

Abstract
One of the most intriguing and important aspects of biological supramolecular materials is its ability to adapt macroscopic properties in response to environmental cues for controlling cellular processes. Recently, bulk matrix stiffness, in particular, stress sensitivity, has been established as a key mechanical cue in cellular function and development. However, stress-stiffening capacity and the ability to control and exploit this key characteristic is relatively new to the field of biomimetic materials. In this work, DNA-responsive hydrogels, composed of semiflexible PIC polymers equipped with DNA cross-linkers, were engineered to create mimics of natural biopolymer networks that capture these essential elastic properties and can be controlled by external stimuli. We show that the elastic properties are governed by the molecular structure of the cross-linker, which can be readily varied providing access to a broad range of highly tunable soft hydrogels with diverse stress-stiffening regimes. By using cross-linkers based on DNA nanoswitches, responsive to pH or ligands, internal control elements of mechanical properties are implemented that allow for dynamic control of elastic properties with high specificity. The work broadens the current knowledge necessary for the development of user defined biomimetic materials with stress stiffening capacity.
Introduction
Nature has developed sophisticated biological materials that are highly sensitive to various chemical and physical cues for tuning biological processes.1 One such material is the extracellular matrix (ECM), the three-dimensional (3D) polymeric hydrogel network that surrounds cells and translates environmental cues to intracellular biochemical processes. Because the mechanical properties of the ECM are highly correlated with cellular development, behavior and disease, the development of artificial hydrogels that mimic the ECM and its associated mechanical properties has attracted considerable recent interest.2 Hydrogels composed of synthetic polymers are often preferred over hydrogels, reconstituted from natural sources due to their controllable and reproducible chemical and physical properties.3 However, in contrast to natural hydrogels, synthetic hydrogels lack one unique important property: the ability to stress stiffen, that is, to become stiffer when deformed. This nonlinear behavior prevents large deformations, protects cells from rupture at high stress and is thought to play a major role in cell differentiation and migration.4 For migration, spreading, and differentiation, cells directly interact with the ECM by pulling and pushing on it, which besides nonlinear elastics also calls for continuous adaptation of the matrix topology mediated by the sensitive interplay between different types of cross-linkers, in which the onset of stress stiffening plays a major role as well.5 We recently developed a synthetic hydrogel, composed of networks of bundled polyisocyanopeptide (PIC) polymers functionalized with oligo(ethylene glycol) tails that stress-stiffens in the same biological relevant regime as natural biopolymer networks.6 Using these biomimetic hydrogels we were able to show that not only the stress-stiffening capacity but also the onset of stress stiffening is important for controlling stem cell fate in 3D biomimetic scaffolds.7 To extend the versatility and controllability of this synthetic hydrogel material we incorporated functional DNA elements to develop a DNA-responsive hydrogel with tunable stiffness by conjugating short oligonucleotides that can be cross-linked using bridging DNA oligonucleotides.8 The DNA-cross-linked PIC hydrogel network forms a stable biomimetic hydrogel with mechanical properties similar to the PIC and natural hydrogels, including the stress-stiffening capacity.
Typically, a hydrogel with biologically relevant stiffness is composed of either bundled semiflexible polymer networks (e.g., collagen, fibrin, PIC) or densely cross-linked flexible polymers (e.g., polyethylene glycol, hyaluronic acid). The PIC polymers in the DNA-responsive PIC hydrogels do not form tight bundles; therefore, stress stiffening of the DNA-PIC hydrogels appears to result from a combination of the elastic properties and semiflexibility of both the PIC polymers and DNA cross-linkers. In this work, we further investigate the influence of cross-linker flexibility with the aim to capture the interplay between the polymer and cross-linker stiffness and expand our DNA-functionalized PIC hydrogel toolbox with dynamic functionalities that are responsive to pH or ligand binding and act as internal control elements. We engineered DNA cross-linked PIC hydrogels with DNA cross-linkers of different length and stiffness and incorporated DNA nanoswitches that can be controlled by pH or ligand binding to selectively tune the hydrogel stiffness and particularly the onset of stress stiffening. The functional modules of the DNA nanoswitches are based on the contraction and relaxation of a pH-responsive DNA i-motif9 and triple helix10 and a ligand-responsive thrombin aptamer11 that act as molecular switches between tight and relaxed states of the cross-linker. We demonstrate that the molecular structure and flexibility of the DNA cross-linker is critically important for the elastic properties and stress-stiffening profile. Thus, the DNA-responsive hydrogel serves as a platform for the design of biomimetic soft hydrogels with controllable elastic properties, which can be readily tuned by proper choice of the DNA cross-linker and interacting agent.
Experimental Section
Synthesis of Azide-Functionalized Polyisocyanopeptides
Azide-appended polyisocyanopeptides functionalized with tetra ethylene glycol side chains were synthesized as described previously.8,12 Functional azide monomer (N3) and nonfunctional methoxy monomer (OMe) were mixed in a 1:30 ratio in freshly distilled toluene, after which Ni(ClO4)2·6H2O catalyst (dissolved in 9/1 toluene/ethanol) was added. The mixture was stirred for 2–3 days before it was precipitated 3-fold in di-isopropyl ether. A catalyst/monomer ratio of 1/10000 was used to obtain polymers with a length of approximately 335 nm.
Functionalization of DNA with DBCO-PEG4-NHS
DNA oligonucleotides (Table 1) were obtained from Integrated DNA Technologies in desalted-only grade and purified in house using preparative denaturing polyacrylamide gel electrophoresis (PAGE; 20%).8 The NH2-modified DNA (500 μM) was reacted with the heterobifunctional reagent DBCO-PEG4-NHS (1.25 mM, Jena Bioscience) in 50 mM borate buffer pH 8.5 at 25 °C for 2 h. The product was purified using Illustra Microspin G-25 columns (GE Healthcare). The final yield of DNA-DBCO was determined by UV absorption at 260 nm (DNA/DBCO) or 309 nm (DBCO).
Table 1. DNA Sequences Used in This Study.
| single-stranded DNA | DNA sequence (5′–3′) |
|---|---|
| DNA A | NH2-TTT TTT TCA ACA TCA GT |
| DNA B | CTG ATA AGC TAT TTT TT-NH2 |
| DNA C | TAG CTT ATC AGA CTG ATG TTG A |
| DNA AB | NH2-TTT TTT TCA ACA TCA GT CTG ATA AGC TAT TTT TT-NH2 |
| DNA LC (i-motif) | TAG CTT ATC AG CCC CT AAC CCC TAA CCC CTA ACC CC A CTG ATG TTG A |
| DNA LC′ (LC complement) | GGG GTT AGG GGT TAG GGG TTA GGG G |
| DNA THA (triple helix) | 5′-NH2-TTT TTT CCC CTC TCC TCC TTC TTT TCT CTC TAT TAT CTC TCT TTT CTT |
| DNA THB (triple helix) | CCT CCT CTC CCC TTT TTT-NH2-3′ |
| DNA THC (triple helix) | GGG GAG AGG AGG AAG AAA AGA GAG A |
| DNA TB (thrombin aptamer) | TAG CTT ATC AGT GGT TGG TGT GGT TGG TTA CTG ATG TTG A |
Synthesis of the DNA-Functionalized PIC Polymer
Stock solutions of the PIC polymer were prepared by dissolving 5 mg mL–1 of azide-functionalized PIC polymer (13.9 mM monomer) in Milli-Q water. DBCO-functionalized DNAs were mixed with the azide-functionalized PIC polymer in a 1:50 ratio (125 μM DNA-DBCO/6.25 mM PIC monomer) in phosphate buffered saline (PBS, pH 7.4) and incubated at 25 °C for 12 h. The yield of the conjugation reaction was determined by monitoring the absorbance of DBCO before and after the reaction (Figure S1). As the yield of the reaction was ∼90–95%, the polymer conjugate was used without further purification.
Hydrogel Preparation
PIC-DNA A and PIC-DNA B or PIC-DNA THA and PIC-DNA THB conjugates were mixed in 1:1 ratio at 4 °C in PBS, 10 mM MgCl2, pH 7.4. To induce gel formation, complementary DNA oligonucleotides were added in desired concentrations. The total polymer concentration was 2 mg mL–1 for all experiments corresponding to ∼55 μM of the respective conjugated DNAs. For the thrombin mediated hydrogel 0.5 equiv of cross-linker DNA TB was mixed with PIC-DNA A and PIC-DNA B at 4 °C in PBS, 10 mM MgCl2, pH 7.4, to form a precross-linked PIC mixture, and subsequently mixed with thrombin. Thrombin was obtained as a lyophilized powder from human plasma (Sigma-Aldrich) and dissolved in 30 μL of PBS at a concentration of 25 μM prior to addition to the precross-linked PIC mixture.
Rheology Measurements
Time-sweep oscillatory rheology measurements were carried out with a stress-controlled rheometer (Discovery HR-1, TA Instruments) using parallel plate geometry (40 mm diameter; aluminum) and a 200 μm gap. All measurements were performed in a temperature-controlled environment with oil sealing to minimize sample evaporation. Sample volumes were 300 μL. The lag time between hydrogel preparation and start of the measurements was approximately 5 min. Linear rheology measurements were performed at fixed frequency (1 Hz) and strain (2%). For the variable temperature rheology experiments, the temperature was ramped at 1 °C min–1. The nonlinear stress-stiffening profiles were investigated using a previously described prestress protocol.5b A small oscillatory stress δσ was superposed on a constant prestress and the resultant oscillatory strain (δγ) was recorded in a frequency sweep experiment (0.1–10.0 Hz). The differential modulus K′ (δσ/δγ) was extracted at a specific applied prestress (σ) and frequency (1 Hz). The critical stress is defined as the value where σ increases by at least 10% over its original value.
Circular Dichroism (CD) Measurements
CD spectra were recorded using a JASCO J-810 circular dichroism spectropolarimeter and a quartz cuvette with a path length of 1 mm (PerkinElmer). All samples were prepared in PBS, 10 mM MgCl2. The PIC concentrations were adjusted to 0.2 mg mL –1. The optical chamber of the CD spectrometer was deoxygenated with dry nitrogen for 30 min before use and kept under nitrogen atmosphere during experiments. For each spectrum, five scans were accumulated and automatically averaged.
Results and Discussion
Effect of Cross-Linker Flexibility
We prepared the DNA-functionalized PIC hydrogels using a previously published protocol8 (Figure 1). Briefly, azide-functionalized tetraethylene glycol PIC polymers are used to conjugate two different single-stranded DNAs (ssDNA A and ssDNA B) that can be cross-linked using different complementary ssDNAs (Table 1). The DNA density on the PIC polymers is tuned by the initial azide and DNA concentrations. Using a 1:30 azide-appended monomer to non-azide-appended monomer ratio, the average distance of the azide groups on the PIC polymers is 3.5 nm and using an efficiency of ∼90% azide-DBCO coupling (Figure S1) the statistical average distance between the ssDNAs becomes 6 nm. The average length of the synthesized PIC polymers was 335 nm with thus approximately 50 ssDNA molecules conjugated to a single PIC polymer.
Figure 1.

Preparation of DNA responsive PIC hydrogels. The azide-functionalized PIC polymer is conjugated with ssDNA using DBCO and strain-promoted click chemistry. Two separate batches of PIC polymers conjugated with DNA A or PIC-DNA B are mixed together with complementary cross-linker DNAs to form the hydrogel.
The thermoresponsive PIC polymer forms hydrogels by hydrophobic interaction of the tetra-ethylene glycol moieties with a lower critical solution temperature (LCST) of 37 °C in PBS.6 DNA-cross-linked PIC hydrogels are therefore prepared by mixing PIC-DNA A and PIC-DNA B with stoichiometric amounts of complementary DNA below the LCST of PIC. Mixing in DNA C generates a 22 base pair (bp) double-stranded (ds) DNA cross-link leaving six single-stranded (ss) nucleotides on either end. Rheological analysis shows that at 30 °C the hydrogel forms within 1 h (Figure 2a) and remains stable as shown by a broad range frequency sweep (Figure S2).8 The gel stiffness of the hydrogel is measured using a well-established prestress protocol where the hydrogel is subjected to a small oscillatory stress (δσ), resulting in a small oscillatory strain (δγ) at a constant prestress (σ). The gel stiffness, described by the differential modulus K′ = δσ/δγ shows a linear regime at low σ where the stiffness is constant and K′ equals the plateau modulus G0 = 45 Pa (Figure 2b and Table 2). Stress stiffening results in a nonlinear regime at higher stress, setting in at a critical stress σc = 3.5 Pa, which is a measure of the sensitivity to applied stress. The hydrogel can withstand a shear stress σmax up to 40 Pa beyond which the hydrogel network breaks down. The stiffening index m of the nonlinear regime, which is a measure of the response to applied stress, is 0.8 (K′ ∼ σ0.8). This stiffening index is lower than the value reported and theoretically predicted for the bundled PIC hydrogels and biological semiflexible bundled networks (m = 3/2, F-actin, intermediate filaments), but falls in the range of collagen and fibrin networks and F-actin cross-linked with weak cross-linkers.13 The lower stiffening index can be explained by the extensibility of the DNA-cross-linked hydrogel in which many PIC polymers interact through bridging DNA cross-links and indicates interplay between elastic properties of the PIC polymers and shorter dsDNA cross-links, which both resist strain in the coupled network.
Figure 2.

Rheological analysis of PIC-DNA hydrogels with 0.9 equiv DNA cross-linker at 30 °C in PBS, 10 × 10–3 M MgCl2, pH 7.4. (a) Time evolution of the storage modulus (G′) after addition of cross-linker DNA C (green), LC (blue), or LC + LC′ (orange) to DNA A and DNA B functionalized PIC polymers. (b) Strain stiffening profile of the PIC-DNA hydrogel with cross-linker DNA C. The gel stiffness is represented as the differential modulus K′(δσ/δγ) as a function of stress σ. At low stress, K′ equals G0, the plateau modulus. The nonlinear regime sets in beyond a critical stress σc, where K′ increases with applied stress and finally ruptures at maximum stress σmax. (c) Normalized differential modulus (K′/G0) as a function of stress σ of PIC-DNA hydrogels cross-linked with DNA C (green), LC (blue), or LC + LC′ (orange). (d) K′/G0 as a function of σ of PIC-DNA hydrogels, covalently cross-linked with DNA AB (black) or DNA AB + C (orange).
Table 2. Summary of Mechanical Properties.
| DNA construct |
rheological properties |
|||
|---|---|---|---|---|
| cross-linker sequence | contour length (nm) | G0 (Pa) | σc (Pa) | σmax (Pa) |
| Noncovalent Cross-Linker | ||||
| ds-DNA C | 14 | 45 ± 1.5 | 3.5 ± 0.25 | 40 |
| ss-DNA LC (pH 7.4) | 26 | 34.5 ± 3.7 | 0.65 ± 0.05 | 18 ± 1.15 |
| ds-DNA LC | 19 | 40 ± 2.8 | 2.5 ± 0.25 | 22 |
| Covalent Cross-Linker | ||||
| ss-DNA AB | 16 | 8 | 2.3 | >200 |
| ds-DNA AB/C | 14 | 8.5 | 2.4 | >200 |
| pH Responsive i-Motif | ||||
| ss-DNA LC (pH 5.2) | 16 | 100 ± 6 | 4 ± 0.5 | 30 |
| pH Responsive Triple Helix | ||||
| DNA THC (pH 7.4) | 29 | 7.5 ± 0.5 | 0.65 ± 0.05 | 5.5 ± 0.75 |
| DNA THC (pH 5.2) | 5 | 16 ± 2.7 | 2 ± 0.25 | 5.5 ± 0.75 |
| Thrombin Responsive Aptamer | ||||
| DNA TB (without thrombin) | 23 | 11 | 0.6 | 3 |
| DNA TB (with thrombin) | 16 | 19 | 1.2 | 3 |
To further probe the effect of DNA cross-links with different length and flexibility, a hydrogel was prepared using cross-linker DNA LC that hybridizes with PIC-DNA A and PIC-DNA B using the same base pair interactions as cross-linker DNA C, but contains an additional 25 nt ssDNA in-between. With near stoichiometric amounts of cross-linker LC the hydrogel shows a somewhat lower stiffness, G0 = 35 Pa (Figure 2b). The onset of stiffening is, however, much lower (σc = 0.65 Pa), indicating higher sensitivity to stress. Also, the stiffness profile is markedly different, showing multiple stress regimes (Figure 2c). Starting with the common linear regime, the stiffness initially rises at the critical stress, but is followed by a second linear regime, and finally ends in a second stiffening regime setting in at ∼1.5 Pa. This complex behavior can be explained by the different entropic elasticities of the ssDNA and dsDNA domains in the cross-linker that contribute to the total elasticity of the network as they are extended under applied stress. Introducing a central ssDNA part with a persistence length of 1 nm provides additional softer extensional stiffness to the PIC polymers and dsDNA linkers, which have persistence lengths of 10 and 50 nm, respectively. Thus, we hypothesize that the first linear regime represents entropic elasticity from unbending of the network, typically observed for natural hydrogel networks,5,13 while the second linear regime represents initial entropic stretching of the soft ssDNA, followed by further stretching of extended DNA linkers and PIC polymers in the second nonlinear regime. Similar profiles were recently observed for fibrin fibers, where the multidomain stiffening response was shown to originate from the hierarchal architecture of the fibers.13 Despite sharing the dsDNA binding domains A and B, hydrogels cross-linked with DNA LC rupture at lower stress σmax = 18 Pa, which possibly results from separating the two A and B double-stranded domains by the single-stranded domain. Using the concept of extensional stiffness mediated by DNA cross-linkers as starting point, it should be possible to control the stiffness of the hydrogel network by changing elastic properties of the DNA cross-linker. Indeed, adding complementary ssDNA LC′ to the hydrogel cross-linked with LC, which results in a dsDNA linker with higher persistence length and elasticity, restores the single linear and nonlinear regime with higher G0 and critical stress σc = 2.5 Pa (Figure 2c). Together, these results indicate that the extensional stiffness of the DNA cross-linkers has a major impact on the mechanical properties of the DNA-PIC hydrogel, which can be controlled by changing the molecular structure of the cross-linkers.
Effect of Covalent Cross-Linkers
To further investigate the influence of DNA cross-linker architecture and stiffness, covalent DNA cross-links were introduced. Single 34 nucleotide ssDNAs harboring both the DNA A and DNA B sequences in tandem (DNA AB) with both 5′ and 3′ terminal amino groups functionalized with DBCO were clicked onto the PIC network at 60 °C, that is, above the gelation temperature of the PIC polymers,5a which allows for cross-linking of the polymers in the bundled state. The presence of a ssDNA cross-linked network was confirmed by rheology measurements after cooling the hydrogel down from 60 to 30 °C, showing formation of a stable hydrogel at 30 °C (Figure S3). Subjecting the network covalently cross-linked with ssDNA to the prestress protocol reveals a single linear regime with G0 = 8 Pa and a single nonlinear regime with high response to applied stress (m = 1.5–1.7) that sets in at a critical stress σc of 2.3 Pa (Figure 2d). These values are closer to values reported for PIC polymers, indicating dominant elasticity of the PIC polymers due to higher bundling. By incorporating a covalent DNA cross-linker, however, the hydrogel network is more resistant to stress and can withstand an applied stress over 200 Pa before rupture occurs. We also prepared a hydrogel with a dsDNA cross-linker composed of covalent linker DNA AB annealed to complementary ssDNA C using preannealed DBCO-dsDNA AB/C-DBCO. Surprisingly, annealing complementary DNA C to the covalent linker DNA AB has no effect on the elastic properties as shown by similar G0, σc, and overall curvature (Figure 2d).
Apparently, in the covalently cross-linked system a single-stranded or double-stranded character of the DNA cross-linker does not differentiate the mechanical properties of the hydrogel. This suggests that by cross-linking the PIC polymers in the bundled state, the PIC polymers remain bundled and dominate the elastic properties of the hydrogel. The elastic properties of the covalently cross-linked polymer networks are, however, different from the noncovalently cross-linked networks, which overall adds to the notion that the macroscopic elastic properties are highly sensitive to the molecular structure of the DNA cross-linkers.
pH-Controlled Stress Sensitivity
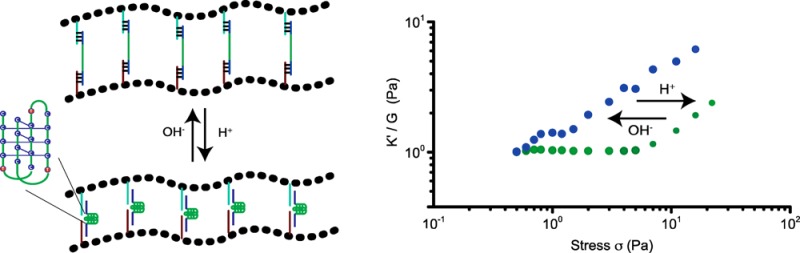
So far we engineered hydrogels, using DNA cross-linkers that can be conveniently varied to tune the mechanical properties of the hydrogel by changing DNA structure and stoichiometry, but harbor no further functionalities that can act as internal control elements. Thus, the next step in adding complexity and functionality to our hydrogel toolbox was to introduce DNA elements that are responsive to external stimuli other than complementary DNAs and switch conformation upon binding to the external stimulant. First, we introduced a pH sensitive DNA cross-linker that can switch conformation by incorporating DNA i-motifs. DNA i-motifs are four-stranded C-rich DNA structures that can self-assemble by forming intercalated, cytosine–cytosine+ base pairs under acidic conditions (pH 5.2), which dissociate into a random coil at pH 7.4 (Figure 3a).3d,9 We bridged PIC-DNA A and PIC-DNA B using the ssDNA i-motif sequence, DNA LC, introduced earlier. Formation of the pH-induced i-motif structure free in solution and incorporated in PIC hydrogel linkers was supported by circular dichroism (CD) measurements (Figures 3b and S4). At pH 5.2, the CD spectrum shows a large positive signal at 288 nm and a small negative signal at ∼260 nm, which is typical for the noncanonical i-motif structure. At pH 7.4 the CD spectrum is shifted with a smaller positive signal at ∼265 nm and a slightly smaller negative signal at ∼250 nm, which is characteristic for a B-form DNA double helix in accordance with disassembly of the i-motif.14 The mechanical properties of the hydrogel network with i-motif DNA cross-linkers are clearly sensitive to pH. As mentioned above, at pH 7.4 the cross-linked polymer network forms a stable hydrogel (G0 = 35 Pa) that starts to stress stiffen at low stress (σc = 0.6 Pa), followed by multiple stress regimes. The hydrogel becomes stiffer by formation of the i-motif upon changing the pH to 5.2. G0 increases more than 2-fold to 100 Pa (Figures 3c and S5) and the critical stress is shifted to 4 Pa (Figure 3d). Folding of the i-motif will reduce the end-to-end distance of the internal DNA sequence to ∼2 nm and thus modify the extensional stiffness of the cross-linker. Therefore, the change in mechanical properties can be attributed to internal stress generated by the contracting DNA i-motif sequence leading to a stiffer DNA cross-linker. As observed for adding complementary sequence LC′, changing the unstructured central ssDNA domain of the linker into a structured entity restores the single linear and nonlinear stress regime. The pH effect on the mechanical properties is fully reversible; the hydrogel relaxes back to the weaker hydrogel upon changing the pH back to 7.4.
Figure 3.

pH-dependent rheological analysis of PIC-DNA cross-linked with 0.9 equiv of DNA LC at 30 °C in PBS, 10 × 10–3 M MgCl2. (a) The i-motif (green trace), incorporated in the DNA cross-linker LC, is in an extended state at pH 7.4 and contracts at pH 5.2; (b) CD spectra to characterize the conformation of the i-motif sequence in the PIC-DNA hydrogel at pH 7.4 (blue) and pH 5.2 (green); (c) Time evolution of storage modulus G′ after addition of DNA cross-linker LC at pH 7.4 (blue) and pH 5.2 (green); (d) K′/G0 as a function of σ at pH 7.4 (blue) and pH 5.2 (green).
To further demonstrate the generality and versatility of the design, a different pH-responsive DNA cross-linker was incorporated that can contract upon lowering the pH using a DNA sequence that folds into a triple helix motif by protonation of cytidines at lower pH (Figure 4a). For our design we used a sequence that can form a triple helix consisting of 52% C-G-C and 48% T-A-T base triplets, and switches conformation between random coil and triple helix with a pKa of 6.5.10 Formation of the DNA triple helix in the network is supported by temperature dependent rheological measurements, which reveal a shift in melting temperature from 45 to 52 °C upon lowering the pH from 7.4 to 5.2 resulting from additional thermodynamic stability from Hoogsteen interactions between the pyrimidine strand and major groove of the DNA double helix (Figure S6). Similar to the cross-linker with i-motif sequence, formation of a stable DNA triple helix at pH 5.2 reduces the end-to-end distance of the central portion of the cross-linker to ∼2 nm, thus, contracting the cross-linker between the PIC polymers. At pH 7.4, the cross-linker consists of a 25bp double helix, connected to a 35nt ssDNA and the stiffness profile of the hydrogel shows multiple stress regimes as observed for the hydrogel network bridged by ssDNA cross-linker LC, with low storage modulus (G0 = 7.5 Pa) and critical stress (σc = 0.65 Pa; Figure 4c). Formation of the DNA triplex stabilizes the hydrogel, as indicated by a 2-fold increase of the storage modulus (G0 = 16 Pa) and results in the collapse of the stiffness profile to a single linear and nonlinear domain, with an upward shift of the critical stress to ∼2 Pa. The lower storage modulus at high pH is in accordance with a lower extensional stiffness of the cross-linker by incorporation of the longer single-stranded and double-stranded sequences controlling the mechanical properties of the hydrogel. These results also support the role of the molecular structure of the cross-linker in controlling the mechanical properties of the hydrogel, which can be internally changed using a switchable DNA cross-linker.
Figure 4.

pH-dependent rheological analysis of PIC-DNA cross-linked with 0.9 equiv of DNA cross-linker THC at 30 °C in PBS, 10 × 10–3 M MgCl2. (a) The triple helix motif (green trace) is in a random extended state at pH 7.4 but forms a stable structure at pH 5.2. (b) Time evolution of storage modulus G′ after addition of DNA cross-linker THC at pH 7.4 (blue) and pH 5.2 (green). (c) K′/G0 as a function of σ at pH 7.4 (blue) and pH 5.2 (green).
Ligand Controlled Stress Sensitivity
To illustrate the versatility of our platform in designing tunable responsive hydrogels with internal control elements, we next engineered a ligand-responsive hydrogel by incorporating a DNA aptamer specific for the human α-thrombin protein.11 The DNA aptamer in complex with thrombin forms a stable quadruplex structure with two G-quartets,15 and can thus act as a molecular switch in response to thrombin. Similar to the i-motif cross-linked hydrogels, we designed a cross-linker (DNA TB) by extending the thrombin aptamer sequence with ssDNA sequences to bridge PIC-DNA A and PIC-DNA B. Cross-linking of the PIC DNA A and B polymers with DNA TB results in formation of a stable hydrogel (G0 = 11 Pa), which, comparable to the results obtained with other cross-linkers with internal unstructured ssDNA sequences, starts to stress stiffen at low stress (σc = 0.6 Pa) and shows multiple stress regimes before rupturing at 3 Pa (Figure 5). Mixing α-thrombin (∼2.5 μM) with the cross-linked hydrogel resolves the multiple stress regimes and stabilizes the hydrogel with G0, increasing nearly 1.5× to 19 Pa, and σc increasing to 1.2 Pa. Taken together, all these results demonstrate that the stiffness and critical stress of the DNA-cross-linked PIC hydrogels can be readily tuned by using switchable internal control elements. The low stiffness of the hydrogels combined with the user-defined adaptability of soft DNA interactions can be particularly suited to address cellular response in 3D matrices, for example, using cell culture grown as spheroids.16 The thermosensitive response of the PIC polymers6 induces polymer bundling resulting in hydrogels with higher stiffness. Therefore, using this material a range from a few Pascal up to a few hundred Pascal can be readily achieved to probe the influence of hydrogel stiffness in 3D matrices.
Figure 5.

Thrombin-dependent rheology of PIC-DNA hydrogels with 0.5 equiv of cross-linker TB. (a) Addition of 2.5 μM thrombin induces formation of a stable quadruplex structure with two G-quartets in the cross-linker. (b) Kinetic rheological measurements of formation of the PIC DNA hydrogel after mixing with DNA cross-linker TB in the absence of thrombin (blue) and stabilization of a preformed PIC-DNA A + B + TB hydrogel in the presence of thrombin (green) at 30 °C in PBS, 10 × 10–3 M MgCl2. (c) K′/G0 as a function of σ in the absence (blue) and presence (green) of thrombin.
Conclusions
The nonlinear stress response of biological supramolecular materials is increasingly recognized as an important adaptive regulation element in cellular function. By exploiting the versatility and programmability of DNA building blocks combined with stress-stiffening PIC polymers we have developed a biomimetic DNA responsive hydrogel toolbox with tunable nonlinear mechanical properties that can be controlled by the structure and flexibility of the DNA cross-linkers. For instance, by using pH sensitive DNA i-motif and DNA triplex motifs as internal control elements, the stress response of the hydrogel can be reversibly tuned with a nearly 2-fold increase in G0 and a shift of nearly 2 Pa in critical stress. The platform is a powerful addition to the development of new adaptive materials with tunable mechanical properties and response by inclusion of the large repertoire of designable responsive DNA cross-linkers that can be varied at will. In particular, access to tunable stress stiffening properties will be instrumental for the development of truly biomimetic materials where the interplay between biochemical and mechanical cues can be precisely tuned. This material has potential in biomedical applications such as tissue engineering or drug delivery and can be further designed to develop self-healing materials or actuators.
Acknowledgments
We acknowledge financial support from NanoNext Projects 08B.05 (S.R.D., H.A.H.) and 3D.12 (R.H, A.E.R.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b00964.
Experiments for determining the amount of DNA-DBCO and PIC Polymer conjugation; Temperature and frequency dependent rheology profiles of DNA-functionalized PIC polymers; CD spectra of free DNA i-motif in solution; Additional rheology data of pH responsive hydrogel; Rheology experiments showing specificity of pH and thrombin effects (PDF).
Author Present Address
† Department of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University, Geert Grooteplein 26, 6525 GA Nijmegen, The Netherlands.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Discher D. E.; Janmey P.; Wang Y. L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310 (5751), 1139–43. 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]; b Discher D. E.; Mooney D. J.; Zandstra P. W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324 (5935), 1673–7. 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Baker B. M.; Chen C. S. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125 (13), 3015–24. 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lutolf M. P.; Hubbell J. A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23 (1), 47–55. 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]; b Tibbitt M. W.; Anseth K. S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103 (4), 655–63. 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Appel E. A.; del Barrio J.; Loh X. J.; Scherman O. A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41 (18), 6195–214. 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- a Stowers R. S.; Allen S. C.; Suggs L. J. Dynamic phototuning of 3D hydrogel stiffness. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (7), 1953–8. 10.1073/pnas.1421897112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li C.; Faulkner-Jones A.; Dun A. R.; Jin J.; Chen P.; Xing Y.; Yang Z.; Li Z.; Shu W.; Liu D.; Duncan R. R. Rapid formation of a supramolecular polypeptide-DNA hydrogel for in situ three-dimensional multilayer bioprinting. Angew. Chem., Int. Ed. 2015, 54 (13), 3957–61. 10.1002/anie.201411383. [DOI] [PubMed] [Google Scholar]; c Jin J.; Xing Y.; Xi Y.; Liu X.; Zhou T.; Ma X.; Yang Z.; Wang S.; Liu D. A triggered DNA hydrogel cover to envelop and release single cells. Adv. Mater. 2013, 25 (34), 4714–7. 10.1002/adma.201301175. [DOI] [PubMed] [Google Scholar]; d Cheng E.; Xing Y.; Chen P.; Yang Y.; Sun Y.; Zhou D.; Xu L.; Fan Q.; Liu D. A pH-triggered, fast-responding DNA hydrogel. Angew. Chem., Int. Ed. 2009, 48 (41), 7660–3. 10.1002/anie.200902538. [DOI] [PubMed] [Google Scholar]; e Murakami Y.; Maeda M. DNA-responsive hydrogels that can shrink or swell. Biomacromolecules 2005, 6, 2927–29. 10.1021/bm0504330. [DOI] [PubMed] [Google Scholar]; f Liu J. Oligonucleotide-functionalized hydrogels as stimuli responsive materials and biosensors. Soft Matter 2011, 7, 6757–67. 10.1039/c1sm05284e. [DOI] [Google Scholar]; g Guvendiren M.; Burdick J. A. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat. Commun. 2012, 3, 792. 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- Storm C.; Pastore J. J.; MacKintosh F. C.; Lubensky T. C.; Janmey P. A. Nonlinear elasticity in biological gels. Nature 2005, 435 (7039), 191–4. 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- a Koenderink G. H.; Dogic Z.; Nakamura F.; Bendix P. M.; MacKintosh F. C.; Hartwig J. H.; Stossel T. P.; Weitz D. A. An active biopolymer network controlled by molecular motors. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (36), 15192–7. 10.1073/pnas.0903974106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gardel M. L.; Nakamura F.; Hartwig J. H.; Crocker J. C.; Stossel T. P.; Weitz D. A. Prestressed F-actin networks cross-linked by hinged filamins replicate mechanical properties of cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (6), 1762–7. 10.1073/pnas.0504777103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wagner B.; Tharmann R.; Haase I.; Fischer M.; Bausch A. R. Cytoskeletal polymer networks: the molecular structure of cross-linkers determines macroscopic properties. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (38), 13974–8. 10.1073/pnas.0510190103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yao N. Y.; Becker D. J.; Broedersz C. P.; Depken M.; Mackintosh F. C.; Pollak M. R.; Weitz D. A. Nonlinear viscoelasticity of actin transiently cross-linked with mutant alpha-actinin-4. J. Mol. Biol. 2011, 411 (5), 1062–71. 10.1016/j.jmb.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lieleg O.; Claessens M. M. A. E.; Bausch A. R. Structure and dynamics of cross-linked actin networks. Soft Matter 2010, 6 (2), 218–225. 10.1039/B912163N. [DOI] [Google Scholar]; f Wachsstock D. H.; Schwarz W. H.; Pollard T. D. Cross-linker dynamics determine the mechanical properties of actin gels. Biophys. J. 1994, 66 (3), 801–9. 10.1016/S0006-3495(94)80856-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kouwer P. H.; Koepf M.; Le Sage V. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; Picken S. J.; Nolte R. J.; Mendes E.; Rowan A. E. Responsive biomimetic networks from polyisocyanopeptide hydrogels. Nature 2013, 493 (7434), 651–5. 10.1038/nature11839. [DOI] [PubMed] [Google Scholar]; b Jaspers M.; Dennison M.; Mabesoone M. F.; MacKintosh F. C.; Rowan A. E.; Kouwer P. H. Ultra-responsive soft matter from strain-stiffening hydrogels. Nat. Commun. 2014, 5, 5808. 10.1038/ncomms6808. [DOI] [PMC free article] [PubMed] [Google Scholar]; c van Buul A. M.; Schwartz E.; Brocorens P.; Koepf M.; Beljonne D.; Maan J. C.; Christianen P. C.; Kouwer P. H.; Nolte R. J.; Engelkamp H.; Blank K.; Rowan A. E. Stiffness versus architecture of single helical polyisocyanopeptides. Chemical Science 2013, 4 (6), 2357–63. 10.1039/c3sc50552a. [DOI] [Google Scholar]
- Das R. K.; Gocheva V.; Hammink R.; Zouani O. F.; Rowan A. E. Stress-stiffening-mediated stem-cell commitment switch in soft responsive hydrogels. Nat. Mater. 2015, 15 (3), 318–25. 10.1038/nmat4483. [DOI] [PubMed] [Google Scholar]
- Deshpande S. R.; Hammink R.; Das R. K.; Nelissen F. H. T.; Blank K. G.; Rowan A. E.; Heus H. A. DNA-Responsive Polyisocyanopeptide Hydrogels with Stress-Stiffening Capacity. Adv. Funct. Mater. 2016, 26 (48), 9075–82. 10.1002/adfm.201602461. [DOI] [Google Scholar]
- a Guo W.; Lu C. H.; Qi X. J.; Orbach R.; Fadeev M.; Yang H. H.; Willner I. Switchable bifunctional stimuli-triggered poly-N-isopropylacrylamide/DNA hydrogels. Angew. Chem., Int. Ed. 2014, 53 (38), 10134–8. 10.1002/anie.201405692. [DOI] [PubMed] [Google Scholar]; b Gehring K.; Leroy J. L.; Gueron M. A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature 1993, 363 (6429), 561–5. 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- a Chen Y.; Lee S. H.; Mao C. A DNA nanomachine based on a duplex-triplex transition. Angew. Chem., Int. Ed. 2004, 43 (40), 5335–8. 10.1002/anie.200460789. [DOI] [PubMed] [Google Scholar]; b Chen Y.; Mao C. Reprogramming DNA-directed reactions on the basis of a DNA conformational change. J. Am. Chem. Soc. 2004, 126 (41), 13240–1. 10.1021/ja045718j. [DOI] [PubMed] [Google Scholar]; c Chen Y.; Mao C. pH-induced reversible expansion/contraction of gold nanoparticle aggregates. Small 2008, 4 (12), 2191–4. 10.1002/smll.200800569. [DOI] [PubMed] [Google Scholar]; d Idili A.; Vallée-Bélisle A.; Ricci F. J. Am. Chem. Soc. 2014, 136 (16), 5836–9. 10.1021/ja500619w. [DOI] [PubMed] [Google Scholar]
- Bock L. C.; Griffin L. C.; Latham J. A.; Vermaas E. H.; Toole J. J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355 (6360), 564–6. 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Eksteen-Akeroyd Z. H.; Jacobs M. H.; Hammink H.; Koepf M.; Lambeck A. J. A.; van Hest J. C. M.; Wilson C. J.; Blank K.; Figdor C. G.; Rowan A. E. Therapeutic Nanoworms: Towards Novel Synthetic Dendritic Cells for Immunotherapy. Chemical Science 2013, 4 (5), 4168–74. 10.1039/c3sc51399h. [DOI] [Google Scholar]
- Piechocka I. K.; Jansen K. A.; Broedersz C. P.; Kurniawan N. A.; MacKintosh F. C.; Koenderink G. H. Multi-scale strain-stiffening of semiflexible bundle networks. Soft Matter 2016, 12 (7), 2145–56. 10.1039/C5SM01992C. [DOI] [PubMed] [Google Scholar]
- Kypr J.; Kejnovska I.; Renciuk D.; Vorlickova M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37 (6), 1713–25. 10.1093/nar/gkp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaya R. F.; Schultze P.; Smith F. W.; Roe J. A.; Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (8), 3745–3749. 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang K.; Cai L.-H.; Lan B.; Fredberg J. J. Hidden in the mist no more. Nat. Methods 2016, 13 (2), 124–5. 10.1038/nmeth.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Steinwachs J.; Metzner C.; Skodzek K.; Lang N.; Thievessen I.; Mark C.; Münster S.; Aifantis K. E.; Fabry B. Three-dimensional force microscopy of cells in biopolymer networks. Nat. Methods 2015, 13 (2), 171–6. 10.1038/nmeth.3685. [DOI] [PubMed] [Google Scholar]; c Nam S.; Hu K. H.; Butte M. J.; Chaudhuri O. Strain enhanced stress relaxation impacts nonlinear elasticity in collagen gels. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (20), 5492–97. 10.1073/pnas.1523906113. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Nagelkerke A.; Bussink J.; Sweep F. C. G. J.; Span P. N. Generation of multicellular tumor spheroids of breast cancer cells: how to go three-dimensional. Anal. Biochem. 2013, 437 (1), 17–9. 10.1016/j.ab.2013.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.