

Abstract
Ultraviolet (UV) light induces mutagenic cyclobutane pyrimidine dimers (CPDs) in nucleosomal DNA that is tightly wrapped around histone octamers. How global‐genome nucleotide excision repair (GG‐NER) processes CPDs despite that this chromatin arrangement is poorly understood. An increased chromatin association of CHD1 (chromodomain helicase DNA‐binding 1) upon UV irradiation indicated possible roles of this chromatin remodeler in the UV damage response. Immunoprecipitation of chromatin fragments revealed that CHD1 co‐localizes in part with GG‐NER factors. Chromatin fractionation showed that the UV‐dependent recruitment of CHD1 occurs to UV lesions in histone‐assembled nucleosomal DNA and that this CHD1 relocation requires the lesion sensor XPC (xeroderma pigmentosum group C). In situ immunofluorescence analyses further demonstrate that CHD1 facilitates substrate handover from XPC to the downstream TFIIH (transcription factor IIH). Consequently, CHD1 depletion slows down CPD excision and sensitizes cells to UV‐induced cytotoxicity. The finding of a CHD1‐driven lesion handover between sequentially acting GG‐NER factors on nucleosomal histone octamers suggests that chromatin provides a recognition scaffold enabling the detection of a subset of CPDs.
Keywords: chromatin remodeling, DNA damage, nucleosomes, skin cancer, UV light
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
Genomic DNA is susceptible to damage caused by a plethora of endogenous or environmental genotoxic agents. In particular, bulky base lesions induced by ultraviolet (UV) light and the consequent accumulation of mutations are the major cause of skin cancer (Mouret et al, 2011; DiGiovanna & Kraemer, 2012; Marteijn et al, 2014). UV irradiation of DNA gives rise to cyclobutane pyrimidine dimers (CPDs) and 6‐4 photoproducts (6‐4PPs) in a ratio of ~3:1 (Kobayashi et al, 2001). The quantitatively predominant CPDs are distributed evenly in chromatin and arise abundantly in nucleosome cores where the DNA is wrapped around histone octamers (Smerdon & Conconi, 1999; Zavala et al, 2014; Han et al, 2016). Nucleotide excision repair (NER) is the versatile process that removes these UV lesions as well as other bulky base adducts elicited by chemical carcinogens or oxygen radicals. Depending on their genomic location, bulky lesions are sensed by two alternative mechanisms. In the template strand of transcribed genes, detection of DNA damage occurs when the elongating RNA polymerase II encounters obstructing lesions (Hanawalt & Spivak, 2008; Vermeulen & Fousteri, 2013). Conversely, global‐genome NER (GG‐NER) detects bulky DNA adducts anywhere in the genome independently of transcription (Sancar, 1996; Hoeijmakers, 2009; Schärer, 2013). Genetic defects in the latter pathway result in the cancer‐prone syndrome xeroderma pigmentosum (XP) with patients being classified into complementation groups (XP‐A through XP‐G) reflecting mutations in distinct repair genes (Friedberg et al, 2006).
The GG‐NER reaction relies on a trimeric complex consisting of XPC, RAD23B (a human homolog of yeast RAD23), and centrin 2 to initially sense the presence of bulky lesions in the DNA double helix (Sugasawa et al, 1998; Araki et al, 2001; Volker et al, 2001). The DNA‐binding function of this initiator complex resides entirely with the XPC subunit that, for the recognition of CPDs, is additionally supported by UV‐damaged DNA‐binding (UV‐DDB) protein, also known as DDB1‐DDB2 heterodimer (Hwang et al, 1999; Wakasugi et al, 2001; Rapic‐Otrin, 2002; Fitch et al, 2003). The XPC subunit mediates recruitment of the transcription factor IIH (TFIIH) complex, which contains the XPD helicase that scans DNA for damage verification (Mathieu et al, 2013) and unwinds the double helix by 20–25 nucleotides around the lesion (Evans et al, 1997; Riedl et al, 2003; Compe & Egly, 2016). The transiently unwound state is then stabilized by XPA in conjunction with replication protein A (RPA) (Li et al, 2015), until the endonucleases XPG and XPF/ERCC1 (a heterodimer of XPF and excision repair cross‐complementing 1) incise the damaged strand on each side of the unwound duplex to remove damaged bases as part of an excised oligonucleotide (Araújo et al, 2000; Reardon & Sancar, 2003; Staresincic et al, 2009). The remaining single‐stranded gap is filled by DNA synthesis and closed by DNA ligation (Moser et al, 2007; Ogi et al, 2010). To allow for repair despite compaction of the DNA substrate in chromatin, this multi‐step process involves the temporary release of histones from damaged DNA (Adam et al, 2016) but how these chromatin rearrangements take place is not yet understood.
Members of distinct families of ATP‐dependent remodelers have been implicated in relaxing histone–DNA interactions to prime chromatin for GG‐NER activity (Czaja et al, 2012; Peterson & Almouzni, 2013). A pioneer study in yeast indicated that switch/sucrose non‐fermenting (SWI/SNF) stimulates GG‐NER activity in transcriptionally silent loci (Gong et al, 2006). In higher eukaryotes, UV‐DDB has been shown to recruit at least three chromatin remodelers, that is, brahma‐related gene 1 (BRG1, a catalytic subunit of SWI/SNF) (Zhang et al, 2009; Zhao et al, 2009), amplified in liver cancer 1 (ALC1) (Pines et al, 2012), and inositol requiring 80 (INO80) (Jiang et al, 2010). In a further study, however, INO80 was not required for chromatin remodeling before initiating GG‐NER activity, but for the restoration of nucleosome repeats after DNA repair (Sarkar et al, 2010). In addition, the mammalian SWI/SNF subunit SNF5 (for sucrose non‐fermenting 5) interacts with XPC protein and its deletion causes UV hypersensitivity (Klochendler‐Yeivin et al, 2006; Ray et al, 2009), although these results were challenged by another study where no effect of SNF5 on UV sensitivity was detected (McKenna et al, 2008). The above reports all suggest that chromatin remodelers facilitate GG‐NER activity although the underlying mechanisms are lacking (Aydin et al, 2014). The basic conundrum remains whether chromatin relaxation precedes DNA lesion detection or vice versa (Rubbi & Milner, 2003). It is also not known if remodelers are recruited to unfold nucleosomes for the access of GG‐NER factors to DNA, or rather promote the assembly of repair complexes, or their release from DNA, after chromatin relaxation by other mechanisms. In addition, previous reports on BRG1 and SNF5 show that chromatin remodelers may assist the GG‐NER reaction by activating cell cycle checkpoints (Ray et al, 2009; Zhang et al, 2013) or that their depletion may lead to apoptosis, which attenuates GG‐NER activity (Gong et al, 2008).
Chromodomain helicase DNA‐binding 1 (CHD1) supports chromatin plasticity crucial for the pluripotency of embryonic stem cells, for transcriptional reprogramming, and for homologous recombination (Gaspar‐Maia et al, 2009; Park et al, 2014; Piatti et al, 2015; Kari et al, 2016). The genomewide range of these known CHD1 functions prompted us to test whether this same remodeler is also involved in chromatin dynamics required for GG‐NER activity. We found that, in UV‐damaged chromatin, CHD1 stimulates the handover between XPC protein and the TFIIH complex at DNA lesion sites. To facilitate this substrate handover, XPC protein recruits CHD1 directly to nucleosome cores indicating that the XPC subunit is able to form lesion recognition intermediates with a subset of damages positioned around histone octamers. The main implication of this unexpected mechanism is that, rather than always representing a barrier impeding the accessibility to damaged DNA, nucleosomes play a scaffolding role in priming specific lesions for the GG‐NER reaction.
Results
CHD1 co‐localizes in chromatin with GG‐NER factors
We tested whether CHD1 translocates to the chromatin of human cells upon exposure to UV‐C light. For that purpose, HeLa cells were UV‐irradiated or mock‐treated and collected after different incubation times. For the detection of chromatin recruitments, the cells were lysed in the presence of 0.3 M NaCl to extract, into the supernatant, free proteins that are not associated with chromatin or only loosely bound to chromatin. The remaining pellet contains chromatin‐bound proteins (Fei et al, 2011). After measuring protein concentrations in each fraction, 25 μg of free proteins and 15 μg of chromatin‐bound proteins were separated by gel electrophoresis and analyzed by immunoblotting. The validity of this approach to monitor UV‐dependent redistributions is demonstrated by the strong relocation to chromatin of DDB2 observed 1 h after irradiation (Fig 1A), in line with the expected association of this recognition subunit with UV lesions (Otrin et al, 1997; Yeh et al, 2012). The immunoblot of Fig 1A also reproduces the well‐described degradation of DDB2 in response to UV exposure (Rapic‐Otrin, 2002), which is apparent after the 3‐ and 6‐h incubation periods. In addition, the UV treatment led to a modest increase in CHD1 in the fraction of chromatin‐bound proteins over the normal presence of this remodeler in the chromatin of unchallenged cells. The immunoblot quantifications using histone H3 as the internal standard revealed that the level of CHD1 in chromatin is increased by ~40% at 1 h after UV irradiation compared to unirradiated controls (Fig 1B).
Figure 1. CHD1 co‐localizes in chromatin with GG‐NER proteins.
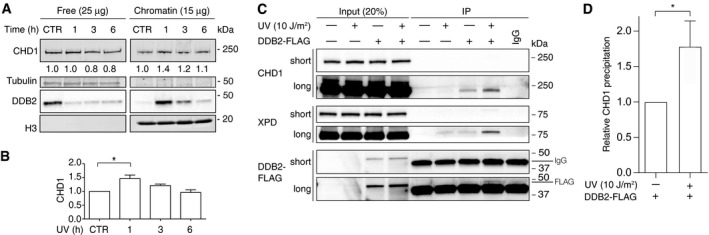
- Chromatin recruitment of DDB2 and CHD1. The chromatin of HeLa cells was salt‐extracted at different times after exposure to UV‐C light (10 J/m2). Free proteins in the supernatant (25 μg per sample) and chromatin‐bound proteins in the pellet (15 μg per sample) were analyzed by gel electrophoresis and immunoblotting. CHD1 (197 kDa) migrates to a position just below the 250‐kDa marker; tubulin and histone H3 were loading controls for the free and chromatin‐associated fraction, respectively. CTR, mock‐treated control cells. Numbers indicate the relative quantity of free CHD1 (normalized to tubulin) and chromatin‐bound CHD1 (normalized to H3), whereby the respective CHD1 levels in unirradiated cells are set to 1.
- Quantification of chromatin‐bound CHD1 normalized to H3 (n = 5 independent experiments). The CHD1 level in the chromatin of unirradiated cells is set to 1.
- Co‐localization of CHD1 with GG‐NER factors in the chromatin of UV‐irradiated cells. HEK293 cells were transfected with a vector for expression of FLAG‐tagged DDB2 and treated with UV light (10 J/m2). After a 1‐h incubation, the chromatin was salt‐extracted and, following fragmentation by MNase digestion, dissolved by sonication. The resulting chromatin fragments were precipitated with anti‐FLAG antibodies, thus exploiting the FLAG tag to isolate nucleoprotein complexes containing DDB2. Input chromatin fractions and immunoprecipitated nucleoprotein complexes were analyzed by blotting with antibodies against CHD1, DDB2, and XPD (short and long exposures are shown). IgG, immunoglobulin G heavy chains interfering with the detection of DDB2‐FLAG.
- Quantified CHD1 levels co‐localizing in fragmented chromatin with NER factors, normalized to the amount of CHD1 in the respective input fractions (n = 3 independent experiments).
To test whether this CHD1 recruited to chromatin upon UV radiation co‐localizes with GG‐NER factors, we transiently transfected HEK293 cells with a construct that drives overexpression of the DDB2 subunit of the UV‐DDB heterodimer fused to the FLAG peptide. HEK293 cells were used for this experiment because of their permissivity to DNA transfections. The purpose of this approach was to exploit the tight binding of DDB2 protein to UV‐damaged DNA and, concomitantly, its transient interactions with the core GG‐NER factors XPC and XPA (Sugasawa et al, 2005; Wakasugi et al, 2009). These two factors, in turn, associate with each other and with the TFIIH complex comprising the XPD helicase (Nocentini et al, 1997; Yokoi et al, 2000; Uchida et al, 2002; Bunick et al, 2006). The DDB2‐FLAG fusion protein was, therefore, used as a molecular bait to isolate short chromatin fragments containing UV lesions and GG‐NER factors and to test whether CHD1 co‐localizes with these nucleoprotein complexes.
After pre‐extraction with 0.3 M NaCl, chromatin was dissected by digestion with saturating amounts of micrococcal nuclease (MNase), which cleaves DNA preferentially in linker segments spacing the nucleosome cores (Fig EV1). Finally, the fragmented chromatin was solubilized by sonication before carrying out precipitations taking advantage of anti‐FLAG antibodies (Fig 1C). This immunoprecipitation of short chromatin fragments from UV‐irradiated cells resulted in the co‐fractionation of both CHD1 and XPD (a core NER subunit). Much less DDB2, CHD1, and XPD were immunoprecipitated from the chromatin of cells that were not subjected to UV radiation or not previously transfected with the DDB2‐FLAG‐expressing construct. The quantification of CHD1 levels in immunoprecipitated complexes, using CHD1 in the respective input fractions as the reference, highlights its redistribution in response to UV radiation (Fig 1D). Collectively, the UV‐dependent CHD1 recruitment to chromatin (Fig 1A) and its co‐localization with short chromatin fragments containing GG‐NER factors (Fig 1C) led to the hypothesis that CHD1 may contribute to GG‐NER activity.
Figure EV1. Chromatin fractionation.
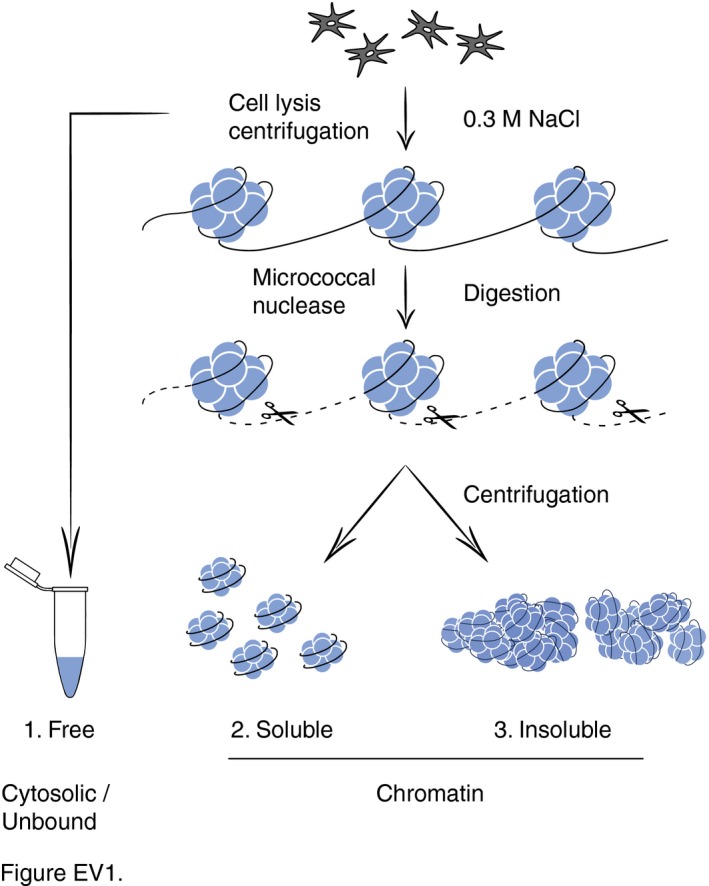
Flow diagram illustrating the chromatin dissection after removal of unbound or loosely bound (free) proteins by salt extraction (0.3 M NaCl). MNase digestion removes linker DNA, thus generating a supernatant of solubilized chromatin proteins and a remaining insoluble pellet strongly enriched in nucleosome cores.
CHD1 is recruited to UV‐damaged nucleosome cores
The observed redistribution of CHD1 protein to chromatin sites containing GG‐NER factors prompted us to use U2OS and HeLa cells to delineate the exact positions to which CHD1 moves after UV radiation. As above, free proteins that are not or only loosely associated with chromatin were removed by salt (0.3 M NaCl) extraction and the remaining chromatin was dissected by incubation with MNase. At a saturating level of this nuclease, the genome is totally converted to short DNA segments of 147 base pairs (Fig 2A). The size of these residual fragments corresponds to the DNA length of nucleosome cores protected from MNase digestion by interactions with histone octamers. Thus, saturating MNase digestions reduce the entire chromatin to nucleosome cores by eliminating all internucleosomal linker DNA segments.
Figure 2. CHD1 is recruited to nucleosome cores upon UV irradiation.
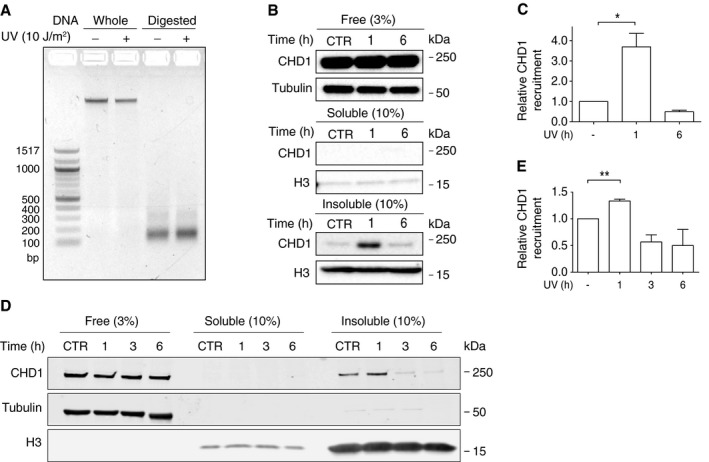
- The chromatin of U2OS cells, untreated or UV‐irradiated (10 J/m2), was fragmented by MNase digestion (4 U/μl). A subsequent agarose gel analysis demonstrates the complete breakdown of internucleosomal linker DNA segments resulting in residual chromatin containing exclusively nucleosome core fragments of 147 base pairs. Whole, undigested chromatin. Digested, MNase‐fragmented chromatin.
- Upon UV irradiation, CHD1 is transiently recruited to nucleosome cores. The chromatin of U2OS cells (harvested 1 or 6 h after the UV pulse) was salt‐extracted and MNase‐digested (4 U/μl) to generate, as shown in the three panels from top down, a fraction of free (or loosely chromatin‐bound) proteins, a fraction of MNase‐solubilized chromatin proteins, and a fraction of condensed and, hence, insoluble nucleosome cores. CTR, unirradiated control. Histone H3 and tubulin serve as the loading standards. The proportion of each fraction loaded onto the gel (3 or 10%) is indicated in parenthesis.
- Quantification of CHD1 recruitment to the insoluble fraction of nucleosome cores of U2OS cells, normalized to the H3 level, following 1 and 6 h after UV irradiation (n = 4 independent experiments). The amount of CHD1 in the nucleosome core fraction of control cells was set to 1.
- Same experiment as in panel (B) using HeLa cells. The UV‐dependent recruitment of CHD1 to the insoluble fraction of nucleosome cores is confirmed but less pronounced than in U2OS cells. The chromatin was analyzed at 1, 3, and 6 h after the UV pulse.
- Quantification of CHD1 recruitment to the insoluble fraction of nucleosome cores of HeLa cells, normalized to the H3 level, following 1, 3, and 6 h after UV irradiation (n = 3 independent experiments). The amount of CHD1 in the nucleosome core fraction of control cells was set to 1.
Analysis of defined proportions of the different fractions from U2OS cells showed that most CHD1 is actually found in the unbound state as free protein extracted with buffer containing 0.3 M NaCl (Fig 2B, top panel). The subsequent MNase digestion of pre‐extracted chromatin generates a soluble supernatant of proteins released from chromatin that also includes some dissociated nucleosome cores containing inter alia histone H3 but no CHD1 protein (Fig 2B, middle panel). However, the vast majority of nucleosome cores remains in a condensed and, hence, insoluble form even after digestion with saturating MNase concentrations (Fig EV1). We observed that a proportion of CHD1 protein is immobilized in this nucleosome core‐enriched fraction upon UV irradiation (Fig 2B, bottom panel). This UV‐dependent recruitment of CHD1 to nucleosome cores is observed around 1 h after UV irradiation but, subsequently, CHD1 disappears from this chromatin localization within 6 h after UV treatment (Fig 2C). A transient recruitment of CHD1 to nucleosome cores, that is, to the insoluble fraction of MNase‐digested chromatin is also observed in HeLa cells (Fig 2D). This UV‐dependent CHD1 recruitment is less pronounced in HeLa than in U2OS cells but follows similar kinetics with an increased occupancy of nucleosome cores detected at 1 h following UV irradiation, but not at the later time points after the UV pulse.
XPC‐dependent recruitment of CHD1 to nucleosomes
Next, different GG‐NER factors were depleted in HeLa cells by transfection with short interfering RNA (siRNA) to understand the mechanism by which CHD1 is relocated to nucleosome cores. The efficiency of each siRNA‐mediated downregulation is demonstrated by immunoblotting (Fig EV2). These depletion experiments revealed that the UV‐dependent recruitment of CHD1 to nucleosome cores, observed 1 h after UV irradiation, was essentially abolished by depletion of XPC protein (Fig 3A). Consistent with this dependence on the XPC subunit, we confirmed as previously reported (Fei et al, 2011) that XPC protein itself effectively binds this same nucleosome core fraction around 1 h after the UV challenge (Fig 3B). Depletion of DDB2, an accessory subunit that is active in the GG‐NER pathway upstream of XPC, does not detectably reduce the UV‐dependent relocation of CHD1 protein to this MNase‐insoluble fraction of nucleosome cores. Similarly, depletion of XPA, a core subunit acting in the GG‐NER pathway downstream of XPC, does not influence the UV‐dependent relocation of CHD1 to nucleosome cores (Fig 3C).
Figure EV2. Efficiency of siRNA‐mediated protein depletions.
- Analysis of CHD1 protein levels after depletion with three different siRNA sequences (16 nM) directed against the CHD1 transcript. Immunoblots of HeLa cell lysates were carried out 2 days after transfection. Tubulin served as the loading control.
- Protein depletion achieved in U2OS cells 2 days after transfection with the indicated siRNA sequences (16 nm).
- Protein depletion achieved in HeLa cells 2 days after transfection with the indicated siRNA sequences (16 nm). Tubulin and histone H3 served as loading controls.
- Protein depletion achieved in HeLa cells 2 days after transfection with siRNA sequences (16 nM) against the indicated chromatin remodelers. Tubulin served as the loading control.
Figure 3. CHD1 is recruited to nucleosome cores by the XPC initiator.
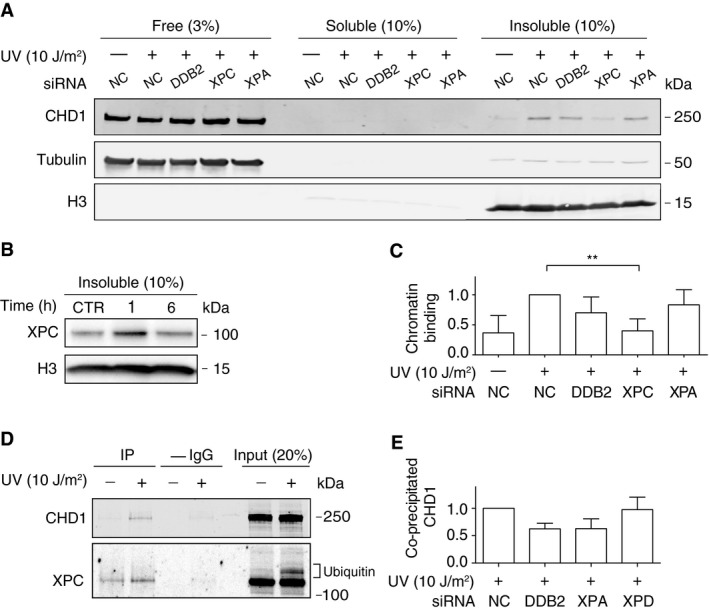
- XPC‐dependent recruitment of CHD1 to nucleosome cores. HeLa cells were siRNA‐transfected as indicated 2 days before irradiation with UV (10 J/m2), or before mock treatment, and incubated for another 1 h. Chromatin was salt‐extracted and MNase‐digested to generate, from left to right, a fraction of free (or loosely chromatin‐bound) proteins, a fraction of solubilized chromatin proteins, and the insoluble fraction of nucleosome cores. NC, non‐coding RNA. Histone H3 and tubulin serve as loading standards.
- Transient association of XPC protein with the insoluble fraction of nucleosome cores of HeLa cells in response to UV irradiation (10 J/m2). CTR, mock‐treated cells.
- Quantification of CHD1 recruitment to the insoluble fraction of nucleosome cores normalized to the level of CHD1 in control reactions with non‐coding RNA (n = 4 independent experiments). The asterisks indicate a significantly lower chromatin binding of CHD1 upon XPC depletion.
- Co‐immunoprecipitation of XPC and CHD1. HeLa cells were left unchallenged or UV‐treated. Following a 1‐h incubation, the chromatin was salt‐extracted and, after MNase fragmentation, dissolved by sonication. Immunoprecipitation was carried out with anti‐XPC antibodies. Input fractions and immunoprecipitated (IP) complexes were analyzed by blotting with antibodies against XPC and CHD1; –IgG, control pull‐down reactions without antibodies. Appendix Fig S3 displays a longer exposure of the same immunoblot showing the low background association of CHD1 with XPC in unchallenged cells.
- HeLa cells were transfected with the indicated siRNA sequences 2 days before UV irradiation. Following a 1‐h incubation, the irradiated cells were processed for immunoprecipitation with anti‐XPC antibodies as in Fig 3D. After immunoblotting, the level of co‐immunoprecipitated CHD1 was quantified and normalized to the amount of immunoprecipitated XPC in each individual sample (n = 3 independent experiments). For a representative blot, see Appendix Fig S4.
The above results indicated that XPC protein mediates the recruitment of CHD1 to UV lesions, implying that XPC and CHD1 interact transiently in UV‐irradiated cells. To test this prediction, HeLa cells were UV‐exposed and, 1 h later, their chromatin was collected, MNase‐fragmented, and solubilized by sonication as described for the experiments of Fig 1C. In this case, however, immunoprecipitations were carried out with anti‐XPC antibodies and control reactions were performed by omitting these antibodies (Fig 3D). The input chromatin samples brought to light the known constitutive association of XPC protein with chromatin even in unchallenged cells and also its expected ubiquitination after UV irradiation (Sugasawa et al, 2005). To avoid unspecific binding, the immunoprecipitation buffer was adapted to contain 1% (vol/vol) of the Triton X‐100 detergent and, therefore, the proportion of antibody‐precipitated XPC protein was low. Nevertheless, this XPC immunoprecipitation resulted in the co‐isolation of CHD1 from the chromatin. The amount of co‐immunoprecipitated CHD1 was higher after UV irradiation, reflecting an extra UV‐dependent recruitment to chromatin not only of XPC but also of CHD1 and lending support to the conclusion that XPC protein is responsible for the co‐localization of CHD1 with GG‐NER sites.
Next, we tested if other NER factors influence this XPC‐CHD1 interaction. Immunoprecipitations with anti‐XPC antibodies were carried out after siRNA transfections to downregulate the expression of DDB2, XPD, or XPA. In these experiments, the amount of co‐isolated CHD1 relative to the immunoprecipitated XPC protein was slightly reduced after downregulation of DDB2 and XPA, but none of these changes were statistically significant (Fig 3E). We conclude that, although DDB2 and XPA are not absolutely needed for the recruitment of CHD1 to chromatin (as shown in Fig 3A and C), the stringent condition used for the XPC immunoprecipitation reveals that these factors may stabilize XPC‐CHD1 interactions.
CHD1 stimulates XPC displacement and recruitment of downstream GG‐NER factors
Due to their larger nuclear surface area in culture, U2OS cells are more amenable than HeLa cells to immunofluorescence analyses. U2OS cells were depleted of CHD1 using siRNA to test by immunofluorescence the impact of this chromatin remodeler on the GG‐NER pathway. This downregulation reduced the level of CHD1 protein by ~80% within 2 days after transfection with siRNA (Fig 4A). However, such a substantial reduction in CHD1 protein did not affect the cell division cycle of unchallenged cells (Appendix Fig S1) and did not trigger any apoptotic responses leading to activation of caspase 3 (Appendix Fig S2). In view of its established role in transcription (Simic et al, 2003; Smolle et al, 2012; Park et al, 2014), it could have been expected that the downregulation of CHD1 may interfere with DNA damage processing by diminishing the expression of repair proteins. However, Fig 4A shows that the CHD1 depletion does not reduce the cellular level of GG‐NER factors like XPB, XPC, or DDB2 measured 2 days after transfection with siRNA. On the contrary, we consistently observed that cells respond to CHD1 depletion with a constitutively increased level of XPC protein (see quantification in Fig 4B). The high molecular weight forms of XPC in the immunoblot of Fig 4A (representing ubiquitinated XPC protein) also indicate that the CHD1 downregulation does not detectably interfere with the UV‐dependent ubiquitination reaction. Reflecting the overall higher XPC level, the amount of ubiquitinated XPC is increased in CHD1‐depleted cells. The CHD1 deficiency similarly does not impair the well‐described proteasomal degradation of DDB2 in response to UV irradiation (Fig 4A and B). The observed upregulation of XPC under conditions of a CHD1 deficiency already takes place at the mRNA level as indicated by quantitative reverse‐transcription PCR measurements (Fig 4C).
Figure 4. Depletion of CHD1 by siRNA treatment.
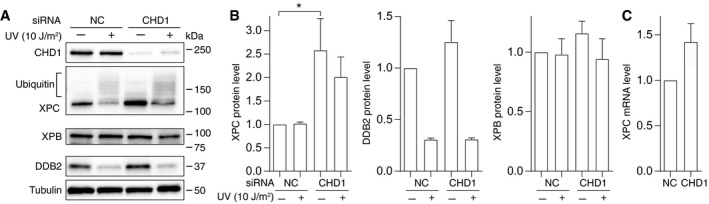
- Effect of CHD1 on NER protein levels. U2OS cells were transfected with non‐coding control RNA (NC) or siRNA against the CHD1 transcript and tested after 2 days. Cells were harvested for analysis 1 h after UV exposure (10 J/m2) or mock treatment. Immunoblots of whole‐cell lysates were carried out with the indicated antibodies. The higher molecular weight forms of XPC protein reflect its ubiquitination by the CRL4DDB2 ligase (Sugasawa et al, 2005). Similarly, the ubiquitin‐dependent degradation of DDB2 is a well‐described response to UV irradiation. Tubulin served as the loading control.
- Quantification of XPC, DDB2, and XPB protein levels determined by immunoblotting 2 days after transfection with siRNA (n = 3–6 independent experiments). The UV‐irradiated samples were analyzed 1 h after treatment, and the XPC amount in control cells was set to 1.
- Increased level of mRNA coding for XPC due to CHD1 depletion 2 days after transfection with siRNA (n = 3 independent experiments).
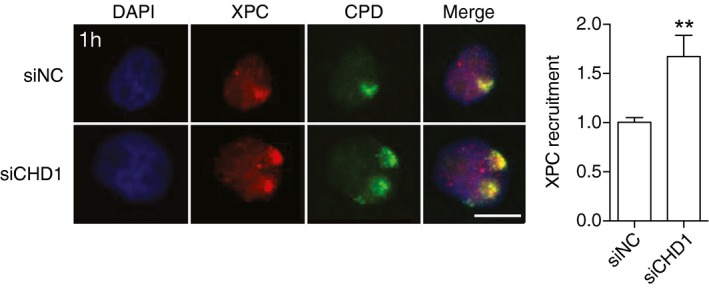
In situ immunofluorescence is recognized as a straightforward tool to monitor the assembly and disassembly of GG‐NER complexes in living human cells (Volker et al, 2001; Fitch et al, 2003). Here, this methodology was used to examine the effect of CHD1 depletion on the recruitment of GG‐NER factors to UV lesions. For that purpose, U2OS cells were irradiated with UV‐C light through the 5‐μm pores of filters to generate local spots of damage containing CPDs. Following 1 or 3 h of incubation, the formaldehyde‐fixed cells were permeabilized and stained with antibodies against CPDs and different GG‐NER proteins. These immunofluorescence studies revealed a differential effect of CHD1 depletion on distinct factors. Upon CHD1 downregulation, the level of the initial damage sensor XPC on spots of CPDs is increased relatively to controls (Fig 5A). This increased accumulation of XPC is statistically significant at 1 h after UV irradiation, and the same response is detected by immunofluorescence analyses of HeLa cells (Fig EV3). This prolonged binding of XPC to lesion sites is not observed upon depletion of the downstream NER factors XPD and XPA (Fig EV4) and, hence, represents a specific reaction to the lack of CHD1.
Figure 5. CHD1 promotes the XPC‐to‐TFIIH handoff.
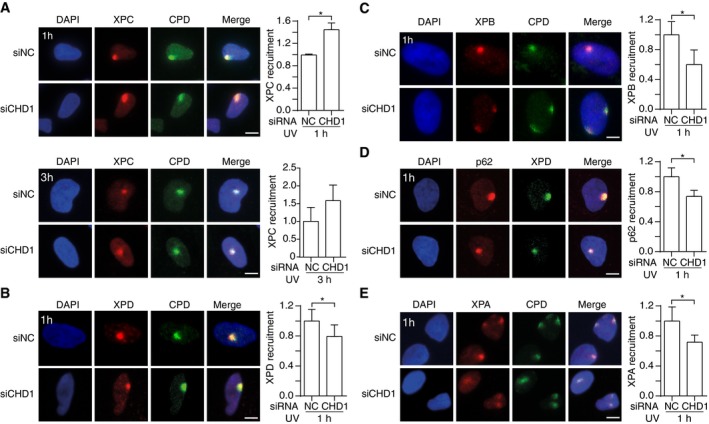
- Accumulation of XPC protein. Representative immunofluorescence images of U2OS cells UV‐irradiated through micropore filters (dose applied to the filter surface: 100 J/m2) to generate local spots of DNA damage. Immunostaining was carried out after 1 or 3 h with antibodies against CPDs and XPC protein. Cells were pretreated with siRNA targeting the CHD1 transcript (siCHD1) or with non‐coding control RNA (siNC). DAPI (4′,6‐diamidino‐2‐phenylindole) was used to stain nuclear DNA. The recruitment of NER subunits was quantified by measuring spot intensities followed by normalization to the nuclear background (n = 6, 100 cells for each experiment). Control values were set to 1.
- Reduced recruitment to UV lesion spots of XPD, a subunit of the TFIIH complex, upon CHD1 depletion. Cells were analyzed 1 h after the UV pulse. The panel shows a representative immunofluorescence image and the quantification over 3 independent experiments with at least 100 cells per experiment.
- Representative image and quantification (n = 3, at least 100 cells per experiment) demonstrating the reduced recruitment to UV lesion spots of XPB (another TFIIH subunit) upon CHD1 depletion. Cells were analyzed 1 h after the UV pulse.
- Representative image and quantification (n = 6, 100 cells per experiment) demonstrating the reduced recruitment to UV lesion spots of p62 (yet another TFIIH subunit) upon CHD1 depletion. Cells were analyzed 1 h after the UV pulse. XPD was used to mark the UV spots, because it was not possible to stain p62 and CPDs simultaneously.
- Representative image and quantification (n = 6, 100 cells per experiment) demonstrating the reduced XPA recruitment to UV lesion spots upon CHD1 depletion. Cells were analyzed 1 h after the UV pulse.
Figure EV3. XPC accumulation in the chromatin of UV‐irradiated HeLa cells.
Representative immunofluorescence images of HeLa cells that were UV‐irradiated (dose applied to filters: 100 J/m2) through micropore filters to generate local spots of DNA damage. Immunostaining was carried out after 1 h with antibodies against CPDs and XPC protein. Cells were pretreated 2 days earlier with siRNA targeting the CHD1 transcript (siCHD1) or with non‐coding control RNA (siNC). DAPI was used to stain nuclear DNA. Scale bar: 10 μm. The recruitment of XPC protein was quantified by measuring spot intensities followed by normalization to the nuclear background. Control values were set to 1. Data are presented as mean ± SEM (n = 3, 100 cells for each experiment). **P ≤ 0.01 (unpaired, two‐tailed t‐test).
Figure EV4. XPC recruitment to local spots of UV damage.
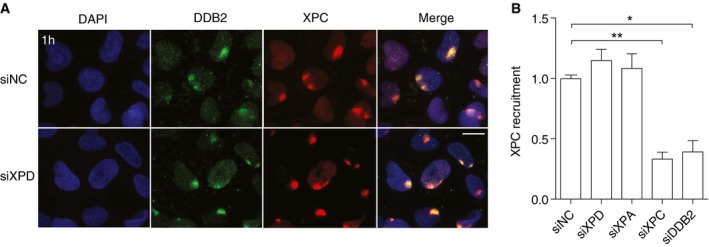
- Representative immunofluorescence images of U2OS cells that were UV‐irradiated (dose applied to filters: 100 J/m2) through micropore filters to generate local spots of DNA damage. Immunostaining was carried out after 1 h with antibodies against DDB2 (as a marker of UV lesions) and XPC proteins. Cells were pretreated 2 days earlier with siRNA targeting the XPD transcript (siXPD) or with non‐coding control RNA (siNC). DAPI was used to stain nuclear DNA. Scale bar: 10 μm.
- The recruitment of XPC protein was quantified by measuring spot intensities followed by normalization to the nuclear background. Control values were set to 1. Cells were pretreated 2 days earlier with siRNA targeting the XPD, XPA, XPC, or DDB2 transcripts, as indicated, or with non‐coding control RNA (siNC). Data are presented as mean ± SEM (n = 3, 100 cells for each experiment). *P ≤ 0.05, **P ≤ 0.01 (unpaired, two‐tailed t‐test).
To quantify protein redistributions, the fluorescence intensity at damaged spots was divided by the background fluorescence measured in each nucleus outside the lesion spots. This procedure ensures that the data demonstrate a truly increased accumulation of XPC protein at lesion sites rather than simply reflecting the higher overall level of this factor following CHD1 depletion. In contrast to the increased accumulation observed for XPC as the pathway initiator, the redistribution of downstream NER factors, assessed 1 h after UV irradiation, is reduced upon CHD1 depletion. A diminished recruitment to UV lesions following CHD1 depletion is observed for the TFIIH subunits XPD (Fig 5B), XPB (Fig 5C) or p62 (Fig 5D), and also for XPA protein (Fig 5E). This differential effect on factor recruitment to UV lesion sites indicates that CHD1 stimulates the coordinated transition from the XPC complex (as the initiator of the GG‐NER pathway) to the follow‐up effector TFIIH and, consequently, to further downstream factors like XPA. In the absence of CHD1, XPC persists on UV lesion sites without being able to hand over the damage to the TFIIH complex. The fact that this prolonged binding of XPC to lesion sites, caused by the lack of CHD1, is not reproduced by depletions of XPD or XPA (Fig EV4) argues against the possibility that this observation represents solely an indirect effect of downstream factors not being recruited efficiently.
CHD1 stimulates CPD excision and reduces UV cytotoxicity
Functional consequences of a CHD1 downregulation (see Fig EV2 for the efficiency of protein depletion) were tested by monitoring the formation and excision of UV lesions in HeLa cells. The lack of CHD1 induced by siRNA treatment does not influence the MNase digestion pattern of chromatin (Fig 6A), indicating that the overall nucleosome assembly is unchanged. Consistent with this maintained chromatin configuration, the initial damage formation (frequency of CPDs and 6‐4PPs) following UV irradiation is not affected by the lack of CHD1 (Fig 6B). However, the excision of CPDs is significantly slowed down upon CHD1 depletion in comparison with the respective excision in control cells transfected with non‐coding RNA. After 24 h of repair incubation, nearly 70% of the initial CPDs were excised in control cells but only 45‐55% of CPDs were repaired in CHD1‐depleted cells. This same inhibitory effect of CHD1 depletion was induced by three different siRNA sequences directed against the CHD1 transcript (Fig 6C). In a side‐by‐side comparison, this reduction in CPD excision after downregulation of CHD1 relative to non‐coding RNA controls was similar to that observed upon depletion of the chromatin remodeler ALC1 as previously reported (Pines et al, 2012) (Fig EV5). In contrast, the CHD1 depletion had no effect on the repair of 6‐4PPs, which are removed from the genome with faster kinetics than CPDs (Fig 6D).
Figure 6. CHD1 stimulates CPD repair.
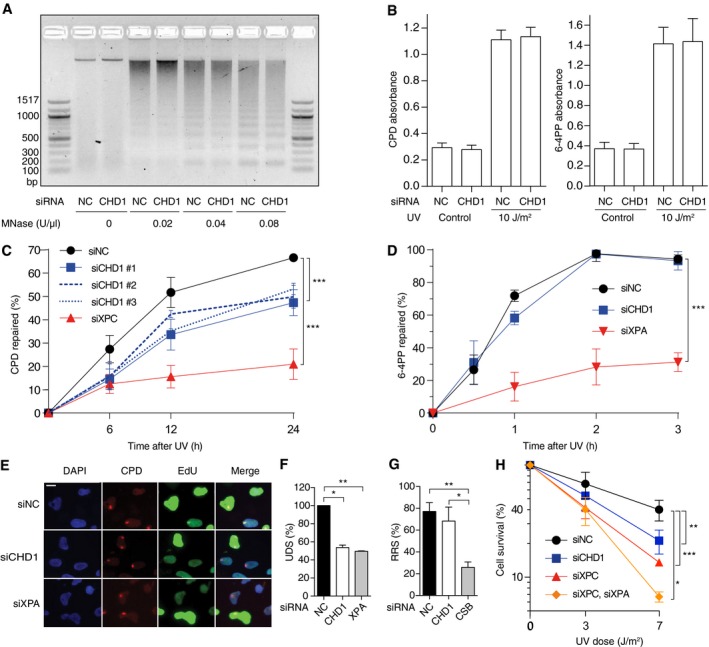
- MNase digestion of the chromatin of CHD1‐depleted HeLa cells in comparison with control cells transfected with non‐coding RNA (NC). Isolated chromatin was incubated with the indicated nuclease concentrations.
- Initial damage formation following UV irradiation (10 J/m2). HeLa cells were transfected with siCHD1 or siNC. Immunoassay absorbance values, providing a measure of UV lesions, were not affected by CHD1 depletions (n = 6, each experiment with four replicates).
- Excision of CPDs in HeLa cells treated with siRNA targeting CHD1 (three different sequences) or XPC, compared to transfections with siNC (n = 6, each experiment with four replicates). The UV dose was 10 J/m2.
- Excision of 6‐4PPs upon treatment with siRNA targeting CHD1 or XPA, in comparison to siNC (n = 3, each experiment with four replicates). The UV dose was 10 J/m2.
- U2OS cells were pretreated with siRNA as indicated and UV‐irradiated through micropore filters 2 days later (dose applied to the filter surface: 100 J/m2). EdU was added after a 2‐h recovery to allow for 6‐4PP excision, and fluorescence reflecting repair synthesis was measured after another 1‐h incubation. Lesion spots were identified by staining with antibodies against CPDs. EdU incorporation was detected by copper‐mediated reaction with the Alexa 488 fluorophore. DAPI was used to visualize nuclear DNA. S‐phase cells displaying an overall bright nuclear EdU signal were excluded from quantifications. Scale bar: 15 ?m.
- Quantification of EdU incorporation reflecting repair synthesis (UDS, unscheduled DNA synthesis) in the damage spots of CHD1‐ or XPA‐depleted U2OS cells normalized to control cells. S‐phase cells were excluded from these evaluations (n = 3 with at least 100 cells per experiment).
- Recovery of RNA synthesis (RSS) assessed by monitoring the nuclear incorporation of EU during 16 h after UV irradiation (10 J/m2) of U2OS cells. Cells were depleted of CHD1 or CSB as indicated (n = 3, 100 cells per experiment). RNA synthesis values, reported as the percentage of unirradiated control cells, show that only the depletion of CSB impairs TC‐NER activity.
- HeLa cells transfected with the indicated siRNA sequences were UV‐irradiated or mock‐treated. Colony survival was quantified 7 days later and expressed as the percentage of controls in a logarithmic scale (n = 3, each experiment with four replicates).
Figure EV5. Comparison between chromatin remodelers.
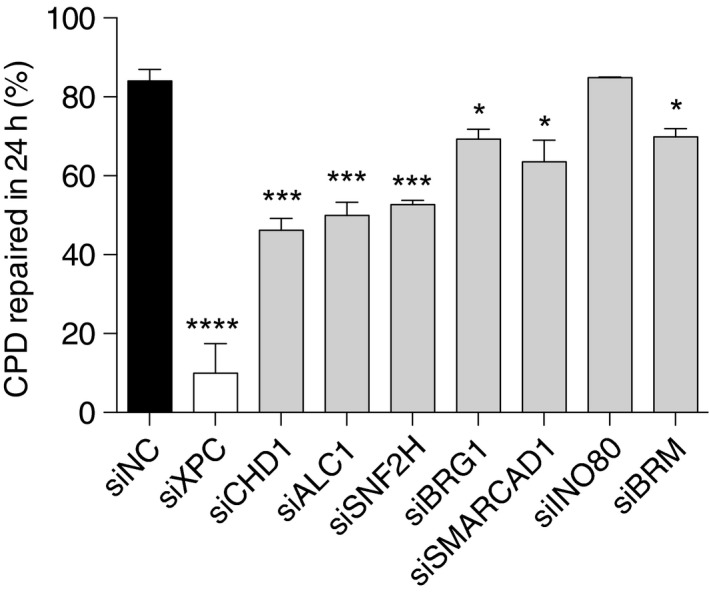
Excision of CPDs in HeLa cells treated with siRNA (16 nM) targeting the indicated chromatin‐remodeling factors, compared to transfection with siNC. The cells were UV‐irradiated (10 J/m2) 2 days after siRNA transfections, and the proportion of excised CPDs was determined after a repair incubation of 24 h. The efficiency of protein downregulation is shown in the immunoblots of Fig EV2A and D. Data are presented as mean ± SEM (n = 5–7 independent experiments with four replicates). ****P ≤ 0.0001, ***P ≤ 0.001, *P ≤ 0.05 (unpaired, two‐tailed t‐test).
To confirm the role of CHD1 in stimulating CPD excision, we monitored the rates of DNA repair patch synthesis. Spots of UV damage were generated in the nuclei of U2OS cells and, for the measurement of repair synthesis elicited specifically by CPDs, these cells were first incubated for 2 h to allow for the removal of 6‐4PPs and then supplemented with the nucleoside analog 5‐ethynyl‐2′‐deoxyuridine (EdU) (Nakazawa et al, 2010) for another 1‐h period. The fluorescence in CPD spots, due to EdU incorporation, revealed that like an XPA deficiency the depletion of CHD1 causes lower levels of DNA repair patch synthesis compared to controls (Fig 6E). The quantification of EdU signals within spots of DNA damage confirmed that DNA repair synthesis takes place in CHD1‐depleted cells at a significantly lower rate than in control cells (Fig 6F). Thus, both CPD excision and unscheduled DNA synthesis are reduced in cells depleted of CHD1, indicating that this remodeler is required for efficient processing of CPDs by the GG‐NER reaction.
Conversely, transcription‐coupled NER (TC‐NER) can be monitored by comparing transcription rates after DNA damage. UV radiation causes a decrease in RNA synthesis, which recovers readily in normal cells due to TC‐NER activity (Nakazawa et al, 2010; Aydin et al, 2014). We globally irradiated U2OS cells, incubated them for 16 h in the presence of 5‐ethynyl uridine (EU) and, thereafter, the EU‐linked fluorescence reflecting RNA synthesis was measured across cell nuclei. As expected, the recovery of RNA synthesis was delayed in cells depleted of Cockayne syndrome group B (CSB) protein required for the TC‐NER reaction. In contrast, the CHD1 depletion did not interfere with this recovery of RNA synthesis (Fig 6G) indicating that, although CHD1 regulates GG‐NER activity, it is not involved in the TC‐NER pathway. Finally, HeLa cell colony assays demonstrate that the reduced rate of CPD repair observed upon CHD1 depletion correlates with significantly lower survival following UV irradiation (Fig 6H).
Discussion
The GG‐NER system needs to process bulky base lesions in condensed chromatin, where genomic DNA is organized in nucleosomes thought to act as physical barriers to damage recognition and repair (Thoma, 2005; Bell et al, 2011; Rodriguez et al, 2015; Adam et al, 2016; Dabin et al, 2016). Being the fundamental repeat unit of chromatin, each nucleosome consists of a core particle containing 147 base pairs of DNA wrapped in superhelical turns ~1.7 times around an octamer of two each of the histone proteins H2A, H2B, H3, and H4. These nucleosome cores are spaced by linker DNA segments of variable lengths (generally 10–70 base pairs) and further interactions with histone H1 promote higher levels of chromatin condensation (Grigoryev, 2012). Within the 147 base pairs of nucleosome core DNA, CPDs arise with a periodicity pattern of 10.3‐nucleotide intervals and are preferentially introduced at sites farthest from the surface of the histone octamer (Gale et al, 1987).
The modulation of chromatin structure by ATP‐dependent remodelers has been identified as one important mechanism that promotes accessibility of the GG‐NER complex to damaged DNA (Ura et al, 2001; Gong et al, 2006; Klochendler‐Yeivin et al, 2006; Ray et al, 2009; Zhang et al, 2009; Zhao et al, 2009; Jiang et al, 2010; Pines et al, 2012). Our present study identifies the chromatin remodeler CHD1 as an accessory GG‐NER factor that facilitates the repair of a subset of CPD lesions located within nucleosome cores. We discovered (i) that a fraction of the cellular CHD1 is recruited to chromatin upon UV irradiation (Fig 1A), (ii) that this extra CHD1 in the chromatin of UV‐irradiated cells co‐localizes with GG‐NER factors (Fig 1C), (iii) that the recruitment of CHD1 to UV damage occurs in a nucleosome core‐enriched fraction (Fig 2), (iv) that this UV‐dependent CHD1 recruitment to nucleosome cores relies on a prior lesion demarcation by XPC protein (Fig 3), (v) that CHD1 stimulates the XPC‐to‐TFIIH handoff at damaged sites (Fig 5) and, therefore, (vi) that the lack of CHD1 slows down the excision of CPDs and enhances the cytotoxicity of UV light (Fig 6). The inhibitory effect of a CHD1 depletion on the repair of CPDs, formed everywhere in the genome, but not on the excision of 6‐4PPs, induced predominantly in linker segments of euchromatin (Gale & Smerdon, 1990; Mitchell et al, 1990; Han et al, 2016), supports the notion that chromatin remodeling by CHD1 is required to ensure the access of GG‐NER factors to DNA damage located within nucleosome cores. Additionally, the measurement of RNA synthesis following UV radiation confirmed that, unlike CSB as an example of TC‐NER factor, CHD1 is not involved in the TC‐NER subpathway (Fig 6G).
Whether CHD1 displaces nucleosomes while facilitating the known interactions between XPC and TFIIH (Yokoi et al, 2000; Uchida et al, 2002) during the GG‐NER process is not known. However, it is of interest to note that such a role has been postulated for CHD1 during transcription initiation. In fact, CHD1 associates with the chromatin template just downstream of transcription start sites at an early stage of transcription (Skene et al, 2014). CHD1 is thereby recruited to the promoter‐proximal nucleosomes of active genes and thought to evict this nucleosome to allow for promoter escape by RNA polymerase II. Skene et al (2014) showed that, in the absence of CHD1 activity, RNA polymerase II remains sequestered on these promoter‐proximal nucleosomes. Our observation that, like RNA polymerase II during transcription initiation, also XPC protein remains sequestered at its binding sites in the absence of CHD1, suggests a similar remodeling activity of CHD1 during initiation of the GG‐NER process. A plausible scenario is that the function of CHD1 in stimulating the XPC‐to‐TFIIH handoff on nucleosomes is facilitated by eviction of the targeted nucleosome.
Although CHD1 co‐localizes in chromatin with DDB2 (Fig 1C), it might be surprising that its recruitment to nucleosomes depends on XPC but apparently not on the accessory DDB2 subunit (Fig 3). Indeed, previous biochemical studies suggested that XPC protein loses the ability to interact with DNA once the substrate is wrapped around histone octamers in nucleosomes (Yasuda et al, 2005), suggesting that preceding DDB2‐induced rearrangements are indispensable before the DNA substrate becomes accessible to the XPC complex (Scrima et al, 2008; Osakabe et al, 2015). It is important in this regard to point out that CPDs are the predominant UV lesions within nucleosome cores (Gale & Smerdon, 1990; Mitchell et al, 1990; Han et al, 2016) and that, when tested in binding assays with naked DNA substrates, the XPC complex is unable to sense the presence of CPDs (Sugasawa, 2001; Hey et al, 2002). If not embedded in chromatin, CPDs appear as non‐distorting lesions that preserve Watson–Crick hydrogen bonding (Kim et al, 1995; Jing et al, 1998; McAteer et al, 1998) and remain, therefore, invisible to the DNA damage‐sensing domains of XPC protein. However, a recent crystal structure of nucleosome cores containing CPD lesions revealed that, unlike their configuration in naked DNA, the two affected pyrimidines do not form proper Watson–Crick hydrogen bonds with the opposite purines and that hydrogen bonds are actually destabilized at one pyrimidine of the CPD lesion (Horikoshi et al, 2016). This substantial local distortion and base pair destabilization detected on nucleosome cores may render the lesions more conducive to recognition by XPC, such that a subset of CPDs in the nucleosome landscape of chromatin becomes amenable to the GG‐NER process even in the absence of DDB2 protein. This scenario may explain our previously reported finding that XPC protein binds to UV lesions in nucleosome core particles but not to UV lesions in internucleosomal linkers in the absence of DDB2 (Fei et al, 2011). In any case, a local distortion of CPDs induced by wrapping around histone octamers may account for the finding of Fig 3 that the XPC‐dependent recruitment of CHD1 to nucleosomes could occur without assistance from the DDB2 subunit. An alternative explanation for this finding is that even trace amounts of DDB2 remaining in the cells after siRNA‐mediated depletion might still be sufficient to load XPC complexes onto these more conducive CPDs located on nucleosome cores.
To summarize, we identified CHD1 as an XPC‐associated remodeler facilitating GG‐NER activity in chromatin. Our findings suggest a scenario by which CHD1 is required for the effective handover of CPD lesions from the XPC initiator to the TFIIH effector when the target lesions are wrapped around the histone octamer of nucleosomes. These findings imply that nucleosomes are not simply an impediment to initiation of the GG‐NER pathway but act as a structural scaffold that, in the presence of CHD1, facilitates the repair of a subset of CPD lesions.
Materials and Methods
Cell lines
HeLa and HEK293 cells (American Type Culture Collection) were grown in Dulbecco's modified Eagle's medium (low‐glucose DMEM; ThermoFisher), U2OS cells (ATCC, cell type certified by STR profiling) in high‐glucose DMEM (Sigma) supplemented with 10% (vol/vol) fetal calf serum (FCS, Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin.
RNA transfections
The following RNAi sequences were used: CHD1#1 (5′‐CAUCAAGCCUCAUCUAAUAtt‐3′) from Ambion; CHD1#2 and CHD1#3 (5′‐AUGCAGAAAUUAGGCGGUUUAtt‐3′ and 5′‐AAGAUUCCGAUGACUCAUCAAtt‐3′) from Qiagen (CHD1 sequence #1 is used unless otherwise stated); DDB2 (5′‐AGGGAUCAAGCAGUUAUUUGA‐3′) from Qiagen; XPA (5′‐GCUACUGGAGGCAUGGCUAtt‐3′) from Qiagen; XPC (5′‐UAGCAAAUGGCUUCUAUCGAA‐3′) from Microsynth and CSB (5′‐GAAGCAAGGUUGUAAUAAAtt‐3′) from Microsynth. The non‐coding control RNA was from Qiagen. Cells were transfected in 10‐cm dishes with siRNA (10 or 16 nM) following the manufacturer's protocol for the Lipofectamine RNAiMAX (Invitrogen) reagent and allowed to incubate for 48 h before starting the experiments.
Determination of mRNA
For gene expression analysis, total RNA from U2OS cells was isolated using an RNase isolation kit (Qiagen) according to the manufacturer's protocol. DNA was removed by DNase I (Qiagen) digestion. RNA concentration was determined in a NanoDrop instrument (Thermo Scientific). One μg RNA from each sample and 3 μg/μl random primers (Invitrogen) were subjected to reverse transcription (Roche) according to manufacturer's protocol. Fifty ng cDNA, 0.5 μl of FAM‐tagged XPC primers (Life Technology), and 0.5 μl of VIC‐tagged GAPDH primers (Life Technology) were applied to qRT–PCR according to manufacturer's protocol (Life Technology, TaqMan Fast Universal PCR Master Mix). The relative gene expression levels are presented as 2−ΔΔCT and normalized to the sample treated with non‐coding siRNA.
Antibodies
The following antibodies, listed according to supplier, were used at the indicated dilutions. Abcam: mouse anti‐DDB2 (ab51017, 1:50 for immunofluorescence, 1:200 for immunoblotting), mouse anti‐XPC (ab6264, 1:1,000 for immunoblotting), mouse anti‐p62 (ab55199, 1:300 for immunofluorescence). Cell Signaling: rabbit anti‐caspase 3 (9501S, 1:1,000 for immunoblotting). Cosmo Bio: mouse anti‐CPD [NMDND001, 1:1,000 for immunofluorescence, 1:5,000 for enzyme‐linked immunosorbent assay (ELISA)], mouse anti‐6‐4PP (NMDND002, 1:1,000 for ELISA). Invitrogen: Alexa Fluor 488 and 594 goat anti‐mouse IgG (1:400 for immunofluorescence). Protein‐Tech: rabbit anti‐CHD1 (20576‐1‐AP, 1:100 for immunoblotting). Santa‐Cruz: mouse anti‐CHD1 (sc‐271626, 1:500 for immunoblotting), goat anti‐H3 (sc‐8654, 1:10,000 for immunoblotting), rabbit anti‐XPB (sc‐293, 1:100 for immunofluorescence), rabbit anti‐XPA (sc‐853, 1:100 for immunoblotting). Sigma: mouse anti‐α‐tubulin (T5168, 1:10,000 for immunoblotting), mouse anti‐FLAG M2 (F3165, 1:1,000 for immunoprecipitation), rabbit anti‐XPC (X1129, 1:100 for immunofluorescence and immunoprecipitation), peroxidase anti‐mouse IgG (1:20,000), peroxidase anti‐rabbit IgG (1:20,000).
UV irradiation
Exposure to UV‐C light was carried out in culture dishes at the indicated doses with a germicidal lamp (wavelength 254 nm) after washing the cells with phosphate‐buffered saline (PBS) and removal of residual buffer. Local damage was generated by irradiation with 100 J/m2 through a 5‐μm polycarbonate filter (Whatman). After UV irradiation, the cells were incubated for the indicated times with fresh culture medium.
Immunoblotting
Cells were treated as indicated, washed with Puck's EDTA (137.0 mM NaCl, 5.4 mM KCl, 5.6 mM glucose, 4.2 mM NaHCO3, 0.7 mM EDTA) and lysed in 100 μl of 1% Triton buffer [150 mM KCl, 25 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 2 mM β‐mercaptoethanol, 5% (vol/vol) glycerol, 1 mM N‐ethylmaleimide, and 1% (vol/vol) Triton X‐100] supplemented with protease inhibitor cocktail (Roche). Protein concentrations were measured by the bicinchoninic acid assay (Pierce). A Laemmli buffer stock [final concentration: 240 mM Tris‐HCl, pH 6.8, 40% (v/v) glycerol, 8% (w/v) sodium dodecyl sulfate (SDS), 0.04% (w/v) bromophenol blue and 5% beta‐mercaptoethanol (v/v)] was added, and the samples were heated to 95°C for 5 min. Fifty μg of sample proteins were separated in 4–20% Criterion TGX Stain‐Free precast gels (Bio‐Rad) for 22 min at 300 V and transferred to PVDF membranes using the Turbo transfer device (Bio‐Rad, 7 min at 5 A). The signals resulting from antibody incubations were analyzed and quantified with the Chemidoc MP Imaging System (Bio‐Rad).
Cell lysis and chromatin digestion
Chromatin was fragmented as described (Fei et al, 2011). Protein synthesis was inhibited by the addition of cycloheximide (100 μg/ml; Sigma) for 30 min prior to UV irradiation. Cells were irradiated with the indicated UV‐C doses and lysed on ice with NP‐40 buffer [25 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM EDTA, 10% (vol/vol) glycerol, 1% (vol/vol) NP‐40, 0.25 mM phenylmethylsulfonyl fluoride, and EDTA‐free protease inhibitor cocktail (Roche)] (Sugasawa et al, 2005). Lysis was carried out for 30 min on a turning wheel. Free proteins not bound to chromatin were recovered in the supernatant after centrifugation (10 min at 15,000 g). The remaining chromatin pellet was resuspended in CS buffer [20 mM Tris–HCl, pH 7.5, 100 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 0.3 M sucrose, and 0.1% (vol/vol) Triton X‐100] (Kapetanaki et al, 2006). This mixture was supplemented with 10‐fold reaction buffer [500 mM Tris–HCl, pH 7.9, 50 mM CaCl2, 0.1 mg/ml bovine serum albumin (New England Biolabs). MNase (0.4 U/μl; New England Biolabs) was added and digestion carried out for 20 min at 37°C. The solubilized constituents were separated from the insoluble pellet by centrifugation (10 min, 15,000 g) after adding EDTA (5 mM final concentration) to stop the reaction.
MNase profiling
U2OS cells were irradiated with UV‐C (10 J/m2) and lysed on ice 1 h later with NP‐40 buffer; the chromatin pellet was resuspended in CS buffer, and the mixture was supplemented with 10‐fold reaction buffer as outlined above. MNase was added in different concentrations, the digestion carried out for 5 min at 37°C and the reaction stopped with 5 mM EDTA. The DNA was extracted from each digested sample (50 μl) by adding 150 μl TE buffer [10 mM Tris–HCl, pH 8.0, 1 mM EDTA] and 200 μl neutral phenol (ThermoFisher). After shaking for 15 min and centrifugation (5 min at 6,000 g), the phenol was discarded and the aqueous phase was washed twice with 200 μl chloroform. The DNA was precipitated with ethanol in the presence of 100 mM sodium acetate, dried, and resuspended in TE buffer. DNA concentrations were determined in the NanoDrop device.
Pull‐down of chromatin‐associated GG‐NER complexes
HEK293 cells were transiently transfected at 80% confluency with plasmid DDB2‐p3XFLAG‐14‐N3 (10 μg) according to the manufacturer's protocol for the FuGENE® HD reagent (Roche), and UV‐irradiated 24 h later. Following another 30 min, the cells were lysed on ice in NP‐40 buffer, the remaining pellet was resuspended in CS buffer and the mixture was supplemented with 10‐fold reaction buffer. The MNase digestion (4 U/μl) was carried out for 20 min at 37°C. The residual insoluble chromatin was recovered by centrifugation (10 min, 16,000 g) and resuspended by sonication on an ice‐water bath (three cycles of 30 s with 30‐s intervals) in CS buffer. Subsequently, nucleoprotein complexes bound to the DDB2‐FLAG prey were purified using 40 μl of anti‐FLAG M2 Affinity Gel (Sigma) in the presence of IP buffer [150 mM NaCl, 50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 1% (vol/vol) NP‐40, 2 mM β‐mercaptoethanol, 5% (vol/vol) glycerol, and all protease inhibitors]. Beads were incubated overnight at 4°C with the sonicated mixture and washed once with TNET [50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% (vol/vol) Triton X‐100, and protease inhibitors] and once with TBS (50 mM Tris‐HCl, pH 7.4, 150 mM NaCl and protease inhibitors). The beads were heated to 95°C for 5 min in 10 μl TBS complemented with 5.0 μl of Laemmli buffer stock. The samples were separated on a 10% (wt/vol) denaturing polyacrylamide gel and transferred to PVDF membranes. Proteins were subsequently detected by immunoblotting.
Pull‐down of XPC protein
After washing on ice with PBS, 10 μl of slurry Protein G sepharose (GE Healthcare) for each sample was washed twice with PBS and then incubated for 45 min at 4°C on a turning wheel with 4 μl anti‐XPC antibodies. After centrifugation (1 min, 100 g), the protein G sepharose was suspended in 100 μl buffer A [0.5 M Tris–HCl, pH 8.0, 20% (vol/vol) glycerol, 300 mM NaCl, 2% (vol/vol) Triton X‐100, 2 mM EDTA, 0,25 mM phenylmethylsulfonyl fluoride and ETDA‐free protease inhibitor cocktail (Roche)], added to 100 μg of fragmented and sonicated chromatin, and incubated for 3 h at 4°C on a turning wheel. The beads were washed twice by centrifugation (2 min, 100 g) in HNTG buffer [20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% (vol/vol) Triton X‐100, and 10% (vol/vol) glycerol] and the samples were analyzed by polyacrylamide gel electrophoresis and immunoblotting.
Immunofluorescence microscopy
Cells were grown on 12‐mm glass coverslips (Thermo Scientific) to 80% confluency and irradiated through filters to induce local spots of UV damage. After the indicated repair times, cells were processed with pre‐extraction buffer [25 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% (vol/vol) Triton X‐100] added for 2.5 min at 4°C. Next, cells were fixed with 4% (wt/vol) paraformaldehyde for 15 min and permeabilized for 20 min with PBS containing 0.1% (vol/vol) Tween 20. For the detection of CPDs and 6‐4 PPs, the DNA was denaturated for 8 min with 0.07 M NaOH in PBS. Following a 30‐min blocking step with PBS containing 20% (vol/vol) FCS, primary antibodies were diluted in PBS containing 5% (vol/vol) FCS and applied for 1 h at 37°C. Washing with PBS–0.1% Tween 20 was followed by incubation with secondary antibodies and DAPI (0.2 μg/ml), diluted in PBS containing 5% FCS, for 1 h at 37°C. Images were taken with a bright field microscope (Leica, 63× oil Plan‐Apochromat, 1.4 numerical aperture oil immersion lens) and analyzed using the ImageJ software. The fluorescence of 100 nuclei was examined, and the accumulation of proteins at UV lesion sites is expressed as the ratio of fluorescence signal intensity in damaged spots relative to the signal intensity of the surrounding nuclear area.
Unscheduled DNA synthesis and RNA synthesis
The synthesis of DNA repair patches was measured by a fluorescence‐based method (Nakazawa et al, 2010). U2OS cells were seeded on 12‐mm coverslips and locally UV‐irradiated. After 2 h, the culture medium was supplemented with 10 mM EdU (Invitrogen) followed by another 1 h of incubation. Cells were washed with PBS, pre‐extracted for 2.5 min, fixed with 4% (wt/vol) paraformaldehyde for 15 min, and permeabilized for 20 min. Antibodies against CPDs were applied as described above. Incorporated EdU was coupled to Alexa Fluor 488 using the Click‐it kit as instructed by the manufacturer (Invitrogen). Images were obtained by microscopy with the Leica instrument and analyzed using the ImageJ software. For quantifications, EdU incorporation was measured in 100 cells by determining fluorescence intensity in the UV‐damaged areas (marked by CPD staining) divided by the background nuclear intensity. S‐phase cells displaying high EdU fluorescence across their entire nucleus were excluded. For the measurement of RNA synthesis, cells were UV‐irradiated globally (10 J/m2) and incubated for 16 h in culture medium supplemented with 100 μM EU (Thermo Scientific). Thereafter, cells were washed, pre‐extracted, fixed, and permeabilized as outlined above. EU incorporation was analyzed in 100 cells per treatment by determining fluorescence intensity of the whole nucleus divided by the background slide intensity.
Quantification of UV lesions
Formation and removal of UV lesions was detected by ELISA as described (Fei et al, 2011). Briefly, whole‐genome DNA was extracted using the DNeasy kit (Qiagen) and denatured by heating to 99°C for 10 min, followed by a 15‐min incubation on ice. A volume of 50 μl per well of denatured DNA (at a concentration of 4 μg/ml for 6‐4PP detection, 200 ng/ml for CPD detection) was distributed into a 96‐well microtiter plate (Greiner) coated with protamine sulfate (Sigma) and dried overnight at 37°C. The DNA‐coated plates were washed five times with PBST [0.05% (vol/vol) Tween 20 in PBS] and blocked with 2% (vol/vol) FBS in PBS at 37°C for 60 min. The antibodies against either 6‐4PPs (64M‐2) or CPDs (TDM‐2) were applied for 30 min (at 37°C). Primary antibodies bound to DNA were recognized by biotin‐labeled F(ab′)2 fragments of anti‐mouse IgG (1:2,000; Invitrogen) added for 30 min at 37°C. After washing the plates, 100 μl of a peroxidase–streptavidin conjugate (1:10,000; Invitrogen) was distributed into each well. The reaction was started by adding 0.5 mg/ml o‐phenylenediamine, 0.007% (vol/vol) H2O2, and citrate‐phosphate buffer (50 mM Na2HPO4, 24 mM citric acid, pH 5.0), stopped with 50 μl of 2 M H2SO4, and monitored by measuring the absorbance at 492 nm in a PLUS384 microplate spectrophotometer (Molecular Devices).
Colony forming assay
HeLa cells treated as indicated were seeded in different dilutions and left for 7 days at 37°C to allow for colony formation. The growing colonies were stained with 0.5% (w/v) crystal violet in 80% ethanol and counted.
Cell cycle analysis
HeLa cells were arrested in G1 by a 24‐h treatment with mimosine (0.5 mM, Sigma). UV‐exposed cells were allowed to recover for the indicated times and labeled with 10 μM EdU for 1 h prior to harvesting. Cell cycle profiles were analyzed using the Life Technologies Click‐iT Edu Alexa Fluor 488 Flow Cytometry Assay kit. Briefly, cells were fixed in 1% (wt/vol) PFA/PBS (Sigma) for 10 min and permeabilized in saponin buffer for 10 min; 200,000 cells were incubated with a mouse anti‐γH2AX antibody (Millipore, 1:2,000) for 1.5 h and with an Alexa Fluor 647 anti‐mouse antibody (Invitrogen A31571, 1:50) for 30 min. EdU was coupled to Alexa Fluor 488 azide for 30 min. Cells were treated with 0.1 mg/ml RNase and DNA was stained with 1 μg/ml DAPI, followed by analysis in a CyAn ADP flow cytometer (Beckman Coulter). Results were analyzed with Flow Jo 10 data analysis software (FLOWJO, LLC).
Statistics
GraphPad Prism 6 was used to perform unpaired, two‐tailed t‐tests as outlined in the figure legends. One‐sample t‐test with a hypothetical value of 1.0 was applied for independent immunoblot assays. P‐values expressed as *P < 0.05, **P < 0.01 and ***P < 0.001 were considered to indicate statistical significance.
Author contributions
PR, CBP, TC, and HN devised and planned the experiments; PR, CBP, TC, and ZG carried out the experiments and analyzed the data; and PR, CBP, and HN wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
This work was supported by the Swiss National Science Foundation (Grant 170111/1), the Velux Foundation (Project 753), and the Swiss Cancer League (2832‐02‐2011). We also acknowledge support by the Center of Clinical Studies.
The EMBO Journal (2017) 36: 3372–3386
References
- Adam S, Dabin J, Chevallier O, Leroy O, Baldeyron C, Corpet A, Lomonte P, Renaud O, Almouzni G, Polo S (2016) Real‐time tracking of parental histones reveals their contribution to chromatin integrity following DNA damage. Mol Cell 64: 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F (2001) Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 276: 18665–18672 [DOI] [PubMed] [Google Scholar]
- Araújo SJ, Tirode F, Coin F, Pospiech H, Syväoja JE, Stucki M, Hübscher U, Egly J‐M, Wood RD (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev 14: 349–359 [PMC free article] [PubMed] [Google Scholar]
- Aydin ÖZ, Vermeulen W, Lans H (2014) ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle 13: 3016–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thomä NH, Schübeler D (2011) Determinants and dynamics of genome accessibility. Nat Rev Genet 12: 554–564 [DOI] [PubMed] [Google Scholar]
- Bunick CG, Miller MR, Fuller BE, Fanning E, Chazin WJ (2006) Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 45: 14965–14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Egly J‐M (2016) Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu Rev Biochem 85: 265–290 [DOI] [PubMed] [Google Scholar]
- Czaja W, Mao P, Smerdon MJ (2012) The emerging roles of ATP‐dependent chromatin remodeling enzymes in nucleotide excision repair. Int J Mol Sci 13: 11954–11973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabin J, Fortuny A, Polo SE (2016) Epigenome maintenance in response to DNA damage. Mol Cell 62: 712–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH (2012) Shining a light on xeroderma pigmentosum. J Invest Dermatol 132: 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD (1997) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J 16: 6559–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Kaczmarek N, Luch A, Glas A, Carell T, Naegeli H (2011) Regulation of nucleotide excision repair by UV‐DDB: prioritization of damage recognition to internucleosomal DNA. PLoS Biol 9: e1001183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch ME, Nakajima S, Yasui A, Ford JM (2003) In vivo recruitment of XPC to UV‐induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem 278: 46906–46910 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, Wood RD (2006) DNA repair: from molecular mechanism to human disease. DNA Repair 5: 986–996 [DOI] [PubMed] [Google Scholar]
- Gale JM, Nissen KA, Smerdon MJ (1987) UV‐induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Biochemistry 84: 6644–6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale JM, Smerdon MJ (1990) UV induced (6‐4) photoproducts are distributed differently than cyclobutane dimers in nucleosomes. Photochem Photobiol 51: 411–417 [DOI] [PubMed] [Google Scholar]
- Gaspar‐Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho‐Santos J, McManus MT, Plath K, Meshorer E, Ramalho‐Santos M (2009) Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 460: 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Fahy D, Smerdon MJ (2006) Rad4‐Rad23 interaction with SWI/SNF links ATP‐dependent chromatin remodeling with nucleotide excision repair. Nat Struct Mol Biol 13: 902–907 [DOI] [PubMed] [Google Scholar]
- Gong F, Fahy D, Liu H, Wang W, Smerdon MJ (2008) Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 7: 1067–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev SA (2012) Nucleosome spacing and chromatin higher‐order folding. Nucleus 3: 493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Srivastava AK, Cui T, Wang Q‐E, Wani AA (2016) Differential DNA lesion formation and repair in heterochromatin and euchromatin. Carcinogenesis 37: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G (2008) Transcription‐coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9: 958–970 [DOI] [PubMed] [Google Scholar]
- Hey T, Lipps G, Sugasawa K, Iwai S, Hanaoka F, Krauss G (2002) The XPC−HR23B complex displays high affinity and specificity for damaged DNA in a true‐equilibrium fluorescence assay. Biochemistry 41: 6583–6587 [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JHJ (2009) DNA damage, aging, and cancer. N Engl J Med 361: 1475–1485 [DOI] [PubMed] [Google Scholar]
- Horikoshi N, Tachiwana H, Kagawa W, Osakabe A, Matsumoto S, Iwai S, Sugasawa K, Kurumizaka H (2016) Crystal structure of the nucleosome containing ultraviolet light‐induced cyclobutane pyrimidine dimer. Biochem Biophys Res Commun 471: 117–122 [DOI] [PubMed] [Google Scholar]
- Hwang BJ, Ford JM, Hanawalt PC, Chu G (1999) Expression of the p48 xeroderma pigmentosum gene is p53‐dependent and is involved in global genomic repair. Proc Natl Acad Sci USA 96: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Wang X, Bao S, Guo R, Johnson DG, Shen X, Li L (2010) INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci USA 107: 2–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Y, Taylor JS, Kao JFL (1998) Thermodynamic and base‐pairing studies of matched and mismatched DNA dodecamer duplexes containing cis‐syn, (6‐4) and dewar photoproducts of TT. Nucleic Acids Res 26: 3845–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanaki MG, Guerrero‐Santoro J, Bisi DC, Hsieh CL, Rapić‐Otrin V, Levine AS (2006) The DDB1‐CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV‐damaged DNA sites. Proc Natl Acad Sci USA 103: 2588–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, Sirma H, Simon R, Will H, Dobbelstein M, Dikomey E, Johnsen SA (2016) Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep 487: e201642352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Soni SD, Arakali AV, Wallace JC, Alderfer JL (1995) Solution structure of a nucleic acid photoproduct of deoxyfluorouridylyl‐(3′‐5′)‐thymidine monophosphate (d‐FpT) determined by NMR and restrained molecular dynamics: structural comparison of two sequence isomer photoadducts (d‐U5p5T and d‐T5p5U). Nucleic Acids Res 23: 1810–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler‐Yeivin A, Picarsky E, Yaniv M (2006) Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol 26: 2661–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Katsumi S, Imoto K, Nakagawa A, Miyagawa S, Furumura M, Mori T (2001) Quantitation and visualization of ultraviolet‐induced DNA damage using specific antibodies: application to pigment cell biology. Pigment Cell Res 14: 94–102 [DOI] [PubMed] [Google Scholar]
- Li C‐L, Golebiowski FMM, Onishi Y, Samara NLL, Sugasawa K, Yang W (2015) Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Mol Cell 59: 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481 [DOI] [PubMed] [Google Scholar]
- Mathieu N, Kaczmarek N, Rüthemann P, Luch A, Naegeli H (2013) DNA quality control by a lesion sensor pocket of the xeroderma pigmentosum group D helicase subunit of TFIIH. Curr Biol 23: 204–212 [DOI] [PubMed] [Google Scholar]
- McAteer K, Jing Y, Kao J, Taylor J‐SS, Kennedy MA (1998) Solution‐state structure of a DNA dodecamer duplex containing a cis‐syn thymine cyclobutane dimer, the major UV photoproduct of DNA. J Mol Biol 282: 1013–1032 [DOI] [PubMed] [Google Scholar]
- McKenna ES, Sansam CG, Cho Y‐J, Greulich H, Evans JA, Thom CS, Moreau LA, Biegel JA, Pomeroy SL, Roberts CWM (2008) Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol 28: 6223–6233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DL, Nguyen TD, Cleaver JE (1990) Nonrandom induction of pyrimidine‐pyrimidone (6‐4) photoproducts in ultraviolet‐irradiated human chromatin. J Biol Chem 265: 5353–5356 [PubMed] [Google Scholar]
- Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LHF, Fousteri MI (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase IIIα in a cell‐cycle‐specific manner. Mol Cell 27: 311–323 [DOI] [PubMed] [Google Scholar]
- Mouret S, Leccia M‐T, Bourrain J‐L, Douki T, Beani J‐C (2011) Individual photosensitivity of human skin and UVA‐induced pyrimidine dimers in DNA. J Invest Dermatol 131: 1539–1546 [DOI] [PubMed] [Google Scholar]
- Nakazawa Y, Yamashita S, Lehmann AR, Ogi T (2010) A semi‐automated non‐radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair 9: 506–516 [DOI] [PubMed] [Google Scholar]
- Nocentini S, Coin F, Saijo M, Tanaka K, Egly JM (1997) DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. J Biol Chem 272: 22991–22994 [DOI] [PubMed] [Google Scholar]
- Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LHF, Yamashita S, Fousteri MI, Lehmann AR (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 37: 714–727 [DOI] [PubMed] [Google Scholar]
- Osakabe A, Tachiwana H, Kagawa W, Horikoshi N, Matsumoto S, Hasegawa M, Matsumoto N, Toga T, Yamamoto J, Hanaoka F, Thomä NH, Sugasawa K, Iwai S, Kurumizaka H (2015) Structural basis of pyrimidine‐pyrimidone (6‐4) photoproduct recognition by UV‐DDB in the nucleosome. Sci Rep 5: 16330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrin VR, McLenigan M, Takao M, Levine AS, Protic M (1997) Translocation of a UV‐damaged DNA binding protein into a tight association with chromatin after treatment of mammalian cells with UV light. J Cell Sci 110: 1159–1168 [DOI] [PubMed] [Google Scholar]
- Park D, Shivram H, Iyer VRV (2014) Chd1 co‐localizes with early transcription elongation factors independently of H3K36 methylation and releases stalled RNA polymerase II at introns. Epigenetics Chromatin 7: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Almouzni G (2013) Nucleosome dynamics as modular systems that integrate DNA damage and repair. Cold Spring Harb Perspect Biol 5: a012658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti P, Lim CY, Nat R, Villunger A, Geley S, Shue YT, Soratroi C, Moser M, Lusser A (2015) Embryonic stem cell differentiation requires full length Chd1. Sci Rep 5: 8007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, Vrieling H, Mullenders L (2012) PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol 199: 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapic‐Otrin V (2002) Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res 30: 2588–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Mir SN, Wani G, Zhao Q, Battu A, Zhu Q, Wang Q‐E, Wani AA (2009) Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol Cell Biol 29: 6206–6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A (2003) Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev 17: 2539–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl T, Hanaoka F, Egly J‐MM (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 22: 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Y, Hinz JM, Smerdon MJ (2015) Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair 32: 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi CP, Milner J (2003) p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 22: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A (1996) DNA excision repair. Annu Rev Biochem 65: 43–81 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Kiely R, McHugh PJ (2010) The Ino80 chromatin‐remodeling complex restores chromatin structure during UV DNA damage repair. J Cell Biol 191: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schärer OD (2013) Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5: a012609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima A, Koníčková R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thomä NH (2008) Structural basis of UV DNA‐damage recognition by the DDB1‐DDB2 complex. Cell 135: 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM (2003) Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22: 1846–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Hernandez AE, Groudine M, Henikoff S (2014) The nucleosomal barrier to promoter escape by RNA polymerase ii is overcome by the chromatin remodeler Chd1. Elife 2014: e02042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon MJ, Conconi A (1999) Modulation of DNA damage and DNA repair in chromatin. Prog Nucleic Acid Res Mol Biol 62: 227–255 [DOI] [PubMed] [Google Scholar]
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL (2012) Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol 19: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand‐Sauthier I, Giglia‐Mari G, Clarkson SG, Vermeulen W, Schärer OD (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 28: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Ng JMY, Masutani C, Iwai S, van der Spek PJ, Eker APM, Hanaoka F, Bootsma D, Hoeijmakers JHJ (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2: 223–232 [DOI] [PubMed] [Google Scholar]
- Sugasawa K (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev 15: 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka KK, Tanaka KK, Hanaoka F (2005) UV‐induced ubiquitylation of XPC protein mediated by UV‐DDB‐ubiquitin ligase complex. Cell 121: 387–400 [DOI] [PubMed] [Google Scholar]
- Thoma F (2005) Repair of UV lesions in nucleosomes – intrinsic properties and remodeling. DNA Repair 4: 855–869 [DOI] [PubMed] [Google Scholar]
- Uchida A, Sugasawa K, Masutani C, Dohmae N, Araki M, Yokoi M, Ohkuma Y, Hanaoka F (2002) The carboxy‐terminal domain of the XPC protein plays a crucial role in nucleotide excision repair through interactions with transcription factor IIH. DNA Repair 1: 449–461 [DOI] [PubMed] [Google Scholar]
- Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F (2001) ATP‐dependent chromatin remodeling facilitates nucleotide excision repair of UV‐induced DNA lesions in synthetic dinucleosomes. EMBO J 20: 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, Fousteri M (2013) Mammalian transcription‐coupled excision repair. Cold Spring Harb Perspect Biol 5: a012625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker M, Moné MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JHJ, van Driel R, van Zeeland AA, Mullenders LHF (2001) Sequential assembly of the nucleotide excision repair factors in vivo . Mol Cell 8: 213–224 [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Shimizu M, Morioka H, Linn S, Nikaido O, Matsunaga T (2001) Damaged DNA‐binding protein DDB stimulates the excision of cyclobutane pyrimidine dimers in vitro in concert with XPA and replication protein A. J Biol Chem 276: 15434–15440 [DOI] [PubMed] [Google Scholar]
- Wakasugi M, Kasashima H, Fukase Y, Imura M, Imai R, Yamada S, Cleaver JE, Matsunaga T (2009) Physical and functional interaction between DDB and XPA in nucleotide excision repair. Nucleic Acids Res 37: 516–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Sugasawa K, Shimizu Y, Iwai S, Shiomi T, Hanaoka F (2005) Nucleosomal structure of undamaged DNA regions suppresses the non‐specific DNA binding of the XPC complex. DNA Repair 4: 389–395 [DOI] [PubMed] [Google Scholar]
- Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, Rapic‐Otrin V (2012) Damaged DNA induced UV‐damaged DNA‐binding Protein (UV‐DDB) dimerization and its roles in chromatinized DNA repair. Proc Natl Acad Sci USA 109: E2737–E2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F (2000) The xeroderma pigmentosum group C protein complex XPC‐HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem 275: 9870–9875 [DOI] [PubMed] [Google Scholar]
- Zavala AG, Morris RT, Wyrick JJ, Smerdon MJ (2014) High‐resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic Acids Res 42: 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Q, Jones K, Patel M, Gong F (2009) The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle 8: 3953–3959 [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen H, Gong M, Gong F, Zhang Q, Jones K, Patel M, Gong F (2013) The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front Oncol 3: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang Q‐EE, Ray A, Wani G, Han C, Milum K, Wani AA (2009) Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin‐remodeling complex. J Biol Chem 284: 30424–30432 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4