

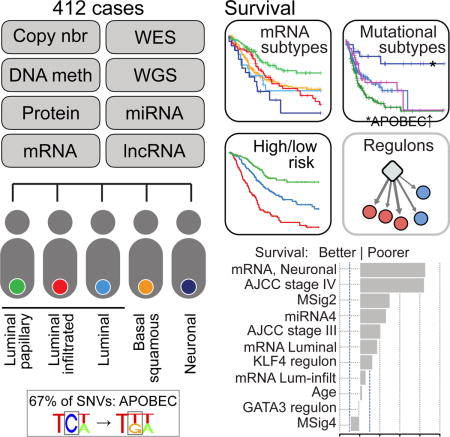
Summary
We report a comprehensive analysis of 412 muscle-invasive bladder cancers characterized by multiple TCGA analytical platforms. Fifty-eight genes were significantly mutated, and the overall mutational load was associated with APOBEC-signature mutagenesis. Clustering by mutation signature identified a high-mutation subset with 75% 5-year survival. mRNA expression clustering refined prior clustering analyses and identified a poor-survival ‘neuronal’ subtype in which the majority of tumors lacked small cell or neuroendocrine histology. Clustering by mRNA, lncRNA, and miRNA expression converged to identify subsets with differential epithelial-mesenchymal transition status, carcinoma-in-situ scores, histologic features, and survival. Our analyses identified 5 expression subtypes that may stratify response to different treatments.
Keywords: APOBEC mutation, basal mRNA subtype, neuronal subtype, DNA methylation, lncRNA transcriptome, luminal mRNA subtype, muscle-invasive bladder cancer, microRNA, neoantigen, regulon
Graphical Abstract
Introduction
Urothelial bladder cancer is a heterogeneous epithelial malignancy that presents most commonly as an exophytic tumor confined to the mucosa or lamina propria. However, 25% of patients have muscle-invasive (MIBC) or metastatic disease at the time of initial diagnosis and have a worse prognosis. We previously reported an integrated genomic analysis of 131 MIBC samples (Cancer Genome Atlas Research Network, 2014a), finding a high somatic mutation rate (median 5.5 per megabase) similar to that of non-small cell lung cancer and melanoma (Lawrence et al., 2013); statistically significant recurrent mutations in 32 genes, including several chromatin regulators; four expression subtypes; recurrent in-frame activating FGFR3–TACC3 fusions; and potential therapeutic targets in 69% of the samples. Here, we report a comprehensive analysis of the full TCGA cohort of 412 MIBC cases. The expanded cohort allowed us to identify: 32 additional significantly mutated genes; that APOBEC-signature mutagenesis is associated with both a high mutation rate and improved clinical outcome; an expression subtype that we term ‘neuronal’; and multiple recurrent translocations that lead to fusion genes. Clustering expression profiles for mRNA, long noncoding RNA, and miRNA further confirmed distinct subsets of MIBC with differential survival.
Demographic, Clinical, and Pathological Data
412 chemotherapy-naive, invasive, high-grade urothelial tumors (T1 [n = 1], T2–T4a, N0–3, M0–1, Tables S1 and S2.1) from 36 tissue source sites (Table S2.2) were re-reviewed by 4 expert genitourinary pathologists, who classified them as pure urothelial or mixed histology (Figure S1, Table S2.3), and assessed immune infiltrates (Table S2.4). 52 (13%) had urothelial carcinoma with variant histology, including 42 squamous, 4 small cell/neuroendocrine, 2 micropapillary, and 4 plasmacytoid. 5 additional tumors that met screening criteria were included: 3 pure squamous cell bladder carcinomas, 1 squamous cell carcinoma of non-bladder origin, and 1 bladder adenocarcinoma. Complete clinical data were available for 406 tumors (Table S1). 35 patients had received prior intravesical immunotherapy with BCG, and 12 had received neoadjuvant chemotherapy (NAC) after tumor acquisition. 230 were alive, 163 had recurred, and 182 had died. The median follow-up was 20.9 months for those alive at last follow-up. At least 122 (67%) deaths were cancer-related. The samples were characterized by clinical data and by 6 molecular profiling platforms (Table S2.5).
Somatic DNA Alterations
Affymetrix SNP6.0 arrays were used to assess somatic copy number alterations (SCNAs). GISTIC analysis identified 34 amplified and 32 deleted genomic regions (q < 0.1, Tables S2.6–8). Many of the focal SCNAs involved genes known to be amplified in bladder cancer, including AHR, BCL2L1, CCND1, CCNE1, E2F3, EGFR, ERBB2, FGFR3, GATA3, KRAS, MDM2, MYCL1, PPARG, PVRL4, SOX4, TERT, YWHAZ and ZNF703 (Cancer Genome Atlas Research Network, 2014a). The most common recurrent (22%) focal deletion (copy number < 1) contained CDKN2A (9p21.3). Recurrent focal deletions in RAD51B (14q24.1) were not observed in the first 131 cases.
Whole-exome sequencing (WES) of 412 tumors and matched normal samples targeted 193,094 exons in 18,862 genes (mean coverage 85X, 79% of target bases > 30X). MuTect identified 131,660 somatic mutations (128,772 single-nucleotide variants [SNVs] and 2,888 indels), with high non-synonymous mutation rates (mean 8.2 and median 5.8 per megabase [Mb]) (Figure 1A). Most mutations were C>G transversions (27%) or C>T transitions (51%). Whole genome-doubling events were found in 221 (54%) tumors (Table S1).
Figure 1. Landscape of mutational signatures, mutations and copy number alterations.
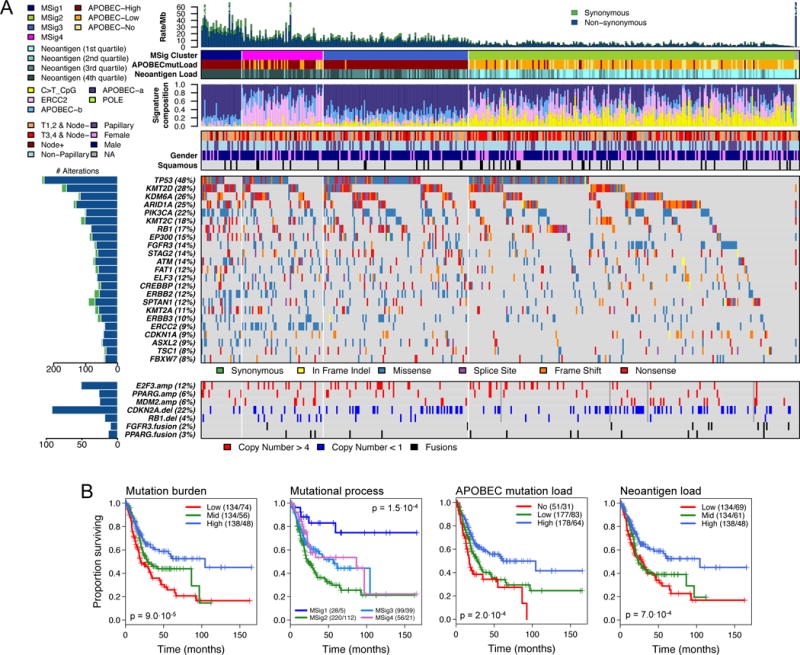
(A) Alteration landscape for 412 primary tumours. Top to bottom: Synonymous and non-synonymous somatic mutation rates, with one ultra-mutated sample with a POLE signature. Mutational signature (MSig) cluster, APOBEC mutation load, and neoantigen load by quartile. Normalized activity of 4 mutational signatures. Combined tumor stage (T1,2 vs. T3,4) and node status, papillary histology, and gender. Somatic mutations for significantly mutated genes (SMGs) with frequency ≥ 7%. Copy number alterations for selected genes, and FGFR3 and PPARG gene fusions.
(B) Kaplan-Meier plots for overall survival (L to R): Overall mutation burden (SNVs); Mutation signature clusters (MSig1–4); APOBEC-mediated mutation load; Neoantigen load;
To identify processes contributing to the high mutation rate, we used Bayesian NMF to identify 5 mutation signatures (Figure S2A). APOBEC-a and APOBEC-b were variants of the hallmark APOBEC mutagenesis signature. A third signature, consisting of C>T transitions at CpG dinucleotides, is likely due to 5-methylcytosine deamination. A fourth, POLE, was present in a single ultra-mutated sample, with > 4000 SNVs and a POLE mutation (P286R). The fifth, ERCC2, had a relatively uniform spectrum of base changes and has been associated with ERCC2 mutations (Kim et al., 2016b).
The APOBEC-a and -b signatures accounted for 67% of all SNVs. Results from an independent method for identifying APOBEC-signature mutations (Roberts et al., 2013) strongly correlated with mutation load assigned to APOBEC-a and -b groups (Figure S2B). The total count of mutations with a stringent APOBEC signature correlated with the remaining mutation burden (r = 0.48, Figure S2C), suggesting that some mutations not assigned to APOBEC-signature mutagenesis were also APOBEC-mediated. As expected (Roberts et al., 2013), levels of APOBEC-signature mutagenesis correlated with expression of APOBEC3A and APOBEC3B (Figure S2D). C>T at CpG and ERCC2 mutation signatures accounted for 20% and 8% of total SNVs, respectively. 64% of all mutations, as well as 62% of APOBEC-a- and 75% of APOBEC-b-signature mutations (likelihood of signature association ≥ 0.7; Methods) (Kasar et al., 2015) were clonal (cancer cell fraction ≥ 0.9), suggesting that more than half of the APOBEC-signature mutation load was likely generated early in bladder cancer development.
Unsupervised clustering of APOBEC-a and -b, ERCC2, and C>T-at-CpG signatures identified four mutational signature clusters, MSig1 to MSig4 (Figure 1; Figure S2E), which were associated with overall survival (Figure 1B, p = 1.4·10−4). Patients with MSig1 cancers (high APOBEC-signature mutagenesis and high mutation burden) showed an exceptional 75% 5-year survival probability. Better survival was also seen in subsets defined by high mutation burden or high APOBEC-signature mutation load (Figure 1B). MSig2 cancers had the lowest mutation rate and poorest 5-year survival (22%). MSig4 cluster samples were enriched in both ERCC2 signature mutations (average contribution 49% vs. 17% in all others, Figure 1A) and ERCC2 mutations (24 out of 39, p = 10−13). ERCC2 signature mutations were highest in smokers with ERCC2 mutations (p = 6.9 × 10−11); for cases with wild type ERCC2, ERCC2 signature mutations were at higher levels in smokers than in non-smokers (Figure S2F).
MutSig 2CV identified 58 significantly mutated genes (SMGs) (q < 0.1; Tables S1 and S2.9). 34 of the 58 had not been identified as SMGs in our earlier analysis (Cancer Genome Atlas Research Network, 2014a); further, 16 of the 34 had not been implicated as cancer SMGs in a recent pan-cancer analysis (Lawrence et al., 2014) (Table S2.9). 7 of the 34 genes were mutated in > 10% of samples: KMT2C (18%), ATM (14%), FAT1 (12%), CREBBP (12%), ERBB2 (12%), SPTAN1 (12%), and KMT2A (11%). Alterations were mutually exclusive between CDKN2A and TP53, CDKN2A and RB1, CDKN2A and E2F3, TP53 and MDM2, FGFR3 and E2F3, and FGFR3 and RB1 (Table S2.10, q < 0.2). Similar analyses showed co-occurrence of alterations in TP53 and RB1, TP53 and E2F3, and FGFR3 and CDKN2A (Table S2.11, q < 0.2). FGFR3 mutations and CDKN2A focal SCNAs co-occurred in 27 (7%) tumors (Table S1), which may be MIBCs that have progressed from non-invasive tumors (Rebouissou et al., 2012). 3 of 4 tumors with plasmacytoid histology had nonsense CDH1 mutations, consistent with a previous report (Al-Ahmadie et al., 2016).
We identified four DNA-based clusters using unsupervised NMF clustering with SMG mutations (Mut) and focal SCNAs (CN) (Figures S2G and H). The four MutCN clusters were characterized by: TP53 and RB1 mutations, SOX4/E2F3 amplification, mutations in chromatin-modifying genes, and FGFR3, KDM6A, and STAG2 mutations.
Neoantigen load was strongly correlated with mutation burden, elevated in the MSig1 cluster (p = 2.9 × 10−12), and associated with survival (p = 5.2 × 10−4; Figure 1B). It was an independent predictor of outcome in addition to age, AJCC tumor stage, and squamous differentiation (p = 8 × 10−4; Table S2.12). Polysolver-based HLA mutation detection identified 21 non-synonymous variants in 19 of 412 tumors (4.6%, Table S2.13). HLA mutations were more common in MSig1 cluster (p = 0.039), suggesting that they may have resulted from APOBEC-signature mutagenesis. HLA mutations were somewhat more common in patients with prior BCG treatment, 4 of 35 (11.4%) vs. 8 of 261 (3.1%) without prior BCG treatment (p = 0.04, Chi-square test), perhaps positively selected in response to immunological pressure.
Using RNA-seq data, we identified 784 gene fusions (Table S2.14). The most common was an intra-chromosomal FGFR3-TACC3 fusion (n = 10). There were 9 cases of an intra-chromosomal translocation ITGB6-LOC100505984, whose functional significance is uncertain. PPARG was involved in 4 TSEN2-PPARG and 2 MKRN2-PPARG fusions, and PPARG expression levels were higher than in samples without such fusions (p = 6 × 10−3). Four of the six PPARG fusions led to mRNA products for which the predicted proteins retained both PPARG’s DNA-binding and ligand-binding domains, suggesting that they were functional (Figure S2I). We searched for similar fusions in RNA-seq data from 30 human bladder cancer cell lines (Table S2.15). In lines 5637 and 1A6 we identified CASC15-PPARG fusions that retained PPARG’s full DNA binding domain; in UC9 we identified an NR2C2-PPARG fusion that retained 75% of PPARG’s ligand-binding domain. PPARG was overexpressed in all three of these cell lines (p < 0.05).
DNA Methylation
Unsupervised clustering using tumor-associated hypermethylated CpG sites or, independently, tumor-associated hypomethylated sites, identified 5 major clusters that were significantly correlated with other data types (Figures S3A and S3B). Of particular interest was a group of tumors with high purity that showed marked loss of DNA methylation (hypomethylation cluster 4, Figure S3B,C). This group significantly overlapped with the DNA hypermethylation cluster 2, which showed a low frequency of DNA hypermethylation (p = 3.2 × 10−8, odds ratio 9.5, Figure S3D). Samples in cluster 4 showed frequent FGFR3 mutation (p = 6 × 10−9) and CDKN2A deletion (p = 4 × 10−13), and had no TP53 or RB1mutations. Further, those tumors belonged to the luminal-papillary mRNA subtype, exhibited papillary histology (p = 5 × 10−12), were almost all node-negative (p = 5 × 10−12), were from younger patients (median age 61 vs. 69; p < 4 × 10−3), and showed better survival (p < 0.05; log-rank test, Figure S3E). DNA hypomethylation appeared more widespread in low-stage, noninvasive urothelial tumors (Wolff et al., 2010). Analysis of hypomethylated CpG sites in this group revealed 12 genes whose hypomethylation was significantly correlated with increased expression (Figure S3F; Table S2.16).
Integrated analysis of DNA methylation and gene expression identified 158 genes that were epigenetically silenced (Table S2.17, Figure S3G). Although some of the silencing events were probably background epigenetic noise, CDKN2A, FAT1, and CASP8 were mutated in some tumors and (mutually exclusively) epigenetically silenced in others (Figure S3H). Silenced genes included latexin (LXN), the only known endogenous carboxypeptidase inhibitor (silenced in 27%), Poly(ADP-ribose) polymerase PARP6 (26%), nicotinate phosphoribosyltransferase (NAPRT) (13%), and SPATC1L (19%). In contrast, we found no evidence for promoter DNA hypermethylation of other classical tumor suppressor genes, including TP53, PTEN, TSC1, TSC2, NF1, NF2, and RB1.
mRNA Expression-Based Molecular Subtypes
Unbiased NMF consensus clustering of RNA-seq data (n = 408) identified five expression subtypes (Figure 2): luminal-papillary (n = 142, 35%), luminal-infiltrated (n = 78, 19%), luminal (n = 26, 6%), basal-squamous (n = 142, 35%), and neuronal (n = 20, 5%). The subtypes were associated with overall survival (p = 4 × 10−4) (Figure S4A). The analysis confirmed the two major luminal and basal transcriptional subtypes identified by TCGA and other groups (Choi et al., 2014b; Damrauer et al., 2014; Sjodahl et al., 2012), while discriminating within those subtypes and identifying luminal and neuronal subtypes (see below). The subtypes were concordant with the four subtypes that we had reported for the 131-tumor subset of the current cohort (Cancer Genome Atlas Research Network, 2014a) (Figure 2; Tables S2.18 and S2.19).
Figure 2. mRNA expression subtypes.
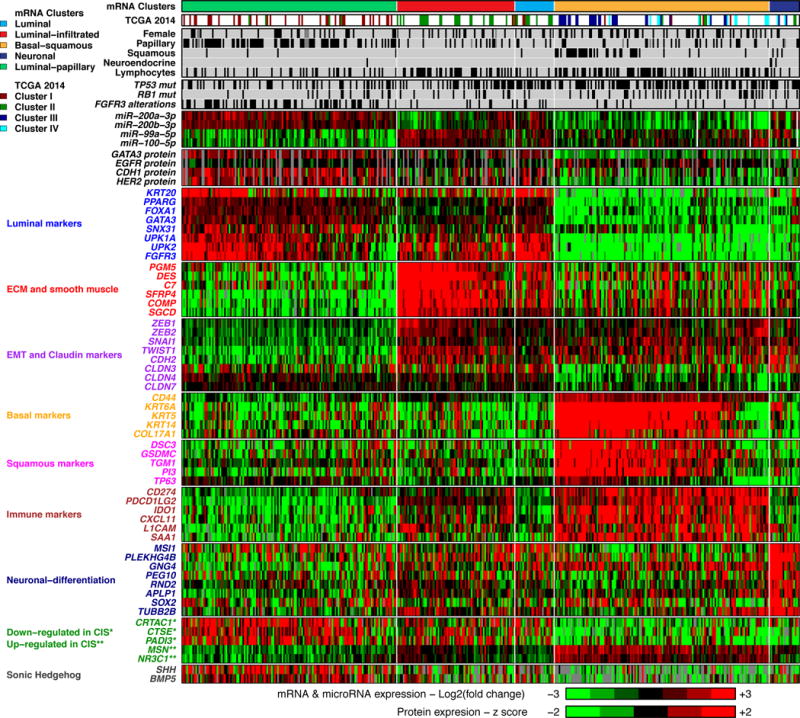
Top, L to R: 5 mRNA expression subtypes: luminal-papillary, luminal-infiltrated, luminal, basal-squamous and neuronal. Covariates: 4 previously reported TCGA subtypes; selected clinical covariates and key genetic alterations; normalized expression for miRNAs and proteins; log2 (fold change against the median expression across samples) for selected genes, for labeled gene sets. Samples within the three luminal subtypes, the basal-squamous subtype, and the neuronal subtype are ordered by luminal, basal, and neuroendocrine signature scores, respectively. Genes that are down-regulated* vs. up-regulated** in CIS.
Most samples in the luminal subtypes showed high expression of uroplakins (UPK2 and UPK1A) and urothelial differentiation markers (FOXA1, GATA3, PPARG) (Figure 2). Although differences in purity appeared to contribute to their separation into different clusters (Figure S4B), each of the three subtypes also showed distinctive expression features with respect to wild type p53, epithelial-mesenchymal transition (EMT), and stromal gene signatures (Figure S4C; Methods: mRNA Expression Profiling: Gene expression signature scores).
The luminal-papillary cluster was enriched in tumors with papillary morphology (58% vs. 20% in other subtypes; p < 10−13), lower stage (T2, 55% vs. 23%; p < 10−8), and higher purity (median 0.84 vs. 0.50 in other luminal subtypes). Several features suggest a dominant role of FGFR3 in 44% of the luminal-papillary tumors: enrichment with FGFR3 mutations (42/57; p < 10−9), amplification (5/5; p = 5 × 10−3), overexpression (4-fold vs. median, 49/67; p < 10−11), and FGFR3-TACC3 fusions (8/10, p = 4 × 10−3). These tumors also had low carcinoma-in-situ (CIS) expression signature scores (Figures S4C and Table S2.20; p < 1016) (Dyrskjot et al., 2004). They retained sonic-hedgehog signaling (SHH, Figure 2). Together, these features suggest that many tumors in this cluster developed from a precursor non-muscle-invasive papillary bladder cancer.
The luminal-infiltrated subtype was distinguished from other luminal subtypes by lower purity (median 0.46 vs. 0.68; p < 10−11), consistent with the presence of lymphocytic infiltrates, and by strong expression of smooth muscle and myofibroblast gene signatures (Figures 2 and S4C). 36 of 45 (80%) of the tumors in this subtype had features similar to an expression subtype that has been associated with chemoresistance and characterized by a wild type p53 signature (Choi et al., 2014b). The wild type p53 signature score was inversely correlated with tumor purity (Pearson r = −0.4; p < .001), suggesting the presence of smooth muscle and fibroblast cells as a driver of the signature. This subtype contained 23 of 24 tumors that we had previously classified as Cluster-II, which was reported to benefit most from anti-PDL1 treatment (Rosenberg et al., 2016), and had an intermediate 5-year survival, comparable to basal-squamous and luminal subtypes (Figure S4A). These tumors had increased expression of several immune markers, including CD274 (PD-L1) and PDCD1 (PD-1) (Figure 2).
The luminal subtype had the highest expression levels of several uroplakins (UPK1A, UPK2) and genes that are highly expressed in terminally differentiated urothelial umbrella cells (KRT20, SNX31) (Figure 2). This suggests that these tumors are derived from intermediate cells that have a transcriptional program that leads to expression of markers characteristic of normal umbrella cells.
The basal-squamous subtype was characterized by high expression of basal and stem-like markers (CD44, KRT5, KRT6A, KRT14) and squamous differentiation markers (TGM1, DSC3, PI3). The subtype included 37 of 45 tumors squamous features (p < 10−11), was enriched in TP53 mutations (p = 5 × 10−3), and was more common in females (33% vs. 21% in other subtypes; p = 0.024). Many tumors in this subtype also showed strong expression of CIS signature genes (Figures 2 and S4A) and loss of SHH signaling (Figure 2), suggesting that they developed from basal cells and CIS lesions. This subtype also showed the strongest immune expression signature, including T-cell markers and inflammation genes (Figure S4C), consistent with relatively low purity (median 0.49) (Figure S4B) and the presence of lymphocytic infiltrates (p < 1 × 10−4). Approximately 20 samples (right portion of this subtype in Figure 2) lacked expression of both basal and squamous markers, but clustered with this subtype because they lacked luminal marker expression and had high immune gene expression.
The neuronal subtype included 3 of 4 with neuroendocrine (NE) histology (p = 5 × 10−3) and an additional 17 tumors that had no histopathologic features suggestive of NE origin. All 20 showed relatively high expression of neuronal differentiation and development genes, as well as typical NE markers (Figures 2 and S4C; p < 10−4). Loss of TP53 and RB1 is a hallmark of small cell NE cancer, and 10 of 20 (50%) samples had mutations in both TP53 and RB1, or TP53 mutation and E2F3 amplification. 17 (85%) of the 20 tumors had alterations in genes in the p53/cell cycle pathway. Notably, this subtype had the poorest survival (p = 4 × 10−4, log-rank test), consistent with the known aggressive phenotype of NE bladder cancers.
As we had previously shown (Cancer Genome Atlas Research Network, 2014a), several proteins (GATA3, EGFR, CDH1, HER2) and miRNAs (miR-200s, miR-99a, miR-100) were strongly differentially expressed among the mRNA subtypes (Figures 2, S4D,E and S5A; Table S2.21, S.22).
Altered Pathways
Many canonical signaling pathways were altered (Figure 3). The p53/Cell Cycle pathway was inactivated in 89% of tumors, with TP53 mutations in 48%, MDM2 amplification (copy number > 4) in 6%, and MDM2 overexpression (>2-fold above the median) in 19%. TP53 mutations were enriched in tumors with genome-doubling events (p < 10−7; Table S2.23), suggesting that loss of TP53 activity facilitates genome doubling (Zack et al., 2013).
Figure 3. Somatic alterations in signaling pathways across mRNA subtypes.
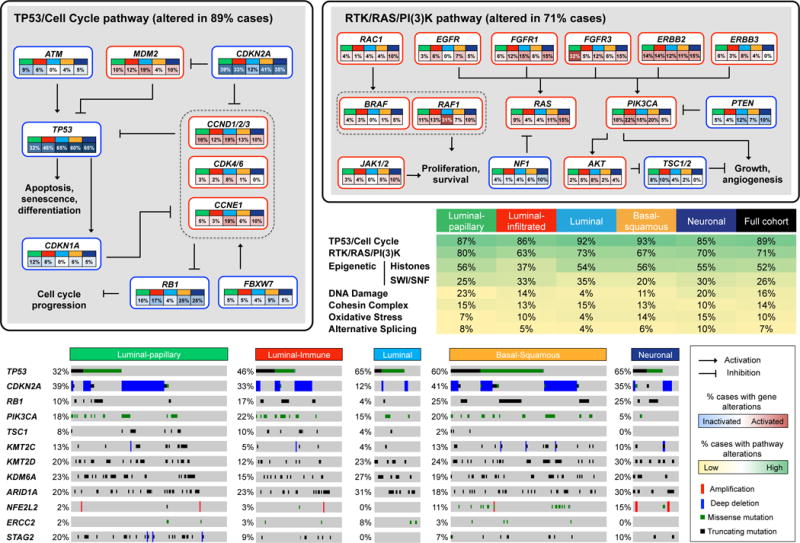
Somatic alterations include mutations and copy-number changes (i.e. deep deletions and high-level amplifications, from GISTIC results). Missense mutations are counted only if they have known oncogenic function based on OncoKB (http://oncokb.org) annotations, or have previously been reported in COSMIC, or occur at known mutational hotspots. The table shows the fraction of samples with alterations in selected signaling pathways. In the pathway diagrams, edges show pairwise molecular interactions; boxes outlined in red denote alterations leading to pathway activation, while boxes outlined in blue denote predicted pathway inactivation. The oncoprint illustrates type and frequency of alteration, as well as patterns of co-occurrence, for selected genes from the pathways highlighted in the table.
RB1 mutations (17%) were mostly inactivating and associated with reduced mRNA levels. CDKN1A mutations (11%) were predominantly inactivating. CDKN2A mutations (7%) and homozygous deletions (22%) were common, as previously described (Williamson et al., 1995).
Alterations in DNA repair pathways included mutations in ATM (n = 57; 14%) and ERCC2 (n = 40; 9%) and deletions in RAD51B (n = 10; 2%). All non-silent ERCC2 mutations were missense, and many mapped within, or within ±10 amino acids of, the conserved helicase domain, suggesting that they impair ERCC2 function and may have dominant negative effects (Van Allen et al., 2014).
The FGFR3, PIK3CA, and RAS oncogenes harbored recurrent hotspot mutations. Most FGFR3 mutations were the known S249C or Y373C, were more frequent in lower-stage tumors (21% in T2 vs. 10% in T3,T4; p = 0.003), and were associated with better survival (p = 0.04). PIK3CA mutations (n = 100; 22%) were more common in the helical domain (E542 and E545; n = 54 total) than in the kinase domain (M1043, H1047; n = 10 total), and were likely due to APOBEC mutagenic activity (Cancer Genome Atlas Research Network, 2017; Roberts et al., 2013). ERBB2 mutations were common at S310 (S280 in the LRG_724t1 transcript) in the extracellular domain (24 of 57, 42%), and were also likely due to APOBEC-signature mutagenesis.
Ten of the 39 SMGs with mutation frequency >5% were in chromatin-modifying or chromatin-regulatory genes: a histone demethylase (KDM6A), histone methyltransferases (KMT2A, KMT2C, KMT2D), histone acetylases (CREBBP, EP300, KANSL1), a member of the SWI/SNF chromatin remodeling complex (ARID1A), and Polycomb group genes (ASXL1, ASXL2). Mutations in these genes were predominantly inactivating (50% frame-shift or nonsense mutations vs. 26% in other SMGs; p = 10−30), strongly suggesting that they are functionally relevant. ARID1A, CREBBP, and KDM6A were also targets of genomic deletion (4.2%, 14.2%, 4.9%, respectively, Table S1).
Noncoding RNAs (lncRNAs and miRNAs) Subdivide mRNA Expression Subtypes
Because lncRNAs can be more specific to biological state than coding RNAs (Nguyen and Carninci, 2016), we calculated transcript abundances for 8167 (Ensembl v82) lncRNAs and processed transcripts. Four unsupervised consensus clusters were associated with purity (p = 2.3 × 10−27), EMT score (p = 9.9 × 10−34), expression of CIS gene sets (p < 1 × 10−39) (Dyrskjot et al., 2004), and 5-year survival (p = 0.015) (Figure 4).
Figure 4. LncRNA expression subtypes.
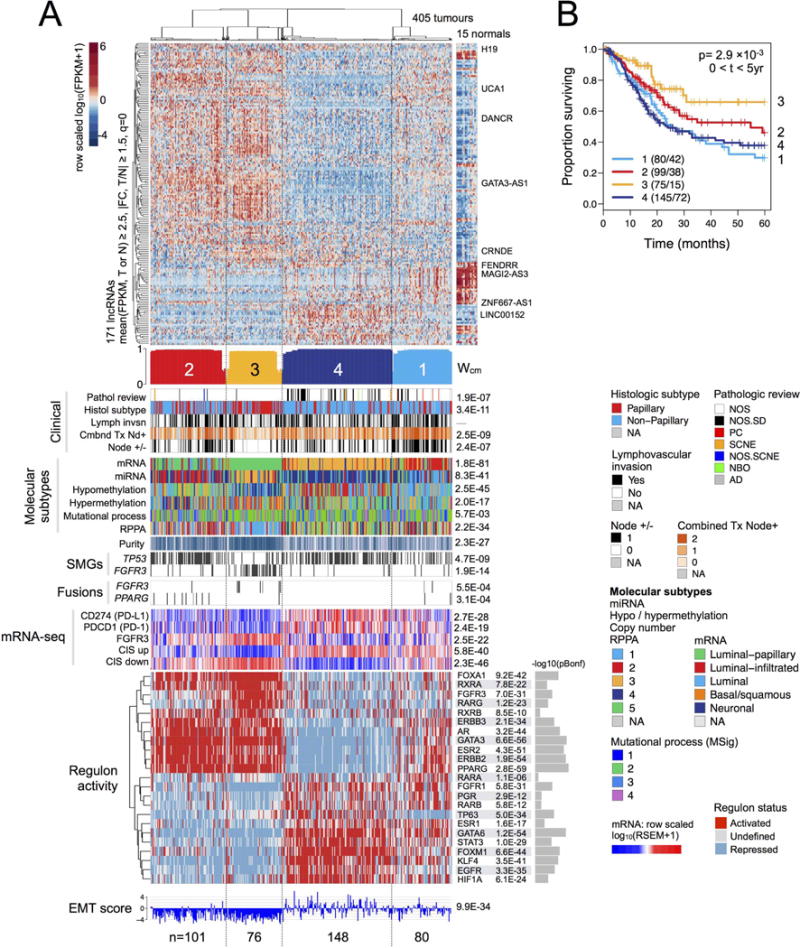
(A) Heatmap and covariates for four unsupervised lncRNA consensus clusters. Top to bottom: normalized abundance heatmap for 171 lncRNAs; profile of silhouette width calculated from the consensus membership heatmap, Wcm; covariates for clinical parameters, molecular subtypes, purity, mutations in TP53 and FGFR3, FGFR3 and PPARG gene fusions; row-scaled mRNA levels for 3 genes; collapsed CIS gene sets (Dyrskjot et al., 2004) (Methods; CIS up = genes up-regulated in CIS; CIS down = genes down-regulated in CIS); row-scaled regulon activity profiles (showing activated, undefined, or repressed status) for 23 regulators; RNA-seq-based EMT scores (Mak et al., 2016). The following p values are Bonferroni-corrected: for mutated genes (for 58 SMGs), gene fusions (for 23 fusions), regulon activity (for 23 regulators), and mRNA-seq (for 12 genes).
(B) A Kaplan-Meier plot for overall 5-year survival according to lncRNA subtype.
The lncRNA clusters were concordant with the mRNA subtypes (p = 2 × 10−81) and further discriminated within them. For example, lncRNA cluster 3 (n = 76), a better-survival subset of the luminal-papillary subtype, was depleted in TP53 mutations but enriched in FGRF3 mutations and fusions. It consisted largely of high-purity, papillary histology, and organ-confined cancers. Levels of many cancer-associated lncRNAs, including DANCR, GAS5, MALAT1, NEAT1, NORAD (LINC00657), and UCA1, were high; others, including ZNF667-AS1 (MORT) and LINC00152 (associated with lower EMT scores), were low (Figure S5B).
For miRNA mature strands, four unsupervised consensus clusters were associated with purity (p = 5 × 10−33), EMT scores (p = 5 × 10−39) (Table S2.24), and 5-year survival (p = 1.7 × 10−3) (Figure 5). They were concordant with subtypes for mRNA (p = 2 × 10−52), lncRNA (p = 2 × 10−45), hypomethylation (p = 5 × 10−30), RPPA (p = 9 × 10−30), and with histological subtype (papillary vs. non-papillary), combined T-stage/Node+, node positive/negative, and CIS gene sets (Dyrskjot et al., 2004). Many cancer-associated miRNAs were differentially abundant across the subtypes (Figure S5C).
Figure 5. MicroRNA expression subtypes.
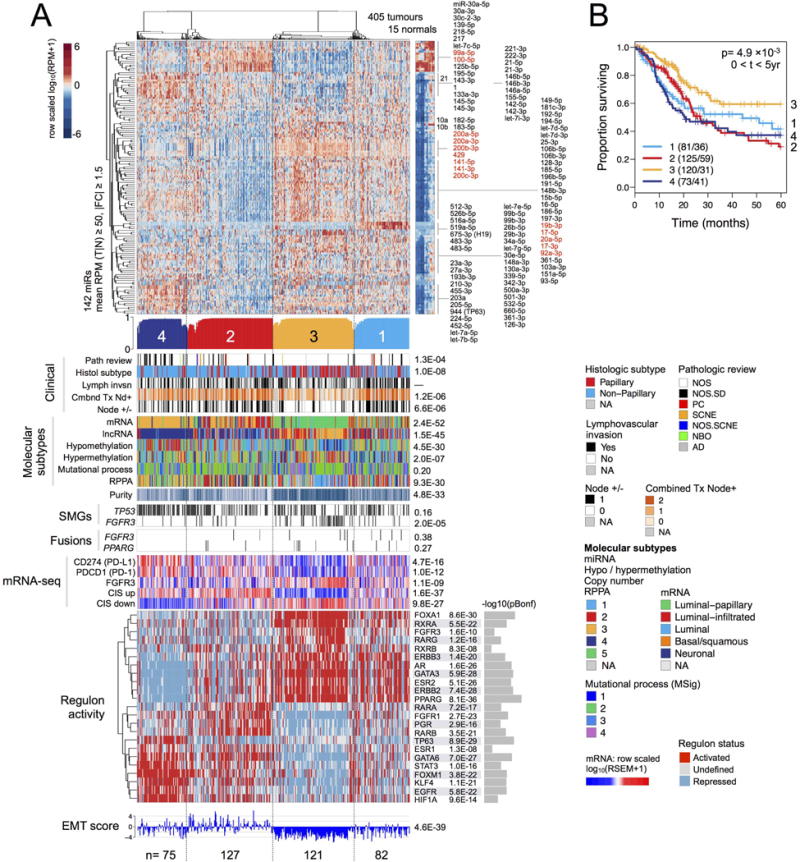
(A) Heatmap and covariates for a 4-cluster unsupervised consensus clustering solution. Top to bottom: Normalized heatmap showing a subset of 142 miRNAs that had a mean RPM ≥ 50 and an absolute value of tumour-vs-normal fold change ≥ 1.5. Profile of silhouette width calculated from the consensus membership heatmap, Wcm, with lower values indicating samples that are atypical cluster members. Covariate tracks for clinical parameters, genomic platform subtypes, purity, mutations in TP53 and FGFR3, and FGFR3 and PPARG gene fusions. Row-scaled regulon activity profiles for 23 regulators that have been associated with bladder cancer. Row-scaled mRNA levels for 12 genes, then for collapsed CIS gene sets (Dyrskjot et al., 2004) (Methods; CIS up = genes up-regulated in CIS; CIS down = genes down-regulated in CIS); and RNA-seq-based EMT scores (Mak et al., 2016). The following p values are Bonferroni-corrected: for mutated genes (for 58 SMGs), gene fusions (for 23 fusions), regulon activity (for 23 regulators), and mRNA-seq (12 genes).
(B) A Kaplan-Meier plot for overall survival data that has been censored at 5 years.
MiRNA subtype 3 was enriched in lncRNA 3 and showed the best survival among the 4 subtypes, consistent with low EMT scores and high miR-200 levels; CD274 (PD-L1) and PDCD1 (PD-1) levels were low. MiRNA subtypes defined subsets within the mRNA subtypes, with miR 4 (n = 75) and miR 2 (n = 127) containing most of the basal/squamous mRNA subtype samples and showing relatively poor survival, consistent with relatively high EMT scores.
Regulon Activity Differences Among RNA Subtypes
To further characterize the molecular differences between RNA-based subtypes, we analyzed activity profiles of 23 candidate ‘regulator’ genes that that have been associated with urothelial cancer (Methods, Table S2.25). By ‘regulator’ we mean a gene whose product induces and/or represses a target gene set, which we refer to as a ‘regulon’ (Castro et al., 2016a). We performed the analysis for this cohort of 412 samples, and also for an independent, mixed non-muscle invasive/MIBC cohort (n = 308) (Sjodahl et al., 2012). In both cohorts, the inferred regulon activity profiles sorted covariates that included histology, mRNA subtype, and EMT score (Figure S5D,E). Segregating by activated vs. repressed profiles identified survival-associated regulators with Kaplan-Meier plots and hazard ratios that were consistent in the two cohorts (Figure S5F,G). This analysis suggested that regulon analysis was robust and biologically relevant.
We then compared regulon activity across RNA-based subtypes, finding that activities varied most strongly for mRNA and lncRNA subtypes, and somewhat less strongly for miRNA subtypes (Figures 4, 5, and S5H). In luminal-papillary cases, 11 regulons were activated. lncRNA subtypes 2 and 3 (Figure 4A) were both associated with the luminal-papillary mRNA subtype and showed similar activation profiles for 9 regulons. Their profiles were consistent with the hypothesis that transcription factors GATA3, FOXA1, and PPARɣ drive luminal cell biology in bladder cancer (Warrick et al., 2016). The better-survival lncRNA 3 differed from lncRNA 2 by having, among other characteristics, an activated regulon for FGFR3 and undefined (i.e. neither activated nor repressed) regulon activity for TP63. Twelve regulons were activated in the basal/squamous cases, which were associated with miRNA clusters 2 and 4 (Figure 5A). The TP63 regulon was generally activated in miR 4 but repressed in miR 2, and the EGFR regulon was largely activated in miR 4 but had variable activity in miR 2. Overall, the analysis implicated certain regulators as important drivers of the differences in expression phenotype among bladder cancer subtypes.
Microbe analysis
We used RNA-seq (n = 408), WES (n = 412), and whole-genome sequence (n = 136) data to identify evidence of infection by HPV (n = 11), HHV4 (n = 6), HHV5 (n = 6), and Polyomavirus (n=1) (Tables S3.1–3.3, S3.7–3.8). For HPV we identified genomic integration in 4 tumors, with breakpoints associated with BCL2L1, SLC2A1-AS1, DEC1, SEC16A, and CCDC68 (Tables S3.4–3.6, 3.9). BK polyoma integration breakpoints were associated with FIGN and LIMA1 genes. For HHV4 and HHV5 we found no evidence for genomic integration. Hence, viral infection may contribute to a small percentage of urothelial carcinomas.
Proteomic Analysis by RPPA
Unsupervised consensus clustering of RPPA proteomic data for 208 antibodies (Table S2.26) and 343 of the 412 tumors resulted in five robust clusters that differed in protein expression profiles, pathway activities, and overall survival (p = 0.019) (Figure S6A,B).
RPPA cluster 1 (epithelial/papillary) showed the best outcomes, a low EMT pathway score (Figure S6C,D), and enrichment with papillary samples (Table S2.27). Cluster 2 (epithelial/intermediate) was intermediate in profile and outcomes. Elevated HER2 expression levels in clusters 1 and 2 suggest that these cases may respond to anti-HER2 directed therapies (e.g. Herceptin, T-DM1).
Cluster 3 (proliferative/low signaling) had a high cell cycle pathway score, principally due to high CYCLINB1 and PCNA expression, with low MAPK, PI3K, and mTOR pathway signaling (Figure S6D). Although cluster 3 tumors showed low levels of signaling, they expressed high levels of EGFR, suggesting them as possible candidates for EGFR inhibitors.
Clusters 4 and 5 had higher EMT pathway scores (Figure S6D) and were enriched with non-papillary and pathologic stage III and IV tumors. Cluster 4 (EMT/hormone signaling) had the worst outcome and relatively high reactive and hormone receptor pathway scores. Cluster 5 (reactive) had high levels of MYH11, HSP70, FIBRONECTIN, COLLAGEN VI, CAVEOLIN1 and RICTOR, as well as remarkably low levels of the proapoptotic mediator BAK, perhaps contributing to the cluster’s poor outcomes. Reactive cancer subtypes showed high levels of proteins that are likely produced in the tumor microenvironment as the result of interactions between cancer cells and cells in the microenvironment, including fibroblasts, as discussed previously for reactive breast cancer subtypes (Cancer Genome Atlas Network, 2012; Dennison et al., 2016).
Integrative Clustering Analysis
We used the Cluster of Cluster Assignments method (COCA) (Hoadley et al., 2014) to integrate and compare the cluster assignments obtained by clustering mRNA, lncRNA, miRNA data independently. The analysis identified overlapping subtype classifications. Although COCA subtypes were largely determined by the mRNA subtypes, lncRNA and miRNA data created finer-grained subdivisions (Figure 6A).
Figure 6. Integrated analysis.
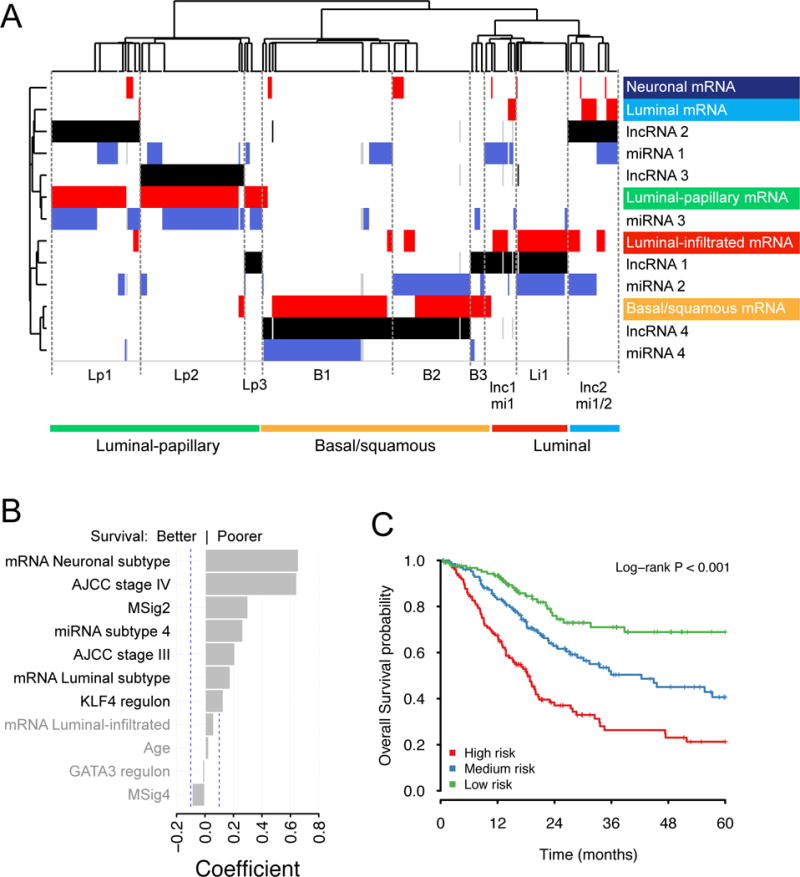
(A) Cluster of cluster assignments analysis (COCA). Unsupervised clustering of subtype calls. Subtype calls for mRNA (red), lncRNA (black), and miRNA (blue) are colored by separate data type. Annotations at the right of and below the heatmap use colors for mRNA subtypes. (B,C) Multivariate Cox analysis for overall survival.
(B) Coefficients (β) from the LASSO-penalized multivariate Cox regression on 15 covariates that were significant (corrected p < 0.05) in univariate survival calculations. Dashed blue lines indicate |β| = 0.1; variables shown in grey text have coefficients with |β| < 0.1.
(C) Kaplan-Meier plot predicted from the cohort, for three tertile risk groups, at 48 months.
Univariate and Multivariate Survival
This rich data set enabled us to do a detailed analysis of clinical and molecular variables for association with overall survival. While follow-up times remain limited, the event rate was high enough that results were informative.
Of 101 covariates analyzed by univariate log-rank tests, 20 had a Benjamini-Hochberg-adjusted p < 0.05 (Table S2.28). We removed 7 with many missing cases, leaving 13 for multivariate Cox regression analysis (Figure S7A). Results of nine candidate penalized methods were approximately equivalent (Figure S7B), we chose LASSO regression (Hutmacher and Kowalski, 2015; Walter and Tiemeier, 2009) to fit a multivariate model. For mRNA, lncRNA, miRNA and MSig subtypes, we set the best-survival subtype as the reference variable.
After filtering regression coefficients at |β| > 0.1, 7 variables representing 4 covariates were retained (Table S2.29, Figure 6B). A coefficient’s sign and magnitude associates a variable with poorer or better survival rates, relative to its reference variable, in the context of the set of regression variables retained in the model. The variables with largest coefficients were AJCC stages III and IV, mRNA neuronal and luminal subtypes, low mutation rate MSig 2, and miRNA subtype 4, which is a subset of basal-squamous cases, and KLF4 regulon activity, all of which were associated with worse survival. The mRNA luminal-infiltrated subtype, age, GATA3 regulon activity, and MSig4 were retained with smaller coefficients (Figure 6B).
The fitted model assigns weights to variables and generates a score for each sample. Thresholding these scores segregated the cohort into predicted risk groups or strata. Tertile thresholds generated three groups that were associated with survival (p < 0.001) (Figure 6C and Table S2.30).
We assessed multivariate Cox regressions that included age and AJCC stage, and subtypes for mRNA, lncRNA, miRNA, or mutational process (MSig), setting the best-survival subtype as the reference. Each molecular covariate had at least one subtype associated with worse survival, independent of age and stage (Figure S7C).
Subtype-stratified potential treatments
In Figure 7, we have integrated results from the multiple platform analyses, and propose therapeutic considerations stratified by expression subtyping. For each subtype we summarize the key drivers, and propose treatment strategies that may be appropriate in multiple clinical scenarios, including peri-operative therapy (neoadjuvant and adjuvant) combined with radical cystectomy, systemic therapy combined with locoregional radiation, or systemic therapy for measurable metastatic disease. We suggest this schema as a framework for prospective hypothesis testing in clinical trials, as well as for validation in ongoing or completed clinical trials that test, or have tested, treatment strategies.
Figure 7.
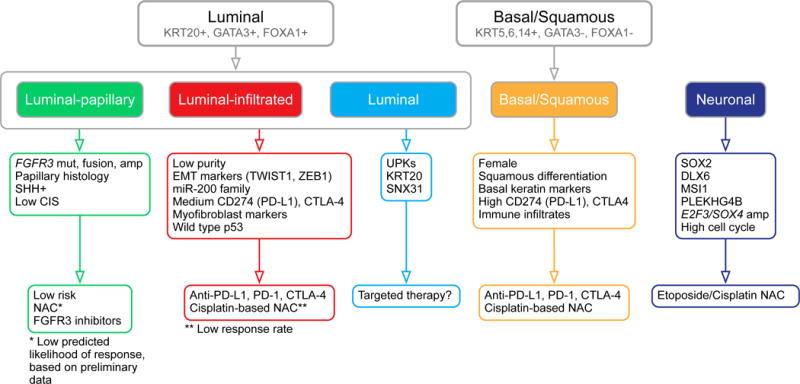
Proposed schema of expression-based, subtype-stratified therapeutic approach as a framework for prospective hypothesis testing in clinical trials. * For luminal-papillary cases, the low predicted likelihood of response is based on preliminary data from (Seiler et al., 2017). See Discussion.
Discussion
Bladder cancer, both non-muscle invasive (NMIBC) and muscle-invasive (MIBC), is a major source of morbidity and mortality worldwide. In the US there will be an estimated 79,000 new cases and 17,000 deaths in 2017 (Siegel et al., 2017). NMIBC occurs mainly as papillary disease with frequent FGFR3 mutations, whereas MIBC has a more diverse mutation spectrum as well as copy-number instability (Balbas-Martinez et al., 2013; Cappellen et al., 1999; Gui et al., 2011; Knowles and Hurst, 2015; van Rhijn et al., 2001).
In the following, we highlight essential findings from the complete cohort of 412 TCGA cases and suggest how these findings may contribute to our understanding of therapeutic possibilities. We identified 34 additional SMGs and 158 genes that are subject to epigenetic silencing, both of which may offer additional potential therapeutic targets, fusion events that implicate PPARG as a key gene in bladder cancer development, and refined subtypes defined by considering both miRNA and lncRNA profiling.
MIBCs show high overall mutation rates similar to those of melanoma and non-small cell lung cancers, and we confirm that these high rates are principally associated with mutation signatures for an endogenous mutagenic enzyme, APOBEC cytidine deaminase (Roberts et al., 2013). Most bladder cancer mutations are clonal, suggesting that APOBEC’s mutagenic activity occurs early in bladder cancer development. A better understanding of the origin and regulation of APOBEC expression and activity in normal bladder could lead to preventive strategies that target APOBEC as a key mutagenic source in bladder cancer.
MSig1’s high mutation burden consisted largely of APOBEC-signature mutations. The subset’s unusually good survival contributes to and correlates with the improved survival of subjects with higher mutational burden and higher neo-antigen load (Figure 1B). We propose that this is due to a natural host immune reaction to the high mutation burden, curbing further tumor growth and metastasis. This hypothesis should be tested in additional bladder cancer cohorts, and the MSig1 subset should be recognized in ongoing clinical trials, including trial of immune checkpoint therapy, as having a much better prognosis than average (see further below).
Chromatin modifier gene mutations are common in bladder cancer and also open potential therapeutic opportunities through rebalancing acetylation and deacetylation, and through other chromatin modifications. Recent studies have identified BRD4-EZH2 chromatin modification as an important growth pathway in bladder cancer, especially in tumors with loss of KDM6A, and shown in preclinical models that the BET inhibitor JQ1 and inhibition of EZH2 have therapeutic benefit (Ler et al., 2017; Wu et al., 2016). Recently, a Phase 2 study of Mocetinostat, a histone deacetylase inhibitor, in patients with locally advanced or metastatic urothelial carcinoma has completed accrual and results are awaited (NCT02236195).
The altered canonical signaling pathways provide multiple opportunities for therapeutic intervention. As one example the p53/Rb pathway is being targeted in a multicenter phase II trial evaluating palbociclib (PD-0332991) in patients with metastatic urothelial carcinoma who have cyclin-dependent kinase inhibitor 2A (CDKN2A) loss and retained retinoblastoma (Rb) expression (NCT02334527).
Our mRNA expression clustering identified the well-known luminal and basal subtypes of bladder cancer and further stratified them into 5 distinct subtypes. Included are two that we did not identify previously, neuronal and luminal, which have recently been corroborated in an independent cohort (Sjodahl et al., 2017). The neuronal subtype (5%) showed, in most cases, no histopathological distinction from other types of MIBC. Nonetheless, it had high levels of TP53 and RB1 mutations, as do small cell carcinomas in other tissues. It had the worst survival of the mRNA expression subtypes, making it important to recognize clinically. The luminal subtype had the highest expression level of uroplakin genes and may have adopted an umbrella cell phenotype. The luminal infiltrated subtype is similar to our previous TCGA subtype II and also similar to a subtype identified by Choi et al (Choi et al., 2014b), is characterized by a mesenchymal expression signature. It appears to be resistant to cisplatin-based chemotherapy and particularly sensitive to immunotherapy with checkpoint inhibitors.
LncRNA and miRNA expression patterns identified survival-related subsets of cases within the mRNA luminal-papillary subtype and basal-squamous subtypes, respectively. Many cancer-associated lncRNAs and miRNAs were differentially abundant among the bladder cancer subtypes. Multivariate regression analyses identified lncRNA and miRNA subtypes as independent predictors of survival.
Our regulon analysis identified the importance of transcriptional driver events in bladder cancer development. In this analysis, regulator activity was associated with survival, as described previously for breast cancer (Castro et al., 2016a). Certain regulon activity profiles varied greatly between the different coding and noncoding gene expression subtypes, suggesting that the regulators are key drivers of those expression subtypes. These findings provide potential targets for intervention, and could be used for subtype discrimination and therapy selection (Castro et al., 2016a).
Integrating RNA subtype classification, pathway information, EMT and CIS signatures, and immune infiltrate analyses leads us to propose a model of mRNA-based expression subtypes that may be associated with unique response to therapies and can be prospectively tested in clinical trials (Figure 7). We note that subsequent therapy was not included in this integrated analysis. Neoadjuvant cisplatin-based chemotherapy is the current standard of care in cisplatin-eligible patients without risk stratification. However, as not all patients derive benefit from chemotherapy, subtype-specific personalized therapies could help to optimize global patient outcome, while preventing unnecessary toxicity to non-responders. The following observations are hypothesis-generating, and thus are not ready to be used for clinical decision making.
The luminal-papillary subtype (35%) is characterized by FGFR3 mutations, fusions with TACC3, and/or amplification; by papillary histology; by active sonic hedgehog signaling; and by low CIS scores. Such cancers have low risk for progression, and preliminary data suggests a low likelihood of response to cisplatin-based neoadjuvant chemotherapy (NAC) (Seiler et al., 2017). The frequency of FGFR3 alterations in luminal papillary tumors suggests that tyrosine kinase inhibitors of FGFR3 may be an effective treatment approach, especially since early phase clinical trials show benefit of pan-FGFR inhibitor agents in FGFR3-selected advanced solid tumors (Karkera et al., 2017; Nogova et al., 2017).
The luminal-infiltrated subtype (19%) is characterized by the lowest purity, with high expression of EMT and myofibroblast markers, and of the miR-200s. It shows medium expression of CD270 (PD-L1) and CTLA4 immune markers. This subtype, corresponding to TCGA subtype II (Cancer Genome Atlas Research Network, 2014a), has been reported to respond to immune checkpoint therapy with atezolizumab in patients with metastatic or unresectable bladder cancer (Rosenberg et al., 2016). Validation of this subtype as a predictive marker for response to immunotherapy is ongoing in multiple clinical trials. Tumors with a luminal-infiltrated subtype may be resistant to cisplatin-based chemotherapy. Clinical trials may therefore be directed to validating this subtype as a negative predictive biomarker for chemotherapy response and for exploring alternative treatment strategies including targeted therapies.
The luminal subtype (6%) shows high expression of luminal markers, as well as KRT20 and SNX31. Due to its novelty, optimal therapy is less not defined. Future trial designs may compare the relative efficacy of either NAC or a therapy targeted to each cancer’s specific mutation profile.
The basal-squamous subtype (35%) is characterized by higher incidence in women, squamous differentiation, basal keratin expression, high expression of CD274 (PD-L1) and CTLA4 immune markers, and other signs of immune infiltration. Both cisplatin-based NAC and immune checkpoint therapy (Sharma et al., 2016) are appropriate therapeutic options, and trials comparing those treatments should be performed.
Finally, the neuronal subtype (5%) is characterized by expression of both neuroendocrine and neuronal genes, as well as a high cell-cycle signature reflective of a proliferative state. The neuronal subtype was recently recognized by others in an independent cohort (Sjodahl et al., 2017). Identifying this subtype currently depends on detecting expression of neuroendocrine/neuronal markers by either mRNA-seq or immunohistochemistry, as they do not exhibit the typical morphologic features associated with neuroendocrine tumors. Etoposide-cisplatin therapy is recommended in neoadjuvant and metastatic settings, as for neuroendocrine neoplasms arising in other sites, but this should also be tested in prospective clinical trials.
Our results suggest that mRNA subtype classification may be possible with a reduced gene set, enabling validation in independent cohorts and informing clinical trial designs that test new personalized therapies. However, additional integrative analyses that include assessment of lncRNAs, miRNAs, and regulon relationships can be expected to refine our subtyping of bladder cancers and aid in the search for optimal personalized targeted therapies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Seth Lerner (slerner@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Tumor and normal whole blood samples were obtained from patients at contributing centers with informed consent according to their local Institutional Review Boards (IRBs, see below). Biospecimens were centrally processed and DNA, RNA, and protein were distributed to TCGA analysis centers. In total, 412 evaluable primary tumors with associated clinicopathologic data were assayed on at least one molecular-profiling platform.
TCGA Project Management has collected necessary human subjects documentation to ensure the project complies with 45-CFR-46 (the “Common Rule”). The program has obtained documentation from every contributing clinical site to verify that IRB approval has been obtained to participate in TCGA. Such documented approval may include one or more of the following:
An IRB-approved protocol with Informed Consent specific to TCGA or a substantially similar program. In the latter case, if the protocol was not TCGA-specific, the clinical site PI provided a further finding from the IRB that the already-approved protocol is sufficient to participate in TCGA.
A TCGA-specific IRB waiver has been granted.
A TCGA-specific letter that the IRB considers one of the exemptions in 45-CFR-46 applicable. The two most common exemptions cited were that the research falls under 46.102(f)(2) or 46.101(b)(4). Both exempt requirements for informed consent, because the received data and material do not contain directly identifiable private information.
A TCGA-specific letter that the IRB does not consider the use of these data and materials to be human subjects research. This was most common for collections in which the donors were deceased.
METHOD DETAILS
Biospecimen Collection; Pathological and Clinical Data
Sample inclusion criteria
Biospecimens were collected from patients diagnosed with muscle-invasive urothelial carcinoma undergoing surgical resection with either transurethral resection or radical cystectomy. No patient had received prior chemotherapy or radiotherapy for their disease. Prior intravesical Bacille Calmette Guerin (BCG) was allowed but not intravesical chemotherapy. Institutional review boards at each tissue source site reviewed protocols and consent documentation and approved submission of cases to TCGA. Cases were staged according to the American Joint Committee on Cancer (AJCC) staging system. Each frozen primary tumor specimen had a companion normal tissue specimen. This could be blood/blood components (including DNA extracted at the tissue source site), adjacent normal tissue taken from greater than 2 cm from the tumor, or both. Specimens were shipped overnight from 36 tissue source sites (TSS) using a cryoport that maintained an average temperature of less than −180°C. Each tumor and adjacent normal tissue specimen (if available) were embedded in optimal cutting temperature (OCT) medium and a histologic section was obtained for review. Each H&E-stained case was reviewed by a board-certified pathologist to confirm that the tumor specimen was histologically consistent with urothelial carcinoma and that the adjacent normal specimen contained no tumor cells. Divergent histologies within the sample could not represent less than 50% of the cancer specimen. Tumor sections were required to contain an average of 60% tumor cell nuclei with equal to or less than 20% necrosis for inclusion in the study, per TCGA protocol requirements.
Sample Processing
RNA and DNA were extracted from tumor and adjacent normal tissue specimens using a modification of the DNA/RNA AllPrep kit (Qiagen). The flow-through from the Qiagen DNA column was processed using a mirVana miRNA Isolation Kit (Ambion). This latter step generated RNA preparations that included RNA <200 nt suitable for miRNA analysis. DNA was extracted from blood using the QiaAmp blood midi kit (Qiagen). Each specimen was quantified by measuring Abs260 with a UV spectrophotometer or by PicoGreen assay. DNA specimens were resolved by 1% agarose gel electrophoresis to confirm high molecular weight fragments. A custom Sequenom SNP panel or the AmpFISTR Identifiler (Applied Biosystems) was utilized to verify tumor DNA and germline DNA were derived from the same patient. Five hundred nanograms of each tumor and normal DNA were sent to Qiagen for REPLI-g whole genome amplification using a 100 μg reaction scale. Only specimens yielding a minimum of 6.9 μg of tumor DNA, 5.15 μg RNA, and 4.9 μg of germline DNA were included in this study. RNA was analyzed via the RNA6000 nano assay (Agilent) for determination of an RNA Integrity Number (RIN), and only the cases with RIN >7.0 were included in this study. A total of 722 bladder urothelial carcinoma cases were received by the BCR and 412 (57%) passed final quality control. Reasons for rejection are described at https://cancergenome.nih.gov/cancersselected/biospeccriteria. Normal controls included peripheral blood (n=392), and/or tumor-adjacent, histologically normal-appearing bladder tissue (n=37).
Pathology Review
All samples were subjected to central review by four urological pathologists (HAA, DEH, BAC, VER), using digitally scanned whole slides of a representative section from a fresh frozen tumor sample submitted for molecular analysis. All samples were systematically evaluated to confirm the histopathologic diagnosis and any variant histology according to the most recent World Health Organization (WHO) classification (Moch et al., 2016). Additionally, all tumor samples were assessed for tumor content (% tumor nuclei), the presence and extent of tumor necrosis and the presence of invasion into muscularis propria. Tumor samples were also evaluated for the presence and extent of inflammatory infiltrate as well as the type of the infiltrating cells in the tumor microenvironment (lymphocytes, neutrophils, eosinophils, histiocytes, plasma cells). Any non-concordant diagnoses among the four pathologists were re-reviewed and resolution achieved after discussion.
Clinical Data
Clinical data were submitted for all cases passing quality control. Patient information was completed immediately following the notification of qualification, and a follow-up submission was required for all living patients one year after the case’s qualification date; follow-up data beyond this were submitted voluntarily. For the work reported here, clinical data for all 412 cases were downloaded from the Genomics Data Commons Data Portal (https://portal.gdc.cancer.gov/) on May 5th, 2017. The majority of the fields included in the dataset used for analysis were found in the patient section of the clinical XML files (e.g. nationwidechildrens.org_clinical.TCGA-HQ-A5ND.xml). This information had been collected during the initial submission from the participating Tissue Source Sites (TSSs). For survival analysis, the follow-up information was also considered, in order to capture each case’s longest number of days to follow-up or death; this information changed survival information for a subset of cases reported in the previous TCGA publication (Cancer Genome Atlas Research Network, 2014a).
Copy Number Analysis
DNA from each tumour or germline sample was hybridized to Affymetrix SNP 6.0 arrays using protocols at the Genome Analysis Platform of the Broad Institute as previously described (McCarroll et al., 2008). Briefly, from raw .CEL files, Birdseed was used to infer a preliminary copy-number at each probe locus (Korn et al., 2008). For each tumour, genome-wide copy number estimates were refined using tangent normalization, in which tumour signal intensities are divided by signal intensities from the linear combination of all normal samples that are most similar to the tumour. This linear combination of normal samples tends to match the noise profile of the tumour better than any set of individual normal samples, thereby reducing the contribution of noise to the final copy-number profile. Individual copy-number estimates then underwent segmentation using Circular Binary Segmentation (Olshen et al., 2004). Segmented copy number profiles for tumour and matched control DNAs were analyzed using Ziggurat Deconstruction, an algorithm that parsimoniously assigns a length and amplitude to the set of inferred copy-number changes underlying each segmented copy number profile, and the analysis of broad copy-number alterations was then conducted as previously described (Mermel et al., 2011). Significant focal copy number alterations were identified from segmented data using GISTIC 2.0 (Mermel et al., 2011). Allelic copy number, regions of homozygous deletions, whole genome doubling and purity and ploidy estimates were calculated using the ABSOLUTE algorithm (Carter et al., 2012a).
DNA Sequencing
DNA sequencing and data processing
Exome capture was performed using Agilent SureSelect Human All Exon 50 Mb according to the manufacturers’ instructions. Briefly, 0.5–3 micrograms of DNA from each sample were used to prepare the sequencing library through shearing of the DNA followed by ligation of sequencing adaptors. All whole exome (WES) and whole genome (WGS) sequencing was performed on the Illumina HiSeq platform. Paired-end sequencing (2 × 101 bp for WGS and 2 × 76 bp for WE) was carried out using HiSeq sequencing instruments; the resulting data was analyzed with the current Illumina pipeline. Basic alignment and sequence QC was done on the Picard and Firehose pipelines at the Broad Institute. Sequencing data were processed using two consecutive pipelines:
Sequencing data processing pipeline (‘Picard pipeline’)
Picard (http://picard.sourceforge.net/) uses the reads and qualities produced by the Illumina software for all lanes and libraries generated for a single sample (either tumor or normal) and produces a single BAM file (http://samtools.sourceforge.net/SAM1.pdf) representing the sample. The final BAM file stores all reads and calibrated qualities along with their alignments to the genome.
Cancer genome analysis pipeline (‘Firehose pipeline’)
- Firehose (http://www.broadinstitute.org/cancer/cga/Firehose) takes the BAM files for the tumor and patient matched normal samples and performs analyses including quality control, local realignment, mutation calling, small insertion and deletion identification, rearrangement detection, coverage calculations and others as described briefly below. The pipeline represents a set of tools for analyzing massively parallel sequencing data for both tumor DNA samples and their patient_matched normal DNA samples. Firehose uses GenePattern (Reich et al., 2006) as its execution engine for pipelines and modules based on input files specified by Firehose. The pipeline contains the following steps:
- Quality control. This step confirms identity of individual tumor and normal to avoid mix-ups between tumor and normal data for the same individual.
- Local realignment of reads. This step realigns reads at sites that potentially harbor small insertions or deletions in either the tumor or the matched normal, to decrease the number of false positive single nucleotide variations caused by misaligned reads.
- Identification of somatic single nucleotide variations (SSNVs) – This step detects candidate SSNVs using a statistical analysis of the bases and qualities in the tumor and normal BAMs, using Mutect (Cibulskis et al., 2013).
- Identification of somatic small insertions and deletions – In this step putative somatic events were first identified within the tumor BAM file and then filtered out using the corresponding normal data, using Indellocator (Ratan et al., 2015)
Mutation significance analysis
Genes with a significant excess of the number of non-synonymous mutations relative to the estimated density of background mutations were identified using MutSig algorithm. MutSig has been used to identify significantly mutated genes (SMGs) in several previous TCGA tumor sequencing projects and has undergone a development path starting from the most basic approach implemented in MutSig 1.0 (Getz et al., 2007) to the current version MutSig 2CV (Lawrence et al., 2014; Lawrence et al., 2013). This study made use of MutSig 2CV to produce a robust list of significantly mutated genes (Table S2.9).
Mutation clonality analysis
We used the ABSOLUTE algorithm (Carter et al., 2012b) with copy number and mutation data to infer purity and ploidy for 400 tumor samples, and estimated the cancer cell fraction (CCF) for each mutation. We classified mutations with CCF ≥ 0.9 as clonal and all other mutations as sub-clonal.
Mutation signature analysis
Mutation signature discovery involves deconvolving cancer somatic mutations, stratified by mutation contexts or biologically meaningful subgroups, into a set of characteristic patterns (signatures), and inferring the contributions of signature activity across samples (Alexandrov et al., 2013). Single nucleotide variants (SNVs) in the 412 samples were classified into 96 base substitution types, i.e. the six base substitutions C>A, C>G, C>T, T>A, T>C, and T>G, within the tri-nucleotide sequence context that includes the bases immediately 5′ and 3′ to each mutated base. Thus the input data for the mutation signature analysis is given as the mutation counts matrix X (96 by N=412), where each element represents an observed mutation count at the context i in the sample j. We applied a Bayesian variant of the non-negative matrix factorization (NMF) with an exponential prior (BayesNMF) (Kim et al., 2016a; Tan and Fevotte, 2013) to enable a de novo signature discovery with an optimal inference for the number of signatures (K*) best explaining the observed X. The mutation count matrix was taken as an input for the BayesNMF and factored into two matrices, W′ (96 by K*) and H′ (K* by N), approximating X by W′H′. All fifty independent BayesNMF runs with a different initial condition for 409 samples converged to the solution of K* = 4, identifying four distinct mutational processes, C>T_CpG, ERCC2, APOBC-b, and APOBEC-a.
To enumerate the number of mutations associated with each mutation signature we performed a scaling transformation, X ~ W′H′ = WH, W = W′U−1 and H = UH′, where U is a K* by K* diagonal matrix with the element corresponding to the 1-norm of column vectors of W′, resulting in the final signature matrix W and the activity matrix H. Note that the kth column vector of W represents a normalized mutability along 96 tri-nucleotide mutation contexts in the kth signature, and the kth row vector of H dictates the number of mutations associated with the kth signature across samples. The MSig clustering analysis for 409 samples was performed using a standard hierarchical clustering in R, with a ‘euclidian’ distance for the signature activity matrix H and a ‘ward.D’ linkage. The number of MSig clusters was chosen by manual inspection.
Using the W and H matrices determined by BayesNMF we annotated each mutation with the probability (likelihood of association) that it was generated by each of the discovered mutational signatures, pms, where ‘m’ denotes a mutation and ‘s’ refers to the signature. Specifically, the likelihood of association to the kth signature for a set of mutations corresponding to the i-th mutation context and j-th sample was defined as [wkhk/Σkwkhk]ij, where wk and hk correspond to the kth column vector and kth row vector of W and H, respectively (Kasar et al., 2015).
Unsupervised clustering of mutations in SMGs and focal SCNAs
We first created a binary event matrix, Q (n by m), comprised of mutations in 53 SMGs and focal SCNAs in the 25 genes that had more than ten SMG mutations and more than ten focal SCNAs across 408 samples. The resulting event matrix was used to compute a consensus matrix, MK, in which the element Mij represents how often both event i and sample j clustered together, with K being the number of clusters, by iterating conventional NMF with Frobenius norm (K * 25) times to approximate Q ~ WH. The cluster membership for event i and sample j was determined by the “maximum association criterion” as i* = max_k [wik] and j* = max_k [hkj] (k = 1 through K). Then the cumulative consensus matrix, M, was computed by summing up all MK with K increasing through 2 to 8, and normalized by the total number of iterations, resulting in the normalized M*. To determine the optimal number of consensus clusters, K*, i.e. that best explain the observed M*, we applied Bayesian non-negative matrix factorization (NMF) with a half-normal prior, finding the best approximation, M* ~ W*H*, where wik in W* (m by K) and hkj in H* (K by m) represents a clustering affinity or an association of the event i and the sample j to the cluster k, respectively. Twelve out of 20 independent BayesNMF runs with different initial conditions converged to the solution of K* = 4, while eight runs converged to the solution of K* = 5. After manual inspection we chose the K* = 4 solution, and reported four MutCN clusters.
Quantitation of Mutagenesis by APOBEC Cytidine Deaminases
The exome-wide prevalence of the APOBEC mutagenesis signature and the enrichment of this signature over its presence expected for random mutagenesis was evaluated with Pattern of Mutagenesis by APOBEC Cytidine Deaminases (P-MACD) analysis pipeline as outlined in (Roberts et al., 2013) and described in detail in Broad Institute TCGA Genome Data Analysis Center (2016): Analysis of mutagenesis by APOBEC cytidine deaminases (P-MACD). Broad Institute of MIT and Harvard (doi:10.7908/C1CC1013). Briefly, analysis is based on previous findings that APOBECs deaminate cytidines predominantly in a tCw motif and that the APOBEC mutagenesis signature is composed of approximately equal numbers of two kinds of changes in this motif – tCw ➔G and tCw ➔T mutations (flanking nucleotides shown in small letters; w=A or T). We calculated on a per sample basis, the enrichment of the APOBEC mutation signature among all mutated cytosines in comparison to the fraction of cytosines that occur in the tCw motif among the +/− 20 nucleotides surrounding each mutated cytosine (“APOBEC_enrich” column in data files). In addition, several other parameters that characterize the prevalence of the APOBEC mutagenesis pattern in a sample and/or that are useful for downstream analyses and comparisons. The main parameter used in this paper was the minimum estimate of the number of APOBEC induced mutations in a sample - “APOBEC_MutLoad_MinEstimate”. It was calculated using the formula: [“tCw➔G+tCw➔T”]x[(“APOBEC_enrich”-1)/“APOBEC_enrich”], which allows estimating the number of APOBEC signature mutations in excess of what would be expected by random mutagenesis. For example, if statistically significant enrichment in a sample would be =2, the minimum estimate of APOBEC-induced mutations would be 50%of total number of APOBEC-signature mutations ([“tCw➔G+tCw➔T”]). Calculated values are rounded to the nearest whole number. “APOBEC_MutLoad_MinEstimate” is calculated only for samples with passing 0.05 FDR threshold for APOBEC enrichment ([“BH_Fisher_p-value_tCw”]=<0.05. Samples with “BH_Fisher_p-value_tCw” value greater than 0.05 receive a value of 0. For some analyses and figures “APOBEC_MutLoad_MinEstimate” parameter was converted into categorical values as follows:
“no”: “APOBEC_MutLoad_MinEstimate”=0
“low”: 0<”APOBEC_MutLoad_MinEstimate” ≤ median of non-zero values in the set of 412 BLCA samples
“high”: “APOBEC_MutLoad_MinEstimate” > median of non-zero values in the set of 412 BLCA samples (median of non-zero values in the set of 412 BLCA samples = 61.5).
Class I HLA mutation and neoantigen analysis
Class I HLA typing and mutation detection
HLA typing and detection of mutations in class I HLA genes (HLA-A/B/C) was performed using Polysolver (Shukla et al., 2015). Briefly, the HLA typing algorithm employs a Bayesian model that first estimates the prior probabilities of different alleles based on the ethnicity of the individual. These probabilities are then updated with a model that takes into account the base qualities and alignments of putative HLA-derived reads against the reference HLA allele database. The alleles for each of the three HLA genes are inferred based on the computed scores in a two-stage process. These inferred HLA alleles serve as the reference for the HLA mutation detection step. Putative HLA reads from the tumor and the germline sample are extracted and aligned to the inferred allele sequences, followed by mutation and insertion/deletion identification with the Mutect (Cibulskis et al., 2013) and Strelka (Saunders et al., 2012) tools respectively.
We used a Chi-square test to assess whether HLA mutations were more common in patients with prior BCG treatment.
Neoantigen prediction
For each patient, we first enumerated a list of all possible 9 and 10-mer peptides bearing somatic mutations, or overlapping open reading frame derived from frameshifting indels or nonstop mutations. These peptides were then evaluated for binding against the patient’s inferred HLA type using the NetMHCpan-3.0 algorithm (Nielsen and Andreatta, 2016). The neoantigen load was defined as the total number of predicted peptide:allele binders with rank percentile score less than or equal to the weak binder threshold (2%). Univariate survival analysis of neoantigen load was evaluated using the Kaplan-Meier method. The effect of neoantigen load in the context of other variables was assessed using the Cox proportional hazards model. The comparison of number of HLA mutations or number of predicted binders between groups (e.g. MSig1 vs MSig2–4 clusters) was performed with two-sided t-tests.
DNA methylation and epigenetic silencing
Assay platform
We used the Illumina Infinium HumanMethylation450 (HM450) DNA methylation platform (Bibikova et al., 2011; Bibikova et al., 2009) to obtain DNA methylation profiles of 412 tumor samples and 21 tumor-adjacent, histologically normal-appearing bladder tissue samples. The HM450 assay analyzes the DNA methylation status of up to 482,421 CpG and 3,091 non-CpG (CpH) sites throughout the genome. It covers 99% of RefSeq genes with multiple probes per gene and 96% of CpG islands from the UCSC database and their flanking regions. The assay probe sequences and information for each interrogated CpG site on Infinium DNA methylation platform are available from Illumina (www.illumina.com).
The DNA methylation score for each assayed CpG or CpH site is represented as a beta (β) value (β = (M/(M+U)) in which M and U indicate the mean methylated and unmethylated signal intensities for each assayed CpG or CpH, respectively. B values range from zero to one, with scores of “0” indicating no DNA methylation and scores of “1” indicating complete DNA methylation. An empirically derived detection p value accompanies each data point and compares the signal intensity with an empirical distribution of signal intensities derived from a set of negative control probes on the array. Any data point with a corresponding p value greater than 0.05 is deemed to not be statistically significantly different from background and is thus masked as “NA” in the Level 3 data packages as described below. Further details on the Illumina Infinium DNA methylation assay technology have been described previously (Bibikova et al., 2011; Bibikova et al., 2009).
Sample and data processing
We performed bisulfite conversion of 1µg of genomic DNA from each sample using the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. We assessed the amount of bisulfite-converted DNA and completeness of bisulfite conversion using a panel of MethyLight-based quality control (QC) reactions as previously described (Campan et al., 2009). All the TCGA samples passed our QC tests and entered the Infinium DNA methylation assay pipeline. Bisulfite-converted DNAs were whole-genome-amplified (WGA) and enzymatically fragmented prior to hybridization to BeadChip arrays as per the Infinium protocol. BeadArrays were scanned using the Illumina iScan technology to produce IDAT files. Raw IDAT files for each sample were processed with the R/Bioconductor package methylumi. TCGA DNA methylation data packages were then generated using the EGC.tools R package which was developed internally and is publicly available on GitHub (https://github.com/uscepigenomecenter/EGC.tools).
TCGA Data Packages
The data levels and the files contained in each data level package are described below and are present on the NCI Genomic Data Commons (https://gdc.cancer.gov).
Level 1 data contain raw IDAT files (two per sample) as produced by the iScan system and as mapped by the Sample and Data Relationship Format (SDRF). These IDAT files were directly processed by the R/Bioconductor package methylumi. We provided a disease-mapping file (BLCA.mappings.csv) in the AUX directory to facilitate this process. Level 2 data contain background-corrected methylated (M) and unmethylated (U) summary intensities as extracted by the R/Bioconductor package methylumi. Detection p values were computed as the minimum of the two values (one per methylation state measurement) for the empirical cumulative density function of the negative control probes in the appropriate color channel. Background correction was performed via normal-exponential deconvolution (Triche et al., 2013). Multiple-batch archives had the intensities in each of the two channels multiplicatively scaled to match a reference sample. The reference sample is defined in each array as the sample having R/G ratio of the normalization control probes closest to 1.0. Level 3 data contain β value calculations with annotations for HGNC gene symbol, chromosome, and genomic coordinates (UCSC hg19, Feb 2009) for each targeted CpG/CpH site on the array. Probes having a common SNP (dbSNP build 135, Minor Allele Frequency >1%) within 10 bp of the interrogated CpG site or having an overlap with a repetitive element (as detected by RepeatMasker and Tandem Repeat Finder based on UCSC hg19, Feb 2009) within 15 bp (from the interrogated CpG site) were masked as “NA” across all samples, and probes with a detection p value greater than 0.05 in a given sample were masked as “NA” on that array. Probes that were mapped to multiple sites in the human genome (UCSC hg19, Feb 2009) were annotated as “NA” for chromosome and 0 for the CpG/CpH coordinate.
We used Level 3 DNA methylation data for the analyses described in this manuscript.
Unsupervised clustering analysis of DNA methylation data
We removed probes which had any “NA”-masked data points and probes that were designed for sequences on X or Y chromosomes or non-CpG sites.
To capture cancer-specific DNA hypermethylation events, we first selected CpG sites that were not methylated in normal tissues (mean β value <0.2). To minimize the influence of variable tumor purity levels on a clustering result, we dichotomized the data using a β value of ≥0.3 to define positive DNA methylation and < 0.3 to specify lack of methylation. The dichotomization not only ameliorated the effect of tumor sample purity on the clustering, but also removed a great portion of residual batch/platform effects that are mostly reflected in small variations near the two ends of the range of β values. We also removed CpG sites that were methylated in leukocytes, a major source of contamination present in a tumor sample (mean β-value >0.2). We then performed consensus clustering with the dichotomized data on 31,249 CpG sites that were methylated in at least 5% of the tumor samples. The optimal number of clusters was assessed based on 80% probe and tumor resampling over 1,000 iterations of hierarchical clustering for K = 2, 3, 4…20 using the binary distance metric for clustering and Ward’s method for linkage as implemented in the R/Bioconductor ConsensusClusterPlus package.
Similarly, in order to investigate subgroups based on cancer-specific DNA hypomethylation, we identified CpG sites that were highly methylated in normal tissues (mean β value >0.8). We dichotomized the data using a β value of <0.7 as a threshold for loss of DNA methylation. We then performed consensus clustering with the dichotomized data on 53,862 CpG sites that showed hypomethylation in at least 10% of the tumors.
Heatmaps were generated to assess clustering results based on the original β values for a subset of the most variably methylated CpG sites across the tumors. The probes were displayed based on the order of unsupervised hierarchal clustering of the β values using the Euclidean distance metric and Ward’s linkage method. Covariate association p values were calculated with Chi-square tests.
Identification of epigenetically silenced genes
We first removed DNA methylation probes overlapping with SNPs, repeats or designed for sequences on X or Y chromosomes or non-CpG sites. The remaining probes were mapped against UCSC Genes using the GenomicFeatures R/Bioconductor package. Probes unmethylated in normal tissues (mean β value <0.2) and located in a promoter region (defined as the 3 kb region spanning from 1,500 bp upstream to 1,500 bp downstream of the transcription start site) were identified. mRNA expression data were log2 transformed [log2(RSEM+1)] and used to assess the gene expression levels associated with DNA methylation changes. DNA methylation and gene expression data were merged by Entrez Gene IDs.
We dichotomized the DNA methylation data using a β value of >0.3 as a threshold for positive DNA methylation and eliminated CpG sites methylated in fewer than 3% of the tumor samples. For each probe/gene pair, we applied the following algorithm: 1) classify the tumors as either methylated (β ≥0.3) or unmethylated (β <0.3); 2) compute the mean expression in the methylated and unmethylated groups; 3) compute the standard deviation of the expression in the unmethylated group. We then selected probes for which the mean expression in the methylated group was less than 1.64 standard deviations from the mean expression of the unmethylated group. We labeled each individual tumor sample as epigenetically silenced for a specific probe/gene pair if: a) it belonged to the methylated group and b) the expression of the corresponding gene was lower than the mean of the unmethylated group of samples. If there were multiple probes associated with the same gene, a sample that was identified as epigenetically silenced at more than half the probes for the corresponding gene was also labeled as epigenetically silenced at the gene level. For each gene, we evaluated resulting silencing call based scatter plot of DNA methylation vs. expression and a heatmap.
We manually examined the list of genes that were significantly mutated. We identified additional genes having evidence for epigenetic silencing at low frequencies. CDKN2A DNA methylation status was assessed based on the probe (cg13601799) located in the p16INK4 promoter CpG island. p16INK4 expression was determined by the log2(RPKM+1) level of its first exon (chr9:21974403-21975038).
The complete list of 158 genes identified as epigenetically silenced is provided in Table S2.17.
Genes upregulated in hypomethylated subtype 4
We used four types of data: 1) β values for 5386 probes for 412 primary tumour samples and 21 adjacent normal samples, 2) RSEM gene-level expression data for 408 primary tumours and 19 adjacent normals, and 3) clinical and molecular data for 412 tumor samples, and 4) pathology review of micrograph images for the adjacent normals, which indicated that we should remove BT-A20U-11, BT-A2LB-11, GD-A2C5-11, and GD-A3OP-11.
Of the 408 tumor samples with RSEM data, 36 were in hypomethylation subtype 4 (‘subtype 4’), and 372 in the other hypomethylation subtypes.
We identified 10368 genes had a mean RSEM abundance of at least 1.0 in each of the tumour groups (subtype 4 vs. other), and an absolute value fold change of at least 1.25 between the two groups. 1863 genes were differentially abundant between the two groups (Benjamini-Hochberg (BH)-corrected p < 0.01, Wilcoxon test), and 436 of the 1863 genes were more abundant in subtype 4, with a fold change of at least 1.5.
We identified 2646 of the 5386 methylation probes that had a fold change more negative than −1.5 between subtype 4 and other samples, i.e. had lower β values in subtype 4. These had BH-corrected Wilcoxon p values < 0.003, and −1/FC ranging from −5.6 to −1.5.
Using the ‘annotations’ from the IlluminaHumanMethylation450kanno.ilmn12.hg19 v0.6.0 R package, we associated 1784 of the 2646 probe IDs with one or more gene symbols. Of these, 681 records had gene symbols that were semicolon-separated lists, e.g. SIRPG;SIRPG;SIRPG, or NCRNA00175;NCRNA00175;COL18A1. We collapsed such lists into unique symbols, and arbitrarily took the last of these symbols, even when the list contained more than one symbol. This associated each probe ID with one RefSeq gene symbol. Because some symbols have more than one associated probe, the 1784 gene symbols contained 1240 unique symbols.
187 unique RefSeq gene symbols were present in both a) the differentially abundant RSEM genes and b) genes associated with differential probes. For each probe-associated gene we retained only the probe with the most significant BH-corrected p value, accepting all annotated relationships of a probe to a CpG island (i.e. Island, N_Shelf, N_Shore, S_Shelf, S_Shore, or OpenSea).
From the 187 RefSeq genes, we identified 39 for which, in hypomethylation subtype 4, the RSEM gene abundance was higher, and the beta value for the associated DNA methylation probe was lower. We then inspected scatterplots of beta vs. RSEM abundance, which included the 14 adjacent normals that were present in both DNA methylation and RSEM datasets, and had passed pathology review. We identified a subset of 12 genes for which DNA hypomethylation in subtype 4 may have resulted in higher RSEM abundance in that subtype.
Statistics
Statistical analysis and data visualization were carried out using the R/Biocoductor software packages (www.bioconductor.org).
mRNA Expression Profiling
mRNA sequencing and expression quantification
RNA was extracted, prepared into mRNA libraries, and sequenced by Illumina HiSeq resulting in paired 50nt reads, and subjected to quality control as previously described (Cancer Genome Atlas Research, 2012). RNA reads were aligned to the hg19 genome assembly using Mapsplice (Wang et al., 2010), and RNA fusion events were automatically detected by MapSplice as previously described. Gene expression was quantified for the transcript models corresponding to the TCGA GAF2.1 (https://gdc-api.nci.nih.gov/v0/data/a0bb9765-3f03-485b-839d-7dce4a9bcfeb), using RSEM (Pasqualucci et al., 2011) and normalized within-sample to a fixed upper quartile. For further details on this processing, refer to GDC Description file under the V2_MapSpliceRSEM workflow (https://gdc-api.nci.nih.gov/legacy/data/cf4559f9-6beb-4bb3-ac43-c99ba6cf7f0f). Data for genes were median-centered across samples for downstream analysis.
Unsupervised mRNA expression clustering
For unsupervised clustering analysis the log2(RSEM) gene expression data for N = 408 samples was pre-processed to determine the most highly expressed and variable 3,347 genes across samples. We removed genes with NA values more than 10% across samples and then selected top 25% most-varying genes by standard deviation of gene expression across samples. The resulting expression matrix R (3347 by 408) was further transformed to the matrix R* of fold changes centered at the median expression. The expression clustering analysis was done by combining BayesNMF (Tan and Fevotte, 2013) with a consensus hierarchical clustering approach, as follows. Using the distance matrix of 1 – C, the element Cij representing the Spearman correlation between the sample i and j across 3347 genes in R*, we first compute a consensus matrix, MK, the element Mij representing how often both samples i and j clustered together, and K being the number of clusters, by iterating a standard hierarchical clustering (K * 500) times with the average linkage option and 80% resampling in sample space. Then the cumulative consensus matrix, M, was computed by summing up all MK with K increasing through 2 to 10, and normalized by the total number of iterations, resulting in the normalized M*. To determine the optimal number of clusters, K*, i.e. that best explain the observed M*, we applied Bayesian non-negative matrix factorization (NMF) with a half-normal prior, finding the best approximation, M* ~ HT H, where hkj in H (K* by N) represents a clustering affinity or an association of the sample j to the cluster k and HT is a transpose of H. Nine out of 20 independent BayesNMF runs with different initial conditions converged to the solution of K* = 5, while 11 runs converged to the solution of K* = 4. After manual inspection we chose the K* = 5 solution, giving rise to the five mRNA expression subtypes: luminal, luminal-infiltrated, basal-squamous, neuronal, and luminal-papillary. We noted that both luminal and luminal-infiltrated clusters merged together to form a single cluster in the K* = 4 solution, indicating that expression patterns in these two clusters were relatively more similar than any other subtypes. The cluster membership for sample j was determined by the “maximum association criterion” as k* = max_k [hkj] (k = 1 through K*). The concordance of the derived expression subtypes was examined in the comparison to those in TCGA marker paper (Cancer Genome Atlas Research Network, 2014a) and other various subtype classifications for 234 TCGA samples (Aine et al., 2015) (Table S2.19).
We selected subtype-specific marker genes in Figure 2 by performing an additional non-negative matrix factorization to the log2(RSEM) gene expression data X with the fixed K* and H* (a column-wise normalization of H) to determine the optimal W (18197 by K*) as X ~ WH*. Note that the element wik in W represents an inferred contribution of the cluster k to the expression of the gene i, i.e. measures an affinity or association of the gene i to the cluster k. The clustering membership of the gene i was determined by the maximum association criterion as k* = max_k [wkj] (k = 1 through K*). We considered the top 1% genes in descending order of wik, with dik ≥ 2.75, where dik refers to the mean expression difference in log2(RSEM) between samples in the cluster k and other samples.
Gene expression signature scores
The raw gene expression signature score in Figure S4 was defined as a mean of log2(RSEM) for the basal markers (CD44, CDH3, KRT1, KRT14, KRT16, KRT5, KRT6A, KRT6B, KRT6C), luminal markers (CYP2J2, ERBB2, ERBB3, FGFR3, FOXA1, GATA3, GPX2, KRT18, KRT19, KRT20, KRT7, KRT8, PPARG, XBP1, UPK1A, UPK2), p53-like markers (ACTG2, CNN1, MYH11, MFAP4, PGM5, FLNC, ACTC1, DES, PCP4), squamous-differentiation markers (DSC1, DSC2, DSC3, DSG1, DSG2, DSG3, S100A7, S100A8), neuroendocrine markers (CHGA, CHGB, SCG2, ENO2, SYP, NCAM1), CIS (carcinoma-in-situ) markers (Dyrskjot et al., 2004), cell-cycle genes (Cuzick et al., 2011), cancer-stem cell markers (CD44, KRT5, RPSA, ALDH1A1I) (Chan et al., 2010), a set of markers known to be associated with EMT (epithelial-mesenchymal; ZEB1, ZEB2, VIM, SNAIL, TWIST1, FOXC2, CDH2), claudin-low markers (CLDN3, CLDN7, CLDN4, CDH1, VIM, SNAI2, TWIST1, ZEB1, ZEB2), and CIT (Cartes d’Identité des Tumeurs) gene sets (Biton et al., 2014). CIT sets included tumour cell component 9; stromal components 3, 8, and 12; and components 5 and 14, which could not be attributed to either tumour or stromal cells. The basal, luminal, p53-like, and claudin-low markers were adapted from (Dadhania et al., 2016), and the squamous, neuroendocrine, and EMT markers were manually chosen based on prior knowledge and literatures. To highlight a differential activity of each signature the raw gene expression signature scores were rank-normalized in Fig. 4c. The CIS signature score was defined as a mean difference of log2(RSEM) between up-regulated and down-regulated genes (Dyrskjot et al., 2004).
miRNAs and RPPAs that were differentially abundant across mRNA subtypes
The 1212 miRNA mature strands were reduced to 303 expressed strands by requiring a mean RPM of at least 10 across n=405 tumor samples. A SAM (samr v2.0) multiclass analysis was run using 1000 permutations, no array centering, a Wilcoxon test statistic, and an FDR output threshold of 0.05. The same settings were used for RPPA normalized abundance data for 195 antibodies and 344 primary tumours.
Detecting somatic gene fusions
A modified version of VirusSeq (Chen et al., 2013) that implements a greedy algorithm with a robust statistical model was used in gene fusion discovery for RNA-Seq data. Specifically, MOSAIK aligner (Lee et al., 2014) was used to align paired-end reads to human genome reference (hg19). A given paired-end read alignment was then quantified in terms of the genomic location (L) of the aligned read pair, the distance (D) between the aligned read pair of the fragment (insert), and the orientation (O) of the read pair. The specific pattern in (L, D, O) space was used as a constraint to define the discordant read pair. For example, a discordant read pair may have an exceptionally long D spanning a region in the reference genome. All discordant reads were then annotated using the genes defined in UCSC refFlat file, and clustered into the ones that support the same fusion event (e.g., FGFR3–TACC3). Finally, each fusion candidate was defined and selected as the discordant read clusters in which a statistical model-based algorithm with greedy strategy was implemented to accurately detect the boundaries of discordant read clusters and in silico fusion junctions. Here, in silico fusion junction is the nucleotide-level genomic coordinate on either side of the gene fusion and is not necessary to be at the ends of known exons. Specifically, the boundary for each discordant read cluster of candidate fusion was estimated on the basis of discordant read mapping locations and orientations with fragment length distribution (e.g., within mean plus three SDs, μ+3*σ) as a constraint of cluster size. The cluster size of discordant reads was measured by using reads’ genomic location excluding introns if mapped reads are located across adjacent exons in a candidate fusion gene. Finally, to help PCR primer design, which facilitates rapid PCR validations, an in silico sequence was generated using the consensus of reads within discordant read clusters for each fusion candidate.
Bladder cancer cell lines
Thirty human bladder cancer cell lines were obtained from the MD Anderson (MDA) Bladder SPORE Tissue Bank. Cell line identities were validated by the MDA Characterized Cell Line Core, using DNA fingerprinting with AmpFlSTR Identifiler Amplification (Applied Biosystems, Foster City, CA). Cell lines were cultured in MEM supplemented with 10% fetal bovine serum, vitamins, sodium pyruvate, L-glutamine, penicillin, streptomycin, and nonessential amino acids at 37°C in 5% CO2 incubator. Total RNA was isolated using a mirVana miRNA isolation kit (ThermoFisher Scientific, Waltham MA). RNA-Seq data was generated with a TruSeq Stranded Total RNA Library Prep Kit, and 76-bp PE reads on an Illumina HiSeq 2500.
Integrative pathway analysis
We evaluated somatic mutations and copy number changes at the gene level, within the context of well-studied signaling pathways. Pathway alteration frequencies were based on the following genes:
TP53/Cell Cycle pathway: ATM, TP53, MDM2, CDKN2A, RB1, CCND1, CDKN1A, PTEN, CCNE1, FBXW7, CDKN1B, CCND1/2/3, and CDK4/6
RTK/RAS/PI3K pathway: PIK3CA, FGFR1/3, ERBB2/3, RAF1, PTEN, TSC1/2, EGFR, AKT1/2, NF1, RAC1, H/N/KRAS, JAK1/2, and BRAF
Histone modification pathway: EP300, CREBBP, KMT2C/D, KDM6A, BAP1, ASXL1/2 and SETD2
SWI/SNF pathway: ARID1A, ARID1B and ARID2
DNA Damage pathway: ERCC2, BRIP1, ATM, BRCA1/2, RAD21/50 and CHEK1
Cohesin complex pathway: STAG1/2, RAD21 and SMC1A/3 (Losada, 2014)
Oxidative stress pathway: NFE2L2, KEAP1, CUL3 and TXNIP
Alternative splicing pathway: RMB10, SF3B1, U2AF1 and CDK12.
Oncogenic relevance was assessed using OncoKB, a knowledgebase for the oncogenic effects of cancer genes that is manually curated by researchers and physicians at Memorial Sloan Kettering (Chakravarty et al., 2017). More precisely, a mutation is counted and included in the diagrams if (1) it has been reported 4 or more times in COSMIC (Forbes et al., 2011), or (2) it has been labeled as oncogenic, or likely oncogenic, in OncoKB.
Amplifications and deep deletions are based on GISTIC calls and indicate somatic alterations in more than half of the baseline gene copies. They are counted and included in the diagrams only if they are labeled as oncogenic, or likely oncogenic, in OncoKB. The actual list of oncogenic and likely oncogenic alterations is regularly updated based on the literature; the most recent version can be retrieved online from the OncoKB public website (www.oncokb.org) or visualized when viewing the data in the cBioPortal (www.cbioportal.org). For known oncogenes, we considered only genetic alterations inferred to be activating; for genes with tumor suppressive roles, only alterations inferred to be inactivating were considered.
Non-coding RNA (lncRNA and miRNA) Sequencing and Analysis
Mapping RNA-seq reads for lncRNAs
RNA sequence reads were aligned to the human reference genome (hg38) and transcriptome (Ensembl v82, September 2015) using STAR 2.4.2a (Dobin et al., 2013). STAR was run with the following parameters: minimum/maximum intron sizes were set to 30 and 500,000, respectively; noncanonical, unannotated junctions were removed; maximum tolerated mismatches was set to 10; and the outSAMstrandField intron motif option was enabled. The Cuffquant command included with Cufflinks 2.2.1 (Trapnell et al., 2013) was used to quantify the read abundances per sample, with fragment bias correction and multiread correction enabled, and all other options set to default. To calculate normalized abundance as fragments per kilobase of exon per million fragments mapped (FPKM), the cuffnorm command was used with default parameters. From the FPKM matrix for the 80 tumor samples, we extracted 8167 genes with “lincRNA” and “processed_transcript” Ensembl biotypes.
miRNA sequencing
We generated miRNA sequencing (miRNA-seq) data from messenger RNA-depleted RNA (Chu et al., 2016). Briefly, we aligned reads to the GRCh37/hg19 reference human genome, assigned read count abundances to miRBase v16 stem-loops and 5p and 3p mature strands, and assigned miRBase v20 mature strand names to MIMAT accession IDs. Note that while we used only reads with exact-match alignments in calculating miRNA abundances, BAM files available from the Genomics Data Commons (https://gdc.cancer.gov/) include all sequence reads.
Unsupervised clustering for lncRNAs, miRNAs
We extracted lncRNAs that were robustly expressed (mean FPKM ≥ 1) and highly variable across the n=80 tumor cohort (≥ 95th FPKM variance percentile) from the matrix of 8167 lncRNAs (above), and identified groups of samples with similar abundance profiles by unsupervised consensus clustering with ConsensusClusterPlus (CCP) 1.20.0 (Wilkerson and Hayes, 2010). Calculations were performed using Spearman correlations, partitioning around medoids (PAM) and 10,000 iterations. From solutions with 2, 3, 4 and 5 clusters we selected a four-cluster solution after assessing consensus membership heatmaps and dendrograms, CCP clustering metrics, Kaplan-Meier (KM) plots, and clustering results from other platforms. To visualize typical vs. atypical cluster members, we calculated a profile of silhouette widths (Wcm) calculated from the consensus membership matrix. To generate an abundance heatmap we identified lncRNAs that had a mean FPKM ≥ 5 and a SAM multiclass (samr 2.0) (Li and Tibshirani, 2013) q ≤ 0.01 across the unsupervised clusters (see differential abundance, below), transformed each row of the matrix by log10(FPKM + 1), then used the pheatmap R package (v1.0.2) to scale and cluster only the rows, using a Pearson distance metric and Ward clustering.
For miRNA mature strand data we used a similar approach. The input was a reads-per-million (RPM) data matrix for the 303 (25% of 1212) most-variant 5p or 3p mature strands, which we transformed by applying log10(RPM+1), then median-centering each miRNA’s record. Using Pearson distances, PAM, and 5000 iterations with a 0.85 random fraction of miRNAs in each iteration, we assessed solutions with between two and eight clusters. After assessing information as for lncRNAs, we focused on a four-cluster solution. As for lncRNAs, to generate a clustering heatmap we first identified miRNAs that were differentially abundant between the unsupervised miRNA clusters using a SAM multiclass analysis (samr 2.0) (Li and Tibshirani, 2013) in R, with a read-count input matrix and a FDR threshold of 0.05. We included miRNAs that had the largest SAM scores and median abundances > 25 RPM. The RPM filtering acknowledged that miRNAs that are more abundant are more likely to be influential (Mullokandov et al., 2012; Thomson and Dinger, 2016). We transformed each row of the matrix by log10(RPM+1), then used the pheatmap R package (v0.7.7 or v1.0.2) to scale and cluster only the rows.
Carcinoma in situ (CIS) signature genes
We used RSEM gene-level data and the pheatmap R package with row scaling to generate heatmaps of normalized expression for 32 ‘up’ and 36 ‘down’ carcinoma in situ (CIS) signature gene sets (Dyrskjot et al., 2004). ‘Up’ genes were (genes are given as in the RSEM file: HGNC symbol | Entrez gene ID): AKR1B10|57016, CALD1|800, CDH11|1009, CLIC4|25932, COL15A1|1306, COL3A1|1281, CXCR4|7852, DCN|1634, DPYSL2|1808, EFEMP1|2202, FLNA|2316, HLA-DQA1|3117, HLA-DQB1|3119, HOXA9|3205, ITM2A|9452, KPNA2|3838, KYNU|8942, LHFP|10186, LUM|4060, LYZ|4069, MAN1C1|57134, MSN|4478, NR3C1|2908, PDGFC|56034, PRG1|23574, RARRES1|5918, S100A8|6279, SGCE|8910, SPARC|6678, TOP2A|7153, TUBB|203068, and UAP1|6675. ‘Down’ genes were: ACSBG1|23205, ANXA10|11199, BBC3|27113, BCAM|4059, BMP7|655, BST2|684, CA12|771, CLCA4|22802, CRTAC1|55118, CTSE|1510, CYP2J2|1573, EEF1A2|1917, ENTPD3|956, FABP4|2167, FGFR3|2261, GRB7|2886, HBG1|3047, HOXA1|3198, HOXB2|3212, INA|9118, ITGB4|3691, IVL|3713, KCNQ1|3784, LAD1|3898, LAMB3|3914, LTBP3|4054, MAPRE3|22924, MST1R|4486, PADI3|51702, PLA2G2A|5320, SOX15|6665, TMPRSS4|56649, TNNI2|7136, TRIM29|23650, UPK2|7379, UPK3B|80761.
To generate compact ‘collapsed’ covariate expression tracks for the ‘up’ and ‘down’ CIS signature gene sets, we calculated Bonferroni-corrected Kruskal-Wallis p values for RSEM gene expression across lncRNA, miRNA and regulon status clusters. For each of the three clustering solutions, for the two gene sets, we used these p values to select a subset of strongly differentially expressed genes. We calculated a profile of the median RSEM expression for each gene subset, across the cluster-ordered cases, and used pheatmap to generate a row-scaled normalized expression track for the median profile. P value-selected gene sets were as follows. For the lncRNA clusters, we used the 21 of 32 ‘up’ genes and 18 of 36 ‘down’ genes that passed a threshold of a Bonferroni-corrected Kruskal p ≤ 1E-15. For the miRNA clusters, we used the 20 ‘up’ genes that passed a Kruskal p ≤ 1E-15 threshold, and the 11 ‘down’ genes that passed Kruskal p ≤ 1E-10.
Covariates associated with unsupervised clusters
We compared unsupervised clusters to clinical and molecular covariates by calculating contingency table association p values using R, with a Chi-square or Fisher exact test for categorical data, and a Kruskal-Wallis test for real-valued data.
Pathology review of adjacent tissue normal samples
After pathology review, four of the 19 adjacent tissue cases were removed from the expression data for lncRNAs and miRNAs: BT-A20U, BT-A2LB, GD-A2C5, and GD-A3OP.
EMT scores from RNA-seq data
The samples were scored based on expression of EMT signature genes (Mak et al., 2016). Briefly, the EMT score for each sample is calculated as the mean expression of epithelial markers subtracted from the mean expression of mesenchymal markers. Higher EMT scores correlate with a more mesenchymal expression pattern.
Regulon analysis
Candidate regulators
We inferred the relative activity of 23 candidate ‘regulator’ genes that had previously been reported as associated with bladder cancer: the steroid hormone receptors ESR1/2, AR and PGR; the nuclear receptors PPARG, three RARs (A/B/G), and three RXRs (A/B/G); the receptor tyrosine kinases ERBB2/3 and FGFR1/3; and the transcription factors FOXA1, FOXM1, GATA3/6, HIF1A, KLF4 and STAT3 and TP63 (Breyer et al., 2016; Choi et al., 2014a; Dadhania et al., 2016; DeGraff et al., 2013; Eriksson et al., 2015; Godoy et al., 2016; Jones et al., 2016; Kardos et al., 2016; Lim et al., 2016). By ‘regulator’ we mean a gene whose product induces and/or represses a target gene set, which we call a ‘regulon’ (Castro et al., 2016a).
Reconstruction of RTN regulons
The BLCA regulator-target associations are inferred using the R package RTN (Castro et al., 2016b), which is extensively described elsewhere for reconstructing regulatory units for transcriptions factors and upstream regulators (Campbell et al., 2016; Castro et al., 2016a; Fletcher et al., 2013). Briefly, gene expression matrices for a set of samples are used to estimate the associations between a regulator and all potential targets. We use two metrics to identify potential regulator-target associations: Mutual Information (MI) and Spearman’s correlation. MI-based inference indicates whether a given regulator is informative of the status of a given target gene, while Spearman’s correlation indicates the direction of the inferred associations. Associations with less than a minimum MI threshold are eliminated by permutation analysis (BH-adjusted p value < 1e-5), and unstable interactions are additionally removed by bootstrapping (n=1000 resamples, consensus bootstrap > 95%), to create a regulatory network. RTN regulons are additionally evaluated by the Data Processing Inequality (DPI) algorithm with tolerance=0.01 (Margolin et al., 2006). Note that MI-based inference computes regulons irrespective of positive or negative associations, and Spearman’s correlation is then used only to assign direction to the predicted regulons. As an optional step, we assessed the stability of the main observations by filtering regulons using the Bioconductor package genefilter v1.56.0 (Gentleman et al., 2016). Feature selection was performed using the coxfilter function on the Sjodahl 2012 cohort (see below) and used to filter genes in the TCGA cohort. Since overfiltering discards both false and true null hypotheses, we also look at the fraction of filtered genes and the total number of observations (i.e. overall results should be stable irrespective of the fractions removed). The list of positively and negatively associated genes in each regulon is provided in Table S2.25.
Regulon activity estimated by two-tailed GSEA
The two-tailed gene set enrichment analysis (GSEA) is described elsewhere (Castro et al., 2016a). Briefly, this approach assesses the skewness of two distributions of a selected gene set in a list of genes that is ranked by a particular phenotype, as follows. The gene set represented for a given regulon is split into positive (A) and negative (B) targets using Spearman’s correlation, and the phenotype corresponds to the gene-wise differential expression observed when comparing a given tumour with the average expression of all tumours in the cohort. The distribution of A and B is then tested by the GSEA statistics in the ranked phenotype, producing independent per-sample enrichment scores (ES), and then differential enrichment scores (dES), which are obtained by subtracting the enrichment score for positive targets (ESA) from that obtained for negative targets (ESB). A large positive dES indicates an induced regulon status while a large negative dES indicates the opposite case. Differential enrichment score values that are near zero (with ESA and ESB distributions skewed to the same side) are assigned as inconclusive. The two-tailed GSEA was performed in R (R-Core-Team, 2012) using the function tni.gsea2 in the RTN package (Castro et al., 2016a).
Regulon activity as readout of clinical and molecular variables
For each case in a cohort we assign an enrichment score (dES) using the two-tailed GSEA approach described above. Then, ordering the cohort’s cases by dES, we can assess how a given regulon is associated with clinical and molecular variables. The cohort cases are also stratified by positive vs. negative dES values, and the stratified cases are used to plot Kaplan-Meier survival curves, with p values calculated using log-rank statistics (Castro et al., 2016a). The survival analysis is performed in R using the functions coxph, survfit and survdiff.
Independent BLCA cohort and transcriptome data
Data used to assess survival statistics in BLCA from an independent cohort are obtained from a large-scale microarray study for n=308 BLCA cases (Sjodahl et al., 2012), which we downloaded from GEO (accession number GSE32894). We use the gene sets in the regulons that we infer from TCGA data to assess the Sjodahl cohort. For each regulon’s gene set, the regulon activity is initially estimated for all tumours of the Sjodahl cohort with the two-tailed GSEA approach, and then the regulon activity is used as readout of clinical and molecular variables, as described above.
Microbial analysis
Microbial screening and genomic integration (British Columbia Cancer Agency)
Microbe analysis consists of two stages: read screening and genomic integration. In the first stage we classify read sequences using a pipeline based on BBT (Release 1.2.10), a fast Bloom filter-based method (Chu et al., 2014). For 48-bp PE RNAseq data, we processed data for 408 tumor samples and 19 tissue normal samples; for 76-bp PE WES data we processed 412 tumor samples and 429 blood or tissue normals; and for 51-bp PE WGS data (high and low pass) we processed 136 tumor samples and 145 blood or tissue normal samples. We ran BBT with a sliding window size (i.e. k-mer length) of 25 bp and a false discovery rate of 0.02. We generated 43 filters from ‘complete’ NCBI genome reference sequences for microbial species that included bacteria, viruses, fungi and protozoa. In a single-pass scan, BBT categorizes each read as matching a filter for human or a single specific microbe, as matching two or more species (multi-match), or as matching none of the filters (no-match). For each filter, we calculated a reads-per-million (RPM) abundance metric (below) and applied a threshold of 0.2 RPM (Cancer Genome Atlas Research Network, 2014b) to identify samples as being positive for specific microbes. For HPV-positive samples, we identified HPV strains with a second BBT run that uses strain-specific filters.
In the second stage of analysis we assessed whether viruses had integrated into the human genome, working only with data sets for which BBT results for HPV, HHV4 or HHV5 were above or close to the 0.2 RPM screening threshold, and with BK-Polyomavirus in DK-A3IT. We performed de novo assembly (Robertson et al., 2010) with ABySS v1.3.4 on each library, using every fourth k-mer value from k=24 to 48 for RNA-seq data, every fourth k-mer from k=52 to 96 for WES data, and k=24, 36 and 48 for WGS data. For HPV analysis we assembled only the reads that BBT had classified as human, HPV match, multi-match, and no-match. We used a similar approach for the HHV4 and HHV5 assemblies. For each library, we merged the contig sets for all k-mer assemblies with Trans-ABySS v1.4.8 to generate a working contig set. We reran BBT on each of these contig sets, applying only human and either HPV or herpes virus filters, identifying the contigs that matched to only a viral filter, or to both the human filter and a viral filter. For HPV-only contigs we confirmed the strain by using BLAT v34 to align each contig to 48 HPV reference sequences. For chimeric multi-match contigs we confirmed the HPV strain, and, for HPV, HHV4 and HHV5, identified integration breakpoints by using BLAT v34 (Kent, 2002) to align each contig to the human GRCh37/hg19 reference sequence, and to 110 HHV4, 22 HHV5, or 48 HPV reference sequences. We retained contig alignments in which the aligned human and viral sequences summed to at least 90% of the contig length, and the human and viral aligned overlapped by less than 50%. We annotated human breakpoint coordinates against RefSeq and UCSC gene annotations (downloaded from the UCSC genome browser on 30-Jun-2013) (Kuhn et al., 2013). Breakpoints that had supporting evidence consisting of at least 3 spanning mate-pair reads or 5 flanking mate-pair reads were considered potential integration sites.
Microbial detection from RNAseq data by PathSeq (The Broad Institute)
PathSeq Microbial Detection
The PathSeq algorithm (Kostic et al., 2011) (https://github.com/ChandraPedamallu/PathSeq) was used to perform computational subtraction of human reads, followed by alignment of residual reads to a combined database of human reference genomes and microbial reference genomes (which includes but is not limited to Human Papillomaviruses (HPV’s), BK Polyomaviruses (BK), Human Herpesviruses (HHV’s)), resulting in the identification of reads mapping to HPV, BK, and HHV genomes in RNA sequencing data.
In brief, for PathSeq human reads were subtracted by first mapping reads to a database of human genomes using BWA (version 0.6.1) (Li and Durbin, 2009), Megablast (version 2.2.23), and Blastn (version 2.2.23) (Altschul et al., 1997). Only sequences with perfect or near perfect matches to the human genome were removed in the subtraction process. To identify HPV/HBK/HHV reads, the resultant non-human reads were aligned with Megablast to a database of microbial genomes that includes multiple HPV, BK and HHV reference genomes. HPV, BK and HHV reference genomes were obtained from the NCBI nucleotide database (downloaded in June 2012).
Subjects were classified as HPV by RNA sequencing if at least 1 HPV read in 1 million human reads were present; otherwise, subjects were classified as HPV-negative. In addition, subjects were classified as BK-positive by RNA sequencing if at least 1 BK reads in 1 million human reads were present; otherwise, subjects were classified as BK-negative. Similar thresholds are used for Human Herpesviruses.
Identification of Human papillomavirus and BK Polyomavirus integration events
An HPV-positive sample was considered integration positive if there were at least 5 spanning read pairs or 10 flanking reads supporting an integration event. In case of HPV-positive, flanking read pairs were defined as having one end of the paired-end read mapped to the HPV genome and its mate pair mapped to the human genome. Spanning reads were defined as having one end of the paired end read spanning the integration junction and its mate pair mapped to either the human or HPV genome. Once HPV reads were obtained, we extracted all pair mates and used Tophat-2.0.84 (Trapnell et al., 2009) with the fusion option enabled to map these paired end reads to a combined database containing the human genome and an HPV genome. Next, spanning reads and flanking reads are identified from the aligned BAM file. Human genes involved in the integration are identified using the breakpoint coordinates against RefSeq and UCSC gene annotations (last modified on 30-Jun-2013) from the UCSC genome browser (Kuhn et al., 2013). A similar approach is followed for identification of BK Polyomavirus integration from RNAseq data.
RPPA protein expression profiling
RPPA experiments and data processing
RPPA lysis buffer was used to extract protein from human tumors and RPPA was performed as described previously (Hennessy et al., 2007; Hu et al., 2007; Liang et al., 2007; Tibes et al., 2006). Frozen tumors were lysed by Precellys homogenization, adjusted to 1 µg/µL concentration as assessed by bicinchoninic acid assay (BCA), boiled with 1% SDS, and manually serial diluted in two-fold of 5 dilutions with lysis buffer. Details on slide preparation, analysis and quantification of spot intensities to generate spot signal intensities (level 1 data), SuperCurve-based QC metric to filter slides with highest QC for each antibody (level 2 data) (Hu et al., 2007), loading control across antibodies for protein measurements (level 3 data) (Gonzalez-Angulo et al., 2011; Hu et al., 2007), and final selection of antibodies for specificity and sensitivity (Hennessy et al., 2010) are given in (Cancer Genome Atlas Research Network, 2014a). In total, 215 antibodies and 343 muscle invasive urothelial/bladder carcinoma (BLCA) samples were used in the analysis, including 109 papillary and 224 non-papillary samples. Forty-two of these 343 samples were squamous cell carcinomas that were mostly non-papillary (40 non-papillary and 2 papillary) samples. RPPA raw data (level 1), SuperCurve nonparameteric model fitting on a single array (level 2), and loading corrected data (level 3) (Ju et al., 2015; Zhang et al., 2009) were deposited at the DCC.
Data normalization
Median centering was performed across all the antibodies for each sample to correct for sample loading differences. These differences could arise because protein concentrations are not uniformly distributed per unit volume of lysate due to several factors such as differences in protein concentrations of large and small cells, differences in the amount of proteins per cell, or heterogeneity of the cells comprising the samples. The expression levels across many different proteins in a sample could be used to estimate differences in the total amount of protein in that sample vs. other samples. Further, subtracting the median protein expression level forces the median value to become zero, allowing for a comparison of protein expressions across samples. These median-centered data were used for the analysis of BLCA samples. Surprisingly, processing similar sets of samples on different slides of the same antibody may result in datasets that have very different means and variances. Neely et al. (Neeley et al., 2009) processed clinically similar ALL samples in two batches and observed differences in their protein data distributions. There were additive and multiplicative effects in the data that could not be accounted by biological or sample loading differences. We observed similar effects when we compared the two batches of bladder tumor protein expression data. A new algorithm, replicates-based normalization (RBN), was therefore developed using replicate samples run across multiple batches to adjust the data for batch effects. The underlying hypothesis is that any observed variation between replicates in different batches is primarily due to linear batch effects plus a component due to random noise. Given a sufficiently large number of replicates, the random noise is expected to cancel out (mean = zero by definition). Remaining differences are treated as systematic batch effects. We can compute those effects for each antibody and subtract them out. Many samples were run in both batches. One batch was arbitrarily designated the “anchor” batch and was to remain unchanged. We then computed the means and standard deviations of the common samples in the anchor batch, as well as the other batch. The difference between the means of each antibody in the two batches and the ratio of the standard deviations provided an estimate of the systematic effects between the batches for that antibody (both location-wise and scale-wise). To cancel out those systematic differences, each data point in the non-anchor batch was adjusted by subtracting the difference in means, then multiplying by the inverse ratio of the standard deviations. Our normalization procedure significantly reduced technical effects, thereby allowing us to merge the datasets from different batches.
RPPA clusters and pathway scores
BLCA samples (n = 343), including 109 papillary and 224 non-papillary samples, were clustered based on 208 antibodies by consensus clustering using the partitioning around medoids algorithm and a Euclidean dissimilarity measure (Wilkerson and Hayes, 2010). The role of cell signaling networks in urothelial carcinomas was illustrated by computing twelve pathway scores similar to those described previously (Akbani et al., 2014).
Kaplan-Meier curves for overall survival
P values are based on the G-rho family of Harrington and Fleming (Xu and Harrington, 2001) tests to evaluate the difference between two or more survival curves.
Carcimoma-in-situ (CIS) gene sets
Carcimoma-in-situ (CIS) gene sets were from (Dyrskjot et al., 2004).
Univariate and Multivariate Survival Analysis
Data preparation and univariate survival analysis
As described in Table S2.28, we recoded values of certain covariates (columns E vs. F), then excluded from the analysis covariates that had many missing values (column C), including CLIN_ajcc_nodes_pathologic_pn and CLIN_ajcc_tumor_pathologic_pt, or that were highly unbalanced between two categories (i.e. one category contained <5% of the cases) (column H). We removed PATH.NOS and PATH_squamous covariates, retaining PATH_Short.Path, because the information in the first two was close to what the latter offered. After excluding all copy number covariates for technical reasons, we had retained 101 covariates. For univariate calculations we used the survdiff function from the R survival v2.40-1 package, and adjusted the log-rank p values with a Benjamini-Hochberg (BH) correction. The univariate analysis identified 18 covariates that were statistically associated with overall survival (BH-adjusted p value < 0.05).
LASSO and Cox regression analysis
As input covariates we used 13 of the 18 that the univariate calculations returned as significant, rejecting five because they had relatively large numbers of missing values:
CLIN_Node_positive_vs_negative (47 cases missing), CLIN_Combined_Tx_Node_positive (64), CLIN_ajcc_nodes_pathologic_pn (47), CLIN_T12_vs_T34 (39), CLIN_ajcc_tumor_pathologic_pt (43). We tested nine types of penalized estimation methods: lasso, adaptive lasso, fused lasso, elastic-net, adaptive elastic-net, SCAD, Snet, MCP, and Mnet assessing the performance of each approach with time-dependent ROC curves (tAUC) (Xiao et al., 2016). When no fitting strategy was significantly better, we choose to use LASSO to fit the final model.
We performed the multivariate Cox regression analysis in R (R Core Development Team, 2016), assuming additive effects. For MSig, mRNA, lncRNA and miRNA subtypes we set, as a reference, a subtype with the best survival (Table S2.29). All LASSO models were fit using the glmnet v2.0-5 and hdnom v4.6 packages (cv.glmnet and hdcox.lasso functions) (Friedman et al., 2010; Xiao et al., 2016). For comparison, we also used a stepwise selection algorithm for model selection (stepAIC function in the R MASS package); stepwise model selection, while widely used, has poor predictive performance compared to modern approaches like LASSO penalized regression (Hutmacher and Kowalski, 2015; Walter and Tiemeier, 2009). We selected the LASSO-penalized Cox model that resulted in minimal prediction error, using Leave-One-Out Cross-Validation (LOOCV), and assessed the stability of the results by bootstrap analysis (n=1000 times).
We determined risk groups from the LASSO model, as follows. The set of regression coefficients is equivalent to a predictive model that is a sum of terms, each of which is a covariate’s coefficient multiplied by the value of that covariate for a case. The cohort is split into training and validation sets in the k-fold cross-validation. The final model is used to estimate a survival probability for each case, at a chosen end-point (e.g. 48 months). The risk groups are then determined from the predicted probabilities; Figure 6C shows low-, medium- and high-risk tertiles. This process, called model calibration, is used to assess how far the model predictions are from actual survival outcomes. For the work reported here, the predictions were made on the samples that were used to build the model; when new cases that have data for the model’s covariates are available, predictions can be made on them using the same approach.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative and statistical methods are noted above according to their respective technologies and analytic approaches.
DATA AND SOFTWARE AVAILABILITY
The data and analysis results are available and can be explored through the Genomic Data Commons (https://gdc.cancer.gov), the Broad Institute GDAC FireBrowse portal (http://gdac.broadinstitute.org), the Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org), and the TCGA publication page (https://tcga-data.nci.nih.gov/docs/publications/). RNA-Seq data for 30 human bladder cancer cell lines are available at the Gene Expression Omnibus (GEO) with the accession GSE97768.
Supplementary Material
Figure S1. Representative micrographs of analyzed tumors. Related to the ‘Demographic, Clinical, and Pathological Data’ section of Methods.
(A–D) Urothelial carcinoma, not otherwise specified and (E–H) urothelial carcinoma with variant histology.
(A) An example of high tumor content with minimal stromal component. The scale bar is 700 µm.
(B) An example of moderate tumor content with stromal and immune cell components. The scale bar is 500 µm.
(C) An example of highly infiltrating tumor with relatively low tumor content, high stromal component and brisk immune cell infiltrate. The scale bar is 200 µm.
(D) Urothelial carcinoma, NOS with evidence of tumor necrosis (*). The scale bar is 300 µm.
(E) Urothelial carcinoma with squamous differentiation with evidence of keratin formation (k). The scale bar is 200 µm.
(F) Another example of urothelial carcinoma with squamous differentiation with evidence intercellular bridges (arrows). The scale bar is 100 µm.
(G) Urothelial carcinoma, micropapillary variant. Characteristically, multiple small clusters of tumor cells are present in clear “lacunar” spaces. The scale bar is 300 µm.
(H) Small cell/neuroendocrine carcinoma of the bladder. Primitive appearing tumor cells with scant cytoplasm and nuclear molding. The scale bar is 200 µm.
Figure S2. Somatic DNA mutational signatures and rearrangements. Related to Figure 1.
(A) Above: Mutational signatures. The spectrum of total SNVs and four mutational signatures in 96 base substitution contexts (mutated pyrimidines and adjacent 5′ and 3′ bases). Note that because the dynamic range for the signatures is large, Y-axis upper limits are different for each signature. Below: signature activities. The number of mutations assigned to each mutational process (Counts) and the normalized contributions across samples. Analysis involved 409 of 412 tumors. We excluded one sample that had a clear POLE signature, and samples that had only 1 and 3 SNVs.
(B,C) Comparison of APOBEC mutation loads inferred using Bayesian NMF, with those obtained using a knowledge-based, experimentally defined Pattern of Mutagenesis by APOBEC Cytidine Deaminases (P-MACD).
(B) Correlation of mutation load assigned to APOBEC mutagenesis by Bayesian NMF (y-axis) and APOBEC mutation load determined by P-MACD (x-axis). P-MACD-defined mutation load is shown with pseudo count of 0.1 to visualize zero values.
(C) Number of mutations with stringent APOBEC signature (tCw to tTw, or tCw to tGw; x-axis) correlates strongly with the number of mutations not conforming to stringent APOBEC signature. DK-A6AW is the outlier ultra-mutated POLE mutant sample.
(D) APOBEC expression vs. levels of APOBEC-signature mutagenesis. Levels of APOBEC-signature mutagenesis were determined for each tumor sample using motif-based analysis (methods: Pattern of Mutagenesis by APOBEC Cytidine Deaminases, P-MACD), and were grouped into No, Low, and High categories. The level of expression of 8 different APOBEC family members are shown as log2(RSEM). mRNA levels of APOBEC3A and APOBEC3B were statistically associated with increased levels of APOBEC-signature mutagenesis. P values are from a Kruskal-Wallis test and were Bonferroni-corrected for multiple testing. APOBEC1 and APOBEC4 are not included because their expression levels were not detectable in most samples.
(E) Unsupervised hierarchical clustering of 409 samples (MSig clusters) based on the number of mutations assigned to four mutational processes.
(F) SNVs, ERCC2 signature mutations, and smoking status, for n = 409 tumor samples (see [A]). Left: Association of overall mutation burden (SNVs) vs. smoking status. Middle: Association of ERCC2 signature mutations vs. smoking status. Right: Association of ERCC2 signature mutations vs. combined status of smoking and ERCC2 mutations. P values are from a Wilcoxon rank-sum test.
(G,H) Mutation/focal Copy Number MutCN clusters based on SMG mutations and focal SCNAs.
(G) Unsupervised Bayesian NMF clustering of mutations in SMGs and focal copy number events (see Methods) identified four DNA-based clusters with differential enrichments of characteristic genetic alterations. Genetic alterations in 53 SMG mutations and 25 focal SCNAs (labels on the left) across 408 samples are indicated by pale blue on a dark blue background..
(H) Normalized strength of association of 78 genetic alterations to the four MutCN clusters.
(I) Protein domain architectures for PPARG fusions. In each of the five schematic fusion diagrams, the left part indicates the 5’ gene, and the right part indicates the location of the fusion breakpoint relative to two PPARG protein domains Top to bottom: MKRN2-PPARG retains both DNA-binding (ZnF C4) and ligand-binding (HOLI) domains; TSEN2-PPARG retains full ZnF_C4 and HOLI domains; TSEN2-PPARG retains partial ZnF_C4 and a full HOLI domain; TSEN2-PPARG retains only a full HOLI domain; TSEN2-PPARG retains only a partial HOLI domain.
Figure S3. DNA methylation subtypes, and genes inactivated by DNA methylation or by multiple mechanisms. Related to Figure 2.
(A,B) Unsupervised clustering using CpG sites that are cancer-specifically hypermethylated (A) or hypomethylated (B). Shown are heatmap representations of DNA methylation profiles of CpG sites with most-variable DNA methylation values (rows) in the 421 tumor samples (columns). Clusters derived by consensus clustering, and selected molecular and clinical features are depicted above each heatmap; heatmaps for DNA methylation profiles of 21 normal-adjacent tissues are to the left and of two leukocyte samples are to the right. For subtypes from other platforms, only those that were statistically associated with the DNA methylation subtypes are shown. CpG sites that are located within gene promoter (PR) or gene body (GB) regions are indicated by the horizontal black bars on the right-hand side of the clustered heatmaps.
(C) Distribution of ABSOLUTE purity across the DNA hypomethylation subtypes.
(D) Concordance between hypermethylation subtypes (rows) and hypomethylation subtypes (columns), shown as a heatmap of odds ratios. A large odds ratio indicates that the sample membership in two clusters is strongly concordant. White vs. red represents low vs. high odds ratios.
(E) Kaplan–Meier analysis comparing patient survival for the five DNA hypomethylation-based clusters.
(F) Twelve genes for which DNA hypomethylation in subtype 4 may contribute to RSEM abundance being higher in that subtype. Relationship of beta to RSEM abundance. Dot colors indicate hypomethylation subtypes, with smaller vs. larger dots reflecting lower/higher ABSOLUTE purity (range 0.09 to 1.0). Squares indicate 14 adjacent tissue normals.
(G) Epigenetically-silenced genes in bladder cancer. Scatter plots showing negative relationships between DNA methylation (x-axis) and gene expression (y-axis) for genes frequently epigenetically silenced in bladder cancer. Each dot represents a tumor sample. Shown are eight genes with biological or clinical interest, as described in the text.
(H) Genes inactivated by multiple mechanisms in bladder cancer. Shown are three significantly mutated genes that are both deleted and epigenetically silenced. For each gene, the scatter plot on the left shows the relationship between DNA methylation (x axis) and gene expression (y axis). The plot on the right displays mutually exclusive occurrence of mutation, deletion, and epigenetic silencing, with the frequency of each alteration shown on the right. Individual tumors are represented by dots that are color-coded by the type of alteration as indicated on the plots.
Figure S4. mRNA expression analysis. Related to Figure 2.
(A) Kaplan-Meier plot for overall survival across the five mRNA expression subtypes, censored at 5 years, with a log-rank p value. LP: luminal-papillary, LI: luminal-infiltrated, L: luminal, BS: basal-squamous, N: neuronal.
(B) Distributions of ABSOLUTE purity estimates for the mRNA subtypes.
(C) Gene expression signature scores for genes sets. For members and literature sources for gene sets, see Methods: mRNA Expression Profiling: Gene expression signature scores. Top: Basal, luminal, wild type p53, squamous-differentiation, and neuroendocrine. Second from top: Carcinoma-in-situ, cell-cycle, cancer-stem cell markers, genes associated with EMT (epithelial-mesenchymal transition), and claudin-low markers. Third from top: CIT (Cartes d’Identité des Tumeurs) gene sets, including tumor cell component 9; stromal components 3, 8 and 12; and components 5 and 14, neither of which could be attributed to tumor or stromal cells. Bottom: Expression levels of selected genes.
(D,E) RPPA antibodies (D) and miRNA mature strands (E) that were differentially abundant across the mRNA subtypes. Barplots show SAM multiclass scores, with higher scores indicating antibodies or miRNAs that were more variable across the subtypes. Text lists give the genes/antibodies and miRNAs with the largest 20 multiclass scores (see Tables S2.17 and S2.18).
Figure S5. Noncoding RNAs and regulons. Related to Figure 4.
(A) miRNAs that were differentially abundant across the messenger RNA subtypes, shown as contrasts from a SAM multiclass analysis, for a subset of miRNAs that were highly ranked as differentially abundant in a SAM multiclass analysis. Subtypes are: P, luminal-papillary. I, luminal-infiltrated. L, luminal. B, basal-squamous. N, neuronal.
(B) lncRNAs that were differentially abundant across the lncRNA subtypes, shown as a heatmap of contrasts from a SAM multiclass analysis for a subset of lncRNAs that were highly ranked as differentially abundant.
(C) miRNAs that were differentially abundant across the miRNA clusters, shown as contrasts from a SAM multiclass analysis, for a subset of miRNAs that were highly ranked as differentially abundant.
(D–G) Regulon analysis for 23 BLCA-associated regulators (see Methods: Regulon analysis: Candidate regulators).
(D) TCGA MIBC cohort (L to R): for the transcription factors GATA3 and GATA6, sorted profiles of differential enrichment score (dES), with a subset of covariates for the score-sorted cohort, and a Kaplan-Meier plot for sample groups that have activated vs. repressed regulon activity or status.
(E) As (D), but for the NMIBC/MIBC Sjodahl cohort (n=308) (Sjodahl et al. 2012), using predicted regulon targets identified in the TCGA cohort.
(F,G) Cox multivariate survival analysis of regulons, with stage, age and gender, assessed in (F) the TCGA cohort and (G) the Sjodahl 2012 cohort, using predicted regulon targets identified in the TCGA cohort. Hazard ratios with 95% confidence intervals.
(H) Statistical significance of variation in regulon activity scores (dES) across mRNA, lncRNA and miRNA subtypes. Barplots show −log10 of Bonferroni-corrected (n=23) Kruskal-Wallis p values.
Figure S6. RPPA proteomics analysis for 343 BLCA samples and 208 antibodies. Related to Figure 2.
(A) Consensus matrix for five unsupervised clusters.
(B) Normalized abundance heatmap. Arrows mark selected proteins that are differentially expressed in certain clusters. Covariate tracks show unsupervised subtypes for mRNA, lncRNA, miRNA and MSig mutational signatures. Collapsed RNA-seq tracks are shown for two carcimoma-in-situ (CIS) gene sets.
(C) Kaplan-Meier curves for overall survival for the five clusters, censored at 5 years.
(D) Distributions of pathway scores, with p values from Kruskal-Wallis tests.
Figure S7. Univariate and multivariate survival analysis. Related to Figure 6.
(A) Kaplan-Meier plots for the 14 covariates that were statistically significant in univariate calculations (BH-corrected log-rank P < 0.05) and were retained for multivariate analysis. The number of curves in each plot corresponds to the number of categories used in univariate calculations (Table S2.27, columns E, Values_Detailed, and F, Values_Recoded). mRNA subtypes 1 to 5 correspond to 1, luminal; 2, luminal-infiltrated; 3, basal-squamous; 4, neuronal; and 5, luminal-papillary. For lncRNA, miRNA, mRNA and Mutational Process, reference variables were the subtype with the best survival.
(B) Time-dependent area-under-the-curve (AUC) curves for nine candidate penalized regression methods, for the full model. For lncRNA, miRNA, mRNA and Mutational Process, the reference variable set was the subtype with the best survival.
(C) Forest plots for multivariate Cox regressions for age, AJCC stage, and mRNA, lncRNA, miRNA and MSig subtypes, with the reference variable for each covariate set to the best-survival subtype. Main effects are shown as hazard ratios with 95% confidence intervals.
Table S1: Master table. Related to STAR methods.
Table S2: Supplemental data. Related to STAR methods.
S2.1: Summary clinical properties of the cohort.
S2.2: Tissue source sites (TSSs)
S2.3: Histology, pathology review
S2.4: Immune infiltrates, pathology review
S2.5: Genomic analysis platforms
S2.6: Amplified genes, GISTIC2 confidence_99
S2.7: Deleted genes, GISTIC2 confidence_99
S2.8: Per-sample copy number gains and losses
S2.9: Significantly mutated genes
S2.10: Mutation exclusivity
S2.11: Mutation co-occurrence
S2.12: Neoantigen load and survival
S2.13: HLA mutations
S2.14: Gene fusions
S2.15: PPARG fusions in 30 human bladder cancer cell lines
S2.16: Hypomethyation subtype 4 genes
S2.17: Epigenetically-silenced genes
S2.18: mRNA subtypes compared to subtypes from other publications (n=234)
S2.19: Subtype comparison reference
S2.20: Per-sample scores for gene expression signatures
S2.21: Proteins that were differentially abundant across the messenger RNA subtypes
S2.22: miRNA mature strands that were differentially abundant across the messenger RNA subtypes
S2.23: SMGs and genome doubling
S2.24: EMT scores calculated from RNA-seq data
S2.25: Genes in 23 regulons
S2.26: RPPA antibodies
S2.27: RPPA subtypes
S2.28: Univariate survival analysis
S2.29: LASSO regression coefficients for multivariate survival analysis
S2.30: Predicted survival
Table S3: Microbe analysis. Related to STAR methods.
S3.1: BBT screening for n=430 (411 tumor samples; 19 normal tissue samples), RPM
S3.2: BBT screening for n=894 (443 tumor samples; 414 blood normal samples; 37 tissue normal samples), RPM
S3.3: BBT screening for n=261 (127 tumor samples; 109 blood normal samples; 25 tissue normal samples), RPM
S3.4: Results of assessing genomic integration with Trans-ABySS in 11 BBT-positive cases
S3.5: Results of assessing genomic integration with Trans-ABySS in 6 BBT-positive cases
S3.6: Results of assessing genomic integration with Trans-ABySS in 6 BBT-positive cases
S3.7: PathSeq RNAseq from 305 primary tumors, RPM
S3.8: PathSeq RNAseq from 305 primary tumors, selected viruses, RPM
S3.9: PathSeq Integration calls for HPV16, 45, 52, 56; HPV6; BK_Polyomavirus
{kind=link}
{kind=link}
Highlights.
Multiplatform analysis informs muscle-invasive bladder cancer subtyping
A framework associating distinct subtyping with therapeutic options
High mutational load is driven mainly by APOBEC-mediated mutagenesis
APOBEC-related mutational signature corresponds to a 75% 5-year survival
Acknowledgments
We are grateful to all of the patients and families who contributed to this study, and for the support of the TCGA Program Office and Steering Committee members Neil Hayes and Paul Spellman for their detailed and thoughtful review of the manuscript. We thank the peer reviewers, whose thoughtful and detailed questions, comments, and requests very substantially improved the manuscript. We appreciate the dedication of Ina Felau for her administrative support throughout this project, and of Lee Ann Chastain for her invaluable organizational skills in final manuscript preparation. This project has been funded in part with federal funds from the U.S. Department of Health and Human Services and through the National Institutes of Health, under various contracts (shown below). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor its member agencies, nor does any mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. US NIH/NCI, TCGA grant U24 CA143866 (A.G.R., K.L.M., S.S., C.C., E.A.G.); US NIH/NCI HHSN 261201000021l, HHSN261201000032I, TCGA Project (B.A.C.); US NIH/NCI HHSN261200800001E (K.E.L., T.M.L.); US NIH/NCI, U24 CA143883 (R.K.); US NIH/NCI, U24 CA143867 (A.D.C., M.M.); US NIH/NCI, U24CA210950, U24CA209851, U01CA168394 (G.B.M., Y.L.); US US NIH/NCI U24 CA199461, CA210949, CA210950; NIH/NCI P50 CA100632; NIH/NCI 4UL1 TR000371 (J.N.W.); NIH/NCI, U24 CA143882 (P.W.L.); US NIH/NCI, P50 CA91846 (B.A.C., D.J.M.; X.S.); NCI CCSG P30 CA016672 (X.S.) for MD Anderson’s Sequencing and Microarray Facility; US NIH/NCI, P30 CA008748 (H.A.); US NIH/NCI, P30 CA016672 (J.Z.); US NIH Intramural Research Program Project, Z1AES103266 (D.A.G.);US NIH/NCI, 1P01CA120964 (D.J.K.); US NIH/NCI R01CA178744 (B.A.C.); US NIH/NCI R01CA155010 (C.J.W.); US NCI PAR-16-025 (S.A.S.); and US DoD Lung Cancer Development (LC150174) (J.N.W.). This work was also partially funded by the Partnership for Bladder Cancer Research, Scott Department of Urology, Baylor College of Medicine (S.P.L); Leukemia and Lymphoma Society Scholar Award (C.J.W.); Mary K. Chapman Foundation (80-107216-19); Michael and Susan Dell Foundation (J.N.W.); National Research Council (CNPq) of Brazil (M.A.A.C.); Pró-Reitoria de Pesquisa/UNICAMP, FAEPE885/16, FAEPE803/16 (B.S.); Federal Agency for Support and Evaluation of Graduate Education (CAPES) of Brazil (V.S.C.). We acknowledge additional TCGA Network funding: U54 HG003273 (R. Gibbs), U54 HG003067 (S. Gabriel), U54 HG003079 (R. Wilson), U24 CA143799 (T. Speed), U24 CA143835 (I. Shmulevich), U24 CA143840 (M. Ladanyi), U24 CA143843 (R Gibbs), U24 CA143845 (G. Getz), U24 CA143848 (D. Hayes), U24 CA143858 (J. Stuart), U24 CA144025 (R. Kucherlapati).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, S.P.L., D.J.K., A.G.R., E.A.G., M.M., D.J.M., J.N.W., H.A., V.E.R., B.A.C.
Data Curation, S.P.L., D.J.K., K.A.H., E.A.G, R.A., J.Z., J.N.W., J.B., H.A., D.E.H., K.M.L., Y.L., T.M.L.
Formal Analysis, D.J.K., D.A.G., L.J.K., G.G., K.A.H., A.G.R., M.A.A.C., B.S.C., V.S.C., S.S., C.C., C.S.P., A.I.O., S.B., R.A., R.S.K., T.H., P.W.L., X.S., J.Z., J.N.W., H.A., F.S., N.S., S.A.S., C.J.W. J.K.
Funding Acquisition, M.M., P.W.L., D.J.M., J.N.W.
Investigation, S.P.L., K.A.H., G.B.M., D.J.M., H.A., D.E.H., B.A.C., K.M.L., T.M.L.
Methodology, S.P.L., E.A.G., M.A.A.C., B.S.C., V.S.C., K.L.M., C.S.P., A.I.O., S.B., R.A., J.B., H.A. J.K.
Project Administration, S.P.L., D.J.K., P.W.L., J.N.W., J.B.
Resources, S.P.L., H.A., V.E.R., B.A.C.
Software, L.J.K., A.G.R., E.A.G, M.A.A.C., B.S., V.S.C., S.S., C.C., C.S.P., S.B., J.N.W.
Supervision, S.P.L., D.J.K., D.A.G., A.D.C., M.A.A.C., B.S.C., K.L.M., M.M., R.A., G.B.M., P.W.L., J.N.W.
Validation, S.P.L., D.J.K., K.L.M., J.N.W., J.B., H.A.
Visualization, D.A.G., A.G.R., E.A.G, M.A.A.C., B.S.C., V.S.C., R.A., R.S.K., T.H., P.W.L., J.N.W., F.S., S.A.S.
Writing original draft, S.P.L., D.J.K., D.A.G., A.D.C., A.G.R., E.A.G, M.A.A.C., K.L.M., C.S.P., R.A., R.S.K., T.H., X.S., J.N.W., H.A., S.A.S. J.K.
Writing review/editing, S.P.L., D.J.K., A.G.R., D.A.G., A.D.C, K.A.H., E.A.G, M.A.A.C, S.B., M.M., R.S.K., G.B.M., P.W.L., J.Z., J.N.W., J.B., H.A., V.E.R.
Potential conflicts of interest
S.P.L. has received investigator initiated research funding from Endo Pharmaceuticals; support for a clinical trial from FKD and Viventia; is a consultant for UroGen, Vaxiion, Nucleix and BioCancell. A.D.C., G.G. and M.M have received research funding from Bayer AG. E.A.G. is now employed by GenomeDx Biosciences. M.M. has no current conflicts; was previously an equity holder in and consultant for Foundation Medicine. G.B.M. is a member of the scientific advisory board and receives research support from AstraZeneca. D.J.M. has stock options in ApoCell, Inc. J.B. has a paid consultancy with Pfizer; has received advisory board and lecture fees from Merck; has received advisory board fees from Genentech; has given uncompensated presentations at Genentech. C.J.W. is a cofounder and advisory board member of Neon Therapeutics. No other conflicts of interest declared.
References
- Aine M, Eriksson P, Liedberg F, Sjodahl G, Hoglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. 2015;5:10957. doi: 10.1038/srep10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ahmadie HA, Iyer G, Lee BH, Scott SN, Mehra R, Bagrodia A, Jordan EJ, Gao SP, Ramirez R, Cha EK, et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat Genet. 2016;48:356–358. doi: 10.1038/ng.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. 2013 doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- Biton A, Bernard-Pierrot I, Lou Y, Krucker C, Chapeaublanc E, Rubio-Perez C, Lopez-Bigas N, Kamoun A, Neuzillet Y, Gestraud P, et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014;9:1235–1245. doi: 10.1016/j.celrep.2014.10.035. [DOI] [PubMed] [Google Scholar]
- Breyer J, Wirtz RM, Laible M, Schlombs K, Erben P, Kriegmair MC, Stoehr R, Eidt S, Denzinger S, Burger M, et al. ESR1, ERBB2, and Ki67 mRNA expression predicts stage and grade of non-muscle-invasive bladder carcinoma (NMIBC) Virchows Arch. 2016;469:547–552. doi: 10.1007/s00428-016-2002-1. [DOI] [PubMed] [Google Scholar]
- Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods Mol Biol. 2009;507:325–337. doi: 10.1007/978-1-59745-522-0_23. [DOI] [PubMed] [Google Scholar]
- Campbell TM, Castro MA, de Santiago I, Fletcher MN, Halim S, Prathalingam R, Ponder BA, Meyer KB. FGFR2 risk SNPs confer breast cancer risk by augmenting oestrogen responsiveness. Carcinogenesis. 2016;37:741–750. doi: 10.1093/carcin/bgw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014a;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014b;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017 doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012a;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012b;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro MA, de Santiago I, Campbell TM, Vaughn C, Hickey TE, Ross E, Tilley WD, Markowetz F, Ponder BA, Meyer KB. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat Genet. 2016a;48:12–21. doi: 10.1038/ng.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro MA, Wang X, Fletcher MN, Meyer KB, Markowetz F. RTN: Reconstruction of transcriptional networks and analysis of master regulators (R/Bioconductor package) 2016b [Google Scholar]
- Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology. 2017;1:1–16. doi: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KS, Volkmer JP, Weissman I. Cancer stem cells in bladder cancer: a revisited and evolving concept. Curr Opin Urol. 2010;20:393–397. doi: 10.1097/MOU.0b013e32833cc9df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yao H, Thompson EJ, Tannir NM, Weinstein JN, Su X. VirusSeq: software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics. 2013;29:266–267. doi: 10.1093/bioinformatics/bts665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Czerniak B, Ochoa A, Su X, Siefker-Radtke A, Dinney C, McConkey DJ. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat Rev Urol. 2014a;11:400–410. doi: 10.1038/nrurol.2014.129. [DOI] [PubMed] [Google Scholar]
- Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014b;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Robertson G, Brooks D, Mungall AJ, Birol I, Coope R, Ma Y, Jones S, Marra MA. Large-scale profiling of microRNAs for The Cancer Genome Atlas. Nucleic Acids Res. 2016;44:e3. doi: 10.1093/nar/gkv808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;12:245–255. doi: 10.1016/S1470-2045(10)70295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadhania V, Zhang M, Zhang L, Bondaruk J, Majewski T, Siefker-Radtke A, Guo CC, Dinney C, Cogdell DE, Zhang S, et al. Meta-analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine. 2016;12:105–117. doi: 10.1016/j.ebiom.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014;111:3110–3115. doi: 10.1073/pnas.1318376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGraff DJ, Cates JM, Mauney JR, Clark PE, Matusik RJ, Adam RM. When urothelial differentiation pathways go wrong: implications for bladder cancer development and progression. Urol Oncol. 2013;31:802–811. doi: 10.1016/j.urolonc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison JB, Shahmoradgoli M, Liu W, Ju Z, Meric-Bernstam F, Perou CM, Sahin AA, Welm A, Oesterreich S, Sikora MJ, et al. High Intratumoral Stromal Content Defines Reactive Breast Cancer as a Low-risk Breast Cancer Subtype. Clin Cancer Res. 2016;22:5068–5078. doi: 10.1158/1078-0432.CCR-16-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Aine M, Veerla S, Liedberg F, Sjodahl G, Hoglund M. Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Med Genomics. 2015;8:25. doi: 10.1186/s12920-015-0101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher MN, Castro MA, Wang X, de Santiago I, O’Reilly M, Chin SF, Rueda OM, Caldas C, Ponder BA, Markowetz F, et al. Master regulators of FGFR2 signalling and breast cancer risk. Nat Commun. 2013;4:2464. doi: 10.1038/ncomms3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- Gentleman R, Carey V, Huber W, Hahne F. genefilter: methods for filtering genes from high-throughput experiments (Bioconductor) 2016 [Google Scholar]
- Getz G, Hofling H, Mesirov JP, Golub TR, Meyerson M, Tibshirani R, Lander ES. Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science. 2007;317:1500. doi: 10.1126/science.1138764. [DOI] [PubMed] [Google Scholar]
- Godoy G, Gakis G, Smith CL, Fahmy O. Effects of androgen and estrogen receptor signaling pathways on bladder cancer initiation and progression. Bladder Cancer. 2016;2:127–137. doi: 10.3233/BLC-160052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Hennessy BT, Meric-Bernstam F, Sahin A, Liu W, Ju Z, Carey MS, Myhre S, Speers C, Deng L, et al. Functional proteomics can define prognosis and predict pathologic complete response in patients with breast cancer. Clinical proteomics. 2011;8:11. doi: 10.1186/1559-0275-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clinical proteomics. 2010;6:129–151. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu YL, Poradosu E, Yu QH, Yu SX, Hall H, Carey MS, Ravoori M, Gonzalez-Angulo AM, Birch R, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. 2007;13:7421–7431. doi: 10.1158/1078-0432.CCR-07-0760. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, He X, Baggerly KA, Coombes KR, Hennessy BTJ, Mills GB. Nonparametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- Hutmacher MM, Kowalski KG. Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol. 2015;79:132–147. doi: 10.1111/bcp.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Felsenstein KM, Theodorescu D. Pharmacogenomics: biomarker-directed therapy for bladder cancer. Urol Clin North Am. 2016;43:77–86. doi: 10.1016/j.ucl.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Liu W, Roebuck PL, Siwak DR, Zhang N, Lu Y, Davies MA, Akbani R, Weinstein JN, Mills GB, et al. Development of a robust classifier for quality control of reverse-phase protein arrays. Bioinformatics. 2015;31:912–918. doi: 10.1093/bioinformatics/btu736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, Iglesia MD, Milowsky MI, Parker JS, Kim WY, et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight. 2016;1:e85902. doi: 10.1172/jci.insight.85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkera JD, Martinez Cardona G, Bell K, Gaffney D, Portale JC, Santiago-Walker A, Moy C, King P, Sharp M, Bahleda R, et al. Oncogenic Characterization and Pharmacologic Sensitivity of Activating Fibroblast Growth Factor Receptor (FGFR) Genetic Alterations to the Selective FGFR Inhibitor Erdafitinib. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-16-0518. [DOI] [PubMed] [Google Scholar]
- Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, Lawrence MS, Kiezun A, Fernandes SM, Bahl S, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun. 2015;6:8866. doi: 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D’Andrea A, Getz G. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016a;48:600–606. doi: 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Tiao G, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D’Andrea AD, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016b;48:600–606. doi: 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WP, Stromberg MP, Ward A, Stewart C, Garrison EP, Marth GT. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One. 2014;9:e90581. doi: 10.1371/journal.pone.0090581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ler LD, Ghosh S, Chai X, Thike AA, Heng HL, Siew EY, Dey S, Koh LK, Lim JQ, Lim WK, et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aai8312. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat Methods Med Res. 2013;22:519–536. doi: 10.1177/0962280211428386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JY, Shao SH, Xu ZX, Hennessy B, Ding ZY, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–U125. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Lim S, Koh MJ, Jeong HJ, Cho NH, Choi YD, Cho do Y, Lee HY, Rha SY. Fibroblast growth factor receptor 1 overexpression is associated with poor survival in patients with resected muscle invasive urothelial carcinoma. Yonsei Med J. 2016;57:831–839. doi: 10.3349/ymj.2016.57.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer. 2014;14:389–393. doi: 10.1038/nrc3743. [DOI] [PubMed] [Google Scholar]
- Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, Rodriguez-Canales J, Wistuba II, et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin Cancer Res. 2016;22:609–620. doi: 10.1158/1078-0432.CCR-15-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006;1:662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeley ES, Kornblau SM, Coombes KR, Baggerly KA. Variable slope normalization of reverse phase protein arrays. Bioinformatics. 2009;25:1384–1389. doi: 10.1093/bioinformatics/btp174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q, Carninci P. Expression Specificity of Disease-Associated lncRNAs: Toward Personalized Medicine. Curr Top Microbiol Immunol. 2016;394:237–258. doi: 10.1007/82_2015_464. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, Schellens JH, Cassier PA, Camidge DR, Schuler M, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol. 2017;35:157–165. doi: 10.1200/JCO.2016.67.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Development Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing); 2016. [Google Scholar]
- Ratan A, Olson TL, Loughran TP, Jr, Miller W. Identification of indels in next-generation sequencing data. BMC Bioinformatics. 2015;16:42. doi: 10.1186/s12859-015-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouissou S, Herault A, Letouze E, Neuzillet Y, Laplanche A, Ofualuka K, Maille P, Leroy K, Riou A, Lepage ML, et al. CDKN2A homozygous deletion is associated with muscle invasion in FGFR3-mutated urothelial bladder carcinoma. J Pathol. 2012;227:315–324. doi: 10.1002/path.4017. [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxford, England) 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Seiler R, Ashab HA, Erho N, van Rhijn BW, Winters B, Douglas J, Van Kessel KE, Fransen van de Putte EE, Sommerlad M, Wang NQ, et al. Impact of Molecular Subtypes in Muscle-invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur Urol. 2017 doi: 10.1016/j.eururo.2017.03.030. [DOI] [PubMed] [Google Scholar]
- Sharma P, Callahan MK, Bono P, Kim J, Spiliopoulou P, Calvo E, Pillai RN, Ott PA, de Braud F, Morse M, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016;17:1590–1598. doi: 10.1016/S1470-2045(16)30496-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nature biotechnology. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Sjodahl G, Eriksson P, Liedberg F, Hoglund M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. 2017;242:113–125. doi: 10.1002/path.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Ferno M, Ringner M, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18:3377–3386. doi: 10.1158/1078-0432.CCR-12-0077-T. [DOI] [PubMed] [Google Scholar]
- Tan VY, Fevotte C. Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans Pattern Anal Mach Intell. 2013;35:1592–1605. doi: 10.1109/TPAMI.2012.240. [DOI] [PubMed] [Google Scholar]
- Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- Tibes R, Qiu YH, Hennessy B, Andreeff M, Miiis GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triche TJ, Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O’Connor KW, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4:1140–1153. doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res. 2001;61:1265–1268. [PubMed] [Google Scholar]
- Walter S, Tiemeier H. Variable selection: current practice in epidemiological studies. Eur J Epidemiol. 2009;24:733–736. doi: 10.1007/s10654-009-9411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JI, Walter V, Yamashita H, Chung E, Shuman L, Amponsa VO, Zheng Z, Chan W, Whitcomb TL, Yue F, et al. FOXA1, GATA3 and PPAR cooperate to drive luminal subtype in bladder cancer: a molecular analysis of established human cell lines. Sci Rep. 2016;6:38531. doi: 10.1038/srep38531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP, Elder PA, Shaw ME, Devlin J, Knowles MA. p16 (CDKN2) is a major deletion target at 9p21 in bladder cancer. Hum Mol Genet. 1995;4:1569–1577. doi: 10.1093/hmg/4.9.1569. [DOI] [PubMed] [Google Scholar]
- Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, Kawashima K, Laird PW, Jones PA, Liang G. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–8178. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang M, Wang L, Luo G, Li Y, Zeng F, et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol Cancer Ther. 2016;15:1029–1042. doi: 10.1158/1535-7163.MCT-15-0750. [DOI] [PubMed] [Google Scholar]
- Xiao N, Xu QS, Li MZ. hdnom: building nomograms for penalized Cox models with high-dimensional survival data. bioRxiv. 2016;065524 [Google Scholar]
- Xu R, Harrington DP. A semiparametric estimate of treatment effects with censored data. Biometrics. 2001;57:875–885. doi: 10.1111/j.0006-341x.2001.00875.x. [DOI] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wei Q, Mao L, Liu W, Mills GB, Coombes K. Serial dilution curve: a new method for analysis of reverse phase protein array data. Bioinformatics. 2009;25:650–654. doi: 10.1093/bioinformatics/btn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative micrographs of analyzed tumors. Related to the ‘Demographic, Clinical, and Pathological Data’ section of Methods.
(A–D) Urothelial carcinoma, not otherwise specified and (E–H) urothelial carcinoma with variant histology.
(A) An example of high tumor content with minimal stromal component. The scale bar is 700 µm.
(B) An example of moderate tumor content with stromal and immune cell components. The scale bar is 500 µm.
(C) An example of highly infiltrating tumor with relatively low tumor content, high stromal component and brisk immune cell infiltrate. The scale bar is 200 µm.
(D) Urothelial carcinoma, NOS with evidence of tumor necrosis (*). The scale bar is 300 µm.
(E) Urothelial carcinoma with squamous differentiation with evidence of keratin formation (k). The scale bar is 200 µm.
(F) Another example of urothelial carcinoma with squamous differentiation with evidence intercellular bridges (arrows). The scale bar is 100 µm.
(G) Urothelial carcinoma, micropapillary variant. Characteristically, multiple small clusters of tumor cells are present in clear “lacunar” spaces. The scale bar is 300 µm.
(H) Small cell/neuroendocrine carcinoma of the bladder. Primitive appearing tumor cells with scant cytoplasm and nuclear molding. The scale bar is 200 µm.
Figure S2. Somatic DNA mutational signatures and rearrangements. Related to Figure 1.
(A) Above: Mutational signatures. The spectrum of total SNVs and four mutational signatures in 96 base substitution contexts (mutated pyrimidines and adjacent 5′ and 3′ bases). Note that because the dynamic range for the signatures is large, Y-axis upper limits are different for each signature. Below: signature activities. The number of mutations assigned to each mutational process (Counts) and the normalized contributions across samples. Analysis involved 409 of 412 tumors. We excluded one sample that had a clear POLE signature, and samples that had only 1 and 3 SNVs.
(B,C) Comparison of APOBEC mutation loads inferred using Bayesian NMF, with those obtained using a knowledge-based, experimentally defined Pattern of Mutagenesis by APOBEC Cytidine Deaminases (P-MACD).
(B) Correlation of mutation load assigned to APOBEC mutagenesis by Bayesian NMF (y-axis) and APOBEC mutation load determined by P-MACD (x-axis). P-MACD-defined mutation load is shown with pseudo count of 0.1 to visualize zero values.
(C) Number of mutations with stringent APOBEC signature (tCw to tTw, or tCw to tGw; x-axis) correlates strongly with the number of mutations not conforming to stringent APOBEC signature. DK-A6AW is the outlier ultra-mutated POLE mutant sample.
(D) APOBEC expression vs. levels of APOBEC-signature mutagenesis. Levels of APOBEC-signature mutagenesis were determined for each tumor sample using motif-based analysis (methods: Pattern of Mutagenesis by APOBEC Cytidine Deaminases, P-MACD), and were grouped into No, Low, and High categories. The level of expression of 8 different APOBEC family members are shown as log2(RSEM). mRNA levels of APOBEC3A and APOBEC3B were statistically associated with increased levels of APOBEC-signature mutagenesis. P values are from a Kruskal-Wallis test and were Bonferroni-corrected for multiple testing. APOBEC1 and APOBEC4 are not included because their expression levels were not detectable in most samples.
(E) Unsupervised hierarchical clustering of 409 samples (MSig clusters) based on the number of mutations assigned to four mutational processes.
(F) SNVs, ERCC2 signature mutations, and smoking status, for n = 409 tumor samples (see [A]). Left: Association of overall mutation burden (SNVs) vs. smoking status. Middle: Association of ERCC2 signature mutations vs. smoking status. Right: Association of ERCC2 signature mutations vs. combined status of smoking and ERCC2 mutations. P values are from a Wilcoxon rank-sum test.
(G,H) Mutation/focal Copy Number MutCN clusters based on SMG mutations and focal SCNAs.
(G) Unsupervised Bayesian NMF clustering of mutations in SMGs and focal copy number events (see Methods) identified four DNA-based clusters with differential enrichments of characteristic genetic alterations. Genetic alterations in 53 SMG mutations and 25 focal SCNAs (labels on the left) across 408 samples are indicated by pale blue on a dark blue background..
(H) Normalized strength of association of 78 genetic alterations to the four MutCN clusters.
(I) Protein domain architectures for PPARG fusions. In each of the five schematic fusion diagrams, the left part indicates the 5’ gene, and the right part indicates the location of the fusion breakpoint relative to two PPARG protein domains Top to bottom: MKRN2-PPARG retains both DNA-binding (ZnF C4) and ligand-binding (HOLI) domains; TSEN2-PPARG retains full ZnF_C4 and HOLI domains; TSEN2-PPARG retains partial ZnF_C4 and a full HOLI domain; TSEN2-PPARG retains only a full HOLI domain; TSEN2-PPARG retains only a partial HOLI domain.
Figure S3. DNA methylation subtypes, and genes inactivated by DNA methylation or by multiple mechanisms. Related to Figure 2.
(A,B) Unsupervised clustering using CpG sites that are cancer-specifically hypermethylated (A) or hypomethylated (B). Shown are heatmap representations of DNA methylation profiles of CpG sites with most-variable DNA methylation values (rows) in the 421 tumor samples (columns). Clusters derived by consensus clustering, and selected molecular and clinical features are depicted above each heatmap; heatmaps for DNA methylation profiles of 21 normal-adjacent tissues are to the left and of two leukocyte samples are to the right. For subtypes from other platforms, only those that were statistically associated with the DNA methylation subtypes are shown. CpG sites that are located within gene promoter (PR) or gene body (GB) regions are indicated by the horizontal black bars on the right-hand side of the clustered heatmaps.
(C) Distribution of ABSOLUTE purity across the DNA hypomethylation subtypes.
(D) Concordance between hypermethylation subtypes (rows) and hypomethylation subtypes (columns), shown as a heatmap of odds ratios. A large odds ratio indicates that the sample membership in two clusters is strongly concordant. White vs. red represents low vs. high odds ratios.
(E) Kaplan–Meier analysis comparing patient survival for the five DNA hypomethylation-based clusters.
(F) Twelve genes for which DNA hypomethylation in subtype 4 may contribute to RSEM abundance being higher in that subtype. Relationship of beta to RSEM abundance. Dot colors indicate hypomethylation subtypes, with smaller vs. larger dots reflecting lower/higher ABSOLUTE purity (range 0.09 to 1.0). Squares indicate 14 adjacent tissue normals.
(G) Epigenetically-silenced genes in bladder cancer. Scatter plots showing negative relationships between DNA methylation (x-axis) and gene expression (y-axis) for genes frequently epigenetically silenced in bladder cancer. Each dot represents a tumor sample. Shown are eight genes with biological or clinical interest, as described in the text.
(H) Genes inactivated by multiple mechanisms in bladder cancer. Shown are three significantly mutated genes that are both deleted and epigenetically silenced. For each gene, the scatter plot on the left shows the relationship between DNA methylation (x axis) and gene expression (y axis). The plot on the right displays mutually exclusive occurrence of mutation, deletion, and epigenetic silencing, with the frequency of each alteration shown on the right. Individual tumors are represented by dots that are color-coded by the type of alteration as indicated on the plots.
Figure S4. mRNA expression analysis. Related to Figure 2.
(A) Kaplan-Meier plot for overall survival across the five mRNA expression subtypes, censored at 5 years, with a log-rank p value. LP: luminal-papillary, LI: luminal-infiltrated, L: luminal, BS: basal-squamous, N: neuronal.
(B) Distributions of ABSOLUTE purity estimates for the mRNA subtypes.
(C) Gene expression signature scores for genes sets. For members and literature sources for gene sets, see Methods: mRNA Expression Profiling: Gene expression signature scores. Top: Basal, luminal, wild type p53, squamous-differentiation, and neuroendocrine. Second from top: Carcinoma-in-situ, cell-cycle, cancer-stem cell markers, genes associated with EMT (epithelial-mesenchymal transition), and claudin-low markers. Third from top: CIT (Cartes d’Identité des Tumeurs) gene sets, including tumor cell component 9; stromal components 3, 8 and 12; and components 5 and 14, neither of which could be attributed to tumor or stromal cells. Bottom: Expression levels of selected genes.
(D,E) RPPA antibodies (D) and miRNA mature strands (E) that were differentially abundant across the mRNA subtypes. Barplots show SAM multiclass scores, with higher scores indicating antibodies or miRNAs that were more variable across the subtypes. Text lists give the genes/antibodies and miRNAs with the largest 20 multiclass scores (see Tables S2.17 and S2.18).
Figure S5. Noncoding RNAs and regulons. Related to Figure 4.
(A) miRNAs that were differentially abundant across the messenger RNA subtypes, shown as contrasts from a SAM multiclass analysis, for a subset of miRNAs that were highly ranked as differentially abundant in a SAM multiclass analysis. Subtypes are: P, luminal-papillary. I, luminal-infiltrated. L, luminal. B, basal-squamous. N, neuronal.
(B) lncRNAs that were differentially abundant across the lncRNA subtypes, shown as a heatmap of contrasts from a SAM multiclass analysis for a subset of lncRNAs that were highly ranked as differentially abundant.
(C) miRNAs that were differentially abundant across the miRNA clusters, shown as contrasts from a SAM multiclass analysis, for a subset of miRNAs that were highly ranked as differentially abundant.
(D–G) Regulon analysis for 23 BLCA-associated regulators (see Methods: Regulon analysis: Candidate regulators).
(D) TCGA MIBC cohort (L to R): for the transcription factors GATA3 and GATA6, sorted profiles of differential enrichment score (dES), with a subset of covariates for the score-sorted cohort, and a Kaplan-Meier plot for sample groups that have activated vs. repressed regulon activity or status.
(E) As (D), but for the NMIBC/MIBC Sjodahl cohort (n=308) (Sjodahl et al. 2012), using predicted regulon targets identified in the TCGA cohort.
(F,G) Cox multivariate survival analysis of regulons, with stage, age and gender, assessed in (F) the TCGA cohort and (G) the Sjodahl 2012 cohort, using predicted regulon targets identified in the TCGA cohort. Hazard ratios with 95% confidence intervals.
(H) Statistical significance of variation in regulon activity scores (dES) across mRNA, lncRNA and miRNA subtypes. Barplots show −log10 of Bonferroni-corrected (n=23) Kruskal-Wallis p values.
Figure S6. RPPA proteomics analysis for 343 BLCA samples and 208 antibodies. Related to Figure 2.
(A) Consensus matrix for five unsupervised clusters.
(B) Normalized abundance heatmap. Arrows mark selected proteins that are differentially expressed in certain clusters. Covariate tracks show unsupervised subtypes for mRNA, lncRNA, miRNA and MSig mutational signatures. Collapsed RNA-seq tracks are shown for two carcimoma-in-situ (CIS) gene sets.
(C) Kaplan-Meier curves for overall survival for the five clusters, censored at 5 years.
(D) Distributions of pathway scores, with p values from Kruskal-Wallis tests.
Figure S7. Univariate and multivariate survival analysis. Related to Figure 6.
(A) Kaplan-Meier plots for the 14 covariates that were statistically significant in univariate calculations (BH-corrected log-rank P < 0.05) and were retained for multivariate analysis. The number of curves in each plot corresponds to the number of categories used in univariate calculations (Table S2.27, columns E, Values_Detailed, and F, Values_Recoded). mRNA subtypes 1 to 5 correspond to 1, luminal; 2, luminal-infiltrated; 3, basal-squamous; 4, neuronal; and 5, luminal-papillary. For lncRNA, miRNA, mRNA and Mutational Process, reference variables were the subtype with the best survival.
(B) Time-dependent area-under-the-curve (AUC) curves for nine candidate penalized regression methods, for the full model. For lncRNA, miRNA, mRNA and Mutational Process, the reference variable set was the subtype with the best survival.
(C) Forest plots for multivariate Cox regressions for age, AJCC stage, and mRNA, lncRNA, miRNA and MSig subtypes, with the reference variable for each covariate set to the best-survival subtype. Main effects are shown as hazard ratios with 95% confidence intervals.
Table S1: Master table. Related to STAR methods.
Table S2: Supplemental data. Related to STAR methods.
S2.1: Summary clinical properties of the cohort.
S2.2: Tissue source sites (TSSs)
S2.3: Histology, pathology review
S2.4: Immune infiltrates, pathology review
S2.5: Genomic analysis platforms
S2.6: Amplified genes, GISTIC2 confidence_99
S2.7: Deleted genes, GISTIC2 confidence_99
S2.8: Per-sample copy number gains and losses
S2.9: Significantly mutated genes
S2.10: Mutation exclusivity
S2.11: Mutation co-occurrence
S2.12: Neoantigen load and survival
S2.13: HLA mutations
S2.14: Gene fusions
S2.15: PPARG fusions in 30 human bladder cancer cell lines
S2.16: Hypomethyation subtype 4 genes
S2.17: Epigenetically-silenced genes
S2.18: mRNA subtypes compared to subtypes from other publications (n=234)
S2.19: Subtype comparison reference
S2.20: Per-sample scores for gene expression signatures
S2.21: Proteins that were differentially abundant across the messenger RNA subtypes
S2.22: miRNA mature strands that were differentially abundant across the messenger RNA subtypes
S2.23: SMGs and genome doubling
S2.24: EMT scores calculated from RNA-seq data
S2.25: Genes in 23 regulons
S2.26: RPPA antibodies
S2.27: RPPA subtypes
S2.28: Univariate survival analysis
S2.29: LASSO regression coefficients for multivariate survival analysis
S2.30: Predicted survival
Table S3: Microbe analysis. Related to STAR methods.
S3.1: BBT screening for n=430 (411 tumor samples; 19 normal tissue samples), RPM
S3.2: BBT screening for n=894 (443 tumor samples; 414 blood normal samples; 37 tissue normal samples), RPM
S3.3: BBT screening for n=261 (127 tumor samples; 109 blood normal samples; 25 tissue normal samples), RPM
S3.4: Results of assessing genomic integration with Trans-ABySS in 11 BBT-positive cases
S3.5: Results of assessing genomic integration with Trans-ABySS in 6 BBT-positive cases
S3.6: Results of assessing genomic integration with Trans-ABySS in 6 BBT-positive cases
S3.7: PathSeq RNAseq from 305 primary tumors, RPM
S3.8: PathSeq RNAseq from 305 primary tumors, selected viruses, RPM
S3.9: PathSeq Integration calls for HPV16, 45, 52, 56; HPV6; BK_Polyomavirus