

ABSTRACT
The latency-associated nuclear antigen (LANA) of the Kaposi's sarcoma-associated herpesvirus (KSHV) performs a variety of functions to establish and maintain KSHV latency. During latency, LANA localizes to discrete punctate spots in the nucleus, where it tethers viral episomes to cellular chromatin and interacts with nuclear components to regulate cellular and viral gene expression. Using highly sensitive tyramide signal amplification, we determined that LANA localizes to the cytoplasm in different cell types undergoing the lytic cycle of replication after de novo primary infection and after spontaneous, tetradecanoyl phorbol acetate-, or open reading frame 50 (ORF50)/replication transactivator (RTA)-induced activation. We confirmed the presence of cytoplasmic LANA in a subset of cells in lytically active multicentric Castleman disease lesions. The induction of cellular migration by scratch-wounding confluent cell cultures, culturing under subconfluent conditions, or induction of cell differentiation in primary cultures upregulated the number of cells permissive for primary lytic KSHV infection. The induction of lytic replication was characterized by high-level expression of cytoplasmic LANA and nuclear ORF59, a marker of lytic replication. Subcellular fractionation studies revealed the presence of multiple isoforms of LANA in the cytoplasm of ORF50/RTA-activated Vero cells undergoing primary infection. Mass spectrometry analysis demonstrated that cytoplasmic LANA isoforms were full length, containing the N-terminal nuclear localization signal. These results suggest that trafficking of LANA to different subcellular locations is a regulated phenomenon, which allows LANA to interact with cellular components in different compartments during both the latent and the replicative stages of the KSHV life cycle.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus (KSHV) causes AIDS-related malignancies, including lymphomas and Kaposi's sarcoma. KSHV establishes lifelong infections using its latency-associated nuclear antigen (LANA). During latency, LANA localizes to the nucleus, where it connects viral and cellular DNA complexes and regulates gene expression, allowing the virus to maintain long-term infections. Our research shows that intact LANA traffics to the cytoplasm of cells undergoing permissive lytic infections and latently infected cells in which the virus is induced to replicate. This suggests that LANA plays important roles in the cytoplasm and nuclear compartments of the cell during different stages of the KSHV life cycle. Determining cytoplasmic function and mechanism for regulation of the nuclear localization of LANA will enhance our understanding of the biology of this virus, leading to therapeutic approaches to eliminate infection and block its pathological effects.
KEYWORDS: KSHV, LANA, activation, cytoplasm, mass spectrometry, migration, viral replication
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus that causes several human malignancies, including Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and AIDS-associated multicentric Castleman disease (MCD) (1). Like all herpesviruses, KSHV has two distinct gene expression patterns that result in either a persistent latent infection or an active lytic infection leading to virus replication. However, latency is considered the default pathway for KSHV infections. Initial studies showed that tumor cells in KS lesions were latently infected as gene expression was very restricted, with two small RNAs, i.e., T0.7 encoding K12 Kaposin and T1.1, a polyadenylated nuclear RNA (PAN), constituting the bulk of KSHV transcripts (3, 4). Subsequent studies in KSHV-infected PEL cell lines revealed the presence of a cluster of latency-associated transcripts downstream of K12 encoding open reading frame 71 (ORF71; v-FLIP), ORF72 (v-cyclin), and ORF73, the latency-associated nuclear antigen (LANA) (5–8). Immunohistochemistry studies showed that LANA was present in the nuclei of KSHV-infected PEL cells, as well as the vast majority of spindled tumor cells in KS lesions (7, 9, 10). Anti-LANA antibodies gave a characteristic punctate staining pattern in the nucleus of latently infected cells, which became a universal marker for KSHV latency.
A wide variety of endothelial, epithelial, fibroblast, and lymphocyte cells are susceptible to KSHV infection, resulting in a typical latent infection with restricted gene expression and accumulation of nuclear LANA (11–14). KSHV infection of endothelial and fibroblast cells induces an early transient expression of ORF50, the replication transactivator (RTA), which is the only viral gene that is necessary and sufficient for KSHV replication (15, 16). This is followed by a brief burst of expression of a subset of lytic genes with immunomodulatory and apoptotic functions, whose transcripts disappear 8 to 24 h postinfection (17). It has been suggested that the transient expression of this subset of genes is important for the establishment and maintenance of latency but does not lead to viral replication, since genes critical for DNA replication, such as ORF9 (the viral DNA polymerase) and ORF59 (the DNA polymerase processivity factor), are absent from the RTA-induced transient gene expression burst. The early spike in RTA induces LANA expression, which then shuts off RTA expression through transcriptional repression of the RTA promoter (13, 18, 19). LANA autoactivates its own promoter, leading to the accumulation of LANA in the nucleus and the establishment of latency in the infected cell (20).
Very few examples of primary KSHV lytic infections have been observed previously. Productive lytic infection has been documented in human endothelial cells and human papillomavirus 16 (HPV16)-immortalized keratinocytes; however, this required high titers of concentrated recombinant KSHV (21, 22). Primary tonsillar B cells grown in a lymphoid aggregate culture were also productively infected by KSHV (23). The majority of KSHV infection studies used chemical inducers such as the phorbol ester tetradecanoyl phorbol acetate (TPA) or sodium butyrate, an inhibitor of histone deacetylase, to reactivate latent KSHV infections and induce lytic replication (24–26). In addition, overexpression of recombinant KSHV ORF50 RTA has been shown to reactivate latent KSHV infections in a variety of cell types (27). The switch from latent to lytic replication after induction is initiated by the immediate-early expression of RTA, which then activates the entire lytic replication cycle, leading to the expression of a cascade of viral genes in an ordered progression resulting in the production of infectious virions.
Early on, it was noted that a small percentage of cells in latently infected cultures expressed markers of lytic KSHV replication. In endothelial cells for example, ∼1% of cells expressed ORF59 lytic marker 48 h after de novo infection, indicating “spontaneous” reactivation of the lytic program of replication (28). Lytic cycle gene expression, including ORF59, ORF26 (major capsid protein), and ORF K2 (vIL-6), has also been detected in a small percentage of cells in latently infected PEL cell cultures and in KS tumor lesions (29–32). Because this lytic gene expression is confined to a small proportion of cells in infected cultures, it has been difficult to determine whether it is due to an active lytic infection in specific cells or whether this represents an additional viral transcription program that could be cell type specific or associated with particular cellular proliferation and/or differentiation states. However, using a model of primary lytic replication, we have shown a direct correlation between the expression of ORF59, the induction of the lytic program of replication, and the production of infectious virions of a primate homolog of KSHV (33). Furthermore, previous studies have shown that an ORF59 deletion mutant of KSHV is defective in virion production, substantiating the critical role of ORF59 in the lytic cycle of replication (34).
We have observed that cell lines showing spontaneous reactivation of KSHV after latent infection, such as HEK293 and Vero cells, have moderate constitutive levels of cellular transcription factors that can directly activate the RTA promoter (13). This level of promoter activity is not sufficient to drive RTA-induced lytic gene expression and viral replication in the majority of infected cells. However, the latently infected cells are poised to allow reactivation of the viral genome and induction of viral replication, and small changes in the status of the cell can induce spontaneous reactivation. Treatment with phorbol esters or sodium butyrate increases the level of cellular transcription factors activating the RTA promoter, resulting in increased RTA expression and induction of viral lytic replication. In contrast, cell lines such as the gastric epithelial cell line (AGS) have minimal constitutive expression of cellular factors that can activate the RTA promoter, and KSHV-infected AGS cells exhibit a deep latent state, with no evidence of spontaneous reactivation (13).
Reactivation of lytic replication by cellular differentiation has been studied in oral epithelial cells infected with the recombinant rKSHV.219 virus, which expresses green fluorescent protein (GFP) from a constitutive cellular EF-1α promoter during typical latent infections and red fluorescent protein (RFP) from the lytic cycle PAN promoter after reactivation (35). Culturing of human oral keratinocytes (HOKs) latently infected with rKSHV.219 in organotypic raft cultures induced the differentiation of infected cells at the apical surface of the stratified epithelial layer, as evidenced by increased expression of the differentiation antigens, involucrin, and keratins 6, 13, 14, and 19. The differentiation of these epithelial cells induced expression of RFP and the late virion glycoprotein K8.1, as well as the production of infectious rKSHV.219 at the epithelial surface (36). In further studies using a submerged direct-plating model of keratinocyte differentiation, differentiated rKSHV.219-infected primary keratinocytes expressed RFP after plating in a suprabasal position onto a confluent epithelial cell culture (37). These studies showed that differentiation and suprabasal positioning in an epithelial layer were sufficient to activate KSHV lytic replication.
In contrast to the latency observed in KS lesions and PEL tumors, KSHV-associated MCD lesions show a high percentage of infected cells expressing early-, intermediate-, and late-stage KSHV proteins, including ORF59, K2 (vIL-6), K8 (K-bZip), K9 (vIRF-1), K10 (vIRF-4), and the K8.1 glycoprotein (30, 31, 38, 39). The elevated expression of KSHV lytic genes suggests that MCD lesions are sites of lytic replication where differentiation signals in a unique B cell subset activate the lytic program of KSHV transcription.
In latent KSHV infections, LANA localizes to punctate foci in the nucleus at sites where LANA tethers viral episomes to host chromatin, ensuring partition of the KSHV genomes to daughter nuclei during mitosis (40, 41). In addition to episome maintenance, LANA regulates viral and cellular gene transcription (20, 42–45), recruits enzymes that affect chromatin remodeling (46–48), promotes cell survival and growth (2, 49–53), and inhibits apoptosis (54, 55). LANA also represses the expression of RTA and other KSHV lytic genes and inhibits the KSHV replication program (18, 53, 56).
LANA is a large protein with three structural domains (57). The serine-proline-rich N terminus contains a complex bipartite nuclear localization signal (NLS), which utilizes both classical and nonclassical pathways for nuclear import (58, 59), a chromatin-binding motif (60), and domains responsible for interaction with various transcription regulators (53, 61). The C-terminal domain is responsible for binding to the terminal repeats of the KSHV genome and tethering the viral episome to host cell chromatin (40). This domain also interacts with various cellular proteins that localize to the nucleus, including p53, pRb, and MeCP2 (54, 62, 63), and nuclear localization is observed when the LANA is expressed as a truncated C-terminal peptide (59, 64). The central region of LANA contains several repetitive acidic regions whose heterogeneity across different KSHV strains results in LANA proteins ranging from 1,003 to 1,162 amino acids in length. Whereas the predicted size of LANA is ∼100 to 120 kDa, the protein migrates as a number of different forms in sodium dodecyl sulfate (SDS) gels with mobilities from 130 to 234 kDa (7, 10). LANA variants have been associated with noncanonical translation initiation (65), noncanonical polyadenylation (66), and caspase modification (67).
In early immunohistochemical studies, LANA staining was detected in the cytoplasm of a subset of cells in MCD lesions (38, 68). Subsequently, isoforms of LANA were detected in cytoplasmic extracts of KSHV-infected HEK293 and BCBL-1 PEL cells (65, 69). Induction of BCBL-1 cells with TPA increased the levels of cytoplasmic LANA isoforms (69), and cytoplasmic forms bound to the innate immune sensor cGMP-AMP synthase (cGAS) and to components of the MRN (Mre11-Rad50-NBS1) repair complex, suggesting a role in antagonizing the innate immune response and thus facilitating lytic reactivation (69, 70). We previously developed ultrasensitive enhancement of fluorescent labeling for confocal microscopy using tyramide signal amplification (TSA) (71, 72). During these studies, we observed strong LANA fluorescence in the cytoplasm of specific cells in different KSHV-infected cultures using a LANA-specific monoclonal antibody. In the present study, we used TSA-enhanced confocal microscopy and other techniques to investigate the connection between cytoplasmic LANA isoforms and KSHV lytic replication. We confirmed the presence of cytoplasmic LANA in lytically active MCD lesions. In addition, we observed a strong correlation between the expression of cytoplasmic LANA and nuclear ORF59 after lytic reactivation of KSHV in latently infected cells, as well as after primary lytic infections of KSHV in differentiated and/or migrating cells. Subcellular fractionation revealed the presence of multiple LANA isoforms in the cytosol, and full-length isoforms containing the N-terminal nuclear localization were detected by mass spectrometry. The presence of full-length isoforms in the cytosol is suggestive of a regulatory process that can block or enhance nuclear localization of LANA, thereby altering the cellular distribution of this critical viral protein during the different lytic and latent programs of KSHV infection.
RESULTS
Cytoplasmic LANA in KSHV-associated MCD lesions.
Previously, two different staining patterns were reported for antibodies to KSHV ORF73 LANA in lymph nodes from patients with MCD. One pattern was similar to that seen in spindled tumor cells in KS lesions with the presence of multiple punctate dots in the nucleus. The other pattern showed diffuse staining in both the cytoplasm and nucleus (38, 68). To confirm this differential localization, we prepared paraffin-embedded sections from a KS skin nodule and two MCD lymph nodes for KSHV LANA localization using monoclonal antibody LN53 (73). In the KS lesion, LN53 specifically localized to nuclei in KS spindle cells, where the brown diaminobenzidine chromogen formed punctate LANA dots (Fig. 1A, arrowheads). In MCD lesions, the LN53 antibody gave two different staining patterns. In some cells, LN53 staining was confined to the nucleus with punctate structures, like the pattern seen in KS lesions (Fig. 1B, arrowheads). In other cells, the LN53 also localized diffusely throughout the cytoplasm (Fig. 1B, arrows). The cytoplasmic staining was confined to individual cells having distinct borders and thus was not due to nonspecific chromogen diffusion. The cells with cytoplasmic LANA staining appeared larger with more intense staining than the adjacent cells with nuclear LANA alone. In most cases, the nuclear distribution of LN53 in cells with cytoplasmic LANA was not in distinct punctate dots as seen in the smaller cells (Fig. 1B, arrowheads), but appeared to be distributed throughout the nucleus (Fig. 1B, arrows). The presence of cytoplasmic LANA in specific cells in the MCD lesions suggests an activation of latent KSHV, which results in altered subcellular trafficking of LANA and the potential for interactions with different cellular components.
FIG 1.

Differential localization of nuclear and cytoplasmic LANA in KS and MCD lesions. Paraffin-embedded tissue sections were prepared for immunohistochemical detection of KSHV LANA protein using anti-LANA monoclonal antibody LN53. (A) KS lesion. The arrowheads indicate spindle-like tumor cells with punctate LANA staining in the nucleus. (B) Montage of different cells from two MCD lesions. The arrowheads indicate small cells with punctate LANA staining in the nucleus, similar to that seen in the KS lesion. The arrows indicate larger cells with LANA localized to the nucleus and cytoplasm. Scale bar, 20 μm.
LANA differentially localizes to the nucleus and cytosol after de novo KSHV infection.
To investigate the bimodal distribution of LANA, we utilized TSA to enhance the fluorescent signal obtained from the LN53 antibody in primary cultures of HOKs and Vero monkey kidney epithelial cells infected with BCBL-1-derived KSHV. Since cellular differentiation of epithelial cells has been shown to activate KSHV latency (36, 37), HOK cells were plated in low-calcium medium (0.03 mM) and then shifted to high-calcium medium (0.15 mM) for 24 h to induce differentiation. Vero cells were chosen since they are commonly used to titer KSHV infections, and a high percentage of infected cells can be obtained (71). The cell cultures were infected with gradient-purified BCBL-1-derived KSHV (1,000 genomes/cell) for 24 h. LANA was localized by confocal immunofluorescence microscopy using the LN53 antibody with TSA, as described previously (71). The position of the nucleus was visualized with the TO-PRO-3 fluorescent stain, which targets the nuclear DNA. In most infected HOKs (Fig. 2A, arrowhead) and Vero cells (Fig. 2B, arrowhead), LANA fluorescence localized to the nucleus in the characteristic punctate pattern, with a conspicuous absence of LANA fluorescence in TO-PRO-free nucleolar spots. In approximately 5% of the cells, however, high levels of LANA fluorescence localized to the cytoplasmic compartment (Fig. 2, arrows). The punctate nuclear LANA staining in adjacent cells is more clearly observed in a higher intensity image of Fig. 2A (see Fig. S1 in the supplemental material). The pattern of LANA fluorescence in the cytoplasm was not uniformly diffuse but appeared fibrillar, suggesting an interaction with cytoskeleton components in the cytoplasm. In some cases, perinuclear LANA staining was observed, in addition to cytoplasmic staining (Fig. 2B, arrow). The accumulation of LANA in the cytoplasm of cells 24 h postinfection (hpi) suggests that nuclear localization of newly synthesized LANA was blocked or inhibited in specific cells during the initial stages of infection.
FIG 2.

Differential localization of nuclear and cytoplasmic LANA in KSHV-infected primary HOK and Vero cell cultures. (A) Primary HOK cultures induced to differentiate using high calcium (0.15 mM) for 24 h and (B) Vero monkey kidney epithelial cell cultures were infected with KSHV. At 24 hpi, the cell cultures were fixed and stained for LANA (green) using the LN53 antibody with TSA enhancement. Cell nuclei were labeled using TO-PRO-3 (blue). The arrowheads indicate cells with punctate nuclear LANA fluorescence, and the arrows indicate cells with diffuse LANA fluorescence in the cytoplasm. Scale bar, 20 μm.
Cells induced to migrate and proliferate preferentially express cytoplasmic LANA after de novo infection.
In the initial infection studies described above, Vero cells had been cultured in a 16-mm spot for confocal immunofluorescence analysis prior to KSHV infection. At 24 hpi, the cultures were stained for LANA using the LN53 antibody with TSA enhancement, and a high percentage of LANA-positive cells was observed across the entire cell culture. At the spot center where the culture was confluent, LANA fluorescence localized only to the nucleus, with the punctate pattern characteristic of a typical latent infection (Fig. 3B, arrowheads). At the culture edge, where cells were expanding onto the cell-free dish surface, we also observed cells with punctate nuclear LANA (Fig. 3A, arrowhead), but a substantial number of edge cells showed a diffuse cytoplasmic LANA fluorescence (Fig. 3A, arrows). These findings indicate that KSHV infection of a high percentage of migrating/proliferating cells at the spot edge did not result in the typical latency state where LANA would only localize to specific compartments within the nucleus to block lytic replication and tether the viral episome to host chromatin.
FIG 3.
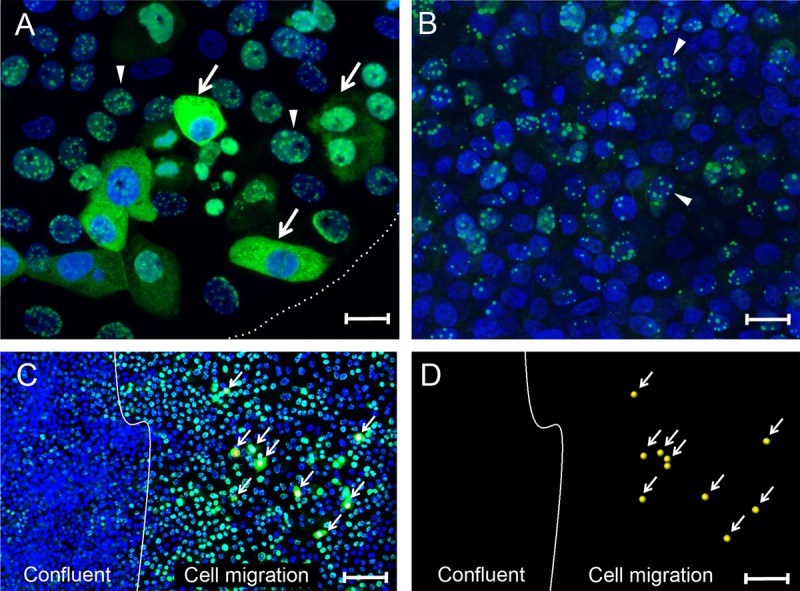
Differential localization of nuclear and cytoplasmic LANA in migrating Vero cells. (A and B) Vero cells plated in a 16-mm “spot” culture were infected with KSHV. At 24 hpi, the cultures were fixed and stained for LANA (green) and nuclei (blue). (A) Sparse edge of the spot culture, delineated with a dotted white line. The arrows indicate cells with diffuse LANA fluorescence in the cytoplasm located at the edge of the cell culture where the cells were expanding into the open area of the plate. The arrowheads indicate adjacent cells with punctate LANA fluorescence in the nucleus. (B) Center of the confluent spot culture. The arrowheads indicate examples of cells with punctate nuclear LANA fluorescence. (C) Confluent Vero cell cultures were scratch-wounded to stimulate cell migration and proliferation. After a 24-h migration period, the cultures were infected with KSHV. At 24 hpi, the cultures were fixed and stained as described above. The white line marks the edge of the confluent culture following wounding and represents the start point of cell migration. The arrows indicate cells in the migration area with diffuse LANA fluorescence in the cytoplasm (marked with a yellow dot in the nucleus). Adjacent cells and cells in the confluent area had punctate LANA fluorescence in the nucleus. (D) The positions of cells with cytoplasmic LANA relative to the wound edge are shown without the blue and green channels. Scale bars: A and B, 20 μm; C and D, 100 μm.
To further assess the correlation between cell migration and cytoplasmic LANA, confluent Vero cell cultures were scratch wounded to induce cell proliferation and migration at the wound edge in order to close the gap prior to establishment of new cell-cell contacts (74–76). In this procedure, the cell monolayer was scraped with a sterile pipette tip to form a gap, and the debris was removed by washing. Cells induced to migrate undergo dramatic morphological alterations, becoming elongated and highly motile with changes in cell surface signaling and adhesion receptors, polarity, and metabolism (77). The Vero cell culture was incubated for 24 h after scratching to allow cell migration and was then infected with KSHV. At 24 hpi, the cell culture was stained for LANA using the LN53 antibody with TSA enhancement. The approximate wound edge separating confluent cells from the newly migrated cells is shown (Fig. 3C and D, white line). The cell density in the confluent region was 53 ± 6 per 100 μm2 (n = 10) compared to 28 ± 6 μm2 within the subconfluent migration zone (n = 20). Although only 40% of the confluent cells showed evidence of KSHV infection with positive LANA staining, 94% of the cells in the migration area were LANA positive (Fig. 3C). The infected cells in the confluent region showed low levels of typical punctate nuclear LANA staining. In contrast, the cells in the migration area showed high levels of punctate nuclear LANA staining, with some cells exhibiting strong cytoplasmic LANA staining (Fig. 3C, arrows). The cells shown in Fig. 3C with cytoplasmic LANA are marked with a yellow dot and are visualized in Fig. 3D. The LANA staining in the subconfluent migration region was more than 3-fold stronger than in the confluent region, with 91 ± 26 compared to 27 ± 15 fluorescent LANA pixels per LANA-positive cell. The morphological and phenotypic changes in cells induced to migrate may have altered the nature of the viral infection, possibly upregulating the cellular receptors that facilitate KSHV entry. However, since the majority (84%) of cells in the migration area did not exhibit cytoplasmic LANA, it is likely that a second activating event in a subset of the migrating cells must play a role in the cytoplasmic accumulation of LANA.
The accumulation of cytoplasmic LANA strongly correlates with the induction of the lytic cycle of replication.
To determine whether the induction of migration could induce a cellular phenotype permissive for KSHV lytic replication, subconfluent Vero cell cultures were infected with BCBL-1-derived KSHV for 24 h, fixed, and then sequentially stained for LANA and ORF59, a DNA polymerase processivity factor that serves as a common marker of KSHV lytic replication. ORF59 is a critical component of the KSHV replication pathway, where it interacts with the DNA polymerase to replicate the KSHV genome (78). As seen previously, the majority of the Vero cells showed a typical latent infection with LANA accumulating in the nucleus in punctate spots, with no evidence of ORF59 fluorescence (Fig. 4A and B, arrowhead). However, a subset of infected Vero cells showed distinct accumulation of LANA in the cytoplasm and ORF59 in the nucleus (Fig. 4A and B, arrows). High-resolution analysis of a cell with cytoplasmic LANA and nuclear ORF59 fluorescence revealed patchy, sometimes fibrillar, concentrations of LANA fluorescence in the cytoplasm, a finding reminiscent of intermediate filament distributions detected by electron microscopy (79). In contrast, the ORF59 staining in the nucleus appeared globular with distinct lobes, which is consistent with the localization of ORF59 in replication compartments during lytic reactivation (80). Cellular DNA (blue) was clearly excluded from these lobes in the periphery of the nucleus. Thus, the accumulation of cytoplasmic LANA in a subset of cells in subconfluent Vero cultures was highly correlated with ORF59 expression and the lytic replication pathway.
FIG 4.
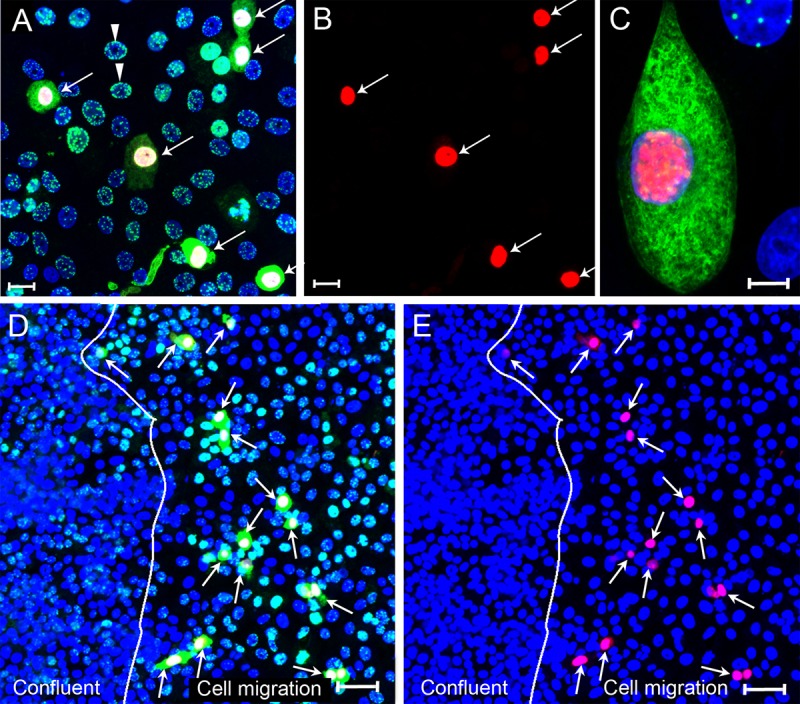
Cytoplasmic LANA correlates with the lytic cycle marker ORF59 after primary KSHV infection. (A to C) Subconfluent Vero cell cultures were infected with KSHV and fixed at 24 hpi. Cells were sequentially stained with mouse anti-ORF59 (red) and rat anti-LANA (LN53) (green) using TSA enhancement, as described in Materials and Methods. The cell nuclei were stained with TO-PRO-3 (blue). (A) Overlay of ORF59, LANA, and TO-PRO-3 fluorescence (red, green, and blue channels). The arrows point to cells with cytoplasmic LANA. All cells with cytoplasmic LANA also expressed nuclear ORF 59. (B) ORF59 fluorescence in panel A (red channel alone). (C) High-resolution image showing both nuclear ORF59 and cytoplasmic LANA fluorescence. (D and E) Confluent Vero cultures were scratch wounded, and cells on the edge of the wound migrated into the open space for 24 h. Cell cultures were infected with KSHV for 24 h, fixed, and sequentially stained for ORF59 and LANA, as described above. (D) Overlay of ORF59, LANA, and TO-PRO-3 fluorescence (red, green, and blue channels). The arrows point to cells with nuclear ORF59 and cytoplasmic LANA fluorescence in the cell migration area. (E) Overlay of ORF59 and TO-PRO-3 fluorescence (red and blue channels). The arrows point to cells with nuclear ORF59 fluorescence. The line demarcating the confluent and cell migration regions is shown. Scale bars: A and B, 20 μm; C and D, 50 μm.
To further study this phenomenon, confluent Vero cultures were scratch wounded as described previously, and cells were allowed to migrate into the open area for 12 h. The wounded cultures were infected with BCBL-1-derived KSHV and, at 24 hpi, the cells were fixed and sequentially stained for LANA and ORF59. As seen in Fig. 3C and D, cells in the confluent region showed a typical latent infection with punctate nuclear LANA and no visible ORF59 staining (Fig. 4D and E). However, a large number of cells with cytoplasmic LANA and nuclear ORF59 fluorescence (Fig. 4D and E, arrows) were observed in the migration region. Previous studies showed that ORF59 is not expressed during a typical latent infection (17) but accumulates in the nuclei of KSHV-infected cells that are induced to replicate KSHV after chemical reactivation (81). Therefore, the nuclear accumulation of ORF59 24 h after infection without artificial induction is indicative of a permissive KSHV infection with the induction of the lytic cycle of replication in a subset of Vero cells induced to migrate. The presence of cytoplasmic LANA suggests that blocking the transport of LANA to its typical nuclear compartments and the accumulation of LANA in the cytoplasm with specific interactions with components, such as the cytoskeleton, could be an integral process in KSHV lytic replication.
Since treatment of cultures undergoing long-term latent KSHV infections with chemical inducers activates the program of lytic replication in a percentage of infected cells, we determined whether reactivation could induce the expression of cytoplasmic LANA. Naturally infected BCBL-1 cells carrying a long-term latent KSHV infection were treated with 20 nM TPA to induce reactivation. After 12 h, the cells were fixed and sequentially stained for LANA and ORF59. At this time point, the majority of cells still showed typical KSHV latency with punctate LANA localized to the nucleus (Fig. 5, arrowhead). However, numerous cells showed evidence of both ORF59 staining in the nucleus and LANA staining in the cytoplasm (Fig. 5, arrows). A fluorescence profile was obtained across a cell in panel A with both nuclear ORF59 and cytoplasmic LANA staining. Although the red ORF59 and blue TO-PRO-3 fluorescence were limited to the cell nucleus, high levels of green LANA staining were detected in the cell cytoplasm (Fig. 5B [red = ORF59, blue = TO-PRO-3, and green = LANA]). Only low levels of LANA were observed in the region of the nucleus.
FIG 5.

Cytoplasmic LANA correlates with ORF59 expression after reactivation of latent KSHV infections. (A) BCBL-1 cells carrying a long-term latent KSHV infection were treated with TPA (20 ng/ml) for 48 h to reactivate KSHV. The cells were fixed and sequentially stained for ORF59, LANA, and cell nuclei, as in Fig. 4. Cells with nuclear ORF59 and cytoplasmic LANA fluorescence (arrows) or punctate nuclear LANA fluorescence (arrowheads) are indicated. (B) Profile of the fluorescence staining across a BCBL-1 cell with cytoplasmic LANA (green-Ch3) and nuclear ORF59 (red-Ch2) in panel A, as well as TO-PRO-3 staining of DNA (blue-Ch1). Scale bars, 20 μm. (C) HPV-immortalized DMVEC cells were infected with KSHV and cultured for 35 days. The cells were fixed and stained sequentially for ORF59 (green) and LANA (orange). The ORF59 and LANA fluorescence are overlaid on a phase-contrast image. The cell with nuclear ORF59 (large arrow, green) also expressed cytoplasmic LANA (small arrow, orange). Punctate LANA and ORF59 fluorescence colocalized (yellow) in the nucleus of this cell. Punctate LANA fluorescence was observed alone in cells lacking ORF59 fluorescence (arrowheads).
The program of lytic replication has also been observed in a small percentage of latently infected endothelial cell cultures. In these cells, nuclear ORF59 expression is detected, indicative of a “spontaneous reactivation” of the latent KSHV genome (28). To determine whether cytoplasmic LANA expression is observed in cells undergoing spontaneous reactivation, we examined KSHV infection of E6/E7 human papillomavirus-immortalized dermal microvascular endothelial cells (DMVECs), since these cells maintain the KSHV genome for extended periods (82). In these cultures, ORF59 expression was initially detected in a low percentage (<1%) of infected cells, but this increased with time up to 5% at 8 weeks postinfection (82). Electron microscopy analysis detected herpesvirus-like structures within the nuclei and cytoplasmic cisternae, indicating that the ORF59-positive cells were productively infected (82). At 35 days postinfection, the KSHV-infected DMVECs were sequentially stained for both LANA (orange) and ORF59 (green). Most infected cells showed punctate nuclear LANA staining, consistent with a latent infection (Fig. 5C, arrowhead). However, ORF59-positive cells with distinct LANA staining in the cytoplasm were also observed (Fig. 5C, large arrow), typically with a spindled morphology (small arrow).
Previous studies have shown that in vitro differentiation of latently infected epithelial cells activates the program of lytic KSHV replication (36, 37). To study the effects of differentiation on the expression of cytoplasmic LANA, primary HOK cell cultures were prepared from gingival biopsy specimens, as described previously (83). The cells were initially grown in low-calcium medium (0.03 mM) and then induced to differentiate in high-calcium medium (0.15 mM) (84, 85). As observed previously, the cultures grew to high density with multiple cell types that were morphologically different (86, 87). Approximately 70% of the cells were small with a diameter of ∼23 μm, while the remainder were noticeably larger (∼65 μm in diameter) often forming an apical layer (Fig. 6A, asterisk). These larger cells have been shown previously to be differentiated, with expression of differentiation markers, including keratins K1, and K10, involucrin, profilaggrin, and keratinocyte transglutaminase (86–88). The primary HOK cultures were infected with BCBL-1-derived KSHV, fixed at 24 hpi, and sequentially stained for ORF59 and LANA. An xy projection revealed that the majority of the small cells were latently infected, showing punctate LANA spots in the nucleus with no evidence of ORF59 expression (Fig. 6B, arrowheads). In contrast, cells with the larger size (∼ 5%) showed high levels of nuclear ORF59 and cytoplasmic LANA expression, indicating that the large cells were permissive for KSHV lytic replication (Fig. 6B, arrow). The intensity of the green LANA fluorescence channel was decreased to not overexpose the cytoplasmic LANA fluorescence. This diminished the intensity of the punctate LANA fluorescence in the nucleus. The punctate nuclear LANA staining in small epithelial cells is more clearly observed in images made using higher-intensity green LANA fluorescence (see Fig. S1 in the supplemental material). An xz projection revealed that the smaller latently infected cells adhered to the dish surface, forming a basal layer of cells, while the larger cells with nuclear ORF59 and cytoplasmic LANA expression were located in an apical position on top of the basal layer (Fig. 6C). These results suggest that the increased calcium levels induced epithelial differentiation, leading to an increase in cell size and migration to the apical surface. The differentiated cells showed a cellular phenotype that was permissive for a lytic KSHV infection, resulting in expression of the ORF59 lytic marker and accumulation of LANA in the cytoplasm.
FIG 6.

Cytoplasmic LANA correlates with ORF59 expression after primary infection of differentiated gingival epithelial cells. (A) Primary HOK cultures were induced to differentiate using high calcium (0.15 mM) for 24 h. Cell nuclei were labeled with TO-PRO-3 (blue). The cell borders are indicated with dotted lines distinguishing large-diameter cells (40 to 70 μm), which have been previously shown to express markers of keratinocyte differentiation (labeled with an asterisk) (87) and small cells (15 to 25 μm) showing an undifferentiated phenotype (unlabeled). (B and C) A differentiated HOK culture was infected with KSHV and, at 24 hpi, the culture was fixed and sequentially stained for ORF59, LANA, and cell nuclei, as described in Fig. 4. Panel B shows an xy projection. The arrow identifies a cell with diffuse LANA fluorescence (green) in the cytoplasm and ORF59 fluorescence (red) in the nucleus. The arrowheads indicate cells with punctate LANA fluorescence in the nuclei lacking ORF59 fluorescence. Panel C is an xz projection showing that the cell with both ORF59 and cytoplasmic LANA fluorescence has migrated onto a basal layer of cells exhibiting punctate nuclear LANA fluorescence. Scale bars, 20 μm.
Overexpression of ORF50/RTA in KSHV-infected Vero cells induces the expression of cytoplasmic LANA and nuclear ORF59.
High levels of KSHV replication can be achieved in vitro by overexpressing the viral replication and transactivator (RTA), encoded by ORF50, which is the only viral gene necessary to initiate virus replication (15, 16). To examine the direct effect of RTA on the expression of cytoplasmic LANA, near-confluent cultures of Vero cells were initially infected with BCBL-1-derived KSHV. At 24 hpi, the cultures were mock infected or superinfected with a baculovirus engineered to express KSHV ORF50/RTA (BacK50) (35). After 12 h, the cultures were fixed and sequentially stained for ORF59 and LANA, as described above. Quantitation of the stained cells is shown in Fig. 7E. Regardless of ORF50 expression, KSHV infection of Vero cells was extremely efficient, with >95% of the cells expressing LANA (Fig. 7B and D, green). In the absence of BacK50, approximately 3.4% of the infected cells expressed ORF59 in the nucleus, indicative of a primary KSHV infection (Fig. 7A, red), and all the ORF59-positive cells showed distinct expression of cytoplasmic LANA (Fig. 7B, green). The ORF59-positive nuclei were rendered as red spheres to more clearly indicate these cells in the background of LANA fluorescence. In the presence of BacK50, 35.6% of the KSHV-infected Vero cells were ORF59 positive, indicating that a high-level reactivation of lytic replication was induced by ectopic expression of RTA (Fig. 7C). In all cases, the ORF59-positive cells showed distinct expression of cytoplasmic LANA (Fig. 7D). Occasionally, cells expressing cytoplasmic LANA were observed with no detectable ORF59 expression (Fig. 7B and D, arrowhead). The latter observation suggests that expression of cytoplasmic LANA precedes the nuclear accumulation of ORF59 during activation of the program of lytic replication.
FIG 7.
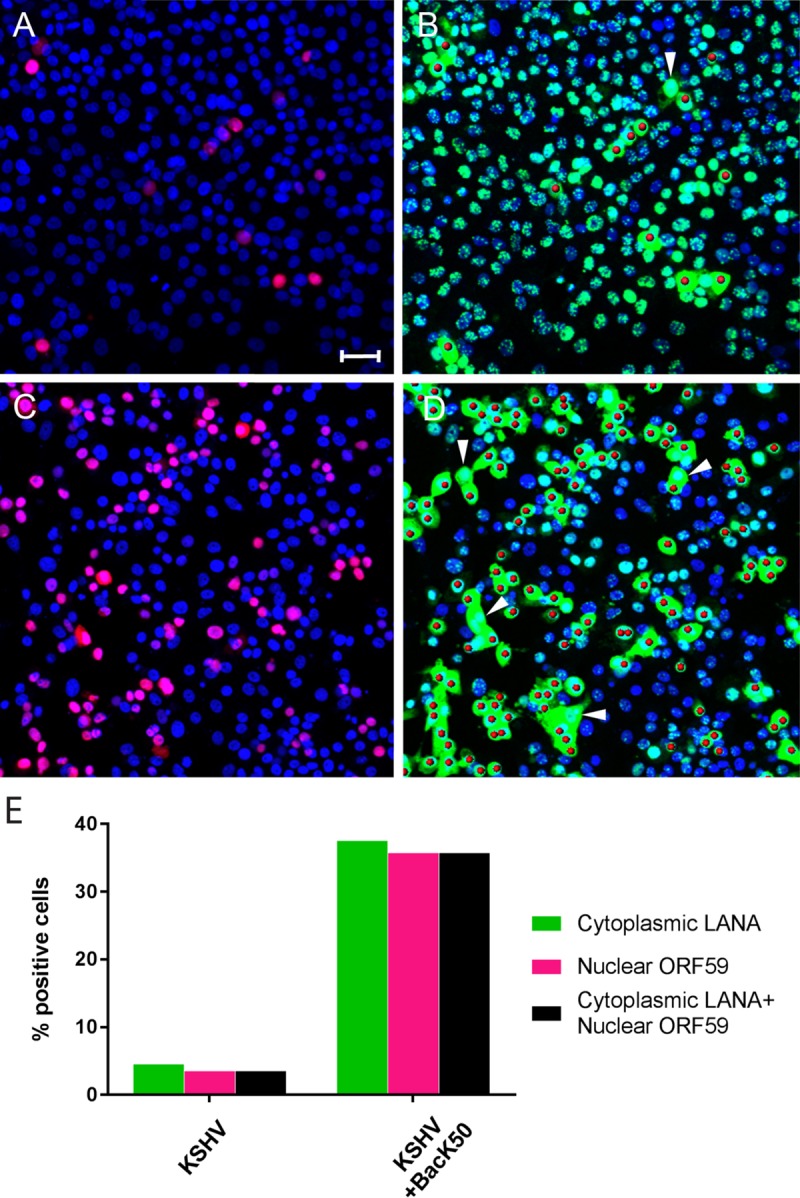
Overexpression of ORF50 RTA in KSHV-infected Vero cells induces cytoplasmic LANA and ORF59 expression. Vero cells were infected with KSHV for 24 h and then were either mock infected (A and B) or superinfected with BacK50 recombinant baculovirus expressing the KSHV ORF50 RTA (6 h) to activate KSHV replication (C and D); the cells were then cultured for an additional 24 h. The cells were fixed and sequentially stained for ORF59, LANA, and cell nuclei, as described in Fig. 4. Panels A and C show the TO-PRO-3 (blue) and ORF59 (red) fluorescence; panels B and D show the TO-PRO-3 (blue) and LANA (green) fluorescence, with the ORF59-positive nuclei rendered as red spheres. The arrowheads indicate cells with cytoplasmic LANA fluorescence and no ORF59 fluorescence. (E) Cell populations expressing cytoplasmic LANA and nuclear ORF59 were quantitated (number of cells analyzed = 375 [KSHV] and 339 [KSHV+BacK50]). Scale bar, 50 μm.
ORF50/RTA is not sufficient to induce cytoplasmic LANA expression and KSHV replication.
We have previously shown that KSHV infection of a gastric epithelial cell line (AGS) results in a basal state of KSHV latency, in which no ORF59-positive spontaneous reactivation is observed (13). This basal state of latency correlates with the absence of active cellular factors, such as SP1, which are necessary for the activation of the ORF50/RTA promoter. Sodium butyrate treatment of AGS cells increases the level of cellular factors responsible for OR50 promoter activity up to the constitutive level seen in Vero and HEK293 cells, but this is not sufficient to induce KSHV replication, in either case (13). AGS cells were infected with the recombinant KSHV strain, rKSHV.219, carrying the puromycin resistance gene (35), and virus-harboring AGS cells were selected using puromycin to establish a high-titer KSHV-infected AGS.219 cell line. A culture of stably infected cells was stained to detect LANA, ORF59, and ORF50/RTA using specific antibodies. Strong expression of LANA with the characteristic punctate pattern was detected in the nuclei of AGS.219 (Fig. 8A); no cytoplasmic LANA was observed, nor did cells express either ORF59 (Fig. 8B) or ORF50 (Fig. 8C). This confirms our previous studies on primary KSHV infection of parental AGS cells and is consistent with a basal state of latency in this cell type (13). Unlike Vero cells (Fig. 7D), when AGS.219 cells were subjected to lytic reactivation protocols (either sodium butyrate treatment or BacK50 transduction) and stained as described above, only punctate nuclear LANA was observed (Fig. 8D and G). Sodium butyrate treatment of AGS.219 cells failed to induce the expression of either ORF59 or ORF50 (Fig. 8E and F). Interestingly, overexpression of ORF50 also failed to induce the expression of ORF59 (Fig. 8H), even though a significant number of superinfected cells expressed high levels of recombinant ORF50 protein (Fig. 8I, arrows). These studies clearly show that KSHV infection of AGS cells is nonpermissive, resulting in a basal latency state that is not responsive to induction by sodium butyrate or ORF50/RTA overexpression. This basal latency state is associated with the absence of cellular transcription factors, such as SP1, that are required for KSHV ORF50/RTA promoter activity (13). In the absence of these cellular factors, neither sodium butyrate nor ORF50/RTA can activate the program of KSHV lytic replication or induce the expression of cytoplasmic LANA.
FIG 8.
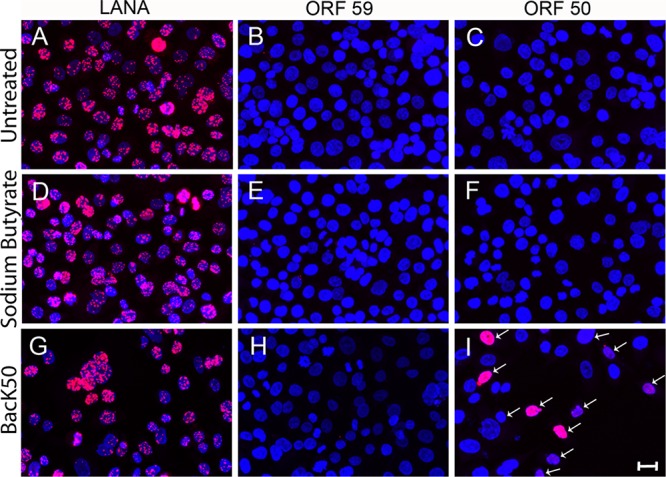
ORF50 RTA or sodium butyrate fails to induce cytoplasmic LANA or ORF59 expression in AGS.219 gastric epithelial cells latently infected with KSHV. The AGS.219 cell line latently infected with recombinant KSHV.219 was either left untreated (A to C) or activated with sodium butyrate (D to F) or BacK50 (G to I) for 6 h. The cells were cultured for an additional 24 h, fixed, and incubated with antibodies to LANA (A, D, and G), ORF59 (B, E, and H), or ORF50 (C, F, and I), followed by TSA enhancement. High levels of punctate, nuclear LANA were detected in both untreated (A) and activated (D and G) cells, but no cytoplasmic LANA was observed. No ORF59 or ORF50 fluorescence was detected in the untreated cultures (B and C, respectively) or the butyrate-activated cultures (E and F, respectively). The BacK50-treated cultured showed ORF50 fluorescence (I) but no ORF59 fluorescence (H). Scale bar, 20 μm.
Detergent extraction of cytoplasmic LANA.
To further analyze the cytoplasmic and nuclear forms of LANA, Vero cells infected with BCBL-derived KSHV and activated with BacK50 were left untreated or treated with 0.1% NP-40 prior to fixation to extract cytoplasmic proteins. In untreated cultures, cells exhibiting either punctate nuclear (arrow) or cytoplasmic (arrowhead) forms of LANA were observed (Fig. 9A), as shown previously. The cells with cytoplasmic LANA also expressed nuclear ORF59 (Fig. 9B and C, arrowhead), while cells with punctate nuclear LANA did not (Fig. 9B and C, arrow). After NP-40 treatment, punctate LANA was still detected in the nucleus of ORF59-negative cells (Fig. 9D to F, arrow). However, in ORF59-positive cells, the cytoplasmic LANA was removed by NP-40 extraction (Fig. 9D to F, arrowhead). These cells still had LANA staining in the nucleus, but this was diffuse rather than punctate (Fig. 9D, arrowhead).
FIG 9.
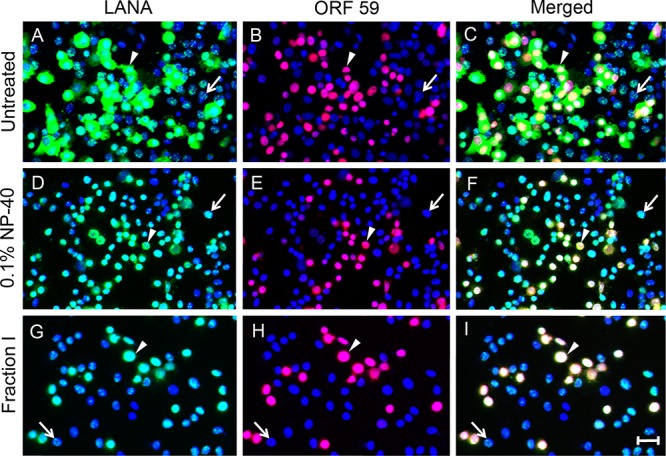
Cytoplasmic LANA is sensitive to mild detergent extraction. Vero cells were infected with KSHV and induced to lytically replicate by superinfection with BacK50 as in Fig. 7 and were either left untreated (A to C) or briefly treated with 0.1% NP-40 (D to F), or the ProteoExtract fraction I extraction buffer (G to I) to solubilize cytoplasmic proteins. The cells were fixed and sequentially stained for LANA, ORF59, and cell nuclei, as in Fig. 4. In the untreated culture (A to C), the arrowheads indicate one of many cells with cytoplasmic LANA (A) and nuclear ORF59 fluorescence (B), merged in panel C. The arrow indicates one of many cells with punctate nuclear LANA (A) and no ORF59 fluorescence (B). After extraction with 0.1% NP-40 or fraction I buffer, the arrowhead indicates one of many cells, in which cytoplasmic LANA was removed (D and G), yet the nuclear ORF59 and LANA remain (E and H; merged in panels F and I). The arrows indicate cells with punctate nuclear LANA (D and G) or no nuclear ORF59 (E and H). Scale bar, 50 μm.
KSHV/BacK50-infected Vero cells were subsequently extracted with a validated cytoplasmic extraction buffer (ProteoExtract subcellular proteome extraction kit, fraction I [Calbiochem]). Similar to 0.1% NP-40 extraction, the fraction I buffer removed the cytoplasmic LANA from the ORF59-positive cells (Fig. 9G to I, arrowhead) but did not alter the punctate nuclear LANA in the ORF59-negative cells (Fig. 9G to I, arrow). Neither buffer extracted nuclear ORF59 (Fig. 9E and H). After extraction, only nuclear LANA was detected in ORF59-positive cells, and this was confined to the nuclear regions containing ORF59 (Fig. 8F and I, arrowhead). A comparison of the intensity of LANA fluorescence revealed a significantly higher level of nuclear LANA in the ORF59-positive cells lytically reactivated by BacK50 (Fig. 9D and G, arrowhead) compared to the unactivated cells harboring a latent infection (Fig. 9D and G, arrow).
Subcellular fractionation of LANA isoforms.
Techniques to isolate proteins in different subcellular fractions have been previously developed (89, 90) and commercialized (ProteoExtract subcellular proteome extraction kit). The fractionation protocol utilizes different detergents and salt concentrations to produce a fraction I (∼35% of total protein) enriched in cytosolic markers, a fraction II (∼50% of total protein) enriched in markers for membrane and organelle proteins, a fraction III (∼5% of total protein) enriched in nuclear proteins, and a fraction IV (∼7 to 10% of total protein) enriched in intermediate filaments, actin, cytoskeleton-associating proteins (89, 90). Vero cells were infected with BCBL-derived KSHV for 24 h and then superinfected with BacK50 expressing recombinant ORF50 for 12 h to induce lytic replication. Infected cultures were then sequentially extracted using the ProteoExtract kit to generate the four different extraction fractions described above. Equivalent amounts of the different fractions were analyzed by Western blotting for the presence of LANA isoforms reacting with the LN53 anti-LANA antibody that targets the internal repeat regions. In cytoplasmic fraction I, four distinct LANA isoforms were detected that migrated with relative molecular masses of 118, 155, 195, and 214 kDa (Fig. 10A); the 118- and 195-kDa bands were the predominant species. Minimal LANA-reactive proteins were detected in the subsequent membrane/organelle fraction II. In nuclear fraction III and cytoskeletal fraction IV, a major LANA isoform migrating with a relative molecular mass of 118 kDa was detected.
FIG 10.

LANA isoforms migrating with high and low relative molecular masses are isolated from the cytosolic fraction of KSHV-infected Vero cells treated with BacK50. Vero cells undergoing a primary KSHV infection (24 hpi) (A to C) and puromycin-selected AGS.219 cells undergoing a long-term latent infection (D) were superinfected with BacK50, as described in Fig. 7. The ProteoExtract fractionation system was used to purify subcellular fractions from the cytosol (lanes I), membrane/organelles (lanes II), nucleus (lanes III), and cytoskeleton (lanes IV) of the infected cells. These fractions were screened by Western blotting for LANA isoforms (A and D) and nuclear lamin β1 (C), as described in Materials and Methods. Dots indicate multiple LANA isoforms ranging from 118- to 230-kDa relative molecular masses in the cytosol fraction I of the induced Vero cells (A) and in the nuclear and cytoskeleton fractions III and IV of the latent AGS.219 cells (D). An asterisk indicates nuclear lamin β1 isoforms in the nuclear and cytoskeleton fractions III and IV of the induced Vero cells (C). (B) Proteins in the cytosol fraction I of the infected Vero cells were further purified on a LANA affinity column for mass spectrometry analysis and screened by Western blotting for LANA isoforms. M, molecular mass standards.
For comparison, the four extracted fractions were analyzed by Western blotting using an antibody for lamin β1, a nuclear protein, which localizes next to the inner nuclear membrane. The major form of lamin β1 (∼70 kDa) was clearly detected in the cytoskeletal fraction IV (asterisk), with multiple minor isoforms in nuclear fraction III (Fig. 10C). No lamin β1 was detected in cytosolic fraction I or the subsequent membrane/organelle fraction II, showing the specificity of the fractionation protocol. In addition, the KSHV-infected AGS.219 cell line was superinfected with BacK50 for 12 h, as described in Fig. 8, and sequentially extracted with the ProteoExtract kit. Equivalent amounts of the different fractions were then analyzed by Western blotting for the presence of LANA isoforms. In cytoplasmic fraction I, a single minor isoform migrating with a relative molecular mass of 118 kDa was detected (Fig. 10D). No LANA was detected in the organelle/membrane fraction II. Major isoforms migrating with relative molecular masses of 100 to 214 kDa were detected in nuclear fraction III, whereas smaller amounts of these isoforms were detected in cytoskeletal fraction IV. In order to detect LANA isoforms in the AGS.219 cytoplasm, the relative amount of extract loaded was increased substantially compared to the Vero cells. Thus, the fraction III lane, which contained the vast majority of the AGS.219 LANA isoforms, was overloaded. Overall, the fractionation analysis demonstrated that the vast majority of LANA in the BacK50-treated AGS.219 cells was nuclear.
Purification and mass spectrometry analysis of cytoplasmic LANA.
Depending on the KSHV strain, the LANA sequence encoded by ORF73 contains 1,003 to 1,162 amino acids, with predicted molecular masses ranging from 103 to 130 kDa, due to the variable presence of repeats in the central portion of the protein (91). However, SDS-PAGE analysis has consistently shown multiple isoforms within the same strain, with higher relative molecular masses of 130 to 250 kDa (7, 10). Originally, the different isoforms were thought to have different molecular masses due to posttranslational modification or proteolytic cleavage, and the higher-than-expected relative molecular masses were believed to be due to aberrant migration in SDS gels of the highly negatively charged stretch of amino acids in the internal repeat region. Subsequently, it was suggested that the decreased sizes of the different LANA isoforms were due to internal ribosome initiation, which generated N-terminally truncated LANA isoforms lacking the NLS, and recombinant LANA isoforms with truncated N termini were shown to accumulate in the cytosol (65).
To characterize the cytoplasmic LANA isoforms detected in KSHV-infected, BacK50-induced Vero cells, we prepared a large quantity of infected and induced Vero cell cultures and used the fraction I buffer of the ProteoExtract kit to obtain a cytoplasmic fraction, as described above. The LANA isoforms were purified on an anti-LANA affinity column, as described in Materials and Methods. Western blot analysis of the purified cytosolic material showed a high level of each of the four LANA isoforms (Fig. 10B), equivalent to that seen in the original extract (Fig. 10A). The purified extract was electrophoresed in a preparative SDS gel and the LANA isoforms were excised in two fractions (Fig. 11A, regions 1 and 2), and submitted for mass spectrometry analysis. Region 1 contained the slowest-migrating LANA isoforms, with relative molecular masses of 155, 195, and 214 kDa, while region 2 contained the fastest-migrating LANA isoform with a relative molecular mass of 118 kDa. Due to the low abundance of the 155- and 214-kDa species, it was not possible to isolate or analyze them separately. Figure 11B shows a schematic of the structure of the 1,117-amino-acid LANA produced by the BCBL-1 KSHV strain used for infection and the position of peptides detected by mass spectrometry analysis.
FIG 11.

Full-length LANA isoforms containing the N-terminal NLS motif are identified in the cytosolic fraction of BacK50-induced KSHV-infected Vero cells. (A) The LANA affinity-purified proteins from the cytosol fraction I of the BacK50-induced, KSHV-infected Vero cells (Fig. 10B) were separated by SDS-gel electrophoresis, and gel regions 1 and 2 containing the different LANA isoforms were analyzed separately by mass spectrometry. (B) A schematic of the BCBL-1 LANA structure (1,117 aa) is shown with N-terminal domain containing the major bipartite NLS (58) and the C-terminal domain separated by a large region of different repeated motifs. The trypsin peptides identified by mass spectrometry in the gel regions 1 and 2 are labeled A-U and are mapped onto the structure. Peptide derivatives are labeled with a prime designation (′) and peptides with modifications are indicated with superscript letter (m, methylation; p, phosphorylation). The number of peptide spectrum matches identified in multiple spectrometry analyses is shown below each peptide. No peptides were detected in the “edd,” “deqq,” and “eqel” repeat regions, which lack obvious trypsin cleavage sites. (C) The positions and sequences of the peptides matching the ORF73 LANA sequence in the BCBL-1 strain of KSHV (NCBI accession no. ADQ57959) used for infection are shown, with the positions of modified amino acids. The confidence level of the spectral calls is indicated (H, high; M, medium; L, low).
In gel region 1, containing the slowest-migrating cytoplasmic LANA isoform, peptides were detected by mass spectrometry across the complete full-length LANA sequence (Fig. 11B and C). The relative number of each detected peptide was similar, ranging from 1 to 5. The N-terminal peptide Am (amino acids 10 to 20), immediately upstream of the bipartite NLS (58), was detected with methylation of arginine, Arg12 (Fig. 11B and C). In addition, N-terminal peptides B (amino acids 33 to 44) and C (amino acids 47 to 55) within and flanking the NLS were detected, as was the C-terminal peptide M (amino acids 1096 to 1117). These results indicate that the cytoplasmic LANA isoforms in this region of the gel were full length and contained the bipartite NLS domain. Additional peptides were detected in the N-terminal and C-terminal regions of LANA, confirming this conclusion (Fig. 11B and C), although the coverage across the entire protein was low. A number of other modified peptides from the N-terminal, central repeat, and C-terminal regions were detected containing methylation of arginines (peptides Gm, Km, and Mmp) and lysines (peptides F′m and F″m) (Fig. 11C). Peptides Gm and Km had methylated arginines in the “epq” domain of the large central repeat region, the only repeat region containing positively charged residues (Fig. 11B). The N-terminal peptide Dp was phosphorylated on serines (Ser186 and Ser187), while the C-terminal peptide Mmp was phosphorylated on threonine Thr960 in addition to methylation on Arg956.
In gel region 2, two instances of peptide Amp, located upstream of the bipartite NLS, were detected with methylation of arginines Arg12 and Arg20 and phosphorylation of serines Ser10 and Ser13 (Fig. 11B and C). Methylation of Arg20 has been previously detected in recombinant LANA overexpressed in 293T cells directed by protein arginine methyltransferase 1 (92). Peptide Fm, containing methylated lysines Lys295 and Lys298, was also detected in the N-terminal region. Additional peptides derived from the “epq” repeat region (peptides G, H, I, and J) and the C-terminal domain (peptides L, N, Q, R, and T) were detected. The C-terminal domain also showed a number of peptides containing phosphorylation of serine and threonine (peptides Mmp, Q/Rp, and R′p) and methylation of arginine (peptides Mmp and Om). Interestingly, more methylated arginine and lysine residues were detected in the slower migrating LANA isoforms in gel region 1 (7 versus 5), while more phosphorylated serine and threonine residues were detected in the faster-migrating isoform in gel region 2 (8 versus 3) (see Fig. S2 in the supplemental material). In summary, our analysis revealed the presence of full-length LANA isoforms in the cytoplasm of Vero cells induced to lytically replicate by BacK50 activation after de novo KSHV infection with posttranslational modifications that could explain differential migration in SDS gels.
DISCUSSION
The accumulation of punctate forms of LANA in the nuclei of KSHV-infected cells is considered to be the general marker of KSHV latency due to its ubiquitous presence in latently infected cells and its functional interactions, which include tethering the KSHV episome to host cell chromatin and blocking expression of the ORF50/RTA protein that is necessary for lytic viral replication. In our study, we demonstrate that LANA isoforms also accumulate in the cytoplasm of KSHV-infected cells that are induced to undergo lytic replication. We have used highly sensitive tyramide signal amplification (TSA) to demonstrate the presence of LANA in the cytoplasm of different cell types using anti-LANA monoclonal antibodies and confocal immunofluorescence microscopy. Although cells latently infected with KSHV and lacking expression of the lytic marker ORF59 exhibit punctate LANA fluorescence in the cell nuclei, infected cells undergoing lytic reactivation by various methods show high levels of nuclear ORF59 expression and high levels of cytoplasmic LANA expression. Since ORF59 expression is highly correlated with viral replication (33, 81, 82), cytoplasmic LANA may play a critical role in establishing an environment conducive to production and transmission of infectious KSHV virions.
We have observed the coexpression of cytoplasmic LANA and nuclear ORF59 during spontaneous reactivation of lytic replication in long-term latently infected endothelial cells, TPA-activation of lytic replication in long-term latently infected PEL cells, and de novo primary lytic infections of both Vero cells induced to migrate and HOK cell cultures induced to differentiate. These phenotypes were observed in cells infected with two strains of KSHV: BCBL-1-derived wild-type virus and recombinant rKSHV.219. We have also confirmed previous reports of cytoplasmic LANA expression in immunoblast-like cells in KSHV-associated MCD lesions (38, 68). MCD lesions have a strong lytic character with a high percentage of infected cells expressing early-, intermediate-, and late-stage KSHV proteins, including ORF59, K2 (vIL-6), K8 (K-bZip), K9 (vIRF-1), K10 (vIRF-4), and the K8.1 glycoprotein (30, 31, 38, 39), thus further correlating cytoplasmic LANA expression and KSHV lytic replication. We attempted to show colocalization of nuclear ORF59 and cytoplasmic LANA in MCD lesions but were unsuccessful in developing techniques for simultaneous retrieval of both LANA and ORF59 antigens in paraffin-embedded MCD lesions. Nevertheless, the strong correlation we observed between cytoplasmic LANA and nuclear ORF59 expression in cultured cells in vitro strongly suggests that the MCD cells expressing lytic antigens also express cytoplasmic LANA.
Cytoplasmic LANA has been reported previously in KSHV-infected BCBL-1 and BC-1 PEL cells (65). In this study by Toptan et al., a clustering of small fluorescent LANA dots was detected in the perinuclear space adjacent to one side of abnormally shaped nuclei. Nuclear and cytoplasmic subcellular fractions prepared from BC-1 cells were probed by Western blotting using an antibody recognizing the internal repeat domain of LANA and an antibody recognizing the N-terminal peptide of LANA (aa 33 to 50). Multiple LANA isoforms with different electrophoretic mobilities were detected in total, nuclear, and cytoplasmic extracts using the internal repeat antibody. Similar isoform patterns were observed in the cytoplasmic and nuclear fractions, with the majority of the different isoforms present in the nuclear fraction. No changes in LANA isoforms were observed after TPA-induction of BC-1 cells. Accurate size estimates for these isoforms were not possible looking at the published gels, but blotting with the N-terminal antibody revealed the presence of the LANA isoform with the highest relative molecular weight in the cytoplasmic fraction. Using truncated recombinant LANA constructs the study explored the possibility that the different electrophoretic mobilities of the LANA isoforms were due to noncanonical translation initiation with the production of truncated proteins lacking the N-terminal region that contains the NLS.
A subsequent study by Zhang et al. followed up on the analysis of cytoplasmic forms of LANA using similar methods (69). Multiple LANA isoforms with electrophoretic mobilities ranging from ∼110 to 230 kDa were detected in BCBL-1 cells. Similar to the Toptan study, all of the isoforms were detected in both cytoplasmic and nuclear fractions, and the majority of the different isoforms were present in the nuclear fraction. Coimmunoprecipitation studies revealed an interaction between cytoplasmic forms of LANA and cGMP-AMP synthase (cGAS), a cytoplasmic DNA sensor, which plays an important role in the host innate immune response through the beta interferon (IFN-β) pathway (93, 94). When BCBL-1 cells were treated with TPA, an increase in all of the LANA isoforms in the cytoplasm was observed, including those with the highest relative molecular weight, suggesting that cytoplasmic isoforms of LANA could facilitate lytic replication by antagonizing cGAS (69). More recently, using immunoprecipitation, cytoplasmic isoforms of LANA were found to bind to Rad50, Mre11, and NBS1, components of the MRN complex, which also functions to sense foreign DNA in the cytoplasm (70). The MRN complex activates the NF-κB pathway in response to cytoplasmic DNA, and cytoplasmic isoforms of LANA antagonize NF-κB activation and NF-κB-dependent suppression of the KSHV lytic cycle.
In our study, BacK50 induction of lytic replication in Vero cells undergoing a primary KSHV infection induced a high level of four major LANA isoforms in the cytoplasm. These cytoplasmic LANA isoforms migrated with relative molecular masses of ca. 118, 155, 195, and 214 kDa and appeared to be newly synthesized, accumulating in the cytoplasm during the 12-h BacK50 activation period. The 118-kDa LANA isoform was the only major isoform detected in the nucleus of these cells. We demonstrated the specificity of the selective ProteoExtract extraction protocol using antibodies to nuclear lamin-β1, which was not detected in the cytoplasmic fraction. In the BacK50-treated AGS.219 cells, the same four LANA isoforms were detected; however, the vast majority of these isoforms were detected in the nuclear fraction. Although we did not observe cytoplasmic LANA fluorescence in the BacK50-treated AGS.219 cells by confocal microscopy, small amounts of the 118-kDa LANA isoform were detected in the cytoplasmic fraction by Western blotting by overloading the SDS-PAGE gel. Our study utilized an immunoprecipitation step to purify cytoplasmic forms of LANA for mass spectrometry analysis, which was similar to that used by Mariggio et al. examining the cytoplasmic LANA interactions with the MRN complex (70). Our mass spectrometry analysis revealed the presence of other proteins coprecipitating with cytoplasmic LANA, which migrated at the same position as LANA in the gel regions that were analyzed. High levels of Rad50 peptides with posttranscriptional modifications, including methylation, phosphorylation, and glycosylation, were detected in gel region 1, suggesting that Rad50 (∼150 kDa) coimmunoprecipitated with cytoplasmic LANA (data not shown).
To obtain sufficient amounts of cytoplasmic LANA to analyze by mass spectrometry, Vero cells were used due to their high reactivation efficiency (95). LANA isoforms were extracted and purified by affinity chromatography using the LN53 monoclonal antibody and preparative gel electrophoresis. Two regions of the gel containing the LANA isoforms were analyzed separately by mass spectrometry. This analysis revealed peptides mapping to the N-terminal and C-terminal domains in both gel regions, suggesting that the 118-, 155-, 195-, and 214-kDa LANA isoforms were full-length proteins with an N-terminal domain containing the bipartite NLS (aa 24 to 47) (58). We did not find any evidence that the different electrophoretic mobilities of the LANA isoforms were due to truncation of LANA at the N or C terminus by mechanisms such as noncanonical translation initiation or caspase cleavage mechanisms, which have been described previously (65, 67).
Because previous studies have shown that LANA undergoes numerous posttranslational modifications (96), we used different strategies to identify phosphorylated, sumoylated, or methylated peptides in the mass spectrometry spectral data of the cytoplasmic isoforms of LANA. Although such analyses are subject to high false discovery rates due to the high number of amino acid combinations yielding peptide sequences that are isobaric to modified peptides of a different sequence, we identified peptide modifications in the cytoplasmic isoforms of LANA, which have been observed previously using different approaches. We detected N-terminal peptides containing methylation of Arg12 and Arg20 in the different cytoplasmic LANA isoforms. Although methylation of Arg12 has not previously been identified, Arg20 has been shown previously to be methylated by protein arginine methyltransferase 1 (92). We detected N-terminal peptides containing phosphorylation of Ser10 and Ser13, flanking the methylated Arg12. Ser10, which is within the chromatin binding domain, was previously shown to be phosphorylated by casein kinase 1 (97). Phosphorylation of the N-terminal (aa 91 to 340) and C-terminal (aa 906 to 1070; BCBL-1 LANA sequence) regions of LANA has been previously detected (98, 99), and our mass spectrometry analysis identified the phosphorylation of serines 186, 187, 1027, 1041, 1051, and 1067 and threonines 960 and 1054 in these regions (see Fig. S2 in the supplemental material). Although the phosphorylation of Ser205 and Ser206 by the PIM kinases has been reported previously (95, 100), we did not detect phosphorylation of these residues in the cytoplasmic LANA isoforms.
By reactivating KSHV-infected cells with BacK50, we were able to induce a high level of cytoplasmic LANA expression for subsequent analysis. As seen in prior studies (65, 69), the highest relative molecular weight LANA isoforms were present in the cytoplasm. In contrast to these studies, however, we determined using mass spectrometry that these isoforms were full-length LANA proteins containing the N-terminal nuclear localization signal and the C-terminal peptide. The presence of full-length LANA isoforms in the cytoplasm after BacK50 activation suggests that nuclear localization of LANA is blocked during the induction of lytic replication in migrating, differentiating, and spontaneously reactivated cells. Taken together, these studies reveal the complexity of LANA expression and function and indicate that our understanding of the role of LANA in the virus life cycle remains incomplete. Studying LANA in different cell types using different methodologies should continue to improve this understanding.
Mitogen-activated protein kinases (MAPKs), including Jun N-terminase kinase (JNK), p38, and Erk, play crucial roles in cell migration through distinct mechanisms (101). Phosphorylation of proteins by these kinases induces cytoskeleton reorganization, maturation of actin-nucleating centers, and the formation of filopodia and lamellipodia during migration. MAPKs have also been shown to play a critical role in the reactivation of KSHV from latency as kinase inhibitors blocked KSHV lytic replication at the early stages of reactivation (102, 103). Furthermore, the N terminus of KSHV LANA is highly susceptible to phosphorylation by MAPK14 and other kinases, which affects both LANA accumulation and function (97). We have shown that the N terminus of LANA contains a bipartite NLS (58), which is flanked by serine and threonine targets of these protein kinases. Phosphorylation of residues within NLSs has been shown to inhibit binding to members of the importin family of nuclear translocation proteins, providing an important regulatory process for altering subcellular trafficking (104). Epstein-Barr virus EBNA1 is a functional homolog of KSHV LANA, and its NLS is closely related to the signal in LANA (58). Both LANA and EBNA1 motifs have a serine immediately adjacent to an arginine in the NLS (Ser29 and Ser383, respectively), which is predicted to bind to the P4 position in the importin α binding site (58). Phosphorylation of the EBNA1 Ser383 downregulates nuclear import of EBNA1 (105). The similarity in the position of Ser29 in KSHV LANA suggests that phosphorylation of this residue could inhibit the nuclear import function of the LANA NLS. We could not determine whether Ser29 was phosphorylated since Ser29 is flanked by numerous arginine and lysine residues, resulting in tryptic peptides too small to identify in our mass spectrometry analysis. In preliminary experiments, we have shown that LANA N-terminal peptides containing a phosphomimetic aspartic acid substitution for Ser29 lose their ability to localize to the nucleus (unpublished observations). Our mass spectrometry analysis detected phosphorylation of Ser10 and Ser13 and methylation of Arg12 and Arg20 adjacent to the bipartite NLS, which could block NLS function and inhibit the nuclear localization of these cytoplasmic LANA isoforms. Although the modification of these residues needs to be confirmed by further analysis, our data suggest that upregulation of the kinases during cellular migration/differentiation could activate pathways critical for KSHV replication and block the nuclear localization of LANA. This would relieve the inhibitory effect of LANA on expression of the KSHV ORF50 replication transactivator, allowing lytic gene expression to proceed, as has been seen for the Pim-1 and Pim-3 kinases (95).
Using confocal immunofluorescence, we observed that cytoplasmic LANA is dispersed across the whole cytoplasm of the infected cell with patchy sometimes fibrillar concentrations of LANA fluorescence, reminiscent of intermediate filament distributions detected by electron microscopy (79). Our subcellular fractionation studies detected high levels of all four major LANA isoforms (118, 155, 195, and 215 kDa) in the cytoplasm. The 118-kDa isoform was also detected not only in the nuclear fraction but also in the cytoskeletal/matrix fraction, which is enriched in intermediate filaments, actin, cytoskeleton-associated factors, and nuclear matrix (89, 90). This suggests that newly synthesized LANA isoforms induced by lytic activation do not interact with the nuclear transport proteins importin α and β but instead localize across the cell cytoplasm in association with cytoskeletal proteins, such as intermediate filaments. The distribution of LANA in the cytoplasm is similar to that seen for cGAS under normal conditions (106), suggesting that full-length LANA may interact with cGAS in association with intermediate filaments.
Our study is the first to report evidence of primary KSHV lytic infections in epithelial cells induced to migrate or differentiate. These primary lytic infections were characterized by the expression of both cytoplasmic LANA and nuclear ORF59 in a large background of latently infected cells with punctate nuclear LANA. Primary epithelial cell cultures induced to differentiate show two cellular morphologies. Most cells are small, undifferentiated basal keratinocytes. Such cells express basal cell markers p63 and basonuclin, lack markers of epithelial cell differentiation, and maintain their proliferative capabilities (107, 108). The remaining cells are large, terminally differentiated keratinocytes, which lose their proliferative potential (87, 109, 110). These cells express markers of differentiation, including involucrin and the keratins K1, K3, and K10 (37, 111, 112). Previous studies observed that KSHV infection of undifferentiated epithelial cells was latent, with punctate nuclear LANA fluorescence and no evidence of lytic gene expression (36). When the latently infected epithelial cells were induced to differentiate by raft culturing or calcium induction, the latent KSHV was activated, leading to the expression of lytic genes and the production of virus (36, 37). Our study extends these findings to show that epithelial cells induced to differentiate are permissive for de novo KSHV lytic infection, characterized by the expression of the ORF59 marker of lytic replication and cytoplasmic LANA 24 h after infection.
Although de novo infection of Vero epithelial cell cultures resulted in latent KSHV infections in the majority of cells, characterized by punctate nuclear LANA and no ORF59 expression, a small variable number of cells were permissive for KSHV infection with high levels of cytoplasmic LANA and nuclear ORF59. Altering the growth conditions to allow cell migration and proliferation resulted in a substantial increase in the number of cells that were permissive for lytic infection. Similarly, within the background of many endothelial cells with long-term latent infections, spontaneous reactivation is observed in a small number of cells resulting in ORF59 expression (28, 113) and virus replication (82). We have shown that this reactivation is characterized by coexpression of nuclear ORF59 and cytoplasmic LANA. This suggests that the Vero and various endothelial cell cultures, while generally displaying a nonpermissive phenotype resulting in a latent infection, can transition to a permissive phenotype through small changes in gene expression induced during migration or proliferation. Thus, these cell types can be considered semipermissive, since at any time a small number of cells are permissive for viral replication with ORF59 expression (i.e., spontaneous reactivation). In such semipermissive cells, lytic replication can be experimentally induced with sodium butyrate or phorbol esters or by ectopic expression of recombinant ORF50/RTA. In contrast, the undifferentiated basal HOK cells and the gastric AGS epithelial cell line, while being susceptible to latent KSHV infections, are strictly nonpermissive for KSHV replication. De novo infection is characterized by the absence of ORF59 and cytoplasmic LANA expression, and long-term infected AGS cells show a basal latency state, in which typical activation events such as sodium butyrate or ORF50/RTA are unable to induce cytoplasmic LANA or nuclear ORF59 expression and viral replication.
We have shown that differential permissiveness of cells to KSHV infection correlates with the constitutive level of active transcription factors, such as SP1, which are necessary to activate the ORF50/RTA promoter (13). In AGS cells, the ORF50/RTA promoter is inactive and sodium butyrate or ORF50/RTA is not sufficient to support KSHV replication in infected cells. In semipermissive cells, such as Vero and HEK293 cells, there are sufficient transcription factors to induce a moderate level of ORF50/RTA promoter activity; however, this is insufficient for widespread KSHV replication (13). Without artificial induction by sodium butyrate, TPA, or ectopic ORF50/RTA, only occasional cells, possibly migrating or proliferating cells, achieve sufficient levels of promoter activity to become permissive for KSHV replication. Our studies suggest that large-scale changes in gene expression associated with calcium-induced terminal epithelial cell differentiation can convert nonpermissive undifferentiated HOK cells into cells that are permissive for KSHV replication. Future electron microscopy studies are needed to confirm KSHV replication and virion production in such cells.
Several differences were apparent between the localization of LANA isoforms in Vero cells undergoing a primary de novo KSHV infection in our study compared to BC-1 and BCBL-1 PEL cells carrying long-term latent KSHV infections in the Toptan and Zhang studies. In our study, the infected Vero cells were extracted after a 24-h KSHV infection and a 12-h BacK50 superinfection. The strong increase in LANA immunofluorescence in the BacK50 activated cells compared to the latent cells indicates that the expression of the majority of the LANA isoforms in these cells occurred during the 12-h activation with ORF50/RTA and thus represents newly synthesized proteins. During this time period, all of the major LANA isoforms with relative molecular masses of 118, 155, 195, and 214 kDa accumulated in the cytoplasm. In contrast, the 118-kDa isoform was the only major isoform translocated to the nucleus. The opposite situation was seen in the BacK50-treated AGS.219 cells, where a large amount of all of the LANA isoforms was detected in the nucleus, and only a small amount of the 118-kDa isoform was observed in the cytoplasm. In these cells, LANA had accumulated in the nucleus over the long-term culture, since no significant increase in LANA fluorescence was observed after BacK50 treatment. It is not clear whether the small amount of the 118-kDa isoform accumulated in the cytoplasm after the BacK50 treatment, since this was below the level of detection by confocal analysis. The situation in the long-term latent infections in AGS.219 cells maintained under selection was similar to that seen in the long-term latent infections in BC-1 and BCBL-1 cells in the Toptan and Zhang studies, where all of the major LANA isoforms were present in the nuclear fraction, while the more rapidly migrating isoforms were the major forms detected in the cytoplasm. The origin of the cytoplasmic LANA isoforms in the long-term latent infections is not clear and could be due to newly synthesized LANA, in which nuclear localization was blocked by phosphorylation, or to previously synthesized LANA that was exported from the nucleus. A major nuclear export pathway uses the CRM1/exportin-1 receptor, which interacts with a leucine-rich nuclear export signal (114). Analysis of the KSHV LANA sequence revealed a leucine-rich motif “33CDLGDDLHLQ42” embedded within the linker region of the bipartite N-terminal NLS, which has strong sequence homology to the nuclear export motifs of P53, PKI, HIV rev, and MDM2. We are currently investigating the function of this potential export signal.
Although most activated cells with cytoplasmic LANA are ORF59 positive, a small number of cells show distinct cytoplasmic LANA without ORF59 expression. This suggests the possibility of a temporal expression of the lytic genes after KSHV reactivation with an early accumulation of cytoplasmic LANA after the induction of ORF50/RTA and the subsequent expression of ORF59 and other lytic genes. This phenomenon was only observed in KSHV-infected cells after inducing reactivation by overexpression of recombinant ORF50/RTA in a baculovirus vector. Since the nuclear localization of LANA is believed to be important for the suppression of the ORF50/RTA promoter and downstream lytic gene expression, the inhibition of LANA nuclear localization and/or the induction of LANA nuclear export may play a critical role in KSHV lytic replication. Interactions with cytoplasmic components, such as intermediate filaments and cGAS, could be other ways this important multifunctional protein contributes to the biology and pathology of KSHV.
MATERIALS AND METHODS
Tissue samples.
Tissue slides from archived diagnostic samples of two MCD cases and one KS case from HIV-infected individuals were obtained from the Department of Laboratory Medicine, University of Washington, under IRB approval. The KS sample was a violaceous nodule in the skin of the foot. The MCD samples were from lymph nodes containing multiple loose aggregates of intermediate-sized lambda-restricted plasmacytoid cells, with no evidence of lymphoma. All samples were positive for KSHV by staining for ORF73 LANA.
Cell lines.
Primary HOK cultures prepared from gingival tissue overlying impacted third molar teeth (83) were kindly provided by B. A. Dale. The cells were grown in keratinocyte basal medium (Cambrex Bioscience) with keratinocyte growth medium supplements in 0.03 mM Ca2+. Differentiation was induced by raising the calcium content to 0.15 mM. African green monkey kidney epithelial cells (Vero) were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; 10% DMEM). The gastric adenocarcinoma epithelial (AGS) cell line (ATCC CRL 1739) was provided by N. Salama. AGS cells contained a persistent parainfluenza virus type 5 infection (115), which was cured using Ribivarin (unpublished results). AGS cells were infected with rKSHV.219 carrying the puromycin resistance gene (35), and virus-harboring AGS cells (AGS.219) were selected and cultured in DMEM with 10% FBS and puromycin (2.5 μg/ml). The BCBL-1 cell line, naturally infected with KSHV, was derived from a pleural effusion lymphoma from a patient with HIV disease (26) and cultured in 10% RPMI containing 1.0 mM HEPES and 0.01% 2-mercaptoethanol. The dermal microvascular endothelial cells (DMVECs) immortalized with human papillomavirus E6/E7 were cultured in endothelial-SFM medium (Gibco BRL) as previously described (82).
Immunological reagents.
Rat anti-LANA monoclonal antibody (LN53) and mouse anti-ORF59 were purchased from Advanced Biotechnologies. Rabbit anti-lamin β1 (ab16048) was purchased from Abcam. Goat anti-mouse IgG-HRP and goat anti-rat IgG-HRP were purchased from Jackson ImmunoResearch. Normal goat serum (NGS) was purchased from Sigma. TSA 488, TSA 594, and TO-PRO-3 were purchased from Invitrogen. Biotinylated anti-rat immunoglobulin and streptavidin-biotin-peroxidase complex were purchased from Vector Laboratories. Goat anti-rabbit 800 and goat anti-rat 800 antibodies were purchased from Thermo-Fisher.
Virus preparations.
KSHV virions from culture medium of TPA-treated BCBL-1 cells were concentrated ∼200-fold on a 50% Opti-Prep cushion, as previously described (72). The virus stock was diluted in DMEM/bovine serum albumin (BSA) and infections were performed at ∼1,000 genomes/cell. The BacK50 recombinant baculovirus expressing the KSHV ORF50 replication transactivator (RTA) was obtained from J. Vieira (35), and infection stocks were purified from SF9 insect cells. The virus titer was determined on Vero cells and used at a multiplicity of infection (MOI) of approximately 20 to 40 to ensure infection of the majority of mammalian cells.
Virus infections and reactivations.
Primary HOK cultures (2.5 × 106) were plated in low-calcium (0.03 mM Ca2+) keratinocyte medium for 24 h and then shifted to high-calcium medium (0.15 mM Ca2+) for 24 h to induce differentiation. The HOK cultures were infected with gradient-purified KSHV for 3 h at 37°C, washed, cultured for an additional 24 h in 0.15 mM Ca2+ medium, and fixed in 8% paraformaldehyde for immunofluorescence staining.
Vero cells were plated and cultured for 2 to 5 days to generate different levels of confluence. In some experiments, confluent (day 5) cultures were scratch-wounded using a Pipetman p1000 pipette tip (Gilson) attached to an aspirator. Cells were allowed to migrate for 12 h prior to KSHV infection. The cultures were equilibrated in DMEM containing 0.2% BSA and infected for 3 to 5 h with gradient-purified KSHV at 37°C. The cultures were washed to remove unbound virus and cultured further for 24 h. Nuclear KSHV LANA was routinely detected by immunofluorescence assay in >90% of the cells. Some cultures were fixed and prepared for antibody localization. Other cultures were superinfected with BacK50 (MOI of 20 to 40) for 6 h to activate KSHV replication (35). The cultures were washed and incubated for an additional 24 h prior to fixation. In some experiments, live KSHV/BacK50 coinfected cells were cooled to 4°C in DMEM and extracted with 0.1% NP-40 in phosphate-buffered saline containing protease inhibitors (Cell Signaling Technologies) for 30 min at 4°C, briefly washed with cold DMEM, and fixed before immunofluorescence staining. Alternatively, the infected Vero cells were treated with extraction buffers from a ProteoExtract subcellular proteome extraction kit to prepare different subcellular fractions (Calbiochem).
AGS.219 cells carrying recombinant rKSHV.219 under puromycin selection were activated either with 6 mM sodium butyrate or BacK50 for 6 h, washed, cultured further for 24 h, fixed, and prepared for antibody localization.
DMVECs immortalized with HPV E6/E7 were infected with BCBL-1-derived KSHV. The KSHV-infected DMVEC cells were fixed at day 35 postinfection to localize LANA and ORF59 (82).
Immunofluorescence.
The virus-infected cultures described above were fixed in 8% paraformaldehyde in phosphate-buffered saline for 30 min at 25°C. Free aldehydes were quenched with 50 mM NH4Cl, and cells were permeabilized with 0.5% NP-40–0.2% Tween 20. Endogenous peroxidase activity was inhibited with 3% H2O2. The cells were incubated with 10% NGS, followed by 10% milk containing 1% NGS (Blotto/NGS) to block nonspecific binding. To detect KSHV-infected cells, the rat anti-LANA monoclonal antibody LN53 was diluted in Blotto/NGS (1:200 to 1:500) and incubated with cells for 16 h. The cells were washed and bound antibody was detected with goat anti-rat IgG-HRP (1:100), followed by a 10-min TSA amplification with TSA 488. For colocalization of ORF59 and LANA, the cells were first incubated with mouse anti-ORF59 (diluted 1:200), a goat anti-mouse HRP, and TSA 594, as described above. The cultures were then treated with 1% H2O2 to quench any peroxidase activity, washed, and blocked with Blotto/NGS as described above. LANA was then detected using a goat anti-rat IgG-HRP secondary antibody and TSA 488, as described above (71). The nuclei were stained with TO-PRO-3. LANA and ORF59 were detected in DMVECs using secondary antibodies conjugated to different Alexa dyes. The cells were analyzed by confocal microscopy.
The amount of LANA fluorescence associated with migrating and stationary KSHV-infected Vero cells was quantitated using the Zeiss LSM software histogram function. The stationary and migrating regions of the wounded culture were divided into 100-μm2 regions of interest (ROIs). The stationary region contained 10 ROIs of 100 μm2 each, while the migrating region contained 20 ROIs. The number of cells in each ROI was determined using Imaris 7.3 (Bitplane). The total number of LANA pixels within the ROI was determined using a fluorescent intensity range of 100 to 255. The mean pixel count for each ROI was determined and divided by the cell number per ROI to estimate an average pixel count per cell for each ROI. In some images, nuclear ORF59 was rendered as spheres using Imaris 7.3 (Bitplane). The confocal images were generated on an LSM 5 Pa system (Zeiss) equipped with 40× 1.3 NA and 63× NA 1.4 objective lenses. All figures were maximum projections complied from 30 to 50 sections with intervals ranging from 0.3 to 0.4 μm and a Z distance of 10 to 14 μm (71).
Immunohistochemistry.
Paraffin-embedded sections from a KS skin lesion and lymph node biopsy specimens with MCD were treated with 1% H2O2 in methanol to block endogenous peroxidase, and antigen retrieval was performed in citrate buffer (pH 6.0) for 20 min with steam. The sections were blocked with 5% donkey serum plus 0.3% Triton, followed by avidin-biotin block (Vector Laboratories) for 1 h. Rat anti-LANA (LN53) was diluted 1:300 and incubated with the sections for 16 h at 4°C, followed by incubation with biotinylated anti-rat immunoglobulin (1:500) for 30 min and a streptavidin-biotin-peroxidase complex for 30 min. The peroxidase activity was visualized using diaminobenzidine (Vector Laboratories).
Cell fractionation and purification of cytoplasmic LANA.
Subconfluent Vero cells were infected with gradient-purified KSHV for 6 h, washed, and incubated for 24 h. The primary KSHV-infected Vero cell culture and a culture of AGS.219 cells containing a long-term infection with rKSHV.219 under puromycin selection were superinfected with BacK50, as described above. The cultures were cooled (4°C) and sequentially treated with a ProteoExtract subcellular proteome extraction kit, as specified by the manufacturer (Calbiochem), yielding cytoplasmic proteins (fraction I), membranes and membrane organelle proteins (fraction II), nuclear protein (fraction III), and cytoskeleton proteins (fraction IV). Fractions I to IV were centrifuged as specified, and aliquots were precipitated using the ProteoExtract protein precipitation kit (Calbiochem). The precipitated proteins were solubilized in reducing sample buffer (NuPAGE; Life Technologies), electrophoresed on a 4 to 12% Bis-Tris gel, and transferred to polyvinylidene difluoride membrane, as described previously (116). The membrane was blocked with Blotto/NGS, incubated with rat anti-LANA (1:700) or rabbit anti-lamin β1 (1:1250) for 16 h, washed, and exposed to goat anti-rat 800 (1:2,000) or goat anti-rabbit 800 (1:5,000) in Odyssey blocking buffer (LI-COR) for 1.5 h, and fluorescence was detected by a LI-COR scanner.
In some experiments cytoplasmic LANA was purified on antibody-conjugated magnetic beads. Briefly, subconfluent Vero cultures (eleven T75 flasks) were coinfected with KSHV and BacK50, as described above, and extracted with ProteoExtract extraction buffer I, as described above, to obtain a fraction I extract containing the cytoplasmic LANA. Rat anti-LANA (100 μg) was coupled to protein A (pA) magnetic beads (1 ml) and then chemically cross-linked with BS3, as specified by the manufacturer (Dynal). The fraction I extract was precleared with pA magnetic beads and then incubated with anti-LANA-magnetic beads for 16 h at 4°C. The beads were washed, and the attached proteins were eluted with triethylamine (1:100) for 5 min. The eluates were brought to neutral pH with 1 M Tris HCl. The fraction I extract was cycled over the anti-LANA-magnetic beads seven times to increase the yield of cytoplasmic LANA. The eluates were pooled and concentrated on an Amicon Ultra-2 10K filter (Millipore), diluted into SDS-mercaptoethanol sample buffer, and electrophoresed on a preparative gel. Two regions of the gel were harvested and submitted for mass spectroscopy analysis.
LC-MS/MS analysis.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed with an Easy-nLC 1000 (Thermo Scientific) coupled to an Orbitrap Fusion mass spectrometer (Thermo Scientific). The LC system configured in a vented format consisted of a fused-silica nanospray needle (PicoTip emitter, 75 μm [inner diameter]; New Objective) packed in-house with Magic C18 AQ 100Å reverse-phase media (Michrom Bioresources, Inc.) (26 cm), and a trap (IntegraFrit capillary; 100 μm [inner diameter]; New Objective) containing Magic C18 AQ 200Å (2 cm). The peptide sample was loaded onto the column and separated using a two-mobile-phase system consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The chromatographic separation was achieved over a 107-min gradient from 2 to 50% B (2 to 7% B for 2 min, 7 to 33% B for 90 min, 33 to 50% B for 10 min, and 50% B for 5 min) at a flow rate of 300 nl/min. The mass spectrometer was operated in a data-dependent MS/MS mode over an m/z range of 400 to 1,500. The mass resolution was set to 120,000. The cycle time was set to 3 s, and the most abundant ions from the precursor scan were selected for MS/MS analysis using 27% normalized HCD collision energy and analyzed with an ion trap. Selected ions were dynamically excluded for 30 s. Peptide masses were compared to theoretical masses of tryptic peptides of proteins in the Uniprot_HUM_031914_cRAP_HHV8_Chlorocebus_Sabeus.fasta using Discoverer version 1.4.1.14 (DBVersion:79). This database included the human and African green monkey protein sequences, as well as the HHV8/KSHV protein sequences and the common Repository of Adventitious Proteins (cRAP). Alternatively, the peptide masses were compared to tryptic peptides of the ORF73 LANA sequence (ADQ57959) of the KSHV strain present in the BCBL-1 cell line, used for the infection studies. Oxidation of methionines, mono-, di-, and trimethylation of lysines and arginines and oxidation of mono-, di-, and triphosphorylation of serines and threonines were set as variable modifications, while carbamidomethylation of cysteines was considered a fixed modification. The minimal peptide length was set to six amino acids; a maximum of two missed cleavages were allowed. Phosphorylation sites were localized using the phosphoRS 3.0 node.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge B. Dale for the kind gift of the primary human oral keratinocyte cultures, N. Salama for the gastric adenocarcinoma epithelial (AGS) cell line, J. Vieira for the rKSHV.219 recombinant virus and BacK50 recombinant baculovirus, and the proteomics core at the Fred Hutchinson Cancer Research Center for the mass spectrometry analysis. We also acknowledge J. Staheli for critical reading of the manuscript.
This study was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under awards DE18927 and DE021954.
The content is solely the responsibility of the authors and does not necessarily represent official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01532-17.
REFERENCES
- 1.Schulz TF, Cesarman E. 2015. Kaposi's sarcoma-associated herpesvirus: mechanisms of oncogenesis. Curr Opin Virol 14:116–128. doi: 10.1016/j.coviro.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Watanabe T, Sugaya M, Atkins AM, Aquilino EA, Yang A, Borris DL, Brady J, Blauvelt A. 2003. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen prolongs the life span of primary human umbilical vein endothelial cells. J Virol 77:6188–6196. doi: 10.1128/JVI.77.11.6188-6196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71:715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong W, Wang H, Herndier B, Ganem D. 1996. Restricted expression of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci U S A 93:6641–6646. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:8309–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J Clin Invest 100:2606–2610. doi: 10.1172/JCI119804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss RA, Talbot SJ. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J Hum Virol 1:19–29. [PubMed] [Google Scholar]
- 8.Talbot SJ, Weiss RA, Kellam P, Boshoff C. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84–94. doi: 10.1006/viro.1999.9672. [DOI] [PubMed] [Google Scholar]
- 9.Katano H, Sato Y, Kurata T, Mori S, Sata T. 1999. High expression of HHV-8-encoded ORF73 protein in spindle-shaped cells of Kaposi's sarcoma. Am J Pathol 155:47–52. doi: 10.1016/S0002-9440(10)65097-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol 71:5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol 77:6474–6481. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackbourn DJ, Lennette E, Klencke B, Moses A, Chandran B, Weinstein M, Glogau RG, Witte MH, Way DL, Kutzkey T, Herndier B, Levy JA. 2000. The restricted cellular host range of human herpesvirus 8. AIDS 14:1123–1133. doi: 10.1097/00002030-200006160-00009. [DOI] [PubMed] [Google Scholar]
- 13.DeMaster LK, Rose TM. 2014. A critical Sp1 element in the rhesus rhadinovirus (RRV) RTA promoter confers high-level activity that correlates with cellular permissivity for viral replication. Virology 448:196–209. doi: 10.1016/j.virol.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renne R, Blackbourn D, Whitby D, Levy J, Ganem D. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol 72:5182–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol 78:3601–3620. doi: 10.1128/JVI.78.7.3601-3620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J Virol 79:7453–7465. doi: 10.1128/JVI.79.12.7453-7465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Verma SC, Cai Q, Robertson ES. 2011. The single RBP-Jκ site within the LANA promoter is crucial for establishing Kaposi's sarcoma-associated herpesvirus latency during primary infection. J Virol 85:6148–6161. doi: 10.1128/JVI.02608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol 78:6585–6594. doi: 10.1128/JVI.78.12.6585-6594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol 77:9738–9749. doi: 10.1128/JVI.77.18.9738-9749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong D, Wu NC, Xie Y, Feng J, Tong L, Brulois KF, Luan H, Du Y, Jung JU, Wang CY, Kang MK, Park NH, Sun R, Wu TT. 2014. Kaposi's sarcoma-associated herpesvirus ORF18 and ORF30 are essential for late gene expression during lytic replication. J Virol 88:11369–11382. doi: 10.1128/JVI.00793-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myoung J, Ganem D. 2011. Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells. Virology 413:1–11. doi: 10.1016/j.virol.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller G, Rigsby MO, Heston L, Grogan E, Sun R, Metroka C, Levy JA, Gao SJ, Chang Y, Moore P. 1996. Antibodies to butyrate-inducible antigens of Kaposi's sarcoma-associated herpesvirus in patients with HIV-1 infection. N Engl J Med 334:1292–1297. doi: 10.1056/NEJM199605163342003. [DOI] [PubMed] [Google Scholar]
- 25.Renne R, Lagunoff M, Zhong W, Ganem D. 1996. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J Virol 70:8151–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 27.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 28.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol 76:2440–2448. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ascherl G, Hohenadl C, Monini P, Zietz C, Browning PJ, Ensoli B, Sturzl M. 1999. Expression of human herpesvirus-8 (HHV-8) encoded pathogenic genes in Kaposi's sarcoma (KS) primary lesions. Adv Enzyme Regul 39:331–339. doi: 10.1016/S0065-2571(98)00019-3. [DOI] [PubMed] [Google Scholar]
- 30.Katano H, Sato Y, Kurata T, Mori S, Sata T. 2000. Apr 10. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 269:335–344. [DOI] [PubMed] [Google Scholar]
- 31.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. J Pathol Mar. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Am J Pathol 156:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturzl M, Wunderlich A, Ascherl G, Hohenadl C, Monini P, Zietz C, Browning PJ, Neipel F, Biberfeld P, Ensoli B. 1999. Human herpesvirus-8 (HHV-8) gene expression in Kaposi's sarcoma (KS) primary lesions: an in situ hybridization study. Leukemia 13(Suppl 1):S110–S112. doi: 10.1038/sj.leu.2401323. [DOI] [PubMed] [Google Scholar]
- 33.Bruce AG, Bakke AM, Gravett CA, DeMaster LK, Bielefeldt-Ohmann H, Burnside KL, Rose TM. 2009. The ORF59 DNA polymerase processivity factor homologs of Old World primate RV2 rhadinoviruses are highly conserved nuclear antigens expressed in differentiated epithelium in infected macaques. Virol J 6:205. doi: 10.1186/1743-422X-6-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDowell ME, Purushothaman P, Rossetto CC, Pari GS, Verma SC. 2013. Phosphorylation of Kaposi's sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J Virol 87:8038–8052. doi: 10.1128/JVI.03460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 36.Johnson AS, Maronian N, Vieira J. 2005. Activation of Kaposi's sarcoma-associated herpesvirus lytic gene expression during epithelial differentiation. J Virol 79:13769–13777. doi: 10.1128/JVI.79.21.13769-13777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seifi A, Weaver EM, Whipple ME, Ikoma M, Farrenberg J, Huang ML, Vieira J. 2011. The lytic activation of KSHV during keratinocyte differentiation is dependent upon a suprabasal position, the loss of integrin engagement, and calcium, but not the interaction of cadherins. Virology 410:17–29. doi: 10.1016/j.virol.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 38.Abe Y, Matsubara D, Gatanaga H, Oka S, Kimura S, Sasao Y, Saitoh K, Fujii T, Sato Y, Sata T, Katano H. 2006. Distinct expression of Kaposi's sarcoma-associated herpesvirus-encoded proteins in Kaposi's sarcoma and multicentric Castleman's disease. Pathol Int 56:617–624. doi: 10.1111/j.1440-1827.2006.02017.x. [DOI] [PubMed] [Google Scholar]
- 39.Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. 1997. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am J Pathol 151:1517–1522. [PMC free article] [PubMed] [Google Scholar]
- 40.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- 41.Cotter MA II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 42.An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. 2005. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem 280:3862–3874. doi: 10.1074/jbc.M407435200. [DOI] [PubMed] [Google Scholar]
- 43.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. 2008. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111:4731–4740. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J Biol Chem 277:27401–27411. doi: 10.1074/jbc.M203489200. [DOI] [PubMed] [Google Scholar]
- 45.Liang D, Hu H, Li S, Dong J, Wang X, Wang Y, He L, He Z, Gao Y, Gao SJ, Lan K. 2014. Oncogenic herpesvirus KSHV Hijacks BMP-Smad1-Id signaling to promote tumorigenesis. PLoS Pathog 10:e1004253. doi: 10.1371/journal.ppat.1004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog 10:e1004240. doi: 10.1371/journal.ppat.1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, Kung HJ, Campbell M, Izumiya Y. 2013. Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J Virol 87:6782–6793. doi: 10.1128/JVI.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shamay M, Krithivas A, Zhang J, Hayward SD. 2006. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc Natl Acad Sci U S A 103:14554–14559. doi: 10.1073/pnas.0604469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bubman D, Guasparri I, Cesarman E. 2007. Deregulation of c-Myc in primary effusion lymphoma by Kaposi's sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 26:4979–4986. doi: 10.1038/sj.onc.1210299. [DOI] [PubMed] [Google Scholar]
- 50.Fakhari FD, Jeong JH, Kanan Y, Dittmer DP. 2006. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest 116:735–742. doi: 10.1172/JCI26190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81:10451–10459. doi: 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu J, Verma SC, Murakami M, Cai Q, Kumar P, Xiao B, Robertson ES. 2009. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (KSHV) upregulates survivin expression in KSHV-associated B-lymphoma cells and contributes to their proliferation. J Virol 83:7129–7141. doi: 10.1128/JVI.00397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verma SC, Borah S, Robertson ES. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol 78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friborg J Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the Lana protein of KSHV protects against cell death. Nature 402:889–894. [DOI] [PubMed] [Google Scholar]
- 55.Katano H, Sato Y, Sata T. 2001. Expression of p53 and human herpesvirus-8 (HHV-8)-encoded latency-associated nuclear antigen with inhibition of apoptosis in HHV-8-associated malignancies. Cancer 92:3076–3084. doi:. [DOI] [PubMed] [Google Scholar]
- 56.Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cherezova L, Burnside KL, Rose TM. 2011. Conservation of complex nuclear localization signals utilizing classical and nonclassical nuclear import pathways in LANA homologs of KSHV and RFHV. PLoS One 6:e18920. doi: 10.1371/journal.pone.0018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J Virol 75:3948–3959. doi: 10.1128/JVI.75.8.3948-3959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong LY, Matchett GA, Wilson AC. 2004. Transcriptional activation by the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J Virol 78:10074–10085. doi: 10.1128/JVI.78.18.10074-10085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krithivas A, Young DB, Liao G, Greene D, Hayward SD. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J Virol 74:9637–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J Virol 84:2318–2330. doi: 10.1128/JVI.01097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med 6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 64.Schwam DR, Luciano RL, Mahajan SS, Wong L, Wilson AC. 2000. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J Virol 74:8532–8540. doi: 10.1128/JVI.74.18.8532-8540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toptan T, Fonseca L, Kwun HJ, Chang Y, Moore PS. 2013. Complex alternative cytoplasmic protein isoforms of the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 generated through noncanonical translation initiation. J Virol 87:2744–2755. doi: 10.1128/JVI.03061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canham M, Talbot SJ. 2004. A naturally occurring C-terminal truncated isoform of the latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus does not associate with viral episomal DNA. J Gen Virol 85:1363–1369. doi: 10.1099/vir.0.79802-0. [DOI] [PubMed] [Google Scholar]
- 67.Davis DA, Naiman NE, Wang V, Shrestha P, Haque M, Hu D, Anagho HA, Carey RF, Davidoff KS, Yarchoan R. 2015. Identification of caspase cleavage sites in KSHV latency-associated nuclear antigen and their effects on caspase-related host defense responses. PLoS Pathog 11:e1005064. doi: 10.1371/journal.ppat.1005064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naresh KN, Rice AJ, Bower M. 2008. Lymph nodes involved by multicentric Castleman disease among HIV-positive individuals are often involved by Kaposi sarcoma. Am J Surg Pathol 32:1006–1012. doi: 10.1097/PAS.0b013e318160ed97. [DOI] [PubMed] [Google Scholar]
- 69.Zhang G, Chan B, Samarina N, Abere B, Weidner-Glunde M, Buch A, Pich A, Brinkmann MM, Schulz TF. 2016. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci U S A 113:E1034–E1043. doi: 10.1073/pnas.1516812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mariggio G, Koch S, Zhang G, Weidner-Glunde M, Ruckert J, Kati S, Santag S, Schulz TF. 2017. Kaposi sarcoma herpesvirus (KSHV) latency-associated nuclear antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLoS Pathog 13:e1006335. doi: 10.1371/journal.ppat.1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garrigues HJ, DeMaster LK, Rubinchikova YE, Rose TM. 2014. KSHV attachment and entry are dependent on αVβ3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate. Virology 464-465: 118–133. doi: 10.1016/j.virol.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garrigues HJ, Rubinchikova YE, Rose TM. 2014. KSHV cell attachment sites revealed by ultrasensitive tyramide signal amplification (TSA) localize to membrane microdomains that are upregulated on mitotic cells. Virology 452-453: 75–85. doi: 10.1016/j.virol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kellam P, Bourboulia D, Dupin N, Shotton C, Fisher C, Talbot S, Boshoff C, Weiss RA. 1999. Characterization of monoclonal antibodies raised against the latent nuclear antigen of human herpesvirus 8. J Virol 73:5149–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fronza M, Heinzmann B, Hamburger M, Laufer S, Merfort I. 2009. Determination of the wound healing effect of Calendula extracts using the scratch assay with 3T3 fibroblasts. J Ethnopharmacol 126:463–467. doi: 10.1016/j.jep.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 75.Liang CC, Park AY, Guan JL. 2007. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 76.Walter MN, Wright KT, Fuller HR, MacNeil S, Johnson WE. 2010. Mesenchymal stem cell-conditioned medium accelerates skin wound healing: an in vitro study of fibroblast and keratinocyte scratch assays. Exp Cell Res 316:1271–1281. doi: 10.1016/j.yexcr.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 77.DeBiasio R, Bright GR, Ernst LA, Waggoner AS, Taylor DL. 1987. Five-parameter fluorescence imaging: wound healing of living Swiss 3T3 cells. J Cell Biol 105:1613–1622. doi: 10.1083/jcb.105.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin K, Dai CY, Ricciardi RP. 1998. Cloning and functional analysis of Kaposi's sarcoma-associated herpesvirus DNA polymerase and its processivity factor. J Virol 72:6228–6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Orenstein JM, Ciufo DM, Zoeteweij JP, Blauvelt A, Hayward GS. 2000. Morphogenesis of HHV8 in primary human dermal microvascular endothelium and primary effusion lymphomas. Ultrastruct Pathol 24:291–300. doi: 10.1080/019131200750035012. [DOI] [PubMed] [Google Scholar]
- 80.Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. 2001. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol 75:1487–1506. doi: 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan SR, Bloomer C, Chandran B. 1998. Identification and characterization of human herpesvirus-8 lytic cycle-associated ORF 59 protein and the encoding cDNA by monoclonal antibody. Virology 240:118–126. doi: 10.1006/viro.1997.8911. [DOI] [PubMed] [Google Scholar]
- 82.Moses AV, Fish KN, Ruhl R, Smith PP, Strussenberg JG, Zhu L, Chandran B, Nelson JA. 1999. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol 73:6892–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krisanaprakornkit S, Weinberg A, Perez CN, Dale BA. 1998. Expression of the peptide antibiotic human beta-defensin 1 in cultured gingival epithelial cells and gingival tissue. Infect Immun 66:4222–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. 1980. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 19:245–254. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- 85.Yuspa SH, Ben T, Hennings H. 1983. The induction of epidermal transglutaminase and terminal differentiation by tumor promoters in cultured epidermal cells. Carcinogenesis 4:1413–1418. doi: 10.1093/carcin/4.11.1413. [DOI] [PubMed] [Google Scholar]
- 86.Liu AY, Destoumieux D, Wong AV, Park CH, Valore EV, Liu L, Ganz T. 2002. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J Investig Dermatol 118:275–281. doi: 10.1046/j.0022-202x.2001.01651.x. [DOI] [PubMed] [Google Scholar]
- 87.Watt FM, Green H. 1981. Involucrin synthesis is correlated with cell size in human epidermal cultures. J Cell Biol 90:738–742. doi: 10.1083/jcb.90.3.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yuspa SH, Kilkenny AE, Roop DR, Strickland JE, Tucker R, Hennings H, Jaken S. 1989. Consequences of exposure to initiating levels of carcinogens in vitro and in vivo: altered differentiation and growth, mutations, and transformation. Prog Clin Biol Res 298:127–135. [PubMed] [Google Scholar]
- 89.Ramsby M, Makowski G. 2011. Differential detergent fractionation of eukaryotic cells. Cold Spring Harbor Protoc 2011:prot5592. doi: 10.1101/pdb.prot5592. [DOI] [PubMed] [Google Scholar]
- 90.Ramsby ML, Makowski GS, Khairallah EA. 1994. Differential detergent fractionation of isolated hepatocytes: biochemical, immunochemical and two-dimensional gel electrophoresis characterization of cytoskeletal and noncytoskeletal compartments. Electrophoresis 15:265–277. doi: 10.1002/elps.1150150146. [DOI] [PubMed] [Google Scholar]
- 91.Gao SJ, Zhang YJ, Deng JH, Rabkin CS, Flore O, Jenson HB. 1999. Molecular polymorphism of Kaposi's sarcoma-associated herpesvirus (Human herpesvirus 8) latent nuclear antigen: evidence for a large repertoire of viral genotypes and dual infection with different viral genotypes. J Infect Dis 180:1466–1476. (Erratum, 180:1756.) [DOI] [PubMed] [Google Scholar]
- 92.Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, Luciw PA, Kung HJ, Izumiya Y. 2012. Protein arginine methyltransferase 1-directed methylation of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen. J Biol Chem 287:5806–5818. doi: 10.1074/jbc.M111.289496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. 2009. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5:e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Campbell M, Izumiya Y. 2012. Posttranslational modifications of Kaposi's sarcoma-associated herpesvirus regulatory proteins: SUMO and KSHV. Front Microbiol 3:31. doi: 10.3389/fmicb.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Woodard C, Shamay M, Liao G, Zhu J, Ng AN, Li R, Newman R, Rho HS, Hu J, Wan J, Qian J, Zhu H, Hayward SD. 2012. Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog 8:e1002972. doi: 10.1371/journal.ppat.1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cha S, Lim C, Lee JY, Song YJ, Park J, Choe J, Seo T. 2010. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi's sarcoma-associated herpesvirus latent replication. Biochem Biophys Res Commun 394:934–939. doi: 10.1016/j.bbrc.2010.03.086. [DOI] [PubMed] [Google Scholar]
- 99.Platt GM, Simpson GR, Mittnacht S, Schulz TF. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J Virol 73:9789–9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. 2006. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 101.Huang C, Jacobson K, Schaller MD. 2004. MAP kinases and cell migration. J Cell Sci 117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 102.Pan H, Xie J, Ye F, Gao SJ. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol 80:5371–5382. doi: 10.1128/JVI.02299-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ. 2008. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154. doi: 10.1016/j.virol.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Harreman MT, Kline TM, Milford HG, Harben MB, Hodel AE, Corbett AH. 2004. Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J Biol Chem 279:20613–20621. doi: 10.1074/jbc.M401720200. [DOI] [PubMed] [Google Scholar]
- 105.Kitamura R, Sekimoto T, Ito S, Harada S, Yamagata H, Masai H, Yoneda Y, Yanagi K. 2006. Nuclear import of Epstein-Barr virus nuclear antigen 1 mediated by NPI-1 (importin α5) is up- and down-regulated by phosphorylation of the nuclear localization signal for which Lys379 and Arg380 are essential. J Virol 80:1979–1991. doi: 10.1128/JVI.80.4.1979-1991.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barrandon Y, Green H. 1985. Cell size as a determinant of the clone-forming ability of human keratinocytes. Proc Natl Acad Sci U S A 82:5390–5394. doi: 10.1073/pnas.82.16.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parsa R, Yang A, McKeon F, Green H. 1999. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Investig Dermatol 113:1099–1105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 109.Dazard JE, Piette J, Basset-Seguin N, Blanchard JM, Gandarillas A. 2000. Switch from p53 to MDM2 as differentiating human keratinocytes lose their proliferative potential and increase in cellular size. Oncogene 19:3693–3705. doi: 10.1038/sj.onc.1203695. [DOI] [PubMed] [Google Scholar]
- 110.De Paiva CS, Pflugfelder SC, Li DQ. 2006. Cell size correlates with phenotype and proliferative capacity in human corneal epithelial cells. Stem cells 24:368–375. doi: 10.1634/stemcells.2005-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fuchs E, Green H. 1980. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 19:1033–1042. doi: 10.1016/0092-8674(80)90094-X. [DOI] [PubMed] [Google Scholar]
- 112.Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. 1989. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol 109:1207–1217. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bruce AG, Barcy S, DiMaio T, Gan E, Garrigues HJ, Lagunoff M, Rose TM. 2017. Quantitative analysis of the KSHV transcriptome following primary infection of blood and lymphatic endothelial cells. Pathogens 6:E11. doi: 10.3390/pathogens6010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060. doi: 10.1016/S0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 115.Young DF, Carlos TS, Hagmaier K, Fan L, Randall RE. 2007. AGS and other tissue culture cells can unknowingly be persistently infected with PIV5; a virus that blocks interferon signaling by degrading STAT1. Virology 365:238–240. doi: 10.1016/j.virol.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 116.Hirano H, Watanabe T. 1990. Microsequencing of proteins electrotransferred onto immobilizing matrices from polyacrylamide gel electrophoresis: application to an insoluble protein. Electrophoresis 11:573–580. doi: 10.1002/elps.1150110708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.