymerase I Transcription by the Tumor Suppressor p53. Mol Cell Biol. 2000;20:5930–5938. doi: 10.1128/mcb.20.16.5930-5938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2015;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene set enrichment analysis comparing GFP cells treated with NCS with GFP cells not treated with NCS. Gene sets with false discovery rate q < 0.2 and enrichment score > 0 (enriched in NCS-treated GFP cells over non-NCS-treated GFP cells) are shown.
Gene set enrichment analysis comparing MYC-GFP cells with GFP cells during the DSB response. Gene sets with false discovery rate q < 0.2 and enrichment score > 0 (enriched in NCS-treated MYC-GFP cells over NCS-treated GFP cells) are shown.