

Summary
Antibodies can impact HIV-1 infection in multiple ways including antibody-dependent cellular cytotoxicity (ADCC), a correlate of protection observed in the RV144 vaccine trial. One of the most potent ADCC-inducing epitopes on HIV-1 Env is recognized by the C11 antibody. Here we present the crystal structure, at 2.9 Å resolution, of the C11-like antibody N12-i3, in a quaternary complex with the HIV-1 gp120, a CD4-mimicking peptide M48U1, and an A32-like antibody, N5-i5. Antibody N12-i3 recognizes an epitope centered on the N-terminal “8th-strand” of a critical β-sandwich, which our analysis indicates to be emblematic of a late entry state, after the gp120 detachment. In prior entry states, this sandwich comprises only 7-strands, with the 8th strand instead pairing with a portion of the gp120 C terminus. The conformational gymnastics of HIV-1 gp120 thus includes altered β-strand pairing – possibly to reduce immunogenicity – though nevertheless still recognized by the human immune system.
eTOC Blurb
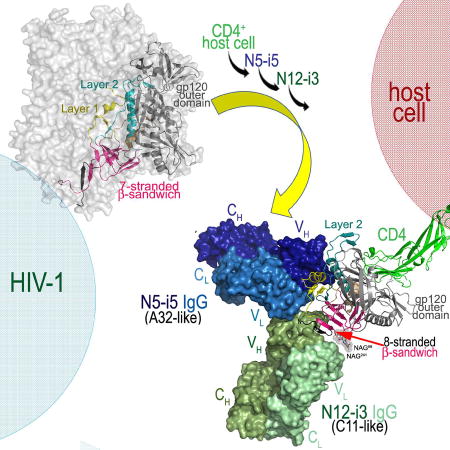
Tolbert at al. describe a crystal structure of Fabs of A32- and C11-like antibody bound to single gp120 subunit. The C11-like antibody, N12-i3 recognizes new 8-stranded β-sandwich structure of gp120 that is formed at the late stage of HIV-1 entry.
Introduction
The native envelope (Env) glycoprotein trimer expressed at the surface of the viral particles or infected cells and its transition state structures induced on the cell surface after receptor/co-receptor binding constitute major epitope targets for potentially protective anti-HIV-1 antibody responses. The extreme genetic diversity of Env, its heavy glycosylation, and conformational masking are effective evasion mechanisms against antibodies targeting epitopes available on Env in its native, un-triggered state ((Moore et al., 2009; Wei et al., 2003). In contrast, Env transition state structures exposed after engagement of the Env trimer with host receptors during viral entry (Guan et al., 2013; Mengistu et al., 2015), or on infected cell surfaces after cis (Veillette et al., 2014) or trans (Finnegan et al., 2001) triggering by CD4 (CD4-induced [CD4i] epitopes) are attractive targets for antibodies with protective potential (reviewed in (DeVico, 2007; Lewis et al., 2017b). These transition state structures harbor the most conserved regions of Env and become temporarily available for antibody recognition, providing CD4i antibodies a limited window of opportunity to counter the infection. Theoretically, antibodies that target Env transition state structures could afford protection by virus neutralization but this is limited strictly to Tier-1 viruses that are not representative of transmitted founder viruses which typically exhibit Tier-2/3 neutralization resistance (Seaman et al., 2010). Recent analyses suggest that CD4i antibodies contribute to protection via Fcγ receptor (FcγR)-mediated effector functions such as antibody-dependent cellular cytotoxicity (ADCC) (Ferrari et al., 2011; Guan et al., 2013; Veillette et al., 2014). Previously, we defined Epitope Cluster A as a potent ADCC target recognized by CD4i mAbs (Guan et al., 2013). Epitope Cluster A maps to the inner domain and the extended N-, C-termini of gp120 ((Acharya et al., 2014; Gohain et al., 2015) and reviewed in (Lewis et al., 2014; Lewis et al., 2017a; Veillette et al., 2016)). Antibodies specific for Cluster A epitopes were shown to mediate the majority of the ADCC activity detected in chronically-infected individuals (Ferrari et al., 2011; Guan et al., 2013; Veillette et al., 2014) as well as being implicated in protection for the clinical RV144 vaccine trial (Bonsignori et al., 2012; Haynes et al., 2012; Tomaras et al., 2013) and our SHIV challenge studies in rhesus macaques (DeVico et al., 2007; Fouts et al., 2015) immunized with a gp120 transition state vaccine (Fouts et al., 2000). Epitope Cluster A is buried at the trimer axis on native Env and inaccessible for antibody recognition until interaction with cell surface CD4. We and others have shown that exposure of these epitopes on the virion spike and on the surface of infected cells strictly depends on the cell surface form of CD4, since triggering with soluble CD4 does not lead to exposure of these epitope targets (Acharya et al., 2014; Mengistu et al., 2015; Ray et al., 2014; Veillette et al., 2014). These data point toward the possibility of an additional energy component required for trimer unfolding and effective presentation of epitopes of the Cluster A region that is provided in vivo by conformational changes in the D3D4 region of cell-surface CD4 (Acharya et al., 2014; Gohain et al., 2015; Veillette et al., 2015). Recent data also indicate that small CD4 mimetic compounds and the CD4-peptide mimetic miniprotein M48U1 (Martin et al., 2003; Van Herrewege et al., 2008) are able to induce the CD4-bound conformation of Env at the surface of HIV-1 infected cells and thus sensitize HIV-1-infected cells to the ADCC killing mediated by Cluster A antibodies provided that anti-co-receptor binding site antibodies are present (Richard et al., 2016; Richard et al., 2015).
We reported previously that the Cluster A region consists of three epitope specificities as defined by ELISA competition for binding to CD4 triggered gp120 with the two prototype mAbs: A32 and C11 (Guan et al., 2013). One subgroup only competes with A32 (A32-like epitopes), the second only competes with C11 (C11-like epitopes), and the third competes with both A32 and C11 (hybrid A32-C11-like epitopes). The existence of these three subregions was confirmed using fluorescence correlation spectroscopy coupled with fluorescence resonance energy transfer (FRET-FCS) (Gohain et al., 2015). Recently, we defined the A32 and the mixed A32-C11 epitopes of Cluster A region at the atomic level (Acharya et al., 2014; Gohain et al., 2015; Tolbert et al., 2016). Both of these epitope regions mapped within the inner domain of gp120 including mobile layers 1 and 2 of constant regions 1 and 2 (C1-C2 region) in CD4-triggered gp120 (Acharya et al., 2014; Gohain et al., 2015; Tolbert et al., 2016). Further, the A32-C11 mixed epitope extended to a few residues in the 7-stranded β-sandwich of the gp120 inner domain (Gohain et al., 2015). Here, we continue with the mapping of the epitope subgroups within the Cluster A region and present the structural signatures of the C11 epitope subregion. We determined the crystal structure of the complex formed between the gp120 coree that has extended N- and C-termini along with the antigen binding fragments (Fab) of two Cluster A antibodies: the A32-like antibody N5-i5 (Guan et al., 2013) and the C11-like antibody N12-i3 (Guan et al., 2013). This structure confirms that these two epitope regions are non-overlapping and defines new features of the gp120 N-terminus not seen previously. When bound to N12-i3, residues 31–42 of the gp120 N-terminus form an 8th β-strand to extend the 7-stranded β-sandwich formed by the gp120 inner domain seen in other Env structures including the PGT151-stabilized Env trimer (Lee et al., 2016), the CD4-trigered Env trimer (Ozorowski et al., 2017), and the gp120 core with N- and C-termini (Finzi et al., 2010; Pancera et al., 2010). These results indicate the importance of Epitope Cluster A as a major ADCC target for HIV-1 as well as identifying a new N-terminal structure that is implicated as an intermediate in a late entry state.
Results
N12-i3 and C11 induce potent ADCC against CD4 positive HIV-1 infected cells
We previously derived mAb N12-i3 from memory B cells (BMem) of a Natural Viral Suppressor, an HIV-1 infected individual who suppressed HIV-1 replication to <400 copies/ml in the absence of antiretroviral therapy (Guan et al., 2009; Sajadi et al., 2009; Sajadi et al., 2007). As reported in (Guan et al., 2013) N12-i3 and C11 constitute the most potent ADCC antibodies of the Cluster A region against CEM-NKr-CCR5 target cells sensitized with gp120 (Gomez-Roman et al., 2006). This ADCC method is designed to detect effector functions of antibodies recognizing transitional epitopes exposed during the earliest stage of viral entry, the interaction of the Env trimer with the host cell receptor, CD4 (Guan et al., 2013). To further characterize whether N12-i3 is also capable of mediating ADCC against target cells productively infected by cell-free virus or cell–cell virus spread (Orlandi et al., 2016) we tested its activity in an RFADCC assay against EGFP-CEM-NKr-CCR5-SNAP cells infected for 5 days with a replication competent HIV-1BaL molecular clone (Figure 1). As shown in Figure 1, N12-i3, similarly to C11, A32 and N5-i5, effectively recognizes and induces the killing of only infected or HIV-1-sensitized cells that have not downregulated cell surface CD4 (respectively p24+high/CD4+ and p24+low/CD4+) and is unable to recognize productively infected cells in which CD4 is downregulated (p24+/CD4− cells). This is in agreement with recent reports indicating that epitopes within the Cluster A region are strictly CD4 inducible and become exposed on HIV-1 infected cells only if cell surface CD4 is still available on the same cell to trigger the Env trimer (Ding et al., 2015; Veillette et al., 2014).
Figure 1. Binding of N12-i3 to EGFP-CEM-NKr-CCR5-SNAP cells infected with BaL infectious molecular clone (IMC).

EGFP-CEM-NKr-CCR5-SNAP were infected with IMC BaL virus and evaluated 5-days post-infection for (A) intracellular p24, cell surface levels of CD4 and (B) the binding of: N12-i3, Cluster A mAbs A32, N5-i5 and C11, and Synagis. Panel A shows the gating strategy for EGFP-CEM-NKr-CCR5-SNAP Bal-infected (Blue gates) and Mock (Red gate) cells, previously defined as GFP+ and L/D dye- cells. For Bal-infected cells two gates were delineated: p24+/CD4- and p24+low/CD4+; one gate was delineated for Mock p24-/CD4+. (C) ADCC using the RFADCC method. The curves shown are normalized for plateau cytotoxicity values using C11.
N12-i3 binds to Env exclusively within the 8-stranded β-sandwich of CD4-triggered gp120 and recognizes a novel epitope formed at the gp120 N-terminus
We have shown previously that A32- and C11-like epitopes constitute two non-overlapping epitope subregions within the Cluster A region (Gohain et al., 2015; Guan et al., 2013). To describe the C11-like epitope for the first time at the atomic level and to precisely define the A32- and C11-like epitope subregions in context of a single Env antigen, we obtained the crystal structure of the complex formed between CD4-mimetic M48U1-triggered N/C-termini-gp12093TH057 coree and antigen binding fragment (Fab) of representative antibody of each class: the A32-like mAb N5-i5 and the C11-like mAb N12-i3. The complex crystals belonged to space group C2 with two copies of the complex present in the asymmetric unit (Figure S1). The structure was solved at a resolution of 2.94 Å and refined to a final R/Rfree of 24.6/30.4%. The data collection and refinement statistics for the structure are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| N12-i3 Fab/N5-i5 Fab- N/C- termini-gp12093TH057 -coree- M48U1 |
|
|---|---|
| Data collection | |
| Wavelength, Å | 0.9795 |
| Space group | C2 |
| Cell parameters | |
| a, b, c, Å | 311.0, 53.3, 223.6 |
| α, β, γ, ° | 90, 128.9, 90 |
| Complex/a.u. | 2 |
| Resolution, (Å) | 50–2.94 (3.0–2.94) |
| # of reflections | |
| Total | 229,966 |
| Unique | 62,153 |
| Rmergb, % | 18.4 (92.3) |
| I/σ | 6.0 (1.1) |
| Completeness, % | 99.8 (99.9) |
| Redundancy | 3.7 (3.8) |
|
| |
| Refinement Statistics | |
| Resolution, Å | 50.0–2.94 |
| Rc, % | 23.1 |
| Rfreed, % | 29.1 |
| # of atoms | |
| Protein | 18,731 |
| Ligand/Ion | 360 |
| Water | 20 |
| Overall B value (Å)2 | |
| Protein | 52.4 |
| Ligand/Ion | 63.3 |
| Water | 27.5 |
| Root mean square deviation | |
| Bond lengths, Å | 0.005 |
| Bond angles, ° | 1.14 |
| Ramachandrane | |
| favored, % | 88.8 |
| allowed, % | 8.0 |
| outliers, % | 3.2 |
Values in parentheses are for highest-resolution shell
Rmerge = Σ | I - <I> | /Σ I, where I is the observed intensity and <I> is the average intensity obtained from multiple observations of symmetry-related reflections after rejections
R = Σ || Fo | - | Fc || /Σ | Fo | , where Fo and Fc are the observed and calculated structure factors, respectively
Rfree = as defined by (Brunger, 1997)
Calculated with MolProbity (Chen et al., 2010)
The structure of N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 coree-M48U1 complex (Figure 2) confirms that N12-i3 and N5-i5 recognize non-overlapping areas of the CD4-triggered gp120. Whereas the A32-like antibody N5-i5 binds to the gp120 Env utilizing the mode of binding and epitope footprint as described previously ((Acharya et al., 2014), Figure S2), the C11-like antibody N12-i3 recognizes a novel epitope, formed almost exclusively through residues of the 7-stranded β-sandwich of the inner domain and the extended N-terminus of gp120 of the C1 and C2 regions. Interestingly, in order to form the N12-i3 epitope, the extended N-terminus of gp120 (residues 31–42) undergoes a structural rearrangement to form a β4-strand that docks parallel to the β0-strand of the 7-stranded β-sandwich (Figure 2, blow up). As a result of the rearrangement, a new 8-stranded β-sandwich structure is formed consisting of the 7-strands contributed by residues of inner domain and the eight strand contributed by residues of the extended N-terminus of gp120. N12-i3 approaches CD4-triggered gp120 and recognizes its cognate epitope within the 8-stranded β-sandwich at an approximately 90° angle as compared to the angle of approach of the A32-like antibody N5-i5. The N12-i3 and N5-i5 modes of binding allow both to bind simultaneously as predicted by competition ELISA, FRET-FCS, and SPR (Gohain et al., 2015; Guan et al., 2013).
Figure 2. Crystal structure of the N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex.

Only complementary determining regions (CDRs) involved in N12-i3 Fab binding are colored and shown. The N-terminus of gp120 (black) forms an eighth strand to the 7-strand β-sandwich (magenta) of the inner domain to create the N12-i3 epitope. See also Table S1 and Figure S1 and S2.
Figure 3A shows the detailed map of N12-i3 and N5-i5 contacts within gp120. The N12-i3 epitope maps in close proximity to the N5-i5 epitope and is formed by residues, as measured by the buried surface area (BSA), of the newly formed 8-stranded β-sandwich structure (residues 31–40 of the extended N-terminus and 42–43, 45, 84–87 (C1 region), 224, 244–246 (C2 region) and 491 (C5 region) of the 7-stranded β-sandwich) with exception of one residue (Q82) that maps to Layer 1 of inner domain (Figure 3). By binding the edge of the β-sheet, at the eighth strand, N12-i3 is able to grasp both sides burying 1693 Å2 total surface area at the interface, the average calculated for two copies of the complex present in asymmetric unit of crystal (Table S1).
Figure 3. N12-i3 epitope footprint.

(A)N12-i3 (green) and N5-i5 (blue) gp120 contact residues mapped as spheres on the gp120 ribbon diagram and epitope footprints plotted on the gp120 surface with layers colored as in Figure 1. gp120 side chain (+) and main chain (−) contact residues colored green for hydrophobic, blue for hydrophilic and black for both as determined by a 5 Å cut off value over the corresponding sequence. Buried surface residues as determined by PISA are shaded. See also Figure S3. (B) Blow up view of the N12-i3 interaction with sugars at positions 88 and 241 (top) and the 8-stranded β-sandwich formation (bottom). CDR L1 and CDR H2 of the N12-i3 Fab are shown with the side chains of CDR L1 potentially contributing to contacts with the sugar at position 241 and the side chains of CDR H2 contributing to stabilization of the Y39YGVP turn of the 8-stranded β-sandwich shown as sticks. (C) Binding of N12-i3 to glycosylated and deglycosylated single-chain gp120BaL-sCD4 complex (FLSC). Surface plasmon resonance sensorgrams and binding kinetics are shown with wild type glycosylated FLSC – red curves and GnT1 cell grown deglycosylated FLSC – blue curves. Program fits to each sensogram are shown with black lines.
The antibody contacts to gp120 are mediated mostly by the heavy chain of N12-i3 (723 Å2 of total BSA of the complex, Table S1, Figure S3) and its protruding CDR H2 that binds to the β4- and β0-strands of the 8-stranded β-sandwich (Figure 2). The CDR H2 contacts to the β-strand are predominantly hydrophobic mediated by I52aPIF and M56P motifs with key interactions provided by the aliphatic ring of F54 that stacks against Y39, L42 and P43 of the gp120 N-terminus stabilizing the Y39YGVP turn (Figure 3B). The turn brings the β4-strand close to the β0-strand and allows the formation of the 8-stranded β-sandwich structure. Although the contacts of N12-i3 to the 8-stranded β-sandwich are mediated mostly by its heavy chain and these interactions are strictly protein-protein contacts, the light chain provides possible contacts to the Env glycan shield. From the light chain, CDR L3 is involved in few stabilizing interactions to the β4- strand, and the CDR L1 is located in close proximity to the truncated sugars molecules, N-acetyl-D-glucosamine (GlcNAc), directly proximal to the asparagine residues at position 88 and 241 (Figure 2). However, in the complex, only GlcNAc attached to N241 is involved in interactions (as accessed by 5Å cutoff) to the side chains of S30 and S31 of CDR L1 (Figure 3B). Since the truncated sugar molecules GlcNAc were introduced to help in co-crystallization studies and generated after the enzyme Endoglycosidase H (Endo H) treatment, we tested if N-linked branched wild-type glycans at positions 88 and 241 contribute to the N12-i3 binding. We tested binding of N12-i3 to the CD4-triggered gp120 expressed in wild-type HEK293 cells allowing production of the N-linked wild-type glycans and the CD4-triggered gp120 with sugars truncated to retain only the GlcNAc. As shown in Figure 3C, the affinity of N12-i3 for Env antigen remains unchanged for both CD4-triggered gp120 antigen preparations, indicating that branched glycans at positions 88 and 241 do not contribute to the N12-i3 binding.
The gp120 N-terminus exists in several conformations that change during viral entry
The gp120 subunit in the N12-i3 bound state assumes a unique conformation, not described previously for any of monomeric gp120 or Env trimers preparations studied in the unbound or the Fab-triggered states. Residues 35–39 of the gp120 N-terminus fold into a β-strand (strand β4) that packs parallel to strand β0 of the inner domain to form a stable 5-stranded β-sheet (strands β4, β0, β7, β5, and β25) in the 8-stranded β-sandwich which also includes strands β3, β1, and β6 (Figure 4A and B). The parallel β4- and β0 assembly is stabilized by a network of the main chain hydrogen bonds (Figure 4A, blow up). In the only other gp120 complex with full N- and C- termini studied in a context of the ternary Fab-CD4-triggerred gp120 complex, 48d Fab-N/C-termini-gp120Hxbc2coree– CD4 complex (PDB: 3JWD, (Pancera et al., 2010)), the N- and C- termini form a short antiparallel β-sheet consisting of strands β4 and β26, respectively, that are separated from the 7-stranded β-sandwich (Figure 4A and B). A similar distribution of secondary structural elements with the β4- and β26- strands packing antiparallel and separated from the 7-stranded β-sandwich is also seen in the membrane associated Env pre-fusion soluble trimer, Fab PGT151-EnvJRFL (PDB: 5FUU, (Lee et al., 2016)) and other soluble SOSIP gp140 trimers. Of note, the β-sheet formed by the N-terminus in the N12-i3 complex (W35VTVY39) is nearly twice the length of that formed in the 48d complex (Y40GV42) and comparable in length to that seen between the N- and C- termini in the Env pre-fusion trimer (W35VTVYY40). In the N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 coree complex the gp120 C-termini is disordered with no interpretable densities found for residues 493–511 of gp120. This indicates that N12-i3 epitope does not involve residues of gp120 C-terminus, and this epitope is likely formed only after the N- and C- termini are separated.
Figure 4. Properties of the N12-i3 epitope.

(A) Comparison of the gp120 conformation in the N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex to the gp120 conformations in the Fab 48d-N/C-termini-gp120Hxbc2coree-CD4 structure (PDB: 3JWD) and in the Env pre-fusion trimer, Fab PGT151-EnvJRFL (PDB: 5FUU). A blow-up view shows a network of hydrogen bonds stabilizing the interaction of β4- and β0-strands. (B) The topology of the inner domain and N/C-termini of gp120 from the available crystals structures of gp120. The N-terminus as seen in the N12-i3 Fab/N5-i5 Fab complex structure is shown in black and the point of the C-terminus truncation is indicated by an arrow over the topology of the inner domain and N/C-termini from the Fab 48d-N/C-termini-gp120Hxbc2coree-CD4 structure (PDB: 3JWD). (C) and (D) Sequence conservation among residues forming the N12-i3 and N5-i5 epitope. Residues colored dark blue differ from the Hxbc2 sequence in a range of 0.2–7% and residues colored red differ in a range of 87–99.9%. Only unique sequences in the database having an equivalent residue at each position were included in the calculated percentage representing approximately 32,000 sequences on average. (C) Ribbon diagram and molecular surface of the gp120 coree from the N12-i3 Fab/N5-i5 Fab complex (left) and gp120/gp41 heterodimer of the Env pre-fusion trimer, Fab PGT151-EnvJRFL (PDB: 5FUU) (right). N12-i3 and N5-i5 epitope footprints and regions of the nascent N12-i3 and N5-i5 epitopes are shown over the molecular surfaces of gp120 coree (solid line) and gp120/gp41 heterodimer, respectively (broken line). (D) Sequence, including variable regions, of gp120JRFL. The N5-i5 and N12-i3 epitope footprints are highlighted with a solid line for N12-i3 and broken line for N5-i5.
The N12-i3 epitope is highly conserved
We have shown previously based on co-crystal structures for A32 and other the A32-like monoclonal antibodies including N5-i5 that the A32-epitope region is highly conserved. Figure 4C and D shows both the N5-i5 and N12-i3 epitope footprints displayed on the surface of M48U1-triggered N/C-termini-gp12093TH057 coree with surface colored according to protein sequence conservation determined using the Los Alamos HIV Sequence Database (www.hiv.lanl.gov). The coloring scheme ranges from most conserved resides shown in blue to the least conserved shown in red. The N12-i3 epitope maps to a highly conserved region and involves residues almost completely conserved such as W35, which is mutated from the consensus in only about 0.34% of the sequences in the HIV Sequence Database Compendium (https://www.hiv.lanl.gov/content/sequence/HIV/COMPENDIUM/compendium.html). Additionally, the most conserved residue at the gp120 N-terminus, G41 (mutated in less than 0.1% of sequences) contributes to the β-turn formation and the G41VP residues immediately following the 8th β-strand are all mutated in less than 0.2% of sequences. The gp120 N-terminal residues 35–45 which form the major anchoring point for N12-i3 are mutated from the consensus in less than 2% of gp120 sequences (less than 0.5% if one excludes residues 38, 39, and 44). The high conservation of this gp120 region most likely results from the fact that it contributes directly to the formation of the gp120/gp41 heterodimer of the Env trimer. As shown in Figure 4C (right panel), regions of the nascent N12-i3 and N5-i5 epitopes are buried at the heterodimer interface and are involved directly in interaction with the gp41 protomer, as previously suggested by mutagenesis (Finzi et al., 2010).
Variable levels of the N12-i3 and N5-i5 epitope become accessible for antibody recognition during Env trimer opening
The Cluster A region consisting of A32- and C11-epitope subregions is buried in native Env trimers on virions or on the surfaces of HIV-1 infected cells (Acharya et al., 2014; Gohain et al., 2015; Guan et al., 2013; Ray et al., 2014; Veillette et al., 2014). It was shown previously that this epitope region becomes exposed and is accessible for antibody recognition only after binding to cell surface CD4 (Mengistu et al., 2015) or on productively infected cells until CD4 is down-regulated by Nef and Vpu (Alsahafi et al., 2015; Ding et al., 2015; Richard et al., 2015; Veillette et al., 2015; Veillette et al., 2014; Veillette et al., 2016). Once exposed, Epitope Cluster A becomes a very potent ADCC target (Ferrari et al., 2011; Guan et al., 2013; Veillette et al., 2014). Recent data reveal also that the cell surface CD4-induced conformational rearrangements can be very effectively mimicked on the infected cell surface by small compound or peptide derived CD4 mimetics that alone or in presence of co-receptor binding site antibodies are capable of effective sensitization of infected cells to Cluster A antibody binding and ADCC killing (Richard et al., 2016; Richard et al., 2015). CD4 mimetics therefore constitute useful tools to the study of the structural rearrangements of the Env trimer which occur upon trimer triggering with cell surface CD4. On the other hand, our structural analysis indicates that the A32- and C11- epitope regions map to the non-overlapping gp120 regions that are buried at the pre-fusion trimer and may become available for antibody recognition at different levels during the CD4-induced opening of the Env trimer. The region of the nascent A32- epitope is nested inside the center of the pre-fusion trimer with multiple residues involved directly in contacts to the upper part of α7 and α9-helix of gp41 ((Acharya et al., 2014) and Figure 4C, right panel). In contrast, residues of the nascent C11 epitope are localized at the bottom of the pre-fusion trimer (close to the viral membrane) with the N-terminus interacting with the gp120 C-terminus, disulfide loop region of the principal immunodominant domain and the β27 strand the gp41 (Figure 4C, right panel). In the trimer, a 3-stranded antiparallel β-sheet is formed, assembled from the β27 strand of gp41 and the β4 and β26 strands of gp120. In order to test if there is any difference in the accessibility of the A32- and C11-subregions during Env trimer opening we tested the ability of antibodies of the A32-subregion: A32 and N5-i5 and the C11-subregion: C11 and N12-i3 to recognize Env targets present at the surface of CEM-Nkr cells infected with a wild-type infectious molecular clone coding for functional Nef and Vpu proteins and ADA Env (48 hours post-infection as described and Experimental Procedures). Staining was performed in the presence or absence of the CD4 mimetic peptide M48U1 which was mixed or not with the co-receptor binding site antibody 17b (Figure 5A and Figure S4). As we reported previously (Richard et al., 2015, Richard et al., 2016) the M48U1 mimetic combined with 17b is able to effectively trigger the conformational rearrangements of Env trimers present on infected cells to expose Cluster A epitopes. However, in the conditions tested (100nM M48U1 and 5 ug/mL 17b) we observed differences in the levels of antibody bound with A32 and the A32-like antibody N5-i5 binding more efficiently than C11 or the C11-like antibody N12-i3. This may indicate that the A32-like epitope region is exposed earlier during the CD4 mimetic-induced opening of Env trimer. Moreover, as shown in Figure 5B, we observed that while a Fab fragment of 17b was sufficient to expose A32 and N5-i5 epitopes within HIV-1JR-FL Env trimers expressed at the surface of 293T cells, it failed to do so for C11 and N12-i3. A 17b Fab 2 or full 17b antibody were absolutely required to expose N12-i3 and C11 epitopes, implying that it is “easier” to expose A32 and N5-i5 epitopes than N12-i3 and C11 epitopes. This suggests that A32 exposure may precede C11 exposure (Figure 5B).
Figure 5. Exposure of A32-like and C11-like epitopes at the surface of HIV-1 infected cells and HIV-1 virions.

(A) CEM.NKr cells were infected with the infectious HIV-1NL-4.3 ADA/GFP molecular clone; 48h post-infection cells were stained with Alexa Fluor 647-conjugated mAbs: 17b, A32, N5-i5, N12-i3 and C11 alone, in the presence of the CD4 mimetic M48U1 (100nM) or together with M48U1 and co-receptor binding site 17b (5µg/ml). Data are the averages of four independent experiments. (B) Binding to HIV-1JR-FL Env trimers expressed at the surface of 293T cells. At 48-hours post-transfection, the exposure of Cluster A epitopes was assessed with Alexa Fluor 647 (AF647)-conjugated A32, N12-i3, or C11 antibodies in the presence or absence of 17b, the 17b Fab-fragment, or the Fab-2 fragment with or without CD4mimetic-M48U1 (100nM). Data are the averages from four independent experiments. Statistical significant was evaluated using paired student t test, * P < 0.05, ** P <0.01, ***P <0.001, **** P < 0.00001; ns, not significant. See also Figure S4 and S5. (C) Effect of M48U1 on the binding of Cluster A mAbs to BaL pseudovirus particles in solution as determined by the FCS assay. The relative fraction of mAb that adopts a lower diffusion coefficient as a result of virion binding is shown. All measurements were performed in triplicate. Average values are shown; error bars indicate standard deviations.
Finally, we tested if the CD4 mimetic M48U1 could be also effective in sensitizing the Env trimers present on pseudovirions for Cluster A antibody binding (Figure 5C). We showed previously (Ray et al., 2014) that triggering of the Env trimers present on pseudovirions with the soluble CD4 (sCD4) is insufficient to expose the Cluster A region. Figure 5C shows the binding of a panel of Cluster A antibodies to pseudovirions expressing the CCR5-tropic HIV-1BaL envelope (a “tier 1” easily neutralized envelope, (Ray et al., 2014) in solution in the presence or absence of serial M48U1 concentrations ranging from 0 to 64 mM by FCS (Figure 5C). In the absence of M48U1, a small fraction of Cluster A mAbs (8 to 15% depending on the mAb) adopted the lower diffusion coefficient (8 µm2/sec) indicative of virion binding. However, this fraction improved with increasing amounts of M48U1, reflecting higher mAb attachment to HIV-1BaL virion particles. The binding signals for A32 and A32-like antibody N5-i5 increased from 16% to 37% and 15% to 31%, respectively, from 0 to 64 mM of M48U1. In contrast, the binding of C11 and N12-i3 only moderately improved reaching 17% to 27% in the presence of the highest amounts of M48U1. Overall, we observed increased binding of Cluster A mAbs to virions treated with M48U1 as compared to previous reports of Cluster A exposure with soluble CD4 (Ray et al., 2014). Our data is also consistent with the data from infected cells and Env trimers expressed at the surface of 293 cells, showing that the A32 subregion is more efficiently accessible for antibody recognition within HIVBaL virions than the C11 subregion upon triggering with a CD4 mimetic. Of note, we also have tested the binding (data not shown) of the Cluster A antibody panel to HIVBaL virions in presence of M48U1 (25 µg/ml) and 17b (10 µg/ml). Under these conditions we observed an increase in the binding of A32 (from 20% to 28% for virions treated with M48U1 alone or for virions treated with M48U1 and 17b in combination) but no noticeable differences in the binding of C11 or N12-i3.
Discussion
FcγR- mediated effector functions such as ADCC are thought to constitute an important component of protective immunity against HIV-1. They trigger clearing of virus particles or virus-infected cells through mechanisms involving interactions of the antibody constant (Fc) region with FcγR on the surface of effector cells including NK cells, monocytes, macrophages, dendritic cells, and neutrophils (Sulica et al., 2001; Takai, 2002). ADCC responses appear relatively early during acute infection and are detectable as early as 21 and 48 days after SIVmac251 (Asmal et al., 2011; Sun et al., 2011) infection in macaques and acute HIV-1 (Pollara et al., 2010) infection in humans. These early ADCC responses, which in general are broadly reactive, precede the appearance of neutralizing antibody responses with similar breadth (Koup et al., 1991; Lyerly et al., 1987). ADCC is thought to eliminate virus-infected cells through a mechanism in which antibodies provide specificity for the release of cytotoxic mediators by linking the infected target to the effector cell.
The non-neutralizing, transitional Cluster A epitopes consisting of A32- and C11- and C11/A32 hybrid- epitopes (Guan et al., 2013) constitute major targets for ADCC responses in both acute and chronic HIV-1 infection as well as those induced by vaccination (Ampol et al., 2012; Bonsignori et al., 2012; Ferrari et al., 2011; Guan et al., 2013; Robinson et al., 2005). Early analyses of the antibody repertoire in patients on HAART during acute infection revealed high frequencies of B cells producing anti-Env antibodies directed at CD4i epitope targets that include the Cluster A region, with more than 47% of cells producing antibodies of this type (Robinson et al., 2005). These studies pointed toward CD4i epitopes as the most immunogenic during acute infection. Recent data are consistent with this early observation and confirm that chronically infected individuals also frequently elicit antibodies specific for this region. Although the majority of recent studies examine CD4i targets in context of the A32 epitope (Ferrari et al., 2011), growing evidence points toward the C11-epitope region as at least equally important, contributing significantly to plasma-mediated ADCC activity of HIV-infected individuals. Our studies of natural virus suppressors who control HIV-1 without therapy have shown high frequencies of long-lived memory B cells for antibodies recognizing the conserved CD4i epitopes of this type (Guan et al., 2013; Guan et al., 2009). Half of a set of four Cluster A antibodies isolated from five HIV-1– infected individuals recognized the C11-epitope region (Guan et al., 2013). Recently Williams and colleagues (Williams et al., 2015) showed, using their novel strategy to isolate antibodies originated from unique B cell, that antibodies similar to C11 are common in HIV-infected individuals and antibodies targeting this epitope can be commonly produced. In this study, C11-like antibodies accounted for between 18–78% of ADCC activity in 9 chronically infected individuals and they were shown to develop from unique B-cell lineages in the same individual within the first 6 months of infection. The short time required for development as well as usual structural features of C11-like antibodies such as relatively short CDR H3s and low levels of somatic mutation of variable regions (Guan et al., 2013; Williams et al., 2015) support a role of the C11 epitope region as a potential vaccine target. It is possible that vaccination strategies designed to induce this type of antibody response will overcome the great challenges associated with the vaccines aimed to induce a broadly neutralizing antibody response. Interestingly, whereas the A32- epitopes were implicated in the protective effect of ADCC responses in humans induced in the RV144 vaccine trial (Bonsignori et al., 2012; Haynes et al., 2012; Tomaras et al., 2013), the vaccination studies in non-human primate (NHP) using the conformationally constrained gp120-CD4 immunogen (FLSC) (Fouts et al., 2000) directly identified C11-like antibodies as a correlate of protection (DeVico et al., 2007; Fouts et al., 2015).
The A32- and C11- epitopes constitute non-overlapping regions of the gp41-inteactive face of gp120. We describe here the crystal structure of the complex formed between Fab of antibody representing each of these two epitope subregion bound to the same antigen: CD4 mimetic-triggered gp120 coree. Our structural analysis confirms that A32- and C11-like antibodies can simultaneously bind one CD4-triggered gp120 subunit occupying proximal epitope surfaces. Whereas our A32-like antibody, N5-i5, binds to discontinuous epitope exclusively within the inner domain mobile layers 1 and 2 (Gohain et al., 2015), the C11-like antibody, N12-i3, attaches at approximately 90° angle and recognizes residues of the newly formed 8-stranded β-sandwich of the inner domain of the C1-C2 region. N12-i3 binds at the top of the sandwich engaging the extended N-terminus (residues 31–42) to add an 8th strand to the 7-stranded β-sandwich structure known to be formed in pre-fusion Env trimers or monomeric gp120 studied in the context of other Fab-triggered states.
The structure of N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 coree-M48U1 complex provides the first look into molecular details of C11-epitope region in absence of structural information of the epitope of the prototype antibody C11. Although the exact epitope footprints of these two antibodies may not be exactly the same, they certainly recognize the same structural elements of CD4-triggered Env. We recently defined the putative contact region of C11 (Gohain et al., 2015) by ELISA, FCS-FRET, SPR competition assays and analyses of residues shown by mutagenesis to be involved in C11 binding (Finzi et al., 2010; Moore et al., 1994). Our studies mapped to the 7-stranded β-sandwich in the gp41-interactive region, immediately adjacent to the A32-like epitope surface (Acharya et al., 2014; Gohain et al., 2015). Our predicted putative binding site overlaps well with N12-i3 epitope with most of residues defined by mutagenesis to form C11 epitope contributing also to the N12-i3-gp12093TH057 coree interface. However, one structural signature of the C11 epitope region was missing at that time and could not be simply predicted based on available structural information: the formation of 8-stranded β-sandwich structure.
The N12-i3 epitope, similar to A32 subregion epitopes, maps to highly conserved regions of gp120 (only approximately 16% of sequences differ from Hxbc2 for residues 31–45 and only approximately 0.4% for residues 35–45). This high degree of conservation among HIV sequences predicts the broad reactivity of antibodies that target this region across HIV clades. Indeed, recently demonstrated strong cross-clade breadth with activity against 10 of 11 envelopes tested, including those from clades A, B, C, A/D and C/D was reported for two C11-like mAb isolated from HIV-specific memory B cells from a subtype A-infected Kenyan woman (Williams et al., 2015).
The 8-stranded β-sandwich structure as seen in the N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex is unique, formed late during CD4-induced Env trimer opening most likely post-detachment of gp41 and gp120 subunits. We and others have shown that soluble CD4 induces a metastable conformation of the trimer in which gp120 protomers are displaced but still remain involved in the gp41 interaction thus incapable of exposure of the A32- and C11-epitope regions (Acharya et al., 2014; Ozorowski et al., 2017; Pancera et al., 2010; Wang et al., 2016). We are able, however, to induce the conformational changes of the viral trimer similar to those induced by cell surface CD4 with the CD4 miniprotein mimetic M48U1 alone or in combination with co-receptor binding site antibody. M48U1 was initially developed to interfere with the initial steps of viral attachment to the CD4 cell receptor thus is capable of virus neutralization (Martin et al., 2003; Van Herrewege et al., 2008). Our earlier structural analysis confirm that M48U1 targets the Phe 43 cavity in a manner similar to that of CD4 but its cyclohexylmethoxyl group reaches deeper into the cavity. The increased surface area buried and improved shape complementarity of this group within the Phe43 pocket was found to contribute to M48U1 effectiveness in binding to HIV Env and CD4 antagonism (Stricher et al., 2008). The same feature most likely also contributes to the efficiency of M48U1 in trimer disassembly and exposure of Cluster A epitope region.
Our analyses of the binding of A32- and C11-like mAbs to the CD4-mimetic-M48U1 (or M48U1/17b mix)-triggered envelope trimers present on the infected cell surface, expressed at the surface of transfected cells or pseudo-viruses indicate that A32 epitope subregion is exposed more easily than the C11 subregion. At any given concentration of M48U1 (or M48U1/17b mix) we observed a better recognition by A32-like antibodies as compared to C11-like antibodies. This indicates the possibility that the A32-epitope is accessible earlier during CD4-induced opening of the HIV-1 Env trimer. Our recent data using confocal and super-resolution microscopy to track the exposure of Cluster A epitopes as they appear on single CCR5-tropic HIV-1 particles bound to target cell indicate that both the A32 and C11 epitope subregions become accessible for antibody recognition very rapidly upon target cell binding (Mengistu et al., 2015). The same level of accessibility of both epitopes was detected five minutes post-attachment of virus to the CD4+ target cells regardless of presence or absence of co-receptor. This indicates that structural rearrangements exposing both epitope structures occur very rapidly, most likely in the co-receptor independent manor.
Based on our data we propose the following model of the HIV-1 Env trimer rearrangements that lead to the exposure of the Cluster A epitopes during the entry process (Figure 6). In the pre-fusion Env trimer both the A32 and C11 subregion are occluded by the interaction between gp120 and gp41 and the N- and C- termini of gp120 clasped by gp41 (Figure 6, conformation from PDB: 5FUU, (Lee et al., 2016)). In this state, the gp120 inner domain is packed against a gp41 protomer preventing it from collapsing into a 6-helix bundle – the driving force behind membrane fusion. In the pre-fusion trimer, the N- and C- termini of gp120 are held by a triple-tryptophan clasp consisting of tryptophans 623, 628, and 623 of gp41 (Pancera et al., 2014). Upon binding of the Env trimer to the cell surface CD4, the inner domain of gp120 undergoes a series of conformation rearrangements with the formation of the bridging sheet and changes to the region of the nascent N5-i5 epitope (conformation from PDB: 5VN3, (Ozorowski et al., 2017)). In this state, the α0-helix and the β1-strand of the N5-i5 epitope are formed and the β1 helix is positioned to assume conformation as seen in the N5-i5-bound state. Although in this Env conformation the N5-i5 epitope is largely formed, it is not accessible for antibody recognition due to occlusion at the trimer interface. At this step, the 7-stranded β-sandwich remains intact with the N-, C-termini occupying the gp41 tryptophan clasp, thus the N12-i3 epitope is not formed. Later in the entry process gp120 protomers are displaced from gp41 allowing the exposure of the A32 epitope subregions (PDB: 3JWD, (Pancera et al., 2010)). In this stage, the N5-i5 epitope is fully formed and exposed for antibody binding but not the N12-i3 epitope as the 8-stranded β-sandwich structure is yet to be assembled. Finally, the N- and C- termini are released from gp41, and are most likely separated, permitting the addition of the 8th β-strand to the gp120 inner domain 7-stranded β-sandwich and formation of the N12-i3 epitope (the conformation of gp120 as observed in the N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex).
Figure 6. Model of the exposure of the Cluster A epitopes during the entry process.

In the pre-fusion Env trimer both epitopes are occluded by the interaction between gp120 and gp41 with the gp120 N and C termini clasped by gp41 (left panel). Binding of CD4 to the outer domain of gp120 induces formation of the co-receptor binding site which includes conformational changes to the gp120 inner domain (panel two from left). These changes to mobile layers 1 and 2 of the inner domain create the N5-i5 epitope and disrupt the gp120-gp41 interaction (panel three from left). Binding of co-receptor or further opening up of the trimer disengage the N-terminus of gp120 from the gp41 clasp. The N-terminus is then free to form the eighth strand of the inner domain β-sandwich creating the N12-i3 epitope (rightmost panel). Figures were assembled using available structures of the Env pre-fusion trimer (Cry-EM structure of cleaved wild type JR-FL Env trimer in complex with PGT151 Fab, PDB: 5FUU), CD4-triggered Env trimer (Cryo-EM structure of a B41 SOSIP Env trimers in complex with CD4 and antibody 17b, PDB: 5VN3) and 48d Fab-N/C-termini-gp120Hxbc2coree–CD4 complex (PDB: 3JWD) on which N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex was superimposed. The bottom panels show the gp120 subunit only.
STAR methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Marzena Pazgier, mpazgier@ihv.umaryland.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
RFADCC and cell-surface staining was performed using EGFP-CEM-NKr-CCR5-SNAP cell line (Orlandi et al., 2016). EGFP-CEM-NKr-CCR5-SNAP cells were grown in R10 medium at 37°C.
METHOD DETAILS
Protein preparation and crystallization
N12-i3 and N5-i5 Fabs and gp120 protein for use in crystallization were prepared as previously described (Acharya et al., 2014; Gohain et al., 2015). N12-i3 only crystallized with gp120 in the presence of both N12-i3 Fab and fab of the A32-like antibody N5-i5. The dual Fab complex was made sequentially by combining gp120, M48U1, and N5-i5 Fab with a slight molar excess of N5-i5 Fab, purification by gel filtration on a Superdex 200 gel filtration column (GE Healthcare) (equilibrated with 5 mM Tris-HCI pH 7.2 and 150 mM sodium chloride), addition of N12-i3 Fab in slight molar excess to the purified complex, and purification again by gel filtration on the same gel filtration column.
Initial crystallization trials were performed in hanging drop crystallization trays with commercially available crystallization screens from Molecular Dimensions and Hampton Research. Crystals from screens were then optimized manually to produce crystals suitable for data collection. The N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex crystals were grown from 15% PEG 2000 MME, 0.1 M Tris-HCl pH 8.0, and 0.1 M potassium chloride, and crystals were flash frozen in liquid nitrogen after a brief soak in crystallization buffer plus cryoprotectant, 15% MPD.
Data collection and structure solution
Data were collected on a PILATUS detector at SSRL beam line 12–2. The N12-i3/N5-i5 Fab complex crystallized in monoclinic space group C2 with cell dimensions a = 311.0 Å, b = 53.3 Å, c = 223.6 Å, and β = 128.9. The N12-i3/N5-i5 complex was solved by molecular replacement using Fab and gp120 coordinates from the N5-i5-gp12093TH057coree-CD4 structure (PDB: 4H8W) with PHASER (McCoy et al., 2007) from the CCP4 suite (Winn et al., 2011). Refinement was done with REFMAC (Murshudov et al., 2011) and PHENIX (Adams et al., 2002). Model building was done using COOT (Emsley and Cowtan, 2004). Data collection and refinement statistics are shown in Table 1.
RFADCC and cell-surface staining
Infection, staining and RFADCC were performed as described previously (Gohain et al., 2015; Orlandi et al., 2016). In summary, EGFP-CEM-NKr-CCR5-SNAP cells were spinoculated for 2 h at 2000 RPM at 12 °C in 96-well U-bottom plate (5 × 105 cells/well) with 240 ng of IMC BaL virus (control cells were incubated without virus), as measured by HIV-1 p24 antigen capture ELISA. As previously described (Gohain et al., 2015) the peak of infection was reached culturing the cells in the presence of Bal IMC at 37°C for 5 days, when EGFP-CEM-NKr-CCR5-SNAP cel ls were harvested, washed twice with R10 medium and divided in two groups: The first group was stained with N12-i3, A32, N5-i5, C11 and Synagis (MedImmune) mAbs labeled with human IgG Zenon-APC kit (Invitrogen), Live/Dead Fixable Near-IR Dead Cell Stain (Molecular Probes) and (eFluor 450)-conjugated mouse anti-CD4 OKT4 mAb (eBioscience) for 30 min at RT. After washing and permeabilizing (Perm-Fix, BD-PharMingen,) the cells were stained with (PE)-conjugated mouse anti-p24 mAb (KC57-RD1; Beckman Coulter, Inc.). After two washes, HIV-1- infected or mock cells were fixed in 2% PFA.
The second group of Mock and Bal-infected EGFP-CEM-NKr-CCR5-SNAP cells was stained with SNAP-Surface Alexa Fluor 647 dye and utilized as targets for the RFADCC (Orlandi et al., 2016). Serial dilutions of N12-i3, A32, N5-i5, C11 and Synagis were incubated with mock and Bal-infected targets for 15 mins at RT and, subsequently with PBMC as effectors. The cells were allowed to interact for 3 hours at 37°C, at t he end of which the cells were washed once with PBS and fixed with 2% PFA. Binding and RFADCC assays were then analyzed with an LSRII Fortessa flow cytometer (BDBiosciences). Data analysis was performed with FlowJo software (Tree Star, Inc., San Carlos, Calif.).
Infection and staining of CEM-NKr cells
The infectious molecular clone NL4.3 coding for GFP, the ADA envelope and functional Nef and Vpu proteins was used as previously described (Veillette et al., 2014). In order to achieve 20% of infection of CEM-NKr cells, the proviral vector and a VSVG-encoding plasmid were co-transfected in 293T cells by standard calcium phosphate transfection. Concentrated virus (over a 20% sucrose cushion) used to infect CEM-NKr cells by spin infection, as described (Veillette et al., 2014). Forty-eight hours post infection, cell-surface staining was performed as previously described and MFI histograms shows signal on live infected populations (Richard et al., 2015; Veillette et al., 2015). Binding of HIV-1-infected cells by Alexa-Fluor 647-conjugated Cluster A mAbs A32, N5-i5, N12–13 and C11 or co-receptor binding site 17b Ab was performed in presence of M48U1 (100 nM) or with equivalent volume of vehicle (PBS). Binding of HIV-1-infected cells with Alexa-Fluor 647-conjugated Abs A32, N5-i5, N12-i3, C11 and 17b was performed alone or in combination with unconjugated 17b (5µg/ml). Detection of GFP+ infected cells was performed as described (Richard et al., 2015). The percentage of infected cells (GFP+) was determined by gating the living cell population based on the viability dye staining (Aqua Vivid, Thermo Fisher Scientific). Samples were analyzed on a LSRII cytometer (BD Biosciences, Mississauga, ON, Canada) and data analysis was performed using FlowJo vX.0.7 (Tree Star, Ashland, OR, USA).
Epitope exposure by flow cytometry
To express the wild-type HIV-1JRFL Env for flow cytometric analysis, a stop codon was introduced into the expression plasmid for the codon-optimized Env, replacing the codon for Gly 711. This modification results in the expression of Env with a deletion of the cytoplasmic tail, which increases the level of Env on the cell surface. Env expression plasmids were transfected into 3×105 293T cells along with a pIRES-GFP vector at a ratio of 2 µg Env to 0.5 µg green fluorescent protein (GFP) expression plasmid. 16 h after transfection, cells were washed with fresh media and Env exposure was evaluated 24 h later. A32, N5-i5, N12-i3, and C11 were used to detect CD4-induced exposure and each of the antibodies was conjugated to an Alexa-Fluor 647 (AF647) probe (Invitrogen). Transfected 293T cells were incubated with either the 2G12 antibody (1 µg/ml), which recognizes the outer domain of gp120, or AF647-A32, AF647-N12-i3, AF647-C11 (4 µg/ml) in the absence or presence of M48U1 (100 nM) or 17b, 17bFab1, and 17bFab2 (5 µg/ml) for 1-hour at room temperature. The 2G12 and 17b antibodies were detected using a goat anti-human antibody coupled to Alexa Fluor 647 (Invitrogen). Antibody binding was detected by gating on GFP-positive cells on an LSRII cytometer (BD Biosciences, Mississauga, ON, Canada). Data analysis was performed using FlowJo vX.0.7 (Tree Star, Ashland, OR, USA).
Fluorescence correlation spectroscopy measurements
FCS analyses were performed similar to previously reported (Gohain et al., 2015; Ray et al., 2014). mAbs A32, N5-i5, C11, N12-i3 and 17b were labeled with Alexa 647 probe with a dye-to-protein ratios of 3 for FCS experiments. HIV-1BaL pseudoviruses were produced by co-transfection of HEK293T cells with an Env-deficient HIV-1 backbone plasmid, pNL4-3-ΔE-EGFP, along with Env expression plasmid pHIV-1-BaL 0.1. All FCS experiments used virus preparations diluted to a 10 µg/ml p24 equivalent. gp120 to p24 antigen ratios were typically 1:10 to 1:50. The 10 µg/ml p24 equivalent value typically corresponded to TCID50/ml values in the range of 200,000 to 650,000 (HIV-1BaL). Pseudoviruses with 10µg/ml p24 equivalent concentrations in 25µl reaction buffer were first treated for 90 min at 37°C with different concentrations (0 to 64 mM) of M48U1 to open up the cluster A epitopes and 100 µg/ml of nonspecific (Calbiochem) IgG1 to block nonspecific binding. This was followed by addition of 5 µg/ml of Alexa Fluor 647-conjugated mAbs. MAbs were allowed to interact with virions for 90 min at 37°C. FCS measurements were performed in a confocal microscope (ISS Q2) with a high-numerical-aperture (NA) water objective (60x; NA 1.2). The excitation source was a Fianium SC-400 super-continuum laser with a NKT super-select AOTF filter. The fluorescence was collected by avalanche photodiodes through a band-pass (650 – 720 nm; Chroma) filter and a B&H SPC-150 card operated in a photon time-tag time-resolved (TTTR) mode. ISS VistaVision software was used to analyze the FCS data to assess the in vitro binding of mAbs to HIV-1 BaL pseudovirus particles by determining the translational diffusion coefficients. The percentage of total mAb (at given test concentrations) that shifts into the more slowly diffusing species which is the virion-bound fraction reflects the relative magnitude of cognate epitope exposure in the target population of virions (Ray et al., 2014).
Surface plasmon resonance (SPR) measurements
Binding affinities of N12-i3 to full length single chain (FLSC) were assessed by surface plasmon resonance on a Biacore T-100 (GE Healthcare) as previously described (Tolbert et al., 2016). Varying concentrations of single-chain gp120BaL-sCD4 (FLSC) (Fouts et al., 2000) either expressed in 293 cells (FLSC with wild type glycosylation) or in GnT1- cells and deglycosylated with Endo Hf (New England Biolabs) (deglycosylated FLSC) were passed over N12-i3 IgG bound to a protein A coupled sensor chip. Binding affinities were calculated using a 1:1 binding model with the BIAevaluation software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Antibody binding analyses
Data are presented as mean and standard deviation of the mean fluorescence intensity over mock (MFI/Mock). Data are the averages from four independent experiments. Statistical significant was evaluated using paired student t test, * P < 0.05, ** P <0.01, ***P <0.001, **** P < 0.00001; ns, not significant.
DATA AND SOFTWARE AVAILABILITY
Accession Numbers
The atomic coordinates for the structures presented in this study have been deposited in the Protein Data Bank under ID code: 5W4L
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human monoclonal N12-i3 | Isolated from patient (Guan et al., 2013) | HIV Database: record number 2973 |
| Human monoclonal N5-i5 | Isolated from patient (Guan et al., 2013) | HIV Database: record number 2971 |
| Human monoclonal A32 | Pazgier lab | HIV Database: record number 660 |
| Human monoclonal C11 | Pazgier lab | HIV Database: record number 707 |
| Human monoclonal 17b | Pazgier lab | HIV Database: record number 658 |
| Human monoclonal 2G12 | Finzi lab | N/A |
| Synagis (Palivizumab) | Medmiune | N/A |
| (eFluor 450)-conjugated mouse anti-CD4 OKT4 mAb | eBioscience | Cat# 48–0048–42 |
| (PE)-conjugated mouse anti-p24 mAb (KC57-RD1) | Beckman Coulter, Inc | Cat# 6604667 |
| Bacterial and Virus Strains | ||
| infectious molecular clone BaL virus | (Orlandi et al., 2016) | IMC Bal virus |
| infectious molecular clone NL4.3 coding for GFP, the ADA envelope and functional Nef and Vpu | (Veillette et al., 2014) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| full length single chain protein | (Fouts et al., 2000) | FLSC |
| M48U1 | (Martin et al., 2003) | CD4-peptide mimetic |
| Endo Hf | New England BioLabs | Cat#P0703S |
| Critical Commercial Assays | ||
| human IgG Zenon-APC kit | Invitrogen | Cat# Z25451 |
| Live/Dead Fixable Near-IR Dead Cell Stain | Molecular Probes | Cat# L10119 |
| Fixation/Permeabilization Solution Kit | BD-Bioscience | Cat# 554714 |
| SNAP-Surface Alexa Fluor 647 | New England BioLabs | Cat# S9136S |
| Aqua Vivid | Thermo Fisher Scientific | Cat# L43957 |
| Deposited Data | ||
| N12-i3 Fab/N5-i5 Fab- N/C-termini-gp12093TH057 -coree-M48U1 complex | This work | 5W4L |
| Fab 48d-N/C-termini-gp120Hxbc2coree-CD4 structure | (Pancera et al., 2010) | 3JWD |
| Fab PGT151-EnvJRFL structure | (Lee et al., 2016) | 5FUU |
| Cryo-EM structure of a B41 SOSIP Env trimers in complex with CD4 and antibody 17b | (Ozorowski et al., 2017) | 5VN3 |
| N5-i5 Fab- gp12093TH057 -coree-d1d2CD4 structure | (Acharya et al., 2014) | 4H8W |
| Experimental Models: Cell Lines | ||
| EGFP-CEM-NKr-CCR5-SNAP cells | (Orlandi et al., 2016) | N/A |
| CEM-NKr cells | The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEM.NKR-CCR5 from Dr. Alexandra Trkola | N/A |
| HEK293T cells | ATCC | ATCC® CRL-3216™ |
| Recombinant DNA | ||
| pIRES-GFP vector | Clontech | Cat#6029–1 |
| pNL4-3-ΔE-EGFP plasmid | (Ray et al., 2014) | N/A |
| pHIV-1-BaL 0.1 plasmid | AIDS Research and Reference Reagent Program, Division of AIDS, NIAID | N/A |
| Software and Algorithms | ||
| FlowJo software | Tree Star,Inc., San Carlos, Calif. | https://www.flowio.com |
| FlowJo vX.0.7 software | Tree Star, Ashland, OR, USA | http://docs.flowio.com/vx/faq/qeneral-faq/tree-star-flowio |
| ISS Vista Vision software | ISS, Inc., Champaign, IL, USA | http://www.iss.com/microscopv/software/vistavision.html |
| PHASER | (McCoy et al., 2007) | http://www.phaser.cimr.cam.ac.uk/index.php/Phaser_Crystallographic_Software |
| CCP4 | (Winn et al., 2011) | http://www.ccp4.ac.uk |
| REFMAC | (Murshudov et al., 2011) | http://www.ccp4.ac.uk/html/refmac5/description |
| PHENIX | (Adams et al., 2002) | https://www.phenix-online.orq |
| Coot | (Emsley and Cowtan, 2004) | http://www2.mrc-lmb.cam.ac.uk/Personal/pemslev/coot/ |
| MolProbity | (Chen et al., 2010) | http://molprobitv.biochem.duke.edu/ |
Highlights.
the C11-like ADCC epitope footprint is defined at the molecular level
the C11 subregion maps to a newly formed gp120 8-stranded β-sandwich
the C11 and A32 epitope subregions are adjacent and non-overlapping
the C11 epitope is formed upon release of the gp120 N-terminus from the Env trimer
Acknowledgments
We thank our IHV colleagues for outstanding support of the studies leading to the ideas presented above, specifically Dr. Anthony L. DeVico for his continued critical insights. This work was supported by NIH grants: NIAID R01 AI116274 to MP, R01AI129769 to MP and AF, NIGMS R01 GM117836 to KR, NIAID P01 AI120756 to GT and the Bill and Melinda Gates Foundation: #OPP1033109 to GKL. This work was partially supported also by a CIHR foundation grant #352417 to A.F. is supported by the Canada Research Chairs program. N.A. is the recipient of a King Abdullah scholarship for higher education from the Saudi Government. S.D. is a recipient of a FRQS postdoctoral Fellowship Award. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Additionally, crystallographic data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of the SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research, by the National Institutes of Health (NIH) National Center for Research Resources, Biomedical Technology Program (P41RR001209), and by the National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: W.D.T, N.G. and M.P. designed, performed research and analyzed the data; N.A., V.V, S.D, K.R and A. F. performed antibody binding experiments and analyzed the data; Ch.O and G.K.L. tested activities of antibodies in an RFADCC assay and analyzed the data; W.D.T and M.P. wrote the paper with all authors providing comments or revisions.
The authors have no conflicts of interest to report.
References
- Acharya P, Tolbert WD, Gohain N, Wu X, Yu L, Liu T, Huang W, Huang CC, Kwon YD, Louder RK, et al. Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. Journal of virology. 2014;88:12895–12906. doi: 10.1128/JVI.02194-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, Finzi A. Nef Proteins from HIV-1 Elite Controllers Are Inefficient at Preventing Antibody-Dependent Cellular Cytotoxicity. Journal of virology. 2015;90:2993–3002. doi: 10.1128/JVI.02973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampol S, Pattanapanyasat K, Sutthent R, Permpikul P, Kantakamalakul W. Comprehensive investigation of common antibody-dependent cell-mediated cytotoxicity antibody epitopes of HIV-1 CRF01_AE gp120. AIDS Res Hum Retroviruses. 2012;28:1250–1258. doi: 10.1089/AID.2011.0346. [DOI] [PubMed] [Google Scholar]
- Asmal M, Sun Y, Lane S, Yeh W, Schmidt SD, Mascola JR, Letvin NL. Antibody-dependent cell-mediated viral inhibition emerges after simian immunodeficiency virus SIVmac251 infection of rhesus monkeys coincident with gp140-binding antibodies and is effective against neutralization-resistant viruses. Journal of virology. 2011;85:5465–5475. doi: 10.1128/JVI.00313-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Pollara J, Moody MA, Alpert MD, Chen X, Hwang KK, Gilbert PB, Huang Y, Gurley TC, Kozink DM, et al. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. Journal of virology. 2012;86:11521–11532. doi: 10.1128/JVI.01023-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. Free R value: cross-validation in crystallography. Methods Enzymol. 1997;277:366–396. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVico A, Fouts T, Lewis GK, Gallo RC, Godfrey K, Charurat M, Harris I, Galmin L, Pal R. Antibodies to CD4-induced sites in HIV gp120 correlate with the control of SHIV challenge in macaques vaccinated with subunit immunogens. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17477–17482. doi: 10.1073/pnas.0707399104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVico AL. CD4-induced epitopes in the HIV envelope glycoprotein, gp120. Current HIV research. 2007;5:561–571. doi: 10.2174/157016207782418560. [DOI] [PubMed] [Google Scholar]
- Ding S, Veillette M, Coutu M, Prevost J, Scharf L, Bjorkman PJ, Ferrari G, Robinson JE, Sturzel C, Hahn BH, et al. A Highly Conserved Residue of the HIV-1 gp120 Inner Domain Is Important for Antibody-Dependent Cellular Cytotoxicity Responses Mediated by Anti-cluster A Antibodies. Journal of virology. 2015;90:2127–2134. doi: 10.1128/JVI.02779-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, et al. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. Journal of virology. 2011;85:7029–7036. doi: 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J Virol. 2001;75:11096–11105. doi: 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell. 2010;37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts TR, Bagley K, Prado IJ, Bobb KL, Schwartz JA, Xu R, Zagursky RJ, Egan MA, Eldridge JH, LaBranche CC, et al. Balance of cellular and humoral immunity determines the level of protection by HIV vaccines in rhesus macaque models of HIV infection. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E992–999. doi: 10.1073/pnas.1423669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouts TR, Tuskan R, Godfrey K, Reitz M, Hone D, Lewis GK, DeVico AL. Expression and characterization of a single-chain polypeptide analogue of the human immunodeficiency virus type 1 gp120-CD4 receptor complex. Journal of virology. 2000;74:11427–11436. doi: 10.1128/jvi.74.24.11427-11436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohain N, Tolbert WD, Acharya P, Yu L, Liu T, Zhao P, Orlandi C, Visciano ML, Kamin-Lewis R, Sajadi MM, et al. Cocrystal Structures of Antibody N60-i3 and Antibody JR4 in Complex with gp120 Define More Cluster A Epitopes Involved in Effective Antibody-Dependent Effector Function against HIV-1. Journal of virology. 2015;89:8840–8854. doi: 10.1128/JVI.01232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman VR, Florese RH, Patterson LJ, Peng B, Venzon D, Aldrich K, Robert-Guroff M. A simplified method for the rapid fluorometric assessment of antibody-dependent cell-mediated cytotoxicity. J Immunol Methods. 2006;308:53–67. doi: 10.1016/j.jim.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, et al. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E69–78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Sajadi MM, Kamin-Lewis R, Fouts TR, Dimitrov A, Zhang Z, Redfield RR, DeVico AL, Gallo RC, Lewis GK. Discordant memory B cell and circulating anti-Env antibody responses in HIV-1 infection. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3952–3957. doi: 10.1073/pnas.0813392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. The New England journal of medicine. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koup RA, Robinson JE, Nguyen QV, Pikora CA, Blais B, Roskey A, Panicali D, Sullivan JL. Antibody-dependent cell-mediated cytotoxicity directed by a human monoclonal antibody reactive with gp120 of HIV-1. Aids. 1991;5:1309–1314. doi: 10.1097/00002030-199111000-00004. [DOI] [PubMed] [Google Scholar]
- Lee JH, Ozorowski G, Ward AB. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science. 2016;351:1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GK, Guan Y, Kamin-Lewis R, Sajadi M, Pazgier M, Devico AL. Epitope target structures of Fc-mediated effector function during HIV-1 acquisition. Current opinion in HIV and AIDS. 2014;9:263–270. doi: 10.1097/COH.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GK, Pazgier M, DeVico AL. Survivors Remorse: antibody-mediated protection against HIV-1. Immunol Rev. 2017a;275:271–284. doi: 10.1111/imr.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GK, Pazgier M, Evans DT, Ferrari G, Bournazos S, Parsons MS, Bernard NF, Finzi A. Beyond Viral Neutralization. AIDS Res Hum Retroviruses. 2017b doi: 10.1089/aid.2016.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly HK, Reed DL, Matthews TJ, Langlois AJ, Ahearne PA, Petteway SR, Jr, Weinhold KJ. Anti-GP 120 antibodies from HIV seropositive individuals mediate broadly reactive anti-HIV ADCC. AIDS Res Hum Retroviruses. 1987;3:409–422. doi: 10.1089/aid.1987.3.409. [DOI] [PubMed] [Google Scholar]
- Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, et al. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengistu M, Ray K, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus envelope glycoprotein gp120 on virions bound to target cells. PLoS Pathog. 2015;11:e1004772. doi: 10.1371/journal.ppat.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Willey RL, Lewis GK, Robinson J, Sodroski J. Immunological evidence for interactions between the first, second, and fifth conserved domains of the gp120 surface glycoprotein of human immunodeficiency virus type 1. Journal of virology. 1994;68:6836–6847. doi: 10.1128/jvi.68.11.6836-6847.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PL, Ranchobe N, Lambson BE, Gray ES, Cave E, Abrahams MR, Bandawe G, Mlisana K, Abdool Karim SS, Williamson C, et al. Limited neutralizing antibody specificities drive neutralization escape in early HIV-1 subtype C infection. PLoS Pathog. 2009;5:e1000598. doi: 10.1371/journal.ppat.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi C, Flinko R, Lewis GK. A new cell line for high throughput HIV-specific antibody-dependent cellular cytotoxicity (ADCC) and cell-to-cell virus transmission studies. J Immunol Methods. 2016;433:51–58. doi: 10.1016/j.jim.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozorowski G, Pallesen J, de Val N, Lyumkis D, Cottrell CA, Torres JL, Copps J, Stanfield RL, Cupo A, Pugach P, et al. Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike. Nature. 2017 doi: 10.1038/nature23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K, Mengistu M, Yu L, Lewis GK, Lakowicz JR, DeVico AL. Antigenic properties of the HIV envelope on virions in solution. Journal of virology. 2014;88:1795–1808. doi: 10.1128/JVI.03048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Pacheco B, Gohain N, Veillette M, Ding S, Alsahafi N, Tolbert WD, Prevost J, Chapleau JP, Coutu M, et al. Co-receptor Binding Site Antibodies Enable CD4-Mimetics to Expose Conserved Anti-cluster A ADCC Epitopes on HIV-1 Envelope Glycoproteins. EBioMedicine. 2016;12:208–218. doi: 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, et al. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E2687–2694. doi: 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Elliott DH, Martin EA, Micken K, Rosenberg ES. High frequencies of antibody responses to CD4 induced epitopes in HIV infected patients started on HAART during acute infection. Hum Antibodies. 2005;14:115–121. [PubMed] [Google Scholar]
- Sajadi MM, Constantine NT, Mann DL, Charurat M, Dadzan E, Kadlecik P, Redfield RR. Epidemiologic characteristics and natural history of HIV-1 natural viral suppressors. J Acquir Immune Defic Syndr. 2009;50:403–408. doi: 10.1097/QAI.0b013e3181945f1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV-1 natural viral suppressors: control of viral replication in the absence of therapy. Aids. 2007;21:517–519. doi: 10.1097/QAD.0b013e328013d9eb. [DOI] [PubMed] [Google Scholar]
- Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. Journal of virology. 2010;84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricher F, Huang CC, Descours A, Duquesnoy S, Combes O, Decker JM, Kwon YD, Lusso P, Shaw GM, Vita C, et al. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J Mol Biol. 2008;382:510–524. doi: 10.1016/j.jmb.2008.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulica A, Morel P, Metes D, Herberman RB. Ig-binding receptors on human NK cells as effector and regulatory surface molecules. Int Rev Immunol. 2001;20:371–414. doi: 10.3109/08830180109054414. [DOI] [PubMed] [Google Scholar]
- Sun Y, Asmal M, Lane S, Permar SR, Schmidt SD, Mascola JR, Letvin NL. Antibody-dependent cell-mediated cytotoxicity in simian immunodeficiency virus-infected rhesus monkeys. Journal of virology. 2011;85:6906–6912. doi: 10.1128/JVI.00326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- Tolbert WD, Gohain N, Veillette M, Chapleau JP, Orlandi C, Visciano ML, Ebadi M, DeVico AL, Fouts TR, Finzi A, et al. Paring Down HIV Env: Design and Crystal Structure of a Stabilized Inner Domain of HIV-1 gp120 Displaying a Major ADCC Target of the A32 Region. Structure. 2016;24:697–709. doi: 10.1016/j.str.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9019–9024. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Herrewege Y, Morellato L, Descours A, Aerts L, Michiels J, Heyndrickx L, Martin L, Vanham G. CD4 mimetic miniproteins: potent anti-HIV compounds with promising activity as microbicides. The Journal of antimicrobial chemotherapy. 2008;61:818–826. doi: 10.1093/jac/dkn042. [DOI] [PubMed] [Google Scholar]
- Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. Journal of virology. 2015;89:545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, et al. Interaction with Cellular CD4 Exposes HIV-1 Envelope Epitopes Targeted by Antibody-Dependent Cell-Mediated Cytotoxicity. Journal of virology. 2014;88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillette M, Richard J, Pazgier M, Lewis GK, Parsons MS, Finzi A. Role of HIV-1 Envelope Glycoproteins Conformation and Accessory Proteins on ADCC Responses. Current HIV research. 2016;14:9–23. doi: 10.2174/1570162x13666150827093449. [DOI] [PubMed] [Google Scholar]
- Wang H, Cohen AA, Galimidi RP, Gristick HB, Jensen GJ, Bjorkman PJ. Cryo-EM structure of a CD4-bound open HIV-1 envelope trimer reveals structural rearrangements of the gp120 V1V2 loop. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E7151–E7158. doi: 10.1073/pnas.1615939113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- Willey S, Aasa-Chapman MM. Humoral immunity to HIV-1: neutralisation and antibody effector functions. Trends Microbiol. 2008;16:596–604. doi: 10.1016/j.tim.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Williams KL, Cortez V, Dingens AS, Gach JS, Rainwater S, Weis JF, Chen X, Spearman P, Forthal DN, Overbaugh J. HIV-specific CD4-induced Antibodies Mediate Broad and Potent Antibody-dependent Cellular Cytotoxicity Activity and Are Commonly Detected in Plasma From HIV-infected humans. EBioMedicine. 2015;2:1464–1477. doi: 10.1016/j.ebiom.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.