

In this study, Kondo et al. performed large-scale CRISPR/Cas9 mutagenesis of “conserved, essential” and “young, RNAi-lethal” genes and confirmed the lethality of conserved genes but not young genes. Additionally, two young gene mutants resulted in spermatogenesis and/or male sterility, indicating that young genes have a preferential impact on male reproductive system function.
Keywords: Drosophila, gene evolution, spermatogenesis, testis
Abstract
Relatively little is known about the in vivo functions of newly emerging genes, especially in metazoans. Although prior RNAi studies reported prevalent lethality among young gene knockdowns, our phylogenomic analyses reveal that young Drosophila genes are frequently restricted to the nonessential male reproductive system. We performed large-scale CRISPR/Cas9 mutagenesis of “conserved, essential” and “young, RNAi-lethal” genes and broadly confirmed the lethality of the former but the viability of the latter. Nevertheless, certain young gene mutants exhibit defective spermatogenesis and/or male sterility. Moreover, we detected widespread signatures of positive selection on young male-biased genes. Thus, young genes have a preferential impact on male reproductive system function.
While it is a tantalizing prospect that newly emerging genes might drive species-specific biology, few functional studies of young genes have been conducted within intact metazoan organisms. We can infer that deeply conserved genes have obligatory activities that underlie their strong signatures of purifying selection. On the other hand, the biological impact of recently emerged genes acting as either RNAs or proteins is less certain. A conservative hypothesis is that new genes are neutral at birth such that, were we able to peer into future generations, most of them would end up being inactivated by random mutational processes. Alternatively, new genes may comprise an evolutionary playground, potentially yielding substantial species-specific functions (Kaessmann 2010). Support for this scenario comes from evidence of positive selection acting on certain young genes, including some expressed in the CNS or gonads (Haerty et al. 2007; Zhang et al. 2010, 2011). However, despite much computational analysis on the catalogs and sequence properties of young genes, there is limited data from in vivo genetic assays, especially within metazoan organisms. Still, individual cases of recently evolved genes with dramatic activities are known. For example, Umbrea/HP6 encodes a young centromeric HP1 homolog that is evolving under positive selection and has important roles in centromere function in Drosophila melanogaster (Ross et al. 2013).
In D. melanogaster, transgenic RNAi experiments were used to estimate that ∼30% of phylogenetically young genes were required for viability (Chen et al. 2010). This was striking given that 25%–30% of all Drosophila genes were estimated to be essential (Miklos and Rubin 1996). The functional impact of young genes might be broader, as some knockdowns yielded viable adults with morphological or behavioral defects (Chen et al. 2010, 2012). These data were taken to support that young genes frequently drive phenotypic diversity (Chen et al. 2013). However, most of these lethal phenotypes were not reproduced with independent RNAi triggers. Instead, we used phylogenomic analysis of spatial gene expression, evolutionary selection analysis, and large-scale CRISPR/Cas9 mutagenesis to demonstrate that newly emerging genes have a selective functional impact on the male reproductive system.
Results and Discussion
The reported high frequency of young, essential genes in Drosophila from RNAi-based assays (Chen et al. 2010) prompted us to investigate whether they had characteristic spatiotemporal expression patterns. Analysis of broad modENCODE profiling data revealed that a majority of young Drosophila genes was strongly biased or restricted to testis and/or the male accessory gland (e.g., Fig. 1A), structures that are dispensable for viability. Moreover, this tissue bias was similar among young loci regardless of their previous RNAi-based classification as essential or viable (Supplemental Figs. 1, 2). Overall, these patterns were consistent with the notion that the male reproductive system is permissive for gene birth, as noted earlier in D. melanogaster (Brown et al. 2014; Mohammed et al. 2014; Zhao et al. 2014) and vertebrates (Kaessmann 2010).
Figure 1.

Young Drosophila genes are strongly biased for expression in the male reproductive system. (A) Example of a young Drosophila gene previously assessed as lethal by RNAi assays (Chen et al. 2010) that exhibits highly restricted expression in the testis, with minor amounts detected in other libraries containing the male reproductive system (e.g., dissected accessory gland, whole male bodies, and unsexed larval/pupal material that contains gonads). (B) We revised the catalog of recently evolved genes in D. melanogaster, doubling their numbers from previous studies. (C) We divided D. melanogaster genes according to evolutionary age and calculated their tissue specificity index (τ) across 93 developmental, tissue, and cell line RNA sequencing (RNA-seq) conditions (195 data sets). As gene age decreases, they tend to become more spatiotemporally restricted. Global median τ (0.61) is indicated for reference; note that the τ distribution is actually highly bimodal (Supplemental Fig. 5). (D) The tissue/stage/cell library of maximal expression (Xmax) was plotted for genes with spatially restricted expression (τ > 0.61) in each evolutionary category. Higher τ of young genes is associated with a prominent trend for expression in the male reproductive system (testis and/or accessory gland). This male restriction is substantially elevated among genes born in the pan-Drosophilid ancestor and increases in cohorts of younger genes born along the lineage toward D. melanogaster.
We studied this further by systematically assessing the tissue specificity of D. melanogaster genes according to their evolutionary ages (Supplemental Table 1). As closely related parental sequences can confound computational inferences of gene age, we employed extensive manual vetting by examining chains and nets from whole-genome alignments for evidence of ortholog synteny (Supplemental Fig. 3). After re-examining previous assignments of gene ages (Chen et al. 2010) and assessing updated modENCODE gene models (Brown et al. 2014), we more than doubled the confident set of evolutionarily young Drosophila genes (Fig. 1B; Supplemental Fig. 4). We then calculated a tissue specificity index (τ) for each gene based on modENCODE and public RNA sequencing (RNA-seq) data from 93 developmental stages, dissected tissues, and cell lines (Supplemental Table 1; Brown et al. 2014; Zhao et al. 2014). These analyses showed that the fraction of tissue-specific genes rose substantially in the Dipteran ancestor and that progressively younger genes were even more likely to have restricted expression (Fig. 1C).
The increased tissue restriction of younger genes might be due to their biased expression in many different individual settings or to a major common setting. To examine this, we compared cohorts of genes of varying ages with comparable tissue specificity. Global analysis showed that Drosophila genes exhibit bimodal τ distribution, reflecting broad and restricted patterns, with a median of 0.614 (Supplemental Fig. 5). Interestingly, genes with a higher τ (>0.614) exhibited distinct tissue bias based on their evolutionary age. In particular, we observed a strong expansion of evolutionarily young tissue-restricted genes whose maximal expression (“Xmax”) is in testis or the accessory gland (Fig. 1D; Supplemental Fig. 6). The expression patterns of individual young genes grouped by evolutionary ages are summarized in Supplemental Figures 7–10.
Some young genes with a high τ were dominantly expressed in male body but not female body or other dissected adult body parts (e.g., Fig. 1D). In addition, many young genes that were restricted to the male reproductive system at adult stages were detected in larvae, pupae, and/or mass-isolated imaginal discs (Supplemental Figs. 6–10). We previously documented that the mass-isolated disc library contains larval gonads and happens to be rich in testis mRNAs and small RNAs (Brown et al. 2014; Wen et al. 2015). Thus, these trends likely reflect developmental expression of male-specific genes in unsexed larvae and pupae. Overall, these data broadly extend previous findings that young Drosophila genes are preferentially expressed in the male reproductive system (Zhang et al. 2010; Assis and Bachtrog 2013).
We next applied the concept of the transcriptome age index (TAI), where gene expression data are combined with evolutionary information to convey the relative age of the transcriptome (Domazet-Loso and Tautz 2010). The TAI of a given tissue represents a weighted expression metric that gives stronger weight to young phylostrata, compensating for the fact that older phylostrata usually harbor a larger number of genes. A high TAI indicates that the transcriptome is evolutionarily young, while a low TAI indicates that the transcriptome is evolutionarily ancient. The testis, accessory gland, and adult male libraries are clear outliers exhibiting the highest TAI, with the imaginal disc/gonad, pupal, and larval libraries comprising a group with the next highest TAI (Supplemental Fig. 11). The lowest TAI class is represented by all cell lines, ovaries, and early embryos (Supplemental Fig. 11). These analyses reinforce the notion that the transcriptome of the male reproductive system is uniquely evolutionarily young among all Drosophila tissues.
These phylogenomic expression studies led us to re-examine the 59 proposed young, essential Drosophila genes, based on RNAi assays (Chen et al. 2010). While a powerful technique, RNAi frequently induces unintended effects (Mohr and Perrimon 2012). This is of concern given that many young genes are recent duplicates and thus may share homology with parental loci. Although 45 of 47 RNAi-induced lethal phenotypes were reported as reproducible (Chen et al. 2010), these tests mostly involved different genomic insertions of the same trigger sequence, which should recapitulate both on-target and off-target effects. Additional collections of RNAi transgenes have been generated subsequently, providing opportunities to assess reproducibility (Perkins et al. 2015).
We compared RNAi data from different collections using the ubiquitous driver actin5C-Gal4 (Zeng et al. 2015). Among the 59 “young, essential” genes, 23 lacked independent trigger transgenes, although five of these yielded viability in a different actin5C-Gal4 test. Of the remaining genes, the vast majority (31 of 36) was viable with ubiquitous expression of an independent RNAi transgene (Supplemental Fig. 12). The concordance of RNAi phenotypes among “young, nonessential” genes was higher, but still, 28 genes induced lethality with a different trigger (Supplemental Fig. 12). Thus, lethality assessment was highly influenced by the RNAi transgene assayed, suggesting off targeting.
Given the ambiguity of RNAi data, we turned to CRISPR/Cas9 as an alternative strategy to disrupt gene function. To test whether CRISPR/Cas9 can reliably yield strong loss-of-function mutants, we randomly chose 21 known essential genes on chromosome 2L. We recovered frameshift alleles for each that truncate the majority of their coding potential (Supplemental Table 3). With the exception of one gene (two of two viable alleles of BACE), the 33 alleles of the other 20 genes all recapitulated the expected lethality (Table 1). Further tests showed that CRISPR BACE mutants lacked protein and exhibited age-dependent glial degeneration (Supplemental Fig. 13), recapitulating known BACE mutants. Thus, BACE is in fact likely a viable mutant (D Kretzschmar, pers. comm.). Overall, this new collection of mutants strongly validates our procedure for evaluating essential genes using gene editing in Drosophila and highlights this platform for forward genetic screening.
Table 1.
Summary of 45 D. melanogaster genes subjected to CRISPR/Cas9 mutagenesis
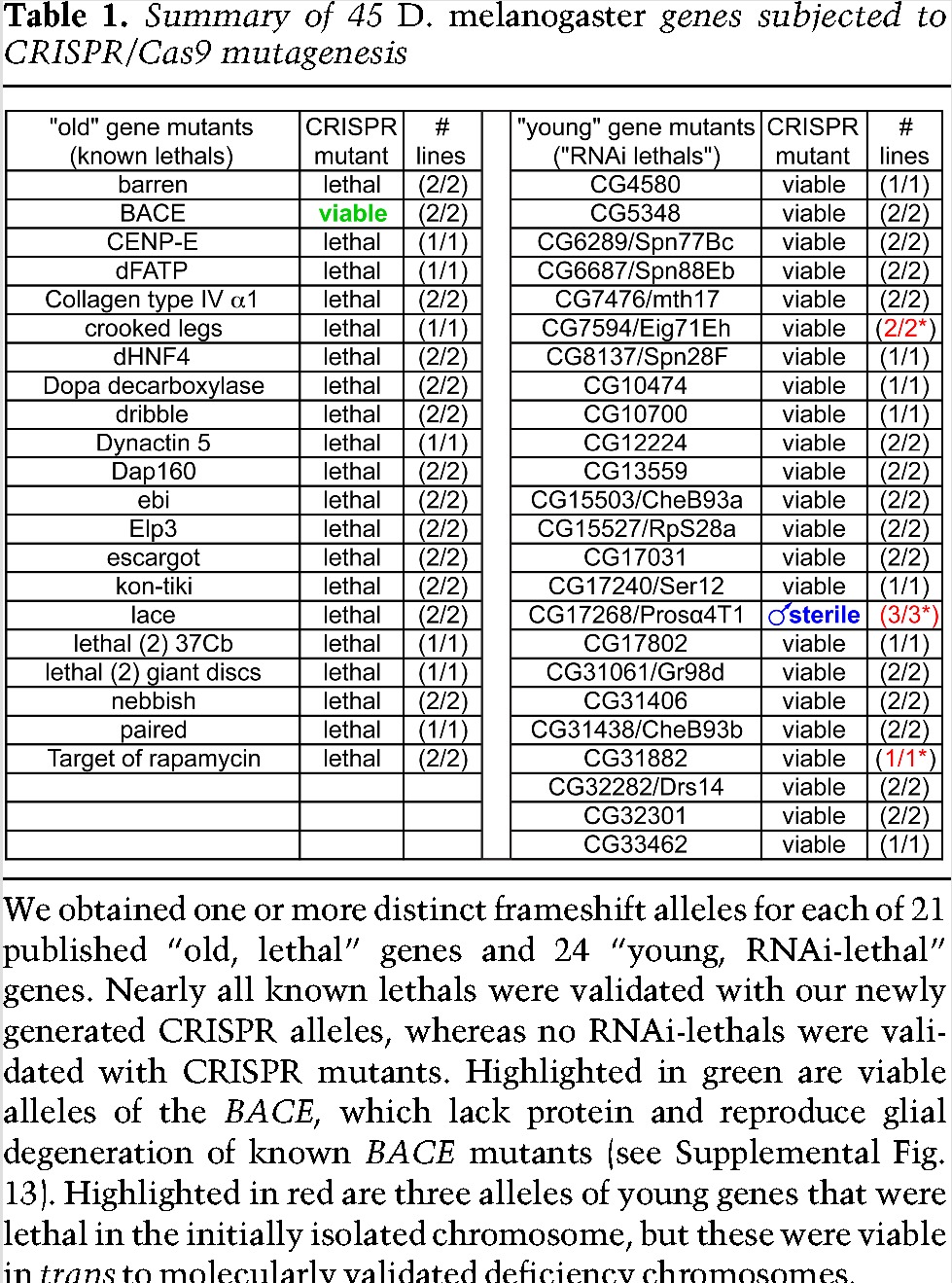
With these data in mind, we mutated 24 recently evolved genes reported as essential, again usually recovering multiple distinct frameshift alleles (Table 1; Supplemental Table 4). In stark contrast to the notion that these are young, essential genes, we found that all of these mutants were viable as homozygotes or hemizygotes. Given that our extensive collection of control mutants recapitulates known lethals, these analyses do not support the notion of frequent, essential, young genes (Table 1).
We sought other evidence of adult-specific phenotypes. None of our young gene mutants exhibited overt morphological defects, unlike in RNAi assays (Chen et al. 2010), and all were female fertile. Still, our expression analyses suggested that the male reproductive system deserved closer attention. While certain homozygous mutants putatively exhibited male fertility defects, some of these did not recapitulate over a cognate deficiency (Fig. 2A,B; see also the Materials and Methods). This indicated the importance of ruling out second site aberrations even with CRISPR/Cas9 mutagenesis.
Figure 2.
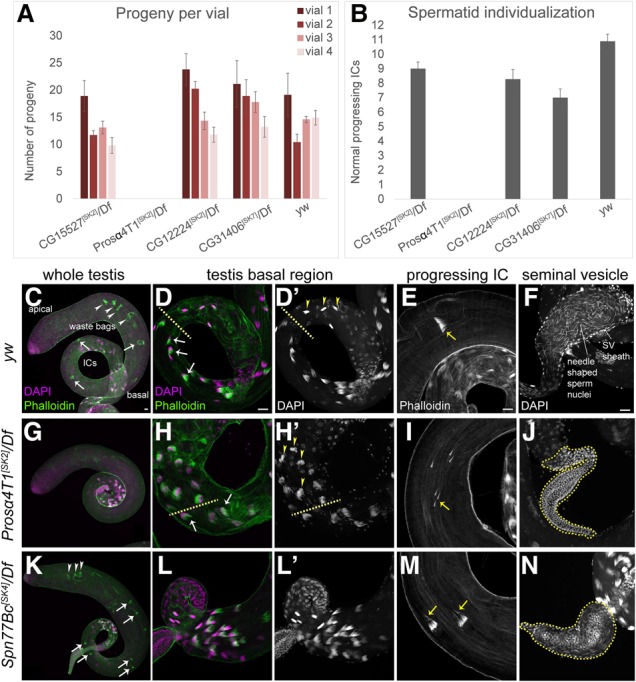
Mutants of selected young Drosophila genes exhibit spermatogenesis defects. (A) Fertility tests of selected mutant alleles that showed male fertility defects as homozygotes. Of these, only CG17268/Prosα4T1 recapitulated sterility in the hemizygous condition. All mutants were induced in a yw background, which was used as a control. (B) Quantification of progressing individualization complexes (ICs) in testes of the indicated genotypes. Consistent with their sterile phenotype, Prosα4T1 hemizygotes lack ICs. (C–N) Cytological analysis of various aspects of spermatogenesis. (C–F) Wild-type testes (yw). (C,D) The condensed nuclei (DAPI, magenta) of the differentiating spermatids are clustered in the basal region. ICs form around the nuclei (D; phalloidin, green; arrows), progress along the sperm tails (C,E; phalloidin; arrows) toward the apical region, and become waste bags (C; arrowheads). Following successful individualization, mature sperm are coiled in the base of the testis (D′; arrowheads) and transferred to the seminal vesicles (F). (G–J) Prosα4T1 mutants. (G) ICs and waste bags are absent. (H,I) Spermatid nuclei are properly condensed and ICs are formed (H; arrows), but progressing ICs are fully dispersed (I; arrows). (H′) Mature sperm nuclei appear disorganized in the coiling region (arrowheads). (J) The seminal vesicle is devoid of mature sperm, and only the round nuclei of the seminal vesicle wall are evident. (K–N) Spn77Bc mutants. Nuclear organization and IC formation are normal (K,L), but Spn77Bc mutants exhibit high frequency of disorganized ICs with lagging actin cones (M) and reduced seminal vesicles (N). Bars, 20 µm.
However, mutants of the young gene CG17268/Prosα4T1 (Supplemental Fig. 3) revealed a striking defect. Three different frameshift alleles (SK2, SK11, and SK13) were completely male sterile in bulk crosses as both homozygotes and hemizygotes; quantification across 10 individual Prosα4T1[SK2]/Df males is shown in Figure 2A. To understand their basis, we surveyed testis morphology and sperm differentiation. We did not observe gross defects in the stem cell niche, in spermatogonial mitosis, or in cytokinesis accompanying the meiotic divisions (Supplemental Fig. 14). Instead, Prosα4T1 mutants exhibited aberrant spermatid differentiation. In wild type, the condensed nuclei of sister spermatids are bundled together in the basal region of the testis following flagellar elongation (Fig. 2C,D). Actin-rich individualization complexes (ICs), containing one actin cone for each nucleus in the germline cyst, form around the nuclei, travel down the flagella toward the apical region (Fig. 2E), and become waste bags (Fig. 2C). Following individualization, mature sperm are then deposited into the seminal vesicle (Fig. 2F). Prosα4T1 hemizygotes initially formed normal nuclear bundles and ICs and incorporated protamines into sperm chromatin (Fig. 2G,H; Supplemental Fig. 14). However, its ICs became completely dispersed during migration (Fig. 2I), and the nuclei of maturing sperm exhibited aberrant morphology in the coiling region, correlating with failed individualization (Fig. 2, cf. D/D′ and H/H′, arrowheads). Consequently, the seminal vesicles of Prosα4T1 mutants were devoid of mature sperm (Fig. 2J). We documented a similar suite of phenotypes in Prosα4T1[SK2] homozygous testes (Supplemental Fig. 14).
Since some fertile mutants might exhibit spermatogenesis defects, we screened the remaining mutants for cytological aberrations and male fertility. We did not reproducibly observe subfertility or spermatogenesis defects among most other mutants of young genes. However, we found that CG6289/Spn77Bc is required for normal spermatid individualization. Western blotting confirmed that these mutants strongly reduced Spn77Bc protein (Supplemental Fig. 15A). Its loss of function was not as severe as with Prosα4T1 in that well-defined ICs and waste bags were formed and nuclear dynamics appeared normal (Fig. 2K,L); hub organization and meiotic cytokineses were also normal (Supplemental Fig. 15B–G). Still, one-third of individualization structures in Spn77Bc[SK4] hemizygotes contained dispersed actin cones (Fig. 2M), and the deposition of mature sperm in the seminal vesicle was compromised (Fig. 2N). We confirmed these spermatogenesis phenotypes in independent Spn77Bc alleles as well as in a hemizygous combination. Further cytological analyses are shown in Supplemental Figure 15, H–S, and quantitative analyses are shown in Supplemental Figure 15T. Thus, even though mutation of Spn77Bc did not compromise overall fertility (Supplemental Fig. 15U), loss of this recently emerged gene has a detectable impact on spermatogenesis.
We sought additional evidence that the dominant trend of gene birth in the male reproductive system might have a functional impact. Although most ancient genes evolve by purifying negative selection, some male reproductive genes exhibit signatures of positive selection, indicating their active participation in functional networks that are under pressure for evolutionary change (Haerty et al. 2007; Assis and Bachtrog 2013). To determine relative rates of divergence in our expanded catalogs of young genes, we estimated the dN/dS ratio between D. melanogaster and Drosophila simulans orthologs, a measure of the relative rate of amino acid-changing nucleotide substitutions. Old genes with preferential expression in the testis or accessory gland exhibited low dN/dS, as expected, indicating sustained purifying selection, but progressively younger cohorts of male-biased genes exhibited increasingly higher dN/dS values that rose far above the genome average (Supplemental Fig. 16A,B). The excess of nonsynonymous divergent sites within young male-biased genes is consistent with positive selection. However, young genes expressed outside the testis and young genes expressed broadly also exhibited elevated dN/dS (Supplemental Fig. 16C–E).
As increased dN/dS does not robustly distinguish between positive selection and reduced constraint, we conducted further analysis using an unpolarized McDonald-Kreitman framework to estimate the direction of selection (DoS) metric (Stoletzki and Eyre-Walker 2011) across different age categories of genes. DoS measures natural selection on recent time scales by contrasting nonsynonymous and synonymous polymorphisms to nonsynonymous and synonymous divergence. The DoS metric has an expected value of zero under neutrality, takes negative values under negative selection, and takes positive values under positive selection. The genome average for our data is slightly less than zero (−0.04) (Supplemental Fig. 16F). DoS statistics indicate that among genes highly restricted to the male reproductive system, ancient loci are subject to purifying selection, whereas progressively younger genes that originated within the Drosophilid phylogeny exhibit DoS values greater than zero, demonstrating positive selection (Fig. 3A). Such evolutionary behavior is not seen with young, tissue-restricted genes expressed elsewhere (Fig. 3B) or with broadly expressed genes of various ages (Supplemental Fig. 16G,H). Overall, these data indicate that many young, testis/accessory gland-restricted genes are not simply neutrally evolving loci but encode genetic functions that are under active positive selection.
Figure 3.
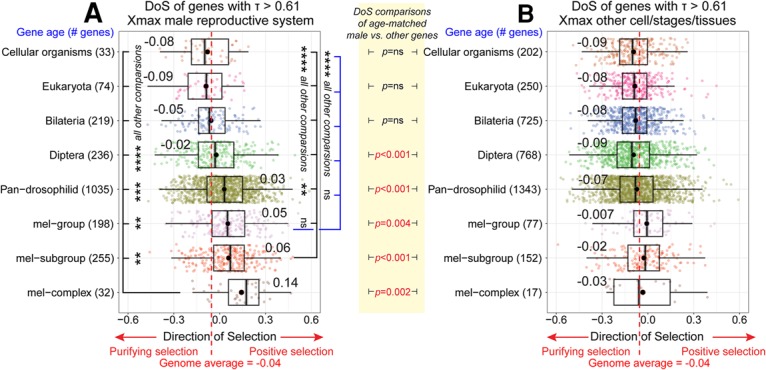
Positive selection on young genes restricted to the male reproductive system. (A,B) DoS tests of genes with tissue-restricted genes (τ > 0.61) (see Fig. 1C) were calculated for loci in the male reproductive system (A) or in other tissues/stages/cell types (B). Positive DoS values indicates adaptive selection, whereas negative DoS values indicate purifying selection. The genome average DoS (−0.04) is marked with a red dotted line. (A) Among testis/accessory gland-restricted genes, a trend for positive DoS is robust among genes born in the pan-Drosophilid ancestor and increases progressively with younger age groups. Wilcoxon signed-rank tests demonstrate that frequencies of positive selection are greatest among cohorts of younger genes. (ns) Not significant; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (B) Among genes expressed in other settings, the average DoS is negative for all age groups and only above the genome average for mel group and mel subgroup young genes. Wilcoxon signed-rank tests show that DoS values between groups of the same age category are much higher among genes restricted to the male reproductive system.
In this study, we present expression-based and genetic evidence that young genes infrequently harbor essential functions in Drosophila. Instead, we observed that young genes exhibit a propensity for dominant expression in the male reproductive system (Fig. 1), have a functional impact on spermatogenesis (Fig. 2), and evolve under net positive selection (Fig. 3). The testis has long been recognized as a tissue that is permissive for gene birth (Zhao et al. 2014), including for protein-coding, putative long noncoding, and even small RNA genes in D. melanogaster (Brown et al. 2014; Mohammed et al. 2014; Zhao et al. 2014; Wen et al. 2016). Notably, recent work showed that knockouts of some young testis noncoding RNAs also reveal spermatogenesis defects (Wen et al. 2016). Moreover, the Drosophila testis expresses specialized versions of several housekeeping complexes, including the proteasome (Belote and Zhong 2009), basal transcription (Hiller et al. 2004), and translational machinery (e.g., Fig. 1A; Baker and Fuller 2007). This links with the observation that duplicate copies of genes tend to be expressed in testis and have signatures for neofunctionalization (Assis and Bachtrog 2013), a principle that we extend in this study. Strikingly, we show that even a newly emerged testis-specific subunit of a housekeeping machine (Prosα4T1) has become indispensable for male reproduction.
The testis appears to be a setting whose chromatin is not only permissive for gene emergence but also subject to unique selective pressures that may foster gene neofunctionalization, adaptive evolution, and speciation (Kaessmann 2010). Although we did not identify critical functions for other young gene knockouts, it is possible that other sensitized or quantitative assays may reveal effects. With these first broad functional assessments in an invertebrate, it will be of great interest to see whether recently emerged genes have comparable functional attributes in vertebrates.
Materials and methods
Analysis of Drosophila gene ages
We re-examined previous catalogs of evolutionarily recent genes (Chen et al. 2010, 2012) and updated these using additional genes from D. melanogaster release r6.13 using a multistage pipeline and extensive manual vetting of young genes for contiguity in University of California at Santa Cruz Genome Browser chains and net alignments (see the Supplemental Material). A summary of D. melanogaster gene ages is in Supplemental Table 1.
Analysis of Drosophila spatial expression preference
We mapped 189 D. melanogaster modENCODE poly(A)+ RNA-seq data sets (Sequence Read Archive [SRA] PRJNA75285) and six additional “Raleigh” testis RNA-seq data sets (SRA PRJNA210329) to the r6.13 version of the D. melanogaster genome. A summary of D. melanogaster expression data (FPKM [fragments per kilobase per million mapped reads]) across all RNA-seq data sets is in Supplemental Table 1. Spatial expression preference was calculated using the tissue specificity index (Yanai et al. 2005). Further details are in the Supplemental Material.
TAI
The premise of the TAI was formulated by Domazet-Loso and Tautz (2010), who used gene expression data to quantify the conservation of genes that contribute to the transcriptome during a specific stage or biological process. This is achieved by transcriptome indices (Drost et al. 2016), where, in principle, any transcriptome data set can be combined with evolutionary information. In the equation below, the TAI of the tissue s is defined as the weighted mean of the phylostratum rank ps of gene i by the expression value e in the transcriptome of tissue s, where n is the total number of genes in the analysis:
Selection tests
We performed two selection tests using D. melanogaster population genomic data. Using D. simulans as an outgroup, we constructed 12,618 gene alignments for the longest transcript per gene (data and alignment methods are in the Supplemental Material). We measured relative rates of divergence (dN/dS) for gene alignments between D. melanogaster and D. simulans using a custom PERL script. dN/dS is a ratio of the number of nonsynonymous (replacement) substitutions per nonsynonymous site to the number of synonymous (silent) substitutions per synonymous site. In addition, we performed unpolarized McDonald-Kreitman tests (McDonald and Kreitman 1991) and estimated the DoS statistic. The DoS statistic is defined by the following equation, where DN are nonsynonymous substitutions, DS are synonymous substitutions, PN are nonsynonymous polymorphisms, and PS are synonymous polymorphisms:
Positive DoS values indicate directional or positive selection, while negative DoS values indicate purifying selection; zero indicates neutral evolution. Selection patterns on genes categorized into different age groups were then compared using the DoS metric. McDonald-Kreitman tests limit the availability of recently emerged genes in the “mel-only” category for DoS analysis due to the requirement of D. simulans as an outgroup for McDonald-Kreitman tests. PERL scripts to perform dN/dS and McDonald-Kreitman tests are available at https://github.com/Lai-Lab-Sloan-Kettering.
Analysis of RNAi phenotypes
We collected catalogs of RNAi phenotypes from several genome-wide studies (Dietzl et al. 2007; Perkins et al. 2015; Zeng et al. 2015), analyzing the Vienna Drosophila Resource Center (VDRC) “KK” and “GD” collections, the Harvard Transgenic RNAi Project (TRiP) collection, and the National Institute of Genetics (NIG) RNAi collection. We parsed them for their agreement of viability/lethality calls across different insertions of the same RNAi trigger and for the agreement of different triggers against the same gene using the ubiquitous driver Act5C-Gal4 and summarized information from other Gal4 drivers (Supplemental Table 2).
CRISPR/Cas9-mediated mutagenesis
We used the transgenic Cas9/guide RNA (gRNA) system (Kondo and Ueda 2013) to perform mutagenesis in the yw background as described in detail in the Supplemental Material. A full accounting of the mutagenesis pipeline, gRNA sequences, mutant alleles, and predicted mutant proteins are in Supplemental Tables 3 and 4.
Testis immunostaining
Adult testes from 0- to 4-d-old males were fixed in 5% formaldehyde/PBS and stained overnight with anti-α-spectrin antibody (1:25; Developmental Studies Hybridoma Bank, 3A9) or anti-cleaved caspase 3 (1:100; Cell Signaling Technology, 9664) in PBS/0.1% Triton X-100 (PBX) + 5% normal donkey serum. The next day, the testes were washed three times in PBX and incubated with Alexa fluor 488-conjugated donkey anti-mouse or anti-rabbit (1:1000; Molecular Probes), rhodamine–phalloidin (1:200; Sigma-Aldrich), and DAPI (1:4000; Roche).
Western blotting
We used rabbit CG6289/Spn77Bc (1:1000), rabbit anti-dBACE (1:2000), and mouse anti-tubulin (1:500) in standard Western blotting procedures. Details are in the Supplemental Material.
Supplementary Material
Acknowledgments
We are very grateful to Doris Kretzschmar for sharing unpublished characterization of BACE mutants. We thank Sayani Sen for assistance with fly husbandry, and Mariana Wolfner, the Bloomington Stock Center, and the Developmental Studies Hybridoma Bank for fly stocks and antibodies. S.K. was supported by the Mochida Memorial Foundation for Medical and Pharmaceutical Research. A.S. was supported by National Institutes of Health (NIH) R01-GM102192. Work in J.S.’s laboratory was supported by NIH R15-HD080511. Work in E.C.L.’s group was supported by the NIH (R01-NS083833 and R01-GM083300) and Memorial Sloan-Kettering Core Grant P30-CA008748.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.303131.117.
References
- Assis R, Bachtrog D. 2013. Neofunctionalization of young duplicate genes in Drosophila. Proc Natl Acad Sci 110: 17409–17414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CC, Fuller MT. 2007. Translational control of meiotic cell cycle progression and spermatid differentiation in male germ cells by a novel eIF4G homolog. Development 134: 2863–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, Zhong L. 2009. Duplicated proteasome subunit genes in Drosophila and their roles in spermatogenesis. Heredity 103: 23–31. [DOI] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May G, Stoiber M, Duff M, Booth B, Park S, Suzuki A, Wan K, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang YE, Long M. 2010. New genes in Drosophila quickly become essential. Science 330: 1682–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Spletter M, Ni X, White KP, Luo L, Long M. 2012. Frequent recent origination of brain genes shaped the evolution of foraging behavior in Drosophila. Cell Rep 1: 118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Krinsky BH, Long M. 2013. New genes as drivers of phenotypic evolution. Nat Rev Genet 14: 645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. 2007. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448: 151–156. [DOI] [PubMed] [Google Scholar]
- Domazet-Loso T, Tautz D. 2010. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468: 815–818. [DOI] [PubMed] [Google Scholar]
- Drost HG, Bellstadt J, O'Maoileidigh DS, Silva AT, Gabel A, Weinholdt C, Ryan PT, Dekkers BJ, Bentsink L, Hilhorst HW, et al. 2016. Post-embryonic hourglass patterns mark ontogenetic transitions in plant development. Mol Biol Evol 33: 1158–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, Wong A, Ravi Ram K, Sirot LK, Levesque L, Artieri CG, Wolfner MF, Civetta A, et al. 2007. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics 177: 1321–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Chen X, Pringle MJ, Suchorolski M, Sancak Y, Viswanathan S, Bolival B, Lin TY, Marino S, Fuller MT. 2004. Testis-specific TAF homologs collaborate to control a tissue-specific transcription program. Development 131: 5297–5308. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. 2010. Origins, evolution, and phenotypic impact of new genes. Genome Res 20: 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Ueda R. 2013. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics 195: 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Miklos GL, Rubin GM. 1996. The role of the genome project in determining gene function: insights from model organisms. Cell 86: 521–529. [DOI] [PubMed] [Google Scholar]
- Mohammed J, Bortolamiol-Becet D, Flynt AS, Gronau I, Siepel A, Lai EC. 2014. Adaptive evolution of testis-specific, recently-evolved, clustered miRNAs in Drosophila. RNA 20: 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Perrimon N. 2012. RNAi screening: new approaches, understandings, and organisms. Wiley Interdiscip Rev RNA 3: 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins LA, Holderbaum L, Tao R, Hu Y, Sopko R, McCall K, Yang-Zhou D, Flockhart I, Binari R, Shim HS, et al. 2015. The transgenic RNAi project at Harvard Medical School: resources and validation. Genetics 201: 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross BD, Rosin L, Thomae AW, Hiatt MA, Vermaak D, de la Cruz AF, Imhof A, Mellone BG, Malik HS. 2013. Stepwise evolution of essential centromere function in a Drosophila neogene. Science 340: 1211–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. 2011. Estimation of the neutrality index. Mol Biol Evol 28: 63–70. [DOI] [PubMed] [Google Scholar]
- Wen J, Duan H, Bejarano F, Okamura K, Fabian L, Brill JA, Bortolamiol-Becet D, Martin R, Ruby JG, Lai EC. 2015. Adaptive regulation of testis gene expression and control of male fertility by the Drosophila harpin RNA pathway. Mol Cell 57: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen K, Yang L, Xiong T, Di C, Ma D, Wu M, Xue Z, Zhang X, Long L, Zhang W, et al. 2016. Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res 26: 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659. [DOI] [PubMed] [Google Scholar]
- Zeng X, Han L, Singh SR, Liu H, Neumuller RA, Yan D, Hu Y, Liu Y, Liu W, Lin X, et al. 2015. Genome-wide RNAi screen identifies networks involved in intestinal stem cell regulation in Drosophila. Cell Rep 10: 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE, Vibranovski MD, Krinsky BH, Long M. 2010. Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res 20: 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE, Landback P, Vibranovski MD, Long M. 2011. Accelerated recruitment of new brain development genes into the human genome. PLoS Biol 9: e1001179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Saelao P, Jones CD, Begun DJ. 2014. Origin and spread of de novo genes in Drosophila melanogaster populations. Science 343: 769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.