

Abstract
Protein tyrosine phosphatases (PTPs) are a large family of 107 signaling enzymes that catalyze the hydrolytic removal of phosphate groups from tyrosine residues in a target protein. The phosphorylation status of tyrosine residues on proteins serve as a ubiquitous mechanism for cellular signal transduction. Aberrant function of PTPs can lead to many human diseases, such as diabetes, obesity, cancer, and autoimmune diseases. As the number of disease relevant PTPs increases, there is urgency in developing highly potent inhibitors that are selective towards specific PTPs. Most current efforts have been devoted to the development of active site-directed and reversible inhibitors for PTPs. This review summarizes recent progress made in the field of covalent inhibitors to target PTPs. Here, we discuss the in vivo and in vitro inactivation of various PTPs by small molecule-containing electrophiles, such as Michael acceptors, α-halo ketones, epoxides, and isothiocyanates, etc. as well as oxidizing agents. We also suggest potential strategies to transform these electrophiles into isozyme selective covalent PTP inhibitors.
Graphical Abstract
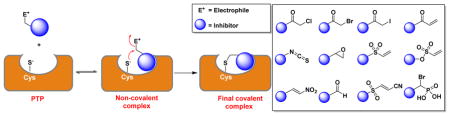
Chemical strategies for covalent inhibition of protein tyrosine phosphatases.
Introduction
Tyrosine phosphorylation of proteins plays an important role in a variety of cellular processes such as proliferation, migration, and apoptosis. The tyrosine phosphorylation status of a target protein is controlled by the balanced and opposing action of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs).1 PTKs transfer the γ-phosphoryl group (PO32−) from ATP to the tyrosine residue on a target protein, whereas PTPs remove the phosphate group to generate the dephosphorylated target protein and an inorganic phosphate.1 Dysregulation of both PTKs and PTPs are associated with pathological processes such as cancer, diabetes, obesity, and autoimmune disorders.2 PTKs are a family of 90 enzymes encoded by the human genome.3–5 Currently, over 20 small molecule inhibitors targeting PTKs have already been approved by FDA to treat various cancers, and many other inhibitors are currently under clinical development.2,6 Of the ~107 PTPs encoded by the human genome, at least 37 have been associated with human cancer and approximately an equal number of PTPs are involved in oncogenic and tumor suppressor activities.7,8 However, no single drug targeting PTPs has yet been commercialized. PTPs are extremely difficult targets for drug discovery, as over two decades of intense efforts by academia and the pharmaceutical industry generated only a small number of potent and selective PTP inhibitors with robust in vivo activity.9–11 The vast majority of PTP inhibitors reported in the literature are phosphotyrosine (pTyr) mimetics that occupy an active-site and extend into the surrounding region to gain selectivity. Several highly potent two-site binding small molecules have been reported as inhibitors of PTPs.12–16 Recently, allosteric inhibition strategies have generated promising small molecule inhibitors that are potent and selective towards some of the disease relevant PTPs. Chen et al. discovered a highly potent (IC50 = 0.071 μM), selective and orally bioavailable inhibitor SHP099 for SHP2, the first oncogenic phosphatase in the PTP family.17–19 SHP2 in its basal state exists in an autoinhibited closed conformation due to an interaction between its N-terminal Src homology 2 (SH2) domain and the PTP domain. Binding of phosphotyrosine (pTyr) ligands to SH2 domains disrupts the autoinhibitory interaction that leads to activation of the phosphatase.20 SHP099 inhibits SHP2 by stabilizing it in an auto-inhibited conformation. Trodusquemine (MSI-1436) was characterized as a selective and non-competitive inhibitor of PTP1B,21 a negative regulator of insulin and leptin signaling.22–25 MSI-1436 binds to the C-terminal segment of PTP1B that allosterically locks PTP1B in an inactive conformation. More recently, strategies to selectively target oxidized PTPs were also reported. By utilizing a double mutant form of PTP1B (CASA) as an antigen that mimics the active site Cys oxidized form of PTP1B, a single chain of variable fragments (scFvs) was developed to recognize and stabilize an oxidized PTP1B in the inactive state inside a cell.26 Protein oligomerization is also known to play an important regulatory role in the activity of some PTPs. Compound 43 was discovered by in silico library screening as an oligomerization inhibitor of phosphatase of regenerating liver 1 (PRL1).27 Compound 43 inhibits PRL1 trimerization without affecting catalytic activity, and also blocks PRL1-induced cell proliferation and migration through reduced ERK1/2 and Akt activity.
The strategy of using an inhibitor with a reactive group (often an electrophile) to covalently modify a target protein is gaining interest among the research community. Covalent inhibitors have several advantages, such as low dose requirement for achievable high potency, prolonged duration of action (due to covalent bond formation), and resistance to mutations.28,29 However, off-target interactions that are also long lasting can have toxic side effects. For instance, covalent modification of cytochrome P450 by electrophilic reagents leads to severe side effects.30 Though the covalent inhibition strategy has, in general, been avoided by the pharmaceutical industry due to a widely perceived increase in toxicity from covalent drugs, there are many examples of blockbuster drugs, such as aspirin, penicillin, omeprazole, and clopidogrel, that exert their pharmacological effects through covalent modification of their respective target proteins. Indeed, clopidogrel (to prevent cardiovascular diseases), lansoprazole, and esomeprazole (proton pump inhibitors) constituted three of the ten top-selling drugs in the United States in 2009.31 Currently, there are approximately 42 approved covalent drugs to treat diseases such as infections, cancer, and cardiovascular.28,32 In fact, there is no correlation between thiol conjugate formation and the in vivo toxicity of approved covalent drugs.33 Recent successes of covalent kinase inhibitors has prompted a renewed interest in developing covalent inhibitors for drug discovery.34 Given the requirement of the active site Cys for PTP catalysis, PTPs represent prime targets for covalent inhibition.35 Some aspects of covalent inhibition and activity based probes targeting PTPs have been covered by two recent mini-reviews.36,37 Here, we provide a comprehensive summary and an update on recent progress on efforts to develop covalent inhibitors (both reversible and irreversible) to target PTPs.
Classification of PTPs
PTPs are Cys dependent phosphatases with a conserved active site motif C(X)5R that can be broadly divided into four groups based on their structure and substrate specificity.7 Group I consist of classical PTPs (38 members) that are specific for phosphotyrosine (pTyr). The classical PTPs are strictly tyrosine-specific and are further subdivided into two categories based on their subcellular localization: intracellular PTPs (e.g., PTP1B, SHP, HePTP, STEP, etc.) and receptor-like PTPs (e.g., CD45, LAR, PTPα, PTPβ, PTPσ, etc.). Group II contains dual-specific phosphatases (DSPs, 61 members) that can hydrolyze phosphate from tyrosine, serine/threonine, phosphatidylinositol and glycogen. Thus, the DSPs (VH1-like enzymes) are the most diverse group in terms of substrate specificity and can be divided into several subgroups that share less sequence identity with each other. The DSP family of PTPs contains highly specialized types of phosphatases; members of this family include phosphatases of regenerating liver (PRLs), mitogen-activated protein kinase phosphatases (MKPs), myotubularins, phosphatase and tensin homologue (PTEN) type phosphatases, atypical DSPs, CDC14s, and slingshots. Group III consists only one member, the low-molecular-weight phosphotyrosine phosphatase (LMW-PTP). In humans, this class is represented by an 18 kDa tyrosine-specific low molecular weight phosphatase. The activity of LMW-PTP is regulated by reversible oxidation of cysteine residues and the tyrosine phosphorylation status.38 Group IV PTPs are represented by three phosphatases: CDC25A, CDC25B, and CDC25C (CDC refers to cell division cycle). These three cell cycle regulators dephosphorylate cyclin-dependent kinases (CDKs) at their inhibitory N-terminal Thr/Tyr motifs.39 This dephosphorylation leads to the activation of these CDKs to drive cell cycle progression.40 Overexpression of CDC25B has been correlated to the malignancy of tumors.39,41 All PTPs are cysteine-based enzymes that catalyze a dephosphorylation reaction in which an active-site cysteine thiolate (RS−) acts as a nucleophile and attacks the phosphorus center of the substrate phosphoryl group.35
Mechanism of PTP catalysis
All PTPs share a common catalytic domain of approximately 250 amino acids made of a central parallel β–sheet with flanking α-helices containing a β-loop-α-loop that encompasses the PTP signature motif (I/V)HCXXGXXR(S/T), also called the catalytic loop or P-loop.42 This loop consists of 11 amino acid residues, of which cysteine and arginine are critical for catalytic activity. Key residues essential for PTP catalysis are depicted in the structure of the catalytic domain of PTP1B (Fig. 1). The thiolate group of the invariant active site cysteine (Cys215 in PTP1B) functions as the attacking nucleophile to initiate phosphate monoester hydrolysis. The active site arginine (Arg221 in PTP1B) is a highly conserved residue among PTPs and is essential for phosphotyrosine substrate binding; it also stabilizes the transition state by forming a pair of hydrogen bonds with the phosphate oxygen atoms.43,44 The WPD loop (tryptophan, proline, and aspartic acid) is also a conserved loop in PTPs and is a flexible surface loop. Crystallographic studies revealed that the WPD loop can take either an open or closed conformation.44,45 A unique aspartic acid located in the WPD loop (Asp181 in PTP1B) acts a general acid/base catalyst in the dephosphorylation reaction. The pTyr loop contains a tyrosine residue (Tyr46 in PTP1B) that defines the depth of the active site cleft and contributes to the specificity of pTyr-containing substrates.46
Fig. 1.
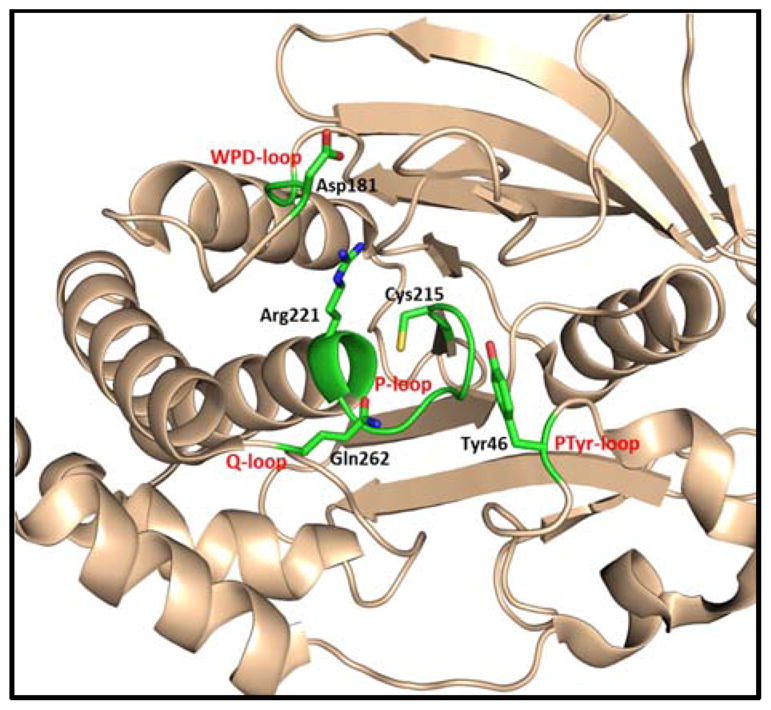
X-ray crystal structure of PTP1B showing the key residues for catalytic activity.
PTPs exert their catalytic activity by using a nucleophilic cysteine located at the bottom of the active site. This active-site cysteine residue predominantly exists as a thiolate (RS−) at physiological pH, due to its unusually low pKa of ~4.5.47,48 The dianionic phosphotyrosine (pTyr) residue of the substrate binds to the positively charged active site of PTP by an array of hydrogen bonds with the guanidine side chain of arginine in the P-loop and backbone amide residues. Nucleophilic attack of cysteine thiolate onto the phosphorus atom of the substrate phosphoryl group leads to formation of a phospho-cysteine intermediate (Fig. 2).49–51 In this step, a conserved aspartic acid residue in the WPD loop acts as a general acid that protonates the tyrosyl-leaving group. 52,53 In the next step, an activated water molecule attacks at the phospho-cysteine intermediate (catalyzed by the same conserved Asp in the WPD loop as a general base); this leads to release of inorganic phosphate and regenerates the cysteine thiolate.46 This step is mediated by the conserved glutamine (Gln262 in PTP1B) present in the Q-loop, which coordinates with a water molecule by hydrogen bonding interactions.54
Fig. 2.

The general mechanism of the PTP catalyzed dephosphorylation reaction.
Role of PTPs in diseases
PTPs were once considered as housekeeping enzymes that function with no or little substrate specificity.55 However, over the past two decades research has demonstrated the essential roles of PTPs in many human diseases. PTP1B was the first PTP purified to homogeneity, and experiments by Elchebly et al. and Klaman et al. on PTP1B knockout mice revealed its role in insulin and leptin signaling pathways.24,25,56,57 The PTP1B knockout mice displayed higher insulin sensitivity, lower glucose levels, and resistance to obesity with a high fat diet. Moreover, increased PTP1B activity and expression in adipose tissue and skeletal muscle of obese insulin-resistant human subjects were also reported.58,59 Other PTPs, including TC-PTP, LMW-PTP, and PTP-MEG2, have also been implicated as negative regulators of insulin and/or leptin signaling.60–62 Consequently, there is great interest in developing inhibitors to target these PTPs as apeutic agents for type 2 diabetes and obesity. More recently, Krishnan et al. showed that inhibition of PTP1B may reverse the symptoms of Rett Syndrome (RTT), a neurological disorder similar to that of Huntington’s disease and cystic fibrosis.63 Src-homology 2 domain-containing phosphatase 2 (SHP2), encoded by the Ptpn11 gene, promotes signaling downstream from cytokine and growth factor receptors and upstream of Ras. Germline mutations in SHP2 were linked to Noonan and LEOPARD syndromes. Activating mutations of SHP2 have been found to be associated with multiple cancers, such as lung cancer, breast cancer, colon cancer, prostate cancer, leukemia, neuroblastoma, glioblastoma, and melanoma.16,64,65
CDC25 family phosphatases play critical roles in eukaryotic cell cycle control. They promote activation of cyclin-dependent kinases (CDKs) by removing the inhibitory phosphate(s) in CDKs.66 Overexpression of CDC25 phosphatases were found in a number of human cancers, including head, neck, breast, colon, lung, gastric, pancreatic, and neuroblastoma.67 CDC25 overexpression was observed at all stages of tumorigenesis, indicating their roles throughout tumor development. STriatal-enriched protein tyrosine phosphatase (STEP) is a neuron-specific phosphatase with two alternative spliced isoforms (STEP46 and STEP61). STEP regulates the activity of mitogen-activated protein kinase (MAPK) family members ERK1/2 and p38, the tyrosine kinases Fyn, and Pyk2, and the glutamate receptor GluN2B subunit of NMDARs.68 Genetic reduction of STEP improves cognition in Alzheimer’s disease (AD) mouse models, thus identifying STEP as a promising therapeutic target for the treatment of AD.69 The phosphatases of regenerating liver (PRLs) are a subfamily of three dual specificity phosphatases. All three PRLs, PRL1/2/3 promote cell proliferation, migration, invasion, tumor growth, and metastasis.70 PRL3 is the most studied of the three PRLs and it is overexpressed in liver metastatic tissue from colon cancer, but not in normal colon tissue. PRL-3 was also found to be overexpressed in many other human cancer types, such as melanomas, gastric, ovarian, breast, acute myeloid leukemia, and Hodgkin’s lymphoma.70 CD45 (lymphocyte common antigen), a class I receptor-like PTP, promotes antigen receptor signaling in lymphocytes.71 CD45 and CD45-associated molecules play essential roles in T cell and B cell receptor signaling. Due to its important role in immune signaling pathways, CD45 has been linked to many autoimmune diseases.2 For instance, a single-nucleotide polymorphism (C77G) in exon 4 of CD45 is associated with increased incidence of multiple sclerosis, HIV, systemic sclerosis, and autoimmune hepatitis.72 Lymphoid tyrosine phosphatase (LYP), encoded by the Ptpn22 gene, is associated with a wide spectrum of autoimmune disorders such as, rheumatoid arthritis, type 1 diabetes, systemic lupus erythematosus, and Graves’ disease.73 A hallmark of cancer cells is their ability to evade apoptosis. RNA interference (RNAi) screens to identify anti-apoptosis genes in HeLa cells revealed 28 human PTP genes as positive regulators of cell survival, and 4 PTPs as cell death phosphatases.74 This study demonstrated the regulatory role of PTP activity in maintaining cellular homeostasis. As described above, dysregulation of PTP activity has been linked to many human diseases, such as cancer, diabetes/obesity, autoimmune disorders, neurological diseases, and infectious diseases. Therefore, a majority of the members of PTP family are molecular targets for drug discovery.
Kinetic parameters for covalent inhibitors
Covalent inhibitors work by forming a covalent bond with a reactive functional group in the target enzyme. Consequently, the kinetics for enzyme inactivation by a covalent inhibitor is not an equilibrium process as with reversible inhibition. The potency of a reversible inhibitor can be measured by its IC50 value.75 The IC50 is the concentration of the inhibitor at which 50% of the target enzyme is inhibited. The Cheng-Prusoff equation can be used to convert IC50 into an inhibition constant Ki.76 Unlike reversible inhibitors, where inhibition is only concentration dependent, covalent inhibitors show both time and concentration dependent target inhibition (a covalent bond formation is a time-dependent event). If sufficient time is available, covalent inhibitors can completely neutralize the target rather than go to equilibrium (Fig. 3). As a result, potency and selectivity of a covalent inhibitor cannot be determined by IC50 values; instead, the rate of inactivation of the target (kinact) and inhibition constant (KI) are preferred. Most often, the bimolecular rate constant (kinact/KI), is chosen to rank the potency of covalent inhibitors.28,75,77 Compared to IC50 determination, obtaining kinact/KI for covalent inhibitors is more laborious and involves two steps. In a binding assay, the extent of inhibition is measured over time at different inhibitor concentrations. The observed rate constant (kobs) for each concentration is obtained from the slope of a semi-logarithmic plot of inhibition vs time. The kobs values are re-plotted against inhibitor concentration and fitted to a hyperbolic equation (Equation 1) to determine KI and kinact values.78 in cases where it is difficult to derive both Ki and kinact (low concentrations of inhibitors), one can obtain the bimolecular rate constant via a kobs versus [inhibitor] plot.
Fig. 3.

Comparison of traditional non-covalent and covalent interactions between an enzyme and an inhibitor. A) Interaction of traditional non-covalent reversible inhibitor with an enzyme. B) Interaction of covalent inhibitor with an enzyme.
| Equation 1 |
Covalent PTP inhibitors
Many compounds have been reported that are capable of inactivating the PTPs. We classified these PTP covalent inhibitors into eight categories based on the reacting (warhead) groups present in the molecule for covalent modification of active site residues of the PTPs.
1. Oxidizing agents
Due to the highly nucleophilic nature of the catalytic cysteine residue in PTPs, the active site thiol group is prone to oxidation that causes either transient or permanent loss of phosphatase activity in cells. Reactive oxygen species (ROS), such as superoxide anion (O2•−) and hydrogen peroxide (H2O2) produced during a variety of signaling events, are capable of modulating PTP activity by oxidative inactivation.47 This inactivation involves conversion of the catalytic cysteine residue to a sulfenic acid, a sulfenyl amide, or a disulfide (Fig. 4).47,79–81 Oxidation of the active site thiol group to sulfenic acid (SOH) or sulfenyl amide is a reversible event, but its over-oxidation to sulfinic acid (SO2H) or sulfonic acid (SO3H) causes permanent loss of PTP catalytic activity. In the case of PTP1B and PTPα, the sulfenic acid quickly loses a water molecule to form a relatively stable cyclic sulfenyl amide moiety (Scheme 1).82–84 Reaction of this sulfenyl amide with cellular thiols regenerates the enzyme activity via formation of a mixed disulfide.85 On the other hand, oxidation of active site thiol of CDC25C,86 PRL1,87 PTEN80 and LMW-PTP88 results in the formation of a disulfide bond with a neighboring cysteine residue. This redox chemistry of PTPs by ROS serves as a regulatory mechanism under certain physiological conditions. Increased levels of cellular oxidants, in response to growth factors, triggers the tyrosine phosphorylation dependent signaling by transient inactivation of members of the PTP family.89 For instance, during insulin signaling events, a burst of H2O2 causes inactivation of PTP1B via oxidation of the active site cysteine thiolate (Cys 215) to a sulfenyl amide moiety. Reaction with cellular thiols then slowly regenerates the active enzyme that terminates cellular responses to insulin.82
Fig. 4.

X-ray crystal structures of oxidized forms of PTP1B, P-loop residues shown in ball and stick and WPD loop in red. A) Sulfenic acid form (pdb code: 1oet).83 B) Sulfenyl amide form (pdb code: 1oes).83 C) Sulfinic acid form (pdb code: 1oeu).83 D) Sulfonic acid form (pdb code: 1oeo).82
Scheme 1.

Redox regulation of protein tyrosine phosphatases.
In vitro analysis of hydrogen peroxide-mediated inactivation of PTPs revealed that hydrogen peroxide is a sluggish inactivator of PTPs with rate constants of 10–20 M−1 s−1. LaButii et al. reported peroxymonophosphate (2−O3POOH) as an exceptional inactivator of PTP1B with KI = 6.6 × 10−7 M and kinact = 0.043 s−1.90 As a result, peroxymonophosphate (kinact/KI = 65,553 M−1 s−1) is approximately 7,000 times more potent than hydrogen peroxide, an endogenous regulator of PTP1B.91 Like hydrogen peroxide, peroxymonophosphate is also an active-site directed and thiol reversible inactivator of PTP1B. In a recent study, Meng & Zhang showed that hydroxyl radical could also be a viable oxidizing agent for the PTP active site under physiological conditions.92 Importantly, second-order rate constants of hydroxyl radical-mediated inactivation of PTPs are 2–3 orders of magnitude higher than those mediated by hydrogen peroxide under identical experimental conditions.
In an attempt to discover organic compounds that could modulate PTP1B activity by redox mechanism, Bhattacharya et al. screened a set of organic hydroperoxides (1–6) against PTP1B (Fig. 5).93 Peracetic acid 1 showed time-dependent inactivation of PTP1B, which is consistent with covalent modification of PTP1B. The site of covalent modification was confirmed by competition experiments with phosphate ion. The presence of phosphate ion caused approximately 40% decrease in the inactivation power of 1, indicating the active-site directed modification of the enzyme. Further analysis revealed the reversible nature of the inactivation, where 73% of initial activity was restored when 1 modified PTP1B was treated with low molecular weight thiol. Compounds 5 and 6 showed superior inactivation kinetics compared to 1, which may be attributed to the non-covalent interaction of aromatic rings with pTyr binding residues such as Tyr46 and Phe182 prior to covalent modification. tert-butyl hydroperoxide 2 and cumene hydroperoxide 3 showed no inactivation of PTP1B.90 Steric bulk of the tertiary group could be preventing these two compounds from penetrating the deep active-site pocket of PTP1B.
Fig. 5.

Chemical structures of organic hydroperoxides used for PTP1B inactivation. 93
Nitric oxide (NO) produced in biological systems is known to play important functional roles in numerous processes in the cardiovascular, immune, and nervous systems.94 Post-translational modifications through S-nitrosylation of protein thiols by nitric oxide is well documented.95,96 Caselli et al. conducted in vivo and in vitro experiments to investigate the role of nitric oxide on LMW-PTP and Yersinia PTP. In the case of LMW-PTP, mass spectrometric analysis of tryptic fragments confirmed oxidation of Cys12 and Cys17 to form a disulfide bond.97 In vivo inhibition of phosphatases by NO was also observed in RAW 264.7 murine macrophages. These studies confirm the ability of NO to inactivate PTPs and suggests a possible role of nitric oxide in the redox regulation of cellular processes involving PTPs.98
Takakura et al. investigated the effects of biological oxidants such as peroxynitrite, and S-nitrosoglutathione (GSNO) on the activity of three human PTPs, CD45, PTP1B and LAR.99 All these PTPs were irreversibly inactivated by peroxynitrite in less than 1 s with IC50 values of less than 0.9 μM. The bimolecular reaction rates for peroxynitrite mediated inactivation of PTP1B, LAR, and CD45, were 2.2 × 107, 2.3 × 107, and 2.0 × 108 M−1s−1, respectively. These high reaction rates suggest that PTPs could be inactivated by peroxynitrite even in the presence of thiol reagents. Incubation of peroxynitrite-inactivated LAR with 20 mM DTT recovered only about 10% of the original activity; similar results were observed with PTP1B and CD45, indicating the oxidation of active site thiol to sulfinic acid (OSO2H) or even to sulfonic acid (OSO3H) which cannot be reduced by biological thiols such as DTT.47 Conversely, incubation of LAR with 1 mM of GSNO for 30 min showed 50% of inhibition and the inhibition was completely reversed by DTT and β-mercaptoethanol. These results demonstrate that any PTP with sequence CX5R in the active-site could be oxidized by peroxynitrite in cells, causing an increase in tyrosine phosphorylation and thereby impacting tyrosine phosphorylation dependent signaling events.
In an another study, Ming and co-workers demonstrated low-molecular weight S-nitrosothiols such as GSNO, S-nitroso-captopril, S-nitroso-N-acetyl-D,L-penicillamine (SNAP), and S-nitrosylated human serum albumin as time and concentration dependent inhibitors of mammalian PTP1B and Yersinia PTP (YopH) (Fig. 6).100 These four S-nitrosothiols exhibited kinact/KI values ranging from 37 to 113 M−1 min−1 and 66 to 613 M−1 min−1 against PTP1B and YopH, respectively (Table 1). Moreover, both single-S-nitrosylated and poly-S-nitrosylated human serum albumin showed second order rate constants of 472 and 1,188 M−1 min−1, against YopH, respectively. This indicates a possible role of in vivo inactivation with a variety of cysteine-dependent enzymes by S-nitrosylated albumin. In an another instance, Barrios research group reported oxidative inactivation of LYP with sodium nitroprusside (SNP) (Fig. 6) and a novel peptide-based nitrosating reagent, Ac-ARLIEDNE(HcyNO)TAREG-NH2 (where Hcy-NO = S-nitrosohomocysteine).101 The SNP inactivated LYP with an equilibrium binding constant (KI) of 27.4 μM, an inactivation rate constant (kinact) of 0.383 min−1, and mixed-type inactivation kinetics. This peptide showed competitive LYP inactivation kinetics with a KI of 7.00 μM and kinact of 0.0472 min−1. Inactivation of LYP by SNP resulted through the formation of a LYP-NO adduct, whereas inactivation by the peptide did not result in formation of an LYP-NO adduct. Based on these observations, it was proposed that general NO donors nitrosate any free cysteine residue while the active-site directed peptide selectively nitrosates the active site cysteine, resulting in formation of a disulfide bond between the active site cysteine residue and a neighboring cysteine.
Fig. 6.

Structures of various nitroso compounds used for inactivation of PTPs.100
Table 1.
Kinetic parameters reported for the inactivation of PTP1B and YopH by various S-nitrosothiols.100
| Inhibitor | Enzyme | kinact (min−1) | KI (mM) | kinact/KI (M−1 min−1) |
|---|---|---|---|---|
| GSNO | PTP1B | 0.046 | 0.466 | 99 |
| YopH | 0.038 | 0.573 | 66 | |
| SNAP | PTP1B | 0.109 | 0.969 | 113 |
| YopH | 2.09 | 3.41 | 613 | |
| S-NO captopril | PTP1B | 0.108 | 1.040 | 104 |
| YopH | 0.124 | 0.848 | 146 | |
| Glucose-SNAP-2 | PTP1B | 0.111 | 3.014 | 37 |
| YopH | 0.084 | 1.152 | 73 |
2. Phosphates and phosphonates
A general strategy to develop inhibitors for PTPs is to design a nonhydrolyzable phosphonate moiety that mimics the phosphate group of pTyr. However, replacement of the phenolic oxygen atom with a non-polar methylene group may decrease affinity for the active site and also reduce water solubility.102 To address these issues, medicinal chemists replaced the hydrogens on the α-carbon with halogen atoms, to make the phosphonate an effective electronic isostere of the phosphate group. Chen et al. synthesized phosphonodifluoromethyl phenylalanine (F2Pmp) as a nonhydrolyzable pTyr mimetic. Presence of the difluoromethyl group increased the affinity of a F2Pmp-containing peptide over a Pmp-containing counterpart by 1,000-fold towards PTP1B. This increased affinity may be due to hydrogen bonding interactions of the two fluorine atoms, similar to the phosphate ester oxygen in pTyr.11,103,104
In 1996, Taylor et al. examined α-halobenzylphosphonates as possible inactivators of PTPs. Compounds 7, 8, and 9 (Fig. 7) were screened for inhibition against Yop51 PTP from Yersinia enterocolitica. A direct comparison of these compounds revealed that bromide 9 is a superior inactivator compared to chloride 8 and fluoride 7, which showed modest and no inactivation, respectively. Decrease in the inactivation rate of bromide 9 in the presence of p-nitrophenyl phosphate (substrate) or arsenate (known competitive inhibitor of the enzyme) confirmed the active site directed inactivation (Fig. 7).105 Later, Tulsi et al. prepared L-phosphonobromomethylphenylalanine (BrPmp) derivatives 10 and 11 as irreversible inhibitors of PTP CD45. The Kitz–Wilson analysis estimated the rate of irreversible inhibition (kinact) of CD45 as 0.05 min−1 for both 10 and 11, and KI values of 40 μM and 16 μM, respectively. These results indicate that tripeptide 11 was more potent than BrPmp alone, suggesting additional interactions of the peptide side chains to improve enzyme binding.106
Fig. 7.

Structures of phosphate and phosphonate based covalent inhibitors.
In 2004, Zhang and co-workers utilized the α-bromobenzylphosphonate (α-BBP) moiety to design activity-based probes to facilitate global analysis of PTPs.107 They synthesized probes 12 and 13, where α-BBP acts as a PTP-specific trapping device and a linker with a biotin tag for capture and visualization of PTP activity. Two possible mechanisms were proposed for the action of these probes against PTPs (Fig. 8). In one of the mechanisms, the active site cysteine thiolate attacks the phosphorus atom of the probes to yield a S-P covalent bond (Fig. 8A). In another mechanism, the general acid/base Asp residue (note the Cys is not in a position to make this attack based on its structure) attacks the carbon bearing the bromine atom in an SN2 fashion to form a stable ester linkage (Fig. 8B). Site-directed mutagenesis experiments indicated that a covalent adduct was formed between α-BBP and PTP involving the active site Cys residue, consistent with mechanism A in figure 8. These α-BBP-based agents showed broad reactivity against a set of PTPs in an active site-directed, irreversible fashion. Interestingly, these probes are inert toward serine/threonine, acid or alkaline phosphatases, and serine or cysteine proteases, validating their utility as broad spectrum mechanism-based probes. The selectivity of these agents may be due to the structural resemblance between α-BBP and phosphotyrosine and the juxtaposition of the electrophilic group to the active-site nucleophile. Kumar et al. incorporated rhodamine tags in place of the biotin into α-BBP derivatives to detect PTPs in various cancer cell lines.108 Rhodamine probes showed reactivity and selectivity profiles similar to the biotin probes against PTPs and demonstrated 1,000 fold more sensitivity than the biotin probes.
Fig. 8.

Proposed mechanism for covalent modification of PTPs by α-bromophosphonates.
4-Difluoromethylphenyl bis(cyclohexylammonium) phosphate 14 (Fig. 7), was reported as a mechanism-based inactivtor of human prostatic acid phosphatase and SHP protein tyrosine phosphatase.109 This 4-difluoromethylphenyl phosphate (DFPP) derivative is inert itself, but upon hydrolysis of the phosphate group leads to the spontaneous elimination of a fluoride ion, which generates a highly reactive electrophilic quinone methide group. This quinone methide can be attacked by nucleophiles in an active-site to give stable Michael addition products (Fig. 9). The inactivation of human prostatic acid phosphatase and SHP by 14 followed pseudo first order kinetics. The inactivation rate constants for human prostatic acid phosphatase reported as kinact = 0.15 min−1 and KI = 1 mM. This quinone methide approach has been used by several research groups to design PTP activity based probes,36,110,111 although it should be noted that 4-fluromethylaryl phosphate is not specific for PTPs, as it also reacts with other phosphatases such as prostatic acid phosphatase and Ser/Thr phosphatase calcineurin.109,112,113
Fig. 9.

Proposed mechanism of inactivation of PTPs by 4-difluoromethylphenyl phosphate (DFPP).
Cyclic phosphate ester 2-Methoxy-4H-1,3,2-benzodioxaphosphor-2-one, also known as salioxon, is a covalent inhibitor of esterases (Fig. 7). Due to its structural resemblance with phenyl phosphate, which is a substrate of PTPs, it was expected to inactivate PTPs. Salioxon showed irreversible inhibition of PTP1B, YPTP1, and SHP-1, although the compound is a rather weak inactivator of these PTPs.114 Unfortunately, no kinetic or mechanistic studies of PTP inhibition by salioxon were reported. Salioxon is an interesting example of a nonhydrolyzable pTyr mimetic, and may be modified at phenyl ring and/or cyclic phosphate group to generate more potent and selective inactivators for PTPs.
3. α-halo carbonyls
Alpha halo carbonyl compounds present an electrophilic center at the α-carbon atom that can be attacked by nucleophilic residues in proteins.48,115 Pot & Dixon first reported iodoacetate as an inactivator of the cytoplasmic PTP domain of LAR, a receptor-like PTP.115 Inactivation of LAR by iodoacetate was time dependent and the loss of catalytic activity followed pseudo-first order kinetics. Surprisingly, the structurally similar iodoacetamide (Fig. 10) showed no inhibition against LAR.116 Zhang and Dixon (1993) investigated iodoacetate vs iodoacetmide as inactivators of the Yersinia PTP and rationalized the difference in reactivity between the two compounds.48 According to their findings, iodoacetate exhibits selective and stoichiometric labeling of active site cysteine (Cys403) of Yersinia PTP. In addition, iodoacetate showed 940-fold more reactivity toward Yersinia PTP compared to iodoacetamide. This differential reactivity correlates with the positively charged nature of the residues within the active-site of the enzyme.
Fig. 10.

Chemical structures of α-halo ketones used as inactivators of PTPs.
In 1999, Pei and co-workers reported α-haloacetophenone derivatives as photocleavable covalent inhibitors of PTPs.117,118 The authors envisioned the α-haloacetophenone moiety as a pTyr mimetic, where the electron-rich halogen atom could mimic the phosphate group, and the phenyl ring could provide hydrophobic interactions with the active site of a PTP similar to that of the phenyl ring in a natural substrate. Such a binding would place the highly electrophilic α-carbon of the α-haloacetophenone next to the catalytic cysteine thiolate of the PTP for an SN2 reaction. This would result in loss of phosphatase activity via the formation of a stable thioether linkage. In this study, compounds 15, 16, 17, and 18 (Fig. 10) were assayed in an inactivation experiment against PTP1B, SHP1, the catalytic domain of SHP1 (SHP1 ΔSH2), and LAR. All four compounds showed time and concentration dependent inactivation of the PTPs tested. Inhibitor 15 showed the highest affinity, with kinact values of 0.57 and 0.40 min−1, and KI values of 42 and 43 μM for PTP1B and SHP1(ΔSH2), respectively. The α-bromo derivative 17 is 13-fold more potent than its α-chloro counterpart 18; this may be due to either the smaller size of the chlorine atom in α-chloroacetyl group making it a less effective pTyr mimetic compared to the large bromine atom or the presence of a better leaving group Br in 17. MALDI mass spectrometric analysis of the trypsin digested fragments of 17, inactivated SHP1, and kinetic analysis of C453S mutant experiments were consistent with the covalent attachment of 17 to the active-site Cys 435 of SHP1. Western-blot analysis of B cell phosphoproteins showed increased pTyr levels in 17-treated B cells. To improve potency, selectivity, and membrane permeability of the α-haloacetophenone core, the same research group undertook a SAR study.118 They prepared over twenty bromo- and chloro- acetophenone derivatives and tested their efficacy against SHP1 and PTP1B. Compounds with altered electronic properties of the phenyl ring showed no significant improvement in potency against PTPs. However, compounds 19 and 20, bearing a tripeptide (Gly-Glu-Glu) group attached via a peptide bond, showed highest potency against SHP1 and PTP1B. Analogue 19 exhibited a KI of 2.8 mM, kinact of 1.2 min−1, and specificity constant (kinact/KI) of 4.3×105 M−1 min−1 against PTP1B. It also showed 25-fold improved potency against SHP1 compared to the parent analogue 14. While inhibitor 20 has a kinact/KI value of 3.6×104 M−1 min−1 and 666 M−1 min−1 against PTP1B and SHP1, respectively, showing a 54-fold selectivity toward PTP1B. The inactivation by these agents quickly reversed upon irradiation of UV light at 350 nm (Fig. 11). For example, approximately 80% of original activity was recovered by the irradiation of 17 modified SHP-1 for ~15 min (Fig. 11). These molecules could provide a novel class of photo-reversible PTP inhibitory agents for controlling cellular signaling processes.
Fig. 11.

Inactivation of PTPs by α-haloacetophenones.
4. α,β-unsaturated compounds
The Michael addition is the most commonly used reaction to achieve covalent modification of proteins. Typically, functional groups that undergo this addition reaction include α,β-unsaturated carbonyls, vinyl sulfones, vinyl sulfonates, quinones, alkynyl amides, propargylic acid derivatives, and cyanoacrylates.119,120 The Michael addition of cysteine thiol to these functional groups is a reliable way to selectively modify cysteine residues in target proteins.121. Seiner et al. reported acrolein as a potent time-dependent inactivator of PTP1B (kinact = 0.02 s−1 and KI = 2.3 × 10−4 M).122 The authors determined percentage enzyme activity that remained for a series of structurally related aldehydes against PTP1B (Fig. 12). When incubated with 225 nM of PTP1B, acrolein, crotonaldehyde, 3-methyl-2-butenal, glyoxal, and propanal (all 500 μM, for 10 min) showed 4%, 83%, 93%, 95%, and 93% of remained enzyme activities, respectively. This suggests that the double bond in acrolein is essential for the inactivation of PTP1B. In addition, steric bulk at the alkene terminus weakens the inactivating properties of these compounds. Together, these results support an inactivation mechanism involving Michael addition of the sulfur atom of active site cysteine 215 to the β-carbon of acrolein (Scheme 2).
Fig. 12.
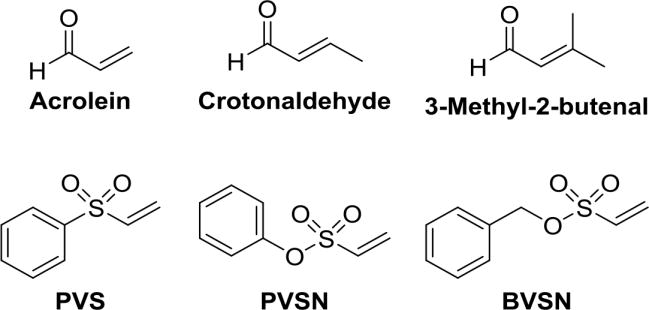
Structures of α,β-unsaturated compounds used for covalent modification of PTPs. 122, 123
Scheme 2.

The proposed mechanism of inactivation of PTP1B by acrolein.122
In an effort to design highly selective activity based probes towards PTPs, Liu et al. screened aryl vinyl sulfonates and sulfones against several PTPs (Fig. 12).123 Phenyl vinyl sulfone (PVS) and phenyl vinyl sulfonate (PVSN) inactivated the YopH PTP in a time- and concentration-dependent manner. These compounds showed comparable kinetics, modest affinity with KI= 290 μM, kinact = 0.061 min−1 (PVSN), and KI = 350 μM, kinact = 0.08 min−1 (PVS) against YopH. Similar kinetic results were observed with other PTPs such as PTP1B, PRL1, VHR, HePTP, DEP-1, and the low molecular weight PTP. Mass spectrometric and X-ray crystal structure analysis of PVS- and PVSN-modified YopH revealed covalent modification of active site Cys403 via a Michael addition of the thiolate of Cys403 onto the β-carbon of the vinyl group (Fig. 13). The bond lengths for the C-S bonds (thioether) between Cys403 of YopH and PVS and PVSN are 1.82 and 1.73 Å, respectively, which are close to the typical C-S bond length of ~1.8 Å. X-ray crystal structures of PVS and PVSN modified YopH are very similar. Due to the presence of an extra oxygen atom, the SO2 in PVSN moves 0.7 Å deeper into the active site; as a result, the H-bonding pattern varies between the two complexes. However, the total number of hydrogen bonds in both structures remains the same. PVSN showed no inactivation against acid or alkaline phosphatases. It was 240–700-fold less effective in inactivating the cysteine proteases, papain and cathepsin B compared to HePTP and VHR. The authors have developed activity-based probes using the PVSN moiety to profile PTP activity in complex proteome. They demonstrated the highly selective nature of these probes towards labeling PTPs from a complex E. coli proteome. These probes have several advantages over α-bromobenzylphosphonates, including higher affinity for the PTP active site, resistance to solvolysis, and increased cell-permeability.
Fig. 13.

X-ray crystal structures of PVS (left) and PVSN (right) complexes with YopH. 123
A screen of the NIH molecular libraries small molecules repository (MLSMR) identified compounds 21 and 22 as covalent inhibitors of LYP (Fig. 14).124 Kinetic analysis of these two compounds is consistent with covalent capture of the enzyme, with nanomolar KI and reciprocal millisecond kinact values, indicating the best kinact/KI ratios compared to other reported covalent LYP inhibitors. The anti-inflammatory compound BAY-11-7082 (Fig. 14) was recently reported as a potent, irreversible inhibitor of PTPs.125 Molecular modeling studies and mass spectrometry analysis of BAY-11-7082 modified PTP1B, revealed covalent modification of the enzyme via Michael addition reaction between Cys215 thiolate and C2 carbon of the inhibitor (Fig. 15). Most importantly, this compound showed 100-fold selectivity toward classical PTPs over DUSPs. BAY-11-7082 caused an increase in tyrosine phosphorylation in RAW 264 macrophages, similar to the effects of vanadate, a general PTP inhibitor. These results indicate that many known reported biological effects of BAY-11-7082 in part may be due to its inhibition potential of PTP activity. The proposed mode of action for BAY-11-7082 is similar to that of PVSN and PVS.
Fig. 14.

Structures of electrophilic compounds with activity as covalent PTP inhibitors.
Fig. 15.

Proposed mechanism for the inactivation of PTP1B by BAY-11-7082.
Natural products serve as good starting points for the development of new drugs. Currently, several natural product derivatives have been reported as inhibitors of various disease relevant PTPs.126–129 Ueda and co-workers identified 4-isoavenaciolide (4iA) (Fig. 14), a natural product isolated from fungal strain, as an irreversible inhibitor of a dual-specificity protein phosphatase, VHR (vaccinia H1 related) with an IC50 of 1.4 μM.130 Mass spectrometry analyses of 4iA modified VHR fragments confirmed the covalent attachment of two molecules of 4-iA to one molecule of VHR at two different sites: Cys124 in the catalytic domain and Cys171 on the α6 helix positioned surface domain. Due to the presence of a reactive exo-methylene group in 4-iA, it was proposed that 4-iA inhibits VHR through a Michael addition of the catalytic cysteine (Cys124) thiol. Interestingly, 4-iA inhibited only dual-specific phosphatases and tyrosine phosphatases and showed no inhibition against serine/threonine phosphatases (PP1 and PP2A). These results indicate that 4-iA is a cysteine-targeting inhibitor of protein phosphatases with a common HCX5RS/T motif in the catalytic site.
Menadione (Vitamin K3) (Fig. 14), a naphthoquinone derivative, was shown to have antiproliferative activity towards a variety of human cells. This compound shows lower levels of toxicity compared to other quinone-based chemotherapeutic agents.131 Menadione was found to inactivate CDC25A by covalent modification at the active site.132 Therefore, Choi and co-workers screened a group of hydrophilic naphthoquinone derivatives against CDC25A. Analogue 2,3-dichloro-5,8-dihydroxynapthoquinone 23 (Fig. 14) was the most potent of the compounds tested. Flow cytometry revealed a cell-cycle delay at the G1/S phase transition, confirming a decrease in the CDC25A activity by 23. Later, Ham et al. synthesized a sulfone analogue of naphthoquinone 24 (Fig. 14) and tested it against a panel of PTPs.133 Compound 24 inactivated PTP1B with a dissociation constant KI of 3.5 μM and an inactivation rate constant kinact of 2.2 × 10−2 sec−1, while no inactivation was observed against CDC25A, CDC25B, CDC25C, YopH, and LAR (up to 40 μM). Dialysis and competitive experiments confirmed the active site directed irreversible inactivation of PTP1B by compound 24.
Trans-β-nitrostyrene (TBNS) and its derivatives were found to be slow-binding, reversible inhibitors of PTP1B, SHP1, and YopH.134 In the absence of any added free thiols, compound 25 (TBNS) (Fig. 16) exhibited potent inhibition of PTP1B and YopH with an IC50 of 2.5 μM and 5 μM, respectively. It showed no inactivation against alkaline phosphatase. A structure-activity relationship of TBNS was established through the evaluation of a small collection of derivatives of TBNS. All the derivatives 26–32 (except 29) (Fig. 16) inactivated PTP1B with IC50 values ranging from 2.7 μM to 28 μM in the absence of added thiols. Interestingly, compound 29 showed no significant inhibition of PTP1B and SHP1, even at higher concentrations up to 600 μM. This suggests the necessity of C=C bond in TBNS, C=C maintains a rigid planar geometry that is critical for binding and/or positioning the NO2 group for nucleophilic attack by the active site thiolate. Analogue 33, which has a tripeptide (Gly-Glu-Glu) at the para position of TBNS, showed slightly improved potency with as IC50 of 1.4 μM against PTP1B. UV-vis spectroscopy, site-directed mutagenesis, and heteronuclear single-quantum correlation (HSQC) NMR spectroscopy studies suggested a mechanism in which TBNS fits into the active site, in a manner similar to that of pTyr, followed by nucleophilic attack of Cys-215 thiolate on to the nitro group in TBNS to form a covalent thiol-reversible adduct (Fig. 17).
Fig. 16.

Chemical structures of PTP inactivators with nitro group.
Fig. 17.

Reversible covalent modification of PTPs by TBNS derivatives.
5. Carbonyl compounds
Peptidyl aldehydes are known to inhibit serine and cysteine proteases via a covalent, reversible mechanism.135 These aldehydes react with active site nuclephiles in proteases to form a covalent hemiacetal or hemithioacetal linkage that mimics the transition state of a normal enzyme reaction. This tetrahedral hemiactal or hemithioacetal intermediate can collapse to regenerate the free enzyme. As a result, the enzyme-inhibitor complex is in equilibrium with free inhibitor and free enzyme. By taking advantage of this reversible covalent nature of the reaction between aldehydes and protein nucleophiles, medicinal chemists have developed a number of highly specific aldehyde-based inhibitors for proteases. Interestingly, calpeptin, a potent dipeptide aldehyde inhibitor of calpain, showed modest reactivity over several PTPs.136,137 This observation led Pei et al. to scereen a set of aldehydes against PTPs such as PTP1B, SHP1, and VHR.138 Among the aldehydes tested (34–36) (Fig. 18), 4-carboxycinnamaldehyde 36 showed the highest activity against PTPs, with IC50 values of 230 and 970 μM against SHP1 and PTP1B, respectively. Interestingly, it showed no inhibition against (up to 2 mM) VHR, which may be due to a wider, shallower active-site of VHR compared to other two PTPs.139 Early studies by Moran et al. demonstrated that a weakly active cinnamic acid can be converted into a highly potent inhibitor 37 (Fig. 18) for PTP1B (KI = 79 nM) by introducing the tripeptide (Gly-Glu-Glu) at the para position of cinnamic acid.140 In an attempt to improve potency and selectivity of 36, Pei et al. used Moran’s strategy and attached the tripeptide GEE at the para position of cinnamaldehyde to synthesize 38 (Cinn-GEE). As expected, aldehyde 38 showed improved potency against all three PTPs (e.g., KI = 5.4 μM against PTP1B). The mechanism of inhibition was investigated using 1H-13C HSQC NMR spectroscopy of 13C labelled Cinn-GEE and site-directed mutagenesis. HSQC of 13C labeled aldehyde carbon containing Cinn-GEE showed a single cross-peak at δ 9.64 (1H) and δ 201 (13C), whereas the PTP1B/Cinn-GEE complex showed three distinct cross-peaks at δ 7.6–7.8 (1H) and 130–137 (13C). Mutation of the catalytic cysteine to alanine (C215A) showed no effect on the crosspeaks, but mutation of active-site arginine to alanine (R221A) abolished all three cross-peaks. Similar experiments with 13C labeled benzylic carbon containing a Cinn-GEE/PTP1B complex revealed a change in the hybridization from sp2 to sp3 for the benzylic carbon. This is consistent with a mechanism involving the formation of an initial noncovalent (E•I) complex, followed by a covalent imine adduct (E-I) formation between the guanidine group of the active-site Arg221 and aldehyde of the inhibitor (Fig. 19). Conjugate addition at the β-carbon by an unidentified nucleophile (could be β-mercaptoethanol or a water molecule in buffer or protein side chain) produced the enamine (E-I*).
Fig. 18.

Structures of aldehyde based inhibitors used for inactivation of PTPs. 138, 140, 141
Fig. 19.

Proposed mechanism for the reaction of PTP with cinnamaldehyde derivatives (RSH = mercaptoethanol).
In an another instance, Shim and co-workers screened a set of over thirty formylchromone derivatives against PTP1B.141 Their search revealed TP33 and TP34 (Fig. 18) as the most potent and selective inhibitors for PTP1B. Both TP33 and TP34 showed 500, 30, and >10-fold selectivety over LAR, TC-PTP, and SHP1 respectively. Molecular modeling studies of TP34 on PTP1B placed the aldehyde carbon atom 3.4 Å away from the sulfur atom of Cys215 and the nitrogen atom of Arg221, suggesting the posibility of forming S–C bond (thioether) or N=C bond (imine). The S atom was oriented in the right position to the carbonyl of aldehyde allowing a nucleophilic attack, whereas Arg221 was not in a proper orientation for imine formation. These observations favor the formation of thioether linkage over imine, suggesting a mechanism different from the one reported by Pei and co-workers.
6. Isothiocyanates
Dietary isothiocyanates (ITCs), such as allyl isothiocyanate (AITC) and phenethyl isothiocyanate (PEITC) (Fig. 20A), have anti-carcinogenic activities and are of potential use in the chemoprevention of cancer.142–144 More recently, Lewis et al. reported thiol reversible inactivation of PTP1B and SHP2 with various isothiocyanates.145 Results in the case of PTP1B confirmed a mechanism involving covalent, thiol reversible, modification of the active site cysteine residue. Second-order inactivation rate constants for inactivation of SHP2 by AITC were reported as 16 and 9 M−1 s−1 in the Tris and glutarate buffer systems, respectively. The presence of active site directed inhibitors, such as phosphate and arsenate, decreased the inactivation rates at which SHP2 was inactivated by AITC or sulforapane, indicating covalent modification of the enzyme’s active site (Fig. 20B). PTP1B dephosphorylates Tyr-490 of TrkA in PC12 cells, suggesting its key regulatory role in NGF signaling. Over-expression of PTP1B in PC12 cells resulted in inhibition of neuritogenesis due to the dephosphorylation of TrkA. 6-methylsulfinylhexyl isothiocyanate (6-HITC), the major ITC compound in wasabi, improved neuritogenesis in PC12 cells by inhibiting PTP1B.146 Unfortunately, a detailed mechanism of regulation of PTP1B by 6-HITC was not reported. These results suggest that 6-HITC may serve as a potential lead compound in the development of drugs to treat neurodegenerative diseases.
Fig. 20.

A) Structures of various isothiocyanates used by Lewis et al. and Shibata et al. as inactivators of PTPs. B) Proposed mechanism for the reaction between isothiocyanates and cysteine residues in proteins.
7. Epoxides
Due to their strained three-membered ring structures, epoxides (oxiranes) offer reactive electrophilic carbon atoms that can be attacked by a variety of nucleophiles. In order to identify the active site residues of low molecular weight PTP, Zhang and co-workers conducted inactivation experiments using epoxides such as phosphomycin, 1,2-epoxy-3-(p-nitrophenoxy)propane (EPNP), and (R)-(−) and (S)-(+)-2-(benzyloxymethyl)-oxiranes (Fig. 21).147 EPNP and (R)-(−) and (S)-(+)-2-(benzyloxymethyl)-oxiranes acted as active-site directed irreversible covalent inactivators, whereas phosphomycin was a simple competitive inhibitor of the enzyme. These observations were consistent with the importance of a hydrophobic moiety on the substrate for efficient binding. An investigation into the mechanism of inactivation by EPNP revealed covalent binding of Cys62 and Cys145 residues on the low molecular weight PTP. Since these two Cys residues are away from the active site, the mode of inhibition by EPNP may be allosteric.
Fig. 21.

Chemical structures of epoxides used to inactivate PTPs.
In an attempt to further develop epoxides as PTP inactivators, Park et al. tested styrene oxide against a panel of five PTPs: the catalytic domain of human SHP1 (SHP1-cat), a yeast PTP1 (YPTP1), a Yersinia PTP (YopH), the cytosolic PTP domain of CD45 (CD45-cyto), and human PTP1B.148 In the case of PTP1B and SHP1-cat, over 80% of inhibition was observed when the enzyme was preincubated with 20 mM styrene oxide for 10 min in a p-nitrophenyl phosphate (pNPP) based assay. Further experiments with PTP1B at various preincubation times and inhibitor concentrations confirmed the time and concentration-dependent inhibition. Preincubation of 2 mM of compounds 39, 40, 41, and 42 (Fig. 21) for 10 min with PTP1B showed 85%, 81%, 68%, and 29% of remaining activity, respectively. Compound 42 with a naphthalene ring showed improved inhibition, but poor solubility prevented reliable measurement of kinetic data at higher concentrations. Later, Dana and coworkers synthesized a small set of sulfonyloxiranyl derivatives based on molecular modeling studies.149 Reaction of compounds 43–47 (Fig. 21) with PTP1B under pseudo first order kinetic conditions ([43] ≫ [PTP1B]) resulted in a time dependent loss of phosphatase activity. The covalent nature of inactivation was further confirmed by dilution, competition, and MALDI-TOF mass spectrometry experiments.150 Epoxides 43, 44, and 45 showed very similar reactivity; epoxide 46 showed slightly more reactivity, whereas 47 showed no reactivity against PTP1B. The lack of reactivity of aliphatic analog 47 with PTP1B indicated the importance an aryl moiety for effective PTP1B binding. In human A549 lung cancer cell lines, compound 39 caused increase in intracellular tyrosine phosphorylation levels in a dose-dependent manner, suggesting a cell-permeable nature of this class of compounds.
8. Other PTP inactivation mechanisms
In addition to the above discussed covalent inhibitors, several other endogenous and exogenous chemicals have also been reported as inactivators of PTPs. Recently, new covalent allosteric inhibition of PTPs was also reported where covalent modification of cysteines present in pockets close to the active site led to loss of phosphatase activity.151 Fluorogenic reagent 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole (ABDF, compound 48) (Fig. 22) was reported as an allosteric inhibitor of PTP1B.152 Tryptic digestion followed by mass spectrometry analysis confirmed covalent modification of Cys121 in PTP1B. The authors reasoned that disruption of the active-site, due to the covalent modification of Cys121, was responsible for the loss of phosphatase activity. Cys 121 is a conserved residue among various PTPs, and ABDF inactivates closely related TC-PTP and LAR, but not CD45, showing some degree of sequence specificity toward inactivation. Surprisingly, ABDF reacts selectively with only cysteine 121, leaving unmodified five other cysteines, including the highly reactive active site Cys215 in PTP1B. In addition, ABDF caused a dose-dependent increase in the level of insulin receptor phosphorylation in CHO-hIR cells through PTP1B inhibition. Similar allosteric inhibition of PTP1B was observed with 1,2-naphthoquinone 49 (Fig. 22).153 MALDI-TOF mass spectrometry analysis and mutation experiments revealed that compound 49 mediated PTP1B inactivation was mainly due to covalent modification of Cys-121 in the enzyme and some binding interactions to His-25 and Cys-215. These two examples, together with that of EPNP and LWM-PTP,147 illustrate that targeting cysteine residues present in pockets outside an active site may also be exploited in designing inhibitors for PTPs.
Fig. 22.

Chemical structures of PTP inactivators.
The benzopentatiepin analogue 8-(trifluoromethyl)-1,2,3,4,5-benzopentathiepin-6-amine hydrochloride (TC-2153) was identified as a reversible covalent inhibitor of STriatal-Enriched protein tyrosine Phosphatase (STEP).154 In vitro selectivity experiments on full length PTPs revealed that TC-2153 is more potent on isoforms of STEP, STEP46 (IC50 = 57 nM) and STEP61 (IC50 = 93 nM), compared to highly related PTPs, PTP-SL (IC50 = 220 nM), and HePTP (IC50 = 363 nM). It showed even greater selectivity over SHP2 (IC50 = 6,896 nM) and PTP1B (IC50 = 723 nM). In cell-based assays, TC-2153 increased tyrosine phosphorylation of STEP substrates ERK1/2, Pyk2, and GluN2B. Kinetic constants kinact (0.0176 s−1), KI (115 nM), and kinact/KI (153,000 M−1s−1) for TC-2153 against STEP were reported.68 TC-2153 treatment restored BDNF expression and improved cognitive and motor functions in phencyclidine-treated mice through inhibition of STEP61.
The seleninic acid group (RSeO2H) is a bioisostere for carboxylic acid (RCO2H), due to its similar structural and protolytic properties.155 Monoanionic seleninate is also considered as a potential bioisosteric match for the phosphate group.156,157 In 2008, Abdo et al. synthesized compound 50 (Fig. 22) with the seleninate acting as a phosphate mimic in pTyr.158 Incubation of 50 with YopH, PTP1B, VHR, and VHX resulted in a time and concentration dependent loss of enzyme activity. Kinetic, mutational, and mass spectrometric analytical data together revealed 50 as an irreversible inhibitor of the PTPs tested. YopH inactivation by 50 showed saturation kinetics and exibited values for the binding constant KI and the inactivation rate constant ki of 44.5 μM and 0.243 min−1, respectively. Further analysis with X-ray crystalographic structure data of PTP1B complexed with 50, confirmed the covalent modification of PTP1B via a selenosulfide bond between the sulfur atom of Cys215 and the selenium in 50 (Fig. 23). Selenosulfide bonds can be readily cleaved by the action of thiols (DTT) in aqueous solution.159 Interestingly, the selenosulfide linkage present in modified YopH is stable even at millimolar concentrations of DTT, suggesting the inaccesible nature of this bond in the deep active-site of YopH.
Fig. 23.

X-ray crystal structure of 50 complexed with PTP1B.158 The figure was made using PyMol.
In 2010, Piovan et al. screened a series of twelve hypervalent selenium (IV) and tellurium (IV) compounds (organoselenanes and organotelluranes) as potential inactivators of PTPs.160 All these compounds (Fig. 24) inactivated PTP1B and YopH in a time and concentration dependent manner. The inactivation is most likely due to the covalent modification of the active site cysteine thiol by the chalcogen atom. Kinetic analysis of these compounds (51–62) revealed organotelluranes as being more potent than organoselenanes for inactivating PTP1B and YopH. SAR studies showed no obvious contributions from halogens or stereochemistry at the benzylic position on inactivation power of these organochalcogenanes. Organotellurane 57 showed an equilibrium binding constant KI of 1.9 ± 0.17 mM and an inactivation rate constant kinact of 17.2 ± 0.9 min−1. Though these compounds are weak inactivators of the PTPs tested, potent and selective inhibitors could be developed by introducing specificity determinants into the aryl ring.
Fig. 24.

The chemical structures of organoselenanes and organotelluranes.160
Heavy metals have been noted as covalent inhibitors of various PTPs. Kim and co-workers examined the regulatory roles of heavy metals ions such as Mg2+, Mn2+, Ca2+, Co2+, Fe3+, Zn2+, Cu2+, and Cd2+ on human dual specificity protein phosphatase VHR in vitro.161 Among the metal ions examined, only Fe3+, Cu2+, Zn2+, and Cd2+ inactivated VHR, and Cu2+ ion demonstrated the most potent inactivation. The addition of excess EDTA diminished Fe3+, Zn2+, and Cd2+ mediated inactivation of VHR, whereas additional DTT was required to restore the activity of Cu2+-inactivated VHR. These two observations suggest that inactivation by Fe3+, Zn2+, and Cd2+ occurred largely from simple binding of these metal ions to certain residues of VHR; however, the Cu2+ mediated inactivation was largely due to the oxidative property of Cu2+. Additionally, Cu2+ inactivated other PTPs such as SHP1 and PTP1B. In a recent study, Karvar et al. demonstrated Au(I)-phosphine complex, [Au((CH2CH2CN)2PPh)Cl], as a selective LYP inhibitor both in vitro and in cells.162 The complex 63 showed an IC50 of 3.5 μM against LYP, and over 14 fold selectivity against PTP-PEST, HePTP, and CD45. Mode of inhibition studies on LYP, the LYP mutant C129/131S, and PTP-PEST suggested the coordination of Au(I) by both the active site cysteine (Cys227) as well as either Cys129 or Cys231 as a potential mechanism for selective LYP inhibition. This is consistent with previous reports on competitive and thiol reversible inhibition of PTPs by Au(I) complexes.163,164 In an interesting approach, Bishop and co-workers introduced non-natural allosteric-inhibition sites in PTPs through protein engineering; these sites assist to sensitize target PTPs to selective inhibition by FlAsH (fluorescein arsenical hairpin binder) and other biarsenical compounds (Fig. 25).165 They designed these sites based on naturally occurring cryptic allosteric site present in a wild-type SHP2 catalytic domain that is sensitive to inhibition by biarsenicals even in the absence of engineering.166,167 To improve interactions between SHP2’s allosteric site and FlAsH, they introduced a cysteine residue at position 368 (V368C mutant), because valine 368 is not conserved among classical PTPs, and it also contains a solvent-accessible side chain. As expected, V368C SHP2’s demonstrated increased affinity of the protein/FlAsH interaction through an additional covalent interaction between the thiol of cysteine 368 and arsenic. Moreover, the authors successfully introduced these allosteric-inhibition sites into other PTPs including PTP1B, HePTP, PTPH1, and FAP1 from different PTP subfamilies, to generate biarsenical responsive PTPs.
Fig. 25.

Chemical structures of Au(I) complex and FlAsH.
Dithiolethiones are known for their chemopreventive activity.168,169 Bhattacharyya et al. demonstrated that the dithiolethione analogue oltipraz inactivated PTPs via covalent, but thiol-reversible, modification of catalytic cysteine residues.170 The proposed mechanism involves the nucleophilic attack of thiolate of Cys215 followed by a series of thiol disulfide exchange reactions which then leads to the covalent modification of Cys215 of the PTP1B (Fig. 26). It was shown that the UV irradiation of PTP-overexpressing cells leads to partial inactivation of several PTPs. Four PTPs (SHP1, RPTPα, RPTPσ, and DEP-1), were partially inactivated by UV irradiation at 355 nm. In the case of RPTPα, UV irradiation leads to the generation of a substrate-trapping configuration that is similar to the C433S mutant.171
Fig. 26.

The proposed mechanism of inactivation of PTP1B by oltipraz.
As mentioned in the previous section, oxidation of PTPs by various endogenous chemicals produce unique electrophilic groups such as sulfenic acid and sulfenyl amide, which could be captured by nucleophilic reagents to completely knock down the phosphatase activity of PTPs. For instance, Kate Carroll’s group recently reported dimedone derivatives as covalent inhibitors of the sulfenic acid forms of YopH and PTP1B (Fig. 27A).172–174 Classical tyrosine phosphatases, PTP1B and PTPα when exposed to biological oxidants, form an electrophilic sulfenyl amide via a sulfenic acid intermediate. Chemical model studies conducted by Shieu et al. and Ruddraraju et al. showed that the sulfenyl amide moiety in PTPs could be covalently modified by 1,3-dicarbonyl compounds to generate stable adducts (Fig. 27B).175–177 These adducts were shown to be stable under high concentrations of thiols (e.g., 50 mM DTT at 45 °C for 6 h). In an another study, Parson et al. reported thiol reversible covalent capture of sulfenyl amide by sulfone stabilized carbon nucleophiles such as β-ketosulfones, β-disulfones, and β-cyano sulfones.178 Both sulfenic acid and sulfenyl amide present a unique electrophilic sulfur atom for selective covalent modification of PTPs by a suitable nucleophile. Taken together, these results suggest that the above mentioned nucleophilic warhead groups, when incorporated into a high affinity binding module, may generate compounds that exploit non-covalent interactions to facilitate selective irreversible capture of oxidized PTPs. The ability to target differentially oxidized cysteine residues on PTPs may afford further means of achieving selectivity and offer an alternative strategy to modulate PTP activity in cells.
Fig. 27.

Covalent capture of oxidized forms of PTPs. A) Covalent modification of sulfenic acid by 1,3-diketone derivatives. B) Covalent capture of sulfenyl amide model compound by various carbon nucleophiles.
Conclusion
Given the diverse regulatory roles played by PTPs and their implications in human diseases, such as cancer, diabetes, and neurological disorders, PTPs are highly important drug targets. But, developing potent, selective, and active site-directed small molecule inhibitors (with drug-like properties) to target PTPs is challenging, mainly due to the highly conserved and cationic nature of the active sites of various PTPs. However, several research groups have successfully generated active site-directed small molecule inhibitors with high potency and selectivity as well as robust in vivo activity to target disease-relevant PTPs.10, 11 Currently, there is also renewed interest among investigators to develop allosteric and covalent inhibitors to modulate PTP activity. Allosteric inhibition strategy has generated a few successful small molecule inhibitors in the case of PTP1B and SHP2. The covalent inhibition strategy was successful in the case of PTKs drug discovery and has generated some promising small molecules for selective inactivation of PTKs.34 But, no selective inactivators have been reported for an individual PTP. Covalent inhibitors have several advantages over traditional non-covalent inhibitors; low doses are needed to show the desired pharmaceutical effect, generate stable and long-lived ties, and are resistant to mutations.179 Although off-target interactions are a major concern, a careful and systematic approach could lead to a drug candidate that addresses the above-mentioned challenges in PTP drug discovery. We have summarized the efforts undertaken by researchers to design covalent inhibitors for selective inactivation of PTPs. Some of these inhibitors demonstrated class specific inhibition; however, none of them showed selectivity toward an individual PTP. We believe that an important future direction for the field is to develop selective covalent PTP inhibitors for functional interrogation, target validation, and therapeutic development.
One approach to install specificity for covalent inhibition of PTPs is to design bifunctional molecules that consist of a specificity determinant and a functional warhead capable of generating a covalent linkage with the PTP of choice. The former serves as a template by binding to the target PTP, thereby allowing the latter to undergo covalent modification of an active site residue. Although the active site structures are highly conserved among various PTPs, the surfaces of these proteins are remarkably diverse, a property that may be responsible for substrate recognition, regulatory mechanisms such as oligomerization, and association with other regulatory proteins.180 Therefore, targeting non-conserved surface residues for achieving selectivity using the exo-affinity labelling technique is also a viable strategy for the inactivation of individual PTPs.181 This strategy involves a combination of an inhibitor and an electrophile, where an inhibitor binds to the active site and the electrophile extends out of the active-site and covalently modifies a non-conserved surface residue on the target PTP. Covalent modification of post-translationally modified PTPs, such as oxidized or nitrosylated forms, also has great potential for future drug discovery research. The success of covalent inactivation of oxidized forms of PTPs depends on a better understanding of mechanisms leading to selective PTP oxidation in a cellular environment. The strategy of introducing an electrophile onto a highly potent and selective inhibitor of a particular target PTP, based on X-ray crystal structure studies or docking studies, could provide selective and potent inactivators of PTPs. Computational tools such as CovalentDock,182 GOLD,183 CovDock-VS,184 Autodock,185 Glide,186 CovDock,187 etc. are available to aid researchers to design covalent inhibitors with drug-like properties.188 Covalent inhibitors can also be used to develop chemical and biological tools, such as activity-based probes (ABPs), for functional assignment of PTPs at the proteome level in both normal and diseased states.
Acknowledgments
We gratefully acknowledge the financial support of NIH Grants CA69202 and CA207288.
Biographies

Kasi V. Ruddraraju was born in Andhra Pradesh, India. He received his Master’s degree in organic chemistry from Osmania University in 2007. Then he joined Albany molecular research Inc. (AMRI) as a research scientist. He moved to the United States in 2011 to pursue his PhD degree in organic chemistry under Dr. Kent Gates at the University of Missouri-Columbia. His PhD thesis research involved design, synthesis, and structure activity relationship (SAR) studies of covalent inhibitors for protein tyrosine phosphatase 1B (PTP1B). Currently, he is working as a post-doctoral research associate for Dr. Zhong-Yin Zhang in the department of medicinal chemistry and molecular pharmacology at Purdue University. His current research involves design and synthesis of therapeutic agents that target protein tyrosine phosphatases (PTPs) to treat diabetes and various cancers.

Dr. Zhong-Yin Zhang is Distinguished Profesor of Medicinal Chemistry, Robert C. and Charlotte P. Anderson Chair in Pharmacology, and Head of the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University. Research in his laboratory spans the disciplines of chemistry and biology with an emphasis on the structure and function of protein tyrosine phosphatases (PTPs), roles of PTP in normal physiology and pathological conditions, and the design and synthesis of PTP inhibitors as chemical probes to interrogate PTP function and as novel therapeutics for the treatment of cancer, diabetes and obesity, autoimmune disorders, neurodegenerative and infectious diseases. He obtained his Ph.D. in Chemistry from Purdue University and completed his postdoctoral training at the University of Michigan. Before returning to Purdue, he was Robert A. Harris Professor and Chairman of the Department of Biochemistry and Molecular Biology at Indiana University School of Medicine.
References
- 1.Hunter T. Cell. 1987;50:823–829. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- 2.He RJ, Yu ZH, Zhang RY, Zhang ZY. Acta Pharmacol Sin. 2014;35:1227–1246. doi: 10.1038/aps.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai M, Terzaghi W, Wang H. Plant Signaling & Behavior. 2013;8:e22508. doi: 10.4161/psb.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson SA, Hunter T. Nat Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 5.Robinson DR, Wu YM, Lin SF. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 6.Cohen P, Alessi DR. ACS Chem Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 8.Julien SG, Dubé N, Hardy S, Tremblay ML. Nature reviews Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 9.Combs AP. J Med Chem. 2010;53:2333–2344. doi: 10.1021/jm901090b. [DOI] [PubMed] [Google Scholar]
- 10.He R, Zeng LF, He Y, Zhang S, Zhang ZY. FEBS J. 2013;280:731–750. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang ZY. Acc Chem Res. 2017;50:122–129. doi: 10.1021/acs.accounts.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen K, Keng YF, Wu L, Guo XL, Lawrence DS, Zhang ZY. J Biol Chem. 2001;276:47311–47319. doi: 10.1074/jbc.M106568200. [DOI] [PubMed] [Google Scholar]
- 13.Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Proc Natl Acad Sci U S A. 1997;94:13420–13425. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xin Z, Oost TK, Abad-Zapatero C, Hajduk PJ, Pei Z, Szczepankiewicz BG, Hutchins CW, Ballaron SJ, Stashko MA, Lubben T, Trevillyan JM, Jirousek MR, Liu G. Bioorg Med Chem Lett. 2003;13:1887–1890. doi: 10.1016/s0960-894x(03)00302-0. [DOI] [PubMed] [Google Scholar]
- 15.He R, Yu ZH, Zhang RY, Wu L, Gunawan AM, Zhang ZY. ACS Med Chem Lett. 2015;6:1231–1235. doi: 10.1021/acsmedchemlett.5b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng LF, Zhang RY, Yu ZH, Li S, Wu L, Gunawan AM, Lane BS, Mali RS, Li X, Chan RJ, Kapur R, Wells CD, Zhang ZY. J Med Chem. 2014;57:6594–6609. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, Dobson JR, Deng Z, Fei F, Firestone B, Fodor M, Fridrich C, Gao H, Grunenfelder D, Hao HX, Jacob J, Ho S, Hsiao K, Kang ZB, Karki R, Kato M, Larrow J, La Bonte LR, Lenoir F, Liu G, Liu S, Majumdar D, Meyer MJ, Palermo M, Perez L, Pu M, Price E, Quinn C, Shakya S, Shultz MD, Slisz J, Venkatesan K, Wang P, Warmuth M, Williams S, Yang G, Yuan J, Zhang JH, Zhu P, Ramsey T, Keen NJ, Sellers WR, Stams T, Fortin PD. Nature. 2016;535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 18.Chan RJ, Feng GS. Blood. 2007;109:862–867. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostman A, Hellberg C, Bohmer FD. Nature reviews Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 20.Tonks NK. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 21.Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, Jensen MR, Gauss CM, Page R, Blackledge M, Muthuswamy SK, Peti W, Tonks NK. Nat Chem Biol. 2014;10:558–566. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim Y-B, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. Developmental Cell. 2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 23.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Developmental Cell. 2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 24.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 26.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai Y, Yu ZH, Liu S, Zhang L, Zhang RY, Zeng LF, Zhang S, Zhang ZY. Cancer Res. 2016;76:4805–4815. doi: 10.1158/0008-5472.CAN-15-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauer RA. Drug Discov Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Hagel M, Niu DQ, St Martin T, Sheets MP, Qiao LX, Bernard H, Karp RM, Zhu ZD, Labenski MT, Chaturvedi P, Nacht M, Westlin WF, Petter RC, Singh J. Nature Chemical Biology. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- 30.Grimm SW, Einolf HJ, Hall SD, He K, Lim HK, Ling KHJ, Lu C, Nomeir AA, Seibert E, Skordos KW, Tonn GR, Van Horn R, Wang RW, Wong YN, Yang TJ, Obach RS. Drug Metabolism and Disposition. 2009;37:1355. doi: 10.1124/dmd.109.026716. [DOI] [PubMed] [Google Scholar]
- 31.Singh J, Petter RC, Baillie TA, Whitty A. Nature reviews Drug discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 32.Mah R, Thomas JR, Shafer CM. Bioorg Med Chem Lett. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Gan J, Ruan Q, He B, Zhu M, Shyu WC, Humphreys WG. Chemical Research in Toxicology. 2009;22:690–698. doi: 10.1021/tx800368n. [DOI] [PubMed] [Google Scholar]
- 34.Singh J, Petter RC, Kluge AF. Curr Opin Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- 35.Zhang ZY. Prog Nucleic Acid Res Mol Biol. 2003;73:171–220. doi: 10.1016/s0079-6603(03)01006-7. [DOI] [PubMed] [Google Scholar]
- 36.Krishnamurthy D, Barrios AM. Curr Opin Chem Biol. 2009;13:375–381. doi: 10.1016/j.cbpa.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 37.Dana D, Kumar S. Chemistry & Biology Interface. 2012;2:1–11. [Google Scholar]
- 38.Alho I, Costa L, Bicho M, Coelho C. Tumour Biol. 2013;34:1979–1989. doi: 10.1007/s13277-013-0784-1. [DOI] [PubMed] [Google Scholar]
- 39.Kristjansdottir K, Rudolph J. Chemistry & biology. 2004;11:1043–1051. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Karlsson-Rosenthal C, Millar JB. Trends in cell biology. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Trope CG, Flørenes VA, Suo Z, Nesland JM, Holm R. BMC Cancer. 2010;10:233. doi: 10.1186/1471-2407-10-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neel BG, Tonks NK. Current opinion in cell biology. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 43.Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, Dixon JE. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 44.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 45.Barford D, Flint AJ, Tonks NK. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 46.Tonks NK. FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 47.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 48.Zhang ZY, Dixon JE. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 49.Guan KL, Dixon JE. J Biol Chem. 1991;266:17026–17030. [PubMed] [Google Scholar]
- 50.Cho H, Krishnaraj R, Kitas E, Bannwarth W, Walsh CT, Anderson KS. Journal of the American Chemical Society. 1992;114:7296–7298. [Google Scholar]
- 51.Denu JM, Lohse DL, Vijayalakshmi J, Saper MA, Dixon JE. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:2493–2498. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pannifer AD, Flint AJ, Tonks NK, Barford D. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 53.Zhang ZY, Wang Y, Dixon JE. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:1624–1627. doi: 10.1073/pnas.91.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y, Wu L, Noh SJ, Guan KL, Zhang ZY. J Biol Chem. 1998;273:5484–5492. doi: 10.1074/jbc.273.10.5484. [DOI] [PubMed] [Google Scholar]
- 55.Tonks NK. The FEBS journal. 2013;280:346–378. doi: 10.1111/febs.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tonks NK, Diltz CD, Fischer EH. J Biol Chem. 1988;263:6722–6730. [PubMed] [Google Scholar]
- 57.Tonks NK, Diltz CD, Fischer EH. J Biol Chem. 1988;263:6731–6737. [PubMed] [Google Scholar]
- 58.Ahmad F, Considine RV, Goldstein BJ. J Clin Invest. 1995;95:2806–2812. doi: 10.1172/JCI117985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahmad F, Azevedo JL, Cortright R, Dohm GL, Goldstein BJ. Journal of Clinical Investigation. 1997;100:449–458. doi: 10.1172/JCI119552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang ZY, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T. Cell Metab. 2011;14:684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho CY, Koo SH, Wang Y, Callaway S, Hedrick S, Mak PA, Orth AP, Peters EC, Saez E, Montminy M, Schultz PG, Chanda SK. Cell Metab. 2006;3:367–378. doi: 10.1016/j.cmet.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 62.Maccari R, Ottana R. J Med Chem. 2012;55:2–22. doi: 10.1021/jm200607g. [DOI] [PubMed] [Google Scholar]
- 63.Krishnan N, Krishnan K, Connors CR, Choy MS, Page R, Peti W, Van Aelst L, Shea SD, Tonks NK. The Journal of Clinical Investigation. 2015;125:3163–3177. doi: 10.1172/JCI80323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou XD, Agazie YM. Cell Death Differ. 2008;15:988–996. doi: 10.1038/cdd.2008.54. [DOI] [PubMed] [Google Scholar]
- 65.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, Confalonieri S, Quarto M, Hu G, Balwierz PJ, Pachkov M, Elledge SJ, van Nimwegen E, Stadler MB, Bentires-Alj M. Nat Med. 2012;18:529–537. doi: 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- 66.Aressy B, Ducommun B. Anticancer Agents Med Chem. 2008;8:818–824. doi: 10.2174/187152008786847756. [DOI] [PubMed] [Google Scholar]
- 67.Gasparotto D, Maestro R, Piccinin S, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Cancer Res. 1997;57:2366–2368. [PubMed] [Google Scholar]
- 68.Xu J, Chatterjee M, Baguley TD, Brouillette J, Kurup P, Ghosh D, Kanyo J, Zhang Y, Seyb K, Ononenyi C, Foscue E, Anderson GM, Gresack J, Cuny GD, Glicksman MA, Greengard P, Lam TT, Tautz L, Nairn AC, Ellman JA, Lombroso PJ. PLoS biology. 2014;12:e1001923. doi: 10.1371/journal.pbio.1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ. Proceedings of the National Academy of Sciences. 2010;107:19014–19019. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rubio T, Köhn M. Biochemical Society Transactions. 2016;44:1305–1312. doi: 10.1042/BST20160146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hermiston ML, Xu Z, Weiss A. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- 72.Dawes R, Hennig B, Irving W, Petrova S, Boxall S, Ward V, Wallace D, Macallan DC, Thursz M, Hill A, Bodmer W, Beverley PCL, Tchilian EZ. Journal of Medical Genetics. 2006;43:678–684. doi: 10.1136/jmg.2005.040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vang T, Miletic AV, Bottini N, Mustelin T. Autoimmunity. 2007;40:453–461. doi: 10.1080/08916930701464897. [DOI] [PubMed] [Google Scholar]
- 74.MacKeigan JP, Murphy LO, Blenis J. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 75.Krippendorff BF, Neuhaus R, Lienau P, Reichel A, Huisinga W. Journal of biomolecular screening. 2009;14:913–923. doi: 10.1177/1087057109336751. [DOI] [PubMed] [Google Scholar]
- 76.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 77.Strelow JM. Journal of biomolecular screening. 2016 doi: 10.1177/1087057116656596. [DOI] [PubMed] [Google Scholar]
- 78.Kitz R, Wilson IB. The Journal of biological chemistry. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 79.den Hertog J, Groen A, van der Wijk T. Arch Biochem Biophys. 2005;434:11–15. doi: 10.1016/j.abb.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 80.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 81.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 82.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 83.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 84.Defelipe LA, Lanzarotti E, Gauto D, Marti MA, Turjanski AG. PLoS Comput Biol. 2015;11:e1004051. doi: 10.1371/journal.pcbi.1004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parsons ZD, Gates KS. Biochemistry. 2013;52:6412–6423. doi: 10.1021/bi400451m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Savitsky PA, Finkel T. Journal of Biological Chemistry. 2002;277:20535–20540. doi: 10.1074/jbc.M201589200. [DOI] [PubMed] [Google Scholar]
- 87.Sun JP, Wang WQ, Yang H, Liu S, Liang F, Fedorov AA, Almo SC, Zhang ZY. Biochemistry. 2005;44:12009–12021. doi: 10.1021/bi0509191. [DOI] [PubMed] [Google Scholar]
- 88.Caselli A, Marzocchini R, Camici G, Manao G, Moneti G, Pieraccini G, Ramponi G. Journal of Biological Chemistry. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 89.Truong TH, Carroll KS. Biochemistry. 2012;51:9954–9965. doi: 10.1021/bi301441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Denu JM, Tanner KG. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 91.LaButti JN, Gates KS. Bioorg Med Chem Lett. 2009;19:218–221. doi: 10.1016/j.bmcl.2008.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meng FG, Zhang ZY. Biochimica et biophysica acta. 2013;1834:464–469. doi: 10.1016/j.bbapap.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bhattacharya S, Labutti JN, Seiner DR, Gates KS. Bioorg Med Chem Lett. 2008;18:5856–5859. doi: 10.1016/j.bmcl.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rosselli M, Keller PJ, Dubey RK. Hum Reprod Update. 1998;4:3–24. doi: 10.1093/humupd/4.1.3. [DOI] [PubMed] [Google Scholar]
- 95.Hess DT, Stamler JS. The Journal of Biological Chemistry. 2012;287:4411–4418. doi: 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. The Journal of Biological Chemistry. 2013;288:26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Caselli A, Camici G, Manao G, Moneti G, Pazzagli L, Cappugi G, Ramponi G. Journal of Biological Chemistry. 1994;269:24878–24882. [PubMed] [Google Scholar]
- 98.Caselli A, Chiarugi P, Camici G, Manao G, Ramponi G. FEBS letters. 1995;374:249–252. doi: 10.1016/0014-5793(95)01120-4. [DOI] [PubMed] [Google Scholar]
- 99.Takakura K, Beckman JS, MacMillan-Crow LA, Crow JP. Archives of Biochemistry and Biophysics. 1999;369:197–207. doi: 10.1006/abbi.1999.1374. [DOI] [PubMed] [Google Scholar]
- 100.Xian M, Wang K, Chen XC, Hou YC, McGill A, Chen X, Zhou B, Zhang ZY, Cheng JP, Wang PG. Biochemical and biophysical research communications. 2000;268:310–314. doi: 10.1006/bbrc.2000.2117. [DOI] [PubMed] [Google Scholar]
- 101.Karver CE, Ahmed VF, Barrios AM. Bioorganic & Medicinal Chemistry Letters. 2011;21:285–287. doi: 10.1016/j.bmcl.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 102.Downey AM, Cairo CW. MedChemComm. 2014;5:1619–1633. [Google Scholar]
- 103.Zhang YL, Yao ZJ, Sarmiento M, Wu L, Burke TR, Zhang ZY. Journal of Biological Chemistry. 2000;275:34205–34212. doi: 10.1074/jbc.M004490200. [DOI] [PubMed] [Google Scholar]
- 104.Chen L, Wu L, Otaka A, Smyth MS, Roller PP, Burke TR, Jr, den Hertog J, Zhang ZY. Biochem Biophys Res Commun. 1995;216:976–984. doi: 10.1006/bbrc.1995.2716. [DOI] [PubMed] [Google Scholar]
- 105.Taylor WP, Zhang ZY, Widlanski TS. Bioorg Med Chem. 1996;4:1515–1520. doi: 10.1016/0968-0896(96)00144-7. [DOI] [PubMed] [Google Scholar]
- 106.Tulsi NS, Downey AM, Cairo CW. Bioorg Med Chem. 2010;18:8679–8686. doi: 10.1016/j.bmc.2010.09.040. [DOI] [PubMed] [Google Scholar]
- 107.Kumar S, Zhou B, Liang F, Wang WQ, Huang Z, Zhang ZY. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7943–7948. doi: 10.1073/pnas.0402323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kumar S, Zhou B, Liang F, Yang H, Wang WQ, Zhang ZY. Journal of Proteome Research. 2006;5:1898–1905. doi: 10.1021/pr050449x. [DOI] [PubMed] [Google Scholar]
- 109.Wang QP, Dechert U, Jirik F, Withers SG. Biochemical and biophysical research communications. 1994;200:577–583. doi: 10.1006/bbrc.1994.1487. [DOI] [PubMed] [Google Scholar]
- 110.Kalesh KA, Tan LP, Lu K, Gao L, Wang J, Yao SQ. Chem Commun (Camb) 2010;46:589–591. doi: 10.1039/b919744c. [DOI] [PubMed] [Google Scholar]
- 111.Lo LC, Pang TL, Kuo CH, Chiang YL, Wang HY, Lin JJ. Journal of Proteome Research. 2002;1:35–40. doi: 10.1021/pr015506a. [DOI] [PubMed] [Google Scholar]
- 112.Myers J, Widlanski T. Science. 1993;262:1451–1453. doi: 10.1126/science.8248785. [DOI] [PubMed] [Google Scholar]
- 113.Born TL, Myers JK, Widlanski TS, Rusnak F. Journal of Biological Chemistry. 1995;270:25651–25655. doi: 10.1074/jbc.270.43.25651. [DOI] [PubMed] [Google Scholar]
- 114.Cho H, Park J, Yang D, Ham SW. Bulletin of the Korean Chemical Society. 2000;21:515–517. [Google Scholar]
- 115.Pot DA, Dixon JE. The Journal of biological chemistry. 1992;267:140–143. [PubMed] [Google Scholar]
- 116.Pot DA, Woodford TA, Remboutsika E, Haun RS, Dixon JE. Journal of Biological Chemistry. 1991;266:19688–19696. [PubMed] [Google Scholar]
- 117.Arabaci G, Guo XC, Beebe KD, Coggeshall KM, Pei D. Journal of the American Chemical Society. 1999;121:5085–5086. [Google Scholar]
- 118.Arabaci G, Yi T, Fu H, Porter ME, Beebe KD, Pei D. Bioorganic & Medicinal Chemistry Letters. 2002;12:3047–3050. doi: 10.1016/s0960-894x(02)00681-9. [DOI] [PubMed] [Google Scholar]
- 119.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J. Nat Chem Biol. 2015;11:525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chalker JM, Bernardes GJ, Lin YA, Davis BG. Chem Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- 122.Seiner DR, LaButti JN, Gates KS. Chemical research in toxicology. 2007;20:1315–1320. doi: 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu S, Zhou B, Yang H, He Y, Jiang ZX, Kumar S, Wu L, Zhang ZY. Journal of the American Chemical Society. 2008;130:8251–8260. doi: 10.1021/ja711125p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ahmed VF, Bottini N, Barrios AM. ChemMedChem. 2014;9:296–299. doi: 10.1002/cmdc.201300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Krishnan N, Bencze G, Cohen P, Tonks NK. The FEBS journal. 2013;280:2830–2841. doi: 10.1111/febs.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jiang CS, Liang LF, Guo YW. Acta Pharmacol Sin. 2012;33:1217–1245. doi: 10.1038/aps.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carr G, Berrue F, Klaiklay S, Pelletier I, Landry M, Kerr RG. Methods. 2014;65:229–238. doi: 10.1016/j.ymeth.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 128.Bialy L, Waldmann H. Angewandte Chemie International Edition. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 129.Liu S, Yu Z, Yu X, Huang SX, Luo Y, Wu L, Shen W, Yang Z, Wang L, Gunawan AM, Chan RJ, Shen B, Zhang ZY. Chem Biol. 2011;18:101–110. doi: 10.1016/j.chembiol.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ueda K, Usui T, Nakayama H, Ueki M, Takio K, Ubukata M, Osada H. FEBS letters. 2002;525:48–52. doi: 10.1016/s0014-5793(02)03065-x. [DOI] [PubMed] [Google Scholar]
- 131.Nutter LM, Cheng AL, Hung HL, Hsieh RK, Ngo EO, Liu TW. Biochem Pharmacol. 1991;41:1283–1292. doi: 10.1016/0006-2952(91)90099-q. [DOI] [PubMed] [Google Scholar]
- 132.Ham SW, Park HJ, Lim DH. Bioorganic Chemistry. 1997;25:33–36. [Google Scholar]
- 133.Ham SW, Park J, Lee SJ, Kim W, Kang K, Choi KH. Bioorganic & Medicinal Chemistry Letters. 1998;8:2507–2510. doi: 10.1016/s0960-894x(98)00411-9. [DOI] [PubMed] [Google Scholar]
- 134.Park J, Pei DH. Biochemistry. 2004;43:15014–15021. doi: 10.1021/bi0486233. [DOI] [PubMed] [Google Scholar]
- 135.Sarubbi E, Seneci PF, Angelastro MR, Peet NP, Denaro M, Islam K. FEBS Lett. 1993;319:253–256. doi: 10.1016/0014-5793(93)80557-b. [DOI] [PubMed] [Google Scholar]
- 136.Schoenwaelder SM, Burridge K. Journal of Biological Chemistry. 1999;274:14359–14367. doi: 10.1074/jbc.274.20.14359. [DOI] [PubMed] [Google Scholar]
- 137.Schoenwaelder SM, Petch LA, Williamson D, Shen R, Feng GS, Burridge K. Curr Biol. 2000;10:1523–1526. doi: 10.1016/s0960-9822(00)00831-9. [DOI] [PubMed] [Google Scholar]
- 138.Fu H, Park J, Pei D. Biochemistry. 2002;41:10700–10709. doi: 10.1021/bi0258748. [DOI] [PubMed] [Google Scholar]
- 139.Yuvaniyama J, Denu JM, Dixon JE, Saper MA. Science. 1996;272:1328–1331. doi: 10.1126/science.272.5266.1328. [DOI] [PubMed] [Google Scholar]
- 140.Moran EJ, Sarshar S, Cargill JF, Shahbaz MM, Lio A, Mjalli AMM, Armstrong RW. Journal of the American Chemical Society. 1995;117:10787–10788. [Google Scholar]
- 141.Shim YS, Kim KC, Lee KA, Shrestha S, Lee KH, Kim CK, Cho HJ. Bioorganic & Medicinal Chemistry. 2005;13:1325–1332. doi: 10.1016/j.bmc.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 142.Gerhauser C. Curr Opin Clin Nutr Metab Care. 2013;16:405–410. doi: 10.1097/MCO.0b013e328362014e. [DOI] [PubMed] [Google Scholar]
- 143.Hecht SS. J Cell Biochem Suppl. 1995;22:195–209. doi: 10.1002/jcb.240590825. [DOI] [PubMed] [Google Scholar]
- 144.Singh SV, Singh K. Carcinogenesis. 2012;33:1833–1842. doi: 10.1093/carcin/bgs216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lewis SM, Li Y, Catalano MJ, Laciak AR, Singh H, Seiner DR, Reilly TJ, Tanner JJ, Gates KS. Bioorg Med Chem Lett. 2015;25:4549–4552. doi: 10.1016/j.bmcl.2015.08.065. [DOI] [PubMed] [Google Scholar]
- 146.Shibata T, Nakahara H, Kita N, Matsubara Y, Han C, Morimitsu Y, Iwamoto N, Kumagai Y, Nishida M, Kurose H, Aoki N, Ojika M, Uchida K. Journal of neurochemistry. 2008;107:1248–1260. doi: 10.1111/j.1471-4159.2008.05686.x. [DOI] [PubMed] [Google Scholar]
- 147.Zhang ZY, Davis JP, Van Etten RL. Biochemistry. 1992;31:1701–1711. doi: 10.1021/bi00121a018. [DOI] [PubMed] [Google Scholar]
- 148.Donghwan Park YSS, Chul Kim Ki, Park Junseo, Yang Dongki, Cho Hyeongjin. Journal of the Korean Chemical Society. 2002;46:296–300. [Google Scholar]
- 149.Dana D, Das TK, Kumar I, Davalos AR, Mark KJ, Ramai D, Chang EJ, Talele TT, Kumar S. Chemical biology & drug design. 2012;80:489–499. doi: 10.1111/j.1747-0285.2012.01437.x. [DOI] [PubMed] [Google Scholar]
- 150.Sarmiento M, Zhao Y, Gordon SJ, Zhang ZY. J Biol Chem. 1998;273:26368–26374. doi: 10.1074/jbc.273.41.26368. [DOI] [PubMed] [Google Scholar]
- 151.Punthasee P, Laciak AR, Cummings AH, Ruddraraju KV, Lewis SM, Hillebrand R, Singh H, Tanner JJ, Gates KS. Biochemistry. 2017;56:2051–2060. doi: 10.1021/acs.biochem.7b00151. [DOI] [PubMed] [Google Scholar]
- 152.Hansen SK, Cancilla MT, Shiau TP, Kung J, Chen T, Erlanson DA. Biochemistry. 2005;44:7704–7712. doi: 10.1021/bi047417s. [DOI] [PubMed] [Google Scholar]
- 153.Iwamoto N, Sumi D, Ishii T, Uchida K, Cho AK, Froines JR, Kumagai Y. The Journal of biological chemistry. 2007;282:33396–33404. doi: 10.1074/jbc.M705224200. [DOI] [PubMed] [Google Scholar]
- 154.Xu J, Kurup P, Baguley TD, Foscue E, Ellman JA, Nairn AC, Lombroso PJ. Cell Mol Life Sci. 2016;73:1503–1514. doi: 10.1007/s00018-015-2057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stuhr-Hansen N, Ebert B, Krogsgaard-Larsen P, Kehler J. Organic Letters. 2000;2:7–9. doi: 10.1021/ol991093w. [DOI] [PubMed] [Google Scholar]
- 156.Rye CS, Baell JB. Curr Med Chem. 2005;12:3127–3141. doi: 10.2174/092986705774933452. [DOI] [PubMed] [Google Scholar]
- 157.Elliott TS, Slowey A, Ye Y, Conway SJ. MedChemComm. 2012;3:735–751. [Google Scholar]
- 158.Abdo M, Liu S, Zhou B, Walls CD, Wu L, Knapp S, Zhang ZY. J Am Chem Soc. 2008;130:13196–13197. doi: 10.1021/ja804489m. [DOI] [PubMed] [Google Scholar]
- 159.Besse D, Siedler F, Diercks T, Kessler H, Moroder L. Angewandte Chemie International Edition in English. 1997;36:883–885. [Google Scholar]
- 160.Piovan L, Wu L, Zhang ZY, Andrade LH. Org Biomol Chem. 2011;9:1347–1351. doi: 10.1039/c0ob01050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim JH, Cho H, Ryu SE, Choi MU. Arch Biochem Biophys. 2000;382:72–80. doi: 10.1006/abbi.2000.1996. [DOI] [PubMed] [Google Scholar]
- 162.Karver MR, Krishnamurthy D, Bottini N, Barrios AM. Journal of Inorganic Biochemistry. 2010;104:268–273. doi: 10.1016/j.jinorgbio.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Krishnamurthy D, Karver MR, Fiorillo E, Orrú V, Stanford SM, Bottini N, Barrios AM. Journal of Medicinal Chemistry. 2008;51:4790–4795. doi: 10.1021/jm800101w. [DOI] [PubMed] [Google Scholar]
- 164.Karver MR, Krishnamurthy D, Kulkarni RA, Bottini N, Barrios AM. Journal of Medicinal Chemistry. 2009;52:6912–6918. doi: 10.1021/jm901220m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chio CM, Yu X, Bishop AC. Bioorg Med Chem. 2015;23:2828–2838. doi: 10.1016/j.bmc.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang XY, Bishop AC. Journal of the American Chemical Society. 2007;129:3812–3813. doi: 10.1021/ja069098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chio CM, Lim CS, Bishop AC. Biochemistry. 2015;54:497–504. doi: 10.1021/bi5013595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kensler TW, Groopman JD, Sutter TR, Curphey TJ, Roebuck BD. Chem Res Toxicol. 1999;12:113–126. doi: 10.1021/tx980185b. [DOI] [PubMed] [Google Scholar]
- 169.Maxuitenko YY, Libby AH, Joyner HH, Curphey TJ, MacMillan DL, Kensler TW, Roebuck BD. Carcinogenesis. 1998;19:1609–1615. doi: 10.1093/carcin/19.9.1609. [DOI] [PubMed] [Google Scholar]
- 170.Bhattacharyya S, Zhou H, Seiner DR, Gates KS. Bioorg Med Chem. 2010;18:5945–5949. doi: 10.1016/j.bmc.2010.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gross S, Knebel A, Tenev T, Neininger A, Gaestel M, Herrlich P, Bohmer FD. Journal of Biological Chemistry. 1999;274:26378–26386. doi: 10.1074/jbc.274.37.26378. [DOI] [PubMed] [Google Scholar]
- 172.Leonard SE, Garcia FJ, Goodsell DS, Carroll KS. Angewandte Chemie. 2011;50:4423–4427. doi: 10.1002/anie.201007871. [DOI] [PubMed] [Google Scholar]
- 173.Seo YH, Carroll KS. Bioorganic & Medicinal Chemistry Letters. 2009;19:356–359. doi: 10.1016/j.bmcl.2008.11.073. [DOI] [PubMed] [Google Scholar]
- 174.Reddie KG, Seo YH, Muse WB, Iii, Leonard SE, Carroll KS. Molecular bioSystems. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ruddraraju KV, Hillebrand R, Barnes CL, Gates KS. Acta Crystallogr E Crystallogr Commun. 2015;71:741–743. doi: 10.1107/S2056989015010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ruddraraju KV, Parsons ZD, Llufrio EM, Frost NL, Gates KS. J Org Chem. 2015;80:12015–12026. doi: 10.1021/acs.joc.5b01949. [DOI] [PubMed] [Google Scholar]
- 177.Shiau TP, Erlanson DA, Gordon EM. Org Lett. 2006;8:5697–5699. doi: 10.1021/ol062077j. [DOI] [PubMed] [Google Scholar]
- 178.Parsons ZD, Ruddraraju KV, Santo N, Gates KS. Bioorg Med Chem. 2016;24:2631–2640. doi: 10.1016/j.bmc.2016.03.054. [DOI] [PubMed] [Google Scholar]
- 179.Smith AJT, Zhang X, Leach AG, Houk KN. Journal of medicinal chemistry. 2009;52:225–233. doi: 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Baker BR, Patel RP. Journal of Pharmaceutical Sciences. 1964;53:714–720. doi: 10.1002/jps.2600530703. [DOI] [PubMed] [Google Scholar]
- 182.Ouyang X, Zhou S, Su CT, Ge Z, Li R, Kwoh CK. J Comput Chem. 2013;34:326–336. doi: 10.1002/jcc.23136. [DOI] [PubMed] [Google Scholar]
- 183.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, Murray CW. J Med Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 184.Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O. J Chem Inf Model. 2014;54:1941–1950. doi: 10.1021/ci500175r. [DOI] [PubMed] [Google Scholar]
- 185.Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Expert Opin Drug Discov. 2010;5:597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 187.Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, Harder E. J Chem Inf Model. 2014;54:1932–1940. doi: 10.1021/ci500118s. [DOI] [PubMed] [Google Scholar]
- 188.Kumalo HM, Bhakat S, Soliman ME. Molecules. 2015;20:1984–2000. doi: 10.3390/molecules20021984. [DOI] [PMC free article] [PubMed] [Google Scholar]