

Abstract
Over the past decade, the Rosetta biomolecular modeling suite has informed diverse biological questions and engineering challenges ranging from interpretation of low-resolution structural data to design of nanomaterials, protein therapeutics, and vaccines. Central to Rosetta’s success is the energy function: a model parameterized from small molecule and X-ray crystal structure data used to approximate the energy associated with each biomolecule conformation. This paper describes the mathematical models and physical concepts that underlie the latest Rosetta Energy Function, REF15. Applying these concepts, we explain how to use Rosetta energies to identify and analyze the features of biomolecular models. Finally, we discuss the latest advances in the energy function that extend capabilities from soluble proteins to also include membrane proteins, peptides containing non-canonical amino acids, small molecules, carbohydrates, nucleic acids, and other macromolecules.
Keywords: Rosetta, molecular modeling, energy function, force field, structure prediction, design
Graphical Abstract
Introduction
Proteins adopt diverse three-dimensional conformations to carry out the complex mechanisms of life. Their structures are constrained by the underlying amino acid sequence1 and stabilized by a fine balance between enthalphic and entropic contributions to non-covalent interactions.2 Energy functions that seek to approximate the energy of these interactions are fundamental to computational modeling of biomolecular structures. The goal of this paper is to describe the energy calculations used by the Rosetta macromolecular modeling program:3 we explain the underlying physical concepts, mathematical models, latest advances, and application to biomolecular simulations.
Energy functions are based on Anfinsen’s hypothesis that native-like protein conformations represent unique, low-energy, thermodynamically stable conformations.4 These folded states reside in minima on the energy landscape, and they have a net favorable change in Gibbs free energy, which is the sum of contributions from both enthalpy (ΔH) and entropy (TΔS) relative to the unfolded state. To follow these heuristics, macromolecular modeling programs require a mathematical function that can discriminate between the unfolded, folded, and native-like conformations. Typically, these functions are a linear combination of terms that compute energies as a function of various degrees of freedom.
The earliest macromolecular energy functions combined a Lennard-Jones potential for van der Waals interactions5–7 with harmonic torsional potentials8 that were parameterized using force constants from vibrational spectra of small molecules.9–11 These formulations were first applied to investigating the structures of hemolysin,12 trypsin inhibitor,13 and hemoglobin14 and have now diversified into a large family of commonly used energy functions such as AMBER,15 DREIDING,16 OPLS,17 and CHARMM.18,19 Many of these energy functions also rely on new terms and parameterizations. For example, faster computers have enabled the derivation of parameters from ab initio quantum calculations.20 The maturation of X-ray crystallography and NMR protein structure determination methods has enabled development of statistical potentials derived from per-residue, inter-residue, secondary-structure, and whole structure features.21–28 Additionally, there are alternate models of electrostatics and solvation, such as a Generalized Born approximation of the Poisson-Boltzmann equation29 and polarizable electrostatic terms that accommodate varying charge distributions.30
The first version of the Rosetta energy function was developed for proteins by Simons et al.31 Initially, it used statistical potentials describing individual residue environments and frequent residue-pair interactions derived from the Protein Databank (PDB).32 Later, the authors added terms for packing of van der Waals spheres, hydrogen bonding, secondary-structure, and van der Waals interactions to improve the performance of ab initio structure prediction.33 These terms were for low-resolution modeling, meaning that the scores were dependent on only the coordinates of the backbone atoms and that interactions between the side chains were treated implicitly.
To enable higher resolution modeling, in the early 2000s, Kuhlman et al.34 implemented an all-atom energy function that emphasized atomic packing, hydrogen bonding, solvation, and protein torsion angles commonly found in folded proteins. This energy function first included a Lennard-Jones term35, a pairwise additive implicit solvation model,36 a statistically-derived electrostatics term, and a term for backbone-dependent rotamer preferences.37 Shortly after, several terms were added, including and an orientation-dependent hydrogen bonding term38 in agreement with electronic structure calculations.39 This combination of traditional molecular mechanics energies and statistical torsion potentials enabled Rosetta to reach several milestones in structure prediction and design including accurate ab initio structure prediction.40 hot-spot prediction,41,42 protein—protein docking,43 small molecule docking,44 and specificity redesign45 as well as the first de novo designed protein backbone not found in nature46 and the first computationally designed new protein—protein interface.47
The Rosetta energy function has changed dramatically since it was last described in complete detail by Rohl et al.48 in 2004. It has undergone significant advances ranging from improved models of hydrogen bonding49 and solvation,50 to updated evaluation of backbone51 and rotamer conformations.52 Along the way, these developments have enabled Rosetta to address new biomolecular modeling problems including refinement of low-resolution X-ray structures and use of sparse data,53,54 and the design of vaccines,55 biomineralization peptides,56 self-assembling materials,57 and enzymes that perform new functions.58,59 Instead of arbitrary units, the energy function is now also fitted to estimate energies in kcal/mol. The details of the energy function advances are distributed across code comments, methods development papers, application papers, and individual experts, making it challenging for Rosetta developers and users in both academia and industry to learn the underlying concepts. Moreover, members of the Rosetta community are actively working to generalize the all-atom energy function for use in different contexts60,61 and for all biomolecules including RNA,62 DNA,63,64 small-molecule ligands,65,66 non-canonical amino acids and backbones,67–69 and carbohydrates,70 further encouraging us to reexamine the underpinnings of the energy function. Thus, there is a need for an up-to-date description of the current energy function.
In this paper, we describe the new default energy function, called the Rosetta Energy Function 2015 (REF15). Our discussion aims to expose the physical and mathematical details of the energy function required for rigorous understanding. In addition, we explain how to apply the computed energies to analyze structural models produced by Rosetta simulations. We hope this paper will provide critically needed documentation of the energy methods as well as an educational resource to help students and scientists interpret the results of these simulations.
Computing the total Rosetta energy
The Rosetta energy function approximates the energy of a biomolecule conformation. This quantity, called ΔEtotal, is computed from a linear combination of energy terms Ei which are calculated as a function of geometric degrees of freedom, θ, chemical identities, aa, and scaled by a weight on each term, w, as shown in Eq. 1.
| (1) |
Here, we explain the Rosetta energy function term by term. First, we describe energies of interactions between non-bonded atom-pairs important for atomic packing, electrostatics, and solvation. Second, we explain empirical potentials used to model hydrogen- and disulfide-bonds. Next, we explain statistical potentials used to describe backbone and side-chain torsional preferences in proteins. After, we explain a set of terms that accommodate features not explicitly captured yet important for native structural feature recapitulation. Finally, we discuss how the energy terms are combined into a single function used to approximate the energy of biomolecules. For reference, items in the fixed width font are names of energy terms in the Rosetta code. The energy terms are summarized in Table 1.
Table 1.
Summary of terms in the REF15 energy function for proteins.
| Term | Description | Weight | Units | Ref. |
|---|---|---|---|---|
| fa_atr | Attractive energy between two atoms on different residues separated by distance, d | 1.0 | kcal/mol | [5,6] |
| fa_rep | Repulsive energy between two atoms on different residues separated by distance, d | 0.55 | kcal/mol | [5,6] |
| fa_intra_rep | Repulsive energy between two atoms on the same residue, separated by distance, d | 0.005 | kcal/mol | [5,6] |
| fa_sol | Gaussian exclusion implicit solvation energy between protein atoms in different residues | 1.0 | kcal/mol | [36] |
| lk_ball_wtd | Orientation-dependent solvation of polar atoms assuming ideal water geometry | 1.0 | kcal/mol | [50,71] |
| fa_intra_sol | Gaussian exclusion implicit solvation energy between protein atoms in the same residue | 1.0 | kcal/mol | [36] |
| fa_elec | Energy of interaction between two non-bonded charged atoms separated by distance, d | 1.0 | kcal/mol | [50] |
| hbond_lr_bb | Energy of short range hydrogen bonds | 1.0 | kcal/mol | [38,49] |
| hbond_sr_bb | Energy of long range hydrogen bonds | 1.0 | kcal/mol | [38,49] |
| hbond_bb_sc | Energy of backbone-side chain hydrogen bonds | 1.0 | kcal/mol | [38,49] |
| hbond_sc | Energy of side chain to side chain hydrogen bonds | 1.0 | kcal/mol | [38,49] |
| dslf_fa13 | Energy of disulfide bridges | 1.25 | kcal/mol | [49] |
| rama_prepro | Probability of backbone ϕ,ψ angles given amino acid type | 0.45 kcal/mol/kT | kT | [50,51] |
| p_aa_pp | Probability of amino acid identity given backbone ϕ,ψ angles | 0.4 kcal/mol/kT | kT | [51] |
| fa_dun | Probability that a chosen rotamer is native-like given backbone ϕ,ψ angles | 0.7 kcal/mol/kT | kT | [52] |
| omega | Backbone-dependent penalty for cis ω dihedrals that deviate from 0° and trans ω dihedrals that deviate from 180° | 0.6 kcal/mol/AU | Arbitrary Units (AU) | [72] |
| pro_close | Penalty for an open proline ring and proline ω bonding energy | 1.25 kcal/mol/AU | Arbitrary Units | [51] |
| yhh_planarity | Sinusoidal penalty for non-planar tyrosine χ3 dihedral angle | 0.625 kcal/mol/AU | Arbitrary Units | [49] |
| ref | Reference energies for amino acid types | 1.0 kcal/mol/AU | Arbitrary Units | [1,51] |
Terms for atom-pair interactions
van der Waals interactions are short-range attractive and repulsive forces that vary with atom-pair distance. Whereas attractive forces result from the cross-correlated motions of electrons in neighboring non-bonded atoms, repulsive forces occur because electrons cannot occupy the same orbitals by the Pauli exclusion principle. To model van der Waals interactions, Rosetta uses the Lennard-Jones (LJ) 6–12 potential5,6 which calculates the interaction energy of atoms i and j in different residues given their summed atomic radii σi,j,a atom-pair distance, di,j, and the geometric mean of well depths, εi,j (Eq. 2). The atomic radii and well depths are derived from small molecule liquid phase data optimized in context of the energy model.50
| (2) |
Rosetta splits the LJ potential at the function’s minimum (di,j = σi,j) into two components that can be weighted separately: attractive ( fa_atr) and repulsive ( fa_rep). By decomposing the function this way, we can alter component weights without changing the minimum-energy distance or introducing any derivative discontinuities. Many conformational sampling protocols in Rosetta take advantage of this splitting by slowly increasing the weight of the repulsive component to traverse rugged energy landscapes and to prevent structures from unfolding during sampling.73
The repulsive van der Waals energy, fa_rep, varies as a function of atom-pair distance. At short distances, atomic overlap results in strong forces that lead to large changes in the energy. The steep term can cause poor performance in minimization routines and overall structure prediction and design calculations.74,75 To alleviate this problem, we weaken the repulsive component by replacing the term with a softer linear term when d ≤ 0.6 σi,j. The term is computed using the atom-type specific parameters mi,j and bi,j which are fit to ensure derivative continuity at d = 0.6σi,j After the linear component, the function transitions smoothly to the 6–12 form until di,j = σ, where it reaches zero and remains zero (Eq. 3; Fig. 1A).
Figure 1. Van der Waals and electrostatics energies.

Comparison between pairwise energies of non-bonded atoms computed by Rosetta and the form computed by traditional molecular mechanics force fields. Here, the interaction between the backbone nitrogen and carbon are used as an example. (A) Lennard-Jones van der Waals energy with well-depths εNbb = 0.162 and εCbb = 0.063 and atomic radii rNbb = 1.763 and rCbb = 2.011 (red) and Rosetta fa_rep (blue). (B) Lennard-Jones van der Waals energy (red) and Rosetta fa_atr (blue). As the atom-pair distance approaches 6.0 Å, the fa_atr term smoothly approaches zero and deviates slightly from the original Lennard-Jones potential. (C) Coulomb electrostatics energy with a dielectric constant ε = 10, and partial charges pNbb = −0.604 and qCbb = 0.090 (red) compared with Rosetta fa_elec (blue). The fa_elec model is shifted to reach zero at the cutoff distance 6.0 Å.
| (3) |
Rosetta also includes an intra-residue version of the repulsive component, fa_intra_rep, with the same functional form as the fa_rep term (Eq. 3). We include this term because the knowledge-based rotamer energy ( fa_dun, below) under-estimates intra-residue collisions.
The attractive van der Waals energy, fa_atr has a value of −εi,j when di,j = 0 and then transitions to the 6–12 potential as the distance increases (Eq. 4; Fig. 1B). For speed, we truncate the LJ term beyond 6.0 Å where the van der Waals forces are small. To avoid derivative discontinuities, we use a cubic polynomial function, fpoly(di,j) after 4.5 Å to transition the standard Lennard-Jones functional form smoothly to zero. These smooth derivatives are necessary to ensure that bumps do not accumulate in the distributions of structural features at inflections points in the energy landscape during conformational sampling with gradient-based minimization (Sheffler 2006, Unpublished).
| (4) |
All three terms are multiplied by a connectivity weight to exclude the large repulsive energetic contributions that would otherwise be calculated for atoms separated by fewer than four chemical bonds (Eq. 5). This weight is common to molecular force fields that assume covalent bonds are not formed or broken during a simulation. Rosetta uses four chemical bonds as the “crossover” separation when transitions from zero to one (rather than the three chemical bonds used by traditional force fields) to limit the effects of double-counting due to knowledge-based torsional potentials.
| (5) |
The comparison between Eq. 2 and the modified LJ potential (Eq. 3–4) is shown in Fig. 1A and Fig. 1B.
Electrostatics
Non-bonded electrostatic interactions arise from forces between fully and partially charged atoms. To evaluate these interactions, Rosetta uses Coulomb’s Law with partial charges originally taken from CHARMM and adjusted via a group optimization scheme (Table S3).50 Coulomb’s law is a pairwise term commonly expressed in terms of the distance between atoms i and j (di,j), dielectric constant ε, partial atomic charges for each atom qi and qj, and Coulomb’s constant, C0 = 322 Å kcal/mol e−2 (with e being the elementary charge) (Eq. 6).
| (6) |
To approximate electrostatic interactions in biomolecules, we modify the potential to account for the difference in dielectric constant between the protein core and solvent-exposed surface.76 Specifically, we substitute the constant ε in Eq. 6 with a sigmoidal function ε(di,j) that increases from εcore = 6 to εsolvent = 80 when the atom-pair distance is between 0 Å and 4 Å (Eq. 7–8):
| (7) |
| (8) |
As with the van der Waals term, we make several heuristic approximations to adapt this calculation for simulations of biomolecules. To avoid strong repulsive forces at short distances, we replace the steep gradient with the constant Eelec(dmin) when di,j < 1.45 Å. Next, since the distance-dependent dielectric assumption results in dampened long-range electrostatics, for speed we truncate the potential at dmax = 5.5 Å and we shift the Coulomb curve by subtracting a term to shift the potential to zero at dmax (Eq. 9).
| (9) |
We use cubic polynomials, and to smooth between the traditional form and our adjustments while avoiding derivative discontinuities. The energy is also multiplied by the connectivity weight, (Eq. 5). The final modified electrostatic potential is given by Eq. 10 and compared to the standard form in Fig. 1C.
| (10) |
Solvation
Native-like protein conformations minimize the exposure of hydrophobic side chains to the surrounding polar solvent. Unfortunately, explicitly modeling all the interactions between solvent and protein atoms is computationally expensive. Instead, Rosetta represents the solvent as bulk water based upon the Lazaridis—Karplus (LK) implicit Gaussian exclusion model.36 Rosetta’s solvation model has two components: an isotropic solvation energy, called fa_sol, that assumes bulk water is uniformly distributed around the atoms (Fig. 2A) and an anisotropic solvation energy, called lk_ball_wtd, that accounts for specific waters nearby polar atoms that form the solvation shell (Fig. 2B).
Figure 2. A two component Lazaridis-Karplus solvation model.

Rosetta uses two energy terms to evaluate the desolvation of protein side chains: an isotropic ( fa_sol) and anisotropic ( lk_ball_wtd) term. (A) and (B) demonstrate the difference between isotropic and anisotropic solvation of the NH2 group by CH3 on the asparagine side chain. The contours vary from low energy (blue) to high energy (yellow). The arrows represent the approach vectors for the pair potentials shown in C-E. In the bottom panel, we compare fa_sol, lk_ball and lk_ball_wtd energies for the solvation of the NH2 group on asparagine for three different approach angles: (C) in line with the 1HD2 atom, (D) along the bisector of the angle between 1HD1 and 1HD2 and (E) vertically down from above the plane of the hydrogens (out of plane).
The isotropic (Lazaridis-Karpus) model36 is based on the function fdesolv that describes the energy required to desolvate (remove contacting water) an atom i when approached by a neighboring atom j. In Rosetta, we exclude Lazaridis-Karplus’ ΔGref term because we implement our own reference energy (discussed later). The energy of the atom-pair interaction varies with separation distance di,j, experimentally determined vapor-to-water transfer free energies ΔGfree, summed atomic radii σi,j, correlation length λ, and atomic volume of the desolvating atom Vj (Eq. 11).
| (11) |
At short distances, fa_rep prevents atoms from overlapping; however, many protocols briefly down-weight or disable the fa_rep term. To avoid scenarios where fdesolv encourages atom-pair overlap in the absence of fa_rep, we smoothly increase the value of the function to a constant at close distances when the van der Waals spheres overlap (di,j = σi,j). At large distances, the function asymptotically approaches zero; therefore, we truncate the function at 6.0 Å for speed. We also transition between the constants at short and long distances using distance-dependent cubic polynomials and with constants c0 = 0.3 Å and c1 = 0.2 Å that define a window for smoothing. The overall desolvation function is given by Eq. 12.
| (12) |
The total isotropic solvation energy (Eq. 13), fa_sol, is computed as a sum including atom j desolvating atom i and vice-versa and scaled by the previously-defined connectivity weight.
| (13) |
Rosetta also includes an intra-residue version of the isotropic solvation energy, fa_intra_sol, with the same functional form as the fa_sol term (Eq. 13).
A recent innovation (2016) is the addition of an energy term ( lk_ball_wtd) to model the orientation-dependent solvation of polar atoms. This anisotropic model increases the desolvation penalty for occluding polar atoms near sites where waters may form hydrogen bonding interactions. For polar atoms, we subtract off part of the isotropic energy of Eq. 13 and then add the anisotropic energy to account for the position of the desolvating atom relative to hypothesized water positions.
To compute the anisotropic energy, we first calculate the set of ideal water sites around atom i, 𝒲i = {νi1, νi2,…}. This set contains 1 to 3 water sites, depending on the atom type of atom i. Each site is 2.65 Å from atom i and has an optimal hydrogen-bond geometry, and we consider the potential overlap of a desolvating atom j with each water. The overlap is considered negligible until the van der Waals sphere of the desolvating atom j (radius σj) touches the van der Waals sphere of the water at site k (radius σw), and then the term smoothly increases over a zone of partial overlap of approximately 0.5 Å. Thus, for each water site, k, with coordinates νj,k, we compute an occlusion measure to capture the gap between the hypothetical water and the desolvating atom j (Eq. 14), using the offset Ω = 3.7 Å2 to Table provide the ramp-up buffer.
| (14) |
Next, we find the soft minimum of over all water sites in 𝒲i by computing the log-average:
| (15) |
Then, and Ω are used to compute a damping function flkfrac (Eq. 16) that varies from zero when the desolvating atom is at least a van der Waals distance from any preferred water site to one when the desolvating atom overlaps a water site by more than ~ 0.5 Å.
| (16) |
We calculate the anisotropic energy of desolvating a polar atom Elk_ball by scaling the desolvation function gdesolv by the damping function flkfrac and an atom-type specific weight waniso that is typically ~0.7 (Eq. 17). The amount of isotropic solvation energy subtracted is gdesolv multiplied by wiso, where wiso is an atom-type specific weight typically ~0.3 (Eq. 18; the total weight on the isotropic contribution through both fa_sol and lk_ball_wtd terms is thus ~0.7). The isotropic and anisotropic components are then summed to yield a new desolvation function, hdesolv (Eq. 19).
| (17) |
| (18) |
| (19) |
Like fa_sol, the energy of desolvating atom i by atom j and then j by i are summed to yield the overall lk_ball_wtd energy (Eq. 20) but only counting the desolvation of polar, hydrogen-bonding heavy atoms (O,N) defined as the set ℘. Fig. 2 shows a comparison between fa_sol, the lk_ball term (Eq. 17), and the sum of fa_sol and lk_ball_wtd for the example of an asparagine NH2 desolvated from three different approach angles. As the approach angle varies, the sum of lk_ball_wtd and fa_sol creates a larger desolvation penalty when waters sites are occluded and a smaller penalty otherwise, relative to the fa_sol term alone.
| (20) |
Hydrogen bonding
Hydrogen bonds are partially covalent interactions that form when a nucleophilic heavy atom donates electron density to a polar hydrogen.77 At short ranges (< 2.5 Å), they exhibit geometries that maximize orbital overlap.78 The interactions between hydrogen bonding groups are also partially described by electrostatics. While this hybrid covalent-electrostatic character is complex, it is crucial for capturing the structural specificity that underlies protein folding, function, and interactions.
Rosetta calculates the energy of hydrogen bonds using fa_elec and a hydrogen bonding model that evaluates energies based on the orientation preferences of hydrogen bonds found in high-resolution crystal structures.38,49 To derive this model, we curated intra-protein polar contacts from ~8,000 high resolution crystal structures (Top8000 dataset79) and identified features using adaptive density estimation. We then empirically fit the functional form of the energy such that the Rosetta-generated polar contacts mimic the distributions from Top8000. The resulting hydrogen bonding energy is evaluated for all pairs of donor hydrogens, H, and acceptors, A, as a function of four degrees of freedom (Fig. 3A): (1) the distance between the donor and acceptor, dHA (2) the angle formed by the donor, acceptor, and donor-heavy atom, θAHD (3) the angle formed by the acceptor’s parent atom (“base”) B, acceptor, and the donor, θBAH and (4) the torsion, ϕB2BAH, formed by the donor, acceptor, and two subsequent parent atoms B and B2. (Fig. 3A). B, the parent atom of A, is the first atom on the shortest path to the root atom (e.g. Cα). The B2 atom of A is the parent atom of B (e.g., the sp2 plane is defined by B2, B, and A). For convenience, the hydrogen bonding energy is subdivided into four separate terms: long range backbone hydrogen bonds ( hbond_lr_bb), short range backbone hydrogen bonds ( hbond_sr_bb), hydrogen bonds between backbone and side chain atoms ( hbond_bb_sc), and hydrogen bonds between side chain atoms ( hbond_sc).
Figure 3. Orientation-dependent hydrogen bonding model.

(A) Degrees of freedom evaluated by the hydrogen bonding term: acceptor—donor distance, dHA, angle between the base, acceptor and hydrogen θEAH, angle between the acceptor, hydrogen, and donor, θAHD, and dihedral angle corresponding to rotation around the base—acceptor bond, ϕB2BAH. (B) Lambert-azimuthal projection of the energy landscape for an sp2 hybridized acceptor.49 (C) energy landscape for an sp3 hybridized acceptor. Example energies for the histidine imidazole ring acceptor hydrogen bonding with a protein backbone amide: (D) energy vs. the acceptor—donor distance, (E) energy vs. the acceptor-hydrogen-donor angle, (F) energy vs. the base-acceptor—hydrogen angle, .
To avoid over-counting, side-chain to backbone hydrogen bonds are excluded if the backbone group is already involved in a hydrogen bond. For speed, the component terms have simple analytic functional forms (Fig. 3B–F; Supporting Information Eq. S1–7). The term is also multiplied by two atom-type specific weights, WH and WA, that account for the varying strength of hydrogen bonds. The overall model is given by Eq. 21 where the term depends on the orbital hybridization of the acceptor, ρ. Finally, the function is also smoothed with f(x) (Eq. 22) to avoid derivative discontinuities and ensure that edge-case hydrogen bonds are considered.
| (21) |
| (22) |
Disulfide bonding
Disulfide bonds are covalent interactions that link sulfur atoms in cysteine residues. Typically, in Rosetta, we rely on a tree-based kinematic system3,80 to keep bond lengths and angles fixed so that we may sample conformation space changing only torsions. For this reason, we do not generally need terms that evaluate bond-length and bond-angle energetics. However, with disulfide bonds and proline (below), the extra bonds cannot be represented with a tree (since a tree graph is acyclic), and thus must be treated explicitly. Thus, disulfide bonds are a special case of inter-residue covalent contact that requires a representation with more degrees of freedom. To evaluate disulfide bonding interactions, Rosetta identifies pairs of cysteines that have covalent bonds linking the Sγ atoms. Then, Rosetta computes the energy of these interactions using an orientation-dependent model called dslf_fa13.49 The model was derived by curating intra-protein disulfide bonds from Top8000 and identifying features using kernel density estimates. For speed, the feature distributions are modeled using skewed Gaussian functions and a mixture of 1, 2, and 3, von Mises functions (Supporting Information Eq. S8–11).
The overall disulfide energy is computed as a function of six degrees of freedom (Fig. 4) that map to four component energies. First, the geometry of the sulfur-sulfur distance dSS is evaluated by . Second, the angle formed by either Cβ1 or Cβ2 with S—S bond is evaluated by . Third, the dihedral formed by either Cα1Cβ1 or Cα2Cβ2 with the S—S bond is evaluated by . Finally, the dihedral formed by Cβ1,Cβ2 and the S—S bond is evaluated by . The complete disulfide bonding energy evaluated for all S-S pairs is given by Eq. 23.
Figure 4. Orientation-dependent disulfide bonding model.

(A) Degrees of freedom evaluated by the disulfide bonding energy: sulfur—sulfur distance, dSS, angle between the β-carbon and two sulfur atoms, θCSS, dihedral corresponding to rotation about the α -Carbon and sulfur bond ϕCαS, and dihedral corresponding to rotation about the S—S bond ϕSS. (B) , (C) (D) (E) .
| (23) |
Terms for Protein Backbone and Side Chain Torsions
Rosetta evaluates backbone and side-chain conformations in torsion space to greatly reduce the search domain and increase computational efficiency. Traditional molecular mechanics force fields describe torsional energies in terms of sines and cosines which have at times performed poorly at reproducing the observed backbone-dihedral distributions in unstructured regions.81 Instead, Rosetta uses several knowledge-based terms for torsion angles that are fast approximations of quantum effects and more accurately model the preferred conformations of protein backbones and side-chains.
Ramachandran
To evaluate backbone ϕ and ψ angles, we defined an energy term called rama_prepro based on Ramachandran maps for each amino acid, using torsions from 3,985 protein chains with a resolution ≤ 1.8 Å, R-factor ≤ 0.22 and sequence identity ≤ 50%.82 Amino acids with low electron density (in the bottom 25th percentile of each residue type) were removed from the data set. The resulting ~581,000 residues were used in adaptive kernel density estimates52 of Ramachandran maps with a grid step of 10° for both ϕ and ψ. Residues preceding proline are also treated separately because they exhibit distinct ϕ,ψ preferences due to steric interactions with the proline’s Cδ.83 The energy, called rama_prepro, is then computed by converting the probabilities to energies at the grid points via the inverted Boltzmann relation84 (Eq. 24; Fig 5). The energies are then evaluated using bicubic interpolation. The Supporting Information includes a detailed discussion of why interpolation is performed on the backbone torsional energies rather than the probabilities (Fig. S3, Eqs. S12–13).
Figure 5. Backbone torsion energies.

The backbone-dependent torsion energies are demonstrated for the lysine residue. (A) The ϕ angle is defined by the backbone atoms Ci–1 – N – Ca – C and the ψ angle is defined by N – Ca – C – Ni+1. (B) rama_prepro energy of lysine without a proline at i-1. (C) rama_prepro energy of lysine with a proline at i-1. (D) p_aa_pp energy of lysine.
| (24) |
Backbone design term
Rosetta also computes the likelihood of placing a specific amino acid side chain given an existing ϕ,ψ backbone conformation. This term, called p_aa_pp represents the propensity of observing an amino acid relative to the other 19 canonical amino acids.85 The knowledge-based propensity, P(aa|ϕ,ψ) (Eq. 25) was derived using the adaptive kernel density estimates for P(ϕ,ψ|aa) and Bayes’ rule. The equation for p_aa_pp is given in Eq. 26 (Fig. 5D).
| (25) |
| (26) |
Side-chain conformations
Protein side chains mostly occupy discrete conformations (rotamers) separated by large energy barriers. To evaluate rotamer conformations, Rosetta derives probabilities from the 2010 backbone-dependent rotamer library (dunbrack.fccc.edu/bbdep2010), which contains the frequencies, means, and standard deviations of individual χ angles for each χ angle k of each rotamer of each amino acid type.52 The probability has three components: (1) observing a specific rotamer given the backbone dihedral angles (2) observing specific χ angles given the rotamer and (3) observing the terminal χ angle distribution, which is either Gaussian-like or continuous when the terminal χ angle is sp2 hybridized (Eq. 27). Here, T represents the number of rotameric χ angles + 1.
| (27) |
The 2010 rotamer library distinguishes between rotameric and non-rotameric torsions. A torsion is rotameric when the third of the four atoms defining the torsion is sp3 hybridized (i.e. preferring ~60°, ~180° and ~−60°, with steep energy barriers between the wells), If the last χ torsion is rotameric, probability p(χT|ϕ,ψ,rot,aa) is fixed at one. On the other hand, a torsion is non-rotameric if its third atom is sp2 hybridized: the library describes its probability distribution continuously, instead. The category of semi-rotameric amino acids with both rotameric and non-rotameric dihedrals encompasses eight amino acids: Asp, Asn, Gln, Glu, His, Phe, Tyr, and Trp.86
The probability of each rotamer p(rot|ϕ,ψ,aa) is derived from the same dataset as the Ramachandran maps described above. The probabilities were identified using adaptive kernel density estimation and the same dataset is used to estimate the mean and standard deviation for each χ dihedral in the rotamer, and μχk and σχk, as functions of the backbone dihedrals, allowing us to compute a probability for the χ values using Eq. 28.
| (28) |
This formulation is reminiscent of the Gaussian distribution, except that it is missing the normalization coefficient of (2πσχk(ϕ,ψ|rot,aa))−1/2. After taking the log of this probability, the term resembles Hooke’s law where the spring constant is given by .
The full form of fa_dun is given by Eq. 29 as a sum over all residues r. The difference between the rotameric- and semi-rotameric models is also shown in Fig. 6.
Figure 6. Energies for side-chain rotamer conformations.
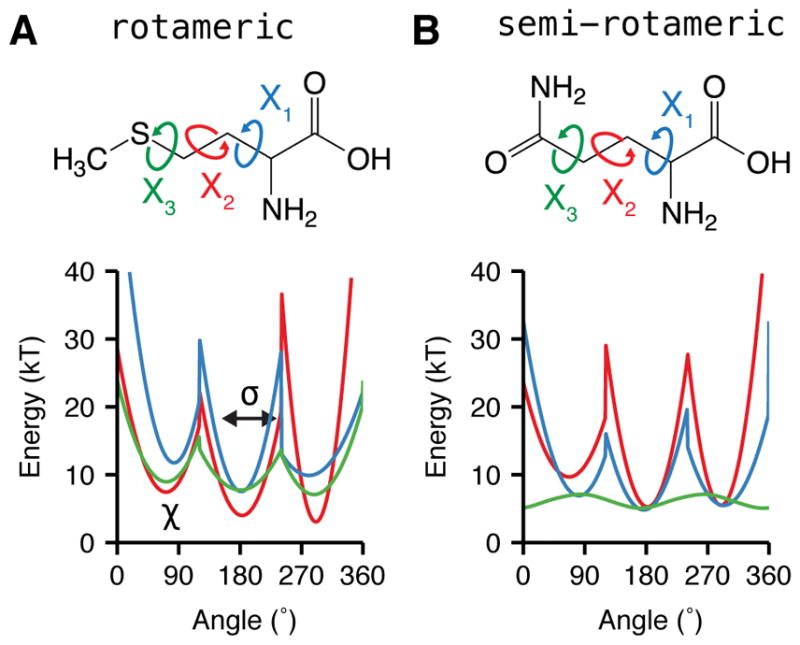
The Dunbrack rotamer energy, fa_dun, is dependent on both the ϕ and ψ backbone torsions and the χ side-chain torsions. Here, we demonstrate the variation of fa_dun when the backbone is fixed in an α-helical conformation with ϕ = −57° and ψ = −47°, and the χ values can vary. χ1 is shown in blue, χ2 shown in red and χ2 shown in green. (A) χ-dependent Dunbrack energy of methionine with an sp3-hybridized terminus (B) χ-dependent energy of glutamine with an sp2-hybridized χ2 terminus. χ1, χ2 and χ2 of methionine and χ1 and χ2 of glutamine express rotameric behavior while χ2 of the latter expresses broad non-rotameric behavior.
| (29) |
The energy from –ln(P(rotr|ϕr,ψr,aar)) is computed using bicubic-spline interpolation; P(χTr,r|ϕr,ψr,rotr,aar) is computed using tricubic-spline interpolation. To save memory, μχk(ϕr,ψr,rotr,aar), and σχk(ϕr,ψr,rotr,aar) are computed using bilinear interpolation, though this has the effect of producing derivative discontinuities at the (ϕ,ψ) grid boundaries. These discontinuities, however, do not appear to produce noticeable artifacts.51
Terms for special case torsions
Peptide bond dihedral angles, ω, remain mostly fixed in a cis- or trans- conformation and depend on the backbone ϕ and ψ angles. Since the electron pair on the backbone nitrogen donates electron density to the electrophilic carbonyl carbon, the peptide bond has partial double bond character. To model this barrier to rotation, Rosetta implements a backbone-dependent harmonic penalty centered near 0° for cis and 180° for trans (Fig. 7A). This energy, called omega, is evaluated on all peptide bonds in the biomolecule (Eq. 30). The means and standard derivations of ω, μω and σω, respectively, are backbone (ϕ,ψ) dependent, as given by kernel regressions of ω on ϕ and ψ.72
Figure 7. Special case torsion energies.

Rosetta implements three additional energy terms to model torsional degrees of freedom with acute preferences. (A) Omega torsion corresponding to rotation about C-N (B) Proline secondary omega torsion corresponding to rotation about C-N related to the C-δ in the ring. (C) Tyrosine terminal χ torsion. (D) Omega energy (E) Proline closure energy (F) Tyrosine planarity energy.
| (30) |
Most Rosetta protocols only search over simple torsions within chains and rigid-body degrees of freedom between chains. However, proline’s side chain requires special treatment because its ring cannot be represented by a kinematic tree.87 Therefore, Rosetta implements a proline closure term, called pro_close (Fig. 7B). There are two components to this energy, shown in Eq. 31. First, there is a torsional potential that operates on the dihedral formed by Or-1–Cr-1–Nr–Cδ,r, called given the observed mean μ ω ′ and standard deviation σ ω ′, where i is the residue index. This term keeps the Cδ atom in the peptide plane. Second, to ensure correct geometry for the two hydrogens bound to Cδ, we build a virtual atom, Nv, off Cδ whose coordinate is controlled by χ2 (Fig. 7B). The pro_close term seeks to align the virtual Nv atom, directly on top of the real backbone nitrogen. The N–Cδ–Cγ bond angle and the N–Cδ bond length are restrained to their ideal values.
| (31) |
Tyrosine also requires special treatment for its χ3 angle because the hydroxyl hydrogen prefers to be in the plane of the aromatic ring.88 To enforce this preference, Rosetta implements a sinusoidal penalty to model the barrier to a χ2 angle that deviates from planarity. This tyrosine hydroxyl penalty is called yhh_planarity (Eq. 32; Fig. 7C).
| (32) |
Terms for modeling non-ideal bond lengths and angles
Cartesian bonding energy
Recently, modeling Cartesian degrees of freedom during gradient-based minimization has been shown to improve Rosetta’s ability to refine low-resolution structures determined by X-ray crystallography and cryo-electron microscopy,53 as well as its ability to discriminate near-native conformations in the absence of experimental data.89 These data suggest that capturing non-ideal bond lengths and angles can be important for accurate modeling of minimum-energy protein conformations. To accommodate, Rosetta now allows these “non-ideal” angles and lengths to be included as additional degrees of freedom in refinement and includes a Cartesian-minimization mode where atom coordinates are explicit degrees of freedom in optimization.
To evaluate the energetics of non-ideal bond lengths, angles and planar groups, an energy term called cart_bonded represents the deviation of these degrees of freedom from ideal using harmonic potentials (Eq. 32–34). Here, di is a bonded-atom-pair distance with di,0 as its ideal distance, θi is a bond angle with θi,0 as its ideal angle, and ϕi is a bond torsion or improper torsion with ϕi,0 as its ideal value and ρi as its periodicity. The ideal bond lengths and angles90,91 were selected based on their ability to rebuild side chains observed in crystal structures (Kevin Karplus & James J. Havranek, unpublished); they were subsequently modified empirically.51 The spring constants for the angle and length terms are from CHARMM32.19 Finally, all planar groups and the Cβ “pseudo-torsion” are constrained using empirically derived values and spring constants:
| (33) |
| (34) |
| (35) |
The function fwrap(x,y) wraps x to the range [0,y). To avoid double counting in the case of Ecart_torsion, the spring constant ki,torsion is zero when the torsion ϕi is being scored by either the rama or fa_dun terms.
Terms for Protein Design
Design reference energy
The terms above are sufficient for comparing different protein conformations with a fixed sequence. However, protein design simulations compare the relative stability of different amino acid sequences given a desired structure to identify models that exhibit a large free energy gap between the folded and unfolded states. Explicit calculations of unfolded state free energies are computationally expensive and error prone. Rosetta therefore approximates the relative energies of the unfolded state ensembles using an unfolded state reference energy, called ref.
Rosetta calculates the reference energy as a sum of individual constant unfolded state reference energies, , for each amino acid, aai (Eq. 36).1
| (36) |
The values are empirically optimized by searching for values that maximize native sequence recovery (discussed below) during design simulations on a large set of high-resolution crystal structures.50,51 During design, this energy term helps normalize the observed frequencies of the different amino acids. When design is turned off, the term contributes a constant offset for a fixed sequence.
Bringing the energy terms together
The Rosetta energy function combines all the terms using a weighted linear sum to approximate free energies (Table 1). Historically, we adjust the weights and parameters to balance the energetic contribution from each term. This balance is important because the van der Waals, solvation, and electrostatics energies partially capture torsional preferences and overlap can cause errors as a result of double counting atomic or residue specific contributions.92 More recently, we fix physics-based terms with weights of 1.0 and perturb other weights and atomic-level parameters using a Nelder-Mead93 scheme to optimize agreement of Rosetta calculations with small-molecule thermodynamic data and high-resolutions structural features.50 The energy function parameters have evolved over the years by optimizing the performance of multiple scientific benchmarks (Table 2).50,51,94 These benchmarks were chosen to test recovery of native-like structural features, ranging from individual hydrogen bond geometries to thermodynamic properties and interface conformations. In addition, and more recently, Song et al.,95 Conway et al.96 and O’Meara et al.46 have fit intra-term parameters to recover features of the experimentally determined folded conformations. An in-depth review of energy function benchmarking can be found in Leaver-Fay et al.97 Table S3 lists the Rosetta database files containing the current full set of physical parameters for each score term.
Table 2.
Common energy function benchmarking methods
| Test | Description | Ref. |
|---|---|---|
| Sequence Recovery | Percentage of the native sequence recovered after backbone redesign | [1,51] |
| Rotamer Recovery | Percentage of native rotamers recovered after full repacking | [51] |
| ΔΔG Prediction | Prediction of free energy changes upon mutation | [98] |
| Loop Modeling | Prediction of loop conformations | [99] |
| High-resolution refinement | Discrimination of native-like decoys upon refinement of ab initio protein models | [100] |
| Docking | Prediction of protein-protein, protein-peptide, or protein-ligand interfaces | [44,101–103] |
| Homology Modeling | Structure prediction incorporating homologous information from templates | [104] |
| Thermodynamic properties | Recapitulation of thermodynamic properties of protein side-chain analogues | [17] |
| Recapitulation of Xtal structure geometries | Recapitulation of features (e.g. atom-pair distance distribution) from high-resolution protein crystal structures | [50] |
Energy Function Units
Initially, Rosetta energies were expressed in a generic unit, called the Rosetta Energy Unit (REU). This choice was made because some original Rosetta energy terms were not calibrated with experimental data, and the use of statistical potentials convoluted interpretation of the energy. Over time, the physical meaning of Rosetta energies has been extensively debated within and outside the community, and several steps have been taken to clarify interpretation. The most recent energy function (REF15) was parameterized on high resolution protein structures and small molecule thermodynamic parameters that were measured in kcal/mol.50 The optimization data show a strong correlation between the experimental data and values predicted by Rosetta (ΔΔG upon mutation, R = 0.994; small molecule ΔHvap; Fig. S2). As a result, Rosetta energies are now a stronger approximation of energies in units of kcal/mol. Therefore, as is standard practice for molecular force fields such as OPLS, CHARMM, and AMBER, we now also express energies in kcal/mol.
Energies in action: Using individual energy terms to analyze Rosetta models
Rosetta energy terms are mathematical models of the physics that governs protein structure, stability, and association. Therefore, the decomposed relative energies of a structure or ensemble of structures can expose important details about the biomolecular model. Now that we have presented the details of each energy term, we here demonstrate how energies can be applied to detailed interpretations of structural models. In this section, we discuss two common structure calculations: (1) estimating the free energy change (ΔΔG) of mutation98 and (2) modeling the structure of a protein-protein interface.102
ΔΔG of mutation
The first example demonstrates how Rosetta can be used to estimate and rationalize thermodynamic parameters. Here, we present an example ΔΔG of mutation calculation for the T193V mutation in the RT-RH derived peptide bound to HIV-1 protease (PDB 1kjg, Fig. 8A).105 The details of this calculation are provided in the Supporting Information.
Figure 8. Structural model of the HIV-1 protease bound to the T4V mutant RT-RH derived peptide.

(A) Structural model of the native HIV-1 peptidase (teal and dark blue), bound to the native peptide (gray) superimposed onto the T4V mutant peptide (magenta). (B) Contributions greater than + 0.1 kcal/mol to the ΔΔG of mutation for T4V. The remaining contributions are: dslf_fa13 = 0 kcal/mol, hbond_lr_bb = −0.09 kcal/mol, hbond_bb_sc = −0.05, hbond_sc = −0.0104, fa_intra_rep = 0.01, fa_intra_sol = −0.07, and yhh_planarity = 0. (C) Hydrophobic patch of residues surrounding position four on the RT-RH peptide.
Rosetta calculates the ΔΔG of the T193V mutation to be −4.95 kcal/mol, and the experiment105 measured −1.11 kcal/mol. Both the experiment and calculation reveal that T193V is stabilizing: yet, these numbers alone do not reveal which specific interactions are responsible for the stabilization. To investigate, we used various analysis tools accessible in PyRosetta106 to identify important energetic contributions to the total ΔΔG. First, we decomposed the ΔΔG into individual energy terms and observe the balance of terms, both favorable and unfavorable, that sum to the total (Fig. 8B). To decompose the most favorable term, Δ fa_sol, we used the print_residue_pair_energies function to identify residues that interact with the mutation site (in this case, residue 4) to produce a nonzero residue pair solvation energy. With the resulting table, we found a hydrophobic pocket around the mutation site formed by residues V27, I45, G46, and I80 on HIV peptidase and residue F194 on the peptide made a large (> 0.05 kcal/mol) and favorable contribution to the change in solvation energy (Fig. 8C).
We further investigated this result on the atomic level with the function print_atom_pair_energy_table by generating atom-pair energy tables (Supporting Information) for residues 5, 27, 45, 46, and 80 against both threonine and valine at residue 193 (Example for residue 80 in Table 3). Here, we find that the specific substitution of the polar hydroxyl on threonine with nonpolar alkyl group on valine stabilizes the peptide in the hydrophobic protease pocket. This result is consistent with chemical intuition and demonstrates how breaking down the total energies can provide insight into characteristics of the mutated structures.
Table 3.
Change in atom pair energies between I80 and T4 versus V4 in kcal/mol
| T193→ V193 Atoms | I80 Atoms | |||
|---|---|---|---|---|
| CB | CG1 | CG2 | CD1 | |
| N | 0.000 | 0.000 | 0.000 | 0.000 |
| CA | 0.000 | 0.000 | 0.000 | 0.004 |
| C | 0.000 | 0.000 | 0.000 | 0.008 |
| O | 0.000 | 0.000 | 0.000 | −0.010 |
| CB | 0.000 | 0.054 | 0.000 | −0.002 |
| OG1 → CG1 | 0.008 | −0.054 | −0.316 | −0.398 |
| CG2 → CG2′ | 0.000 | 0.000 | 0.001 | 0.020 |
Protein-protein docking
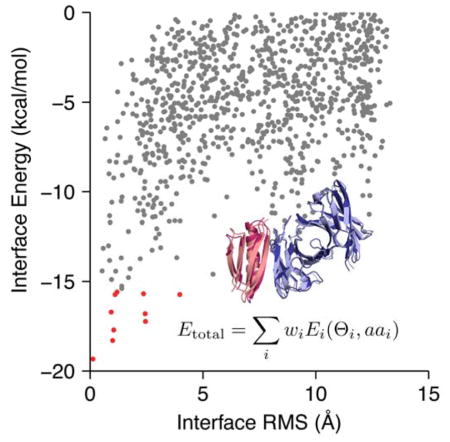
The second example shows how the Rosetta energies of an ensemble of models can be used to discriminate between models and investigate the characteristics of a protein–protein interface. Below, we investigate docked models of West Nile Virus envelope protein and a neutralizing antibody (PDB 1ztx; Fig. 9A).107 Calculation details can be found in the Supporting Information.
Figure 9. Using energies to discriminate docked models of West Nile Virus and the E16 neutralizing antibody.

(A) Comparison of the native E16 antibody (purple) docked to the lowest RMS model of the West Nile Virus envelope protein and several other random models of varying energy to show sampling diversity (gray, semi transparent). (B) Change in the interface energy relative to the unbound state versus RMS to native. Models at low RMS to the native interface have a low overall interface energy due to favorable van der Waals contacts, electrostatic interactions, and side-chain hydrogen bonds, as reflected by the Δ fa_atr, Δ fa_elec, and Δ hbond_sc energy terms.
To evaluate the docked models, we examine the variation of energies as a function of the root mean squared deviation (RMS) between the residues at the interface in each model and the known structure. For our calculation, interface residues are residues with a Cβ atom less than 8.0 Å away from the Cβ of a residue in the other docking partner. The plot of energies against RMS values is called a funnel plot and is intended to mimic the funnel-like energy landscape of protein folding and binding.
Like the previous example, we decompose the energies to yield information about the nature of interactions at the interface. Here, we observed significant changes in the following energy terms upon interface formation relative to the unbound state: fa_atr, fa_rep, fa_sol, lk_ball_wtd, fa_elec, hbond_lr_bb, hbond_bb_sc, and hbond_sc (Fig. 9B). Change in the Lennard-Jones energy upon interface formation is due to the introduction of atom-atom contacts at the interface. As more atoms come into contact near the native conformation (RMS→0), the favorable, attractive energy ( fa_atr) decreases whereas the unfavorable, repulsive energy (Δ fa_rep) increases. Change in the isotropic solvation energy ( fa_sol) is positive (unfavorable), indicating that upon interface formation, polar residues are buried. Balancing the desolvation penalty, the change in polar solvation energy ( lk_ball_wtd) and electrostatics ( fa_elec) is negative due to polar contacts forming at the interface. Finally, the three hydrogen bonding energies ( hbond_lr_bb, hbond_bb_sc, and hbond_sc) reflect the formation of backbone–backbone, backbone–side-chain, and side-chain–side-chain hydrogen bonds at the interface.
Discussion
The Rosetta energy function represents our collaboration’s ongoing pursuit to model the rules in nature that govern biomolecular structure, stability, and association. This paper summarizes the latest version which brings together fundamental physical theories, statistical mechanical models, and observations of protein structures. This work represents almost 20 years of interdisciplinary collaboration in the Rosetta community, which in turn builds on and incorporates decades of work outside the community.
After 20 years, we have improved physical theories, structural data, representations, experiments, and computational tools; yet, energy functions are far from perfect. Compared to the first torsional potentials, energy functions are also now vastly more complex. There are countless ways to arrive at more accurate energy functions. Here, we discuss grand challenges specific to development of the Rosetta energy function in the coming decade.
Modeling biomolecules other than proteins
The Rosetta energy function was originally developed to predict and design protein structures. A clear artifact of this goal is the energy function’s dependence on statistical potentials derived from protein X-ray crystal structures. Today, the Rosetta community also pursues goals ranging from design of synthetic macromolecules to predicting interactions and structures of other biomolecules such as glycoproteins and RNA. Accordingly, an active research thrust is to generalize the all-atom energy function for all biomolecules.
Many of the physically-derived terms (e.g. van der Waals) have already been made compatible with non-canonical amino acids and non-protein biomolecules (Table S5). Recently, Bhardwaj, Mulligan & Bahl et al.69 adapted the rama_prepro, p_aa_pp, fa_dun, pro_close, omega, dslf_fa13, yhh_planarity and ref terms to be compatible with mixed-chirality peptides. Several of Rosetta’s statistical potentials are validated against quantum mechanical calculations for evaluating for non-protein models (Table 4). Early work by Meiler & Baker44 on Rosetta Ligand introduced new atom and residue types for non-protein residues. The first non-protein-energy terms were added by Havranek et al.108 and Yu et al.109 who modified the hydrogen bonding potential to capture planar hydrogen bonds between protein side chains and nucleic acid bases. Renfrew et al.67,110 added molecular mechanics torsions and Lennard-Jones terms to model proteins with non-canonical amino acids, oligosaccharides, β -peptides, and oligo-peptoids.68 Labonte et al 70 implemented Woods’ CarboHydrate-Intrinsic (CHI) function111,112 which evaluates glycan geometries given the axial-equatorial character of the bonds. Das et al. added a set of terms to model Watson-Crick base pairing, π - π interactions in base stacking, and torsional potentials important for predicting and designing RNA structures.62,113–115 Bazzoli & Karanicolas116 recently developed a new polar solvation model that evaluates the penalty associated with displacing waters in the first solvation shell. In addition, Combs et al. tested a small molecule force field based on electron orbital models.117 Many of these terms are presented in detail in the Supporting Information.
Table 4.
New energy terms for biomolecules other than proteins
| Biomolecule | Term | Description | Unit | Ref. |
|---|---|---|---|---|
| Non-Canonical Amino Acids | mm_lj_intra_rep | Repulsive van der Waals energy between two atoms from the same residue | kcal/mol | [67] |
| mm_lj_intra_atr | Attractive van der Waals energy between two atoms from the same residue | kcal/mol | [67] | |
| mm_twist | Molecular mechanics derived torsion term for all proper torsions | kcal/mol | [67] | |
| unfolded | Energy of the unfolded state based on explicit unfolded state model | AU* | [67] | |
| split_unfolded_1b | One-body component of the two-component reference energy, lowest energy of a side chain in a dipeptide model system | AU | In SI | |
| split_unfolded_2b | Two-body component of the two-component reference energy, median two-body interaction energy based on atom type composition | AU | In SI | |
| Carbohydrates | sugar_bb | Energy for carbohydrate torsions | kcal/mol | [70] |
| DNA | gb_elec | Generalized Born model of the electrostatics energy | kcal/mol | [108] |
| RNA | fa_stack | π-π stacking energy for RNA bases | kT | [114] |
| stack_elec | Electrostatic energy for stacked RNA bases | kT | [115] | |
| fa_elec_rna_phos | Electrostatic energy ( fa_elec) between RNA phosphate atoms | kT | [62] | |
| rna_torsion | Knowledge-based torsional potential for RNA | kT | [62] | |
| rna_sugar_close | Penalty for opening an RNA sugar | kT | [62] |
AU, arbitrary units
Expanding Rosetta’s chemical library brings new challenges. Currently, there are separate energy function for various types of biomolecules. Typically, these functions mix physically-derived terms from the protein energy function with molecule-specific statistical potentials, custom weights, and possibly custom atomic parameters. If nature only uses one energy function, why do we need so many? Some discrepancies may result from features that we do not model explicitly, such as π - π n-π* and cation-π interactions. Efforts to converge on a single energy function will therefore pose interesting questions about the set of universal physical determinants of biomolecular structure.
Capturing the intra- and extra-cellular environment
Rosetta traditionally models the solvent surrounding the protein using the Lazaridis-Karplus (LK) model, which assumes a solvent environment made of pure water. In contrast, biology operates under various conditions influenced by pH, redox potential, temperature, solvent viscosity, chaotropes, kosmotropes, and polarizability. Therefore, modeling more details of the intra- and extra-cellular environment would enable Rosetta to identify structures important in different biological contexts.
Currently, Rosetta includes two groups of energy terms to model alternate environments (Table 5). Kilambi et al.118 implemented a method to account for pH by including a term called e_pH that calculates the likelihood of a protein side chain’s protonation state given a user specified pH; it requires the inclusion of both protonated and deprotonated side chains during side-chain rotamer packing. This model can predict pKa values with an RMS error under 1 unit,118 and it improves protein-protein docking, especially in acidic or basic conditions.60 The accuracy of this model is limited by the distance-dependent Coulomb approximation and sensitivity to fine backbone rearrangements.
Table 5.
Energy terms for structure prediction in different contexts
| Context | Term | Description | Unit | Ref. |
|---|---|---|---|---|
| Membrane Environment | fa_mpsolv | Solvation energy dependent on the protein orientation relative to the membrane | kcal/mol | [119,122] |
| fa_mpenv | One-body membrane environment energy dependent on the protein orientation relative to the membrane | kcal/mol | [119,122] | |
| pH | e_pH | Likelihood of side chain protonation given a user-specified pH | kcal/mol | [118] |
In addition, Rosetta implements Lazaridis’ Implicit Membrane Model (IMM) for modeling proteins in a lipid bilayer enviornment.36,119,120 The IMM terms provide a fast approximation of the nonpolar hydrocarbon core of the lipid bilayer and have been successfully applied to membrane protein folding,121 docking, and early design tasks.61 This continuum model has a fixed thickness, omitting the detailed chemistry at the membrane interface and any dynamic bilayer rearrangements.
The origin of energy models: top-down versus bottom-up development
Traditionally, energy functions are developed using a bottom-up approach: experimental observables serve as building blocks to parameterize physics-based formulas. The advent of powerful optimization techniques and artificial intelligence recently empowered the top-down category where numerical methods are used to derive models and/or parameters. Top-down approaches have been used to solve problems in various fields including structural biology and bioinformatics. Recently, top-down development was also applied to optimizing the Lennard-Jones, Lazaridis-Karplus, and Coulomb parameters in the Rosetta energy function (parameters in Table S4–S6).50,93
Top-down approaches have enormous potential to improve the accuracy of biomolecular modeling because more parameters can vary and the objective function can be minimized with more benchmarks. These approaches also introduce new challenges. With any computer-derived models, there is a risk of over-fitting as validation via structure prediction datasets reflect observable states, whereas simulations are intended to predict features of states that experiments cannot yet observe. Computer-derived parameters also introduce a unique kind of uncertainty. Consider the following scenario: the performance of scientific benchmarks improves as physical atomic parameters are perturbed away from the measured experimental values. As there is less physical-basis for parameters, are the predictions and interpretations still meaningful?
Top-down development will also provide power to develop more complicated energy functions. Currently, the Rosetta energy function advances by incrementally addressing weaknesses: with each new paper, we modify analytic formulas, add corrective terms, and adjust weights. As this paper demonstrates, the energy function is significantly more complicated than the initial theoretical forms. Given this complexity increase, an interesting approach to leverage the power of top-down development would be to simplify and subtract terms to evaluate individual benefits.
A highly interdisciplinary endeavor
The Rosetta energy function has advanced rapidly due to the Rosetta Community: a highly-interdisciplinary collaboration between scientists with diverse backgrounds located in over 50 labs around the world. The many facets of our team enable us to probe different aspects of the energy function. For example, expert computer scientists and applied mathematicians have implemented algorithms to speed up calculations. Dedicated software engineers maintain the code and maintain a platform for scientific benchmark testing. Physicists and chemists develop new energy terms that better model the physical rules found in nature. Structural biologists maintain a focus on created biological features and functions. We look forward to leveraging this powerful interdisciplinary scientific team as we head into the next decade of energy function advances.
Conclusion: A living energy function
For the first time since 2004,48 we have documented all of the mathematical and physical details of the Rosetta all-atom energy function highlighting the latest upgrades to both the underlying science and the speed of calculations. In addition, we illustrated how the energies can be used to analyze output models from Rosetta simulations. These advances have enabled Rosetta’s achievements in biomolecular structure prediction and design over the past fifteen years. Still, the energy function is far from complete and will continue to evolve long after this publication. Thus, we hope this document will serve as an important resource for understanding the foundational physical and mathematical concepts in the energy function. Furthermore, we hope to encourage both current and future Rosetta developers and users to understand the strengths and shortcomings of the energy function as it applies to the scientific questions they are trying to answer.
Supplementary Material
Acknowledgments
Funding Sources
RFA is funded by a Hertz Foundation Fellowship and an NSF Graduate Research Fellowship. JRJ and JJG are funded by NIH GM-078221. ALF, JJG and BK are funded by NIH GM-73141. MJO is funded by NSF GM-114961. PDR and RB are funded by the Simons Foundation. MVS and RLD are funded by NIH GM-084453 and NIH GM-111819. MSP is funded by NSF BMAT 1507736. JWL is funded by NIH F32-CA189246. KK is funded by an NSF Graduate Research Fellowship and an SGF Galiban Fellowship. DB, HP and VKM are funded by NIH GM-092802. TK is funded by NIH GM-110089 and GM-117189.
Development of the Rosetta energy function would not be possible without the entire Rosetta Commons collaboration: a community of scientists, engineers, and software developers that have worked together for almost 20 years. We estimate that hundreds of scientists from the 50 institutions in the Rosetta Commons have made minor and major contributions to the advancement of the all-atom energy function. When writing this paper, it was impossible to compile a complete list of energy function contributors. Instead, our author list reflects a small subset of the historical contributors who provided text, figures, and expertise needed to write a comprehensive, complete, and accurate description of the current energy function formulation. We also want to give special recognition to the early pioneers of the Rosetta energy function who are not co-authors: Carol Rohl, Kim Simons, Charlie Strauss, Ingo Ruczinski, William Sheffler, Jens Meiler, Ora Schuler-Furman, James Havranek, and Ian Davis.
We also acknowledge individuals that contributed to assembling the manuscript. We thank Sergey Lyskov for development of the benchmark server that enables continuous and transparent energy function testing. We thank Morgan Nance, Henry Lessen, and Rocco Moretti for helpful comments on the manuscript.
Footnotes
In Rosetta, σi,j has the same definition as the variable in CHARMM.
Author Contributions
Wrote the manuscript: RFA, JRJ, ALF, TK, BK, JJG
Analysis Scripts and Examples: RFA, JRJ, MSP, JJG
Writing, verifying, and/or contributing figures on protein energy terms: ALF, MJO, FPD, HP, MVS, PB, RLD, TK, DB, BK, JJG
Writing, verifying, and/or contributing figures on non-protein energy terms: PDR, KK, VKM, JWL, RB, RD
Supporting Information File 1: Alford_etal_RosettaEnergyFunction_SupportingInfo.pdf
The supporting information file contains a description of changes to the Rosetta energy function since 2000; data describing the calibration of Rosetta energies in kcal/mol; additional details of energy terms and details on smoothing of statistical terms; energy terms for D-amino acids, non-canonical amino acids, carbohydrates, and nucleic acids; and methods describing example energy calculations.
Supporting Information File 2: atom_pair_energy_protocol_capture.tar.gz
A protocol capture within an interactive Python notebook demonstrating the usage of the print_atom_pair_energy_table function.
This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Kuhlman B, Baker D. Native Protein Sequences Are close to Optimal for Their Structures. Proc Natl Acad Sci USA. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson JS. The Anatomy and Taxonomy of Protein Structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 3.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman KW, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y-EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods in enzymology. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anfinsen CB. Principles That Govern the Folding of Protein Chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 5.Lennard-Jones J. On the Determination of Molecular Fields I: From the Variation of Viscosity of a Gas with Temperature. R Soc London, Ser A, Contain Pap a Math Phys Character. 1924;106:441–462. [Google Scholar]
- 6.Lennard-Jones J. On the Determination of Molecular Fields II: From the Variation of Viscosity of a Gas with Temperature. R Soc London, Ser A, Contain Pap a Math Phys Character. 1924;106:464–477. [Google Scholar]
- 7.Levitt M, Lifson S. Refinement of Protein Conformations Using a Macromolecular Energy Minimization Procedure. J Mol Biol. 1969;46(2):269–279. doi: 10.1016/0022-2836(69)90421-5. [DOI] [PubMed] [Google Scholar]
- 8.Urey HC, Bradley CA. The Vibrations of Pentatonic Tetrahedral Molecules. Phys Rev. 1931;38(11):1969–1978. [Google Scholar]
- 9.Westheimer F. Calculation of the Magnitude of Steric Effects. Steric Eff Org Chem. 1956:523–555. [Google Scholar]
- 10.Lifson S, Warshel A. Consistent Force Field for Calculations of Conformations, Vibrational Spectra, and Enthalpies of Cycloalkane and N-Alkane Molecules. J Chem Phys. 1968;49(11):5116–5129. [Google Scholar]
- 11.Warshel A, Lifson S. Consistent Force Field CalculationsII, Crystal Structures, Sublimation Energies, Molecular and Lattice Vibrations, Molecular Conformations, and Enthalpies of Alkanes. J Chem Phys. 1970;53(2):582–594. [Google Scholar]
- 12.Levitt M. Energy Refinement of Hen Egg-White Lysozyme. J Mol Biol. 1974;82(3):393–420. doi: 10.1016/0022-2836(74)90599-3. [DOI] [PubMed] [Google Scholar]
- 13.Gelin BR, Karplus M. Sidechain Torsional Potentials and Motion of Amino Acids in Porteins: Bovine Pancreatic Trypsin Inhibitor. Proc Natl Acad Sci USA. 1975;72(6):2002–2006. doi: 10.1073/pnas.72.6.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levinthal C, Wodak SJ, Kahn P, Dadivanian AK. Hemoglobin Interaction in Sickle Cell Fibers, I: Theoretical Approaches to the Molecular Contacts. Proc Natl Acad Sci USA. 1975;72(4):1330–1334. doi: 10.1073/pnas.72.4.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1996;118(9):2309–2309. [Google Scholar]
- 16.Mayo SL, Olafson BD, Goddard WA. DREIDING: A Generic Force Field for Molecular Simulations. J Phys Chem. 1990;94(26):8897–8909. [Google Scholar]
- 17.Jorgensen WL, Tirado-Rives J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J Am Chem Soc. 1988;110(6):1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 18.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 19.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H. COMPASS: An Ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds. J Phys Chem B. 1998;102(38):7338–7364. [Google Scholar]
- 21.Tanaka S, Scheraga HA. Model of Protein Folding: Inclusion of Short-, Medium-, and Long-Range Interactions. Proc Natl Acad Sci USA. 1975;72(10):3802–3806. doi: 10.1073/pnas.72.10.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka S, Scheraga HA. Model of Protein Folding: Incorporation of a One-Dimensional Short-Range (Ising) Model into a Three-Dimensional Model. Proc Natl Acad Sci USA. 1977;74(4):1320–1323. doi: 10.1073/pnas.74.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazawa S, Jernigan RL. Residue-Residue Potentials with a Favorable Contact Pair Term and an Unfavorable High Packing Density Term, for Simulation and Threading. J Mol Biol. 1996;256(3):623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 24.Wilmanns M, Eisenberg D. Three-Dimensional Profiles from Residue-Pair Preferences: Identification of Sequences with Beta/alpha-Barrel Fold. Proc Natl Acad Sci USA. 1993;90(4):1379–1383. doi: 10.1073/pnas.90.4.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones DT, Taylor WR, Thornton JM. A New Approach to Protein Fold Recognition. Nature. 1992;358(6381):86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 26.Bowie JU, Lüthy R, Eisenberg D. A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure. Science. 1991;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 27.Sippl MJ Calculation of Conformational Ensembles from Potentials of Mean Force. An Approach to the Knowledge-Based Prediction of Local Structures in Globular Proteins. J Mol Biol. 1990;213(4):859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 28.Skolnick J, Kolinski A. Simulations of the Folding of a Globular Protein. Science. 1990;250(4984):1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- 29.Bashford D, Case DA. Generalized Born Models of Macromolecular Solvation Effects. Annu Rev Phys Chem. 2000;51(1):129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 30.Warshel A, Mitsunori K, Pisliakov A. Polarizable Force Fields: History, Test Cases, and Prospects. J Chem Theory Comput. 2007;3(6):2034–2045. doi: 10.1021/ct700127w. [DOI] [PubMed] [Google Scholar]
- 31.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of Protein Tertiary Structures from Fragments with Similar Local Sequences Using Simulated Annealing and Bayesian Scoring Functions. J Mol Biol. 1997;268(1):209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simons KT, Ruczinski I, Kooperberg C, Fox BA, Bystroff C, Baker D. Improved Recognition of Native-like Protein Structures Using a Combination of Sequence-Dependent and Sequence-Independent Features of Proteins. Proteins. 1999;34(1):82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 34.Kuhlman B, Baker D. Native Protein Sequences Are close to Optimal for Their Structures. Proc Natl Acad Sci USA. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neria E, Fischer S, Karplus M. Simulation of Activation Free Energies in Molecular Systems. J Chem Phys. 1996;105(5):1902–1921. [Google Scholar]
- 36.Lazaridis T, Karplus M. Effective Energy Function for Proteins in Solution. Proteins. 1999;35(2):133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 37.Dunbrack RL, Cohen FE, Cohen FE. Bayesian Statistical Analysis of Protein Side-Chain Rotamer Preferences. Protein Sci. 1997;6(8):1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kortemme T, Morozov AV, Baker D. An Orientation-Dependent Hydrogen Bonding Potential Improves Prediction of Specificity and Structure for Proteins and Protein-Protein Complexes. J Mol Biol. 2003;326(4):1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 39.Morozov AV, Kortemme T, Tsemekhman K, Baker D. Close Agreement between the Orientation Dependence of Hydrogen Bonds Observed in Protein Structures and Quantum Mechanical Calculations. Proc Natl Acad Sci USA. 2004;101(18):6946–6951. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradley P, Misura KMS, Baker D. Toward High-Resolution de Novo Structure Prediction for Small Proteins. Science. 2005;309(5742):1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 41.Kortemme T, Baker D. A Simple Physical Model for Binding Energy Hot Spots in Protein-Protein Complexes. Proc Natl Acad Sci USA. 2002;99(22):14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kortemme T, Kim DE, Baker D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci STKE. 2004;2004(219):pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 43.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-Protein Docking with Simultaneous Optimization of Rigid-Body Displacement and Side-Chain Conformations. J Mol Biol. 2003;331(1):281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 44.Meiler J, Baker D. ROSETTALIGAND: Protein-Small Molecule Docking with Full Side-Chain Flexibility. Proteins Struct Funct Bioinforma. 2006;65(3):538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 45.Kortemme T, Joachimiak LA, Bullock AN, Schuler AD, Stoddard BL, Baker D. Computational Redesign of Protein-Protein Interaction Specificity. Nat Struct Mol Biol. 2004;11(4):371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 46.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science. 2003;302(5649):1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 47.Chevalier BS, Kortemme T, Chadsey MS, Baker D, Monnat RJ, Stoddard BL. Design, Activity, and Structure of a Highly Specific Artificial Endonuclease. Mol Cell. 2002;10(4):895–905. doi: 10.1016/s1097-2765(02)00690-1. [DOI] [PubMed] [Google Scholar]
- 48.Rohl CA, Strauss CEM, Misura KMS, Baker D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 49.O’Meara MJ, Leaver-Fay A, Tyka MD, Stein A, Houlihan K, DiMaio F, Bradley P, Kortemme T, Baker D, Snoeyink J, Kuhlman B. Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J Chem Theory Comput. 2015;11(2):609–622. doi: 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park H, Bradley P, Greisen P, Liu Y, Kim DE, Baker D, DiMaio F. Simultaneous Optimization of Biomolecular Energy Function on Features from Small Molecules and Macromolecules. J Chem Theory Comput. 2016;12(12):6201–6212. doi: 10.1021/acs.jctc.6b00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leaver-Fay A, O’Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, Gray JJ, Kortemme T, Richardson JS, Havranek JJ, Snoeyink J, Baker D, Kuhlman B. Scientific Benchmarks for Guiding Macromolecular Energy Function Improvement. Methods Enzymol. 2013;523:109–143. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapovalov MV, Dunbrack RL. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure. 2011;19(6):844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DiMaio F, Song Y, Li X, Brunner MJ, Xu C, Conticello V, Egelman E, Marlovits TC, Cheng Y, Baker D. Atomic-Accuracy Models from 4.5-Å Cryo-Electron Microscopy Data with Density-Guided Iterative Local Refinement. Nat Methods. 2015;12(4):361–365. doi: 10.1038/nmeth.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vortmeier G, DeLuca SH, Els-Heindl S, Chollet C, Scheidt HA, Beck-Sickinger AG, Meiler J, Huster D. Integrating Solid-State NMR and Computational Modeling to Investigate the Structure and Dynamics of Membrane-Associated Ghrelin. PLoS One. 2015;10(3):e0122444. doi: 10.1371/journal.pone.0122444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal V, Connell MJ, Stevens E, Schroeter A, Chen M, Macpherson S, Serra AM, Adachi Y, Holmes MA, Li Y, Klevit RE, Graham BS, Wyatt RT, Baker D, Strong RK, Crowe JE, Johnson PR, Schief WR. Proof of Principle for Epitope-Focused Vaccine Design. Nature. 2014;507(7491):201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masica DL, Schrier SB, Specht EA, Gray JJ. De Novo Design of Peptide−Calcite Biomineralization Systems. J Am Chem Soc. 2010;132(35):12252–12262. doi: 10.1021/ja1001086. [DOI] [PubMed] [Google Scholar]
- 57.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D. Accurate Design of Co-Assembling Multi-Component Protein Nanomaterials. Nature. 2014;510(7503):103–108. doi: 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science. 2010;329(5989):309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf C, Siegel JB, Tinberg C, Camarca A, Gianfrani C, Paski S, Guan R, Montelione G, Baker D, Pultz IS. Engineering of Kuma030: A Gliadin Peptidase That Rapidly Degrades Immunogenic Gliadin Peptides in Gastric Conditions. J Am Chem Soc. 2015;137(40):13106–13113. doi: 10.1021/jacs.5b08325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kilambi KP, Reddy K, Gray JJ. Protein-Protein Docking with Dynamic Residue Protonation States. PLoS Comput Biol. 2014;10(12):e1004018. doi: 10.1371/journal.pcbi.1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alford RF, Koehler Leman J, Weitzner BD, Duran AM, Tilley DC, Elazar A, Gray JJ. An Integrated Framework Advancing Membrane Protein Modeling and Design. PLoS Comput Biol. 2015;11(9):e1004398. doi: 10.1371/journal.pcbi.1004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das R, Karanicolas J, Baker D. Atomic Accuracy in Predicting and Designing Noncanonical RNA Structure. Nat Methods. 2010;7(4):291–294. doi: 10.1038/nmeth.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thyme SB, Baker D, Bradley P. Improved Modeling of Side-Chain--Base Interactions and Plasticity in Protein--DNA Interface Design. J Mol Biol. 2012;419(3–4):255–274. doi: 10.1016/j.jmb.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joyce AP, Zhang C, Bradley P, Havranek JJ. Structure-Based Modeling of Protein: DNA Specificity. Brief Funct Genomics. 2015;14(1):39–49. doi: 10.1093/bfgp/elu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemmon G, Meiler J. Rosetta Ligand Docking with Flexible XML Protocols. Methods Mol Biol. 2012;819:143–155. doi: 10.1007/978-1-61779-465-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Combs SA, DeLuca SL, DeLuca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J. Small-Molecule Ligand Docking into Comparative Models with Rosetta. Nat Protoc. 2013;8(7):1277–1298. doi: 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Renfrew PD, Choi EJ, Bonneau R, Kuhlman B. Incorporation of Noncanonical Amino Acids into Rosetta and Use in Computational Protein-Peptide Interface Design. PLoS One. 2012;7(3):e32637. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drew K, Renfrew PD, Craven TW, Butterfoss GL, Chou F-C, Lyskov S, Bullock BN, Watkins A, Labonte JW, Pacella M, Kilambi KP, Leaver-Fay A, Kuhlman B, Gray JJ, Bradley P, Kirshenbaum K, Arora PS, Das R, Bonneau R. Adding Diverse Noncanonical Backbones to Rosetta: Enabling Peptidomimetic Design. PLoS One. 2013;8(7):e67051. doi: 10.1371/journal.pone.0067051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, Huang P-S, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D. Accurate de Novo Design of Hyperstable Constrained Peptides. Nature. 2016;538(7625):329–335. doi: 10.1038/nature19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Labonte JW, Aldof-Bryfogle J, Schief WR, Gray JJ. Residue-Centric Modeling and Design of Saccharide and Glycoconjugate Structures. J Comput Chem. 2017;38(5):276–287. doi: 10.1002/jcc.24679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yanover C, Bradley P. Extensive Protein and DNA Backbone Sampling Improves Structure-Based Specificity Prediction for C2H2 Zinc Fingers. Nucleic Acids Res. 2011;39(11):4564–4576. doi: 10.1093/nar/gkr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berkholz DS, Driggers CM, Shapovalov MV, Dunbrack RL, Karplus PA, Karplus PA. Nonplanar Peptide Bonds in Proteins Are Common and Conserved but Not Biased toward Active Sites. Proc Natl Acad Sci USA. 2012;109(2):449–453. doi: 10.1073/pnas.1107115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popovic Z, Baker D, Players F. Algorithm Discovery by Protein Folding Game Players. Proc Natl Acad Sci USA. 2011;108(47):18949–18953. doi: 10.1073/pnas.1115898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grigoryan G, Ochoa A, Keating AE. Computing van Der Waals Energies in the Context of the Rotamer Approximation. Proteins Struct Funct Bioinforma. 2007;68(4):863–878. doi: 10.1002/prot.21470. [DOI] [PubMed] [Google Scholar]
- 75.Dahiyat BI, Mayo SL. Probing the Role of Packing Specificity in Protein Design. Proc Natl Acad Sci USA. 1997;94(19):10172–10177. doi: 10.1073/pnas.94.19.10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warshel A, Russell ST. Calculations of Electrostatic Interactions in Biological Systems and in Solutions. Q Rev Biophys. 2009;17(3):283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 77.Hubbard RE, Kamran Haider M, Hubbard RE, Kamran Haider M. Encyclopedia of Life Sciences. John Wiley & Sons, Ltd; Chichester, UK: 2010. Hydrogen Bonds in Proteins: Role and Strength. [Google Scholar]
- 78.Li X-Z, Walker B, Michaelides A. Quantum Nature of the Hydrogen Bond. Proc Natl Acad Sci USA. 2011;108(16):6369–6373. [Google Scholar]
- 79.Richardson JS, Keedy DA, Richardson DC. Biomolecular Forms and Functions: A Celebration of 50 Years of the Ramachandran Map. World Sci. Publ. Co. Pte. Ltd; Singapore: 2013. pp. 46–61. [Google Scholar]
- 80.Wang C, Bradley P, Baker D. Protein–Protein Docking with Backbone Flexibility. J Mol Biol. 2007;373(2):503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 81.Ho BK, Thomas A, Brasseur R. Revisiting the Ramachandran Plot: Hard-Sphere Repulsion, Electrostatics, and H-Bonding in the Alpha-Helix. Protein Sci. 2003;12(11):2508–2522. doi: 10.1110/ps.03235203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang G, Dunbrack RL. PISCES: A Protein Sequence Culling Server. Bioinformatics. 2003;19(12):1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 83.Ting D, Wang G, Shapovalov M, Mitra R, Jordan MI, Dunbrack RL. Neighbor-Dependent Ramachandran Probability Distributions of Amino Acids Developed from a Hierarchical Dirichlet Process Model. PLoS Comput Biol. 2010;6(4):e1000763. doi: 10.1371/journal.pcbi.1000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finkelstein AV, Badretdinov AY, Gutin AM. Why Do Protein Architectures Have Boltzmann-like Statistics? Proteins Struct Funct Genet. 1995;23(2):142–150. doi: 10.1002/prot.340230204. [DOI] [PubMed] [Google Scholar]
- 85.Shortle D. Propensities, Probabilities, and the Boltzmann Hypothesis. Protein Sci. 2003;12(6):1298–1302. doi: 10.1110/ps.0306903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lovell SC, Word JM, Richardson JS, Richardson DC. The Penultimate Rotamer Library. Proteins. 2000;40(3):389–408. [PubMed] [Google Scholar]
- 87.MacArthur MW, Thornton JM. Influence of Proline Residues on Protein Conformation. J Mol Biol. 1991;218(2):397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 88.McDonald IK, Thornton JM. Satisfying Hydrogen Bonding Potential in Proteins. J Mol Biol. 1994;238(5):777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 89.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of Backbone Bond Geometry Improves Protein Energy Landscape Modeling. Protein Sci. 2014;23(1):47–55. doi: 10.1002/pro.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hall MB. Insight II, version 12000. Accelyris Inc; 2005. [Google Scholar]
- 91.Engh RA, Huber R. IUCr Accurate Bond and Angle Parameters for X-Ray Protein Structure Refinement. Acta Crystallogr Sect A Found Crystallogr. 1991;47(4):392–400. [Google Scholar]
- 92.Renfrew PD, Butterfoss GL, Kuhlman B. Using Quantum Mechanics to Improve Estimates of Amino Acid Side Chain Rotamer Energies. Proteins Struct Funct Bioinforma. 2007;71(4):1637–1646. doi: 10.1002/prot.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barton RR, Ivey JS. Nelder-Mead Simplex Modifications for Simulation Optimization. Manage Sci. 1996;42(7):954–973. [Google Scholar]
- 94.ÓConchúir S, Barlow KA, Pache RA, Ollikainen N, Kundert K, O’Meara MJ, Smith CA, Kortemme T. A Web Resource for Standardized Benchmark Datasets, Metrics, and Rosetta Protocols for Macromolecular Modeling and Design. PLoS One. 2015;10(9):e0130433. doi: 10.1371/journal.pone.0130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song Y, Tyka M, Leaver-Fay A, Thompson J, Baker D. Structure-Guided Forcefield Optimization. Proteins. 2011;79(6):1898–1909. doi: 10.1002/prot.23013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conway P, DiMaio F. Improving Hybrid Statistical and Physical Forcefields through Local Structure Enumeration. Protein Sci. 2016;25(8):1525–1534. doi: 10.1002/pro.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leaver-Fay A, O’Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, Gray JJ, Kortemme T, Richardson JS, Havranek JJ, Snoeyink J, Baker D, Kuhlman B. Scientific Benchmarks for Guiding Macromolecular Energy Function Improvement. Methods Enzymol. 2013;523:109–143. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kellogg EH, Leaver-Fay A, Baker D. Role of Conformational Sampling in Computing Mutation-Induced Changes in Protein Structure and Stability. Proteins. 2011;79(3):830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mandell DJ, Coutsias EA, Kortemme T. Sub-Angstrom Accuracy in Protein Loop Reconstruction by Robotics-Inspired Conformational Sampling. Nat Methods. 2009;6(8):551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tyka MD, Keedy DA, André I, DiMaio F, Song Y, Richardson DC, Richardson JS, Baker D. Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. J Mol Biol. 2011;405(2):607–618. doi: 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hwang H, Vreven T, Janin J, Weng Z. Protein-Protein Docking Benchmark Version 4.0. Proteins Struct Funct Bioinforma. 2010;78(15):3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ. Benchmarking and Analysis of Protein Docking Performance in Rosetta v3.2. PLoS One. 2011;6(8):e22477. doi: 10.1371/journal.pone.0022477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raveh B, London N, Zimmerman L, Schueler-Furman O. Rosetta FlexPepDock Ab-Initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS One. 2011;6(4):e18934. doi: 10.1371/journal.pone.0018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Song Y, DiMaio F, Wang RY-R, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-Resolution Comparative Modeling with RosettaCM. Structure. 2013;21(10):1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Altman MD, Nalivaika EA, Prabu-Jeyabalan M, Schiffer CA, Tidor B. Computational Design and Experimental Study of Tighter Binding Peptides to an Inactivated Mutant of HIV-1 Protease. Proteins Struct Funct Bioinforma. 2007;70(3):678–694. doi: 10.1002/prot.21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: A Script-Based Interface for Implementing Molecular Modeling Algorithms Using Rosetta. Bioinformatics. 2010;26(5):689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. Structural Basis of West Nile Virus Neutralization by a Therapeutic Antibody. Nature. 2005;437(7059):764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Havranek JJ, Duarte CM, Baker D. A Simple Physical Model for the Prediction and Design of Protein–DNA Interactions. J Mol Biol. 2004;344(1):59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 109.Chen Y, Kortemme T, Robertson T, Baker D, Varani G. A New Hydrogen-Bonding Potential for the Design of Protein-RNA Interactions Predicts Specific Contacts and Discriminates Decoys. Nucleic Acids Res. 2004;32(17):5147–5162. doi: 10.1093/nar/gkh785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Renfrew PD, Craven TW, Butterfoss GL, Kirshenbaum K, Bonneau R. A Rotamer Library to Enable Modeling and Design of Peptoid Foldamers. J Am Chem Soc. 2014;136(24):8772–8782. doi: 10.1021/ja503776z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nivedha AK, Thieker DF, Makeneni S, Hu H, Woods RJ. Vina-Carb: Improving Glycosidic Angles during Carbohydrate Docking. J Chem Theory Comput. 2016;12(2):892–901. doi: 10.1021/acs.jctc.5b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nivedha AK, Makeneni S, Foley BL, Tessier MB, Woods RJ. Importance of Ligand Conformational Energies in Carbohydrate Docking: Sorting the Wheat from the Chaff. J Comput Chem. 2014;35(7):526–539. doi: 10.1002/jcc.23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Das R, Baker D. Automated de Novo Prediction of Native-like RNA Tertiary Structures. Proc Natl Acad Sci USA. 2007;104(37):14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sripakdeevong P, Kladwang W, Das R. An Enumerative Stepwise Ansatz Enables Atomic-Accuracy RNA Loop Modeling. Proc Natl Acad Sci USA. 2011;108(51):20573–20578. doi: 10.1073/pnas.1106516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chou F-C, Kladwang W, Kappel K, Das R. Blind Tests of RNA Nearest-Neighbor Energy Prediction. Proc Natl Acad Sci USA. 2016;113(30):8430–8435. doi: 10.1073/pnas.1523335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bazzoli A, Karanicolas J. “Solvent Hydrogen-Bond Occlusion”: A New Model of Polar Desolvation for Biomolecular Energetics. J Comput Chem. 2017 doi: 10.1002/jcc.24740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Combs S. Identification and Scoring of Partial Covalent Interactions in Proteins and Protein Ligand Complexes. Vanderbilt University; 2013. [Google Scholar]
- 118.Kilambi KP, Gray JJ. Rapid Calculation of Protein pKa Values Using Rosetta. Biophys J. 2012;103(3):587–595. doi: 10.1016/j.bpj.2012.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Barth P, Schonbrun J, Baker D. Toward High-Resolution Prediction and Design of Transmembrane Helical Protein Structures. Proc Natl Acad Sci USA. 2007;104(40):15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass Membrane Protein Structure Prediction Using Rosetta. Proteins. 2006;62(4):1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Y, Barth P. Evolutionary-Guided de Novo Structure Prediction of Self-Associated Transmembrane Helical Proteins with near-Atomic Accuracy. Nat Commun. 2015;6(7196):1–12. doi: 10.1038/ncomms8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lazaridis T. Effective Energy Function for Proteins in Lipid Membranes. Proteins Struct Funct Genet. 2003;52(2):176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.